

Abstract
| Draft Endorsed by the FEEDAP Panel | 22 March 2017 |
| Submitted for public consultation | 6 April 2017 |
| End of public consultation | 31 May 2017 |
| Adoption by the FEEDAP Panel | 26 September 2017 |
| Entry into force | 1 May 2018 |
Abstract
This guidance document is intended to assist the applicant in the preparation and the presentation of an application, as foreseen in Article 7.6 of Regulation (EC) No 1831/2003, for the authorisation of additives for use in animal nutrition. It specifically covers the assessment of the safety for the target species.
Keywords: guidance, safety, target species
Short abstract
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2017.EN-1304/full
Background and Terms of Reference
Regulation (EC) No 1831/20031 establishes the rules governing the Community authorisation of additives for use in animal nutrition. Moreover, Regulation (EC) No 429/20082 provides detailed rules for the implementation of Regulation (EC) No 1831/2003 as regards the preparation and the presentation of applications and the assessment and the authorisation of feed additives.
The Panel on Additives and Products or Substances used in Animal Feed (FEEDAP Panel) has adopted a series of guidance documents which aim at complementing Regulation (EC) No 429/2008 to support applicants in the preparation and submission of technical dossiers for the authorisation of additives for use in animal nutrition according to Regulation (EC) No 1831/2003.
The European Food Safety Authority (EFSA) asked its FEEDAP Panel to:
identify from the current guidance documents, those that need to be updated, taking into consideration the most recent scientific developments and the experience gained in the assessment of feed additives;
update the guidance documents in need of revision accordingly; this activity can be conducted in different rounds of activities on the basis of the priorities identified and on the feasibility of the revision according the resources available;
taking into account the sensitivity and the relevance of some of the guidance documents under revision and the entity of the revision itself (e.g. substantial or not), consider initiatives like preparatory info‐sessions or public consultations of the draft guidance documents. The relevant comments received in either step will have to be considered and addressed if appropriate in the final version of the guidance documents.
The first of the terms of reference was addressed by a statement of the EFSA FEEDAP Panel (2016), in which it was identified the need to update most of the guidance documents that it produced and set priorities for this update.
This output addresses the second and third terms of reference with regard to the update of the guidance documents dealing with the assessment of the safety of feed additives for the target species. This guidance document underwent a public consultation (EFSA, 2017).
Scope of the guidance
This guidance document is part of a series of documents intended to assist the applicant in the preparation and the presentation of its application for authorisation of a feed additive, as foreseen in Article 7.6 of Regulation (EC) No 1831/2003. This document does not substitute for the obligation of an applicant to comply with the requirements of Regulation (EC) No 1831/2003 and its implementing rules.
Applicants should justify the omission from the dossier of any data or any deviations from the requirements detailed in this guidance.
1. Introduction
Studies involving animals should respect the rules on animal welfare laid down by European Union (EU) legislation, particularly those listed in Directive 63/2010/EU, and they should not be repeated if available elsewhere.3 The use of methods refining or replacing the tests using experimental animals or reducing the number of animals used in these tests shall be encouraged. Such methods must provide the same level of assurance as the methods they aim to replace.
For certain additives, safety for the target animals can be presumed without the need for specific information. For all other additives, safety for the target animals can be assessed by extensive literature searches for studies on target animals, toxicity data (either existing or new) from repeated dose studies in laboratory animals or tolerance studies in target animals.
2. Additives for which safety can be presumed without additional studies
For the following additives, safety for the target animals can be presumed without the need for additional studies:
additives for which no exposure of the target animals to the active substance (or related substances, including contaminants) will occur. Appropriate analytical data to determine that the active substance (or related substances) is not detectable in feed at the time of feeding should be provided.
silage additives where it can be demonstrated that the active substance(s) and/or agent(s) occur as normal constituents of silage and use of the additive does not substantially increase their concentration compared to silage prepared without use of the additive (i.e. where there is no substantial change in exposure).
microorganisms satisfying the requirements of the qualified presumption of safety (QPS) approach to safety assessment (EFSA, 2007; EFSA BIOHAZ Panel, 2017), or when its biology is sufficiently well known to allow pathogenic/toxigenic strains to be excluded by direct testing, provided that all other added components of the additive do not raise safety concerns.
nutritional additives assessed and authorised following the provisions of Regulation (EC) No 1831/2003.
-
nutritional additives not already authorised:
-
1
– when the active substance is sufficiently purified. A product will be considered as sufficiently purified if the unidentified fraction would not contribute to more than 1% (in dry matter (DM)) when the inclusion rate does not exceed 1,000 mg additive/kg complete feed. Higher inclusion rates would need a higher degree of purity (e.g. 10,000 mg additive/kg feed would correspond to 0.1%).
-
2
– when the additive is produced by fermentation using a production organism that (i) satisfies the requirements of the QPS approach to safety assessment or (ii) is a genetically modified microorganism (GMM) for which the recipient strain is considered by EFSA to qualify for the QPS approach to safety assessment and for which the molecular/genetic characterisation does not give rise to concern.
-
1
3. Extensive literature search for studies with target animals
An extensive literature search may provide information on the safety of the feed additive under the proposed conditions of use. The analysis of these data must establish that the active substance(s)/agent(s) in literature studies is (are) identical to that under application or, if not, would still allow conclusions on the additive under application to be made; for additives produced by fermentation, identity includes the production strain. For additives consisting of a mixture, the extensive literature search should cover all the components of the mixture. The concentration of the active substance/agent in feed should preferably exceed or at least cover that proposed in the application. The target species covered in the literature search should be relevant to the application. Application level, replicates, duration, and zootechnical and clinical endpoints measured should allow a conclusion on the absence of adverse effects. This may be achieved by the consideration of data from a number of independent studies. The literature search should also cover all available toxicological endpoints including genotoxicity.
