

Abstract
The EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS) provides a scientific opinion re‐evaluating the safety of glutamic acid–glutamates (E 620–625) when used as food additives. Glutamate is absorbed in the intestine and it is presystemically metabolised in the gut wall. No adverse effects were observed in the available short‐term, subchronic, chronic, reproductive and developmental studies. The only effect observed was increased kidney weight and increased spleen weight; however, the increase in organ weight was not accompanied by adverse histopathological findings and, therefore, the increase in organ weight was not considered as an adverse effect. The Panel considered that glutamic acid–glutamates (E 620–625) did not raise concern with regards to genotoxicity. From a neurodevelopmental toxicity study, a no observed adverse effect level (NOAEL) of 3,200 mg monosodium glutamate/kg body weight (bw) per day could be identified. The Panel assessed the suitability of human data to be used for the derivation of a health‐based guidance value. Although effects on humans were identified human data were not suitable due to the lack of dose–response data from which a dose without effect could be identified. Based on the NOAEL of 3,200 mg monosodium glutamate/kg bw per day from the neurodevelopmental toxicity study and applying the default uncertainty factor of 100, the Panel derived a group acceptable daily intake (ADI) of 30 mg/kg bw per day, expressed as glutamic acid, for glutamic acid and glutamates (E 620–625). The Panel noted that the exposure to glutamic acid and glutamates (E 620–625) exceeded not only the proposed ADI, but also doses associated with adverse effects in humans for some population groups.
Keywords: glutamic acid, E 620, sodium glutamate, E 621, potassium glutamate, E 622, calcium glutamate, E 623, ammonium glutamate, E 624, magnesium glutamate, E 625
Summary
Following a request from the European Commission, the EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS) was asked to re‐evaluate the safety of glutamic acid (E 620), monosodium glutamate (E 621), monopotassium glutamate (E 622), calcium diglutamate (E 623), monoammonium glutamate (E 624) and magnesium diglutamate (E 626) when used as food additives. The Panel was not provided with a newly submitted dossier and based its evaluation on previous evaluations and reviews, additional literature that became available since then and the data available following a public call for data. The Panel noted that not all original studies on which previous evaluations were based were available for this re‐evaluation.
Glutamic acid–glutamates (E 620–625) are authorised food additives in the European Union (EU) according to Annex II of Regulation (EC) No 1333/2008 and specific purity criteria have been defined in the Commission Regulation (EU) No 231/2012.
The Scientific Committee on Food (SCF) established a group acceptable daily intake (ADI) ‘not specified’ in 1990. In the latest evaluation by the Joint FAO/WHO Expert Committee on Food Additives (JECFA) in 2006, the previously established group ADI ‘not specified’ for glutamic acid and its salts was maintained.
In 2015, the Panel evaluated a new production method using the genetically modified Corynebacterium glutamicum EA‐12 strain for l‐glutamic acid as a food additive and concluded that there was no safety concern from the change in the production method of these food additives.
Glutamate is absorbed in the intestine and it is presystemically metabolised in the gut wall. Evidence was limited for increased brain glutamate concentration by even high dose monosodium glutamate (MSG) ingestion (10 g) via the oral route by diet.
Glutamic acid and its salts had a low acute toxicity. Short‐term and subchronic studies showed no effects of MSG treatment up to doses of roughly 5,000 mg/kg body weight (bw) per day (short‐term studies) and 5,250 mg/kg bw per day in one limit dose test and 2,700 mg/kg bw per day in a study performed following the OECD Guideline 408. In studies with protocols according to OECD, performed to fulfil regulatory requirements, increased kidney weight and increased spleen weight was found in rats in chronic studies and studies to investigate reproductive toxicity at doses of 939 mg/kg bw per day in males and 1,039 mg/kg bw per day in females and above. However, the increase in organ weight was not accompanied by adverse histopathological findings. Hence, the Panel considered the extent of increased kidney and spleen weight as not adverse. In these studies, the no observed adverse effect level (NOAEL) was the highest dose tested.
The Panel considered the genotoxicity data set sufficiently robust to evaluate the genotoxicity of MSG and cover by read‐across the limited or missing data for glutamic acid and the other salts. On this basis the Panel considered that glutamic acid (E 620), monosodium glutamate (E 621), monopotassium glutamate (E 622), calcium diglutamate (E 623), monoammonium glutamate (E 624) and magnesium diglutamate (E 625) did not raise concern with regards to genotoxicity when used as food additives.
In three 2‐year studies in rats, no increased tumour rate was observed up to the highest doses tested. The only observed effect was a significant increase in relative kidney weights in both sexes in the study by Shibata et al. (1985) which, however, was not considered as adverse effect. Thus, the NOAELs identified by the Panel from the three 2‐year studies were the highest doses tested. There was no indication of carcinogenicity.
No adverse effects were observed in reproductive and developmental toxicity studies. Because no effects were noted at the highest doses tested none of the studies could be considered for the derivation of a health‐based guidance value (HBGV).
In a developmental neurotoxicity study group delayed early swimming development, diminished rearing frequency in the open field, altered active avoidance acquisition and extinction and prolonged day‐2 passive avoidance retention were observed in the high‐dose group of 5% of MSG. The Panel considered the effects as adverse and identified a NOAEL of 3.4% of MSG (equal to 3,700 mg/kg bw per day in males and 3,200 mg/kg bw per day in females) based on the absence of neurobehaviour effects.
The Panel noted that direct exposure of glutamic acid–glutamates (E 620–625) to neonates (young infants) is not in the scope of the re‐evaluation of glutamates as food additives and, therefore, the histopathological lesions of the central nervous system (CNS) and behavioural changes observed in neonatal animals are not relevant for the assessment of glutamic acid–glutamates (E 620–625).
The Panel assessed the suitability of human data to be used for the derivation of a HBGV. The MSG symptom complex was rarely seen when doses below 3,000 mg (corresponding to 42.9 mg/kg bw per day using a body weight of 70 kg) were ingested but a clear dose–response relationship could not be established. Blood pressure increases were seen in the only dose of 150 mg/kg tested in an experimental study. From epidemiological studies, no dose–response could be derived for this endpoint. Insulin increase was seen with the only dose tested of 10,000 mg MSG in healthy subjects. Although effects on humans were identified, human data were not suitable to derive an HBGV due to the lack of dose–response data from which a dose without effect could be identified.
Considering the available animal studies, the Panel decided to use the NOAEL of a neurodevelopmental toxicity study (3,200 mg MSG/kg bw per day) as reference point to derive an ADI. Applying the default uncertainty factor of 100 to the NOAEL of 3,200 mg/kg bw per day, the Panel derived an ADI of 32 mg MSG/kg bw per day or 27.8 mg glutamate/kg bw per day. The value of 27.8 mg glutamate/kg bw per day was rounded to derive a group ADI of 30 mg/kg bw per day, expressed as glutamic acid.
The Panel noted that the ADI is below the doses which have been associated with the MSG symptom complex (> 42.9 mg/kg bw per day), headache (85.8 mg/kg bw per day), blood pressure increase (150 mg/kg bw per day) and also insulin increase (> 143 mg/kg bw per day) in humans.
To assess the dietary exposure to glutamic acid–glutamates (E 620–625) from their use as food additives, the exposure was calculated based on different exposure scenarios. As glutamic acid–glutamates (E 620–E 625) are authorised to be used in a wide range of foods, the Panel did not identify brand loyalty to specific food categories and therefore selected the refined non‐brand loyal scenario as the most relevant exposure scenario for the safety evaluation of these food additives; it is assumed that the population will very likely be exposed long‐term to the food additives present at the mean reported use or analysed levels in processed food.
The Panel noted that in the non‐brand‐loyal scenario, the mean exposure exceeded the dose which has been associated with the MSG symptom complex (> 42.9 mg/kg bw per day) in toddlers and children, and the high exposure (95th percentile) in infants, toddlers, children and adolescents. The main food contributing to the mean exposure in the non‐brand‐loyal scenario were fine bakery wares for toddlers, children, adolescents, adults and for elderly, and soups and broths for infants. The main contributing food categories for which industry reported use data were fine bakery wares for toddlers, children, adolescents and adults, and for infants soups and broths. For the elderly, both these food categories were important sources of exposure. Other relevant food contributors for which use levels were available were sauces, meat and meat products and seasoning and condiments.
Considering additional exposure from food categories, which may contain glutamic acid–glutamate due to natural presence and for which analytical data were available, the ADI was exceeded at the mean and high (95th percentile) level by all population groups, except for the mean exposure in elderly. The Panel noted that in this exposure assessment scenario exposure in infants, toddlers and children at the high (95th percentile) level was higher than the doses which have been associated with adverse effects in humans such as the MSG symptom complex (> 42.9 mg/kg bw per day), headache (85.8 mg/kg bw per day), blood pressure increase (150 mg/kg bw per day) and insulin increase (> 143 mg/kg bw per day).
Considering the food supplements consumers only scenario, the ADI was exceeded at the mean level in children, and at the high (95th percentile) level for all population groups.
The Panel considered overall that the uncertainties identified would, in general, result in an overestimation of the exposure to glutamic acid–glutamates (E 620–625) from their use as food additives according to Annex II in both the maximum level and refined exposure scenarios.
The Panel noted that considering only food categories authorised according Annex II, in the refined non‐brand‐loyal exposure assessment scenario, exposure estimates exceeded not only the proposed ADI but also doses which were associated with adverse effects in humans such as the MSG symptom complex for all population groups at the high level (95th percentile) but not at the mean. The high exposure in infants deserves special attention and further evaluation of the sources.
The Panel also noted that in the refined non‐brand‐loyal exposure assessment scenario, when considering all the available data for glutamic acid–glutamate, which include all their identified sources in the diet – food additive use, natural presence and addition as nutrient – exposure estimates largely exceeded the proposed ADI for all population groups at the high level (95th percentile) and at the mean (except for the elderly) as well as the doses, which in humans, are associated with adverse effects such as the MSG symptom complex.
The Panel recommended that:
the European Commission considers revising the maximum permitted levels, in particular, in food categories contributing the most to the overall exposure to glutamic acid and its salts: fine bakery wares, soups and broths, sauces, meat and meat products, seasoning and condiments and food supplements.
the European Commission considers revising the current limits for toxic elements – arsenic and lead – in the EU specifications for glutamic acid (E 620) and lead in EU specifications for monosodium glutamate (E 621), monopotasium glutamate (E 622), calcium diglutamate (E 623), monoammonium glutamate (E 624) and magnesium diglutamate (E 625) in order to ensure that they will not be a significant source of exposure to those toxic elements in food.
1. Introduction
The present opinion document deals with the re‐evaluation of glutamic acid (E 620), monosodium glutamate (E 621), monopotassium glutamate (E 622), calcium diglutamate (E 623), monoammonium glutamate (E 624) and magnesium diglutamate (E 626) when used as food additives.
Glutamic acid is occurring as one of the amino acids that are constituents of peptides and proteins but it is also occurring in a free form. This assessment of glutamic acid–glutamates (E 620–625) as food additives deals only with glutamic acid in its free form.
1.1. Background and Terms of Reference as provided by the European Commission
1.1.1. Background as provided by the European Commission
Regulation (EC) No 1333/20081 of the European Parliament and of the Council on food additives requires that food additives are subject to a safety evaluation by the European Food Safety Authority (EFSA) before they are permitted for use in the European Union (EU). In addition, it is foreseen that food additives must be kept under continuous observation and must be re‐evaluated by EFSA.
For this purpose, a programme for the re‐evaluation of food additives that were already permitted in the European Union before 20 January 2009 has been set up under Regulation (EU) No 257/20102. This Regulation also foresees that food additives are re‐evaluated whenever necessary in light of changing conditions of use and new scientific information. For efficiency and practical purposes, the re‐evaluation should, as far as possible, be conducted by group of food additives according to the main functional class to which they belong.
The order of priorities for the re‐evaluation of the currently approved food additives should be set on the basis of the following criteria: the time since the last evaluation of a food additive by the Scientific Committee on Food (SCF) or by EFSA, the availability of new scientific evidence, the extent of use of a food additive in food and the human exposure to the food additive taking also into account the outcome of the Report from the Commission on Dietary Food Additive Intake in the EU3 of 2001. The report ‘Food additives in Europe 20004’ submitted by the Nordic Council of Ministers to the Commission, provides additional information for the prioritisation of additives for re‐evaluation. As colours were among the first additives to be evaluated, these food additives should be re‐evaluated with a highest priority.
In 2003, the Commission already requested EFSA to start a systematic re‐evaluation of authorised food additives. However, as a result of adoption of Regulation (EU) 257/2010, the 2003 Terms of References are replaced by those below.
1.1.2. Terms of Reference as provided by the European Commission
The Commission asks EFSA to re‐evaluate the safety of food additives already permitted in the Union before 2009 and to issue scientific opinions on these additives, taking especially into account the priorities, procedures and deadlines that are enshrined in the Regulation (EU) No 257/2010 of 25 March 2010 setting up a programme for the re‐evaluation of approved food additives in accordance with the Regulation (EC) No 1333/2008 of the European Parliament and of the Council on food additives.
1.2. Information on existing authorisations and evaluations
Glutamic acid–glutamates (E 620–625) are authorised food additives in the EU according to Annex II of Regulation (EC) No 1333/2008 and specific purity criteria have been defined in the Commission Regulation (EU) No 231/20125.
According to Commission Regulation (EC) No 609/20136, l‐glutamic acid is included in the list of amino acids authorised to be added to food for special medical purposes and total diet replacement for weight control.
Glutamic acid and its sodium, potassium, calcium, ammonium and magnesium salts, used as food additives, have previously been evaluated by the SCF in 1990 (SCF, 1991). The SCF in its report stated that they were aware of a series of studies on monosodium glutamate (MSG), but did not give any details or included any primary study reports in the reference list. The SCF concluded the following: ‘The Committee established a group ADI “not specified” on the basis of the data provided and in view of the large normal dietary intake of glutamates’.
Glutamic acid and its sodium, potassium, calcium, ammonium and magnesium salts were evaluated by Joint FAO/WHO Expert Committee on Food Additives (JECFA) in 1970 and 1973 when a group acceptable daily intake (ADI) of 0–120 mg/kg body weight (bw) per day expressed as glutamic acid was established (JECFA, 1971, 1974). In 1987, JECFA re‐evaluated glutamic acid and its sodium, potassium, calcium, ammonium and magnesium salts and established a group ADI ‘not specified’ (JECFA, 1987, 1988). In 2004, JECFA evaluated a series of amino acids and related substances, including glutamic acid, as flavouring agents (JECFA, 2005, 2006a). The Committee reviewed new studies on glutamic acid and MSG published since the last evaluation in 1987 and concluded by maintaining the previously established group ADI ‘not specified’ for glutamic acid and its salts (JECFA, 2006a).
The EFSA ANS Panel evaluated a new production method of glutamic acid where l‐glutamic acid is produced by fed‐batch fermentation using carbon sources of vegetal origin (sucrose). While formerly fermentation was done by Corynebacterium glutamicum strain 2256, the new production method used the genetically modified Corynebacterium glutamicum EA‐12 strain. The process for producing the glutamates was mainly unchanged. The Panel concluded that there was no safety concern from the change in the production method of these food additives (EFSA ANS Panel, 2015).
Glutamic acid and MSG have been registered under the REACH Regulation 1907/20067 (ECHA, Available online: https://echa.europa.eu/brief-profile/-/briefprofile/100.000.267, accessed on 9/5/2017).
Glutamic acid and MSG are permitted in cosmetic products (European Commission database‐CosIng8).
2. Data and methodologies
2.1. Data
The Panel on Food Additives and Nutrient Sources added to Food (ANS) was not provided with a newly submitted dossier. EFSA launched public calls for data,9 , 10 to collect information from interested parties.
The Panel based its assessment on information submitted to EFSA following the public calls for data, information from previous evaluations and additional available literature up to April 2017. Attempts were made at retrieving relevant original study reports on which previous evaluations or reviews were based. However, not always these were available to the Panel.
The EFSA Comprehensive European Food Consumption Database (Comprehensive Database11) was used to estimate the dietary exposure.
The Mintel's Global New Products Database (GNPD) is an online resource listing food products and compulsory ingredient information that should be included in labelling. This database was used to verify the use of glutamic acid–glutamates (E 620–625) as food additives in food products.
2.2. Methodologies
This opinion was formulated following the principles described in the EFSA Guidance on transparency with regard to scientific aspects of risk assessment (EFSA Scientific Committee, 2009) and following the relevant existing guidance documents from the EFSA Scientific Committee.
The Panel assessed the safety of glutamic acid–glutamates (E 620–625) as food additives in line with the principles laid down in Regulation (EU) 257/2010 and the relevant guidance documents: Guidance on submission for food additive evaluations by the SCF (2001).
When the test substance was administered in the feed or in the drinking water, but doses were not explicitly reported by the authors as mg/kg body weight (bw) per day based on actual feed or water consumption, the daily intake was calculated by the Panel using the relevant default values as indicated in the EFSA Scientific Committee Guidance document (EFSA Scientific Committee, 2012) for studies in rodents or, in the case of other animal species, by JECFA (2000). In these cases, the daily intake is expressed as equivalent. When in human studies in adults (aged above 18 years), the dose of the test substance administered was reported in mg/person per day, the dose in mg/kg bw per day was calculated by the Panel using a body weight of 70 kg as default for the adult population as referred to by the EFSA Scientific Committee Guidance document (EFSA Scientific Committee, 2012).
Dietary exposure to glutamic acid–glutamates (E 620–625) from their use as food additives was estimated combining individual food consumption data available within the EFSA Comprehensive European Food Consumption Database with the maximum permitted levels (MPLs) and reported use levels submitted to EFSA following a call for data. Different scenarios were used to calculate exposure (see Section 3.3.1). Uncertainties in the exposure assessment were identified and discussed.
3. Assessment
3.1. Technical data
3.1.1. Identity of the substance
Glutamic acid (E 620)
According to Commission Regulation (EU) No 231/2012, it is identified as:
Chemical name: l‐Glutamic acid, l‐2‐amino‐pentanedioic acid
EINECS Number: 200‐293‐7
Chemical formula: C5H9NO4
Molecular weight: 147.13 g/mol
Solubility: It is sparingly soluble in water and practically insoluble in ethanol or ether.
Physical description: l‐Glutamic acid occurs as white crystals or crystalline powder.
The Panel noted that the name of the food additive ‘E 620 glutamic acid’ does not specify the isomer that is authorised; however from the chemical name provided in the Regulation, it is clear that only l‐glutamic acid is authorised.
The corresponding CAS number for l‐glutamic acid is 56‐86‐0 (SciFinder12, software). Its structural formula is presented in Figure 1.
Figure 1.
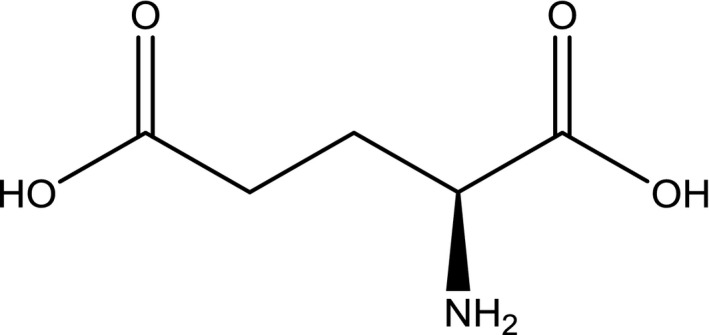
Structural formula of l‐glutamic acid
Synonyms: l‐Glutamic acid; l‐α‐aminoglutaric acid; l‐(+)‐glutamic acid and (S)‐2‐amino‐pentanedioic acid).
Octanol/water partition coefficient (log Po/w) of < –4 (NOTOX, 2010a [Documentation provided to EFSA n. 16]). The reported pKa values ranges are 2.16 (at 25°C), 9.58 and 4.15 (at 25°C) for the α‐carboxyl group, the α‐ammonium ion and the side chain group, respectively (Haynes, 2012).
Monosodium glutamate (E 621)
According to Commission Regulation (EU) No 231/2012 it is identified as:
Chemical name: monosodium l‐glutamate monohydrate
EINECS Number: 205‐538‐1
Chemical formula: C5H8NaNO4 · H2O
Molecular weight: 187.13 g/mol
Solubility: It is freely soluble in water and practically insoluble in ethanol or ether.
Physical description: Monosodium l‐glutamate occurs as white, practically odourless crystals or crystalline powder.
The Panel noted that the EINECS Number provided in the Commission Regulation (EU) No 231/2012 corresponds to the anhydrous form (EC inventory, Available online: https://echa.europa.eu/information-on-chemicals/ec-inventory, accessed on 9/5/2017) while the substance authorised as a food additive is the monohydrate. The structural formula of monosodium glutamate is presented in Figure 2.
Figure 2.
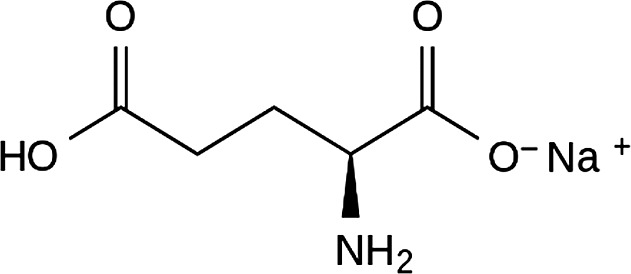
Structural formula of monosodium l‐glutamate anhydrous
CAS number: 6106‐04‐3 for the monohydrate form (SciFinder12, software); no EINECS Number for this CAS number (EC inventory, online).
