

Abstract
EFSA was asked by the European Commission to update the Scientific Opinion on methodological principles and scientific methods to be taken into account when establishing Reference Points for Action (RPAs) for non‐allowed pharmacologically active substances in food of animal origin. This guidance document presents a simple and pragmatic approach which takes into account both analytical and toxicological considerations. The RPA shall be based on the reasonably achievable lowest residue concentration that can unequivocally be determined by official control laboratories, i.e. the reasonably achievable lowest decision limit (CCα). The aim is to check whether this concentration is low enough to adequately protect the consumers of food commodities that contain that substance. The proposed step‐wise approach applies toxicological screening values (TSVs), based on genotoxic potential, pharmacological activity, as well as other effects of the substance. The highest dietary exposure corresponding to the reasonably achievable lowest CCα for the substance has to be estimated and compared with the TSV. Where equal to or lower than the TSV, the reasonably achievable lowest CCα can be accepted as the RPA. If higher, the sensitivity of the analytical method needs to be improved. In the case where no further analytical improvements are feasible within a short to medium time frame, a substance‐specific risk assessment should be considered. This also applies when the potential adverse effects do not allow use of the decision tree, as for high potency carcinogens, inorganic substances or compounds with allergenic effects or causing blood dyscrasias. The CONTAM Panel concluded that RPAs should be food matrix independent. RPAs cannot be applied to non‐edible matrices, which are also monitored for non‐allowed pharmacologically active substances.
Keywords: reference point for action (RPA), toxicological screening value (TSV), decision limit (CCα), non‐allowed pharmacologically active substances
Summary
Following a request from the European Commission, the European Food Safety Authority (EFSA) published in 2013 a Scientific Opinion from the EFSA Panel on Contaminants in the Food Chain (CONTAM Panel) on methodological principles and scientific methods to be taken into account when establishing Reference Points for Action (RPAs) for non‐allowed pharmacologically active substances present in food of animal origin. In 2018, EFSA received a request from the European Commission for an update of this scientific opinion. In particular, the update should:
address the issues discussed by the CONTAM Panel at its 83rd plenary meeting (i.e. whether only the intended use of substances should be considered for the allocation to group II and III substances, whether skin sensitisations should be used as an exclusion criterion, how the criterion specifying that ‘Group I substances are excluded from the guidance document when there is evidence that the related TSV may not be adequately health protective', should be evaluated and whether inorganic substances should be excluded from the guidance document);
consider rewording certain parts of the opinion to avoid any misunderstanding;
reflect that analytical methods used for non‐allowed pharmacologically active substances should be as sensitive as possible, in order to allow the establishment of RPAs at concentrations as low as possible;
define the relationship between the reasonably achievable lowest limit of quantification (RALLOQ) and the decision limit (CCα). Since the CCα of the analytical method needs to be set at or below the RPA, this value is of relevance for determining which concentration for the RPA is analytically achievable.
According to Regulation (EC) No 470/20091, RPAs may be established for non‐allowed pharmacologically active substances when it is deemed necessary to ensure the functioning of controls for food of animal origin that is imported or placed on the market. The RPA shall be based on the lowest residue concentration that can unequivocally be determined by official control laboratories. When residues of such non‐allowed substances are detected at or above the RPA, the food is considered not to comply with Community legislation, and should be removed from the market. However, even where residues of such substances are detected below the RPA, the competent authority shall carry out investigations to determine whether there has been illegal administration of a non‐allowed pharmacologically active substance and, where relevant, shall apply the appropriate penalties.
This guidance document presents a simple and pragmatic approach which takes into account both analytical and toxicological considerations when establishing RPAs but this approach does not replace a full risk assessment. In the previous opinion, a RALLOQ was identified. However, in the area of pharmacologically active substances, the CCα is applied rather than the limit of quantification (LOQ). Therefore, the RALLOQ was replaced by the reasonably achievable lowest CCα. Consequently, the question to define the relationship between CCα and RALLOQ is no longer applicable. The RPA has to be set at the reasonably achievable lowest CCα that can unequivocally be determined by official control laboratories. In order to determine whether the reasonably achievable lowest CCα for the respective substance is low enough to adequately protect the consumer, consideration of the toxic potential and pharmacological activity of the substance is needed. As the substances of concern are non‐allowed, it is likely that the toxicological information on these substances is limited and/or that they have properties inappropriate for authorised substances. For substances that fall under the pesticide regulatory framework, maximum residue limits (MRLs) for animal derived products have already been established and these substances are consequently not subject of this guidance.
Substances which are genotoxic are of concern because they may also be carcinogenic or cause germ cell mutations. Based on analysis of the potency of a large number of carcinogens, the EFSA Scientific Committee (SC) has identified a threshold of toxicological concern (TTC) value of 0.0025 μg/kg body weight (bw) per day for potentially genotoxic compounds as a level of human exposure that would be of low concern from a public health point of view, provided that compounds designated as high potency carcinogens are excluded. The CONTAM Panel decided to use this TTC value of 0.0025 μg/kg bw per day as a toxicological screening value (TSV) for non‐allowed pharmacologically active substances for which there is either direct evidence of genotoxicity or insufficient evidence to conclude that it is not genotoxic. In these cases, the substances are referred to in this guidance as Group I substances.
The database underlying the TTC concept only contains a small number of pharmacologically active substances and, therefore, other TTC values could not be used in this guidance. Non‐allowed pharmacologically active substances could have pharmacological or toxicological properties that might to some extent be comparable with those of allowed substances. Therefore, the CONTAM Panel assessed the acceptable daily intakes (ADIs) for veterinary pharmacologically active substances established by the European Medicines Agency (EMA). In Commission Regulation (EU) No 37/20102, these substances are classified therapeutically as: (a) agents acting on the central/autonomic nervous system, (b) agents acting on the reproductive system, (c) corticoids/glucocorticoids, (d) anti‐infectious agents (antibiotics/antiseptics/chemotherapeutics), (e) anti‐inflammatory agents (antidiarrhoeal and intestinal anti‐inflammatory agents/nonsteroidal anti‐inflammatory agents) and (f) antiparasitic agents (agents acting against endoparasites/ectoparasites/protozoa). In addition, the EMA has also established ADIs for a group of substances having a pharmacological activity different from the classes mentioned above. This group, designated as ‘Other’, comprised substances such as analgesics, diuretics and sedatives.
The ADIs are based on the no‐observed‐effect levels (NOELs) that are most relevant for the safety assessment, and take into account the appropriate uncertainty factors. For agents acting on the nervous system and reproductive system, as well as corticoids/glucocorticoids, the ADIs are based on pharmacological or toxicological effects. For the other compounds, the ADIs are based on pharmacological, antimicrobial or toxicological effects. The CONTAM Panel decided to include only compounds with specific effects (e.g. agonistic or antagonistic) on receptors in the nervous system (e.g. α‐ and β‐adrenergic) in the group of agents acting on the nervous system, and only agents with an effect on the progesterone or prostaglandin F2α receptor in the group acting on the reproductive system. No substances acting on the oestrogen, androgen or thyroid receptor were identified in the available database. The ADIs for pharmacologically active substances acting on the nervous system and reproductive system and for corticoids/glucocorticoids were comparable and clearly lower than the ADIs for the other groups. Therefore, these three classes should be treated separately when establishing a TSV. Since the ADIs for these substances are comparable, the Panel decided to group these three classes and to use the lowest ADI of 0.0042 μg/kg bw per day (see Table 1) as the TSV for non‐allowed pharmacologically active substances acting on specific receptors in the nervous or reproductive system, or being corticoids/glucocorticoids (referred to as Group II in this guidance).
Table 1.
The distribution of acceptable daily intake (ADIs) (μg/kg bw per day) for different classes of veterinary pharmacologically active substances
| Parameter | Acceptable daily intakes (ADI) (μg/kg bw per day) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Group IIa | Group IIIb | ||||||||
| Nervous system | Reproductive system | Corticoids/glucocorticoids | Group II combined | Anti‐infective | Anti‐inflammatory | Antiparasitic | ‘Other’ | Group III combined | |
| n | 7 | 11 | 5 | 23 | 54 | 12 | 58 | 29 | 153 |
| Min | 0.0042 | 0.010 | 0.015 | 0.0042 | 0.40 | 0.22 | 0.25 | 1 | 0.22 |
| P25 | 0.04 | 0.073 | 0.015 | 0.04 | 3.5 | 1.25 | 3.0 | 10 | 3.75 |
| P50 | 0.10 | 0.20 | 0.04 | 0.16 | 7.1 | 7.15 | 8.5 | 30 | 10 |
| P75 | 0.55 | 0.57 | 0.16 | 0.30 | 23.1 | 10 | 19 | 100 | 30 |
| Max | 2.00 | 1.2 | 0.20 | 2.00 | 115 | 500 | 420 | 1,650 | 1,650 |
bw: body weight; max: maximum; min: minimum; n: number; P: percentile of ADIs expressed as μg/kg bw per day.