Relevant information sources should be searched in a structured manner. The applicant should make reasonable efforts to locate all sources of relevant information and provide reasons for the selection of such sources. Bibliographic databases (including at least agricultural/aquacultural and medical/veterinary databases) which record documents such as journals, reports, conference proceedings and books should be searched. In addition, the search should consider sources other than bibliographic databases, such as reference lists of full‐text journal articles (e.g. reviews), websites of conferences or organisations.
Applicants should follow the recommendations of the ‘Technical manual for performing electronic literature searches in food and feed safety’ when performing the searches and documenting its outcome. Moreover, applicants are encouraged to refer to Appendix D of the ‘Tools for critically appraising different study designs, systematic review and literature searches’ for assessing the quality of the search.
The search methodology must be documented and reported in detail to ensure transparency and enable the evaluation and replication of the strategy. The following must be reported:
For database searches:
-
1
– the name of the database and the service provider used;
-
2
– the date of the search and the date range searched;
-
3
– any limits placed on the search such as language or publication status;
-
4
– the full search strategy (all terms and set combinations) and the number of records retrieved.
For sources other than bibliographic databases:
-
Websites and journal table of contents
-
1
– the name of the resource (i.e. website name, the journal name in case of searching in specific tables of contents);
-
2
– the URL (uniform resource locator, the internet address);
-
3
– the date on which the search was conducted and the date range of the search, or the dates, volumes and issues in the case of table of contents;
-
4
– the method of searching, e.g. browsing, using the search engine or scanning tables;
-
5
– any limits applied to the search (e.g. publication types);
-
6
– the search terms used and the number of relevant summary records or full‐text documents retrieved.
-
1
-
References lists
-
1
– the bibliographic details of the documents whose reference lists were scanned;
-
2
– the number of relevant bibliographic references retrieved.
-
1
The extensive literature search should cover at least the last 20 years. The list of relevant references included should be compiled in a reference management software and provided in.RIS format. Copies of the relevant papers should be provided. The applicant must ensure that terms and conditions asserted by any copyright holder of publications or information submitted to EFSA are fully satisfied. The applicant should consult with copyright licensing authorities (i.e. at national level) for guidance on purchasing copyright licenses to reproduce any publications provided to EFSA. The applicant remains solely responsible and liable for obtaining all necessary authorisations and rights to use, reproduce and share the publications provided to EFSA.
If safety for one species/category is derived from literature studies and extrapolation to other species/categories is required, the same principles as described under Section 5.7 should be followed.
4. Toxicity data from repeated dose studies in laboratory animals
For all additives with the exception of viable microorganisms, safety for target animals can be derived from toxicological studies with oral administration in laboratory animals. These data should allow establishing a lowest no observed adverse effect level (NOAEL) or a benchmark dose level (BMDL) (e.g. BMDL5 for continuous data, BMDL10 for quantal data). Ideally, toxicological studies should follow either the latest Organisation for Economic Cooperation and Development (OECD) protocols or those in force at the time the study was made.
To derive a safe daily dose in the target species (mg/kg body weight (BW)), the NOAEL or BMDL, expressed in mg/kg BW, is divided by an uncertainty factor of 100 (to cover intra‐ and interspecies variation).4 The maximum safe concentration in feed (mg/kg complete feed, ‘as is basis’) is obtained by dividing this safe daily dose by the default feed intake (FI; expressed as a g DM per kg BW, Table 1). The resulting value (mg additive/g DM feed) is multiplied by 1,000 to express the feed concentration per kg complete feed and multiplied by 0.88 (or 0.945 for milk replacer for veal calves) to transform it to as is basis (assuming 88% DM for complete feed and 94.5% for milk replacers).
Table 1.
Default values for daily feed intake scaled to body weight (g dry matter (DM)/kg body weight) for the main animal species/categories
| Animal category | Default values daily feed intake (g DM/kg body weight) | Values derived from | |
|---|---|---|---|
| Body weight (kg) | Feed intake (kg DM/day) | ||
| Chicken for fattening | 79 | 2 | 0.158 |
| Laying hen | 53 | 2 | 0.106 |
| Turkey for fattening | 59 | 3 | 0.176 |
| Piglet | 44 | 20 | 0.88 |
| Pig for fattening | 37 | 60 | 2.20 |
| Sow lactating | 30 | 175 | 5.28 |
| Veal calf (milk replacer) | 19 | 100 | 1.89 |
| Cattle for fattening | 20 | 400 | 8.0 |
| Dairy cow | 31 | 650 | 20.0 |
| Sheep/goat | 20 | 60 | 1.2 |
| Horse | 20 | 400 | 8.0 |
| Rabbit | 50 | 2 | 0.1 |
| Salmon | 18 | 0.12 | 0.0021 |
| Dog | 17 | 15 | 0.250 |
| Cat | 20 | 3 | 0.060 |
| Ornamental fish | 5 | 0.012 | 0.000054 |
DM: dry matter.
Maximum safe concentration in feed = ((NOAEL/100)/FI) × 1,000 × 0.88
The default values of feed intake in Table 1 are derived from estimated values of body weight and derived feed intake of the animals at the end of a tolerance study. For animals not listed in Table 1, the applicant should propose the default values.
For additives intended to be used in water, safe concentrations in water can be derived from the safe concentrations in feed (see Section 2.5.1 of the Guidance on the identity, characterisation and conditions of use of feed additives).
If specific toxicological data are not available, the thresholds of toxicological concern (TTC)5 could be applied to flavouring additives only for which a Cramer structural class can be assigned. Assignation to a Cramer class is made using the OECD toolbox6 or other commercial software. The ‘maximum acceptable feed concentrations’ are derived from the thresholds of the TTC approach and based on the default values of feed intake shown in Table 1. Substances in Cramer class I would result in a maximum acceptable concentration in complete feed (mg/kg feed) between 0.3 and 1.5, for Cramer class II between 0.1 and 0.5 and for Cramer class III between 0.02 and 0.08.