Synonyms: Monosodium l‐glutamate monohydrate; sodium glutamate monohydrate; l‐glutamic acid, sodium salt, monohydrate (1:1:1); l‐glutamic acid monosodium salt monohydrate.
Monopotassium glutamate (E 622)
According to Commission Regulation (EU) No 231/2012, it is identified as:
Chemical name: Monopotassium l‐glutamate monohydrate
EINECS Number: 243‐094‐0
Chemical formula: C5H8KNO4 · H2O
Molecular weight: 203.24 g/mol
Solubility: It is freely soluble in water and practically insoluble in ethanol or ether.
Physical description: monopotassium l‐glutamate occurs as white, practically odourless crystals or crystalline powder.
The Panel noted that the EINECS Number provided in the Commission Regulation (EU) No 231/2012 corresponds to the anhydrous form while the substance authorised as a food additive is the monohydrate. The structural formula of monopotassium glutamate is presented in Figure 3.
Figure 3.
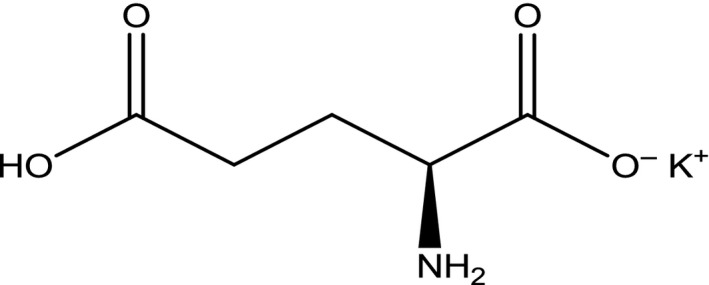
Structural formula of monopotassium l‐glutamate anhydrous
Synonyms: Monopotassium l‐glutamate monohydrate; monopotassium glutamate monohydrate; l‐glutamic acid, potassium salt, monohydrate (1:1:1)
CAS number: 6382‐01‐0 for the monohydrate form (SciFinder12, software); no EINECS Number for this CAS number (EC inventory, online).
Calcium diglutamate (E 623)
According to Commission Regulation (EU) No 231/2012, it is identified as:
Chemical name: Monocalcium di‐l‐glutamate
EINECS Number: 242‐905‐5
Chemical formula: C10H16CaN2O8 · nH2O (n = 0, 1, 2 or 4)
Molecular weight: 332.32 g/mol (anhydrous)
Solubility: It is freely soluble in water and practically insoluble in ethanol or ether.
Physical description: Calcium diglutamate occurs as white, practically odourless crystals or crystalline powder.
The Panel noted that the EINECS Number provided in the Commission Regulation (EU) No 231/2012 corresponds to the CAS number 19238‐49‐4 (EC inventory, online) that refers to a calcium salt where the ratio of calcium molecule is unknown (C5H9NO4 · X Ca) (SciFinder12, software).
The EINECS and CAS numbers for the anhydrous form are 227‐838‐1 and 5996‐22‐5, respectively. The CAS number for calcium diglutamate dihydrate is 129412‐85‐7 and for calcium diglutamate tetrahydrate 69704‐19‐4. The structural formula for calcium diglutamate is presented in Figure 4.
Figure 4.
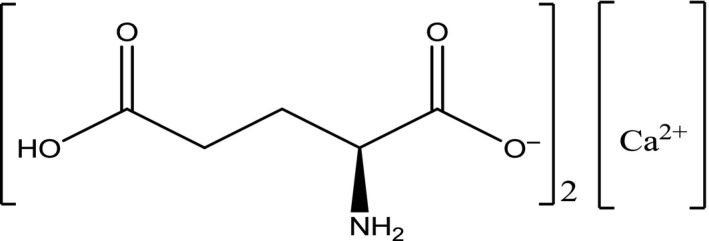
Structural formula of calcium di‐l‐glutamate anhydrous
Synonyms (anhydrous form): Calcium di‐l‐glutamate; calcium glutamate; l‐glutamic acid, calcium salt (2:1).
Monoammonium glutamate (E 624)
According to Commission Regulation (EU) No 231/2012, it is identified as:
Chemical name: Monoammonium l‐glutamate monohydrate
EINECS Number: 231‐447‐1
Chemical formula: C5H12N2O4 · H2O
Molecular weight: 182.18 g/mol
Solubility: It is freely soluble in water and practically insoluble in ethanol or ether.
Physical description: Monoammonium l‐glutamate occurs as white, practically odourless crystals or crystalline powder.
The Panel noted that the EINECS Number provided in the Commission Regulation (EU) No 231/2012 corresponds to the anhydrous form (EC inventory, online) while the substance authorised as a food additive is the monohydrate.
CAS numbers: 139883‐82‐2 for the monohydrate form and 7558‐63‐6 for the anhydrous form (SciFinder12, software). The structural formula for monoammonium glutamate is presented in Figure 5.
Figure 5.
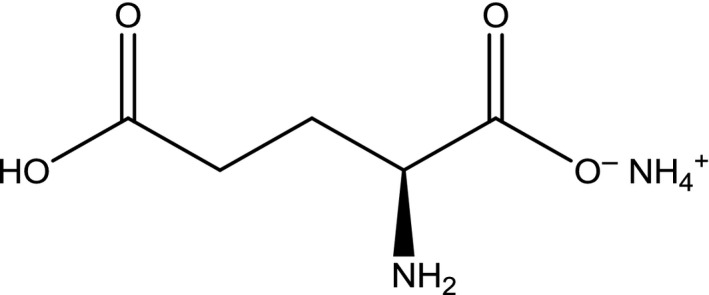
Structural formula of monoammonium l‐glutamate anhydrous
Synonyms: monoammonium l‐glutamate monohydrate; ammonium glutamate monohydrate; l‐glutamic acid, ammonium salt, monohydrate (1:1:1).
Magnesium diglutamate (E 625)
According to Commission Regulation (EU) No 231/2012, it is identified as:
Chemical name: Monomagnesium di‐l‐glutamate tetrahydrate
EINECS Number: 242‐413‐0
Chemical formula: C10H16MgN2O8 · 4 H2O
Molecular weight: 388.62 g/mol
Solubility: It is very soluble in water and practically insoluble in ethanol or ether.
Physical description: Magnesium di‐l‐glutamate occurs as white, practically odourless crystals or crystalline powder.
The Panel noted that the EINECS Number provided in the Commission Regulation (EU) No 231/2012 corresponds to the CAS number 18543‐68‐5 (EC inventory, online) that refers to an anhydrous compound (SciFinder12, software).
The CAS number 110011‐84‐2 refers to the anhydrous l‐glutamic magnesium salt (2:1) (SciFinder12, software). The structural formula of magnesium diglutamate is presented in Figure 6.
Figure 6.
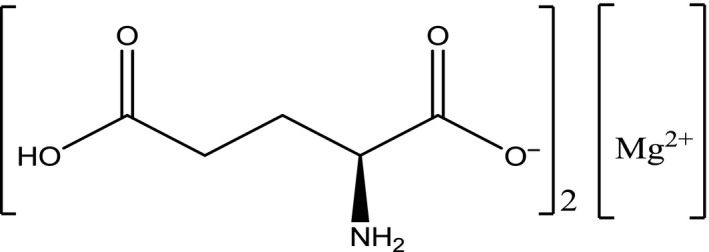
Structural formula of magnesium di‐l‐glutamate anhydrous
Synonyms (anhydrous form): Magnesium di‐l‐glutamate; magnesium glutamate; l‐glutamic acid, magnesium salt (2:1).
3.1.2. Specifications
The specifications for glutamic acid (E 620), monosodium glutamate (E 621), monopotassium glutamate (E 622), calcium diglutamate (E 623), monoammonium glutamate (E 624) and magnesium diglutamate (E 626) used as food additives as defined in the Commission Regulation (EU) No 231/2012 and by JECFA (2006b) are listed in Tables 1, 2, 3, 4, 5–6.
Table 1.
Specifications for glutamic acid (E 620) according to Commission Regulation (EU) No 231/2012 and JECFA (2006b)
| Commission Regulation (EU) No 231/2012 | JECFA (2006b) | |
|---|---|---|
| Definition | ||
| Assay | Not less than 99.0% and not more than 101.0% on the anhydrous basis | Not less than 99.0% on the dried basis |
| Solubility | Sparingly soluble in water; practically insoluble in ethanol or ether | Sparingly soluble in water; practically insoluble in ethanol or ether |
| Description | White crystals or crystalline powder | Colourless or white crystals or crystalline powder |
| Identification | ||
| Test for glutamic acid (by thin layer chromatography) | Passes test | Passes test |
| Specific rotation | [α]D 20 between + 31.5° and + 32.2° (10% solution (anhydrous basis) in 2N HCl, 200 mm tube) | [α]D 20 between + 31.5° and + 32.2° (10% (w/v) soln in 2N HCl) |
| pH | 3.0–3.5 (saturated solution) | 3.0–3.5 (saturated solution) |
| Purity | ||
| Loss on drying | Not more than 0.2% (80°C, 3 h) | Not more than 0.2% (80°C, 3 h) |
| Sulphated ash | Not more than 0.2% | Not more than 0.2% |
| Chloride | Not more than 0.2% | Not more than 0.2% |
| Pyrrolidone carboxylic acid | Not more than 0.2% | Passes test |
| Arsenic | Not more than 2.5 mg/kg | Not more than 3 mg/kg |
| Lead | Not more than 1 mg/kg | Not more than 1 mg/kg |
Table 2.
Specifications for monosodium glutamate (E 621) according to Commission Regulation (EU) No 231/2012 and JECFA (2006b)
| Commission Regulation (EU) No 231/2012 | JECFA (2006b) | |
|---|---|---|
| Definition | ||
| Assay | Not less than 99.0% and not more than 101.0% on the anhydrous basis | Not less than 99.0% on the dried basis |
| Solubility | Freely soluble in water; practically insoluble in ethanol or ether | Freely soluble in water; sparingly soluble in ethanol; practically insoluble in ether |
| Description | White, practically odourless crystals or crystalline powder | White, practically odourless crystals or crystalline powder |
| Identification | ||
| Test for glutamic acid (by thin layer chromatography) | Passes test | Passes test |
| Test for sodium | Passes test | Passes test |
| Specific rotation | [α]D 20 between + 24.8° and + 25.3° (10% solution (anhydrous basis) in 2N HCl, 200 mm tube) | [α]D 20 between + 24.8° and + 25.3° (10% (w/v) solution in 2N hydrochloric acid) |
| pH value | 6.7–7.2 (5% solution) | 6.7–7.2 (1 in 50 solution) |
| Purity | ||
| Loss on drying | Not more than 0.5% (98°C, 5 h) | Not more than 0.5% (98°C, 5 h) |
| Chloride | Not more than 0.2% | Not more than 0.2% |
| Pyrrolidone carboxylic acid | Not more than 0.2% | Passes test |
| Lead | Not more than 1 mg/kg | Not more than 1 mg/kg |
Table 3.
Specifications for monopotassium glutamate (E 622) according to Commission Regulation (EU) No 231/2012 and JECFA (2006b)
| Commission Regulation (EU) No 231/2012 | JECFA (2006b) | |
|---|---|---|
| Definition | ||
| Assay | Not less than 99.0% and not more than 101.0% on the anhydrous basis | Not less than 99.0% on the dried basis |
| Solubility | Freely soluble in water; practically insoluble in ethanol or ether | Freely soluble in water; slightly soluble in ethanol |
| Description | White, practically odourless crystals or crystalline powder | – |
| Identification | ||
| Test for glutamic acid (by thin layer chromatography) | Passes test | Passes test |
| Test for potassium | Passes test | Passes test |
| Specific rotation | [α]D 20 between + 22.5° and + 24.0° (10% solution (anhydrous basis) in 2N HCl, 200 mm tube) | [α]D 20 between + 22.5° and + 24.0° (10% (w/v) solution in 2N HCl) |
| pH value | 6.7–7.3 (2% solution) | 6.7–7.3 (1 in 50 solution) |
| Purity | ||
| Loss on drying | Not more than 0.5% (98°C, 5 h) | Not more than 0.2% (80°C, 5 h) |
| Chloride | Not more than 0.2% | Not more than 0.2% |
| Pyrrolidone carboxylic acid | Not more than 0.2% | Passes test |
| Lead | Not more than 1 mg/kg | Not more than 1 mg/kg |
Table 4.
Specifications for calcium diglutamate (E 623) according to Commission Regulation (EU) No 231/2012 and JECFA (2006b)
| Commission Regulation (EU) No 231/2012 | JECFA (2006b) | |
|---|---|---|
| Definition | ||
| Assay | Not less than 98.0% and not more than 102.0% on the anhydrous basis | Not less than 98.0% and not more than 102.0% on the anhydrous basis |
| Solubility | Freely soluble in water; practically insoluble in ethanol or ether | Freely soluble in water |
| Description | White, practically odourless crystals or crystalline powder | White, practically odourless crystals or crystalline powder |
| Identification | ||
| Test for glutamic acid (by thin layer chromatography) | Passes test | Passes test |
| Test for calcium | Passes test | Passes test |
| Specific rotation | [α]D 20 between + 27.4° and + 29.2° (for calcium diglutamate with n = 4)(10% solution (anhydrous basis) in 2N HCl, 200 mm tube) | [α]D 20 between + 27.4° and + 29.2° (10% (w/v) solution in 2N hydrochloric acid) |
| Purity | ||
| Water | Not more than 19.0% (for calcium diglutamate with n = 4) (Karl Fischer) | Not more than 19% (Karl Fischer method) |
| Chloride | Not more than 0.2% | Not more than 0.2% |
| Pyrrolidone carboxylic acid | Not more than 0.2% | Passes test |
| Lead | Not more than 1 mg/kg | Not more than 1 mg/kg |
Table 5.
Specifications for monoammonium glutamate (E 624) according to Commission Regulation (EU) No 231/2012 and JECFA (2006b)
| Commission Regulation (EU) No 231/2012 | JECFA (2006b) | |
|---|---|---|
| Definition | ||
| Assay | Not less than 99.0% and not more than 101.0% on the anhydrous basis | Not less than 99.0% on the dried basis |
| Solubility | Freely soluble in water; practically insoluble in ethanol or ether | Freely soluble in water |
| Description | White, practically odourless crystals or crystalline powder | White, practically odourless crystals or crystalline powder |
| Identification | ||
| Test for glutamic acid (by thin layer chromatography) | Passes test | Passes test |
| Test for ammonium | Passes test | Passes test |
| Specific rotation | [α]D 20 between + 25.4° and + 26.4° (10% solution (anhydrous basis) in 2N HCl, 200 mm tube) | [α]D 20 between + 25.4° and + 26.4° (10% (w/v) solution in 2N HCl) |
| pH | 6.0–7.0 (5% solution) | 6.0–7.0 (1 in 20 solution) |
| Purity | ||
| Loss on drying | Not more than 0.5% (50°C, 4 h) | Not more than 0.5% (50°C, 4 h) |
| Sulphated ash | Not more than 0.1% | Not more than 0.1% |
| Pyrrolidone carboxylic acid | Not more than 0.2% | Passes test |
| Lead | Not more than 1 mg/kg | Not more than 1 mg/kg |
Table 6.
Specifications for magnesium diglutamate (E 625) according to Commission Regulation (EU) No 231/2012 and JECFA (2006b)
| Commission Regulation (EU) No 231/2012 | JECFA (2006b) | |
|---|---|---|
| Definition | ||
| Assay | Not less than 95.0% and not more than 105.0% on the anhydrous basis | Not less than 95.0% and not more than 105.0% on the anhydrous basis |
| Solubility | Very soluble in water; practically insoluble in ethanol or ether | Very soluble in water; insoluble in ethanol |
| Description | Odourless, white or off‐white crystals or powder | Odourless, white or off‐white crystals or powder |
| Identification | ||
| Test for glutamic acid (by thin layer chromatography) | Passes test | Passes test |
| Test for magnesium | Passes test | Passes test |
| Specific rotation | [[α]D 20 between + 23.8° and + 24.4° (10% solution (anhydrous basis) in 2N HCl, 200 mm tube) | [α]D 20 between + 23.8° and + 24.4° (10% (w/v) solution in 2N HCl) |
| pH | 6.4–7.5 (10% solution) | 6.4–7.5 (1 in 10 solution) |
| Purity | ||
| Water | Not more than 24% (Karl Fischer) | Not more than 24% (Karl Fischer Method) |
| Chloride | Not more than 0.2% | Not more than 0.2% |
| Sulphates | – | Not more than 0.2% |
| Pyrrolidone carboxylic acid | Not more than 0.2% | Passes test |
| Lead | Not more than 1 mg/kg | Not more than 1 mg/kg |
The Panel noted that, according to the EU specifications for glutamic acid (E 620), impurities of the toxic elements arsenic and lead are accepted up to concentrations of 2.5 and 1 mg/kg, respectively, and for the salts of glutamic acid (E 621–625) lead is accepted up a concentration of 1 mg/kg. Contamination at those levels could have a significant impact on the exposure to these metals, for which the intake is already close to the health based guidance values or benchmark doses (lower confidence limits) established by EFSA (EFSA CONTAM Panel, 2009, 2010, 2012, 2014).
The Panel noted that data on bacterial contamination are relevant for the specifications to assure that the bacteria (as well as their products) used in the production process were removed.
3.1.3. Manufacturing process
Glutamic acid is widely distributed as an amino acid in living organisms and was originally extracted, isolated and purified as monosodium glutamate from natural sources (Ault, 2004; Sano, 2009). According to information in these reports, chemical synthesis was the method of choice for production of monosodium glutamate, but presently all glutamate manufacturers seemed to use the fermentation method. In this method glutamic acid is synthesised in large amounts by coryneform bacteria which require biotin for growth. When the selected microorganisms are cultured with carbohydrates and ammonia and under biotin‐limiting conditions they release the l‐form of glutamic acid into the culture medium. At the end of the fermentation, l‐glutamic acid is purified from the culture medium. l‐Glutamic acid may be converted to the monosodium salt by the addition of sodium hydroxide (Sano, 2009).
EFSA evaluated a process for the production of l‐glutamic acid produced by fermentation using the genetically modified C. glutamicum EA‐12 strain obtained from strain 2256 (EFSA ANS Panel, 2015). The modified strain produces l‐glutamic acid more efficiently compared to the ‘standard’ biosynthetic/metabolic pathway present in C. glutamicum described above. Details of the process were described in EFSA opinion evaluating the safety of the change in the production method for the production of l‐glutamic acid (E 620), monosodium‐l‐glutamate (E 621), monopotassium l‐glutamate (E 622), calcium di‐l‐glutamate (E 623), monoammonium l‐glutamate (E 624) and magnesium di‐l‐glutamate (E 625).
In the processes described above, glutamic acid is produced by fed‐batch fermentation using carbon sources of vegetal origin (sucrose), nitrogen sources, salts and vitamins. According to the new manufacturing process (EFSA ANS Panel, 2015), there are no differences in the qualitative composition of the medium between the new and the conventional process except that the quantity of the raw materials used have been adapted, due to the particularity of the new strain.
l‐Glutamic acid is produced in two consecutive production steps: first acid is produced and subsequently purified and extracted for the production of l‐glutamic acid (E 620), monosodium l‐glutamate (E 621) and other glutamate salts (i.e. monopotassium l‐glutamate (E 622), calcium di‐l‐glutamate (E 623), monoammonium l‐glutamate (E 624) and magnesium di‐l‐glutamate (E 625).
3.1.4. Methods of analysis in food
The Panel considered that in the absence of hydrolysis step in the analysis of food samples, the method of analysis can measure only free glutamic acid/glutamate and not glutamic acid contained in the peptides or proteins.
No analytical method is available to differentiate between added and natural occurring amino acids and especially glutamic acid/glutamate (Drauz et al., 2000). For analysis of the total free glutamic acid/glutamate in food, a number of methods of analysis are described in published literature. Those methods are based on high‐performance liquid chromatography (HPLC) and ion‐exchange chromatography (IEC), in which amino acids are extracted from food and a simultaneous analysis of all amino acids is performed. Other reported methods that separate amino acids in food are other chromatographic methods (paper, thin‐layer and gas) and electrophoresis. The separated amino acids are identified by various spectroscopic methods. Enzymatic and microbiological assays have also been used for the analysis of glutamic acid/glutamate in food (Maga, 1983).
An HPLC‐based method has been used by Sporns (1982) where after a clean‐up with acid resin, glutamic acid is measured in croutons, chicken and soy sauce (refractive index detection, with a limit of quantification (LOQ) of 0.15 g/kg expressed as MSG and recovery rates in accordance with internationally accepted performance criteria (Bravenboer et al., 1995)). An advantage of this method is that it can also determine the reaction product pyroglutamate, an information which is useful in order to conclude on the total added amount of glutamic acid/glutamate to food. Montaño et al. (2000) have measured amino acids, including glutamic acid, in olive brine with a precolumn derivatisation using 9‐fluorenylmethylchloroformate (UV detection, with recovery rates in accordance with internationally accepted performance criteria (Bravenboer et al., 1995)). IEC was used by Coppola et al. (1975) to separate MSG in food products, which was subsequently fluorometrically determined with fluorescamine (with an LOQ of 0.5 g/kg expressed as MSG and recovery rates in accordance with internationally accepted performance criteria).