Group II: substances acting on the nervous system, the reproductive system and corticoids/glucocorticoids as set in Commission Regulation (EU) No 37/2010.
Group III: anti‐infective, anti‐inflammatory, antiparasitic substances as set in Commission Regulation (EU) No 37/2010 and ‘Other’ pharmacologically active substances.
For the remaining classes of non‐allowed pharmacologically active substances, and the ‘other’ non‐allowed pharmacologically active substances grouped together, the lowest ADI of 0.22 μg/kg bw per day, was selected as the TSV. This TSV applies to substances without activities falling in the previous two groups (herein referred to as Group III).
The CONTAM Panel noted that if there is information available that a non‐allowed pharmacologically active substance causes blood dyscrasias (such as aplastic anaemia), causes allergy (excluding skin sensitisation), is a high potency carcinogen, or is an inorganic substance, TSVs based on the procedure described above may not be sufficiently health protective and such substances are considered to be outside the scope of this guidance document. For such substances a substance‐specific risk assessment is required.
The CONTAM Panel considered whether RPAs should be set for different food matrices (edible tissues or products). Setting values for all possible substance/matrix combinations was considered impractical, and different values assigned to each combination would give a false impression of precision of the RPA. Therefore, the RPAs should be food matrix independent and should take into account the overall intake of food of animal origin.
Based on the reasonably achievable lowest CCα, the potentially highest exposure should be estimated, across different food matrices and age groups. For the establishment of an RPA, the highest estimated intake has to be compared with the TSV. If this intake is equal to or lower than the TSV, then the reasonably achievable lowest CCα can be accepted as the RPA. If higher, then the sensitivity of the analytical method needs to be improved. In the case where no further analytical improvements are feasible within a short to medium time frame, a substance‐specific risk assessment should be considered, based on available toxicological data.
In this Opinion, the CONTAM Panel illustrates the applicability and the impact of the proposed methodology to establish RPAs for a number of non‐allowed pharmacologically active substances.
The CONTAM Panel emphasises that this is a simple and pragmatic approach and this guidance does not replace a full risk assessment. The CONTAM Panel recognises the uncertainties in deriving the TSVs. Overall, however, this is likely to be a conservative approach.
The CONTAM Panel noted that non‐edible matrices are also monitored for non‐allowed pharmacologically active substances. Such monitoring includes, for example, analysis of shells of shrimps, or monitoring of urine, eyes or hair in livestock animals. RPAs are only applicable to food commodities and are not appropriate for non‐edible matrices.
The CONTAM Panel also identified circumstances where the European Commission might consider it appropriate to consult EFSA for a substance‐specific risk assessment. Situations where this may be appropriate are (i) where the estimated intake derived using the reasonably achievable lowest CCα is higher than the TSV and it is not feasible to lower the CCα of the analytical method, (ii) substances causing blood dyscrasias (aplastic anaemia), causing allergy (excluding skin sensitisation), that are high potency carcinogens or inorganic substances, which are outside the scope of this guidance document, or (iii) where experimental data become available indicating that use of the relevant TSV may not be adequately health protective and re‐categorisation of the substance to another group is not possible.
1. Introduction
1.1. Background and Terms of Reference as provided by the requestor
1.1.1. Background
On 22/01/2011 the Commission sent a mandate to EFSA (ares 2011 198565), requesting for an EFSA opinion on the methodological principles and scientific methods to be taken into account when establishing reference points for action. In response to this request, EFSA published in 2013 a scientific opinion on guidance on methodological principles and scientific methods to be taken into account when establishing Reference Points for Action (RPAs) for non‐allowed pharmacologically active substances present in food of animal origin.
In May 2016, EFSA received in the context of Article 31 of Regulation (EC) No 178/2002, a mandate on dyes in aquaculture and RPAs. EFSA was requested to evaluate whether a series of dyes are covered by the guidance document and to apply a specific part of the guidance document to the substances that are covered. This mandate resulted in the first extensive application of the guidance document and EFSA consulted the CONTAM Panel on the following points:
whether only the intended use of substances should be considered for the allocation to group II and III substances;
whether skin sensitisations should be used as an exclusion criterion;
how the criterion specifying that ‘Group I substances are excluded from the guidance document when there is evidence that the related toxicological screening value (TSV) may not be adequately health protective’, should be evaluated;
whether inorganic substances should be excluded from the guidance document.
At the 83rd plenary meeting, the CONTAM Panel gave direction to EFSA in order to finalise the scientific report on dyes in aquaculture, which was published in July 2017. However, an update of the guidance document is warranted to clarify these points.
Furthermore, also during the discussions on the proposal for the Commission Implementing Regulation on RPAs for non‐allowed pharmacologically active substances in food of animal origin, the need for certain amendments has arisen.
1.1.2. Terms of Reference
In accordance with Art 29 (1) of Regulation (EC) No 178/2002, the European Commission asks the European Food Safety Authority for an update of the scientific opinion on guidance on methodological principles and scientific methods to be taken into account when establishing Reference Points for Action (RPAs) for non‐allowed pharmacologically active substances present in food of animal origin. In particular, the update should:
address the issues discussed by the CONTAM Panel at its 83rd plenary meeting (see above)3;
in order to ensure a correct interpretation of the concept of RPA, it is appropriate to consider rewording certain parts of the opinion to avoid any misunderstanding;
reflect that analytical methods used for non‐allowed pharmacologically active substances should be as sensitive as possible, in order to allow the establishment of RPAs at concentrations as low as possible;
define the relationship between the reasonably achievable lowest limit of quantification (RALLOQ) and the decision limit (CCα). Since the CCα of the analytical method needs to be set at or below the RPA, this value is of relevance for determining which concentration for the RPA is analytically achievable.
1.2. Interpretation of the Terms of Reference
In order to address the issue as to whether only the ‘intended use’ of substances should be considered for the allocation to group II and III substances, the EFSA Panel on Contaminants in the Food Chain (CONTAM Panel) decided that additional criteria regarding the allocation of substances to these groups are required in this guidance. To improve the clarity of the guidance, the CONTAM Panel decided that for group I substances criteria on genotoxicity should be added.
1.3. Supporting information for the assessment
As a result of the treatment of food‐producing animals with veterinary medicinal products (VMPs), residues of pharmacologically active substances contained in VMPs can be present in animal products intended for human consumption. In accordance with Regulation (EC) No 470/2009,1 these VMPs may only be placed on the market if the residues in animal products do not pose any harm to the consumer. Pharmacologically active substances fulfilling this condition are classified as ‘Allowed substances’ in Table 1 of Regulation (EU) No 37/20102. All other pharmacologically active substances are considered as ‘non‐allowed substances’ and a specific subgroup of these non‐allowed substances is the group of ‘Prohibited substances’ which is listed in Table 2 of the same Regulation or which are mentioned in Directive 96/22.4 The substances in Table 2 of Regulation (EU) No 37/2010 are substances for which no maximum residue limit (MRL) was established. This could be due to the fact that based on the available data no safe limit could be identified, or because a final conclusion concerning human health with regard to residues of a substance could not be established given the lack of scientific information. The substances referred to in Directive 96/22 are substances having hormonal or thyrostatic5 action and beta‐agonists, which are prohibited to be used in stock farming.
Table 2.
Comparison of the estimated intake based on hypothetical reasonably achievable lowest CCα value with the TSV for a selected number of non‐allowed pharmacologically active substances
| Substance | Group | CCα (μg/kg)a | Selected food matrix | Maximum acute estimated dietary exposure (μg/kg bw per day) | TSV (μg/kg bw per day) |
|---|---|---|---|---|---|
| Malachite green and leucomalachite green | Ib | 2 | Mammalian meat | 0.033 | 0.0025 |
| Mabuterol | IIc | 0.1 | Milk | 0.018 | 0.0042 |
| Ibuprofen | IIId | 10 | Milk | 1.8 | 0.22 |
bw: body weight; CCα: decision limit; TSV: toxicological screening value.
Hypothetical reasonably achievable lowest CCα.
Group I: compounds for which there is no information on genotoxicity or when there is evidence that the substance is genotoxic, except high potency carcinogens.
Group II: substances acting on the nervous system, the reproductive system or corticoids/glucocorticoids.
Group III: remaining substances.
Regulation (EC) No 470/2009 stipulates that for non‐allowed pharmacologically active substances a RPA may be established when it is deemed necessary to ensure official controls for food of animal origin. The RPA shall be based on the reasonably achievable lowest level that can unequivocally be determined by official control laboratories designated in accordance with Regulation (EC) No 882/20046. When residues of such non‐allowed substances are detected at or above the RPA, the food is considered not to comply with Community legislation. However, even where residues of such substances are detected below the RPA, the competent authority shall carry out investigations to determine whether there has been illegal administration of a non‐allowed pharmacologically active substance and, where relevant, shall apply the appropriate penalties.
