

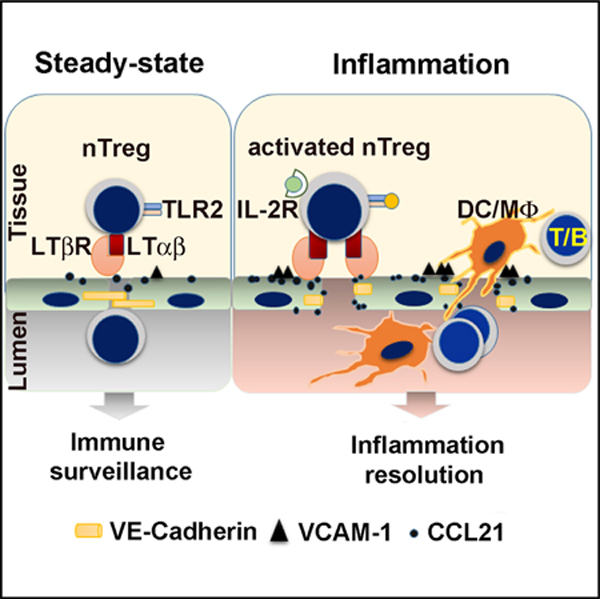
SUMMARY
Regulatory T cells (Tregs) express high levels of cell surface lymphotoxin alpha beta (LTα1β2) to activate the LT beta receptor (LTβR) on the lymphatic endothelial cells (LECs), modulating LEC adhesion molecules, intercellular junctions, and chemokines. We demonstrate a role for Tregs through this pathway to condition the permissiveness of lymphatic endothelia for transendothelial migration (TEM), thus gating leukocyte traffic. Human Tregs share the same property with murine Tregs. Activation of TLR2 on Tregs during inflammation specifically augments LTα1β2-LTβR signaling, which further enhances the permissiveness of LECs to facilitate TEM. The conditioning of endothelia may promote the resolution of inflammation by directing leukocytes out of tissues to lymphatic vessels and draining lymph nodes (dLNs). Thus, Tregs interact with lymphatic endothelia under homeostasis and inflammation and dictate endothelial permissiveness and gating mechanisms for subsequent leukocyte migration through endothelial barriers.
In Brief
Piao et al. demonstrate that Tregs condition lymphatic endothelial cells (LECs) to be more permissive for the transendothelial migration (TEM) of other leukocytes. Activation of Toll-like receptor 2 on Tregs during inflammation specifically augments Treg LTα1β2 expression to intensify LTβR signalling in LECs for enhanced immune cell TEM.
Graphical Abstract:
INTRODUCTION
The lymphotoxin alpha beta (LTα1β2) complex is located on the surface of activated lymphocytes. LTα1β2 is well characterized and crucial for lymphatic organ development and orchestration of immune responses. As it is member of the tumor necrosis factor (TNF) superfamily, surface LTα1β2 expression is maintained once induced by activation (Browning et al., 1997; Chiang et al., 2012; English et al., 1991). Importantly, LTα1β2 is found to be preferentially expressed and used by regulatory T cells (Tregs) for afferent lymphatic migration (Brinkman et al., 2016). Tregs express higher levels of LTα1β2 than naive or activated CD4 T cells and engage the LT beta receptor (LTβ)R on lymphatic endothelial cells (LECs) for afferent lymphatic transendothelial migration (TEM) (Piao et al., 2018). LTα1β2 expression on Tregs and LTβR expression on LECs are important for the suppressive function of Tregs to migrate to draining lymph nodes (dLNs) and enhance islet allograft survival (Brinkman et al., 2016; Zhang et al., 2009).
The two subunits of LTα1β2 form a transmembrane heterotrimer that interacts with the LTβR. Dysregulated expression or deficiency of any of the subunits has been linked to autoimmunity and inflammation. Cytokines or protein ligands that regulate LTα1β2 expression have been described in naive T cells (Schneider et al., 2004). However, the differential expression and regulation of LTα1β2 in Tregs or different immune cell subsets during inflammatory responses have not been well characterized. Although the induction of LTα1β2 in T cells by several distinct signals, including protein kinase C (PKC)-mediated Ets (E26 transformation-specific), nuclear factor κB (NF-κB) (p65/Rel), and Egr-1 (early growth response protein 1)/Sp1 (specific protein 1) promoter activation has been reported (Kuprash et al., 1996; Voon et al., 2004), functionally relevant inducers have not been clearly identified.
LTβR is widely expressed in blood and LECs, intestinal epithelial cells, dendritic cells (DCs), and lymph node (LN) stromal cells (Schneider et al., 2004). In LECs, LTβR regulates leukocyte afferent lymphatic TEM (Piao et al., 2018). Although LTβR signals to both classical and non-classical NF-κB pathways, in LECs it predominantly signals by both a constitutive and ligand-driven non-classical NF-κB-inducing kinase (NIK) pathway. Constitutive NIK activation in LECs is required for TEM and implicates its importance for all immune cell lymphatic recirculation. LTα1β2/LTβR ligand-driven NIK signaling on LECs triggers increased expression of migration molecules and chemokines, such as CCL21 or CXCL12, resulting in enhanced leukocyte TEM across LECs. Because Tregs preferentially and directly engage LTβR-NIK signaling, we investigated the downstream influence of Treg on LECs to regulate and gate the migration of other immune cells.
Tregs constitutively express the interleukin 2 receptor α chain (IL-2Rα; CD25) and rely on IL-2 for Foxp3 induction and Treg differentiation and maintenance (Sakaguchi et al., 2008). Other signals are likely important for peripheral Treg induction, activation, and function. Of note, Toll-like receptor 2 (TLR2) signaling affects Treg expansion and function (Liu et al., 2006; Nyirenda et al., 2011; Sutmuller et al., 2006). Paired with TLR1 or TLR6 to form heterodimeric receptor complexes on the cell surface, TLR2 recognizes lipoproteins from diverse microbial sources (Akira et al., 2006) and signals through TIRAP/MyD88 (TIR domain-containing adaptor protein/myeloid differentiation primary response gene 88) to activate mitogen-activated protein kinases (MAPKs) and NF-κB. Several endogenous TLR2 ligands have also been described, including hyaluronan (HA) (Krüger et al., 2010; Tesar et al., 2006), heat shock protein (HSP) 60, HSP Gp96 (Mkaddem et al., 2009), and high-mobility group box 1 (HMGB1) (Matsuoka et al., 2010). HA and HMGB1 are released from transplanted pancreatic islets and promote islet rejection by inducing inflammatory cytokine secretion by immune cells through TLR2 and TLR4 (Bollyky et al., 2012; Kruger et al., 2010€ ). Tregs, conventional T cells, and activated CD4 all reportedly express functional TLR2; yet, the effects of TLR2 on T cell immunity and suppression are diverse, and many reports are even contradictory (Komai-Koma et al., 2004; Liu et al., 2006; Nyirenda et al., 2011). Thus, the precise roles and mechanisms of TLR2 ligands on different T cell subsets and early graft rejection remain incompletely defined.
Here, we observed that IL-2R signals enhanced Treg LTα1β2 expression. In addition, TLR2 signaling specifically augmented IL-2-induced LTα1β2 expression on activated Treg. Enhanced LTα1β2 expression enabled Treg to condition the permissiveness of afferent LECs by LTβR signaling. Conditioned endothelia facilitated TEM and dLN trafficking of other leukocytes, including T cells, B cells, and myeloid cells. These interactions revealed a mechanism for Tregs to regulate endothelial gating and, thus, lymphoid trafficking from tissues to lymphatics and dLNs.
RESULTS
Tregs Express Higher LTα1β2 Than Other CD4 T Cells
LTα1β2 is expressed in activated T cells, although which signaling pathways lead to increased LTα1β2 expression has not been reported (Brinkman et al., 2016; Browning et al., 1997). We analyzed surface LTα1β2 expression on activated T cell subsets stimulated with IL-2 or anti-CD3 monoclonal antibody (mAb). IL-2 led to a dose-dependent increase of LTα1β2 on in-vitro-stimulated induced Tregs (iTregs) (Figure 1Ai), whereas activation by anti-CD3 ligation caused a modest LTα1β2 increase, which was further enhanced by anti-CD28 mAb co-stimulation or by stimulation together with IL-2 (Figure 1Aii). Notably, blocking IL-2R with anti-CD25 blocking mAb markedly diminished anti-CD3-activated LTα1β2 expression (Figure 1Aiii), suggesting CD3-mediated LTα1β2 is mostly and indirectly induced by CD3-triggered IL-2 secretion. Analysis of various other CD4 T cell subsets indicated that Tregs, especially iTregs, expressed the highest levels of LTα1β2 (Figure 1B).
Figure 1. LTα1β2 Is Induced by IL-2R and TCR Signaling in Tregs.
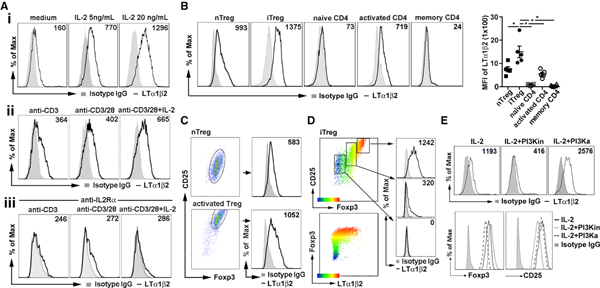
(A) Flow cytometry analysis of LTα1β2 expression on fluorescence-activated cell sorting (FACS)-sorted Foxp3-GFP+CD4+ iTregs treated with indicated dose of IL-2 (i); anti-CD3 (5 μg/mL) alone, together with anti-CD28 (1 μg/mL), or IL-2 (10 ng/mL) (ii); and 2 μg/mL anti-CD25 pretreatment prior to TCR activation as in (ii), for 16 h (iii).