Krishna Veni et al. (2010) applied a high‐performance thin‐layer chromatography (HPTLC) for the detection of glutamate in food products by using ninhydrin for colour development. An option for quantification by using a scanner was also proposed. Gas chromatography (GC coupled with flame ionisation detector (FID) and mass spectrometric (MS) detector) was used by Conacher et al. (1979) to determine glutamate in soups after a derivatisation into its trimethyl silyl ether with a limit of detection (LOD) of 0.05% equal to 500 mg/kg expressed as MSG. Lau and Mok (1995) used liquid chromatography combined with a conductivity detector, in order to determine MSG in soup, baby food, sauces and pistachios after a clean‐up with an ion‐exchange column and its conversion into glutamic acid, with a LOQ of 70 mg/kg expressed as MSG.
Official method AOAC 970.37 (AOAC, 2000) uses potentiometric titration (pH) after retention of glutamic acid on a chromatographic column to measure glutamic acid/glutamate content in food. No information is given on method performance.
The utilisation of a rotating bioreactor containing enzymes and continuous‐flow/stopped‐flow/continuous‐flow operation equipped with an amperometer has been used to measure MSG in food (Janarthanan and Mottola, 1998). Glutamic acid was deaminated by glutamic dehydrogenase in the presence of β‐NAD+ and the potentiometric estimation of the liberated ammonia was used to estimate the glutamic acid content (Nikolelis, 1987). The same reaction, but in combination with colour development and spectrophotometric measurement, is described in Raugel (1999). Vahjen et al. (1991) developed an enzyme electrode with 1,1′‐dimethylferrocene (DMF) as an electron mediator and the method equivalence with an internationally applied method was demonstrated. Basu et al. (2006) have used a similar biosensor with an LOD sufficient to detect as low as 2 mg glutamic acid/kg of food sample. Yılmaz and Karakuş (2011) have developed a potentiometric glutamate biosensor, where ammonium ions produced after an enzymatic reaction were determined potentiometrically and the method equivalence with an internationally applied enzymatic method was demonstrated for chicken bouillon. Udomsopagit et al. (1998) and Mizutani et al. (1998) developed flow injection analysis combined with an enzymic sensor, and presented the method equivalence with other internationally applied enzymatic methods. Isa and Ghani (2009) have developed a solid state glutamate sensor in combination with flow injection analysis and an LOD of 0.008 mol equal to 1.4 g/kg of food sample. Kampha et al. (2004) and Afraa et al. (2013) used an enzymatic method combined with a spectrophotometric measurement for l‐glutamate in food. In the latter publication (Afraa et al., 2013), the method performance was demonstrated to be in line with internationally accepted performance criteria.
Chapman and Zhou (1999) applied microplate‐based fluorometric methods for the enzymatic determination of l‐glutamate in food samples based on cycling enzymes, by demonstrating a LOD of 0.5 pmol/well equal to 0.935 g/soup sample.
Other methods of analysis include the use of an optical biosensor for l‐glutamate measurement by using an optical fibre spectrophotometer (Muslim et al., 2012) with a LOD of 5 μmol in the diluted sample equal to 1.87 mg/kg, isotachophoresis where glutamic acid/glutamate are extracted with water and then inserted into an instrument for tachophoretic separation, with a LOD of 10 mg/kg (Kenndler et al., 1980); and the use of a poly(methylmethacrylate) chip, and on‐column conductivity detection sensors, leading to a separation via isotachophoresis with limited report on method performance (Bodor et al., 2001).
3.1.5. Reaction and fate in food
At high temperatures, glutamic acid like other amino acids can react with available sugars via the Maillard reaction to produce a wide range of compounds not further specified (Yoshida, 1978 as referred to by FASEB, 1995).
MSG in canned food was recovered at 93–100% following canning (124°C for 30 min, pH 5) (Nguyen and Sporns, 1985). The canned food contained various concentrations of phosphates, salt, casein, glucose and potato starch. MSG was thus found to be very stable.
The role of pH (in the range of 0–14), temperature, time, oxygen and nitrogen on the stability of solutions at 1 and 5 g/L of glutamic acid and MSG was evaluated (Gayte‐Sorbier et al., 1985). The amount of glutamic acid was measured during storage at room temperature without atmospheric contact for 24 h. In acidic conditions, small changes began after 3 days of storage. In alkaline conditions, small changes began after 15 days of storage. The greatest change occurred at a pH of 3 where 15% of glutamic acid had disappeared and converted into pyroglutamic acid. In the presence of oxygen, the conversion was faster and larger, whereas the presence of nitrogen did not influence the conversion. Storage at 4°C caused better preservation than storage at room temperature under the same conditions. Changes in the amount of glutamic acid followed boiling for 60 min in various pH values have been studied. The greatest change occurred at a pH of 2 where 17% of glutamic acid disappeared. When glutamic acid was autoclaved, the conversion was greater and faster than that observed in boiling. Under all conditions, the results with MSG were similar to the results with glutamic acid.
A mixture of soybean oil, sugar and MSG was heated at temperatures between 100 and 170°C for different time periods. When MSG and sugar in water were heated at temperatures close to 100°C, 2,5‐dimethyl pyrazine and methyl pyrazine were detected after 160 min. Samples containing MSG, oil and sugar produced 2‐pyrrolidone when heated above 140°C for 40 min. The samples containing soybean oil, sugar and MSG produced 23 different pyrazines when heated at 170°C after 40 min (Wu et al., 2000).
The concentration of natural occurring free glutamic acid in royal jelly (0.46 g/kg) showed no significant difference during storage for up to 10 months at −18°C, 4°C and 25°C (Liming et al., 2009).
Cooking of grape must for 30 h caused an almost linear reduction of natural occurring glutamic acid (Montevecchi et al., 2011). As was already demonstrated before by Nunes and Cavalheiro (2007), glutamic acid was converted into pyroglutamic acid.
In a study with unpasteurised green olives in brine, MSG was extensively degraded (> 75% degradation) after 54 weeks of storage with a higher degradation rate in glass bottles compared with plastic pouches. In the presence of potassium sorbate, MSG was also considerably degraded in olives packed in plastic pouches (> 50% degradation), but hardly degraded in glass bottles. The results indicated that MSG degradation in olives is due to the action of both lactic acid bacteria and yeasts, with the formation of γ‐aminobutyric acid as the major end‐product (De Castro et al., 2014).
In the study by Lee et al. (2014a,b), it has been demonstrated that in the presence of high levels of dissolved hydrogen (i.e. a high redox state) the oxidative decomposition of glutamic acid is reduced at elevated temperatures.
3.2. Authorised uses and use levels
Maximum levels of glutamic acid–glutamates (E 620–625) have been defined in accordance with Annex II to Regulation (EC) No 1333/2008 on food additives, as amended. In this document, these levels are named MPLs.
Currently, glutamic acid–glutamates (E 620–625) are authorised at quantum satis (QS) in salt substitutes (FCS 12.1.2), seasonings and condiments (FCS 12.2.2) and are included in the Group I of food additives at the MPL of 10,000 mg/kg or 10,000 mg/L.
Table 7 summarises foods that are permitted to contain glutamic acid–glutamates (E 620–625) and the corresponding MPLs as set by Annex II to Regulation (EC) No 1333/2008.
Table 7.
MPLs of glutamic acid–glutamates (E 620–625) in foods according to the Annex II to Regulation (EC) No 1333/2008
| Food category number | Food category name | E‐number/group | Restrictions/exception | MPL (mg/L or mg/kg as appropriate) |
|---|---|---|---|---|
| 01.3 | Unflavoured fermented milk products, heat‐treated after fermentation | Group I | 10,000a | |
| 01.4 | Flavoured fermented milk products including heat‐treated products | Group I | 10,000a | |
| 01.6.3 | Other creams | Group I | 10,000a | |
| 01.7.1 | Unripened cheese excluding products falling in category 16 | Group I | Except mozzarella | 10,000a |
| 01.7.5 | Processed cheese | Group I | 10,000a | |
| 01.7.6 | Cheese products (excluding products falling in category 16) | Group I | 10,000a | |
| 01.8 | Dairy analogues, including beverage whiteners | Group I | 10,000a | |
| 02.2.2 | Other fat and oil emulsions including spreads as defined by Council Regulation (EC) No 1234/2007 and liquid emulsions | Group I | 10,000a | |
| 02.3 | Vegetable oil pan spray | Group I | 10,000a | |
| 03 | Edible ices | Group I | 10,000a | |
| 04.2.1 | Dried fruit and vegetables | Group I | 10,000a | |
| 04.2.2 | Fruit and vegetables in vinegar, oil, or brine | Group I | 10,000a | |
| 04.2.4.1 | Fruit and vegetable preparations excluding compote | Group I | 10,000a | |
| 04.2.5.4 | Nut butters and nut spreads | Group I | 10,000a | |
| 04.2.6 | Processed potato products | Group I | 10,000a | |
| 05.1 | Cocoa and chocolate products as covered by Directive 2000/36/EC | Group I | Only energy‐reduced or with no added sugar | 10,000a |
| 05.2 | Other confectionery including breath freshening microsweets | Group I | 10,000a | |
| 05.3 | Chewing gum | Group I | 10,000a | |
| 05.4 | Decorations, coatings and fillings, except fruit‐based fillings covered by category 4.2.4 | Group I | 10,000a | |
| 06.2.2 | Starches | Group I | 10,000a | |
| 06.3 | Breakfast cereals | Group I | 10,000a | |
| 06.4.2 | Dry pasta | Group I | Only gluten free and/or pasta intended for hypoproteic diets in accordance with Directive 2009/39/EC | 10,000a |
| 06.4.4 | Potato Gnocchi | Group I | Except fresh refrigerated potato gnocchi | 10,000a |
| 06.4.5 | Fillings of stuffed pasta (ravioli and similar) | Group I | 10,000a | |
| 06.5 | Noodles | Group I | 10,000a | |
| 06.6 | Batters | Group I | 10,000a | |
| 06.7 | Pre‐cooked or processed cereals | Group I | 10,000a | |
| 07.1 | Bread and rolls | Group I | Except products in 7.1.1 and 7.1.2 | 10,000a |
| 07.2 | Fine bakery wares | Group I | 10,000a | |
| 08.3.1 | Non‐heat‐treated meat products | Group I | 10,000a | |
| 08.3.2 | Heat‐treated meat products | Group I | Except foie gras, foie gras entier, blocs de foie gras, Libamáj, libamáj egészben, libamáj tömbben | 10,000a |
| 08.3.3 | Casings and coatings and decorations for meat | Group I | 10,000a | |
| 09.2 | Processed fish and fishery products including molluscs and crustaceans | Group I | 10,000a | |
| 09.3 | Fish roe | Group I | Only processed fish roe | 10,000a |
| 10.2 | Processed eggs and egg products | Group I | 10,000a | |
| 11.2 | Other sugars and syrups | Group I | 10,000a | |
| 12.1.2 | Salt substitutes | E 620–625 | Quantum satis | |
| 12.2.2 | Seasonings and condiments | E 620–625 | Quantum satis | |
| 12.3 | Vinegars | Group I | 10,000a | |
| 12.4 | Mustard | Group I | 10,000a | |
| 12.5 | Soups and broths | Group I | 10,000a | |
| 12.6 | Sauces | Group I | 10,000a | |
| 12.7 | Salads and savoury‐based sandwich spreads | Group I | 10,000a | |
| 12.8 | Yeast and yeast products | Group I | 10,000a | |
| 12.9 | Protein products, excluding products covered in category 1.8 | Group I | 10,000a | |
| 13.2 | Dietary foods for special medical purposes defined in Directive 1999/21/EC (excluding products from food category 13.1.5) | Group I | 10,000a | |
| 13.3 | Dietary foods for weight control diets intended to replace total daily food intake or an individual meal (the whole or part of the total daily diet) | Group I | 10,000a | |
| 13.4 | Foods suitable for people intolerant to gluten as defined by Regulation (EC) No 41/2009 | Group I | Including dry pasta | 10,000a |
| 14.1.2 | Fruit juices as defined by Directive 2001/112/EC and vegetable juices | Group I | Only vegetable juices | 10,000a |
| 14.1.3 | Fruit nectars as defined by Directive 2001/112/EC and vegetable nectars and similar products | Group I | Only vegetable nectars | 10,000a |
| 14.1.4 | Flavoured drinks | Group I | 10,000a | |
| 14.1.5.2 | Other non‐alcoholic beverages | Group I | Excluding unflavoured leaf tea; including flavoured instant coffee; | 10,000a |
| 14.2.3 | Cider and perry | Group I | 10,000a | |
| 14.2.4 | Fruit wine and made wine | Group I | 10,000a | |
| 14.2.5 | Mead | Group I | 10,000a | |
| 14.2.6 | Spirit drinks as defined in Regulation (EC) No 110/2008 | Group I | Except whisky or whiskey | 10,000a |
| 14.2.7.1 | Aromatised wines | Group I | 10,000a | |
| 14.2.7.2 | Aromatised wine‐based drinks | Group I | 10,000a | |
| 14.2.7.3 | Aromatised wine‐product cocktails | Group I | 10,000a | |
| 14.2.8 | Other alcoholic drinks including mixtures of alcoholic drinks with non‐alcoholic drinks and spirits with less than 15% of alcohol | Group I | 10,000a | |
| 15.1 | Potato‐, cereal‐, flour‐ or starch‐based snacks | Group I | 10,000a | |
| 15.2 | Processed nuts | Group I | 10,000a | |
| 16 | Desserts excluding products covered in categories 1, 3 and 4 | Group I | 10,000a | |
| 17.1b | Food supplements supplied in a solid form including capsules and tablets and similar forms, excluding chewable forms | Group I | 10,000a | |
| 17.2b | Food supplements supplied in a liquid form | Group I | 10,000a | |
| 17.3b | Food supplements supplied in a syrup‐type or chewable form | Group I | 10,000a | |
| 18 | Processed foods not covered by categories 1–17, excluding foods for infants and young children | Group I | 10,000a |
MPL: maximum permitted level.
10,000 mg/kg individually or in combination, expressed as glutamic acid.
FCS 17 refers to food supplements as defined in Directive 2002/46/EC of the European Parliament and of the Council excluding food supplements for infants and young children.
3.3. Exposure data
3.3.1. Reported use levels or data on analytical levels of glutamic acid–glutamates (E 620–625)
Most food additives in the EU are authorised at a specific MPL. However, a food additive may be used at a lower level than the MPL. Therefore, information on actual use levels is required for performing a more realistic exposure assessment, especially for those food additives for which no MPL is set and which are authorised according to QS.
In the framework of Regulation (EC) No 1333/2008 on food additives and of Commission Regulation (EU) No 257/2010 regarding the re‐evaluation of approved food additives, EFSA issued public calls13 for occurrence data (usage level and/or concentration data) on glutamic acid–glutamates (E 620–625). In response to this public call, information on use levels and analytical data on glutamic acid–glutamates (E 620–625) in foods was made available to EFSA by industry and Member States.
Summarised data on reported use levels in foods provided by industry
Industry provided EFSA with use levels (n = 43) of glutamic acid–glutamates (E 620–625) as food additives in foods for 15 out of the 67 food categories in which glutamic acid–glutamates (E 620–625) are authorised.
Information on use levels of glutamic acid–glutamates (E 620–625) in foods was made available to EFSA by FoodDrinkEurope (FDE), Asociación Española de Exportadores e Industriales de Aceitunas de Mesa (ASEMESA), AVIKO and Specialised Nutrition Europe (SNE).
Appendix A lists the use levels of glutamic acid–glutamates (E 620–625) in foods as reported by industry.
Summarised data on concentration levels in food submitted by Member States
The Panel assumed that reported concentrations of glutamic acid/glutamate in foodstuff do not usually include glutamic acid which is contained in proteins and peptides.
In total, 32,671 analytical results were reported to EFSA by six countries: the Czech Republic (n = 2), Germany (n = 31,758), Spain (n = 304), the United Kingdom (n = 296), Hungary (n = 302) and Luxemburg (n = 9). These data were mainly for meat products (FCS 8.3.1 and 8.3.2; n = 10,557), composite foods (FCS 18; n = 8,652), soups and broths (FCS 12.5; n = 4,250), sauces (FCS 12.6; n = 2,055), seasonings and condiments (FCS 12.2.2; n = 1,925), and potato‐, cereal‐, flour‐ or starch‐based snacks (FCS 15.1; n = 1,098). Foods were sampled between 2000 and 2016, and the majority of them (96%) were analysed the year that they were collected.
Out of the total of 32,671 analytical results, 981 were excluded due to inappropriate sampling strategy (i.e. suspect sampling).
Out of the remaining 31,690 analytical results, 30,805 results were related to foods to which the addition of glutamic acid–glutamates (E 620–625) as food additives is authorised according to Annex II, and 885 results related to food categories for which direct addition of glutamic acid–glutamates is not authorised according to Annex II. After excluding 1,528 results above the MPLs set for the added amounts, 30,162 results were considered in the exposure assessment (i.e. 29,277 results on glutamic acid–glutamates (E 620–625) used as food additives and 885 results related to food categories for which direct addition of glutamic acid–glutamates is not authorised according to Annex II). The occurrence of glutamic acid–glutamate in food may be due to its natural presence,14 to its addition as nutrient according to Regulation (EU) Nr 609/2013 (Annex I) or to the addition of glutamic acid–glutamates (E 620–625) as food additives. The Panel considered that those food categories for which direct addition of glutamic acid–glutamates is not authorised according to Annex II are very likely to contain glutamic acid–glutamate derived from natural sources. The Panel noted the use of yeast extracts as a source of glutamate (Lukondeh et al., 2003; Amorim et al., 2016; FDA, online).
The analytical data on glutamic acid–glutamate in food categories for which a direct addition of glutamic acid–glutamates (E 620–625) is authorised according to Annex II were mainly for non‐heat‐treated processed meat (FCS 08.3.1, n = 5,447), heat‐treated processed meat (FCS 08.3.2, n = 4,679), soups and broths (FCS 12.5, n = 3,888), sauces (FCS 12.6, n = 1,872), and seasonings and condiments (FCS 12.2.2, n = 1,336).
The 885 analytical results related to the food categories for which a direct addition of glutamic acid–glutamates is not authorised according to Annex II were mainly for fruit juices and nectars (FCS 14.1.2, n = 500, glutamic acid–glutamates (E 620–625) are authorised in vegetable juices only), non‐gluten‐free pasta (FCS 6.4, n = 204, glutamic acid–glutamates (E 620–625) are authorised in gluten‐free pasta only), foods for infants and young children (FCS 13.1, 13.1.1/2/3, 13.1.5.1, n = 108), butter (FCS 2.2.1, n = 57), rice (FCS 6.1, n = 4) and honey (FCS 11.3, n = 3). These analytical data were used only for a separate exposure scenario considering all available data, as described in Section 3.3.1.
Considering the analytical results related to food categories for which a direct addition of glutamic acid–glutamates (E 620–625) is authorised according to Annex II, 11% were left‐censored: either not quantified (< LOQ) in 1,522 samples or not detected (< LOD) in 1,713 samples. As 789 analytical data were reported as MSG, a conversion factor of 0.87 (see Section 3.1.1) was applied to convert those data into glutamic acid. Complete information on the methods of analysis (e.g. validation) was not made available to EFSA, but all samples were derived from accredited laboratories.
Overall, for the scenario considering the exposure to glutamic acid–glutamates (E 620–625) as food additives, the Panel considered 29,277 analytical data (i.e. only occurrence levels of food categories for which a direct addition of glutamic acid–glutamates (E 620–625) is authorised according to Annex II and which did not exceed the relevant MPL for added amount). For this scenario, data were available for 45 out of 67 food categories in which glutamic acid–glutamates (E 620–625) are authorised (Appendix B).
For the exposure scenario considering all available data, the Panel considered 31,690 analytical data (i.e. 29,277 results considering glutamic acid–glutamates (E 620–625) as food additives, 885 results related to food categories for which direct addition of glutamic acid–glutamates is not authorised according to Annex II, and 1,528 results above the MPLs for the added amount). For this scenario, data were available for 46 out of 67 food categories in which glutamic acid–glutamates (E 620–625) are authorised and for 12 food categories for which direct addition of glutamic acid–glutamates is not authorised according to Annex II.
Appendix B lists the analytical results of glutamic acid–glutamate in food categories for which a direct addition of glutamic acid–glutamates (E 620–625) is authorised according to Annex II as reported by Member States, after the exclusion of analytical results above the MPLs for the added amount.
Appendix C lists all analytical results of glutamic acid–glutamate as reported by Member States, including values above the MPLs for the added amount and food categories for which direct addition of glutamic acid–glutamates (E 620–625) is not authorised according to Annex II.
3.3.2. Summarised data extracted from the Mintel's Global New Products Database
The Mintel GNPD is an online database which monitors product introductions in consumer packaged goods markets worldwide. It contains information of over 2 million food and beverage products of which more than 900,000 are or have been available on the European food market. Mintel started covering EU's food markets in 1996, currently having 20 out of its 28 member countries and Norway presented in the Mintel GNPD.15
For the purpose of this Scientific Opinion, the Mintel GNPD16 was used for checking the labelling of products containing glutamic acid–glutamates (E 620–625) within the EU's food products as the Mintel GNPD shows the compulsory ingredient information presented in the labelling of products.