In order to guarantee a high level of protection of health, the Regulation states that the Commission shall apply a risk assessment based on methodological principles as well as scientific methods in consultation with EFSA (Article 19(3)) and, where appropriate, submit a request to EFSA for a risk assessment as to whether the RPA is adequate to protect human health (Article 19(2)).
In 2011, the European Commission asked EFSA for an opinion on the methodological principles and scientific methods to be taken into account when establishing RPAs. In particular, the opinion should:
Define the relevant methodological principles and scientific criteria to be taken into account when establishing RPAs for non‐allowed pharmacologically active substances present in food of animal origin for which an MRL is not available or cannot be laid down using other procedures in EU legislation to protect public health;
Indicate whether RPAs should differ in function of the matrix tested, and if so, define criteria to be applied;
Propose criteria in which cases it would be appropriate to submit to EFSA a request for a risk assessment as to whether RPAs for specific substances are adequate to protect human health.
Following this request, the CONTAM Panel adopted in 2013 a scientific opinion on guidance on methodological principles and scientific methods to be taken into account when establishing RPAs for non‐allowed pharmacologically active substances present in food of animal origin (EFSA CONTAM Panel, 2013). This guidance document presented a simple and pragmatic approach to deal with residues of non‐allowed pharmacologically active substances in food. This approach took into account both analytical and toxicological considerations in the establishment of RPAs for non‐allowed pharmacologically active substances. However, it should be noted that the guidance document does not replace a full risk assessment. The current Scientific Opinion is an update of the guidance published in 2013. Section 2.1 gives an overview of the main changes.
1.3.1. Minimum Required Performance Limit (MRPL) and Reference Points for Action
In the late 1980s and 1990s, the analysis of non‐allowed pharmacologically active substances in products of animal origin was often performed with different limits of detection being applied between Member States (MS) and even within one MS. As a consequence, the results of the investigations were often not comparable leading to an unequal treatment of food producers. Prominent examples are the determination of chloramphenicol and nitrofuran metabolites in shrimps or clenbuterol in calves. In order to ensure the quality and especially the comparability of the analytical results generated by laboratories approved for official residue control, the EU Commission deemed it necessary to set strict requirements for analytical methods to be used for official control purposes. In this respect, the concept of routine methods and reference methods was superseded by a criteria approach, in which performance criteria and procedures for the validation of screening and confirmatory methods were established. The rules for the analytical methods to be used in the testing of official samples taken pursuant to Council Directive 96/23/EC7 and the common criteria for the interpretation of analytical results of official control laboratories for such samples are specified in Commission Decision of 14 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results (Decision 2002/657/EC). As an important tool to ensure a harmonised implementation of Council Directive 96/23/EC, the Commission progressively established MRPLs for analytical methods for substances for which no permitted limit has been established and in particular for those substances whose use is not allowed, or is specifically prohibited, in the Community.
According to Annex I, point 1.18 of Decision 2002/657/EC8, ‘Minimum required performance limit means minimum content of an analyte in a sample, which at least has to be detected and confirmed. It is intended to harmonise the analytical performance of methods for substances for which no permitted limit has been established’.
Decision 2002/657/EC lays down MRPLs in various matrices for chloramphenicol, nitrofuran metabolites, medroxyprogesterone acetate and the sum of malachite green and leucomalachite green. The MRPLs were adopted as the standard of performance ensuring effective control of Community legislation when testing samples for the presence of certain non‐allowed substances.
However, MRPLs correspond to the average limit above which the detection of a substance or its residues could be construed as being methodologically meaningful. With the ongoing improvement of analytical equipment and methodology, a number of samples were identified that showed concentrations of non‐allowed substances below the MRPLs. These findings often caused trade problems, because analytical results below and above the MRPLs were treated differently, leading either to acceptance or rejection of food lots, especially concerning imports from Third Countries.
In order to establish a harmonised approach for the control of residues of non‐allowed substances in food of animal origin imported into the Community, the Commission enacted Decision 2005/34/EC9 laying down harmonised standards for the testing for certain residues in products of animal origin imported from Third Countries. This Decision lays down the RPAs for residues of substances for which MRPL values have been established in accordance with Decision 2002/657/EC, when analytical tests on imported consignments of products of animal origin confirm the presence of such residues, and the action to be undertaken after such confirmation.
Art. 3.1 of Decision 2005/34/EC stipulates: ‘Where results of analytical tests are at or above the MRPLs laid down in Decision 2002/657/EC, the consignment concerned shall be considered non‐compliant with Community legislation’.
Art. 3.5 of Decision 2005/34/EC states:
Where the results of analytical tests on products are below the MRPLs laid down in Decision 2002/657/EC, the products will not be prohibited from entering the food chain. The competent authority shall retain a record of the findings in case of recurrence. Where the results of analytical tests on products from the same origin show a recurrent pattern indicating a potential problem related to one or several prohibited or unauthorised substances, including for instance the recording of four or more confirmed results below the reference points for action for the same substance in imports from a particular origin within a period of 6 months, the competent authority shall inform the Commission and the other Member States in the Standing Committee on the Food Chain and Animal Health. The Commission shall bring the matter to the attention of the competent authority of the country or countries of origin and shall make appropriate proposals.
Following this Decision, the analytically driven MRPLs originally derived for harmonisation of analytical methods became RPAs for checking compliance of products imported from Third Countries with EU legislation.
However, this Decision regulated only imports from Third Countries and did not apply to food produced within the Community. As a number of products of animal origin originating from MS were found to contain non‐allowed substances below and above the MRPLs, the European Commission and the MS agreed to apply the approach laid down in Decision 2005/34/EC, with the necessary changes, also to food of animal origin produced within the Community. This implies in particular that the MRPLs set according to Commission Decision 2002/657/EC shall also be used as reference points for action. This approach, moreover, means that any detection of substances whose use is not authorised in the Community shall be followed by an investigation into the source of the substance in question and application of appropriate enforcement measures, in particular aiming at the prevention of recurrence in the case of documented illegal use (SANCO‐E.2(04)D/52192710).
2. Assessment
2.1. Main changes compared to the opinion published in 2013
Following the request received from the European Commission to update the scientific opinion on guidance on methodological principles and scientific methods to be taken into account when establishing RPAs for non‐allowed pharmacologically active substances present in food of animal origin, the following changes were made:
Information from the background and terms of reference of the 2013 opinion has been integrated in Section 1.3.
The RALLOQ is replaced by the reasonably achievable lowest CCα.
The concept of toxicologically based limit of quantification (TBLOQ) has been removed from the guidance. Instead, the dietary intake is calculated based on the reasonably achievable lowest CCα and compared with the TSV.
Additional text has been added to Section 2.3.1 to clarify that skin sensitisation is not an exclusion criterion for this guidance.
Changes were implemented in Sections 2.2.2 and 2.3 to better explain the grouping of the substances and when a substance can be excluded from group I and II.
The lowest acceptable daily intake (ADI) rather than the 5th percentile was selected as the TSV for group III substances.
Since the previous opinion, the European Medicines Agency (EMA) published ADIs of additional substances. These were included in the database used to develop and update this guidance.
Clarification has been included that substances that fall under the pesticide regulatory framework are not subject of this guidance.
It has been clarified that the guidance is not applicable to inorganic substances.
Following update of the guidance for establishing the safety of additives for the consumer (EFSA FEEDAP Panel, 2017), the CONTAM Panel revised the text on food consumption considerations in Section 2.2.3.
The present opinion is a consolidated version of the 2013 opinion and these changes.
2.2. Considerations for an updated procedure to establish RPAs according to the framework of Regulation (EC) No 470/2009
The aim of establishing a RPA for non‐allowed pharmacologically active substances is to define an analytical concentration in food of animal origin that can be determined by official control laboratories and which is low enough to adequately protect the consumers of food commodities that contain the respective substance. For this purpose, both analytical and toxicological considerations are required, and these considerations can be made independently of each other.
2.2.1. Analytical considerations
As the RPA will be applied to non‐allowed or prohibited pharmacologically active substances (see Section 1.3) used in animal husbandry, it has to be set at the reasonably achievable lowest level that can unequivocally be determined by official control laboratories. This level is termed ‘decision limit (CCα)’ and defined in Commission Decision 2002/657/EC as follows: ‘Decision limit (CCα) means the limit at and above which it can be concluded with an error probability of α that a sample is non‐compliant’. The Commission Decision 2002/657/EC also lays down how the decision limit has to be established for substances for which no permitted limit has been set, and which performance criteria have to be met by the analytical methods. For setting a RPA, for a particular substance, information is needed on the performance of the analytical methods applied by the various official control laboratories for the confirmatory analysis of the substance. In this respect, the European Union Reference Laboratories (EU‐RLs), are designated in accordance with Regulation (EC) No 882/2004, as well as the corresponding National Reference Laboratories (NRLs) have a specific responsibility as these should contribute to a high quality and uniformity of analytical results. The duties and responsibilities of the EU‐RLs and NRLs are laid down in Articles 32 and 33 of Regulation (EC) No 882/2004.