(B) LTα1β2 expression of FACS-sorted Foxp3-GFP+CD4+ iTregs, Foxp3-GFP+CD44lowCD4+ natural Tregs (nTregs), Foxp3-GFP-CD44lowCD4+ naive CD4, Foxp3-GFP-CD44highCD4+ memory CD4 T cells, or IL-2 + anti-CD3/28-stimulated Foxp3-GFP-CD4+-activated CD4 from Foxp3-GFP reporter mice. Quantitative difference expressed as geometric mean fluorescence intensity (MFI) ± SEM for specific staining minus isotype immunoglobulin G (IgG) MFI.
(C) LTα1β2 expression of Foxp3-GFP+ CD4+nTregs sorted from Foxp3-GFP reporter mice, before and after stimulation with plate-bound anti-CD3 (5 μg/mL), anti-CD28 (1 μg/mL), and IL-2 (20 ng/mL) for 16 h.
(D) LTα1β2 expressed in iTreg subsets stratified by CD25 and Foxp3 expression.
(E) LTα1β2, CD25, and Foxp3 expression in iTregs treated with 500 nM PI3K inhibitor (PI3Ki) or 500 nM PI3K activator (PI3Ka) for 16 h.
Data are representative of 3 (A, C, and D) and 5 (B) independent experiments. *p < 0.05 by one-way ANOVA (B).
Freshly isolated unstimulated-thymus-derived natural Tregs (nTregs) expressed less LTα1β2 than iTregs; however, anti-CD3-activated nTreg expressed comparably high levels of CD25, Foxp3, and LTα1β2 as iTreg (Figure 1C). LTα1β2 was highly expressed on the CD25highFoxp3high iTreg fraction, at an intermediate level on CD25intFoxp3int, and minimally expressed on the CD25lowFoxp3 non-Treg portion (Figure 1D). Foxp3 expression has been linked to the T cell receptor (TCR)-phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway (Sauer et al., 2008), and inhibition of PI3K or Akt signaling increases Foxp3 expression. To determine if the LTα1β2 expression was directly dependent on Foxp3 or CD25 expression, we treated iTregs with a PI3K p110α inhibitor (PI3Ki) or pan-PI3K activator (PI3Ka). PI3Ki-treated Tregs showed enhanced Foxp3 and decreased CD25 and LTα1β2. PI3Ka treatment decreased Foxp3, enhanced LTα1α1b2, and caused no change in CD25 (Figure 1E). Together, the data suggested LTα1β2 was positively regulated by PI3K/Akt signaling, and LTα1β2 expression was not correlated directly with Foxp3 or CD25 expression.
IL-2R Activation Signals through NF-κB, and MAPK Pathways Promote LTα1β2 Expression in Tregs
By blocking downstream signaling molecules, we examined IL-2-mediated NF-κB and MAPK activation pathways for effects on Treg LTα1β2 expression. Blocking NF-κB activation abolished IL-2-driven LTα1β2 expression in freshly isolated nTregs (Figures 2A and 2Bi). Blocking the MAPKs c-Jun NH2-terminal kinase (JNK) and extracellular-signal-regulated kinase (ERK) inhibited LTα1β2 expression to a lesser extent. Immune blot analysis of the Treg cell subsets revealed that nTreg had delayed NF-κB and minimal JNK activation by IL-2 stimulation compared to iTregs (Figure 2C), indicating a specific role for the NF-κB pathway in LTα1β2 expression. In iTregs isolated after 5 days of stimulation culture, blocking ERK activity abolished IL-2mediated LTα1β2 increases, whereas NF-κB blockade had a less inhibitory effect on LTα1β2 expression (Figures 2A and 2Bii). Similar blocking patterns to iTregs were observed in nTregs activated by 5 days of culture with anti-CD3/28 and IL-2 (Figures 2A and 2Biii), with a stronger inhibition of expression of LTα1β2 by blocking MAPK than blocking NF-κB. NF-κB and MAPK activations were induced by IL-2 stimulation in iTregs and activated nTregs, with a similar pattern (Figure 2C), suggesting that these cells shared IL-2-stimulated NF-κB and ERK pathways for enhancing LTα1β2 expression.
Figure 2. IL-2 and TCR Signaling through NF-κB and MAPK Promote LTα1β2 Expression in T Cell Subsets.
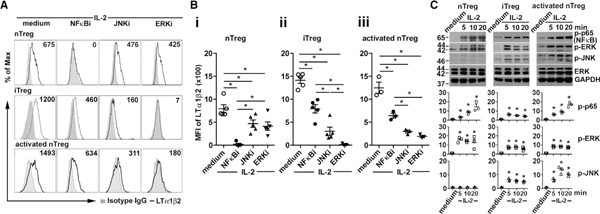
(A) Flow cytometry analysis of LTα1β2 expression on Foxp3-GFP+CD25+CD4+ nTregs, iTregs, or activated nTregs treated with IL-2 (20 ng/mL) together with NF-κB inhibitor (NFkBi; 5 μM), JNK inhibitor (JNKi; 2 μM), or ERK inhibitor (ERKi; 5 μM) at 37°C for 16 h.
(B) △MFI fold changes of LTα1β2 of nTregs (i), iTregs (ii), and activated nTregs (iii). Five (i and ii) or 3 (iii) independent experiments are shown. *p < 0.05 by one-way ANOVA.
(C) Immunoblots for NFκB and MAPKs activation in FACS-sorted nTregs, iTregs, and activated nTregs stimulated with IL-2 (20 ng/mL) as indicated. Representative blot shown. The relative band intensities (normalized to GAPDH, mean ± SEM) from three independent experiments shown under the panels. *p < 0.05 by one-way ANOVA.
Tregs Facilitate Migration of Other Leukocytes Across LECs by LTα1β2-LTβR
LTα1β2 activates LTβR signaling on LECs to increase CCL21 and VCAM-1, which are crucial for Treg TEM (Piao et al., 2018). We also demonstrated that the LTβR-NIK pathway in LECs is constitutively active and that constitutive activation is required for LEC TEM of other non-Treg leukocyte subsets, such as CD4 T cells and DCs. These observations lead us to question whether Tregs, by stimulating LTβR, directly regulated the TEM of other leukocytes across LECs. LEC layers were incubated with various T cell subsets for 3, 6, and 16 h; T cells were washed away; and then a second subset of leukocytes were applied to the LEC layers and permitted to migrate toward CCL19 for 3 h. The results showed that freshly isolated wild-type (WT) nTreg but not LTα−/−nTreg or naive CD4, augmented other CD4, CD8, and immature bone marrow dendritic cell (imBMDC) migration across the LECs (Figures 3Ai–3Aiii). WT iTreg, but not LTα−/−iTreg or activated non-Treg CD4, also enhanced other CD4, CD8, and imBMDC TEM (Figures 3Bi–3Biii). WT iTreg also promoted B cell, neutrophil, and peritoneal macrophage TEM (Figures S1Ai–S1Aiii). The influence of activated nTreg was also tested. Like iTreg, activated WT nTreg but not LTα−/−nTreg enhanced other CD4 TEM (Figure 3C). This conditioning or influence of Tregs on LECs was effective as early as 6 h after the iTreg-LEC interaction (Figures S1Bi and S1Bii), with a minimum of 0.5 × 105 of iTregs added to the LEC cultures (Figures S1Ci and S1Cii). nTreg as well as naive non-Treg CD4 did not affect LEC viability, as shown by the (3-(4, 5-Dimethylthiazol-2-yl)-2, 5 diphenyl tetrazolium bromide) MTT viability assay (Figures S1Di–S1Diii). Thus, migration promotion was not due to non-specific effects on LEC viability. Strikingly, the enhanced migration was dependent on nTreg or iTreg expression of LTα (Figures 3A and 3B) and LEC expression of LTβR (Figures 3D and 3E). The influence of Treg on LEC TEM regulation was further confirmed in vivo under both homeostatic and inflammatory conditions with the footpad migration assay. Injection of iTregs in the un-inflamed (Figure 3F) or inflamed (Figure 3G) footpads followed by the transfer of carboxyfluorescein succinimidyl ester (CFSE)-labeled non-Treg CD4 or CD8 T cells into the same footpad 1 h later promoted T cell migration into popliteal dLN. In contrast, injection of activated WT non-Treg CD4 or LTα−/−Tregs did not promote migration in vivo (Figures 3F and 3G). Importantly, as for mouse Tregs, only human Tregs facilitated other CD4 TEM across human LECs (Figure 3H), emphasizing the commonality of essential functions of Tregs for immune cell migration.
Figure 3. Tregs Promote CD4, CD8, and DC TEM by LTα1β2-LTβR.
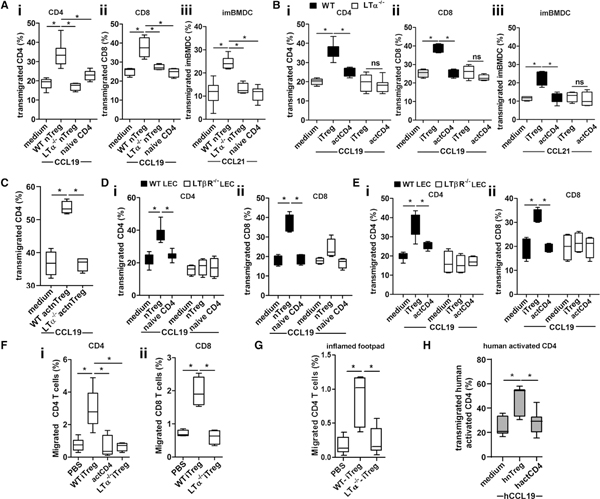
(A) A total of 1 × 105 wild-type (WT) or LTα−/−nTregs (Foxp3-GFP+CD25+CD4+) or naive CD4 (Foxp3-GFP-CD25-CD4+) were incubated with LEC layers in Boyden chambers for 16 h, T cells were removed by washing, and chambers were then loaded with 2 × 105 naive CD4 (i) or CD8 (ii) T cells or 1 × 105 BMDCs (iii) and migrated toward 50 ng/mL CCL19 (CD4 and CD8) or 200 ng/mL CCL21 (BMDC) for 3 h.