According to the Mintel GNPD, glutamic acid–glutamates (E 620–625) or glutamic acid used as a nutrient are labelled on 9,440 food products published in this database between 2012 and 2017.
Appendix D presents the percentage of the food products labelled with glutamic acid–glutamates (E 620–625) or glutamic acid as a nutrient out of the total number of food products per food subcategory according to the Mintel GNPD food classification. Glutamic acid–glutamates (E 620–625) or glutamic acid as a nutrient were reported mainly in composite dishes, such as instant noodles (60%) and dry soups (27%). The presence of glutamic acid–glutamates (E 620–625) or of glutamic acid used as a nutrient was also reported in baby formula (0–6 months, 3 out of 267 products), baby snacks (0–4 years, 1 out of 321 products), and butter (8 out of 1,395 products), despite direct addition of glutamic acid–glutamates (E 620–625) is not authorised according to Annex II in these food categories. Foods for special medical purposes for infants and young children were considered in the present exposure assessment in the scenario with all available data (as described in Section 3.3.1), i.e. 108 analytical data on FCS 13.1, 13.1.1/2/3, 13.1.5.1. The overall percentage of food products labelled with glutamic acid–glutamates (E 620–625) or glutamic acid used as a nutrient, considering the food subcategories with at least one food to which these food additives were added according to the label, was 3.3%. Composite foods labelled with glutamic acid–glutamates (E 620–625) in the Mintel GNPD contain glutamic acid–glutamates (E 620–625) very likely as carry‐over of their ingredients.
3.3.3. Food consumption data used for exposure assessment
EFSA Comprehensive European Food Consumption Database
Since 2010, the EFSA Comprehensive European Food Consumption Database (Comprehensive Database) has been populated with national data on food consumption at a detailed level. Competent authorities in the European countries provide EFSA with data on the level of food consumption by the individual consumer from the most recent national dietary survey in their country (cf. Guidance of EFSA on the ‘Use of the EFSA Comprehensive European Food Consumption Database in Exposure Assessment’, (EFSA, 2011a)). New consumption surveys recently17 added in the Comprehensive database were also taken into account in this assessment.18
The food consumption data gathered by EFSA were collected by different methodologies and thus direct country‐to‐country comparisons should be interpreted with caution. Depending on the food category and the level of detail used for exposure calculations, uncertainties could be introduced owing to possible subjects’ underreporting and/or misreporting of the consumption amounts. Nevertheless, the EFSA Comprehensive Database represents the best available source of food consumption data across Europe at present.
Food consumption data from the following population groups: infants, toddlers, children, adolescents, adults and the elderly were used for the exposure assessment. For the present assessment, food consumption data were available from 33 different dietary surveys carried out in 19 European countries (Table 8).
Table 8.
Population groups considered for the exposure estimates of glutamic acid–glutamates (E 620–625)
| Population | Age range | Countries with food consumption surveys covering more than 1 day |
|---|---|---|
| Infants | From more than 12 weeks up to and including 11 months of age | Bulgaria, Denmark, Finland, Germany, Italy, UK |
| Toddlers | From 12 months up to and including 35 months of age | Belgium, Bulgaria, Denmark, Finland, Germany, Italy, Netherlands, Spain, UK |
| Childrena | From 36 months up to and including 9 years of age | Austria, Belgium, Bulgaria, Czech Republic, Denmark, Finland, France, Germany, Greece, Italy, Latvia, Netherlands, Spain, Sweden, UK |
| Adolescents | From 10 years up to and including 17 years of age | Austria, Belgium, Cyprus, Czech Republic, Denmark, Finland, France, Germany, Italy, Latvia, Spain, Sweden, UK |
| Adults | From 18 years up to and including 64 years of age | Austria, Belgium, Czech Republic, Denmark, Finland, France, Germany, Hungary, Ireland, Italy, Latvia, Netherlands, Romania, Spain, Sweden, UK |
| The elderlya | From 65 years of age and older | Austria, Belgium, Denmark, Finland, France, Germany, Hungary, Ireland, Italy, Romania, Sweden, UK |
The terms ‘children’ and ‘the elderly’ correspond, respectively, to ‘other children’ and the merge of ‘elderly’ and ‘very elderly’ in the Guidance of EFSA on the ‘Use of the EFSA Comprehensive European Food Consumption Database in Exposure Assessment’ (EFSA, 2011a).
Consumption records were codified according to the FoodEx classification system (EFSA, 2011b). Nomenclature from the FoodEx classification system has been linked to the food categorisation system (FCS) as presented in Annex II of Regulation (EC) No 1333/2008, part D, to perform exposure estimates. In practice, the FoodEx food codes were matched to the FCS food categories.
Food categories considered for the exposure assessment of glutamic acid–glutamates (E 620–625)
The 67 food categories in which the use of glutamic acid–glutamates (E 620–625) are authorised according to Annex II were selected from the nomenclature of the EFSA Comprehensive Database (FoodEx classification system), at the most detailed level possible (up to FoodEx Level 4) (EFSA, 2011b).
Some food categories or their restrictions/exceptions are not referenced in the EFSA Comprehensive Database and could therefore not be taken into account in the present estimate. This may have resulted in an underestimation of the exposure. This was the case for 10 food categories (Appendix E). The food categories which were not taken into account are described below (in ascending order of the FCS codes):
01.6.3 Other creams
01.7.6 Cheese products (excluding products falling in category 16)
02.3 Vegetable oil spray
06.6 Batters
06.7 Pre‐cooked or processed cereals
08.3.3 Casings and coatings and decorations for meat
14.2.4 Fruit wine and made wine
14.2.5 Mead
14.2.7.2. Aromatised wine‐based drinks
14.2.7.3. Aromatised wine‐product cocktails.
For the following food categories, the restrictions/exceptions which apply to the use of glutamic acid–glutamates (E 620–625) could not be taken into account, and therefore the whole food category was considered in the exposure assessment. This applies to three food categories and may have resulted in an overestimation of the exposure:
07.1 Bread and rolls, except products in 07.1.1 and 07.1.2
08.3.2 Heat‐treated meat products, except foie gras, foie gras entier, blocs de foie gras, Libamáj, libamáj egészben, libamáj tömbben
9.3 Fish roe, only processed fish roe
For the remaining food categories, the refinements considering the restrictions/exceptions as set in Annex II to Regulation No 1333/2008 were applied. Considering that the food category 18 (Processed foods not covered by categories 1–17, excluding foods for infants and young children) is extremely unspecific (e.g. composite foods), the foods belonging to this food category were reclassified, where possible, under food categories in accordance to their main component. Therefore, food category 18 is not taken into account as contributor to the total exposure estimates. In addition, food categories 13.2, 13.3 and 13.4 were also not considered in exposure assessment (as explained in Section 3.4.1).
For the refined exposure scenario, 12 additional food categories (FCS 01.3, 04.2.5.4, 05.3, 05.4, 06.4.2, 11.2, 14.1.3, 14.1.5.2, 14.2.3, 14.2.6, 14.2.7.1 and 14.2.8) were not taken into account because no concentration data were provided for these food categories to EFSA. The Panel noted that with the exception of pasta (gluten‐free) (FCS 06.4.2, one product out of 10,113 products in the Mintel GNPD) and fermented milk products (FCS 01.3, two products out of 2,892 products in the Mintel GNPD) the remaining 10 food categories were not reported to contain glutamic acid–glutamates (E 620–625) in the Mintel GNPD.
For the scenario considering all available data (as described in section 3.3.1), besides the food categories in which glutamic acid–glutamates (E 620–625) are authorised as food additives (Annex II to Regulation No 1333/2008), 10 additional food categories for which analytical data were reported and in which glutamic acid–glutamate might occur naturally or were used other than as food additives, were considered to estimate the exposure. These included butter (FCS 02.2.1), whole, broken, or flaked grain (FCS 06.1), non‐gluten‐free pasta (FCS 06.4), honey (FCS 11.3), foods for infants and young children (FCS 13.1.1, 13.1.2, 13.1.3, 13.1.5.1), and fruit juices and fruit nectars (FCS 14.1.2, 14.1.3).
Overall, in the regulatory maximum level exposure assessment scenario considering glutamic acid–glutamates (E 620–625) authorised as food additives according to Annex, 50 food categories were included (Appendix E). In the refined exposure assessment scenario considering glutamic acid–glutamates (E 620–625) authorised as food additives according to Annex II, 38 food categories were included (Appendix E) whereas in the refined exposure assessment scenario with all available data (except the ones mentioned in Section 3.3.1), 51 food categories were included (Appendix E).
3.4. Exposure estimate
3.4.1. Exposure to glutamic acid–glutamates (E 620–625) from their use as food additives
The Panel estimated chronic exposure to glutamic acid–glutamates (E 620–625) for the following population groups: infants, toddlers, children, adolescents, adults and the elderly. Dietary exposure to glutamic acid–glutamates (E 620–625) was calculated by multiplying glutamic acid–glutamates (E 620–625) concentrations for each food category (Appendix E) with their respective consumption amount per kilogram of body weight for each individual in the Comprehensive Database. The exposure per food category was subsequently added to derive an individual total exposure per day. These exposure estimates were averaged over the number of survey days, resulting in an individual average exposure per day for the survey period. Dietary surveys with only 1 day per subject were excluded as they are considered as not adequate to assess repeated exposure.
The exposure was estimated for all individuals per survey and per population group, resulting in distributions of individual exposure per survey and population group (Table 8). On the basis of these distributions, the mean and 95th percentile of exposure were calculated per survey and per population group. The 95th percentile of exposure was only calculated for those population groups where the sample size was sufficiently large to allow this calculation (EFSA, 2011a). Therefore, in the present assessment, 95th percentile of exposure for infants from Italy and for toddlers from Belgium, Italy and Spain were not estimated.
Exposure assessment to glutamic acid–glutamates (E 620–625) was carried out by the ANS Panel based on two sets of concentration data: (1) MPLs as set down in the EU legislation (defined as the regulatory maximum level exposure assessment scenario); (2) reported use levels and/or analytical results (not exceeding the MPLs as added amounts) for food categories for which direct addition of glutamic acid–glutamates (E 620–625) is authorised according to Annex II to Regulation (EC) No 1333/2008 defined as the refined exposure assessment scenario.
These scenarios do not consider the intake of food supplements which is covered in an additional scenario detailed below.
Regulatory maximum level exposure assessment scenario
The regulatory maximum level exposure assessment scenario is based on the MPLs as set for the added amount in Annex II to Regulation (EC) No 1333/2008 (Table 7). Although the use of glutamic acid–glutamates (E 620–625) in FCS 12.1.2 is authorised at QS as E 620–625, the level of 10,000 mg/kg (considered as Group I) was used in the MPL scenario. Glutamic acid–glutamates (E 620–625) is also authorised at QS in FCS 12.2.2, the reported usage level of 209,000 mg/kg was used in the MPL scenario. MPLs expressed in weight and used in this exposure scenario are listed in Appendix E.
The Panel considered the exposure estimates derived with this scenario as the most conservative as it is assumed that the population group will be exposed to glutamic acid–glutamates (E 620–625) present in food at the MPL/maximum reported use levels over a longer period of time.
Appendix E summarises the concentration levels of glutamic acid–glutamates (E 620–625) used in the regulatory maximum level exposure assessment scenario.
Refined exposure assessment scenario
The refined exposure assessment scenario is based on use levels reported by food industry and analytical results reported by Member States. This exposure scenario can consider only food categories for which these data were available to the Panel.
In the refined exposure assessment scenarios, the concentration levels considered by the Panel were extracted from the whole data set (i.e. reported use levels and analytical results). To consider left‐censored analytical data (i.e. analytical results < LOD or < LOQ), the substitution method as recommended in the ‘Principles and Methods for the Risk Assessment of Chemicals in Food’ (WHO, 2009) and the EFSA scientific report ‘Management of left‐censored data in dietary exposure assessment of chemical substances’ (EFSA, 2010) was used. In the present opinion, analytical data below LOD or LOQ were assigned half of LOD or LOQ, respectively (medium bound (MB)). Subsequently, per food category, the mean or median, whichever is highest, MB concentration was calculated. It should be noted that the use of medium bound (MB) LOD/LOQ values (half of LOD or LOQ) in the exposure assessment, may have resulted in either an overestimation, where glutamic acid‐glutamate (as added or natural occurring) was not present, or an underestimation, where the concentration was between the MB and LOQ/LOD value, although the analytical method was not able to detect or quantify it.
Appendix E summarises the concentration levels of glutamic acid–glutamates (E 620–625) used in the refined exposure assessment scenario. Based on the available data set, the Panel calculated two refined exposure estimates based on different model populations excluding exposure via food supplements:
-
The brand‐loyal consumer scenario: It was assumed that a consumer is exposed long‐term to glutamic acid–glutamates (E 620–625) present at the maximum reported use/analytical level for one food category. This exposure estimate is calculated as follows:
-
–
Combining food consumption with the maximum of the reported use levels or the 95th percentile of the analytical results, whichever was highest or available, for the main contributing food category at the individual level.
-
–
Using the mean of the typical reported use levels or the mean of analytical results, whichever was highest or available, for the remaining food categories.
-
–
The non‐brand‐loyal consumer scenario: It was assumed that a consumer is exposed long‐term to glutamic acid–glutamates (E 620–625) present at the mean reported use/analytical levels in food. This exposure estimate is calculated using the mean of the typical reported use levels or the mean of analytical results for all food categories.
The Panel noted that for this exposure scenario, the analytical levels of glutamic acid/glutamate present due to natural occurrence or added for purposes others than as food additives have been considered as added food additives.
Specific exposure assessment scenario: Food supplements consumers only scenario
Glutamic acid–glutamates (E 620–625) are authorised in the food category 17 food supplements as defined in Directive 2002/46/EC19, excluding food supplements for infants and young children. As exposure via food supplements may deviate largely from that via food, and that the number of food supplement consumers may be low depending on populations and surveys, this additional scenario was calculated in order to reflect additional exposure to glutamic acid–glutamates (E 620–625) from food supplements compared to the exposure to the food additives excluding these sources.
This scenario was estimated assuming that consumers only of food supplements were exposed to glutamic acid–glutamates (E 620–625) present at the maximum reported use level on a daily basis via consumption of food supplements. For the remaining food categories (38/67), the mean of the typical reported data was used.
As food category 17 does not include food supplements for infants and toddlers (Regulation (EC) No 1333/2008), exposure to glutamic acid–glutamates (E 620–625) from food supplements was not estimated for these two population groups.
Appendix E summarises the concentration levels of glutamic acid–glutamates (E 620–625) used in this specific exposure assessment scenario.
3.4.2. Dietary exposure to glutamic acid–glutamates (E 620–625)
Table 9 summarises the estimated exposure to glutamic acid–glutamates (E 620–625), considering only food categories listed in Annex II to Regulation (EC) No 1333/2008 (Table 7), in six population groups according to the different exposure scenarios. Detailed results per population group and survey are presented in Appendix F.
Table 9.
Summary of dietary exposure to glutamic acid–glutamates (E 620–625) from their use as food additives in the regulatory maximum level exposure assessment scenario and in the refined exposure scenarios, in six population groups (minimum–maximum across the dietary surveys in mg/kg bw per day expressed as glutamic acid)
| Infants (12 weeks–11 months) | Toddlers (12–35 months) | Children (3–9 years) | Adolescents (10–17 years) | Adults (18–64 years) | The elderly (≥ 65 years) | |
|---|---|---|---|---|---|---|
| Regulatory maximum level exposure assessment scenario | ||||||
| Mean | 31–91 | 105–363 | 91–274 | 40–125 | 36–87 | 33–80 |
| 95th percentile | 71–236 | 256–534 | 172–484 | 80–235 | 68–165 | 63–132 |
| Refined estimated exposure assessment scenario | ||||||
| Brand‐loyal scenario | ||||||
| Mean | 11–31 | 23–86 | 15–68 | 6–42 | 8–30 | 8–26 |
| 95th percentile | 26–111 | 47–126 | 27–139 | 12–91 | 15–64 | 14–56 |
| Non‐brand‐loyal scenario | ||||||
| Mean | 5–19 | 19–51 | 9–47 | 3–27 | 5–19 | 4–17 |
| 95th percentile | 20–65 | 35–88 | 17–93 | 7–59 | 9–42 | 9–38 |
From the regulatory maximum level exposure assessment scenario, mean exposure to glutamic acid–glutamates (E 620–625) from their use as food additives ranged from 31 mg/kg bw per day in infants to 363 mg/kg bw per day in toddlers. The 95th percentile of exposure to glutamic acid–glutamates (E 620–625) from their use as food additives ranged from 63 mg/kg bw per day in the elderly to 534 mg/kg bw per day in toddlers.
From the refined estimated exposure scenario, considering concentration levels not exceeding the MPLs of the added amount for food categories listed under Annex II to Regulation No 1333/2008, in the brand‐loyal scenario, mean exposure to glutamic acid–glutamates (E 620–625) ranged from 6 mg/kg bw per day in adolescents to 86 mg/kg bw per day in toddlers. The 95th percentile of exposure to glutamic acid–glutamates (E 620–625) ranged from 12 mg/kg bw per day in adolescents to 139 mg/kg bw per day in children. In the non‐brand‐loyal scenario, mean exposure to glutamic acid–glutamates (E 620–625) ranged from 3 mg/kg bw per day in adolescents to 51 mg/kg bw per day in toddlers. The 95th percentile of exposure to glutamic acid–glutamates (E 620–625) ranged from 7 mg/kg bw per day in adolescents to 93 mg/kg bw per day in children.
For the food supplements consumers only, in the brand‐loyal scenario, mean exposure to glutamic acid–glutamates (E 620–625) ranged from 4 mg/kg bw per day in adolescents to 42 mg/kg bw per day in children. The 95th percentile of exposure ranged from 7 mg/kg bw per day in adolescents to 72 mg/kg bw per day in children.
The most important contributors to the total mean exposure for glutamic acid–glutamates (E 620–625), for the regulatory maximum level exposure assessment scenario, were: bread and rolls for children, adolescents, adults and the elderly; soups and broths for infants; and flavoured fermented milk products for toddlers. For the brand‐loyal and non‐brand‐loyal refined scenario, the main contributors were fine bakery wares for toddlers, children, adolescents and adults; and soups and broths for the infants. For the elderly, the most important contributors were soups and broths in the brand‐loyal scenario and fine bakery wares in the non‐brand loyal scenario.
The main contributing food categories for which industry reported use data were fine bakery wares for toddlers, children, adolescents, adults, and soups and broths for infants. The main contributors for the elderly were soups and broths and fine bakery wares. Other relevant food contributors for which use levels were available were sauces, meat and meat products and seasoning and condiments. The food categories and their contribution to the exposure to glutamic acid–glutamates (E 620–625) are presented in Appendix G.
3.4.3. Exposure to glutamic acid–glutamates using all available data (as described in Section 3.3.1)
The analytical levels provided by the Member States reflect the levels of glutamic acid–glutamate in foods, whatever their origin (food additives, nutrient or natural presence). Analytical data were reported also for food categories for which direct addition of glutamic acid–glutamates (E 620–625) is not authorised (Annex II to Regulation (EC) No 1333/2008).
The exposure estimated using all available data (as described in Section 3.3.1) will very likely reflect more closely the total amount of free glutamic acid–glutamate ingested through the diet. Therefore, the Panel calculated the exposure to glutamic acid–glutamates considering all food categories for which data were available in a separate scenario defined as general population scenario using all available data (as described in Section 3.3.1). In this case, also all analytical results including the ones above the MPLs for the added amounts were considered for the assessment. The refined non‐brand loyal scenario was selected as the most relevant exposure scenario using all available data. The Panel noted that free glutamic acid–glutamates are present in a wide range of foods, making it unlikely that brand loyalty will result in higher exposure in the general population. Appendix E summarises the levels of glutamic acid–glutamate used in the exposure assessment scenario.
Table 10 summarises the estimated exposure to free glutamic acid–glutamates using all available data (as described in Section 3.3.1) in six population groups (Table 8). Detailed results per population group and survey are presented in Appendix F.
Table 10.
Summary of dietary exposure to free glutamic acid–glutamates using all available data (as described in Section 3.3.1) following the approach for the refined non‐brand‐loyal exposure scenario in six population groups (min–max across the dietary surveys in mg/kg bw per day)
| General population non‐brand loyal scenario using all available data | ||||||
|---|---|---|---|---|---|---|
| Infants (12 weeks–11 months) | Toddlers (12–35 months) | Children (3–9 years) | Adolescents (10–17 years) | Adults (18–64 years) | The elderly (≥ 65 years) | |
| Mean | 83–198 | 30–158 | 22–97 | 12–47 | 9–32 | 8–28 |
| 95th percentile | 256–489 | 76–429 | 38–259 | 21–127 | 18–88 | 17–80 |
From the non‐brand‐loyal estimated exposure scenario considering all available data (as described in Section 3.3.1), mean exposure to glutamic acid–glutamate ranged from 8 mg/kg bw per day in the elderly to 198 mg/kg bw per day in infants. The 95th percentile of exposure ranged from 17 mg/kg bw per day in the elderly to 489 mg/kg bw per day in infants.