The CCα for a substance is method dependent and consequently may differ from laboratory to laboratory. It is the task of the EU‐RL/NRL network to collate the different decision limits and make a proposal on the reasonably achievable lowest CCα for the substance.
In the previous opinion, a RALLOQ was identified. However, in the area of pharmacologically active substances the CCα is applied rather than the LOQ. Therefore, the RALLOQ was replaced by the reasonably achievable lowest CCα. Consequently the question to define the relationship between CCα and RALLOQ is no longer applicable.
2.2.2. Toxicological considerations
In order to determine whether the reasonably achievable lowest CCα for the available analytical method is low enough to ensure that there is no health concern for the consumer, consideration of the toxic potential and pharmacological activity of the substance is needed. As the substances of concern are non‐allowed, it is likely that the toxicological information on these substances is limited and/or that they have properties inappropriate for authorised substances.
The CONTAM Panel considered the applicability of the concept of Threshold of Toxicological Concern (TTC), which uses Cramer classes (EFSA Scientific Committee, 2012), as the basis for the derivation of TSVs for non‐allowed pharmacologically active substances for which a threshold mechanism can be assumed. The CONTAM Panel noted that some groups of substances that are the subject of this guidance document (e.g. inorganic substances, steroids11 – see EFSA Scientific Committee, 2012) are excluded from the TTC approach. In addition, the database underlying the TTC approach only contains a small number of pharmacologically active substances. However, pharmacological effects due to exposure via food are considered as adverse. Therefore, the Panel concluded that the TTC concept, with the use of Cramer classes, is not applicable in a general approach for deriving TSVs for non‐allowed pharmacologically active substances in the framework of the establishment of RPAs.
However, an exception was made for the TTC value for genotoxic substances which are of particular concern because they also may be carcinogenic or cause germ cell mutations. The EFSA Scientific Committee (SC) has explored substances with a structural alert for genotoxicity regarding their possible human health risks (EFSA Scientific Committee, 2012). First, high potency carcinogens that would give the highest calculated risks were identified. Then animal bioassay data on over 500 known genotoxic and non‐genotoxic carcinogens were considered. Based on the carcinogenic potency of the substances and mathematical modelling of risks, a TTC value of 0.0025 μg/kg bw per day was established for compounds which are potentially genotoxic (EFSA Scientific Committee, 2012). This value should be sufficiently conservative to be used for substances with genotoxic properties as a level of human exposure that would be of low concern from a public health point of view, provided that compounds designated as high potency carcinogens are excluded.
The CONTAM Panel decided to use this TTC value of 0.0025 μg/kg bw per day as a TSV for non‐allowed pharmacologically active substances for which there is either direct evidence of genotoxicity or insufficient evidence to conclude that they are not genotoxic. In these cases, the substances are referred to in this guidance as Group I substances.
Non‐allowed pharmacologically active substances could have pharmacological or toxicological properties that might to some extent be comparable with those of allowed veterinary pharmacologically active substances, as evaluated by the EMA. Therefore, the CONTAM Panel assessed the ADIs established by the EMA for allowed substances.
These ADIs are based on the no‐observed‐effect levels (NOELs) that are most relevant for the safety assessment, taking into account relevant uncertainty factors. The CONTAM Panel used a database of published ADIs for 177 veterinary pharmacologically active substances to develop and update this guidance. These substances can be grouped into the following therapeutic classes in accordance with Commission Regulation (EU) No 37/2010:
agents acting on the central/autonomic nervous system;
agents acting on the reproductive system;
corticoids/glucocorticoids;
anti‐infectious agents (antibiotics/antiseptics/chemotheurapeutics);
anti‐inflammatory agents (antidiarrheal and intestinal anti‐inflammatory agents/nonsteroidal anti‐inflammatory agents);
antiparasitic agents (agents acting against endoparasites/ectoparasites/protozoa).
In addition, EMA also established ADIs for a group of substances having a pharmacological activity different from the classes mentioned above. This group, designated as ‘Other’ in this guidance document, comprised among others, analgesics, diuretics and sedatives. The CONTAM Panel noted that a low ADI value for alfacalcidol of 0.002 μg/kg bw per day was reported in this group. The EMA concluded that there is no need to establish an MRL for this synthetic vitamin D3 analogue, used for the prevention of milk fever in dairy cows at the end of pregnancy, as inter alia it is rapidly absorbed, extensively metabolised and completely excreted. Given the clinical indication and toxicokinetic properties of this substance, the CONTAM Panel concluded that it is not representative of the type of non‐allowed pharmacologically active substances that might be present as residues in food, and this substance was removed from the database. Therefore, in total, 176 veterinary pharmacologically active substances were used (Appendix B).
Three types of effects are evaluated by EMA for each compound in order to derive an ADI, i.e. pharmacological effects, antimicrobial effects and toxicological effects. The lowest ADI is used as the overall ADI, meaning that the type of effect underlying this overall ADI varies per compound. For agents acting on the nervous system and reproductive system, as well as corticoids/glucocorticoids, the resulting ADIs were based on pharmacological or toxicological effects. For the other compounds, the ADIs are based on one of these three effects. The CONTAM Panel decided to only include compounds with agonistic or antagonistic effects on receptors12 in the nervous system (e.g. α‐ and β‐adrenergic) in the group of agents acting on the nervous system. Similarly, only agents with an effect on the progesterone or prostaglandin F2α receptor were included in the group of agents acting on the reproductive system. This was done independently of whether the ADI was based on pharmacological or toxic effects, since the binding to a receptor may have been involved in the mode of action. No substances acting on the oestrogen or androgen receptor were identified in the database. Neither was this the case for compounds interacting with the thyroid receptor.
The distributions of ADIs for all 176 veterinary pharmacologically active substances together and separately for the different classes are shown in Figure 1 and in Table 1. The distribution of the ADIs for all 176 substances is wide, ranging from 0.0042 to 1,650 μg/kg bw per day. From Figure 1, it is obvious that the ADIs for three classes of pharmacologically active substances, those acting on the nervous or reproductive system and the corticoids/glucocorticoids, are comparable and clearly lower than the ADIs for the other groups. Therefore, these three classes should be treated separately when establishing a TSV. Since the ADIs for these substances are comparable, the Panel decided to group these three classes and to use the lowest ADI of 0.0042 μg/kg bw per day (see Table 1) as the TSV for non‐allowed pharmacologically active substances acting on specific receptors in the nervous or reproductive system, or being corticoids/glucocorticoids (group II).
Figure 1.
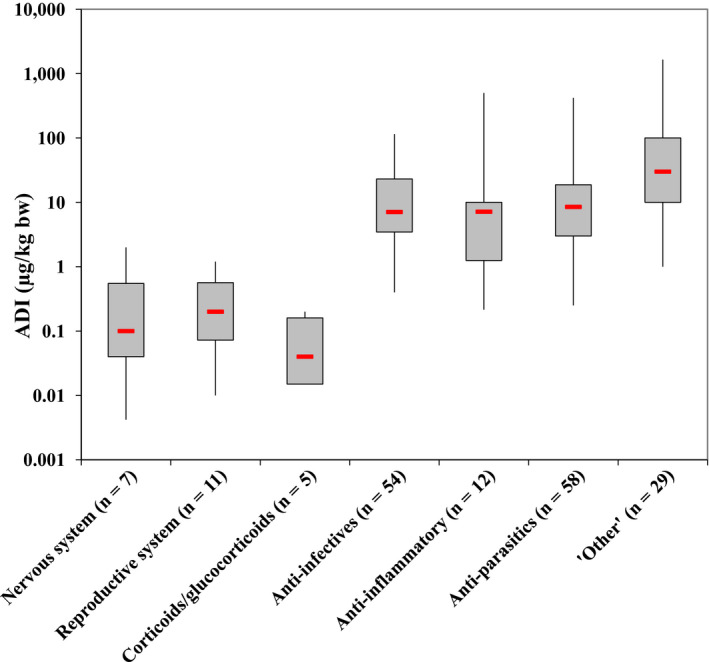
Distribution of acceptable daily intakes (ADIs) established by the European Medicines Agency for 176 allowed veterinary pharmacologically active substances
- The boxes in the figure represent the range from the lower to the upper quartile; the red lines indicate the median.
It was noted that the ADIs for the group of ‘Other’ substances are comparable with those of the anti‐infective, anti‐inflammatory and antiparasitic substances. The CONTAM Panel therefore decided to combine these substances together with the ‘Other’ substances, referred to as Group III in this guidance. This group includes many compounds where effects on the nervous or reproductive system were the result of a toxic rather than pharmacological action via a receptor. Examples are a number of pesticides (avermectins, organophosphates and pyrethroids) that already have established MRLs in animal derived products and could as such be exempted from the grouping. However, the CONTAM Panel decided to include them in group III in order to cover also such neurotoxic effects in the derivation of a TSV. The lowest ADI for this combined group, being 0.22 μg/kg bw per day, was selected as the TSV to be used for substances not falling into Groups I or II.