5. Tolerance studies in target animals
If safety for the target species cannot be established at the maximum proposed concentration by the methods described above, then in vivo studies in the relevant target species/categories are required. The number of tolerance studies required in different animal species/categories is described in Section 5.7.
The aim of the tolerance study is to provide a limited evaluation of short‐term toxicity and a margin of safety7 of the additive to the target animals. It is recommended to combine the tolerance study with one of the efficacy trials, whenever possible. If this is done, then all experimental groups should be included in the statistical analysis.
Studies should be performed and documented according to appropriate quality standards and should respect the rules on animal welfare laid down by EU legislation, particularly those listed in Directive 63/2010/EU. Trials should be compliant with the criteria established by a recognised, externally audited, quality assurance scheme (e.g. good laboratory practice (GLP) in accordance with Directive 2004/10/EC). Evidence should be provided that the work was done by qualified personnel using appropriate facilities and equipment and responsible to a named study director. Studies conducted outside the European Union must follow the same quality standards.
5.1. Test item
Tolerance studies should be based on the additive(s) for which application is made, except in cases where a concentrated form of the additive(s) is recommended for testing (e.g. enzymes and microorganisms). Any other deviations because of practical or other considerations should be justified. A certificate of analysis of the test item used in the study should be provided. The additive could be administered via feed or water for drinking depending on the conditions of use. The concentration of the active substance(s) or agent(s) in the feedingstuffs/water should be confirmed by analysis.
5.2. Experimental groups
The design of a tolerance test includes a minimum of three groups:
-
1
– a control group
The diet/water of the control group should normally not contain the additive tested.8 However, in case of additives for which a nutritional requirement exists, the control group should receive the additive at the lowest requirement level (e.g. according to the National Research Council (NRC), Gesellschaft für Ernährungsphysiologie (GfE) or other national data).
-
2
– a use‐level group
The diet/water of the use‐level group should normally contain the additive at the highest recommended concentration. For those additives for which a maximum concentration is not recommended, the highest typical use level should be used.
-
3
– an overdose group with a multifold of the use‐level.
When the multifold concentration tested is:
-
1
– ≥ 100, test animals should be routinely monitored for visual evidence of clinical signs, performance characteristics and product quality where relevant.
-
2
– 10 to < 100: in addition to the above, haematology and blood chemistry as described in Section 5.5 and other parameters likely to be related to the biological properties of the additive.
-
3
– ≤ 10: in addition to the above, gross pathology and histopathology if relevant, as described in Section 5.5. The study should be designed in such a way that a margin of safety for the additive can be estimated. It is recognised that in some cases, ethical and practical considerations will prevent performing necropsy in certain species/categories (e.g. pets, dairy cows, sows, horses).
Where relevant, higher concentration of the active substance or agent can be obtained by omitting or reducing the amount of carrier. For fermentation products, the ratio of active agent(s)/substance(s) to the other fermentation products must remain the same as in the additive.
The setting conditions (e.g. temperature, light exposure) should be the same for the various groups including housing, husbandry and diet/water administration.
5.3. Animals
Animals used should be healthy and preferably from a homogeneous group. Housing and husbandry conditions should be adequate for the purpose of the study and conform to animal welfare regulations. Preventive treatments with antibiotics/antimicrobials before the start of the trial should be avoided. The acceptability of trials in which animals are treated with antibiotics/antimicrobials during the course of the study will depend on a variety of factors, including the number of animals treated, duration of the treatment, distribution between experimental groups and severity of the disease. The acceptability of these studies will be assessed on a case‐by‐case basis. Any therapeutic/preventive treatments should not interact with the proposed mode of action of the additive and should be recorded individually. Studies with an abnormally high mortality in the control group will not be accepted. This would be judged against European industry standards.
For food‐producing animals, the conditions of the study should be such that optimal performance as described for the breed (e.g. performance standards of breeder companies) could be reached. The higher the zootechnical performance of the animals in a given physiological stage, the more sensitive the endpoint(s) would be to adverse situations. Therefore, it is recommended to use in studies with:
-
1
– chickens for fattening: only male birds
-
2
– laying hens: birds in the first third of the laying period
-
3
– dairy cows: high‐yielding animals in the first third of the lactation period
-
4
– growing pigs: weaned piglets of both sexes
-
5
– cattle: weaned male bovines at the beginning of the fattening period
-
6
– salmonids: juvenile phase.
These recommendations are indicative of the most sensitive conditions for the assessment of safety for the target animals. The applicant may deviate from these recommendations provided that adequate justifications are submitted. The use of both sexes implies separate penning and statistical evaluation (with the exception of piglets).
The recommended age/weight for the different species/categories at the start of the study is detailed in Section 5.6.
5.4. Statistical considerations
5.4.1. Design of the experiment
The experimental unit is the smallest entity to which a given treatment is applied. If animals are penned in groups and all the animals in the pen share the same feed source (and feed intake is not measured individually), then the experimental unit for all parameters is the pen, not the individual animal.
Experimental units allocated to the various experimental groups should not differ in a systematic way. Therefore, a recognised method of randomisation should be used to allocate treatments to the experimental unit (e.g. pen, animal). A randomised block design should be preferably used to control for experimental settings like location within facilities. The same design is also recommended in case of large experiments to ensure concurrency in measurements/determination of endpoints across treatments. Other designs might be also appropriate, in which case the applicant should justify the rationale for the design chosen.
In case of a significant variability across animals of factors which could influence the outcome of the study, animals should be stratified before being randomly allocated to pens/cages/treatments. These factors might include initial body weight, age, stage of lactation, milk yield, parity and egg production.
A proper method for randomisation should be used in order to allow allocation concealment (no a priori knowledge of group assignment). In practice, the randomisation process must ensure that investigator cannot influence the allocation of units to the various groups. It is recommended to implement blinding of the caregivers and investigators, where possible, for instance using a proper codification of the treatment to be administered.