(B) WT or LTα−/−iTregs or activated non-Treg CD4 (actCD4) cells were incubated with LECs and washed, and then chambers were loaded for CD4 (i), CD8 (ii), and imBMDC (ii) TEM as in (A).
(C) WT or LTα−/−nTregs (Foxp3-GFP+CD25+CD4+) were stimulated in vitro with plate-bound anti-CD3 (5 μg/ml), anti-CD28 (1 μg/mL), IL-2 (20 ng/mL), and TGFβ1 (10 ng/mL) for 3 days and harvested, and 1 × 105 Tregs were added to LEC layers in Boyden chambers. After 16 h, Tregs were removed by washing, and chambers then loaded with 2 × 105 purified non-Treg CD4 T cells, which were migrated toward CCL19 for 3 h.
(D and E) WT or LTβR−/−LECs were incubated with 1 × 105 nTregs (D) or iTregs (E) for 16 h and washed, and then chambers loaded for TEM with non-Treg CD4 (i) or CD8 (ii) as in (A).
(F) A total of 1 × 106 iTregs or activated non-Treg CD4 T cells injected into hind footpads of WT C57BL/6J mice 1 h before injection of 2 × 106 CSFE-labeled non-Treg CD4 (i) or CD8 (ii) T cells. After 16 h, popliteal dLNs were analyzed for migrated cells by flow cytometry.
(G) Hind footpads of WT C57BL/6J mice intradermally injected (i.d.) with LPS (1 μg/10 μL PBS/footpad i.d.). After 3 h, 5 × 105 WT or LTα−/−iTregs was transferred to the footpads. After another 1 h, 2 × 106 CSFE-labeled non-Treg CD4 T cells was transferred to the same footpads. Sixteen hours later, CD4 T cell migration to the draining popliteal dLN was assessed.
(H) Activated human naive Tregs (hnTreg) or activated non-Treg CD4 (hactCD4) T cells were incubated with human dermal LEC layers in Boyden chambers for 16 h; T cells were removed by washing, and chambers were then loaded with 3 × 105 human activated non-Treg CD4 for 3-h TEM as in (A). (A–H) Representative of 3 independent experiments; quadruplicate wells for each condition. Data are presented as mean ± SEM, *p < 0.05 by one-way ANOVA.
Treg Engage LTα1β2-LTβR Pathways to Induce VCAM-1 and CCL21 on LECs and Lymphatic Vessels
Co-culture of LECs with WT iTregs, but not LTα−/−iTregs for 16 h induced strong NIK activity (Figure 4A). WT iTregs, but not LTα−/−iTregs, also increased LEC expression of VCAM-1 and CCL21 (Figure 4B) and decreased expression of tight junction proteins vascular endothelial (VE)-cadherin between LECs (Figure 4B). These data showed that Treg modulation of VCAM-1, CCL21, and VE-cadherin were LTα-dependent. Similarly, human naive Tregs expressed higher levels of LTα1β2 than effector CD4 T cells (Figure S2A) and regulated the expression of VCAM-1, CCL21, and VE-cadherin in human LECs (Figure S2B). In vivo Tregs induced increased CCL21 and VCAM-1 and decreased VE-cadherin expression on lymphatic vessels in an LTα-dependent manner (Figures 4C–4E). In concordance with the footpad migration assay (Figure 3F), intradermally injected WT iTregs migrated into the lymphatic vessel lumen from the tissue, whereas LTα−/−iTregs migrated much less efficiently into the lymphatics (Figure 4C). Thus, Treg LTα1β2 conditioned LECs to license TEM through molecular changes of chemokine expression and tight junction structures.
Figure 4. Treg LTα1β2 Activates LEC LTβR by the NIK Pathway to Induce VCAM-1, VE-cadherin, and CCL21.
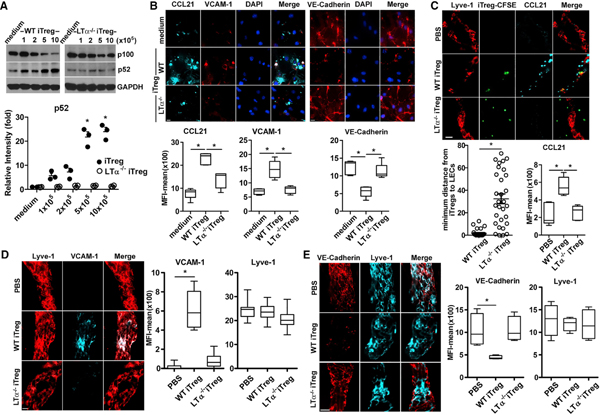
(A) Immunoblot for NIK activation and p100 conversion to p52. Primary LECs incubated with variable numbers of WT or LTα−/−iTregs. After 16 h, cells were lysed for immune blot. Relative band intensities (mean ± SEM) from three independent experiments are shown.
(B) Immunohistochemistry for CCL21, VCAM-1, and VE-cadherin expression in primary LECs incubated with WT or LTα−/−iTreg for 16 h. MFIs of 5 fields are shown as mean ± SEM, *p < 0.05 by one-way ANOVA.
(C–E) Ear whole-mount staining. Pinnae injected with 1 × 106 WT or LTα−/−CSFE-iTregs; after 16 h, ears were collected and stained for Lyve-1 and CCL21 (C), VCAM-1 (D), or VE-cadherin (E). Magnification is 60× (B, D, and E) or 20× (C); scale bar: 14 μm (B, D, and E) or 20 μm (C). Single lymphatic vessels, which are Lyve1+ (C and E). MFIs of 5 Lyve-1+ vessels are shown as mean ± SEM, *p < 0.05 by one-way ANOVA. Representative of 3 independent experiments (A–E). Four ears/two mice in each experiment (C–E).
TLR2 Signaling Preferentially Promotes LTα1β2 Expression and iTreg Migration
TLR2 further stimulates on TCR-activated non-Treg CD4 and CD8 T cells and regulates Treg proliferation and immune suppression (Liu et al., 2006). We tested whether TLR2 stimulation of various T cell subsets influenced LTα1β2 expression. iTregs, as noted above, expressed high levels of LTα1β2, which were markedly increased by IL-2. Importantly, TLR2 activation by either the TLR1/TLR2 agonist Pam3Cys-SK4 (P3C) or the TLR2/TLR6 agonist Pam2CSK4 (P2C) further increased the expression of LTα1β2 on iTregs (Figures 5A, S3A, and S3D). nTreg expression of LTα1β2 also increased in response to IL-2; in contrast, TLR2 stimulation did not enhance LTα1β2 expression (Figure 5A). To elucidate the different responses to TLR2 signaling between Treg subsets, nTregs were stimulated with anti-CD3/28 and IL-2 for 5 days followed by TLR2 stimulation, similar to how iTregs were differentiated from naive CD4. These conditions resulted in a marked increase of LTα1β2 expression by activated nTregs after TLR2 stimulation. Thus, activation of both iTregs and nTregs by IL-2R and TCR determined the response to TLR2 stimulation of LTα1β2 expression. Activated non-Treg CD4 did not demonstrate increased LTα1β2 expression in response to either IL-2 and/or TLR2 stimulation. Naive non-Treg CD4 expressed very little LTα1β2, which was also unaffected by IL-2 or TLR2 stimulation (Figure 5A). Similarly, activated human Tregs but not activated non-Treg CD4 effector T cells expressed high levels of TLR2 (Figure S3E), and TLR2 activation increased LTα1β2 expression on human Tregs but not on effector T cells (Figure S3F).
Figure 5. TLR2 Signaling Promotes iTreg LTα1β2 Expression and TEM.
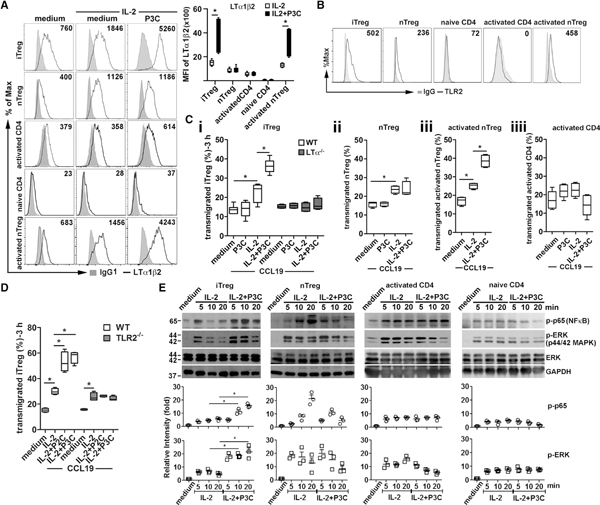
(A) iTregs, nTregs, activated non-Treg CD4, naive non-Treg CD4, and activated nTreg stimulated with IL-2 (20 ng/ml) and/or P3C (500 ng/ml) as indicated. After 16 h, cells were analyzed for LTαβ expression by flow cytometry (left panel). LTα1β2 MFI (right panel) of 5 independent experiments is shown. Mean ± SEM. *p < 0.05 by one-way ANOVA.
(B) Surface TLR2 expression of various T cell subsets analyzed by flow cytometry.
(C) WT or LTα−/−cells with the same stimulation as in (A) assessed for iTreg (i), nTreg (ii), activated nTreg (iii), and activated non-Treg CD4 (iiii) TEM to CCL19 (50 ng/mL).