2.2.3. Matrix and food consumption considerations
RPAs for different matrices
The CONTAM Panel considered whether RPAs should be set for different matrices (edible tissues or products) based on consumption patterns and tissue distribution characteristics of the non‐allowed pharmacologically active substances. Setting values for all possible substance/matrix combinations was considered impractical, and different values assigned to each combination would give a false impression of precision of the RPA. Therefore, the CONTAM Panel concluded that the RPA should be matrix independent and should take into account the overall intake of food of animal origin, assuming that all residues are bioaccessible. In practice, this means that the exposure from the food matrix with the highest consumption should be used to evaluate whether the reasonably achievable lowest CCα is low enough to ensure consumer safety.
Food consumption considerations
To determine whether the reasonably achievable lowest CCα is low enough to adequately protect the consumer of food commodities that contain the respective substance, the corresponding exposure has to be calculated. TSVs are based on the most sensitive relevant effect, which in some instances is an effect arising from acute exposure (e.g. neurotoxicity or developmental effects). Therefore, the CONTAM Panel concluded that it was appropriate to calculate the acute dietary exposure in its approach to derive the estimated intake at the reasonably achievable lowest CCα. Because of the sporadic nature of exposure to residues of non‐allowed pharmacologically active substances, it is unlikely that more than one food containing the same non‐allowed pharmacologically active substance would be consumed on the same day.
In the 2013 opinion, the CONTAM Panel decided that a high consumption figure for dairy products of 1.5 kg for toddlers and 2 kg for adults should be used for non‐allowed pharmacologically active substances that might be applied to animals producing milk for human consumption. Where a substance for which a RPA is needed will not be used in animals producing milk for human consumption (e.g. malachite green), the CONTAM Panel decided that consumption values of 0.2 kg and 0.5 kg per day for toddlers and adults respectively, could be used. These consumption data were derived from default values for consumption of food of animal origin by high consumers, specified in the guidance for establishing the safety of feed additives for the consumer (EFSA FEEDAP Panel, 2012). However, the CONTAM Panel noted that this guidance was updated as of the 1st of May, 2018 (EFSA FEEDAP Panel, 2017). In this updated guidance, no use is made of default values but an on‐line tool (FACE: Feed Additives Consumer Exposure calculator)13 for estimating chronic and acute dietary exposure to residues of feed additives and their metabolites present in food of animal origin is provided. However, this tool can be used to calculate exposure to any substance present in food of animal origin.
The CONTAM Panel advices use of this updated guidance and consequently the FACE tool, instead of the default values. Therefore, when applying this guidance to a specific substance, the reasonably achievable lowest CCα is used as the occurrence value for all available matrices to calculate the ‘maximum highest reliable percentile (HRP)’ for acute exposure. From the calculated acute dietary exposures, the matrix showing the highest acute exposure is selected and the corresponding exposure is used in the evaluation. Only where that matrix can be excluded as potentially containing the non‐allowed pharmacologically active substance (e.g. malachite green in milk), should the matrix showing the next highest intake be used.
A comparison was made between using the default values and the FACE tool. Where the substance can be present in milk, the outcome generated by the FACE tool is comparable with the outcome generated by the default values, for ‘toddlers’ and ‘adults’. However, it was noted that the age group with the highest exposure to substances that might occur in milk is ‘other children’, which was not covered by the default values. Where the substance does not occur in milk, the highest exposure is generated by the FACE tool for ‘other children’, which is comparable with the outcome generated by the default values for ‘toddlers’. Also for ‘adults’, the outcome generated by the FACE tool is comparable with the outcome generated by the default values.
2.2.4. Testing of non‐edible matrices
The CONTAM Panel noted that non‐edible matrices are also monitored for non‐allowed pharmacologically active substances. Such monitoring includes, for example, analysis of shells of shrimps, or monitoring of urine, eyes and hair of livestock animals. RPAs are only applied to food commodities and are not appropriate for non‐edible matrices.
2.3. Procedure for establishing a RPA
A step‐wise approach was developed for the establishment of a RPA for pharmacologically active substances that are not allowed to be used in veterinary medicinal products for food‐producing animals, based on the identified reasonably achievable lowest CCα and the TSV.
2.3.1. Categories of substances excluded from the procedure
For pesticides that may be used as antiparasitic agents, MRLs for animal derived products have already been established. As such, they can be excluded from the guidance.
Since compounds designated as high potency carcinogens are excluded from the TTC approach (EFSA Scientific Committee, 2012) and consequently are not covered by the TTC value of 0.0025 μg/kg bw per day, these substances are excluded from this guidance document. The same applies for inorganic substances, for which also no ADI values are included in the database.
The CONTAM Panel noted that if there is information available that a non‐allowed pharmacologically active substance causes blood dyscrasias (aplastic anaemia) or allergy, a substance specific risk assessment is required, since there are indications that even at very low levels some individuals may be affected. The term ‘Allergy’ refers to Type I – immediate type hypersensitivity reactions that are immunoglobulin E (IgE)‐mediated (like food‐ or respiratory allergies to proteins in particular, and drug allergies), or to Type IV – delayed type hypersensitivity reactions that are T‐cell‐mediated (like skin allergies to small molecules in particular, and called allergic contact dermatitis (ACD) in humans). Both hypersensitivity reactions are characterised by a sensitisation phase (induction of sensitisation to an allergen) and an elicitation phase (the inflammatory response upon repeated exposure to the same allergen).
Sometimes, systemic reactivation (elicitation) of ACD or systemic contact dermatitis (SCD) can occur when individuals with a contact allergy to a certain skin allergen are exposed systemically to the same allergen via exposure routes other than by cutaneous contact, like orally or by injection. In these situations, the systemically administered allergen may reach the skin through the circulatory system. However, only for relatively few individuals has such a reaction been documented. SCD due to oral re‐exposure to drugs has been described by Thyssen and Maibach (2008) and Aquino and Rosner (2018), like for several nonsteroidal anti‐inflammatory drugs such as diclofenac (Lakshmi and Srinivas, 2011) and for corticosteroids such as dexamethasone (Baeck et al., 2009; Beack and Goossens, 2012). These reactions are mainly seen upon medical use of these drugs at relatively high oral doses.
For a non‐allowed pharmacologically active substance, skin sensitisation resulting in ACD may only be induced upon cutaneous contact. In individuals suffering already from contact allergy to a non‐allowed pharmacologically active substance, reactivation of ACD may only occur upon renewed cutaneous contact, whereas after oral exposure, a reactivation of ACD or SCD is unlikely to occur.
Overall, it is concluded that under the intended conditions for use of non‐allowed pharmacologically active substances, a reactivation of ACD or SCD upon oral exposure of individuals with an existing contact allergy to a non‐allowed pharmacologically active substance is unlikely to occur. Therefore, such substances can be assessed by the RPA procedure.
If there is information available under the intended conditions for use that a non‐allowed pharmacologically active substance causes oral or respiratory sensitisation, a substance specific risk assessment remains necessary.
2.3.2. Identification of the TSV
Figure 2 shows the decision tree for assignment of the TSV.
Figure 2.
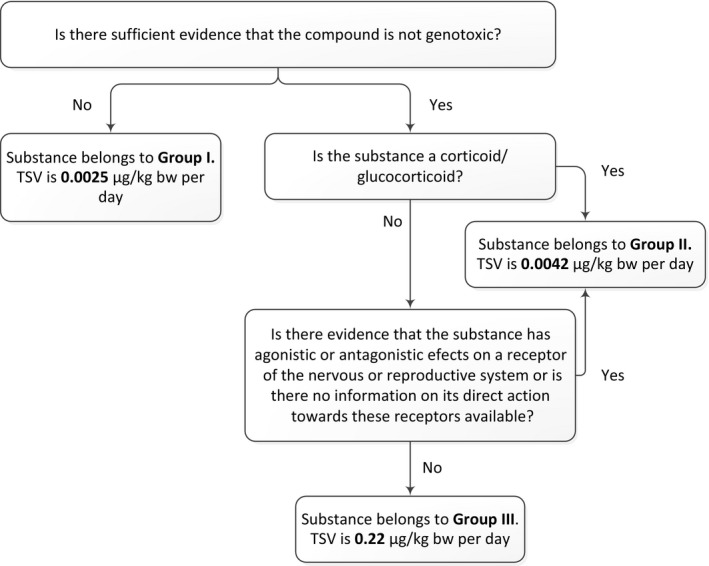
Decision tree for assigning TSVs for non‐allowed pharmacologically active substances14
When there is no information on genotoxicity of the substance, or when there is evidence that the substance is genotoxic, a TSV of 0.0025 μg/kg bw per day should be used. Genotoxic compounds are those that can interact with DNA directly or after metabolic transformation to DNA‐reactive metabolites, or can affect the number or structure of chromosomes. To evaluate the genotoxic potential of a chemical a number of in vitro and in vivo assays are available. According to the Scientific opinion on genotoxicity testing strategies applicable to food and feed safety assessment (EFSA Scientific Committee, 2011), negative results obtained with a basic battery of in vitro tests, comprising a bacterial reverse mutation assay and an in vitro micronucleus assay, are sufficient to conclude that the substance has no genotoxic potential. In the case of positive in vitro results, appropriate in vivo studies (i.e. in vivo mammalian erythrocyte micronucleus test, transgenic rodent assay, in vivo Comet assay) to assess whether the genotoxic potential observed in vitro is expressed in vivo may be performed. According to the Scientific opinion ‘Clarification of some aspects related to genotoxicity assessment’ (EFSA Scientific Committee, 2017), if the results of appropriate and adequately conducted in vivo tests are negative, taking into account other lines of evidence in a weight‐of‐evidence approach, then it can be concluded that the substance is not an in vivo genotoxin.