For the exposure scenario considering all the available data (as described in Section 3.3.1), the most important contributors to the total mean exposure were fine bakery wares for children and adolescents, unripened cheese for toddlers, adults, the elderly, and foods for infants and young children. The food categories and their contribution to the exposure to glutamic acid–glutamate are presented in Appendix G.
3.4.4. Uncertainty analysis
Uncertainties in the exposure assessment of glutamic acid–glutamates (E 620–625) have been discussed above. In accordance with the guidance provided in the EFSA opinion related to uncertainties in dietary exposure assessment (EFSA, 2007), the following sources of uncertainties have been considered and summarised in Table 11.
Table 11.
Qualitative evaluation of influence of uncertainties on the dietary exposure estimate
| Sources of uncertainties | Directiona |
|---|---|
| Consumption data: different methodologies/representativeness/underreporting/misreporting/no portion size standard | +/– |
| Use of data from food consumption survey of a few days to estimate long‐term (chronic) exposure for high percentiles (95th percentile) | + |
| Correspondence of reported use levels and analytical data to the food items in the EFSA Comprehensive Food Consumption Database: uncertainties to which types of food the levels refer to | +/– |
| Uncertainty in possible national differences in use levels of food categories | +/– |
Concentration data:
|
+ +/– |
| The 38/67 food categories which were taken into account in the refined exposure assessment scenarios out of all authorised foods, corresponded to 43–95% of the amount (g of foods by body weight) of food consumption documented in the EFSA Consumption Database | – |
| Food categories selected for the exposure assessment: exclusion of food categories due to missing FoodEx linkage (n = 10/67 total number of food categories) | – |
| Food categories selected for the exposure assessment: inclusion of food categories without considering the restriction/exception (n = 3/67 total number of food categories) | + |
| Regulatory maximum level exposure assessment scenario:exposure calculations based on the MPL according to Annex II to Regulation (EC) No 1333/2008 | + |
Refined exposure assessment scenarios:
|
+/– +/– + − |
+, uncertainty with potential to cause overestimation of exposure; –, uncertainty with potential to cause underestimation of exposure.
As it has been pointed out in section on method of analysis (Section 3.1.4) distinction cannot be done between glutamic acid–glutamate naturally present in food or added as food additive and/or as nutrient in light of the available data.
Overall, the Panel considered that the uncertainties identified would, in general, result in an overestimation of the exposure to glutamic acid–glutamates (E 620–625) as food additives in European countries for the regulatory maximum level exposure scenario. Based on the assumption that the food additives are not used in the food categories in which they are permitted but for which no concentration data were provided by the stakeholders, also the refined scenario resulted in an overestimation of exposure to glutamic acid–glutamates (E 620–625).
Glutamic acid–glutamates (E 620–625) are authorised as a Group I food additive in 66 food categories and has a specific authorised use in 2 food categories (Table 2).20 Since, the majority of food categories correspond to the general Group I food additives authorisation, glutamic acid–glutamates (E 620–625) may not necessarily be used in some of these food categories. The Panel calculated that out of the foods for which a direct addition of glutamic acid–glutamates (E 620–625) is authorised according to Annex II to Regulation (EC) No 1333/2008, 43–95% of the amount of food consumed per population group was reported to potentially contain glutamic acid–glutamates (E 620–625) used as food additives.
Furthermore, the Panel noted that information from the Mintel GNPD (Appendix D) indicated that 71 out of 77 food subcategories, categorised according to the Mintel GNPD nomenclature, in which glutamic acid–glutamates were labelled were included in the current regulatory maximum level and refined exposure assessments. These 71 food subcategories represented approximately 99% of the food products labelled with glutamic acid–glutamates (E 620–625) in the database. For the remaining six food subcategories, representing 1% of the foods labelled with glutamic acid–glutamates, five were not included because glutamic acid–glutamates (E 620–625) are not authorised as food additives (i.e. baby snacks, baby formula, rice, butter, non‐gluten‐free pasta) and one (i.e. meal replacements) because no food consumption data was available.
The Mintel GNPD indicates the possible presence of glutamic acid–glutamates (E 620–625) in most of the food categories considered in the refined assessment of exposure from their use as food additives. Conversely, eight food categories were not found in the Mintel GNPD (i.e. dairy analogues, edible ices, starches, breakfast cereals, potato gnocchi, salt substitutes, protein products, and fruit juices) which are, however, not among the major contributors to the exposure to glutamic acid–glutamates (E 620–625). This might therefore have led to a minor overestimation of the exposure.
The same uncertainties presented above (Section 3.4.1) for the refined exposure to glutamic acid–glutamates (E 620–625) from their use as food additives are also applicable to the refined exposure estimates to glutamic acid–glutamates using all available data.
Given these observations, the Panel overall considered that the uncertainties identified would, in general, result in an overestimation of the exposure to glutamic acid–glutamates (E 620–625) from their use as food additives according to Annex II in both the maximum level and refined exposure scenarios. Exposure to glutamic acid–glutamates via food categories for which a direct addition of glutamic acid–glutamates (E 620–625) is not authorised according to Annex II including fruit juices and nectars, non‐gluten‐free pasta, foods for infants and toddlers, butter, rice and honey resulted in an increase in exposure to glutamic acid–glutamates, especially for infants (Table 10). This was due to the presence of glutamic acid–glutamates in foods for special medical purposes for infants and young children, confirmed by the Mintel GNPD.
3.5. Biological and toxicological data
The Panel is aware that the MSG used as test material in the unpublished studies performed by Hatano Research Institute (2006a,b, 2007 [Documentation provided to EFSA n. 12, 13 and 14]) and Biosafety Research Center (2007a,b,c, 2008 [Documentation provided to EFSA n. 3, 4, 5 and 6]) has been produced by a different manufacturing process than the process evaluated by the Panel in 2015 (EFSA ANS Panel, 2015). No details of the manufacturing process were submitted to EFSA and its assessment is out of the remit of the re‐evaluation of glutamic acid–glutamates (E 620–625) as food additives.
The Panel presumed that the l‐form of glutamate was used in the toxicological studies where this information was not available. Furthermore, the Panel decided to apply a read‐across approach to extrapolate from MSG (E 621) which was used in the toxicological studies to all other glutamates (glutamic acid (E 620), monopotassium glutamate (E 622), calcium diglutamate (E 623), monoammonium glutamate (E 624) and magnesium diglutamate (E 626)) as in the mammalian body MSG is dissociated into glutamate and sodium and the effects of interest are attributed to glutamate.
General overview
Glutamic acid, a non‐essential amino acid, is the most abundant free amino acid in the brain. Glutamate functions as an excitatory neurotransmitter in the central nervous system (CNS). It has an additional function as a link between the redox states of the pyridine nucleotides (NAD+ and NADP+), and as energy source. Glutamate metabolism is closely linked to the Krebs cycle. The reaction glutamate + NAD+ ↔ α‐ketoglutarate + NADH + H+ is catalysed by glutamate dehydrogenase. This mitochondrial reaction, which uses either NAD+ or NADP+ as cofactors, is believed to be at equilibrium and therefore serves to assure that the redox state of both pyridine nucleotides is similar (Krebs and Veech, 1969).
Glutamate also serves as an important potential energy source. When the glucose concentration in the brain is low, the brain mobilises glutamate (Miller et al., 1975). The energy which is made available by metabolising glutamate is similar to the energy provided by glucose.
Nerve impulses trigger the release of glutamate from the pre‐synaptic cell, which in turn binds to the glutamate receptors on the opposing post‐synaptic cell. Neurotransmission is terminated by glutamate uptake by astrocytes.
In studies with very high doses of glutamate, which were administered systemically, brain damage was detected in areas of the brain that were not protected by the blood–brain barrier (BBB) (Price et al., 1981; Olney and Sharpe, 1969). These results supported the concept that over‐stimulation of excitatory amino acid receptors could cause neuronal death (Schwarcz et al., 1984; Albin and Greenamyre, 1992). Subsequently, this hypothesis became a popular pathogenic explanation for neuronal damage observed in acute conditions, e.g. stroke. However, it should be taken into consideration that in such cases glutamate is released within the brain. For example, during an ischemic episode, glutamate is released from brain cells and an excessive concentration of glutamate will be present in the extracellular fluid (ECF) causing extreme excitation of other neurons (Choi et al., 1987; Martin et al., 1994; Castillo et al., 1996; Rothstein, 1996). This in turn may result in the opening of receptor‐coupled ionophores, for example calcium channels. A large influx of calcium associated with impaired intracellular calcium sequestration mechanisms, which activate a host of catabolic enzymes, may ultimately result in neuronal death (Benveniste et al., 1984).
Subcutaneous administration of MSG at doses of 4,000 mg/kg bw and higher in early postnatal period is an acknowledged paradigm to produce cytotoxic effects in the CNS in animals, specifically in rats (Chaparro‐Huerta et al., 2002; Urena‐Guerrero et al., 2003; Shah et al., 2015; Foran et al., 2017)
Besides the role as an excitatory amino acid, MSG is one of the substances possessing the property to evoke the so‐called umami taste. Several types of receptors have been identified for being involved in the signalling: ionotropic receptors (NDMA and kainite receptor) and metabotropic receptors (T1R1 (taste receptor type 1 member 1), T1R3 (taste receptor type 1 member 3) and taste‐specific isoforms of metabotropic glutamate receptors (mGluR), mainly mGluR4 and mGluR1, but also mGluR2 and mGluR3), see review by Stańska and Krzeski (2016). Umami taste has an influence on a plethora of physiological functions. Besides the localisation of the taste receptors on the tongue, throat and upper respiratory tract, it is also situated in the whole gastrointestinal tract (San Gabriel, 2015). By the umami taste, MSG stimulates salivary secretion, gastric hydrochloric acid secretion, pancreatic exocrine secretion and insulin release. In addition, it enhances gastric emptying and propulsions of the colon. By enhancing the taste of food, umami increases on the one hand the appetite, but on the other hand positively influences the reduction of fat mass and increases satiety. Theoretical considerations have linked effects in the gastrointestinal tract to cognitive functioning (Welcome et al., 2015).
Expression of mRNA transcripts for chemosensory receptors (i.e. the 400 ORs, 25 TAS2R bitter taste receptors and the TAS1R umami‐ and sweet‐taste receptor dimers) in a variety of peripheral tissues unrelated to olfaction has been reported. For example, the TAS1R receptor mRNA is expressed in circulating leucocytes, suggesting an involvement in cellular immune response (Malki et al., 2015). Based on experimental findings, a role for bitter‐ and sweet‐taste receptors can also be seen in mediating immune function, and regulating inflammation and antimicrobial activity within the respiratory tract (Workman et al., 2015). Recently, chemosensory cells have also been discovered in the urethra without clear indication of their function (Deckmann and Kummer, 2016).
3.5.1. Absorption, distribution, metabolism and excretion
Absorption
Animals
Glutamate absorption occurs in the small intestine via an active transport system which is sodium dependent (Schultz et al., 1970). Glutamate is metabolised in the gastrointestinal wall to a great extent so that the systemic availability is small. Data from Reeds et al. (1996) showed that pigs nearly completely metabolises glutamate during absorption. Other authors have also shown that extensive gut metabolism in infant pigs limits the systemic availability of even excessive supplemental dietary glutamate (Janeczko et al., 2007). Similar results were reported for other species (Neame and Wiseman, 1958; Windmueller and Spaeth, 1974, 1975; Windmueller, 1982). Burrin and Stoll (2009) reported that the systemic availability in young pigs amounts to 13–17%. In young pigs, absorption from the stomach and metabolism in the gastrointestinal wall have also been shown (Burrin and Stoll, 2009).
Humans
First pass metabolism was investigated in premature infants (delivered at 31 ± 0 week) at postnatal days 10 and 17; the data suggested that MSG is almost entirely removed in its first pass metabolism (Hays et al., 2007).
Distribution
Animals
A quantity of 8,000 mg MSG/kg bw given orally to rats on day 19 of gestation showed a maternal plasma concentration rising from 100 to 1,650 μg/mL whereas no significant changes were measured in plasma glutamic acid of the fetuses (O'Hara et al., 1970 as referred to by JECFA, 1988).
Blood–brain barrier passage
Due to the potential neurotoxic effects of glutamate mechanisms and extent of BBB passage have been widely investigated.
Potential transport mechanisms of glutamate across the blood–brain barrier
Glutamate concentrations in plasma are 30–100 μM, in whole brain 10,000–12,000 μM but only 0.5–2 μM in ECF. These low ECF concentrations are resulting from the uptake of glutamate by neurons, astrocytes and the cells forming the BBB (Hawkins, 2009). The concentration gradient of glutamate across cell membranes in the brain and the specificity of synaptic transmission are maintained by glutamate transporters. The family of brain glutamate transporters consists of five members, the excitatory amino acid transporters 1–5 (EAAT1–5) (Murphy‐Royal et al., 2017). In brain areas that are not contained within the BBB, glutamate uptake can exceed other parts of the brain by 100‐ to 1,000‐fold and once taken up in these areas glutamate can diffuse into other brain areas (Price et al., 1981). Two studies have shown that under special circumstances elevated brain glutamate concentration can be measured following MSG administration.
A quantity of 2,000 mg MSG/kg bw given by gavage in a 20% solution to 7‐day‐old male mice led to an increased glutamate concentration in the nucleus arcuatus of the hypothalamus (Airoldi et al., 1980). In the same investigation, this effect was not seen in adult male mice which may indicate that the age can have an influence on the activity of the transporters responsible for glutamate uptake into the cells forming the BBB.
In rats, a bolus dose of 4,000 mg MSG/kg by gavage caused a significant increase in plasma and extracellular glutamate in the hippocampus and in the hypothalamus. MSG intake with the diet at about the same dose (4,000 mg/kg bw per day) throughout a 21‐day period did not change glutamate concentration in plasma and brain. The authors concluded that an increase in extracellular brain glutamate occurs only after bolus administration of a relatively high dose of MSG, while no changes occur after prolonged intake of MSG in the food (Monno et al., 1995).
Bogdanov et al. (1996) performed a study in male Sprague–Dawley (SD) rats (n = 6–8 per treatment group) to compare the extracellular glutamate levels in the hypothalamic arcuate nucleus following intraperitoneal (i.p) and oral administration. I.p. administration of MSG (single doses: 250, 500, 1,000 and 2,000 mg/kg bw, respectively) caused MSG levels in the hypothalamic arcuate nucleus to increase by 116–1,230%. In contrast in a feeding experiment, consumption of 2,300 mg MSG/kg bw during an 1 h period no changes in glutamate levels were observed. A control group was included in this dietary study.
In addition, in rats (McLaughlan et al., 1970; Walaas and Fonnum, 1978; McCall et al., 1979; Caccia et al., 1982), mice (Schwerin et al., 1950; Airoldi et al., 1980) and neonatal pigs (Stegink et al., 1973a,b), the glutamate level in the brain did not rise after large doses of MSG in the range of 100–4,000 mg MSG/kg bw, even in the presence of large increase of glutamate plasma concentrations.
Humans
Graham et al. (2000) gave 150 mg MSG/kg bw to post‐prandial adults and measured the muscle level at 15–105 min, and found at 45 min muscle glutamate level to be elevated. At 105 min, it was returned to control levels. The authors proposed that resting skeletal muscle was a major sink for the glutamate and metabolises it to aspartate.
Regarding distribution in breast milk, six women were fasted overnight and given MSG in single oral doses of 6,000 mg in water or liquid diet (Stegink et al., 1972; Pitkin et al., 1979). Milk samples were obtained at 1–6 h and blood samples at 0–180 min. Small increases in plasma glutamate were observed, but no changes were found in breast milk.
Raising the plasma concentration of glutamate twofold in four healthy volunteers did not result in increased brain glutamate concentrations (Eriksson et al., 1983). In addition normal glutamate concentrations are almost fourfold the Km of the transport rate into the brain suggesting that the glutamate transport systems are virtually saturated at physiological conditions (Pardridge, 1979).
Metabolism
Glutamate can be metabolised in several organs.
Metabolism of glutamate in the liver leads to glucose, lactate glutamine and other amino acids (Stegink et al., 1983). Glutamate is also metabolised in muscle tissues and is converted to alanine and glutamine.
In the brain, there is evidence that glutamate is predominantly metabolised via transamination to aspartate and not via oxidative deamination (Krebs and Bellamy, 1960; Balazs, 1965; Benjamin and Quastel, 1974; Dennis et al., 1976; Hertz, 1979; Erecińska and Silver, 1990). It has been reported that the majority of glutamate used by the brain is synthesised locally from glutamine and tricarboxylic acid cycle intermediates and from the recycling of brain proteins (Smith, 2000). In the brain, the metabolism of glutamate and three other neurotransmitters, γ‐aminobutyric acid (GABA), glycine and aspartate are tightly linked.
In sheep, Battaglia (2000) have demonstrated that the maternal placental circulation takes up glutamate from the fetoplacental circulation. Uptake of glutamate by the maternal placental circulation and a supply of glutamine to the fetus have also been shown by Hayashi et al. (1978).
Excretion
The major route for amino acid loss is deamination and in general amino acids are only excreted by the kidney to a small extent. Glutamate can be found in the urine of humans (4.3 μg/mg creatinine in females) (Ragginer et al., 2012).
Overall, glutamate is absorbed in the intestine and is metabolised to a great extent in the gut wall. There are several transport systems which can transport glutamate into the brain. After oral administration of glutamate, infant monkeys have a higher plasma concentration as compared to adults a difference which is not observed in humans. In the species investigated (mice, rats, pigs), no increased brain glutamate concentrations were measured following increased plasma glutamate after MSG ingestion from the diet. In rats, administration of 4,000 mg MSG/kg bw by gavage resulted in an increased concentration in the brain whereas the same dose given in the diet had not such an effect which the Panel considered as a consequence of the administration as a bolus dose.
3.5.2. Acute oral toxicity
JECFA (1988) reported for mice oral LD50 values ranged between 12,961 and 19,200 mg/kg bw. The reported oral LD50 value for MSG was 19,900 mg/kg bw in rats. The reported oral LD50 value for l‐glutamic acid was above 2,300 mg/kg bw in rabbits. From the description in JECFA, it is not clear whether it is glutamic acid or one of its salts that have been tested.
An acute toxicity study has been performed as a limit dose test in Wistar rats (Degussa AG, 1983 [Documentation provided to EFSA n. 9]). Five male and five female animals received 5,110 mg glutamic acid/kg. In the observation period none of the animals died or showed signs of toxicity.
Takasaki et al. (1990, only abstract available) reported oral LD50 values for mice and rats were between 7,700 and 8,500 mg/kg bw for monopotassium l‐glutamate, between 13,300 and 18,200 mg/kg bw for calcium di‐l‐glutamate, between 5,900 and 9,100 mg/kg bw for monoammonium l‐glutamate, and between 14,900 and 19,000 mg/kg bw for magnesium di‐l‐glutamate.
The Panel noted that the acute toxicity of glutamic acid and its salts was low.
3.5.3. Short‐term and subchronic toxicity
Short‐term studies
Rats
In a 28‐day study in SD rats, performed as a limit test study, MSG was administered in the diet to 10 males and 10 females per group (CIT, 1997a [Documentation provided to EFSA n. 7]). The study was performed according to Good Laboratory Practice (GLP) and all parameters included in the OECD guideline 407 were tested. There were no adverse effects observed at 4,800 mg/kg bw per day for females and at 5,100 mg/kg bw per day for males, the only doses tested.
In a second 28‐day study in SD rats (CIT, 1997b [Documentation provided to EFSA n. 8]), MSG was administered in the diet to 10 males and 10 females per group. The study was performed according to GLP and following the OECD guideline 407. There was no adverse effect observed at 4,900 mg/kg bw per day for females and at 5,300 mg/kg bw per day for males, the only doses tested.
Dogs
MSG was administered in the diet to beagle dogs for 90 days, four males and four females per dose group (dose: 150, 500 or 1,500 mg/kg bw per day MSG monohydrate) (Biosafety Research Center, 2007b [Documentation provided to EFSA n. 4]). The study was performed according to GLP and following the OECD Guideline 409. The report enumerated clinical signs like vomiting and loose stools/diarrhoea which were transient. Changes in laboratory data were observed which were clinically not relevant and transient. Significant increases in both the absolute weight (+100%) and relative weight (+100%) of the thymus, as compared with the control group, were noted in the high‐dose group of females, which was not accompanied by an increase in mitosis in the cells or other histopathological findings, and were considered by the authors as ‘unspecific lesions’ and not as adverse. No other dose‐dependent and substance‐related adverse effects of oral administration of MSG monohydrate were seen in any of the observations, measurements or examinations in this study. The authors stated that no adverse effects of oral administration of 150, 500 or 1,500 mg/kg bw per day MSG monohydrate to male and female beagle dogs were seen in any of the observations, measurements, or examinations in this study. The authors concluded that the no observed adverse effect level (NOAEL) was 1,500 mg/kg bw per day or more for both sexes. The Panel agreed with the conclusions of the authors.