(D) WT or TLR2−/−iTregs treated and assayed as in (A). Representative of 3 independent experiments with triplicate wells (C and D). *p < 0.05 by one-way ANOVA.
(E) iTregs, nTregs, activated non-Treg CD4, or naive non-Treg CD4 stimulated with IL-2 with or without P3C. After indicated times, cells were lysed and immunoblotted for NF-κB and MAPK activation. Representative blot shown. The relative band intensities (normalized to GAPDH, mean ± SEM) of three independent experiments shown under the panels. IL2 + P3C co-stimulation compared to IL-2 stimulation. *p < 0.05 by one-way ANOVA.
The different responses to the TLR2 ligand raised the possibility that the various CD4 subsets may express different levels of surface TLR2. iTregs and activated nTregs showed high levels of surface TLR2, whereas unstimulated nTregs, naive non-Treg CD4, and activated non-Treg CD4 had lower or minimal cell surface TLR2 (Figure 5B), suggesting that the differential responses to the TLR2 ligand among the T cell subsets was due to differences in the TLR2 expression. Notably, other TLR ligands, such as lipopolysaccharides (LPSs) (TLR4 agonist) and R848 (TLR7 agonist), had no effect on IL-2-stimulated LTα1β2 expression (Figures S3A–S3D), indicating an important role for TLR2 on LTα1β2 expression in Tregs.
These stimulated T cells were assessed for TEM to determine if increased LTα1β2 expression functionally enhanced their migration. IL-2 activation enhanced WT but not LTα−/−iTreg TEM, and WT but not LTα−/−iTreg migration was further enhanced by TLR2 co-stimulation (Figure 5Ci). Correlated with LTα1β2 expression, IL-2 increased resting nTreg migration, whereas TLR2 co-stimulation did not (Figure 5Cii), and TLR2 co-stimulation further increased activated nTreg migration (Figure 5Ciii). Activated non-Treg CD4 migration was not influenced by IL-2 or TLR2 co-stimulation (Figure 5Ciiii). For TLR2−/−iTregs, IL-2 promoted both TLR2−/−and WT TEM; however, TLR2 co-stimulation failed to increase TEM by TLR2−/−iTregs (Figure 5D).
We investigated which signaling pathways TLR2 engaged in T cell subsets stimulated with IL-2 by immunoblotting for classical NF-κB, MAPK, and PI3K-Akt activities. Classical NF-κB (p65) activation and ERK (p44/p42) phosphorylation were induced by IL-2 in all T cell subsets and were further enhanced by TLR2 co-stimulation in iTregs (Figure 5E) and activated nTregs but not in resting nTregs (Figure S3A), which was likely due to the higher levels of surface TLR2 expression in iTregs and activated nTregs (Figure 5B). IL-2 also induced JNK MAPK signaling in all T cell subsets except nTreg (Figures 2C and S4B), whereas TLR2 co-stimulation failed to promote JNK (p54/p46) phosphorylation in any of the T cell subsets. PI3K-Akt (Thr308) signaling was also induced by IL-2, but only in iTregs and activated nTregs (Figures S4A–S4C). Notably, Akt (Thr308) was constitutively activated in activated non-Treg CD4 (Sauer et al., 2008) and was weakly or not activated in nTreg or naive non-Treg CD4. TLR2 co-stimulation increased Akt (Thr308) in both activated nTregs and iTregs (Figures S4A–S4C). Because IL-2R-mediated LTα1β2 expression is controlled by NF-κB and MAPK signaling (Figure 2), together the results indicated that TLR2 augmented classical NF-κB, ERK, and PI3K-Akt (Thr308), but not JNK signaling, by IL-2R for increasing LTα1β2 expression in the Treg subsets.
Pancreatic Islets Mobilize iTregs by Enhancing LT expression
Pancreatic islets express endogenous TLR2 ligands, such as HMGB1 and HA (Matsuoka et al., 2010; Tesar et al., 2006). We hypothesize that transplanted pancreatic islets may trigger TLR2 signaling on local or patrolling Tregs to promote LTα1β2 expression and, thus, mobilize Treg into the dLNs and protect the graft (Zhang et al., 2009). To test this, we challenged iTregs with various doses of allogenic pancreatic islets for 16 h in vitro and found that LTα1β2 expression was dramatically increased (Figure 6A). Blocking with TLR2 inhibitor (2R9) (Piao et al., 2015) abolished the increase. In contrast, for TLR2−/−iTregs, the islets and TLR2 agonists failed to increase LTα1β2 expression (Figure 6B). WT iTreg, but not TLR2−/− iTreg, pre-treated with islets showed a significant increase in TEM in vitro (Figure 6C). To show modulation of LTα1β2 in vivo by islet-derived TLR2 ligands, we co-transferred WT or TLR2−/−nTregs with islets underneath the kidney capsule, and 2 days later, the nTregs within the islets were harvested and analyzed for LTα1β2 expression. Like the in vitro assay, higher LTα1β2 expression was observed on WT nTregs than on TLR2−/−nTregs (Figure 6D). Consistent with the need for increased expression of LTα1β2 to promote migration (Brinkman et al., 2016), WT but not TLR2−/−Tregs prolonged islet graft survival (median survival time: 34 versus 15 days, p < 0.05) (Figure 6E). Furthermore, TLR2−/−Treg migration into dLNs (Figure 6F) was also impaired. To test whether TLR2−/−iTregs had impaired suppressive capability, we analyzed in vitro immune suppression. Consistent with a previous report (Sutmuller et al., 2006), TLR2−/− iTregs retained in vitro immune suppressive function (Figure S5). Thus, islet-derived TLR2 ligands triggered TLR2 signaling on iTregs to enhance LTα1β2 expression, promote migration from the graft to the dLN, and prolong graft survival.
Figure 6. Pancreatic Islets Stimulate Treg LTα1β2 Expression and Migration by TLR2 Activation.
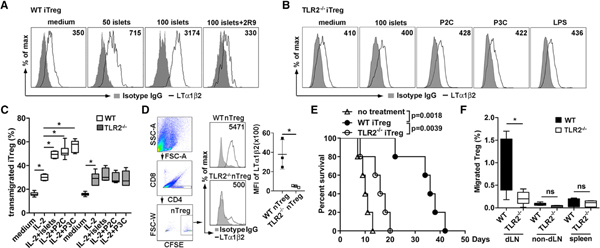
(A) A total of 5 × 105 iTregs from WT C57/BL6J mice were co-cultured with various doses of BALB/c pancreatic islets for 16 h. A total of 20 μM TLR2 inhibitory peptide (2R9) was used as control. Tregs were assessed for LTα1β2 expression by flow cytometry.
(B) A total of 5 × 105 TLR2−/−iTregs stimulated with 100 BALB/c islets, 50 ng/mL P2C, 500 ng/mL P3C, or 100 ng/mL LPS for 16 h prior to LTα1β2 staining.
(C) A total of 5 × 105 WT and TLR2−/−iTregs co-cultured with 100 BALB/c islets for 16 h, and then 2 × 105 of each iTreg assessed for TEM to 50 ng/mL CCL19 for 3 h.
(D) Flow cytometry analysis of LTα1β2 expression on CFSE-labeled WT or TLR2−/−nTregs co-transferred with islets underneath the kidney capsule. Two days after transplantation, the nTregs within the islets were harvested. n = 3 mice per group. Gating strategy and MFI of LTα1β2 expression shown.
(E) Islet survival after co-transfer of WT or TLR2−/−iTregs with transplanted islets. n = 5 mice/group.
(F) Migration of Tregs into the dLNs. After co-transfer of CFSE-labeled WT or TLR2−/−Tregs with transplanted islets, non-dLNs (inguinal LNs), dLNs (peri-renal), and spleen were harvested at day 4 for analysis of percentage of CFSE-Tregs among all CD4 T cells. n = 4 mice/group. Data representative of three independent experiments (A–D). *p < 0.05 by one-way ANOVA (C and F) or by Kaplan-Meier method using the log rank test (E).
DISCUSSION
Here, we showed that iTregs and activated nTregs each directly regulated the responses of LECs, thereby modifying the TEM of other leukocyte subsets. This activity was shared by human Tregs and LECs. With high levels of LTα1β2, transmigrating Tregs engaged LTβR on LECs to modulate endothelial cell responses and structures. The Treg-LEC interaction rapidly induced the cell adhesion molecule VCAM-1, downregulated the cell junction protein VE-cadherin, and promoted the TEM of Tregs and other immune cells, including non-Treg CD4, CD8, B cells, neutrophils, macrophages and immature DCs. Importantly, iTregs and activated nTregs expressed the highest level of TLR2, which shared NF-κB, ERK, MAPK, and PI3K-Akt pathways with IL-2R, and activation of which enhanced LTα1β2 expression, thus efficiently mobilizing immune cell trafficking during inflammation.