In summary, the substance can be excluded from Group I when the results obtained with a basic battery of in vitro tests are negative. In the case of positive results in the in vitro battery of tests the substance may be excluded from Group I when the results of in vivo tests are negative, taking into account other lines of evidence in a weight‐of‐evidence approach. However, if any concerns for genotoxicity remain, exclusion from Group I is not appropriate. Certain compounds induce genotoxic effects via indirect modes of action (i.e. induction of reactive oxygen species, inhibition of DNA repair, topoisomerase inhibitors, tubulin inhibitors, antimetabolites). However, indirect modes of action are not criteria to exclude a compound with genotoxic properties from group I.
When there is evidence that substances have agonistic or antagonistic effects on receptors of the nervous system (e.g. α‐ and β‐adrenergic receptors) or on sex hormone (e.g. progesterone, oestrogen or androgen) receptors or the prostaglandin F2α receptor and thereby cause effects on the reproductive system, they belong to Group II and their TSV is 0.0042 μg/kg bw. Also, substances for which no information of their direct action is available should be considered to belong to Group II.
Only if it can be excluded that compounds belong to group I or II, can they be assigned the TSV of 0.22 μg/kg bw, belonging to Group III (remaining substances).
2.3.3. Establishment of a RPA
Figure 3 shows the decision tree for the establishment of RPAs.
Figure 3.
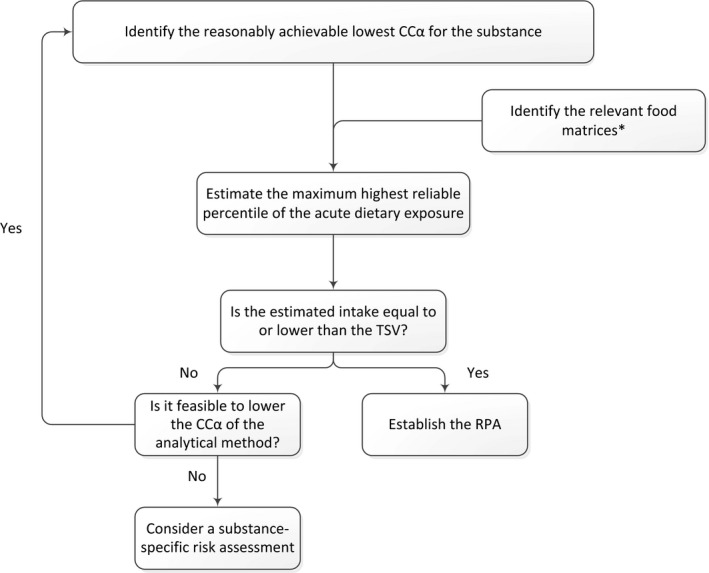
Decision tree for the establishment of a RPA for a non‐allowed pharmacologically active substance14
- *See section 2.2.3 for further details.
For the establishment of a RPA, the estimated intake (see Section 2.2.3) based on the reasonably achievable lowest CCα for the substance has to be compared with the TSV. If the intake is equal to or lower than the TSV, then the reasonably achievable lowest CCα can be accepted as the RPA. If higher, then the sensitivity of the analytical method needs to be improved. In the case where no further analytical improvements are feasible within a short to medium time‐frame, a substance‐specific risk assessment should be considered. Where, in such a situation, toxicological data for the respective non‐allowed pharmacologically active substance are available, these should be taken into consideration.
2.4. Illustration of the methodology to establish a RPA
To illustrate the applicability and the impact of the proposed methodology to establish RPAs, the CONTAM Panel selected a number of non‐allowed pharmacologically active substances that have been detected in food of animal origin over the past years (Appendix A). Based on the relevant characteristics of the substances (e.g. genotoxicity), they were classified into one of the 3 groups (Groups I, II and III), and a TSV was assigned. Based on hypothetical reasonably achievable lowest CCα values, the corresponding intake was estimated and compared with the assigned TSV. This illustration is presented in Table 2.
The hypothetical reasonably achievable lowest CCα of 2 μg/kg for malachite green and leucomalachite green was inserted in the FACE tool to calculate the dietary exposure (see Section 2.2.3). Milk is the food matrix that results in the highest exposure. However, the occurrence of malachite green and leucomalachite green in milk can be excluded, and therefore, the food matrix resulting in the next highest dietary exposure (i.e. mammalian meat) was selected. The highest acute dietary exposure was 0.033 μg/kg bw per day, which represents the maximum HRP calculated across dietary surveys and age groups for mammalian meat. This estimated intake is higher than the TSV of 0.0025 μg/kg for group I, indicating the need for improvement of the sensitivity of the analytical methodology or a substance‐specific risk assessment.
The same approach was followed for mabuterol and ibuprofen. For mabuterol, the presence in milk cannot be excluded and the highest calculated acute dietary exposure was 0.018 μg/kg bw per day, which is higher than the TSV value of 0.0042 μg/kg bw per day. Also for ibuprofen, the presence in milk cannot be excluded and the highest calculated acute dietary exposure was 1.8 μg/kg bw per day, which is higher than the TSV value of 0.22 μg/kg bw per day. This comparison indicates the need for improvement of the sensitivity of the analytical methodology or a substance‐specific risk assessment for both substances.
The CONTAM Panel emphasises that this is a simple and pragmatic approach and this guidance does not replace a full risk assessment. The CONTAM Panel recognises the uncertainties in deriving the TSVs. Overall, this approach is likely to be conservative.
2.5. Proposed criteria for the European Commission to request a risk assessment from EFSA
In some circumstances, the outcome of the proposed methodology to establish RPAs might indicate that it could be appropriate for the European Commission to submit a request to EFSA for a substance‐specific risk assessment. Situations where this may be appropriate are:
Where the estimated intake derived using the reasonably achievable lowest CCα is higher than the TSV and it is not feasible to lower the CCα of the analytical method.
For substances which are outside the scope of this guidance document, such as substances causing blood dyscrasias (aplastic anaemia), causing allergy (excluding skin sensitisation), that are high potency carcinogens or inorganic substances.
Where experimental data become available indicating that use of the relevant TSV may not be adequately health protective.
Abbreviations
- ACD
allergic contact dermatitis
- ADI
acceptable daily intake
- bw
body weight
- CCα
decision limit
- CONTAM Panel
EFSA Panel on Contaminants in the Food Chain
- EMA
European Medicines Agency
- EU‐RL
European Union Reference Laboratory
- FEEDAP Panel
EFSA Panel on Additives and Products or Substances used in Animal Feed
- HRP
highest reliable percentile
- IgE
immunoglobulin E
- LOQ
limit of quantification
- MRL
maximum residue limit
- MRPL
minimum required performance limit
- MS
Member State
- NOEL
no‐observed‐effect level
- NRL
National Reference Laboratory
- RALLOQ
reasonably achievable lowest limit of quantification
- RPA
reference point for action
- SC
EFSA Scientific Committee
- SCD
systemic contact dermatitis
- TBLOQ
toxicologically based limit of quantification
- TSV
toxicological screening value
- TTC
threshold of toxicological concern
- VMP
Veterinary Medicinal Product
Appendix A – Examples of non‐allowed pharmacologically active substances which have been detected in food of animal origin over the past years under the National Residue Control Plans
1.
| Name of the compound | Name of the compound |
|---|---|
| Acepromazine | Malachite green |
| Acid Fast Green B | Mapenterol |
| Azobenzene | Mecarbam |
| Basic blue 26 | Mefenamic acid |
| Boldenone | Megestrol |
| Brillant Green | Melengestrol |
| Bromobuterol | Methylene Blue |
| Carbadox | Methyltestosterone |
| Chloramphenicol | Methylthiouracil |
| Chlorbrombuterol | Methylviolet |
| Chlormadinone | Metronidazole |
| Chlormephos | Nandrolone |
| Chloroform | Naproxen |
| Chlorpromazine | New methylene blue |
| Cimaterol | Nile blue |
| Cimbuterol | Nitenpyram |
| Clencyclohexerol | Nitrofurans (metabolites AMOZ, AHD, SEM, AOZ) |
| Clenpenterol | Olaquindox |
| Clenproperol | Orciprenaline |
| Colchicine | Oxyphenbutazone |
| Cristal Violet | Pararosaniline base |
| Dapsone | Phenylbutazone |
| Dexamethasone | Propiconazole |
| Diclofenac | Propiopromazine |
| Dienestrol | Propylthiouracil |
| Diethylstilbestrol | Pyrazophos |
| Dimetridazole | Quinalphos |
| Erythrosine B | Rhodamine 6G |
| Ethinyloestradiol | Ritodrin |
| Ethoprophos | Ronidazole |
| Ethylviolet | Salbutamol |
| Fenoterol | Salmeterol |
| Formothion | Stanozolol |
| Haloperidol | Tapazole |
| Hexaconazole | Terbutaline |
| Hexestrol | Thiouracil |
| Hydroxymethylclenbuterol | Triazophos |
| Ibuprofen | Tulobuterol |
| Isofenphos | Ultramarine |
| Isofenphos | Zearalanone |
| Mabuterol | Zeranol |
Appendix B – Overview of the allowed pharmacologically active substances and their ADIs used for deriving the TSVs for group II and III
1.