Subchronic toxicity studies
Rats
In a study on effects of different sodium salts on the urinary system, male Fischer 344 rats (10 animals/group) were fed diets containing 0% or 5.83% of MSG (equivalent to 0 and 5,250 mg/kg bw per day) for 10 weeks (Cohen et al., 1995). The study was not in conformity with a regulatory guideline. Histopathological examination after 10 weeks was limited to the stomach, kidneys, and urinary bladder. The food and water consumption in the treated group was comparable to controls. The body weights were significantly decreased in the treated group. Urinalyses revealed a significantly increased sodium level and pH. A significant decrease in urinary creatinine concentration was attributed by the authors to an increased volume of urine even if the increase in volume was not‐significant. An amorphous precipitate was present in the urine of most rats in the treatment group. The incidence of simple hyperplasia of the urothelium in the bladder was significantly increased. Urothelial hyperplasia of the renal papilla was non‐significantly increased in glutamate‐treated animals. As other sodium salts tested concomitantly showed the same effect as for MSG, the Panel considered that these findings were most probably related to the sodium ion rather than to glutamate.
De Groot et al. (1988) studied the effect of a cereal‐based stock diet or a purified casein diet containing 0% or 6% of MSG (equivalent to 0 and 5,400 mg/kg bw per day on the urinary bladder epithelium at different pH of the urine in male albino Wistar rats (10 animals/group). (a) Groups of male rats were fed either stock or purified diets containing 6% of MSG and 1.6% of salt sodium hydrogen carbonate or fed stock diets with 6% of MSG and 1.0% of the ammonium chloride or stock diets with 2.5% of the potassium hydrogen carbonate. Urine was collected during the first 2 h following the beginning of the light period at different days during the study. Histopathological examination after 13 weeks was limited to the kidneys, ureters, urinary bladder, liver, testes, thyroid, adrenals and bone. The food consumption in the treated groups was comparable to controls. The body weights were slightly decreased in all treated groups, but the decrease was only statistically significant in the two groups fed a purified diet with MSG with or without sodium hydrogen carbonate. The relative kidney weight was increased in all treated groups although not reaching statistical significance in the group fed a stock diet with MSG. Urinalyses revealed increased pH values of all groups fed MSG. Rats on the purified diet consistently showed lower urinary pH values than rats on the stock diet. Sodium hydrogen carbonate and potassium hydrogen carbonate increased the pH values, while ammonium chloride decreased the pH values. Epithelial hyperplasia and cysts of the bladder were increased in groups of rats fed stock diet with MSG or potassium hydrogen carbonate. These hyperplastic changes were reduced in the group of rats fed a stock diet with MSG and ammonium chloride, and only occurred to a minimal or slight degree in one animal fed a purified diet with MSG. Urothelial hyperplasia of the renal pelvis or papilla, accompanied by basophilic deposits, was observed in several rats fed MSG and sodium hydrogen carbonate in either of the two diets. The authors concluded that the alkalising properties of MSG were responsible for the urinary bladder hyperplasia observed. The Panel agreed with this conclusion which indicates that the urinary pH might be more important than sodium ion concentration for the observed effect.
MSG was administered in the diet to Sprague–Dawley Crl:CD(SD) rats 20 males and 20 females per dose group (dose: 0, 0.5%, 1.5% or 5% equivalent to 0, 308, 931 and 3,170 mg/kg bw per day for males and 0, 354, 1,066 and 3,620 mg/kg bw per day for females) (Biosafety Research Center, 2007a [Documentation provided to EFSA n. 3]). The study was performed according to GLP and following the OECD Guideline 408. In clinical laboratory tests, an increase in blood urea nitrogen (BUN) was noted in males in the 5% group. This increase was not considered toxicologically significant because the increase in urea was derived from the metabolite of glutamate through urea cycle. Increases in urine sodium concentration and total excretion of sodium in urine were observed which resulted from the sodium in the administered glutamate salt. There were no test substance administration‐related effects on any of the other parameters examined. The NOAEL was identified as the highest dose tested (3,170 mg/kg bw per day for males and 3,620 mg/kg bw per day for females) and the Panel agreed with this NOAEL.
In a 13‐week repeated dose study in rats (TNO, 2014 [Documentation provided to EFSA n. 22]), the oral toxicity of MSG monohydrate21 (purity > 98%) was tested using a protocol according to GLP and following the OECD guideline 408. Doses of 0%, 1%, 2% or 4% MSG monohydrate equal to 0, 700, 1,300 and 2,700 mg/kg bw per day for male and 0, 700, 1,500 and 2,900 mg/kg bw per day for female were tested. There were no clinical signs observed and no animals died. No neurobehavioural findings could be observed, indicating no neurotoxicity up to the dose of 2,700 mg/kg bw per day in males and 2,900 mg/kg bw per day for females. Haematology, clinical chemistry, macroscopic and microscopic examinations showed no noticeable differences between treated groups and controls. In week 12, dose‐related increases in sodium excretion was noted in all treated groups which could be explained by the high sodium intake when administering MSG monohydrate. Other urinary investigations in this study showed no differences between treated animals and control animals. The NOAEL identified by the Panel were the highest doses tested, 2,700 mg/kg bw per day for males and 2,900 mg/kg bw per day for females.
Dogs
Beagle dogs (5 animals/sex per group) were fed diets containing 0%, 2.5%, 5.0% or 10.0% of MSG (equivalent to 0, 625, 1,250 and 2,500 mg/kg bw per day) for 2 years (Owen et al., 1978b). Every 13 weeks each animal was examined ophthalmoscopically and electrocardiographically (ECG). Haematology, blood chemistry and urinalyses were performed initially and at intervals of 13 weeks. From each group 2 animals/sex were killed at 13 weeks and subjected to gross and histopathological examination and organ weight analysis. The remainder of the animals were killed at 104 weeks and subjected to the same examinations. There were no significant differences in clinical signs, food consumption, body weight, ophthalmoscopy, ECG, haematology, blood chemistry, organ weights, mortality and histopathology between controls and treated animals. Urinary volume and sodium excretion were generally higher in the treated than in control dogs. The Panel identified a NOAEL of 2,500 mg MSG/kg bw per day, the highest dose tested.
The Panel further noted that in contrast to the 90‐day study (Biosafety, Research Center, 2007b [Documentation provided to EFSA n. 4]), no increase in thymus weight was reported in this 2‐year study (Owen et al., 1978b), which supports the conclusion of the authors of the 90‐day study that the effects on the thymus weight were ‘unspecific lesions’.
Overall, urothelial hyperplasia of the renal pelvis or papilla was observed in several rats fed MSG at doses of up to 5,400 mg/kg bw per day. The Panel considered that the alkalising effects of MSG were responsible for the urinary bladder hyperplasia observed at this dose. In dogs, the only treatment‐related effects were increased urinary volume and sodium excretion in the 2‐year study. From this study in dogs, the Panel identified a NOAEL of 2,500 mg MSG/kg bw per day, the highest dose tested.
3.5.4. Genotoxicity
In vitro
In the reverse mutation assay using Salmonella Typhimurium TA98, TA100, TA1535, TA1537 and TA1538 strains and Saccharomyces cerevisiae strains, l‐glutamic acid, monopotassium glutamate and monoammonium glutamate were not mutagenic with or without metabolic activation (unpublished reports from (Litton‐Bionetics, 1975a,b, 1977, as referred to by JECFA, 1988).
In the reverse mutation assay using S. Typhimurium TA97 and TA102 strains, glutamic acid was not mutagenic with or without metabolic activation (Fujita et al., 1994, only abstract available). The Panel noted that the study is of limited relevance since only two S. Typhimurium tester strain were used.
In the study by Zeiger et al. (1992), MSG (purity > 81%) was assessed for its mutagenicity in the reverse mutation assay using S. Typhimurium strains TA1535, TA97, TA98 and TA100 up to a maximum concentration of 10,000 μg/plate in water, both in the absence and presence of rat and Chinese hamster liver S9 metabolic activation at 10% and 30% and no mutagenicity was observed. The pre‐incubation method was employed. The Panel noted that the study complies with current OECD Guideline 471 with the exception that tester strains S. Typhimurium TA102 or Escherichia coli WP2uvrA bearing AT mutation were not used. However, since oxidising or cross‐linking properties are not expected, the Panel considered this deviation as no relevant.
In the reverse mutation assay using S. Typhimurium TA98, TA100, TA1535, TA1537 and TA1538, tester strains MSG was not mutagenic with or without metabolic activation (De Flora et al., 1984). The highest concentration assayed was not specifically reported for MSG but as a general approach each test compound was tested up the solubility or toxicity limits. The plate incorporation method was employed. The Panel noted that the study complies with current OECD Guideline 471 with the exception that tester strains S. Typhimurium TA102 or E. coli WP2uvrA bearing AT mutation were not used. However, since oxidising or cross‐linking properties are not expected, the Panel considered this deviation as no relevant.
In the same study (De Flora et al., 1984), MSG was also negative in a DNA‐repair test with E. coli WP2, WP67 and CM871 strains with or without metabolic activation. However, the Panel noted that the relevance of this results was limited since the assay did not receive further validation and is currently not used in the battery of genotoxicity testing for regulatory purposes.
In the study by Ishidate et al. (1984), MSG (purity 99.8%) was assessed for its mutagenicity in the reverse mutation assay using S. Typhimurium strains TA1535, TA1537, TA92, TA94, TA98 and TA100 up to a maximum concentration of 5,000 μg/plate in phosphate buffer, both in the absence and presence of rat liver S9 metabolic activation and no mutagenicity was observed. The pre‐incubation method was employed. The Panel noted that the study complies with current OECD Guideline 471 with the exception that tester strains S. Typhimurium TA102 or E. coli WP2uvrA bearing AT mutation were not used. However, since oxidising or cross‐linking properties are not expected, the Panel considered this deviation as no relevant.
In the reverse mutation assay with S. Typhimurium TA1535, TA1537, TA98, TA100 and E. coli WP2uvrA tester strains, l‐glutamic acid (purity range 99.0–100.5%) was not mutagenic at a concentration range of 100–5,000 μg/plate, both in the absence and presence of rat liver S9 metabolic activation at 10% (v/v) (Notox, 2010b [Documentation provided to EFSA n. 17]). Negative results for l‐glutamic acid were also observed in a separate repeat experiment, at the same concentration range both in the absence and presence of rat liver S9 metabolic activation at 5% (v/v) in S. Typhimurium tester strains TA1535, TA1537, TA98 and in a concentration range finding experiment at a concentration range of 3–5,000 μg/plate both in the absence and presence of rat liver S9 metabolic activation at 5% (v/v) in S. Typhimurium TA100 and E. coli WP2uvrA tester strains. The Panel noted that the study complies with the current OECD Guidelines 471 and was conducted according to GLP.
In the reverse mutation assay with the histidine‐requiring S. Typhimurium strains TA1535, TA1537, TA98, TA100 and the tryptophan‐requiring strain E. coli WP2uvrA, MSG monohydrate21 (purity 98%) was not mutagenic at a concentration range of 62–5,000 μg/plate both in the absence and presence of rat liver S9 metabolic activation and was not toxic to any strain as indicated by the background lawn of the bacterial growth (TNO, 2013b [Documentation provided to EFSA n. 21]). The Panel noted that the study complies with the current OECD Guidelines 471 and was conducted according to GLP.
In a chromosome aberration assay on 242 food additives, MSG (purity 99.8%) was assayed for its clastogenic properties in a Chinese hamster lung (CHL) cell line using three concentrations but only in the absence of S9 metabolic activation (Ishidate et al., 1984). The highest concentration of 10 mg/mL, selected from a cytotoxicity test based on estimation of the 50% growth inhibition, greatly exceeded the highest recommended concentrations in the current OECD Guideline no. 473. Negative results were obtained. However, the Panel noted some shortcomings in the experimental protocol which included the absence of treatment in the presence of S9 metabolic activation, the inclusion of short (3–6 h) treatment and a limited number of metaphases scored. On this basis, the results obtained were considered to be of limited relevance.
In the study by Kim and Yang (1987), MSG was investigated for induction of unscheduled DNA synthesis (UDS) as measured as chemically induced DNA repair and determination of DNA single‐strand breaks using the alkaline elution technique in primary rat hepatocytes cultures. Treatment of cultures were performed for 24 h at concentrations of 1, 5 and 10 mg/mL. Results obtained indicate that MSG did not induce unscheduled DNA synthesis or DNA single‐strand breaks at any of the experimental test points, although the intermediate and higher concentrations employed exceeded the maximum concentration of 2 mg/mL recommended in the genotoxicity assay in vitro in mammalian cells, by the relevant OECD guidelines.
In the study by Xing and Na (1996) assessing the induction of sister chromatid exchanges (SCE) in cultured human lymphocytes, glutamic acid at concentrations of 10, 50 and 100 μg/mL in the absence of metabolic activation, induced a slight though significant increase in the frequency of SCE compared to the concurrent untreated control. The observed increases were modest (the highest increase being 1.43‐fold the untreated concurrent control) and were not concentration‐related. The Panel noted that this last condition was one of the key requirements for a positive outcome of the assay according to the relevant OECD test guideline, TG 479 (1986). In addition, the Panel noted some limitations in the study protocol which include the absence of duplicate culture, positive control and use of metabolic activation. Furthermore, this assay is not included in the current battery of genetic toxicology tests for regulatory purposes.
MSG monohydrate (purity 99.0%), was tested for its potential to induce chromosomal aberrations in a CHL cell line both in the absence and presence of S9 metabolic activation (Hatano Research Institute, 2006a [Documentation provided to EFSA n. 12]). The cells were exposed to concentrations of 0.48, 0.95 and 1.9 mg/mL for 6 h (short‐term treatment) both in the absence and presence of S9 metabolic activation and for 24 h (continuous treatment), only in the absence of metabolic activation. Fixation of cells was performed in all cases at 24 h from the beginning of treatment. The test concentration of 1.9 mg/mL, corresponding to 10 mM represents the highest test concentration recommended by the current OECD guideline no. 473 (2014) and was selected based on the growth inhibition and mitotic reduction values of test compound treated cells compared to the concurrent vehicle control values. The results obtained indicated that MSG monohydrate did not induce any statistically significant increase for both structural chromosomal aberrations and polyploid cells at any concentration assayed in any treatment condition. The Panel noted that the study complies with the current OECD Guideline 473 and was conducted according to GLP.
In a similar study, MSG monohydrate (purity 99.6%) was tested for its potential to induce chromosomal aberrations in a CHL/IU cell line both in the absence and presence of S9 metabolic activation (Hatano Research Institute, 2007 [Documentation provided to EFSA n. 14]). The cells were exposed to concentrations of 0.48, 0.95 and 1.9 mg/mL for 6 h (short‐term treatment) both in the absence and presence of S9 metabolic activation and for 24 h (continuous treatment), only in the absence of metabolic activation. Fixation of cells was performed in all cases at 24 h from the beginning of treatment. The test concentration of 1.9 mg/mL, corresponding to 10 mM represents the highest test concentration recommended by the current OECD guideline no. 473 (2014) and was selected based on the growth inhibition and mitotic reduction values of the test compound treated cells compared to the concurrent vehicle control values. The results obtained indicated that MSG monohydrate did not induce any statistically significant increase for both structural chromosomal aberrations and polyploid cells for the short‐term treatment in the absence and presence of S9 metabolic activation. Statistically significant increases for the induction of structural chromosomal aberrations compared to the concurrent vehicle control were observed at the highest concentration in the 24 h continuous treatment in the absence of S9 metabolic activation. These increases (4%) were judged by the author to be not biologically relevant because low. The Panel agreed with this conclusion and further pointed out that the incidence of structural chromosomal aberrations in the concurrent vehicle control was ‘0’ a condition which rarely occurs. The Panel noted that study complies with the current OECD Guideline 473 and was conducted according to GLP.
MSG monohydrate23 (purity 98%) was evaluated in an in vitro micronucleus assay in human peripheral blood lymphocytes for its ability to induce chromosomal damage or aneuploidy in the presence and absence of rat S9‐mix fraction as an in vitro metabolising system (TNO, 2013a [Documentation provided to EFSA n. 20]). MSG monohydrate was added at 48 h following culture initiation (stimulation by phytohaemagglutinin) either for 4 h in the absence or presence of S9‐mix followed by 20 h recovery, or for 20 h in the absence of S9‐mix followed by a recovery of 28 h. Cytochalasin B (6 μg/mL) was added the start of recovery following 4 or 20 h treatments, in order to block cytokinesis and generate binucleated cells for analysis. For the short (4 h) and extended (20 h) treatments, the final concentrations of the test substance were 3.7, 7.3, 14.6, 29.2, 58,5, 117, 234, 468, 936 or 1,871 μg/mL and 98.3, 197, 393, 492, 614, 768, 960, 1,200, 1,500 or 1,871 μg/mL, respectively. The concentration of 1,871 μg/mL corresponds to 10 mM, the maximum test concentration recommended by the OECD Guideline no. 487 for this assay. Micronuclei were analysed from at least three concentrations for each treatment condition. For 4‐h treatment without and with S9‐mix, the concentrations were 468, 936, and 1,871 μg/mL, and for 24‐h treatment without S9‐mix, the concentrations were 1,200, 1,500 and 1,871 μg/mL. The levels of cytotoxicity (reduction in replication index) at the top concentrations were close to the concurrent solvent controls. Though the reduction of replication index did not reach the recommended 50–60% range of cytotoxicity the assay was considered valid since treatments with the test compound were performed at the maximum concentration of 10 mM recommended by the relevant OECD Guideline. One thousand binucleated cells per culture from two replicate cultures per concentration were scored for micronuclei and the results obtained indicated that MSG monohydrate did not induce micronuclei in cultured human peripheral blood lymphocytes when tested at appropriate concentrations in both the absence and presence of S9‐mix. The Panel noted that the study complies with the current OECD Guideline 487 and was conducted according to GLP.
In vivo
In a dominant lethal test, groups of 12 male albino Charles‐River mice were given MSG once by gavage at doses of 0, 2,700 and 5,400 mg/kg bw (Industrial‐Bio‐test‐Laboratories, 1973, as referred to by JECFA, 1988). Each treated male was mated with three untreated females each week for 6 weeks. The uteri of females were examined following sacrifice at midterm of pregnancy. Compared to controls there were no differences in the number of implantations, resorptions and embryos.
The genotoxicity of MSG was tested in ‘white Swiss mice’ in a series of different tests (Fahmy and Donya, 2001). Single i.p. injections of 800, 1,600 and 3,200 mg MSG/kg bw to five male mice did not induce SCE in bone marrow cells. At least 40 metaphases were examined for each animal. The same doses of MSG given by gavage for 7 days to five mice (male and female but not specified the number of each sex) did not increase the percentage of micronucleated polychromatic erythrocytes (MNPCE) in the bone marrow. At least 300 polychromatic erythrocytes (PCE) were examined for each animal. Several different dosing regimens were used to study the frequency of chromosomal aberrations in mouse bone marrow cells and spermatocytes. Groups of 4–5 mice were administered by gavage 3,200 mg MSG/kg bw per day for 1, 2 and 3 weeks, and 800 and 1,600 mg MSG/kg bw per day for 3 weeks. Other groups were fed 800 mg MSG/kg bw per day in the diet for 1, 2 and 3 months. At least 75 or 80–100 metaphases were examined for each animal. The results obtained indicate that in the bone marrow, the number of aberrant cells induced by MSG was not statistically significantly increased at all treatment conditions or dose‐levels assayed compared to concurrent negative control values when the number of metaphases with chromatid and chromosome gaps was excluded from the analysis. On the contrary, in the spermatocytes statistically significant increases in the frequencies of abnormal metaphases in the form of X–Y and autosomal univalent were observed. The Panel noted that this event does not correspond to structural chromosome aberrations. Sometimes they have been considered as marker of subsequent chromosome mis‐segregation at anaphase I and, consequently, of aneuploidies in secondary spermatocytes. However, direct comparative analyses between metaphase I univalency and metaphase II aneuploidies have disproved this hypothesis (Liang and Pacchierotti, 1988). Furthermore, the Panel noted significant shortcomings in the experimental protocol, which include a limited or insufficient number of cells scored as in the case of the micronucleus test, the analyses of nucleated bone marrow cells instead of normochromatic erythrocytes in the micronucleus test and the inadequate selection of sampling time in the chromosome aberration assay.
MSG monohydrate (purity 99.0%) was assessed for its genotoxicity in an in vivo micronucleus test (Hatano Research Institute, 2006b [Documentation provided to EFSA n. 13]). Groups of five male and five female ICR mice were treated once daily for 3 days at an interval of 24 h by oral gavage with 500, 1,000 and 2,000 mg/kg bw, the latter dose‐level being the limit dose for this test. Animals were sacrificed 24 h after the beginning of treatment. Bone marrow smears were prepared, stained with acridine orange and 2,000 PCE per animal were analysed for the presence of micronuclei. Results obtained indicate that the frequencies of MNPCE did not increase significantly in any test compound treatment group compared to the concurrent vehicle control. The Panel noted that the study was adequately performed according to GLP and met the requirements of the current OECD guideline TG 474 (2014) with the exception that 2,000 PCE per animal, instead of the recommended 4,000 were scored. Furthermore, the Panel noted that despite the absence of any clinical sign and significant reduction of the ratio of immature PCE to mature normochromatic (NCE) erythrocytes the exposure of target organ to test item was ensured since MSG monohydrate was fully absorbed (see Section 3.5.1) and therefore systemically available.