Treg trafficking between inflamed tissues and dLNs is essential for optimal immune suppression (Rudensky and Campbell, 2006; Zhang et al., 2009). Tregs use multiple mechanisms to curb autoimmunity and inflammation by suppressing differentiation, activation, proliferation, and effector function of multiple immune cell types, including non-Treg CD4 and CD8 T cells, B cells, and DCs (Fontenot et al., 2005; Rudensky and Campbell, 2006; Sakaguchi et al., 2008). However, it is uncertain how multiple suppressive mechanisms can be coordinated and deployed to restrict such a large variety of cell types and responses. Tregs are also required for the active regulation of appropriately timed effector cell migration and immune responses (Lund et al., 2008; Rogers et al., 2018). Thus, Tregs have dual roles in suppressing and ensuring appropriate immune responses. We recently demonstrated that Tregs use LTα1β2 to trigger LTβR-NIK signaling in LECs for Treg lymphatic TEM (Brinkman et al., 2016; Piao et al., 2018) and that LTβR-NIK signaling is constitutively active in vivo. The results here suggest that in homeostatic conditions, nTregs equipped with high levels of LTα1β2 patrol tissues and tissue lymphatic vessels and may maintain LEC LTβR constitutive activation. This activity may permit regulatory or effector T cell homing and recirculation to maintain immune surveillance of tissue-derived endogenous or exogenous antigens for suppression of immune responses to the former and induction of immune responses to the latter. During acute inflammation, activated Tregs expressing the highest levels of LTα1β2 are rapidly mobilized and regulate LECs for TEM of a variety of leukocyte subsets. Enhancing TEM may allow rapid antigen presentation between effector T cells and DCs in the dLN. Enhanced TEM of effector cells out of inflamed site may also resolve inflammation and decreased local tissue destruction (Figure S5). Although the fate or stability of Tregs during infection has been challenged because exposure to pro-inflammatory cytokines may convert Tregs into effector T cells (Zhou et al., 2009a, 2009b), their stability under physiologic and inflammatory conditions has been confirmed (Rubtsov et al., 2010) and the Tregs further restrain immunity (Koch et al., 2009). Together, these observations suggest Treg conditioning of endothelial TEM gating might be required at multiple levels of the immune response to regulate systemic trafficking in and out of tissues and lymphoid organs. This also suggests that this single Treg function might be able to regulate a multitude of cell types and functions. These relationships remain to be proven and may require the development of an LTα-flox strain, which does not currently exist.
The transcription factor Foxp3 is the signature marker of immune suppressive Treg (Fontenot et al., 2005), which includes thymic-derived nTregs and peripherally generated iTregs. nTregs and iTregs have been compared, and several differences in gene expression or signal pathways have been described. nTregs are demethylated at Treg-specific promoter and enhancer regions, whereas iTregs are predominately methylated in these regions (Kim et al., 2012), and demethylation correlates with the stability of Foxp3 gene expression (Polansky et al., 2010). We observed that activated nTregs resemble iTregs with respect to Foxp3 and CD25 expression; increased LTα1β2 expression after IL-2, CD3/CD28, and/or TLR2 stimulation; and use of NF-κB-, ERK-, and PI3K-Akt-related intracellular signaling pathways. IL-2/IL-2R induced the greatest increases in LTα1β2 expression, and we clarified that LTα1β2 is mainly induced by IL-2R signals after CD3/CD28 activation. Despite an equal expression of high-affinity IL-2R (Busse et al., 2010; Kim et al., 2001; Sakaguchi et al., 1995), iTregs and activated nTregs had distinct IL-2R signaling patterns compared to activated non-Treg CD4 cells. IL-2 induced transient classical NF-κB and ERK activation in iTregs or activated nTregs but persistent activation in activated non-Treg CD4 (Figure 5E); PI3K-Akt (Thr308) was inducible in iTregs and activated nTregs but was constitutively activated in activated non-Treg CD4; and iTregs favored p54 JNK activation, whereas activated non-Treg CD4 had preferential p46 JNK activation (Figure S4B). Compared to activated T cells, resting naive non-Treg CD4 had weak responses to IL-2, likely due to lower IL-2R expression (Busse et al., 2010; Kim et al., 2001; Sakaguchi et al., 1995). Blocking assays showed that LTα1β2 expression in resting nTregs was controlled by IL-2-activated NF-κB-p65 signaling and in iTregs or activated nTregs by MAPK signaling.
iTregs and nTregs that had been activated in vitro expressed the highest levels of LTα1β2, which engaged LTβR on LECs to then modulate LEC function and structure and promote Treg TEM. We tested the hypothesis that the Treg-LEC interaction and changes in LECs may facilitate the TEM of other immune cells. The results showed that Tregs conditioned LECs so that subsequent migration by non-Treg CD4, CD8, and DC were enhanced, which promoted effective egress out of inflamed tissues. Treg conditioning also mobilized B cell, neutrophil, and macrophage migration, suggesting that Tregs may control leukocyte lymphatic migration during many different inflammatory and immune responses. Treg conditioning of LECs is specific and LT-dependent because LTα−/−Treg failed to provide this activity. LTα1β2 triggered LTβR signaling on LEC-induced VCAM-1 and select chemokines, such as CCL21, CXCL12, and CCL2 (Piao et al., 2018), and decreased the intercellular tight junction protein VE-cadherin between LECs. These observations suggested that Tregs use LTα1β2 to stimulate the chemokine repertoire (including CCL19, CCL21, CXCL12, and CCL2) of LECs, and thus, cells expressing CCR7, CXCR4, or CCR2/4 can be recruited to the proximal lymphatic vessels and transmigrate across the conditioned LECs under the influence of Tregs. Activated Tregs may upregulate other receptors and ligands important for TEM that we have not yet investigated. An analysis of TLR2 surface expression showed that activated Tregs expressed the highest levels of TLR2, and TLR2 activation of Treg, but not activated or naive non-Treg CD4, dramatically increased LTα1β2 expression and further enhanced the ability of Tregs to condition LECs for increased leukocyte TEM. Activation of other TLRs such TLR4 or TLR7 had no such effect, showing the specific role for TLR2 signaling on immune cell migration.
TLR2 reportedly has diverse effects on T cells, including immune suppression, survival, and proliferation (Chen et al., 2009; Liu et al., 2006; Sutmuller et al., 2006). However, TLR2 signaling influence on Treg migration has not yet been reported. Here, we found increased TLR2 expression in activated Tregs, which are likely present during inflammatory events. Increased TLR2 expression correlated with the ability of TLR2 ligands plus IL-2 to induce NF-κB-p65, ERK (p44/42), and PI3K-Akt to increase LTα1β2 expression and regulate migration. Because Treg migration from islets to dLNs is important for allograft protection and survival (Zhang et al., 2009), we tested the role of TLR2 in vivo and proved it was important for mobilizing trafficking and maintaining suppressive function. Islet-derived endogenous ligands triggered TLR2 to increase LTα1β2 expression in vitro and in vivo. TLR2 activated Tregs, then conditioned LECs by LTβR, and licensed them for enhanced TEM of other leukocytes. TLR2-deficient Tregs failed to upregulate LTα1β2, enhance LEC TEM properties, migrate from islets to dLNs, or prolong islet graft survival, suggesting that TLR2 agonists may be tools for regulating Tregs and other immune functions in tissues for protection of the allograft. On the other hand, TLR2 on myeloid cells has also been targeted for anti-inflammatory effects in innate immunity (Meng et al., 2004; Piao et al., 2015). Activation of TLR2 on macrophages or DCs by TLR2 agonists may exaggerate innate immune responses and be directly detrimental to islet integrity so that manipulation of this complex system may be difficult (Leventhal and Schröppel, 2012; Krüger et al., 2010€ ). Nevertheless, the data provide insight into the immune regulatory function of TLR2 agonists for transplantation tolerance.
STAR✶METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jonathan S. Bromberg (jbromberg@som.umaryland.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal strains
Female C57BL/6J (WT, LTβR−/−, LTα−/−) (7–10 weeks), were purchased from The Jackson Laboratory (Bar Harbor, ME). Foxp3GFP mice on a C57BL/6 background were from Dr. A. Rudensky (Memorial Sloan Kettering Cancer Center) (Fontenot et al., 2005). All animal experiments were performed in accordance with Institutional Animal Care and Use Committee approved protocols.
Antibodies and Reagents
Antibodies against phosph-ERK1/2 (Thr202/Tyr204), total ERK1/2, phospho-JNK (Thr183/Tyr185), phospho-p65 (Ser536), phosphor-Akt (Thr308), NFκB-p100/p52, and GAPDH were obtained from Cell Signaling (San Diego, CA). Anti-CD25 (IL-2Rα, clone MAB702) was purchased from R&D Systems (Minneapolis, MN). Lipopolysaccharides (LPS, E. coli O55:B5, L4524), NFκB-IκBα inhibitor BAY11–7082 and ERK inhibitor U0126 were purchased from Sigma-Aldrich (St. Louis, MO); INK inhibitor JNK-IN-8 from Calbiochem (San Diego, CA); PI3K p110α inhibitor PIK2 and activator 740Y-P from Echelon Biosciences Inc. (Salt Lake City, UT) and from Cayman (Ann Arbor, MI), respectively; Pam3CSK4, Pam2CSK4, and R848 (Resiquimod) from InvivoGen (San Diego, CA).
Cells
C57BL/6 mouse (C57–6064L) or human (H-6064L) primary dermal LECs were from Cell Biologics, Inc. (Chicago, IL), and were cultured according to the manufacturer’s instructions in manufacturer-provided mouse endothelial cell medium supplemented with 5% FBS, 2 mM L-glutamine, 100 IU /mL penicillin, vascular endothelial growth factor, endothelial cell growth supplement, heparin, epidermal growth factor, and hydrocortisone.