5.4.2. Sample size
Statistical considerations should be used to determine the size of the sample used to evaluate the potential safety concerns. The setting of the null and alternative hypotheses should be done in light of the problem formulation. Tests for equivalence or non‐inferiority should be done for experiments aiming at demonstrating similarity or non‐inferiority between treated groups and control. Difference testing should be used when the purpose is to confirm superiority or inferiority (i.e. alternative hypothesis stating a difference exists). For difference testing, when only one direction of the effect is biologically relevant, a one‐sided test should be used. A two‐sided test is recommended in all other cases. For the key endpoints, considerations for sample size calculation need to include: (i) the magnitude of the effect that the study is designed to test and its variability; (ii) the expected direction of the effect; (iii) an adequate statistical power and (iv) the confidence level. The magnitude of the effect considered biologically relevant (for difference testing) or the similarity/non‐inferiority range (for equivalence/non‐inferiority testing) should be clearly indicated for each endpoint. Key endpoints include feed intake for all animals and, depending on the production stage, at least one additional endpoint (e.g. body weight gain, egg or milk production, reproductive performance) should be considered. It should be noted that additional key endpoints may arise from toxicological studies or other parameters related to the biological properties of the additive. The highest sample size resulting from the calculations for all key endpoints should be selected. The applicant should justify the selection of the key endpoints.
The Type 2 (β) error measures the risk of non‐detecting an effect (difference)/similarity (equivalence) when it exists. As a guide, it should be lower than or equal to 20% in general, and 25% for experiments with ruminants, minor species, pets and other non‐food‐producing animals. Hence, a power (1‐β) greater than or equal to 80% (75% for ruminants, minor species, pets and non‐food‐producing animals) should always be ensured. Generally, a confidence level of 95% is adopted when testing difference, 90% for testing equivalence. Use of levels below these thresholds should be justified.
5.4.3. Statistical analysis
The statistical analysis should be performed using models that allow comparing treated and control groups whilst controlling for factors that could influence the outcome of the experiment whenever possible. The class of generalised linear mixed models (McCullagh and Nelder, 1989), known as GLMM, offers a suite of methods flexible enough to fit most of the experimental settings. Typically, this type of models includes the treatment and other stratification variables (e.g. age) as fixed factor and blocking factors, if any, as random (e.g. animal/pen location) or as covariates. The response variable is the endpoint under investigation. Under certain conditions, a log or other transformations can be needed in order to linearise the relationship with the explanatory factors. Depending on the type of response variable (i.e. continuous, quantal, binary), different kinds of statistical tests and distributional assumptions could be required. The applicant is requested to assess which one is more appropriate and to provide the rationale of the choice. An indicator of quality of fit should always be provided.
The analysis of variance is one of the simplest models included in the GLMM class. When using this method, a test for group differences should be carried out preferably using the Scheffé, Dunnet, Tukey (Sachs and Hedderich, 2006) or other comparable tests any time multiple comparisons are performed concurrently. Non‐parametric tests may be necessary if only a low number of observations is available, but applicants are encouraged to use sufficient replicates to allow for parametric tests to be performed. When different additives are assessed concurrently using the same control, the statistical evaluation should be done considering only the control and the groups treated with the additive under assessment.
5.5. Endpoints
The endpoints to be measured depend on the design of the tolerance study (see Section 5.2). The minimum required parameters for the different groups of endpoints are listed below and may be augmented on a case‐by‐case basis.
5.5.1. Performance parameters and related parameters
Feed intake, initial and final body weight, body weight gain, feed to gain ratio, water intake for those additives administered via water. Clinical observations (by a veterinarian or under veterinary supervision) including general health status, behaviour, morbidity and mortality (including culling).
In addition,
-
1
– for laying hens: laying rate, egg weight, shell quality, feed to egg mass ratio, egg mass/hen per day
-
2
– for breeding hens: additionally to those required for laying hens, fertility, hatchability and chick viability
-
3
– for dairy animals: milk production (also fat corrected milk), milk composition (total solids, protein, fat, lactose and urea), somatic cell counts, protein, fat and lactose yield
-
4
– for sows: number of piglets born, piglets born alive, litter weight at birth and at weaning, number of piglets weaned, weaning to oestrus interval
-
5
– for fish: specific growth rate (preferably thermal growth coefficient (Jobling, 2003)).
5.5.2. Haematology and clinical chemistry
Generally, samples for haematology and blood chemistry analysis should be taken at the end of the study. Samples should be taken at the start of the trial to establish a baseline in studies involving cattle for fattening, dairy cows, sows, horses, dogs and cats. Samples should be taken from all experimental units and ideally from all animals. However, when total numbers of animals makes it impractical, subsets of animals/pen should be identified for sampling by a random process carried out at the beginning of the study. As a guide, the subset should represent at least 10% of the animals/pen, but in no case less than two animals/pen. Blood samples should not be pooled.
The parameters to be measured should at least include (as appropriate):
Total count for red blood cells, packed cell volume, haemoglobin, mean corpuscular volume, mean corpuscular haemoglobin, mean corpuscular haemoglobin concentration, total and differential counts for leukocytes, platelet counts, prothrombin time and fibrinogen (with the exception of the latter two parameters for fish).
Sodium, potassium, chloride, calcium, phosphate, magnesium, total protein, albumin, globulin, glucose, urea/uric acid (non‐protein nitrogen for fish), cholesterol, creatinine, bilirubin, acute phase proteins, amylase, alanine aminotransferase, aspartate aminotransferase, lactate dehydrogenase, gamma‐glutamyltransferase, alkaline phosphatase and creatine kinase.
Where available, scientific studies reporting reference ranges for the haematology and blood chemistry parameters should be submitted for the species of the tolerance study.