| Substance | Type of overall ADI | Overall ADI (μg/kg bw) |
|---|---|---|
| Agents acting on the nervous system | ||
| Azaperone | Pharmacological | 0.8 |
| Cabergoline | Toxicological | 0.03 |
| Carazolol | Pharmacological | 0.1 |
| Clenbuterol hydrochloride | Pharmacological | 0.0042 |
| Detomidine | Pharmacological | 0.3 |
| Romifidine | Pharmacological | 0.05 |
| Isoxuprine | Pharmacological | 2 |
| Agents acting on the reproductive system | ||
| Alfaprostol | Toxicological | 1 |
| Altrenogest | Pharmacological | 0.2 |
| Azagly‐nafareline | Toxicological | 0.25 |
| Chlormadinone | Pharmacological | 0.07 |
| Cloprostenol | Pharmacological | 0.075 |
| Dinoprost tromethamine | Pharmacological | 0.83 |
| Flugestone acetate | Pharmacological | 0.03 |
| Luprostiol | Toxicological | 0.2 |
| Medroxyprogesterone acetate | Pharmacological | 0.3 |
| Norgestomet | Pharmacological | 0.01 |
| Tiaprost | Toxicological | 1.2 |
| Corticoids | ||
| Beclomethasone dipropionate | Pharmacological | 0.04 |
| Betamethasone | Toxicological | 0.015 |
| Dexamethasone | Toxicological | 0.015 |
| Methylprednisolone | Pharmacological | 0.16 |
| Prednisolone | Pharmacological | 0.2 |
| Anti‐infectious agents | ||
| Acetylisovaleryl‐tylosin | Microbiological | 1.02 |
| Apramycin | Microbiological | 40 |
| Avilamycin | Toxicological | 115 |
| Bacitracin | Microbiological | 3.9 |
| Baquiloprim | Toxicological | 10 |
| Cefacetrile | Microbiological | 3.5 |
| Cefalexin | Microbiological | 54.4 |
| Cefalonium | Microbiological | 15.3 |
| Cefapirin | Microbiological | 2.54 |
| Cefazolin | Microbiological | 10 |
| Cefoperazone | Microbiological | 2.5 |
| Cefquinome | Microbiological | 3.8 |
| Ceftiofur | Microbiological | 20 |
| Chlortetracycline | Microbiological | 3 |
| Clavulanic acid | Toxicological | 50 |
| Colistin | Microbiological | 5 |
| Danofloxacin | Toxicological | 24 |
| Difloxacin | Toxicological | 10 |
| Dihydro‐streptomycin | Toxicological | 25 |
| Doxycycline | Microbiological | 3 |
| Enrofloxacin | Microbiological | 6.2 |
| Erythromycin | Microbiological | 5 |
| Florfenicol | Microbiological | 10 |
| Flumequine | Microbiological | 8.25 |
| Gamithromycin | Toxicological | 10 |
| Gentamicin | Microbiological | 4 |
| Kanamycin | Microbiological | 8 |
| Lincomycin | Microbiological | 10 |
| Marbofloxacin | Microbiological | 4.5 |
| Mecillinam | Microbiological | 23.8 |
| Monensin | Pharmacological | 3.45 |
| Nafcillin | Microbiological | 4.4 |
| Neomycin (including framycetin) | Toxicological | 60 |
| Novobiocin | Microbiological | 1.25 |
| Octenidine dihydrochloride | Toxicological | 0.625 |
| Oxolinic acid | Microbiological | 2.5 |
| Oxytetracycline | Microbiological | 3 |
| Paromomycin | Microbiological | 25 |
| Pirlimycin | Microbiological | 6 |
| Rifaximin | Microbiological | 2 |
| Sarafloxacin | Microbiological | 0.4 |
| Spectinomycin | Microbiological | 40 |
| Spiramycin | Microbiological | 50 |
| Streptomycin | Toxicological | 25 |
| Tetracycline | Microbiological | 3 |
| Thiamphenicol | Microbiological | 2.5 |
| Tiamulin | Toxicological | 30 |
| Tildipirosin | Toxicological | 100 |
| Tilmicosin | Microbiological | 4 |
| Trimethoprim | Microbiological | 4.2 |
| Tulathromycin | Microbiological | 11 |
| Tylosin | Microbiological | 6 |
| Valnemulin | Microbiological | 7.95 |
| Virginiamycin | Microbiological | 21 |
| Anti‐inflammatory agents | ||
| Acetylsalicylic acid | Pharmacological | 8.3 |
| Carprofen | Toxicological | 10 |
| Diclofenac | Toxicological | 0.5 |
| Firocoxib | Toxicological | 0.215 |
| Flunixin | Toxicological | 6 |
| Ketoprofen | Pharmacological | 5 |
| Meloxicam | Toxicological | 1.25 |
| Metamizole | Pharmacological | 10 |
| Paracetamol | Pharmacological | 50 |
| Sodium salicylate | Toxicological | 500 |
| Tolfenamic acid | Toxicological | 10 |
| Vedaprofen | Toxicological | 1.25 |
| Antiparasitic agents | ||
| Abamectin | Toxicological | 2.5 |
| Albendazole | Toxicological | 5 |
| Alphacypermethrin | Toxicological | 15 |
| Amitraz | Toxicological | 3 |
| Amprolium | Toxicological | 100 |
| Azametiphos | Toxicological | 25 |
| Clazuril | Toxicological | 50 |
| Clorsulon | Toxicological | 1 |
| Closantel | Toxicological | 30 |
| Coumafos | Toxicological | 0.25 |
| Cyfluthrin | Pharmacological | 3 |
| Cyhalothrin | Toxicological | 5 |
| Cypermethrin | Toxicological | 15 |
| Cyromazine | Toxicological | 20 |
| Decoquinate | Toxicological | 75 |
| Deltamethrin | Toxicological | 10 |
| Derquantel | Toxicological | 1 |
| Diazinon | Toxicological | 2 |
| Diclazuril | Toxicological | 30 |
| Dicyclanil | Toxicological | 420 |
| Diflubenzuron | Toxicological | 12.4 |
| Doramectin | Toxicological | 0.5 |
| Emamectin | Toxicological | 1 |
| Eprinomectin | Toxicological | 5 |
| Febantel | Toxicological | 7 |
| Fenbendazole | Toxicological | 7 |
| Fenvalerate | Toxicological | 12.5 |
| Fluazuron | Toxicological | 43 |
| Flubendazole | Toxicological | 12 |
| Flumethrin | Toxicological | 1.8 |
| Furalaner | Toxicological | 10 |
| Halofuginone | Toxicological | 0.3 |
| Hexaflumuron | Toxicological | 5 |
| Imidocarb | Toxicological | 10 |
| Ivermectin | Toxicological | 10 |
| Lasalocid | Toxicological | 2.5 |
| Levamisole | Toxicological | 6 |
| Lufenuron | Toxicological | 15 |
| Mebendazole | Toxicological | 12.5 |
| Monepantel | Toxicological | 30 |
| Morantel | Toxicological | 12 |
| Moxidectin | Toxicological | 3 |
| Netobimin | Toxicological | 5 |
| Nitroxinil | Toxicological | 5 |
| Omeprazole | Toxicological | 7 |
| Oxfendazole | Toxicological | 7 |
| Oxibendazole | Toxicological | 60 |
| Oxyclozanide | Toxicological | 30 |
| Permethrin | Toxicological | 10 |
| Phoxim | Toxicological | 3.75 |
| Piperazine | Toxicological | 250 |
| Praziquantel | Toxicological | 170 |
| Rafoxanide | Toxicological | 2 |
| Sisapronil | Toxicological | 1 |
| Teflubenzuron | Toxicological | 10 |
| Thiabendazole | Toxicological | 100 |
| Toltrazuril | Toxicological | 2 |
| Triclabendazole | Toxicological | 1.5 |
| Others | ||
| Bituminosulfonates | Toxicological | 1650 |
| Bromhexine | Toxicological | 5 |
| Bromide (sodium, potassium) | Toxicological | 400 |
| Bronopol | Toxicological | 20 |
| Butafosfan | Toxicological | 600 |
| Butorphanol tartrate | Toxicological | 300 |
| Butylscopolaminium bromide | Pharmacological | 10 |
| Chlorhexidine | Toxicological | 5 |
| Clodronic acid | Toxicological | 50 |
| Dembrexine | Toxicological | 20 |
| Denaverine hydrochloride | Toxicological | 30 |
| Enilconazole | Toxicological | 25 |
| Fenpipramide | Toxicological | 1 |
| Furosemide | Pharmacological | 2.5 |
| Hydrochlorthiazide | Toxicological | 25 |
| Isoeugenol | Toxicological | 75 |
| Melatonin | Pharmacological | 4 |
| Menbutone | Toxicological | 60 |
| 1‐Methyl‐2‐pyrrolidone | Toxicological | 250 |
| Natamycin | Toxicological | 60 |
| Parconazole | Toxicological | 80 |
| Piperonilbutoxide | Toxicological | 200 |
| Policresulen | Toxicological | 1000 |
| Sodium‐2‐fenoxy‐2‐methylpropanoate | Toxicological | 100 |
| Tiludronic acid | Toxicological | 21 |
| Toldimfos | Toxicological | 100 |
| Trichlormethiazide | Toxicological | 5 |
| Vetrabutinehydrochloride | Toxicological | 15 |
| Vincamine | Pharmacological | 9 |
ADI: acceptable daily intake; bw: body weight; TSV: toxicological screening value.