METHOD DETAILS
T Cell Subset, B cell, Neutrophil, Peritoneal Macrophage, and Bone Marrow Derived Dendritic Cell Generation
cCD4, cCD8, and B cells from mouse LNs and spleens were isolated using mouse CD4+, CD8+, and B cell negative selection kits (StemCell Technologies, Inc., Cambridge, MA), and were cultured as previously described (1). Briefly CD4+CD25-Foxp3-GFP-WT or LTα−/−naive CD4 cells with > 98% purity were sorted using a FACS Aria II (BD Biosciences, San Jose, CA). The sorted Foxp3-GFP- naive CD4 T cells (5 × 104) were then cultured in flat-bottom 96-well plates for 5 days at 37 °C in 5% CO2, with IL-2 (20 μg/ ml, eBioscience, San Diego, CA), plate-bound anti-CD3ε mAb (1 μg/ mL, clone 145–2C11, eBioscience), and anti-CD28 mAb (1 μg/mL, clone 37.52, eBioscience) for activated CD4 T cells; and human TGFβ1 (10 μg/ mL, eBioscience) and anti-IL-4 (10 ng/mL, clone 11B11, eBioscience) for induced regulatory T cells (iTreg). At the end of the 5-day culture, the CD4+CD25+Foxp3-GFP+ iTregs and CD4+CD25+Foxp3-GFP- activated CD4 cells were FACS-sorted to > 99% purity. Cells were cultured in complete RPMI 1640 supplemented with 10% FBS, 1 mM sodium pyruvate, 2 mM L-glutamine, 100 IU/mL penicillin, 100 μg /mL streptomycin, non-essential amino acids and 2 × 105 M 2-ME (Sigma-Aldrich). Neutrophils were positively selected with Gr-1FITC (clone RB6–8C5) followed by FITC-selection kit (StemCell Technologies, Inc.) from mouse peripheral blood. Peritoneal macrophages were kind gift of Dr. Stefani Vogel, University of Maryland School of Medicine, Baltimore, MD) (Dobrovolskaia et al., 2003). In brief, 3 mL of 3% sterile fluid thioglycollate (Remel, San Diego, CA) was injected i.p. into wild-type C57/BL6J mice and four days later macrophages were harvested by peritoneal lavage with sterile saline. BMDCs were generated as described (Lutz et al., 1999). Briefly, bone marrow (BM) cells of wild-type mice were treated with 10 ng/ml GM-CSF (R&D Systems) for 10 days in Petri dishes, and the loosely attached cells were collected as immature BMDC (imBMDC). 100 ng/mL LPS was added for 48 hours to induce mature BMDC (mBMDC). CD11c+ DC were purified by CD11c positive selection kit (StemCell Technologies, Inc.).
Primary Murine Skin LEC Purification and Culture
Ears from 5–6-week-old wild-type and LTβR−/−C57BL/6J mice were collected and digested in 4 mg/ml collagenase D (Roche, Indianapolis, IN) at 37°C for 1 hour. The dissociated cells were washed and re-suspended in mouse endothelial cell medium (Cell Biologics, Inc) and plated in 6-well tissue culture plates for 1 weeks until the adherent cells reached confluency. The cells were dissociated with 0.25% trypsin-EDTA (Thermo Fisher Scientific, Waltham, MA) and the LEC were purified with Lyve-1-FITC (Novus Biologicals, Littleton, CO) using anti-FITC positive selection kit (StemCell Technologies, Inc.).
Human T Cell Purification and Culture
Human Treg were enriched from peripheral blood mononuclear cells with anti-CD25 microbeads (Miltenyi Biotec, Bergisch Glad-bach, Germany), and were sorted via FACSAria as nTreg (CD4+CD25highCD127-CD45RA+) and naive CD4 T cells (CD4+CD25-CD127+CD45RA+). The cells were expanded as previously described (Hippen et al., 2011). Briefly, purified nTreg were stimulated with irradiated KT64/86 cells cultured in XVivo-15 (BioWhittaker, Walkersville, MD) media containing 10% human AB serum (Valley Biomedical, Winchester, VA), Pen/Strep (Invitrogen, Carlsbad, CA), N-acetyl cysteine (USP), and recombinant IL-2 (300 IU/mL; Chiron, Emeryville, CA) for 14 days and frozen. When needed, frozen nTreg and naive CD4 were thawed and re-stimulated with anti-CD3/CD28 mAb-Dynabeads (Life Technologies, Carlsbad, CA) at 1:3 (cell to bead) plus recombinant IL-2 (300 U/ml) for 10 days before assay.
MTT Viability Assay
LEC were plated into 24-well tissue culture plates, incubated overnight, and treated with iTreg or naive non-Treg CD4 for 16 hours. The cells were washed and followed by 3-hour incubation with 0.5 mg/mL MTT (3-(4, 5-Dimethylthiazol-2-yl)-2, 5 diphenyl tetrazolium bromide) (Sigma-Aldrich). 50 μL DMSO was added to cells before reading OD at 540 nm.
In Vitro Suppression Assays
CD4+CD25+ iTreg and CD4+CD25- non-Treg T cells were FACS-sorted from the indicated strains. Treg were labeled with eFluor 670 (Molecular Probes, Eugene, OR). CFSE (Life Technologies)-labeled responder CD4+CD25- T cells (5 × 104 cells per well) were cultured with or without Treg at responder:Treg ratios of 1:0, 1:1, 2:1, 4:1 and 8:1 with irradiated (800 rad) syngeneic T cell-depleted splenocytes (5 × 104 cells per well) in 96-well U-bottom plates (Corning) and anti-CD3ε mAb (1 μg /mL, clone 145–2C11, eBioscience) for 3 days. Cells were collected, and cell division was measured by assessing relative CSFE dilution with an LSR Fortessa cytometer (Beckton Dickinson).
Flow Cytometry
For detection of LTα1β2, T cells were incubated with MOPC21(BioExCel, West Lebanon, NH) or mouse LTβRIg (recognizing both human and mouse LTα1β2, gift from Biogen Idec, Cambridge, MA) at 2 μg /mL for 60 minutes at 37 °C in HBSS with 2% FBS. Cells were washed, and then stained for 45 minutes at 4 °C with anti-CD16/32 (clone 93, eBioscience), followed with BV421-rat anti-mouse IgG1 (Biolegend) APC-eflour780-anti-mouse CD4 (GK1.5, eBioscience), and PE-anti-mouse CD25 (PC61.5, eBioscience); cells were then washed and fixed with 4% paraformaldehyde and run on an LSR Fortessa flow cytometer (BD Biosciences). Surface TLR2 staining used anti-mouse or human TLR2–APC (clone T2.5, BioLegend). Results were analyzed with FlowJo 8.7 (Treestar).
Immunoblotting
Cells were lysed in buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 10 mM NaF, 2 mM Na3VO4, 1 mM EDTA, 1 mM EGTA, 0.5% Triton X-100, 0.1 mM DTT, 1 mM PMSF and protease inhibitor cocktail (Roche). Protein in the cell extract was quantified using protein quantification kit (Bio-Rad, Philadelphia, PA) and 10 μg total protein was run on Novex WedgeWell 4%–20% Tris-Glycine Mini Gels (Invitrogen) and transferred to an Immobilon-P membrane (Bio-Rad). Membranes were probed with indicated antibodies. Quantification of blots was performed with ImageJ from the National Institutes for Health. Relative intensity of blots was normalized to housekeeping gene GAPDH and presented as fold induction to non-stimulation.
Lymphatic Transendothelial Migration
Transmigrations across endothelial cells were described previously (Brinkman et al., 2016; Xiong et al., 2017). Briefly the inverted transwell insert (24-well, Corning International) with 5 μm pore size was coated with 0.2% (w/v) gelatin (Bio-Rad) for 1 hour at 37 °C before loading with 1.0 × 105 primary skin LEC in 100 μL LEC medium. After 2 days, the LEC cell layers were treated with various conditions as noted in the text and figure legends prior to adding 2 × 105 T cells or DCs migrating cells in 100 μL to the upper chamber of transwell plate while the lower chamber contained mouse or human CCL19 or CCL21 (50 ng/mL or 200 ng/mL respectively, R&D systems). All cells or reagents were prepared in IMDM containing transferrin and 0.5% (w/v) fatty acid-free BSA (Gemini, West Sacramento, CA). T cells that migrated to the lower chamber after 3 hours at 37 °C were counted.
Immunohistochemistry
Murine or human LEC monolayers were stained for surface VCAM-1, VE-Cadherin or Lyve-1 with rat anti-mouse VCAM-1 (Clone 429, eBioscience) or mouse anti-human VCAM-1 (Clone STA, Biolegend), rat anti-mouse VE-Cadherin (Clone 11D4.1, BD Biosciences) or mouse anti-human VE-Cadherin (Clone BV9, Biolegend), goat-anti-mouse or anti-human CCL21 polyclonal antibody (R&D System), or rabbit anti-mouse Lyve-1 (70R-LR003, Fitzgerald, Acton, MA), then were fixed for 20 min at 4 °C with 4% (w/v) paraformaldehyde (Affymetrix, Santa Clara, CA), Cells were permeablized with PBS 0.2% (v/v) Triton X-100 (Sigma-Aldrich), and treated with 5% donkey serum for 30 min then incubated with listed primary antibodies for overnight at 4 °C. Samples were incubated with secondary antibodies conjugated with Alexa Fluor 448, 546 or 647 (1:400, Jackson ImmunoResearch, West Grove, PA) for 1 hour at 4 °C and mounted in Prolong Gold with DAPI (Thermo Fisher, P36931). Transwell membranes were transferred onto glass slides and visualized by fluorescent microscopy (Zeiss LSM 510 Meta and LSM5 Duo) with a 60× or 20× objective. Images were analyzed with Volocity version 6.3 software. Fluorescence intensity was measured as Mean Fluorescence Intensity (MFI) over the entire field.
Footpad and Whole-Mount Ear Migration Assays
Mice were anaesthetized and 1 × 106 Foxp3-GFP+CD25+ Tregs or activated Foxp3-GFP-CD25+ no-Tregs were injected intradermally into the footpads or ear pinnae in 20 μL PBS as we previously described. For footpad assays, draining popliteal LNs were collected 12 hours post injection and processed for flow cytometry. For ear pinnae assays, whole-mount ears were collected 16 hours post injection. Sample were fixed for 10 minutes at room temperature with 4% paraformaldehyde, then permeabilized with 0.5% Triton X-100 for 30 min at 4°C; Samples were blocked for 1 hour in 5% donkey serum in PBS containing 0.1% Triton X-100 and incubated with listed antibodies overnight at 4 °C. Anti-Lyve-1 (1:100, Fitzgerald 70R-LR003), anti-CCL21 (1:100, R&D Systems), or anti-VCAM-1 (1:100, eBioscience 429). secondary antibodies were conjugated to Alexa Fluor 488, 546 or 647 (1:400, Jackson ImmunoResearch) for 1 hour at 4 °C, mounted in Prolong Gold with DAPI (Thermo Fisher, P36931). Distance of T cells from lymphatic vessels calculated with minimum distance program in Volocity 6.3.