5.5.3. Tissues/organs from necropsy
The following organs and tissues (as appropriate) from all groups should be examined grossly (including weight of the organs) and preserved for microscopic evaluation: liver, kidneys, spleen, adrenal gland, lung, stomach, pancreas, small intestine, colon, caecum, thymus, thyroid gland, heart, intestinal lymph nodes, ovaries/testes. For fish, the following organs should be investigated, kidney, liver, spleen, stomach (where present) and intestinal tract, heart, gonads, gills, bone and eye. Histopathology is normally required only when indicated by findings in the gross pathology.
In all cases, critical endpoints known from the toxicological studies in laboratory animals should be considered. Any adverse effect detected during efficacy trials should also be reported in this section. All deaths should be explained and, if necessary, investigated by gross pathology and histopathology.
5.6. Duration of the tolerance study
The necessary minimum duration of tolerance trials depends on the animal species/category and is reported in Table 2.
Table 2.
Minimum duration of tolerance studies for the main animal species/categories
| Category | Definition of the animal category | Start, from | Duration |
|---|---|---|---|
| Piglets | Young animals having completed the suckling period | Weaning (or not more than 7 days after weaning) |
42 days 35 days if growth rate is ≥ 0.5 kg/day |
| Pigs for fattening | Animals intended for meat production until day of transport to slaughterhouse | 20–35 kg | 42 days |
| Sows | Female animals having been inseminated/mated | From insemination/mating | One cycle: from insemination to the first oestrus after weaning |
| Chickens for fattening | Birds raised for fattening | Hatch | 35 days |
| Laying hens | Productive female birds held for egg production purposes | From 20 weeks of age | 56 days |
| Breeder hens | Female birds held for breeding purposes | From 25 weeks of age | 56 days |
| Turkeys for fattening | Birds raised for fattening | Hatch | 42 days |
| Turkeys for breeding purposes | Female and male birds held for breeding purposes | From 32 weeks of age | 56 days |
| Calves | Calves which are reared for reproduction, veal production or beef production | 1 week of age (for veal production, from 1 to 3 weeks of age) | 42 days |
| Cattle | Bovine animals that have completed the weaning period | Full development of rumination but < 6 months of age | 42 days |
| Cows | Lactating cows | 4 weeks after beginning of lactation | 56 days |
| Lambs/kids | Young animals reared for reproduction or meat production | 1–4 weeks of age | 42 days |
| Sheep/goats | Lactating animals | 4 weeks after beginning of lactation | 56 days |
| Salmon and trout | Growing salmonids |
Trout: 10 Trout: 10 g Salmon: 50 g |
90 days |
| Other fin fish | Growing fin fish | 90 days | |
| Crustaceans | Growing crustaceans | 90 days | |
| Rabbits | Rabbits that are reared for reproduction or meat production | Beginning 1 week after birth | 42 days |
| Breeding does | Does that have become pregnant at least once | From insemination to the end of weaning period (one cycle) | |
| Cats | 28 days | ||
| Dogs | 28 days | ||
| Other non‐food‐producing animals | 28 days |
Tolerance studies for pigs for fattening are not needed if safety for weaned piglets is established.
Tolerance studies for chickens for fattening/reared for laying and turkeys for fattening should normally be performed with 1‐day‐old birds. Tolerance data from chickens or turkeys for fattening are generally taken to include chickens reared for laying or turkeys reared for breeding, respectively.
If calves for rearing and cattle for fattening were applied for, a combined study (28 days for each period) would be considered sufficient.
For minor species not included in the tables above, the duration of the studies should correspond to that of the physiologically related major species listed in Table 3. For all other species, the minimum duration should be 42 days for growing animals and 56 days for adult animals.
Table 3.
Extrapolation of tolerance data from certain species to other physiologically related species
| From | To physiologically related species |
|---|---|
| Chickens for fattening | Other poultry for fattening (e.g. turkeys, ducks, goose, pheasants, quail, guinea fowl, ostrich) and ornamental birds |
| Laying hens | Other birds kept for egg production* (e.g. ducks, goose, pheasants, quail, guinea fowl, ostrich) |
| Pigs | Other Suidae |
| Calves or cattle | Other growing ruminants (e.g. sheep, goat, buffalo) at the corresponding developmental stage |
| Dairy cows | Other dairy ruminants (e.g. goat, sheep, buffalo) |
| Salmon or trout | Ornamental fish |
* Extrapolation to breeders (including turkeys) is only possible if additional data on breeding endpoints are available.
If an additive is applied for a specific and shorter period than that given in the tables above, it should be administered according to the proposed conditions of use. However, the observation period should not be shorter than 28 days and should involve the relevant endpoints (e.g. for sows for reproduction the number of piglets born alive when considering the gestation period, or the number and weight of weaned piglets when considering the lactation period).
5.7. Requirement for tolerance studies
In principle, tolerance tests should provide evidence of the safety of the additive for each of the target species/animal categories for which an application is made. If the application covers only one animal category (as defined in Annex IV of Regulation (EC) No 429/2008), at least one study in this category is required.
When the application covers several target species/categories, it is recognised that it may be unrealistic to expect studies in all potential target species for which application is made, especially when the application is for all animal species. Therefore, interspecies extrapolation of data can be applied. In principle, data can be extrapolated between physiologically similar species (Table 3). The degree to which species are physiologically related is judged predominantly in terms of gastrointestinal function. Similarities in metabolism are also considered. In general, the extrapolation is limited between animals which are kept for the same purpose, i.e. meat production or reproduction (including milk or egg production).
Data from tolerance studies in the individual species/categories listed in column 1 of Table 3 can be used to support the safety for other physiologically related species/categories, provided a sufficient margin of safety (the ratio of tolerated to maximum proposed use level)9 is established for the species/category used for the extrapolation. If the margin of safety is not sufficient, then specific tolerance studies in the relevant species/categories should be provided.