The majority of available data refer to MSG which has been reported to be negative in adequately conducted bacterial reverse mutation assays (Zeiger et al., 1992; De Flora et al., 1984; Ishidate et al., 1984; Notox, 2010b [Documentation provided to EFSA n. 17]; TNO, 2013a [Documentation provided to EFSA n. 20]) in two thoroughly performed and OECD guideline compliant in vitro chromosomal aberration assays in mammalian cells (Hatano Research Institute, 2006a, 2007 [Documentation provided to EFSA n. 12 and 14]) and in a limited study (Ishidate et al., 1984). Negative results were also obtained in an OECD guideline compliant in vitro micronucleus test in cultured human lymphocytes (TNO, 2013b [Documentation provided to EFSA n. 21]) in an UDS test and a DNA single‐strand breaks test in primary rat hepatocyte cultures (Kim and Yang, 1987).
In vivo MSG was also reported to be negative for the induction of chromosomal aberrations in germ cells in a dominant lethal test (Industrial‐Bio‐test‐Laboratories, 1973) and in micronucleus assay in mice (Hatano Research Institute, 2006b [Documentation provided to EFSA n. 13]). MSG was also negative for the induction of SCE and micronuclei in bone marrow cells and chromosomal aberrations in bone marrow cells and spermatocytes (Fahmy and Donya, 2001) though this study bears significant shortcomings.
Similarly, glutamic acid, potassium glutamate and ammonium glutamate were negative in bacterial reverse mutation assays (Litton‐Bionetics, 1975a,b, 1977, as referred to by JECFA, 1988)). The slight non‐dose‐related increase in SCE in cultured human lymphocytes induced by glutamic acid, although statistically significant, was judged by the Panel to be of no biological relevance (Xing and Na, 1996).
No data were available for calcium diglutamate and magnesium diglutamate.
The Panel considered the available in vitro and in vivo data for MSG adequate to evaluate its genotoxicity and to cover by read‐across the limited or missing data for glutamic acid, monopotassium glutamate, calcium diglutamate, magnesium diglutamate and monoammonium glutamate.
Overall, the Panel considered that glutamic acid (E 620), monosodium glutamate (E 621), monopotassium glutamate (E 622), calcium diglutamate (E 623), monoammonium glutamate (E 624) and magnesium diglutamate (E 625) did not raise concern with respect to genotoxicity when used as food additives.
3.5.5. Chronic toxicity and carcinogenicity
Mice
C57 black mice (100 animals in treatment groups, 200 animals in control group) were fed diets containing 0%, 1% or 4% of MSG (equal to 0, 58 and 201 mg/kg bw per day for males and 0, 53 and 183 mg/kg bw per day for females), or 1% or 4% of l‐glutamic acid (equal to 63 and 187 mg/kg bw per day for males and 57 and 168 mg/kg bw per day for females) for 2 years (Ebert, 1979b). Haematology was examined in 10 animals in each group after 30, 150, 215, 280, 370, 530, 664 and 715 days. All survivors were killed after 2 years and examined for macroscopic lesions and histopathology of major organs (trachea, bronchi, lungs, heart, kidneys, spleen, pancreas, submaxillary gland, thyroid, stomach, liver, intestine and skin). There were no significant differences in mortality, haematology, and histology between controls and treated animals. However, because the mortality was very high in all groups with less than 25% survival at 2 years, the Panel decided not to use this study for hazard characterisation.
Rats
CD rats (40 animals/sex per group) were fed diets containing 0%, 1%, 2% or 4% of MSG (equivalent to 0, 450, 900 and 1,800 mg/kg bw per day and 0, 580, 1160 and 2,320 mg/kg bw per day for males and females, respectively) for 2 years (Owen et al., 1978a). Haematology, blood chemistry and urinalyses were performed initially and at 13, 26, 52, 78 and 104 weeks. Serum level of glutamic acid was measured initially and at 26, 52 and 104 weeks. From each group, 10 animals/sex were killed at 12 weeks and subjected to gross and histopathological examination and organ weight analysis. The remainder of the animals were killed at 104 weeks and subjected to the same examinations. The circulating levels of glutamic acid were similar in all groups. There were no adverse significant differences in food consumption, ophthalmoscopy, haematology, blood chemistry, organ weights and mortality between controls and treated animals. Body weight was not affected in females but slightly less in males in the 4% group after 60 weeks. Water consumption, urinary volume and sodium excretion were increased in the 4% group and subepithelial basophilic deposits were observed in the renal pelvis. The tumour incidences were similar in controls and treated animals. The Panel identified a NOAEL of 4% in both sexes (equivalent to 1,800 mg/kg bw per day in males and 2,320 mg/kg bw per day in females), the highest dose tested.
SD rats (35–40 animals/sex in treatment groups, 61–89 animals/sex in control groups) were fed diets containing 0%, 0.1% or 0.4% of MSG (equal to 0, 59 and 133 mg/kg bw per day for males, and 0, 33 and 73 mg/kg bw per day for females), or 0.1% or 0.4% of l‐glutamic acid (equal to 48 and 132 mg/kg bw per day in males and 26 and 73 mg/kg bw per day in females) for up to 2 years (Ebert, 1979a). After 63 days, six animals of each sex from the control group and three animals of each sex from the treatment groups were killed and examined for gross and histological changes. After 214, 275 and 336 days, four females and one male were taken from each group for a multigeneration reproductive test. Haematology was examined after 241, 567 and 681 days. All F0 survivors were killed after 2 years and examined for changes in histology and weight of major organs. There were no significant differences in clinical signs, food consumption, mortality, haematology, organ weights and histopathology between controls and treated animals. The body weights were consistently and dose‐relatedly increased in treated groups compared to controls during the study period, however the differences did not reach statistical significance. The tumour incidences were similar in controls and treated animals. The Panel identified a NOAEL of 0.4% of MSG in both sexes (equivalent to 133 mg/kg bw per day for males and of 73 mg/kg bw per day for females), the highest doses tested.
Fischer 344 rats (50 animals/sex per group) were fed diets containing 0, 0.6, 1.25, 2.5 or 5.0% of MSG (equal to 0, 231, 481, 975 and 1,982 mg/kg bw per day in males, and 0, 268, 553, 1,121 and 2,311 mg/kg bw per day in females) for 2 years (Shibata et al., 1995). Urinalyses were performed on 10 rats of each group after week 1 and months 1, 3, 6, 12, 18 and 24. Haematology was evaluated in all surviving animals at study termination, but creatinine and BUN were not measured A gross pathological examination was made and the brain, heart, liver, spleen, kidneys, urinary bladder, adrenals and gonads were weighed. Extensive histopathological examination was performed on all major organs from animals in the control and high dose groups, as well as from all animals that died or became moribund during the study. Histopathological examination of all other animals was limited to the stomach, liver, kidneys, urinary bladder and gross lesions. There were no significant differences in clinical signs, food consumption, mortality and haematology between controls and treated animals. The final body weight of males in the 5% group was significantly decreased compared to the control group. Urinalysis revealed significantly increased pH and sodium concentrations, and significantly decreased potassium concentrations in both sexes at 2.5% and 5%. The urine volume was occasionally elevated in males in the 5% group. The relative kidney weights were significantly increased (12.7% increase in males and 11.9% increase in females) in both sexes in the 5% group and the relative urinary bladder weights were significantly increased in males in the 5% group. Transitional cell hyperplasia of the renal pelvis associated with moderate or severe chronic nephropathy was increased in males in the 1.25% and 5% groups but not statistically significant, and was not increased in the 2.5% group. The tumour incidences were similar in controls and treated animals. The Panel considered the 10% increase in relative kidney weight as not adverse as the weight changes was not accompanied by histopathological changes and identified a NOAEL of 5% in both sexes (equivalent to 1,982 mg/kg bw per day in males and 2,311 mg/kg bw per day in females), the highest dose tested.
In an assay for the potential tumour‐promoting activity of a series of sodium salts, MSG was given once by gavage to groups of four male F344 rats at doses of 0, 800, 1,600, 2,400 and 3,200 mg/kg bw (Furihata et al., 1989). The activity of ornithine decarboxylase (ODC) in the pyloric mucosa of the stomach was assayed in duplicate on pooled materials. ODC activity increased dose‐related up to 100‐fold with maxima 8 h after administration of MSG. A similar increase was seen when other sodium, potassium, calcium and ammonium salts including sodium chloride were tested. From the results, the authors concluded that the effect was due to the sodium ion and not due to glutamate. The Panel agreed with this conclusion.
In a study on possible promotion of the development of tumours, male Fischer 344 rats (20 animals/sex per group) were fed diets containing no test chemical, 5% of monocalcium di‐l‐glutamate tetrahydrate (equal to 2,514 mg/kg bw per day), 2.5% of monomagnesium di‐l‐glutamate tetrahydrate (equal to 1,259 mg/kg bw per day) or 5% of l‐glutamic acid (equal to 2,568 mg/kg bw per day) for 16 weeks following sequential treatment with three different carcinogens (diethylnitrosamine, N‐methylnitrosourea, and dihydroxydi‐N‐propylnitrosamine) over a 4‐week period (Tamano et al., 1993). Other groups of male Fischer 344 rats serving as negative controls were fed similar diets but without the pre‐treatment with carcinogens. Hyperplasia and tumours were found in many organs in the groups pre‐treated with carcinogens but glutamic acid or its salts did not promote carcinogenesis.
Overall, in three 2‐year studies in rats, no increased tumour rate was observed up to the highest doses tested (1,800 mg/kg bw per day, 133 mg/kg bw per day, 1,982 mg/kg bw per day in males and 2,320 mg/kg bw per day, 73 mg/kg bw per day and 2,311 mg/kg bw per day in females). Hence, the Panel considered MSG as not carcinogenic in rats. The only observed effect was a significant increase in relative kidney weights (12.7% increase in males and 11.9% increase in females)in both sexes in the 5% group in the study by Shibata et al. (1995)) which, however, was not considered as adverse effect because no histopathological changes were found. Thus, the NOAELs identified by the Panel from the three 2‐year studies were the highest dose tested.
3.5.6. Reproductive and developmental toxicity
Reproductive toxicity studies
Mice
In a two‐generation reproduction toxicity study, two strains of mice (IVCS and Swiss albino) were fed diets containing 0%, 2% or 4% (equal to approximately 0, 4,000 or 8,000 mg/kg bw per day) of MSG (Yonetani et al., 1979). The parent generation (2–5 animals/sex per group about 60 days old) were fed the MSG diets from 2 weeks prior to mating until 100 days after delivery. The F1 generation was weaned at 25 days and maintained on test diet until 130 days of age. At 90 days of age, 1 L from each group was selected and sib‐mated to obtain the F2 generation, which was killed at 20 days after birth. Body weight and food intake were recorded regularly throughout the study in all generations. The oestrus cycle was recorded in the parent and F1 generation. Litter size was recorded in the F1 and F2 generation. Vaginal opening was recorded in the F1 generation. Organ weights were recorded and histopathological examinations of major organs (brain, eyes, heart, lungs, liver, spleen, kidneys, adrenals, testes, ovaries and uterus) were carried out in the parent and F1 generation. Day of eye opening and external and skeletal malformations were examined in the F2 generation. No significant differences were noted between dosed and control mice in any of the recorded parameters. The Panel considered this study limited due to the very low number of animals used, the flaws in the study design such as selection of only one F1 litter and mating of siblings to obtain the F2 generation.
In a three‐generation reproduction study, CD‐1 COBS mice were fed diets containing 0%, 1% or 4% of MSG (equal to a 0, 1,500 and 6,000 mg/kg bw per day for males and 0, 1,800 and 7,200 mg/kg bw per day for females) (Anantharaman, 1979). The parent generation (17–33 males/group and 51–99 females/group) were fed MSG diets from 8 to 9 weeks prior to mating until end of lactation. Some of the mice in the F1 generation (116–370 animals/sex per group) were weaned at 4 weeks and maintained on test diet until 36 weeks of age. Others in the F1 generation were mated at 13–14 weeks or 20–21 weeks to obtain two F2 generations (59–229 animals/sex per group) and maintained on test diet until end of lactation. Some of the mice in the F2 generations were weaned at 4 weeks and maintained on test diet until 27–32 weeks of age. Others in the F2 generations were mated at 16 or 32 weeks to obtain two F3 generations (27–110 animals/sex per group) and maintained on test diet until end of lactation. The F3 generations were killed at 0, 3, 14 or 21 days after birth. Body weight and food intake were recorded regularly throughout the study in the F1 and F2 generations. The fertility index, gestation index, viability index, lactation index and litter size was determined in the F1, F2 and F3 generations. Histopathological examination of brain tissue was carried out in the F1 and F3 generations (these results are described in more detail under developmental neurotoxicity). For all of the recorded parental, reproductive and developmental parameters, results were similar in the treated groups and groups fed basal diet. The Panel considered 4% MSG (equal to 6,000 mg/kg bw per day for males and 7,200 mg/kg bw per day for females), the highest doses tested, as the NOAEL for reproductive toxicity.
Glutamic acid was evaluated in the Chernoff/Kavlock developmental toxicity screen together with 54 other chemicals. Pregnant ICR/SIM mice (26–30 animals/group) were given vehicle or glutamic acid (dose level not presented) by gavage on day 8–12 of pregnancy (Seidenberg and Becker, 1987). Maternal mice were weighed on days 7 and 13 of gestation and at the first day after giving birth. The litters and pups were counted and weighed on the day of birth and on day 3. Dead pups were examined for external abnormalities. The uteri of dams not giving birth by gestation day 21 or 22 were examined. According to the authors, no significant differences were found between control and dosed mice in various parameters calculated from the experimental data. The Panel considered this study too limited for being used in hazard characterisation
Rats
SD rats (35–40 animals/sex in treatment groups, 61–89 animals/sex in control groups) were fed diets containing 0%, 0.1% or 0.4% of MSG (equal to 0, 59 and 133 mg/kg bw per day in males, and 0, 33 and 73 mg/kg bw per day in females) or 0.1% or 0.4% of l‐glutamic acid (equal to 48 and 132 mg/kg bw per day in males, and 26 and 73 mg/kg bw per day in females) for up to 2 years (Ebert, 1979a). After 214, 275 and 336 days, four females and one male were taken from each group for a multigeneration reproductive test. Further generations (up to F3–F5) from the original F1 progeny were produced. The only parameters reported are the number of litters, number of offspring and litter size. The authors stated that there were no adverse effects on fertility. The Panel considered this study too limited for the safety assessment as the number for animals used for the reproduction phase of the study was too low and the only reproduction parameters examined were the number of litters and the number of offspring per litter.
A two‐generation reproductive toxicity study was performed according to GLP and following the OECD TG 416 (Biosafety Research Center, 2008 [Documentation provided to EFSA n. 6]). All parameters as mentioned in this guideline were tested including functional tests at weaning. MSG was administered in the diet to Charles‐River rats 30/sex per group at concentrations of 0%, 0.5%, 1.5% or 5%. At 1.5% w/w, the compound intake of F0 parental males and females was 939 and 1,039 mg/kg bw per day, respectively, and for F1 parental males and females 1,305 and 1,422 mg/kg bw per day, respectively. At 5% w/w, the compound intake of F0 parental males and females was 3,131 and 3,496 mg/kg bw per day, respectively, and for F1 parental males and females 4,404 and 4,618 mg/kg bw per day, respectively. No effects have been noted on oestrous cycle and sperm parameters in the parental animals. Reproductive indices were not different from the controls as were offspring viability indices. Litter observations did not indicate a substance related effect. An increased absolute and relative kidney weight was observed in the males and females of the F0 (abs.: 10.1% males, 9.5% females; rel.: 9% males, 14.1% females) and F1 generation (abs.: 9.0% males, 19.7% females; rel.: 10.9% males, 17.8% females) of the 5% group. In addition, the absolute and relative ovary weights of the F1 females were increased of the 5% group. Reduced absolute and relative spleen weights were observed at weaning in both sexes in the F1 generation in the 5% group; this effect was not observed in the F2 generation. No macroscopical and histopathological changes were observed in the kidneys, ovaries and spleen. The authors identified a NOAEL of 1.5% MSG in the diet (equal to 939 and 1,039 mg/kg bw per day in males and females, respectively) for parental toxicity and 5% (equal to 3,131 and 3,496 mg/kg bw per day for males and females, respectively, the highest dose tested) for reproductive and developmental toxicity. The Panel agreed with the NOAEL identified by the authors for reproductive and developmental toxicity; however, the Panel considered that the NOAEL for parental toxicity was 5% (equal to 3,131 and 3,496 mg/kg bw per day for males and females, respectively) the highest dose tested because no histopathological changes were observed in organs in which the weight was increased.
Rabbits
Female New Zealand rabbits (22–24 animals/group) were fed a diet containing 0%, 0.1%, 0.825% or 8.25% of MSG (equivalent to 0, 30, 247 or 2,475 mg/kg bw per day, from 2 to 3 weeks before mating and through the gestation period (JECFA 1988). Male rabbits (16 animals/group) were also dosed for 2–3 weeks before mating. The males showed no treatment‐related abnormalities. The does were subjected to a caesarean section and necropsy on gestation day (GD) 29 or 30. The uterus was examined for resorptions. Offspring were examined for visceral and skeletal anomalies. No significant differences were noted between dosed and control offspring on body weight, food consumption or general behaviour, pups and litter data and no effects were seen in the fetuses. The brain of five male and five female pups of all groups group were checked for neuronal necrosis, but none was found. JECFA identified a NOAEL of 8.25% MSG (equivalent to 2,475 mg/kg bw per day). The Panel agreed with this NOAEL but noted that limited data were available.
Prenatal developmental studies
Rats
Pregnant rats were given MSG orally at doses up to 7,000 mg/kg bw per day from GD 6–15 or GD 15–17 (Khera et al., 1970; as referred to by JECFA, 1988). No differences from controls were reported for the offspring up to the period of weaning. Development to maturity was also normal except that offspring from dams dosed from GD 15–17 during gestation showed impaired ability to reproduce. No further details on this study were available.
Pregnant SD rats (25 animals/group) were fed 0%, 0.5%, 1.5% or 5% MSG in the diet (equal to 0, 302, 898 and 3,019 mg/kg bw per day) from GD 6–20 (Biosafety Research Center, 2007c [Documentation provided to EFSA n. 5]). The study was performed according to GLP and following the US FDA guidelines (2000) (which is similar to OECD TG 414). Clinical signs, body weight and food intake were recorded at regular intervals. On GD 20 the dams were subjected to caesarean section and necropsy. The following organ weights were measured: heart, lungs, liver, kidneys, spleen, adrenals ovaries and uterus (full and empty). The number of corpora lutea, implantations, resorptions, live and dead fetuses, fetal weight and external abnormalities were recorded. Fetuses were examined for visceral and skeletal abnormalities. No maternal or developmental effects were observed. The authors identified a NOAEL of 5% MSG in the diet equal to 3,019 mg/kg bw per day, the highest dose tested. The Panel agreed with this NOAEL.
Rabbits
Female rabbits (9–11 animals/group) were given 0 or 25 mg glutamic acid hydrochloride/kg bw per day or 25 mg MSG/kg bw per day by gavage for 15 days after mating (Yonetani, 1967; as referred to by JECFA, 1988). The conception rate, mean litter size, and nursing rate were determined. The weights of ovaries, adrenal glands, liver, kidneys and spleen were recorded for the dams. In offspring the body weights as well as the weights of testes, the ovaries and adrenal glands were recorded. Offspring were examined for external and skeletal malformations. The body weights were slightly lower in the offspring of dosed dams. No other significant differences were noted between dosed and control offspring. No further details were available. The Panel noted that the only dose tested in this study was low.
Overall, the Panel noted that two dietary reproductive studies and one prenatal developmental study were adequate for hazard characterisation. A three‐generation reproduction toxicity study in mice with a NOAEL of 4% MSG in the diet (equal to 6,000 and 7,200 mg/kg bw per day in males and females, the highest dose tested) (Anantharaman, 1979), a two‐generation reproduction toxicity study in rats with a NOAEL of 5% MSG in the diet (equal to 3,131 and 3,496 mg/kg bw per day for males and females, respectively), the highest dose tested, for parental, reproductive and developmental toxicity (Biosafety Research Center, 2008 [Documentation provided to EFSA n. 6]). In a dietary prenatal developmental toxicity study in rats, a NOAEL of 5% MSG (equal to 3,019 mg/kg bw per day), the highest dose tested, was identified for maternal and developmental toxicity (Biosafety Research Center, 2007c [Documentation provided to EFSA n. 5]). The Panel noted that no reproductive and developmental effects were reported in these studies.
3.5.7. Neurotoxicity
Neurotoxicity in animals from weaning onwards
This section reports mainly studies previously evaluated by JECFA (1988) which the Panel directly cited from the JECFA report and did not re‐evaluate. Studies published after the JECFA evaluation have been assessed and summarised by the Panel.
Mice
‘MSG was administered to 4 weanling mice of each sex ad libitum in the diet or drinking water at levels of 46 g/kg per day or 21 g/kg per day, respectively. No hypothalamic lesions were induced. Plasma glutamic acid levels were doubled by giving MSG at 10% w/v in the diet, but the threshold for neurotoxicity of MSG by dietary administration was not exceeded’ (Heywood et al., 1977 as referred to by JECFA, 1988).