Islet Isolation and Transplantation
BALB/c islets were isolated as previously described (Zhang et al., 2009). Briefly, pancreata were perfused with collagenase P (Roche), harvested, digested, and purified over a discontinuous Ficoll (Sigma-Aldrich) gradient. 400 freshly isolated islets were handpicked and used for T cell stimulation or transplanted beneath the renal capsule of C57BL/6J recipients made diabetic (blood glucose > 300 mg/dl) by intraperitoneal injection of 200 mg/kg streptozotocin (Sigma-Aldrich) at least 1 week before transplant. Blood glucose < 150 mg/dl after transplantation was considered engraftment, and > 300 mg/dl was considered diabetic or graft rejection as appropriate.
QUANTIFICATION AND STATISTICAL ANALYSIS
Numerical data are presented as mean ± SEM. Asterisks mark data statistically different from the controls, with p values noted in the figure legends. A p value of < 0.05 was considered significant for one-way ANOVA and Student’s t tests using Prizm 5 (La Jolla, CA). The number of replicates is noted in the figure legends.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit-anti-NFκB122 p100/p52 | Cell Signaling Technology | Cat# 4882P; RRID:AB_10828354 |
| anti-phospho-NF-κB p65 (Ser536) (93H1) | Cell Signaling Technology | Cat#3033; RRID:AB_331284 |
| Rabbit-anti-GAPDH (clone 14C10) | Cell Signaling Technology | Cat# 2118; RRID:AB_561053 |
| Anti-phospho-JNK (Thr183/Tyr185) (81E11) | Cell Signaling Technology | Cat#4668; RRID:AB_2307320 |
| Anti-phospho-ERK (D13.14.4E) | Cell Signaling Technology | Cat#4370; RRID:AB_2315112 |
| Rabbit-anti-p44/42 MAPK (D137F5) | Cell Signaling Technology | Cat#4695; RRID:AB_390779 |
| Rabbit-anti-phospho-Akt (Thr308) (244F9) | Cell Signaling Technology | Cat#4056; RRID:AB_331163 |
| Anti-CD3ε (145–2C11) | eBioscience | Cat# 14–0031-86; RRID:AB_467051 |
| Anti-CD28 (CD28.2) | eBioscience | Cat# 16–0289-81; RRID:AB_468926 |
| Rat-anti-mouse LTβR (eBio38) | eBioscience | Cat# 16–5671-82; RRID:AB_763451 |
| Anti-rat IgG1 (RG11/39.4) | BioXCell | Cat# BE-0250; RRID: NA |
| Rat-anti-mouse VCAM-1 (429) | eBioscience | Cat# 553329; RRID:AB_394785 |
| Mouse-anti-human VCAM-1 (Sta) | Biolegend | Cat# 305802; RRID:AB_314558 |
| Rat-anti-mouse VE-Cadherin (BV13) | Biolegend | Cat# 138001; RRID:AB_10570497 |
| Mouse anti-human VE-Cadherin (BV9) | Biolegend | Cat# 348501; RRID:AB_10574464 |
| Anti-Rabbit IgG Alexa Flour 647 | Invitrogen | Cat# 1013785; RRID: NA |
| Anti-Rabbit IgG Alexa Flour 546 | Jackson ImmunoResearch | Cat# 705–546-147; RRID:AB_2340430 |
| Anti-Rabbit IgG Alexa Flour 488 | Jackson ImmunoResearch | Cat# 711–545-152; RRID:AB_2313584 |
| Anti-Rat IgG Alexa Flour 647 | Jackson ImmunoResearch | Cat# 712–605-153; RRID:AB_2340694 |
| Anti-Rat IgG CyTM3 | Jackson ImmunoResearch | Cat# 712–165-153; RRID:AB_2340667 |
| Anti-Goat IgG CyTM3 | Jackson ImmunoResearch | Cat# 705–165-003; RRID:AB_2340411 |
| Anti-Goat IgG Alexa Flour 647 | Jackson ImmunoResearch | Cat# 705–605-003; RRID:AB_2340436 |
| Anti-Mouse IgG Alexa Flour 647 | Jackson ImmunoResearch | Cat# 715–605-150; RRID:AB_2340862 |
| Anti- CXCL12 | eBioscience | Cat# 14–7992-81; RRID:AB_468513 |
| Anti-CD3e | Abcam | Cat#ab49943; RRID:AB_868900 |
| Anti-CD11c (HL3) | BD Bioscience | Cat# 553863; RRID:AB_395099 |
| Anti-mouse CCL21 polyclonal Goat IgG | R&D Systems | Cat# AF457; RRID:AB_207208 |
| Anti-human CCL21polyclonal Goat IgG | R&D Systems | Cat# AF366; RRID:AB_355327 |
| Rabbit-anti-Lyve-1 | Fitzgerald | Cat# 70R-LR003; RRID:AB_1287923 |
| Anti-CD16/32 (93) | eBioscience | Cat# 14–0161-82; RRID:AB_467133 |
| Anti-CD4-APC-eflour780 (RM4–5) | eBioscience | Cat# 47–0042-82; RRID:AB_1272183 |
| Rat-anti-CD44-APC (IM7) | eBioscience | Cat# 17–0441-82; RRID:AB_469390 |
| Rat-anti-CD25-PE (PC61.5) | eBioscience | Cat# 12–0251-83; RRID:AB_465608 |
| Rat-anti-VCAM-1-Alexa Flour 647(429) | eBioscience | Cat# 14–1061-82; RRID:AB_467419 |
| Anti-mouse IgG1-BV421 (RMG1–1) | Biolegend | Cat# 406616; RRID:AB_2562234 |
| Anti-human or mouse TLR2 (T2.5) | Abcam | Cat# ab16894; RRID: AB_443530 |
| Rat-anti-LTβR-PE (3C8) | eBioscience | Cat# 12–5671-82; RRID: NA |
| Rat-anti-mouse Gr-1-FITC (RB6–8C5) | Biolegend | Cat#108406; RRID: AB_313371 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LTβRIg | Biogen Idec | gift |
| 4H-isoquinoline-1,3-dione (NIKi) | Enamine LLC | Cat# EN300–15352 |
| BAY-11–7082 | Sigma-Aldrich | Cat# B5556–10MG |
| U0126 | Sigma-Aldrich | Cat# 662005–1MG |
| JNK-IN-8 | Calbiochem | Cat# 1410880–22-6 |
| PI3K p110α inhibitor (PIK2) | Echelon Biosciences Inc. | Cat#B-0304 |
| PI3K activator (740Y-P) | Cayman | Cat# 22598 |
| Lipopolysaccharides (LPS, E. coli O55:B5) | Sigma-Aldrich | Cat# L4524–5MG |
| Pam3CSK4 | InvivoGen | Cat # tlrl-pms |
| Pam2CSK4 | InvivoGen | Cat # tlrl-pm2s-1 |
| Resiquimod (R848) | InvivoGen | Cat# tlrl-r848–5 |
| Recombinant mouse M-CSF | R&D Systems | Cat# 416-ML-050 |
| Recombinant CCL19 | R&D Systems | Cat# 440-M3–025 |
| Recombinant CCL21 | R&D Systems | Cat# 457–6C-025 |
| human TGF-β1 | eBioscience | Cat# 14–8348-62 |
| Recombinant mouse IL-2 | eBioscience | Cat# 14–8021-64 |
| 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliym bromide (MTT) | Sigma-Aldrich | Cat# M2128–250MG |
| Carboxyfluorescein succinimidyl ester (CFSE) | Invitrogen | Cat# C34570 |
| Rat Ig G1 (MOPC21) | BioXCell | Cat# BE0083 |
| Collagenase D | Roche | Cat# 11088866001 |
| Collagenase P | Roche | Cat# 11213857001 |
| Critical Commercial Assays | ||
| mouse CD11c positive selection kit | StemCell Technologies, Inc. | Cat# 18758 |
| mouse FITC positive selection Kit | StemCell Technologies, Inc. | Cat# 17682 |
| mouseCD4+ T cell isolation kit | StemCell Technologies, Inc. | Cat# 19852 |
| mouse CD8+ T cell isolation kit | StemCell Technologies, Inc. | Cat# 19853 |
| mouse B cell isolation kit | StemCell Technologies, Inc. | Cat# 19854 |
| Experimental Models: Cell Lines | ||
| Primary mouse dermal LEC | Cell Biologics, Inc. | Cat# C57–6064 L |
| Primary human dermal LEC | Cell Biologics, Inc. | Cat# H-6064 L |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J WT | The Jackson Laboratory | JAX: 000664 |
| Mouse: B6.129S2-Ltatm1Dch/J | The Jackson Laboratory | JAX: 002258 |
| Mouse: C57BL/6J Foxp3GFP | (Fontenot et al., 2005) | N/A |
| Software and Algorithms | ||
| Volocity 6.3 | Quorum Technologies | http://quorumtechnologies.com/ |
| FlowJo 8.7 | FlowJO, LLC | https://www.flowjo.com |
| Prism 5 | GraphPad Software, Inc | https://www.graphpad.com |
Highlights.