-
1
– If the application is for all poultry/avian species, then tolerance studies should be provided with chickens for fattening and laying hens. In order to cover species for breeding, an additional limited study in breeding hens considering only performance endpoints (see Section 5.5) should be submitted.
-
2
– If the application is for all Suidae, then tolerance studies should be submitted for weaned piglets and sows.
-
3
– If the application is for all ruminant species, then tolerance studies should be submitted in cattle for fattening and dairy cows.
-
4
– If the application is for all fish, then tolerance studies should be submitted in a salmonid (salmon or trout) and another species (e.g. carp, sea bream, sea bass). If the application includes crustaceans, then an additional study in shrimp would be required.
Extrapolation of the conclusions of tolerance studies can also be done to non‐physiologically similar species provided studies are done with a set of different species and a sufficient margin of safety can be established in all the species tested. If the margin of safety is not sufficient in all the different species, a limited extrapolation to other species may still be possible, but limited between the species/categories listed in Table 3, as described above.
-
1
– If the application covers two food‐producing animal species (e.g. pigs and poultry), then the requirement would be limited to a total of three tolerance studies including both species and covering growing and reproductive animals. The recommendations for the use of certain animal categories in tolerance studies (Section 5.3) should be taken into consideration.
-
2
– If the application is for all pets and non‐food‐producing animals, tolerance studies would be required for cats, dogs and a third species (e.g. a laboratory animal).
-
3
– If the application is for horses, it may be possible to derive tolerance from studies with cattle (cattle for fattening or dairy cows) and pigs (pigs for fattening or sows).
-
4
– If the application is for all terrestrial animal species, tolerance studies should be provided with at least chickens for fattening, piglets and dairy cows.
-
5
– If the application is for all animal species, tolerance studies should be provided in at least salmonids, chickens for fattening, piglets and dairy cows.
For certain types of additives, the requirements for tolerance studies above may be modified:
-
1
– For nutritional additives where a tolerance study is required, target animal safety data can be derived from one study in a target species or laboratory animal.
-
2
– For silage additives for which tolerance studies are required, it is usually sufficient to restrict tolerance to a ruminant species, normally the dairy cow. Studies involving other species are required only when the nature of the ensiled material makes it more appropriate for use with non‐ruminants or when there are particular concerns when treated silage is used for categories other than adult ruminants (e.g. moist corn for pigs or fish silage for fur animals).
5.8. Reporting
For each tolerance study, a study report should be submitted describing the objectives, materials and methods, results and conclusions. The initial protocol should be included; any deviations from the protocol should be clearly indicated and justified in the final report. The reports should include the individual data in digital format and detailed results including descriptive statistics, statistical tests and model outcomes. Reports should start with a trial protocol data sheet (Appendix A) followed by the full study report. International units should be used to express the results.
It is recommended that the study report follows the structure detailed below and contains the following information. Applicants are encouraged to follow the recommendations of the EFSA guidance on statistical reporting.
Title: The title should provide a concise and clear description of the study, including the type of study, the product under assessment and animal species/category.
Summary: The summary should include the objectives, a description of the design and methods, the main results and the conclusions of the study.
Objectives: The objectives of the study should be clearly described.
Materials and methods: Methods, apparatus and materials used, details of the species, breed or strain of the animals, their number and the conditions under which they were housed and fed. In particular, the following should be recorded and reported:
Ethical statement
-
1
Indicate compliance with national or institutional guidelines for the care and use of animals.
Animals, housing and husbandry
-
2
Animals: species (for aquatic species intended for human consumption: identification should be made by their colloquial name followed in parenthesis by the Latin binomial), breed, age (and size/length for aquatic species), initial body weight, sex, identification procedure, physiological stage and general health.
-
3
Husbandry conditions: feeding and rearing conditions (pen/tank size, stocking density, temperature, lighting); for aquatic species water quality including water flow rate, water temperature and salinity, where relevant.
-
4
Diets: description of manufacture, form and quantitative composition of the diet(s) in terms of ingredients used, relevant nutrients (calculated and analysed values) and energy (digestible, metabolisable or net).
Study design
-
5
Study location, dates and responsible individuals.
-
6
Study duration.
-
7
The type of design of the study (e.g. factorial, stratified, crossover).
-
8
Experimental groups: number of treatment and control groups, numbers of replicates (experimental unit) per group and number of animals per replicate.
-
9
The experimental unit (e.g. individual animal, pen) should be indicated.
-
10
The basis for the different measurements (e.g. individual animal, pen) should be indicated for each parameter measured.
-
11
Rationale for the selection of the number of animals/replicates used (sample size calculation). Power analysis should be provided.
-
12
Steps taken to minimise bias including randomisation and blinding (see Section 5.1.1 of the EFSA guidance on statistical reporting (EFSA, 2014)).
-
13
Test item: intended concentration of the active substance(s) or agent(s) in the feedingstuffs.
Experimental procedures
-
14
The procedures carried out to the different experimental groups should be detailed. These should include the parameters/endpoints measured, indicating when and how they were measured and information on the methods of analysis.
-
15
The health of the animals should be monitored, morbidity and mortality (including culling) recorded.
-
16
The methodology to correct feed to gain ratio for mortality (including culling) should be reported.
Statistical methods
-
17
The result of the power analysis should be reported.
-
18
The methods to perform statistical analysis should be stated, including those used to identify outliers and handle missing data. If any relevant data points are excluded from the model (e.g. outliers), a justification should be given.
-
19
Describe any methods used to assess whether the data met the assumptions of the statistical approach.
Results: Results of the study should be presented for all endpoints considered in the study. Tables should be used to summarise the results from treatments. For all endpoints which are measured on individual animals in a pen, a summary parameter of the endpoint in the experimental unit should be used (e.g. mean for continuous measurements such as body weight, median and counts for quantal measurements such as severity of an outcome or mortality). Summary parameters should always be adjusted for losses (mortality/culling). The distribution of losses within the treatment groups should be assessed to avoid the risk of introducing a bias.