Suggested citation: EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) , Knutsen HK, Alexander J, Barregård L, Bignami M, Brüschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl‐Kraupp B, Hogstrand C, Nebbia CS, Oswald IP, Petersen A, Rose M, Roudot A‐C, Schwerdtle T, Vollmer G, Vleminckx C, Wallace H, Filipič M, Fürst P, O'Keeffe M, Penninks A, Van Leeuwen R, Baert K and Hoogenboom LR, 2018. Guidance update: methodological principles and scientific methods to be taken into account when establishing Reference Points for Action (RPAs) for non‐allowed pharmacologically active substances present in food of animal origin. EFSA Journal 2018;16(7):5332, 25 pp. 10.2903/j.efsa.2018.5332
Requestor: European Commission
Question number: EFSA‐Q‐2018‐00114
Panel members: Jan Alexander, Lars Barregård, Margherita Bignami, Beat Brüschweiler, Sandra Ceccatelli, Bruce Cottrill, Michael Dinovi, Lutz Edler, Bettina Grasl‐Kraupp, Christer Hogstrand, Laurentius (Ron) Hoogenboom, Helle Katrine Knutsen, Carlo Stefano Nebbia, Isabelle P Oswald, Annette Petersen, Martin Rose, Alain‐Claude Roudot, Tanja Schwerdtle, Christiane Vleminckx, Günter Vollmer and Heather Wallace.
Acknowledgements: The CONTAM Panel acknowledges all European competent institutions and other stakeholders that supported the data collection for the Comprehensive European Food Consumption Database.
Adopted: 14 June 2018
Notes
Regulation (EC) No 470/2009 of the European Parliament and of the Council of 6 May 2009 laying down Community procedures for the establishment of residue limits of pharmacologically active substances in foodstuffs of animal origin, repealing Council Regulation (EEC) No 2377/90 and amending Directive 2001/82/EC of the European Parliament and of the Council and Regulation (EC) No 726/2004 of the European Parliament and of the Council. OJ L 152, 16.6.2009, p. 11–22.
Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. OJ L 15, 20.1.2010, p. 1–72.
Council Directive 96/22/EC of 29 April 1996 concerning the prohibition on the use in stock farming of certain substances having a hormonal or thyrostatic action and of ß‐agonists, and repealing Directives 81/602/EEC, 88/146/EEC and 88/299/EEC. OJ L 125, 23.5.1996, p. 3–9.
A thyrostatic reduces (or stabilises) the production of thyroid hormones.
Regulation (EC) No 882/2004 of the European Parliament and of the Council of 29 April 2004 on official controls performed to ensure the verification of compliance with feed and food law, animal health and animal welfare rules. OJ L 191, 28.5.2004, p. 1–59.
Council Directive 96/23/EC of 29 April 1996 on measures to monitor certain substances and residues thereof in live animals and animal products and repealing Directives 85/358/EEC and 86/469/EEC and Decision 89/187/EEC and 91/664/EEC. OJ L 125, 23.5.1996, p. 10–32.
Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. OJ L 221, 17.8.2002, p. 8–36.
Commission Decision 2005/34/EC laying down harmonised standards for the testing for certain residues in products of animal origin imported from third countries. OJ L 16, 20.1.2005, p. 61–63.
A steroid is a type of organic compound, either natural or synthetic, containing a characteristic arrangement of 17 carbon atoms in four cycloalkane rings that are joined to each other, including three cyclohexane rings and one cyclopentane ring. Steroids vary by the functional groups attached to this four‐ring core and by the oxidation state of the rings.
Receptor: a structural protein molecule on the cell surface or within the cytoplasm that binds to a specific factor, such as a hormone, antigen or neurotransmitter (Dirckx, 1997).
Substances causing blood dyscrasias (aplastic anaemia), causing allergy (excluding skin sensitisation), that are high potency carcinogens or inorganic substances are excluded (see Section 2.3.1).
References
- Aquino M and Rosner G, 2018. Systemic contact dermatitis. Clinical Review in Allergy and Immunology, 10.1007/s12016-018-8686-z [DOI] [PubMed] [Google Scholar]
- Baeck M, Marot L, Nicolas JF, Pilette C, Tennstedt D and Goossens A, 2009. Allergic hypersensitivity to topical and systemic corticosteroids: a review. Allergy, 64, 978–994. 10.1111/j.1398-9995.2009.02038.x [DOI] [PubMed] [Google Scholar]
- Beack M and Goossens A, 2012. Systemic contact dermatitis to corticosteroids. Allergy, 67, 1580–1585. 10.1111/all.12041 [DOI] [PubMed] [Google Scholar]
- Dirckx JH, 1997. Stedman's concise medical & allied health Dictionary. Williams & Wilkins, Baltimore, USA. p. 1098. [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2013. Guidance on methodological principles and scientific methods to be taken into account when establishing Reference Points for Action (RPAs) for non‐allowed pharmacologically active substances present in food of animal origin. EFSA Journal 2013;11(4):3195, 24 pp. 10.2903/j.efsa.2013.3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA FEEDAP Panel (EFSA Panel on Additives and Products or Substances used in Animal Feed), 2012. Guidance for establishing the safety of additives for the consumer. EFSA Journal 2012;10(1):2537, 12 pp. 10.2903/j.efsa.2012.2537 [DOI] [Google Scholar]
- EFSA FEEDAP Panel (EFSA Panel on Products or Substances used in Animal Feed), Rychen G, Aquilina G, Azimonti G, Bampidis V, Bastos ML, Bories G, Chesson A, Cocconcelli PS, Flachowsky G, Gropp J, Kolar B, Kouba M, Lopez‐Alonso M, Lopez Puente S, Mantovani A, Mayo B, Ramos F, Saarela M, Villa RE, Wallace RJ, Wester P, Anguita M, Dujardin B, Galobart J and Innocenti ML, 2017. Guidance on the assessment of the safety of feed additives for the consumer. EFSA Journal 2017;15(10):5022, 17 pp. 10.2903/j.efsa.2017.5022 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2011. Scientific Opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379, 69 pp. 10.2903/j.efsa.2011.2379 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2012. Scientific Opinion on Exploring options for providing advice about possible human health risks based on the concept of Threshold of Toxicological Concern (TTC). EFSA Journal 2012;10(7):2750, 103 pp. 10.2903/j.efsa.2012.2750 [DOI] [Google Scholar]
- EFSA Scientific Committee , Hardy A, Benford D, Halldorsson T, Jeger M, Knutsen HK, More S, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Silano V, Solecki R, Turck D, Younes M, Aquilina G, Crebelli R, Gurtler R, Hirsch‐Ernst KI, Mosesso P, Nielsen E, vanBenthem J , Carfi M, Georgiadis N, Maurici D, Parra Morte J and Schlatter J, 2017. Scientific Opinion on the clarification of some aspects related to genotoxicity assessment. EFSA Journal 2017;15(12):5113, 25 pp. 10.2903/j.efsa.2017.5113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmi C and Srinivas CR, 2011. Systemic (allergic) contact dermatitis to diclofenac. Indian Journal of Dermatology, Venereology and Leprology, 77, 536. 10.4103/0378-6323.82424 [DOI] [PubMed] [Google Scholar]
- Thyssen JB and Maibach HI, 2008. Drug‐elicited systemic allergic (contact) dermatitis ‐ update and possible pathomechanisms. Contact Dermatitis, 59, 195–202. 10.1111/j.1600-0536.2008.01367.x [DOI] [PubMed] [Google Scholar]