Tregs modulate lymphatic endothelial cells (LECs) for transmigration of other cells
IL-2R stimulates NFκB and MAPK pathways to promote lymphotoxin expression on Tregs
Tregs stimulate LEC LTβR to increase VCAM-1 and CCL21 and decrease VE-cadherin
TLR2 signaling on activated Tregs promotes lymphotoxin expression and Treg migration
ACKNOWLEDGMENTS
This work was supported by NIH grant RO1 AI062765 to J.S.B., the Maryland Living Legacy Foundation to J.S.B. and W.P., R01 AI126596 and R01 HL141815 to R.A., RO1 CA72669 to B.R.B., and R01 HL11879 and P01 CA 065493 to B.R.B. and K.L.H. We thank Dr. Wei Chao (Department Anesthesiology, UMB) for the TLR2KO mice and Dr. Xiaoxuan Fan from the UMB-Flow Core Facility for his excellent help with flow cell sorting and data analysis.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j. celrep.2019.12.083.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akira S, Uematsu S, and Takeuchi O (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. [DOI] [PubMed] [Google Scholar]
- Bollyky PL, Bogdani M, Bollyky JB, Hull RL, and Wight TN (2012). The role of hyaluronan and the extracellular matrix in islet inflammation and immune regulation. Curr. Diab. Rep. 12, 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman CC, Iwami D, Hritzo MK, Xiong Y, Ahmad S, Simon T, Hippen KL, Blazar BR, and Bromberg JS (2016). Treg engage lymphotoxin beta receptor for afferent lymphatic transendothelial migration. Nat. Commun. 7, 12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning JL, Sizing ID, Lawton P, Bourdon PR, Rennert PD, Majeau GR, Ambrose CM, Hession C, Miatkowski K, Griffiths DA, et al. (1997). Characterization of lymphotoxin-alpha beta complexes on the surface of mouse lymphocytes. J. Immunol. 159, 3288–3298. [PubMed] [Google Scholar]
- Busse D, de la Rosa M, Hobiger K, Thurley K, Flossdorf M, Scheffold A, and Höfer T (2010). Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc. Natl. Acad. Sci. USA 107, 3058–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Davidson TS, Huter EN, and Shevach EM (2009). Engagement of TLR2 does not reverse the suppressor function of mouse regulatory T cells, but promotes their survival. J. Immunol. 183, 4458–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang EY, Kolumam G, McCutcheon KM, Young J, Lin Z, Balazs M, and Grogan JL (2012). In vivo depletion of lymphotoxin-alpha expressing lymphocytes inhibits xenogeneic graft-versus-host-disease. PLoS One 7, e33106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, Ren T, Cody MJ, Michalek SM, Rice NR, and Vogel SN (2003). Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR “homotolerance” versus “heterotolerance” on NF-kappa B signaling pathway components. J. Immunol. 170, 508–519. [DOI] [PubMed] [Google Scholar]
- English BK, Weaver WM, and Wilson CB (1991). Differential regulation of lymphotoxin and tumor necrosis factor genes in human T lymphocytes. J. Biol. Chem. 266, 7108–7113. [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, and Rudensky AY (2005). Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22, 329–341. [DOI] [PubMed] [Google Scholar]
- Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, McKenna DH, Bromberg JS, Levine BL, Riley JL, et al. (2011). Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci. Transl. Med. 3, 83ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HP, Kelly J, and Leonard WJ (2001). The basis for IL-2-induced IL-2 receptor alpha chain gene regulation: importance of two widely separated IL-2 response elements. Immunity 15, 159–172. [DOI] [PubMed] [Google Scholar]
- Kim YC, Bhairavabhotla R, Yoon J, Golding A, Thornton AM, Tran DQ, and Shevach EM (2012). Oligodeoxynucleotides stabilize Helios-expressing Foxp3+ human T regulatory cells during in vitro expansion. Blood 119, 2810–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, and Campbell DJ (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai-Koma M, Jones L, Ogg GS, Xu D, and Liew FY (2004). TLR2 is expressed on activated T cells as a costimulatory receptor. Proc. Natl. Acad. Sci. USA 101, 3029–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger B, Yin N, Zhang N, Yadav A, Coward W, Lal G, Zang W, S Heeger P, Bromberg JS, Murphy B, and Schröppel B (2010). Islet-expressed TLR2 and TLR4 sense injury and mediate early graft failure after transplantation. Eur. J. Immunol. 40, 2914–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuprash DV, Osipovich OA, Pokholok DK, Alimzhanov MB, Biragyn A, Turetskaya RL, and Nedospasov SA (1996). Functional analysis of the lymphotoxin-beta promoter. Sequence requirements for PMA activation. J. Immunol. 156, 2465–2472. [PubMed] [Google Scholar]
- Leventhal JS, and Schröppel B (2012). Toll-like receptors in transplantation: sensing and reacting to injury. Kidney Int. 81, 826–832. [DOI] [PubMed] [Google Scholar]
- Liu H, Komai-Koma M, Xu D, and Liew FY (2006). Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc. Natl. Acad. Sci. USA 103, 7048–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JM, Hsing L, Pham TT, and Rudensky AY (2008). Coordination of early protective immunity to viral infection by regulatory T cells. Science 320, 1220–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz MB, Kukutsch N, Ogilvie AL, Rössner S, Koch F, Romani N, and Schuler G (1999). An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223, 77–92. [DOI] [PubMed] [Google Scholar]
- Matsuoka N, Itoh T, Watarai H, Sekine-Kondo E, Nagata N, Okamoto K, Mera T, Yamamoto H, Yamada S, Maruyama I, et al. (2010). High-mobility group box 1 is involved in the initial events of early loss of transplanted islets in mice. J. Clin. Invest. 120, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G, Rutz M, Schiemann M, Metzger J, Grabiec A, Schwandner R, Luppa PB, Ebel F, Busch DH, Bauer S, et al. (2004). Antagonistic antibody prevents toll-like receptor 2-driven lethal shock-like syndromes. J. Clin. Invest. 113, 1473–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mkaddem SB, Werts C, Goujon JM, Bens M, Pedruzzi E, Ogier-Denis E, and Vandewalle A (2009). Heat shock protein gp96 interacts with protein phosphatase 5 and controls toll-like receptor 2 (TLR2)-mediated activation of extracellular signal-regulated kinase (ERK) 1/2 in post-hypoxic kidney cells. J. Biol. Chem. 284, 12541–12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyirenda MH, Sanvito L, Darlington PJ, O’Brien K, Zhang GX, Constantinescu CS, Bar-Or A, and Gran B (2011). TLR2 stimulation drives human naive and effector regulatory T cells into a Th17-like phenotype with reduced suppressive function. J. Immunol. 187, 2278–2290. [DOI] [PubMed] [Google Scholar]
- Piao W, Shirey KA, Ru LW, Lai W, Szmacinski H, Snyder GA, Sundberg EJ, Lakowicz JR, Vogel SN, and Toshchakov VY (2015). A Decoy Peptide that Disrupts TIRAP Recruitment to TLRs Is Protective in a Murine Model of Influenza. Cell Rep. 11, 1941–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao W, Xiong Y, Famulski K, Brinkman CC, Li L, Toney N, Wagner C, Saxena V, Simon T, and Bromberg JS (2018). Regulation of T cell afferent lymphatic migration by targeting LTβR-mediated non-classical NFκB signaling. Nat. Commun. 9, 3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polansky JK, Schreiber L, Thelemann C, Ludwig L, Krüger M, Baum-grass R, Cording S, Floess S, Hamann A, and Huehn J (2010). Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J. Mol. Med. (Berl.) 88, 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers MC, Lamens KD, Shafagati N, Johnson M, Oury TD, Joyce S, and Williams JV (2018). CD4+ Regulatory T Cells Exert Differential Functions during Early and Late Stages of the Immune Response to Respiratory Viruses. J. Immunol. 201, 1253–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, and Rudensky AY (2010). Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudensky AY, and Campbell DJ (2006). In vivo sites and cellular mechanisms of T reg cell-mediated suppression. J. Exp. Med. 203, 489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, and Toda M (1995). Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1164. [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, and Ono M (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. [DOI] [PubMed] [Google Scholar]
- Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O’Connor E, et al. (2008). T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. USA 105, 7797–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider K, Potter KG, and Ware CF (2004). Lymphotoxin and LIGHT signaling pathways and target genes. Immunol. Rev. 202, 49–66. [DOI] [PubMed] [Google Scholar]
- Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, Joosten LA, Akira S, Netea MG, and Adema GJ (2006). Toll-like receptor 2 controls expansion and function of regulatory T cells. J. Clin. Invest. 116, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesar BM, Jiang D, Liang J, Palmer SM, Noble PW, and Goldstein DR (2006). The role of hyaluronan degradation products as innate alloimmune agonists. Am. J. Transplant. 6, 2622–2635. [DOI] [PubMed] [Google Scholar]
- Voon DC, Subrata LS, Karimi M, Ulgiati D, and Abraham LJ (2004). TNF and phorbol esters induce lymphotoxin-beta expression through distinct pathways involving Ets and NF-kappa B family members. J. Immunol. 172, 4332–4341. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Brinkman CC, Famulski KS, Mongodin EF, Lord CJ, Hippen KL, Blazar BR, and Bromberg JS (2017). A robust in vitro model for trans-lymphatic endothelial migration. Sci. Rep. 7, 1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Schröppel B, Lal G, Jakubzick C, Mao X, Chen D, Yin N, Jessberger R, Ochando JC, Ding Y, and Bromberg JS (2009). Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity 30, 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout S, Jeker LT, and Bluestone JA (2009a). Plasticity of CD4(+) FoxP3(+) T cells. Curr. Opin. Immunol. 21, 281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martıńez-Llordella M, Ashby M, Nakayama M, Rosenthal W, and Bluestone JA (2009b). Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 10, 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.