-
20
Health status of the animals, morbidity and mortality including culling. The timing and prevalence of any unexpected/undesirable incident/effect in individuals or groups. Therapeutic/preventive treatments, if any should be recorded. Likely cause of death should be established by a veterinarian and reported.
-
21
The report should include data from all animals or experimental units involved in the trials. Cases which cannot be assessed due to a lack or loss of data should be reported, and their distribution within the groups of animals indicated.
-
22
Concentration of the active substance(s) or agent(s) in the feedingstuffs should be periodically analysed and reported. A certificate of analysis of the test item used in the study should be provided.
-
23
Report the results for each endpoint measured/analysis carried out, with a measure of precision (e.g. standard error or confidence interval).
-
24
The report should include descriptive statistics plus detailed outcome of any statistical analysis performed for all measured endpoints and each time point.
-
25
The measurement units should be specified for any result reported.
Discussion
-
26
Interpretation of the results, taking into account the study objectives and hypotheses and other relevant studies in the literature.
-
27
Comments on the study limitations including any potential sources of bias, any limitations of the animal model and the imprecision associated with the results.
Conclusions
-
28
The conclusions from the study should be drawn considering the objectives of the study, the hypothesis and the outcome of the study.
Individual data, certificates of analysis
-
29
The individual data should be provided in a form of an electronic database and should be accompanied by a data dictionary containing the description of the variables and the metadata needed to properly analyse them.
-
30
All codes, log and complete outputs for the final statistical analysis (i.e. the results and analysis reported) should be provided in electronic format.
-
31
The report should include the certificates of analysis for the different analysis performed, reports of the veterinary observations/gross pathology/histopathology, haematology/clinical chemistry, etc.
6. Toxicological studies
The toxicological studies available for the active substance(s) should be taken into account when assessing the safety for the target species.
Depending on structural alerts or other toxicological considerations, genotoxicity studies may be required when the additive is intended for use in long living animals (e.g. pets) and reproduction animals (e.g. cows, sows, breeder hens). This could be achieved by reference to published studies.
7. Interactions in vivo
Any known interactions of the additive with feed materials, other approved additives or medicinal products should be documented.
For those additives which exert their activity mainly by binding (e.g. clays), there is the possibility that the availability of crucial nutrients, micronutrients and other additives could also be affected. It is recognised that it is not practical to consider all possible nutrients/additives. Therefore, it is recommended to measure apparent digestibility of crude protein, zinc, retinyl or tocopheryl esters, thiamin or pyridoxine and an ionophore coccidiostat; the latter in the case, the additive is intended to be used in poultry/rabbits. Such studies should be performed with the highest recommended concentration of the additive and could be made in the context of a tolerance/efficacy study. For other additives which may have a negative impact on the absorption of nutrients, a similar approach should be taken.
8. Microbial studies
Studies are only required when:
the tolerance test give an indication of an adverse effect related to digestive tract disturbances; or
an adverse effect on the gut microbiota can otherwise be anticipated; or
the additive shows specific antimicrobial activity at the feed concentration; or
the additive is an ionophoric coccidiostat.
For the details on how to perform the studies, see the Guidance on the characterisation of microorganisms used as feed additives or as production organisms.
Abbreviations
- BMDL10
benchmark dose level 10
- BW
body weight
- DM
dry matter
- FI
feed intake
- GfE
Gesellschaft für Ernährungsphysiologie
- GLMMs
generalised linear models
- GLP
good laboratory practice
- GMMs
genetically modified microorganisms
- NOAEL
no observed adverse effect level
- NRC
National Research Council
- OECD
Organisation for Economic Cooperation and Development
- QPS
qualified presumption of safety
- TTC
threshold of toxicological concern
- URL
uniform resource location
Appendix A – Trial Protocol data sheet
1.
FOR TERRESTRIAL ANIMALS
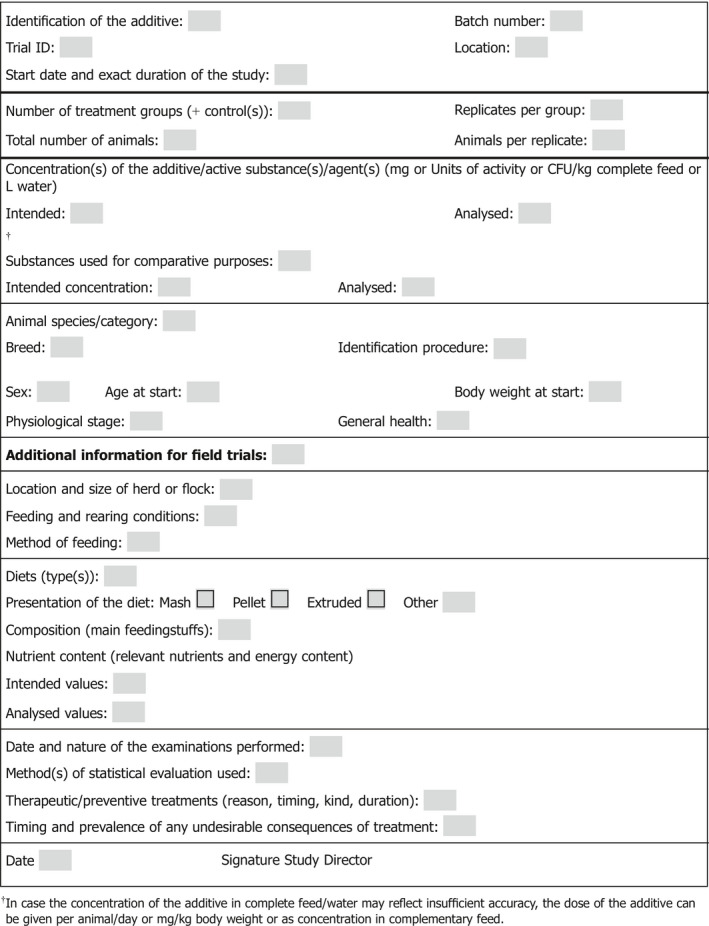
FOR AQUATIC ANIMALS

Suggested citation: EFSA FEEDAP Panel (EFSA Panel on additives and products or substances used in animal feed) , Rychen G, Aquilina G, Azimonti G, Bampidis V, Bastos ML, Bories G, Chesson A, Cocconcelli PS, Flachowsky G, Gropp J, Kolar B, Kouba M, López‐Alonso M, López Puente S, Mantovani A, Mayo B, Ramos F, Saarela M, Villa RE, Wallace RJ, Wester P, Anguita M, Galobart J, Innocenti ML and Martino L, 2017. Guidance on the assessment of the safety of feed additives for the target species. EFSA Journal 2017;15(10):5021, 19 pp. 10.2903/j.efsa.2017.5021
Requestor: EFSA
Question number: EFSA‐Q‐2016‐00553
Panel members: Gabriele Aquilina, Giovanna Azimonti, Vasileios Bampidis, Maria de Lourdes Bastos, Georges Bories, Andrew Chesson, Pier Sandro Cocconcelli, Gerhard Flachowsky, Jürgen Gropp, Boris Kolar, Maryline Kouba, Marta López‐Alonso, Secundino López Puente, Alberto Mantovani, Baltasar Mayo, Fernando Ramos, Guido Rychen, Maria Saarela, Roberto Edoardo Villa, Robert John Wallace and Pieter Wester.
Acknowledgements: The Panel wishes to thank the members of the working group on guidance update for the preparatory work on this scientific output.
Adopted: 26 September 2017
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2017.EN-1304/full
Notes
Regulation (EC) No 1831/2003 of the European Parliament and of the Council of 22 September 2003 on additives for use in animal nutrition. OJ L 268, 18.10.2003, p. 29.
Commission Regulation (EC) No 429/2008 of 25 April 2008 on detailed rules for the implementation of Regulation (EC) No 1831/2003 of the European Parliament and of the Council as regards the preparation and the presentation of applications and the assessment and the authorisation of feed additives. OJ L 133, 22.5.2008, p. 1.
Without prejudice to Article 20 of Regulation (EC) No 1831/2003.
The uncertainty factor may be adjusted to take into account particular metabolic considerations, the nature and quality of the toxicological studies, etc.
JECFA (FAO/WHO, 1996, Food additive series 35, IPCS, WHO Geneva). Review of the Threshold of Toxicological Concern (TTC) approach and development of new TTC decision tree. EFSA supporting publication 2016: EN‐1006. 50 pp. http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2016.EN-1006/pdf
Margin of safety: ratio of the highest tolerated additive concentration in feed and the highest use level proposed.
When the additive modifies the physicochemical properties of the feed, this should be considered in formulating the feed for a control group.
A sufficient margin of safety generally is at least 10, meaning that a concentration of at least 10 times the highest recommended (approved) concentration of the additive was tolerated by relevant species without any adverse effects. However, for some substances (e.g. organic acids, some flavourings, some clays, some coccidiostats), a lower margin of safety may be considered.
References
- EFSA (European Food Safety Authority), 2007. Opinion of the Scientific Committee on a request from EFSA on the introduction of a Qualified Presumption of Safety (QPS) approach for assessment of selected microorganisms referred to EFSA. EFSA Journal 2007;5(12):587, 16 pp. 10.2903/j.efsa.2007.587 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2014. Guidance on Statistical Reporting. EFSA Journal 2014;12(12):3908, 18 pp. 10.2903/j.efsa.2014.3908 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2017. Outcome of the public consultation on the draft guidance on the assessment of the safety of feed additives for the target species. EFSA supporting publication 2017:EN‐1304. 42 pp. 10.2903/sp.efsa.2017.EN-1304 [DOI] [Google Scholar]
- EFSA BIOHAZ Panel (EFSA Panel on Biological Hazards), Ricci A, Allende A, Bolton D, Chemaly M, Davies R, Girones R, Herman L, Koutsoumanis K, Lindqvist R, Nørrung B, Robertson L, Ru G, Sanaa M, Simmons M, Skandamis P, Snary E, Speybroeck N, Ter Kuile B, Threlfall J, Wahlström H, Cocconcelli PS, Klein G (deceased), Prieto Maradona M, Querol A, Peixe L, Suarez JE, Sundh I, Vlak JM, Aguillera‐Gomez M, Barizzone F, Brozzi R, Correia S, Heng L, Istace F, Lythgo C and Fernández Escámez PS, 2017. Scientific Opinion on the update of the list of QPS‐recommended biological agents intentionally added to food or feed as notified to EFSA. EFSA Journal 2017;15(3):4664, 177 pp. 10.2903/j.efsa.2017.4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA FEEDAP Panel (EFSA Panel on Additives and Products or Substances used in Animal Feed), 2016. Statement of the panel: Analysis of the need for an update of the guidance documents. EFSA Journal 2016;14(5):4473, 8 pp. 10.2903/j.efsa.2016.4473 [DOI] [Google Scholar]
- Jobling M, 2003. The thermal growth coefficient (TGC) model of fish growth: a cautionary note. Aquaculture Research, 34, 581–584. 10.1046/j.1365-2109.2003.00859.x [DOI] [Google Scholar]
- McCullagh J and Nelder JA, 1989. Generalized Linear Models, Second Edition. Chapman and Hall/CRC Monographs on Statistics & Applied Probability, London. [Google Scholar]
- Sachs L and Hedderich J, 2006. Angewandte Statistik. Methodensammlung mit R [Taschenbuch]. Springer, Berlin: [Google Scholar]