‘Weanling mice were fasted and deprived of fluids overnight, then given as drinking fluid either 10% MSG; a mixture of 5% MSG, 4.5% monosodium aspartate and 1% aspartame; 5% MSG; 2.5% MSG and 2.5% aspartate; or a choice between 10% MSG and deionised water. The brains were examined about 4 h after start of treatment. All animals ingesting MSG‐containing solutions sustained hypothalamic injury’ (Olney et al., 1980 as referred to by JECFA, 1988).
‘Weanling mice were deprived of water for 14 h overnight and then given solutions of MSG as the sole drinking fluid; 12–180 animals receiving > 3 g MSG/kg bw in drinking water developed lesions of the arcuate nucleus. Fewer lesions were observed if free choice was allowed between water and MSG solutions, if glucose or arginine were also added to the MSG solutions, or if water deprivation occurred during the day’ (Torii and Takasaki, 1983 as referred to by JECFA, 1988). In the weanling period, mice seemed to be more susceptible than other species. In some of the studies, doses showed histopathologically confirmed neurotoxic effects.
Rats
‘Male rats aged 23 days were deprived of water or water and food overnight, and then given aqueous solutions of MSG at concentrations up to 10%. No hypothalamic lesions were found with intakes up to 5 g MSG/kg bw’ (Takasaki and Torii, 1983 referred to by JECFA, 1988).
‘Five groups of 6 weanling rats were given basal diet with added 20% glucose, 20% MSG, 40% MSG, or 17% l‐glutamic acid and sodium equivalent to 20% MSG for 5 weeks and then killed. No specific endocrine or neurological defects were seen’ (Wen et al., 1973 as referred to by JECFA, 1988).
‘No CNS damage was observed in rats which were fed 4% MSG in the diet for 2 years’ (Heywood and Worden, 1979 as referred to by JECFA, 1988).
In a study on behaviour, weaned male Druckrey rats (20 animals/group) were treated orally with 0 or 2,000 mg/kg bw per day of MSG in a 10% aqueous solution for 10 days (Ali et al., 2000). They were experimentally investigated 90 days later for behavioural effects. Locomotor activity (spontaneous and after apomorphine challenge) was monitored as well as cognitive ability. No spontaneous locomotor effects were found, but after apomorphine challenge marked significant decreases in distance travelled, ambulatory and stereotypic times and the number of stereotypic movements with an increase in resting time were observed. Significant decreases in the learning performance were seen in the learning (acquisition) phase without any changes in the extinction and relearning phases.
In a study on behaviour and hormonal changes, adult male SD rats (6 animals/group) were given orally by gavage 0 or 3,000 mg/kg bw per day of MSG in an aqueous solution for 6 days per week for 7 weeks (Seo et al., 2010). As the purpose of the study was to investigate whether MSG administration aggravates behavioural and hormonal changes induced by chronic variable stress, two additional groups were included in the study – one group was exposed to chronic variable stress and one group to a combination of chronic variable stress and 3,000 mg MSG/kg bw per day. Food intake was recorded daily and body weights were recorded twice weekly. An open field test was performed before and after the treatments. After 7 weeks, the adrenal weight and the plasma level of corticosterone were determined. The daily food intake and the body weights were significantly decreased in all treatment groups compared to control. In general, there were inconsistent or no changes in different kinds of activity in the open field test between the control group and the groups exposed to MSG or chronic variable stress. However, for the group exposed to the combination of MSG and chronic variable stress, significant decreases were noted for most kinds of activity. The relative adrenal gland weight and the level of plasma corticosterone were unchanged in the MSG group. However, for the group exposed to the combination of MSG and chronic variable stress, the relative adrenal weight was increased and the level of corticosterone was decreased to a higher degree than exposure to chronic variable stress alone.
Hamsters
‘Groups of male and female hamsters aged 25 days were deprived of water or water and food overnight, then offered solutions containing. 0, 2, 4, 6 or 8% MSG for 30 min. Six hour later the animals were killed and the brains examined; no hypothalamic lesions were seen’ (Takasaki and Torii, 1983 as referred to in JECFA, 1988).
Dogs
‘No CNS damage was observed in dogs which were fed 10% MSG in the diet for 2 years’ (Heywood and Worden, 1979 as referred to by JECFA, 1988). Details of the study are described in Section 3.5.3 (subchronic toxicity studies in dogs)
Neurotoxicity in neonatal animals
This section reports mainly studies previously evaluated by JECFA (1988) which the Panel directly cited from the JECFA report and did not re‐evaluate.
Mice
Neonatal mice aged 10–12 days received single oral doses of MSG at 250, 500, 750, 1,000 or 2,000 mg/kg bw by gavage (n = 7–23 per group). After 3–6 h all treated animals were anaesthetised with chloral hydrate and killed by brain perfusion with glutaraldehyde. Brain damage, as evidenced by necrotic neurons, was evident in arcuate nuclei in 52% of the mice at 500 mg/kg, 81% at 750 mg/kg, 100% at 1,000 mg/kg, and 100% at 2,000 mg/kg. The lesions were identical by both light and electron microscopy to subcutaneous‐produced lesions. No lesions were seen in the control and in the 250 mg/kg group. The number of necrotic neurons increased with the dose. Four animals tested with glutamic acid also developed the same lesions at 1,000 mg/kg bw. Effect additivity was suggested with aspartate as 500 mg/kg MSG given in combination with 500 mg/kg aspartate had the same effect as 1,000 mg/kg MSG or 1,000 mg/kg aspartate (Olney and Ho, 1970).
‘Six 9–10‐day old mice, dosed orally with 10% MSG (2,000 mg/kg), showed characteristic brain lesions’ (Geil, 1970; as referred to by JECFA, 1988).
‘Groups of 10–12‐day old Swiss Webster albino mice, each containing 7–23 animals, were given single oral doses of MSG at levels of 0.25, 0.5, 0.75, 1.0 or 2.0 g/kg. Groups of 2 or 4 mice of the same age were given single oral doses of either 1.0 or 3.0 g/kg L‐glutamic acid or monosodium L‐aspartate or 3.0 g/kg L‐glutamate‐L‐aspartate, MSG, NaCl, L‐glycine, L‐serine, L‐alanine, L‐leucine, D,L‐methionine, L‐phenylalanine, L‐proline, L‐lysine, L‐arginine, or L‐cysteine. The animals were sacrificed after dosing and brains were examined by either light or electron microscopy. The severity of brain damage was estimated by quantifying the pathological changes in the hypothalamus. One g/kg of glutamic acid destroyed approximately the same number of hypothalamic neurons as a comparable dose of MSG. Of the amino acids tested, only aspartate and cysteine produced hypothalamic damage. These amino acids caused both retinal and hypothalamic lesions similar to those found after treatment with MSG’ (Olney and Ho, 1970 as referred to by JECFA, 1988).
‘MSG was administered at dose levels of 1 g/kg to infant mice and 2 and 4 g/kg to infant rats. All the animals developed lesions in the arcuate area of the hypothalamus and median eminence. No evidence of cellular pathology was noted in controls’ (Burde et al., 1971; as referred to by JECFA 1988).
‘MSG, monopotassium glutamate, sodium chloride, and sodium gluconate at 1 g/kg in a 10% w/v solution (and comparable volumes of distilled water), were administered orally and subcutaneously to mice and rats at 3 or 12 days of age and to dogs at 3 or 35 days of age and the animals were killed within 24 h of dosage. Examination of the eyes and of the preoptic and arcuate nuclei of the hypothalamus by two pathologists revealed no dose‐related histomorphological effects in any of the test groups at either of the two ages selected to correspond to the periods before and at the beginning of solid food intake’ (Oser et al., 1971 as referred to by JECFA, 1988).
‘Groups of 3‐ and 12‐day old C57BL/J6 mice, each containing 5 animals, were given single intragastric or single subcutaneous doses of MSG, sodium chloride, sodium gluconate, potassium glutamate (all 10% solutions, doses were 10 mL/kg bw), or water. All animals were killed 24 h after dosing. Microscopic examination of the brains, particularly the ventral hypothalamus, did not show any neuronal necrosis of the hypothalamic arcuate nuclei’ (Oser et al., 1973 as referred to by JECFA, 1988).
‘Neonatal mice were given 0, 2 or 4 g MSG/kg bw by gavage and sacrificed after 20 or 30 min or after 1, 2, 3 or 24 h. At the higher‐dose level, oedema and necrosis of the neurons of the arcuate nucleus were seen after 20 min and preoptic and arcuate nucleus lesions were observed after 30 min. The lesions spread wide with time affecting the tectum and other structures in 2–3 h. Phagocytosis was seen in the arcuate nucleus after 24 h. Primary lesions in neurons were seen at the electron microscope level after 30 min’ (Lemkey‐Johnston and Reynolds, 1972, 1974 as referred to by JECFA, 1988).
Sodium chloride was administered to 3–9‐day old mice as a 10% solution by gavage at doses of sodium equimolar with the sodium in 1–10 g MSG/kg bw. Glutamic acid hydrochloride was administered at 2 and 4 g/kg bw as a 20% (w/v) solution and sucrose (in 80% w/v) was given at dosages equimolar to 4, 8 or 10 g MSG/kg bw. Oedema and pyknotic nuclei were seen in sodium chloride‐treated animals of 5 days of age; no lesions were seen in animals older than 6 days at the time of treatment, nor in mice given sucrose’ (Lemkey‐Johnston et al., 1975 as referred to by JECFA, 1988).
‘Groups of 10‐day old mice were given by gavage 2 or 4 g MSG/kg bw together with 0.63 g NaCl/kg bw or 1.93 g glucose/kg bw. Sodium chloride did not potentiate MSG‐induced brain lesions, while glucose reduced significantly the number of neurotic neurons in the arcuate nucleus. In another experiment, groups of 10‐day old mice were given by gavage 2 g MSG/kg bw alone or together with 1.93 g/kg bw of glucose, fructose, galactose or lactose, or 1.83 g/kg bw of sucrose. The mono‐ and disaccharides reduced significantly the number of neurons affected in the arcuate nucleus’ (Takasaki, 1979 as referred to by JECFA, 1988).
‘Groups of infant mice, 10 days of age, were given by gavage 2 g MSG/kg bw alone or 2 g MSG/kg bw along with 2.3 g arginine HCl/kg bw, 0.2 g leucine/kg bw, or 0.02 units of insulin (prior to gavage). All the additional treatments reduced the extent of injury to the arcuate nucleus relative to MSG alone’ (Takasaki and Yugari, 1980 as referred to by JECFA 1988).
‘Suckling mice were given by gavage 2, 6 or 9 mg MSG/kg bw on days 6–10 postnatally and were killed on day 11. No hypothalamic lesions were noted. Another group given 6 mg MSG/kg bw for 5 days were weaned and observed for 12 months. No hypophagia, obesity, or hyperactivity were noted’ (Wen et al., 1973 as referred to by JECFA 1988).
‘Casein and fibrin hydrolysates were administered to 9–11‐day old mice at doses of 20, 50 or 100 μL/g bw and plasma amino acid levels were determined at intervals. No neuronal damage resulted when plasma glutamate levels were below 24 μmole/dL (normal, 6–10 μmol/dL); the minimal threshold plasma level for neuronal injury was estimated as about 50–52 μmole/dL. Older (25 days old) mice showed a markedly greater ability to metabolise glutamate, which probably accounts for their decreased sensitivity to glutamate’ (Stegink et al., 1974 as referred to by JECFA 1988).
MSG was given by gavage to 8‐day‐old mice at dose levels of 250 or 500 mg/kg bw (Daabees et al., 1985). Plasma glutamate concentrations were determined at each dose level. No animal given glutamate at 250 mg/kg bw developed neuronal necrosis. However, neuronal necrosis developed in three of the ten animals given MSG at 500 mg/kg bw. At this dose level, the peak plasma glutamate concentration was 880 μM; in the 250 mg/kg bw group the peak concentration was 480 μM.
Rats
‘At birth, the rat brain glutamate concentration was about 4 mM and increases over a period of 20 days to the adult value of approximately 10 mM. When a 4 g/kg dose of MSG was given intragastrically, convulsions were seldom observed, and when it occurred only after 90 min. 2 g MSG/kg given intraperitoneally always caused convulsions. When young rats were given 4 g/kg of MSG, monosodium aspartate or glycine, the glutamine level was increased significantly in the brain in all cases, but only MSG and aspartate caused convulsions. D‐Glutamate (4 g/kg), which is not deaminated by the rat, also caused convulsions. These results suggested that the convulsions caused by MSG are not due to liberated ammonia, but rather to the amino acid anion. At 4,000 mg/kg, MSG gave rise to serum concentrations of glutamate of about 70 mM, strongly suggesting osmotic problems’ (Mushahwar and Koeppe, 1971 as referred to by JECFA 1988).
‘An experiment was conducted in which 45 male and 45 female rats were fed 1 or 250 mg/kg MSG daily from 1 to 90 days of age, at which time the animals were killed cells. A comparable control group received only laboratory chow over the same period of time. General clinical observations, body weights, haematologic parameters, and other clinical chemical measurements were within the normal range. At autopsy, organ weights were within the normal range. Histochemical and ultrastructural studies of the hypothalamus and median eminence showed no evidence of repair or replacement of neuronal cells by elements of glial or ependymal’ (Golberg, 1973 as referred to by JECFA, 1988).
Weanling rats, 3 days of age, and 12 days of age were given either single intragastric or single subcutaneous doses of MSG, sodium chloride, sodium gluconate, potassium glutamate (all 10% in solutions, dose: 10 mg/kg bw), or water (n = 5 per group). All animals in these groups were killed 24 h after dosing. In addition, in another experiment, rats (3 days of age) were treated with the same test compounds at the same dose levels (n = 12 per group). Half of each group was sacrificed at 6 h and the other half at 24 h after dosing. Microscopic examination of the brain, particularly the ventral hypothalamus, did not show neuronal necrosis of the hypothalamic arcuate nuclei, except in one rat dosed with 1,000 mg/kg MSG at 3 days of age, and killed 24 h later, which showed an area in the median eminence which contained cells with slight nuclear pyknosis and prominent vacuolation (Oser et al., 1973).
‘Rats given 4 g MSG/kg bw on days 1–10 of life and tested at 50 days in a swimming maze were less able to learn the maze. Glutamate levels in the brain, liver and blood were raised after 10 days, while other amino acid levels were also changed. Structural alterations were probably responsible for the permanent impairment of brain function’ (Berry et al., 1974 as referred to by JECFA, 1988).
Iwata et al. (1979) investigated the effect of age and of the route of administration of MSG on behaviour and histopathology of the brain. Rats (2 days of age) were injected subcutaneously (s.c) with saline, 200 mg MSG/kg bw, or 4,000 mg MSG/kg bw for 10 days (n = 50 per group); one group 10 days of age (n = 50) was injected s.c. with 4,000 mg MSG/kg bw for 10 days; two groups 10 days of age (n = 50 per group) were given saline or 500 mg MSG/kg bw by gavage; and one group 10 days of age (n = 50) was put on an ad libitum diet containing 10% MSG. The animals were observed for up to 9 months. Multiple injections of 4,000 mg MSG/kg bw to neonates caused low grip strength, hypoactivity, changes in spontaneous motor activity, deficit of learning ability and tail mutilation. The same treatment beginning at 10 days of age resulted in only slight behavioural abnormalities later in life or no detectable changes. Adverse effects, in particular behavioural effects were not observed in the groups with subcutaneous injection at doses lower than 4,000 mg/kg bw per day and were more pronounced if the treatment was performed in rats 2 days of age. No effects were observed in the groups of rats treated with oral doses of MSG. Adverse effects were related to histological evidence of injury. (Iwata et al., 1979).
Dogs
‘Groups of 6 pups, 3–4 days of age, were dosed either orally or subcutaneously with 1 g/kg bw of MSG, sodium chloride, sodium gluconate, potassium glutamate or water. Pups were sacrificed at either 3 h, 24 h or 52 weeks after dosing. Other groups of dogs 35 days of age received single doses of test material, and were sacrificed at either 4 or 24 h post‐dosing. Body weights of dogs which were dosed once at 3 days of age and followed for a year, showed no evidence of effects of any treatment. Femur weight, as well as weight of the pituitary gland, ovaries, uterus and mammary glands were similar to controls. Gross and microscopic examinations of these tissues failed to reveal any abnormalities. Extensive microscopic examination of brain tissue of all test animals did not show any treatment‐related changes’(Oser et al., 1973 as referred to by JECFA, 1988).
Monkeys
‘Monkeys, 4‐day old, received single doses of MSG (4 mg/kg in phosphate buffer), either subcutaneously or orally. Animals receiving subcutaneous injections were sacrificed at 3, 24, and 72 h, the one receiving an oral dose at 24 h. No brain lesions were observed’ (Abraham et al., 1971 as referred to by JECFA, 1988).
‘Doses of 250 mg or 1 g/kg MSG were administered orally daily for 30 days to two groups of 3 infant rhesus monkeys starting at 1 day after birth. General clinical observations over a period of 30 days revealed normal growth, development and activity. No changes were observed in the levels of haemoglobin, haematocrit, RBC or WBC counts or reticulocytes. The levels of glucose, urea nitrogen, and serum potassium, calcium, and sodium were within the normal ranges. At autopsy, complete histological, histochemical and ultrastructural investigations of the entire arcuate nuclei and median eminence region failed to reveal any necrotic or damaged neurons’ (Golberg, 1973 as referred to by JECFA, 1988).
‘Three infant monkeys, 5‐day of age, received MSG by stomach tube at a dose of 2 g/kg. Two infant monkeys of 10 days of age, 2 of 20 days of age, and 2 at 40 days of age, received the same treatment. Two animals at 80 days of age received 4 g/kg. One control monkey was included in each group. The animals were observed for 4 h after dosing and then sacrificed. After a period of fixation, a block of tissue was removed from each brain which included the hypothalamus. Serial sections, 10 mm thick, were made in the horizontal plane and examined by light microscopy. No changes that were considered to be associated with the administration of MSG were observed in the hypothalamus of the monkeys’ (HRC, 1971 as referred to by JECFA, 1988).
‘A group of 3‐ to 4‐day old cynomolgus monkeys received either subcutaneously or orally single doses of 1 g/kg bw MSG, sodium chloride, sodium gluconate, or potassium glutamate and were sacrificed 3 or 24 h post‐dosing. Another group of monkeys (3–4 days old) received orally either 4 g/kg MSG or sodium chloride, and were sacrificed at 3, 6 and 24 h post‐dosing (3 and 24 h in the case of sodium chloride‐dosed monkeys). Detailed microscopic examination of the hypothalamus did not show any evidence of MSG induced necrosis or any differences between any of the groups. Examination of the eyes did not reveal any effects due to MSG. Glutamate and glutamine blood levels showed considerable variation in individual values among the animals dosed orally and subcutaneously. Subcutaneous dosing resulted in values an order of magnitude higher than those observed by oral dosing’ (Oser et al., 1973 as referred to by JECFA, 1988).
‘MSG was administered by intragastric intubation at doses of 2 g/kg to 2 monkeys aged 2 days. Two monkeys of similar age were used as controls. Four h after dosing, the animals were sacrificed. Examination of the hypothalamus (bordered rostrally by the optic chiasma and caudally by the pons) by light and electron microscopy did not show changes caused by administration of the test compound. Changes observed by electron microscopy occurred as frequently in control animals as in test animals and appeared to be due to fixation artefacts’ (Heywood et al., 1972, as referred to by JECFA, 1988).
‘Rhesus monkeys (Macaca mulatta) aged between 4 and 80 days were divided into groups by age. Each group contained 3 test animals and 1 control animal. Dosage levels were 2 g/kg for animals up to 44 days old and 4 g/kg for animals up to 80 days of age. The animals were observed for 4 h and then sacrificed. Immediately prior to dosing and prior to sacrifice, serum and plasma samples were obtained for measurement of SGPT, SGOT and plasma glutamic acid concentration. At sacrifice liver samples were obtained for measurement of GPT and GOT. SGPT and SGOT values did not show significant increases over the test period. Plasma glutamic acid was within the normal range. Liver GPT and GOT values were within the normal ranges. Examination of the hypothalamus region and associated structures by light microscopy did not reveal any compound‐related effects’ (Heywood et al., 1971 as referred to by JECFA, 1988).
‘Sixteen infant monkeys (M. mulatta or M. irus) were fasted for 4 h before receiving by stomach tube single doses of a 50% solution of MSG, equivalent to doses of 1, 2 or 4 g/kg bw. Control animals received distilled water. At 6 h post‐dosing the animals were sacrificed and the brains perfused for examination by light and electron microscopy. No morphological differences were observed in the hypothalamic regions of treated and control monkeys. Inadequately fixed tissue had the same appearance as that of a previously‐reported brain lesion in a newborn monkey’ (Reynolds et al., 1971 as referred to by JECFA, 1988).
‘Ten infant squirrel monkeys were fed either a 0, 4.8, 9.1, or 17% (based on dry weight) MSG formula diet for 9 weeks. Three of the test monkeys died. Two died of effects not related to MSG. The third, which was on the 17% diet group, developed convulsive seizures. However, the other 2 animals in this group were unaffected. Clinical observations were made daily, and at the end of the test period the monkeys were sacrificed and the major organs examined microscopically. Sections of the retina and hypothalamus were examined by electron microscopy. No hypothalamic or retinal lesions were observed’ (Wen et al., 1973 as referred to by JECFA, 1988).
‘An infant cynomolgus monkey and an infant brush monkey were fed 0.1% MSG formula for one year. Daily observations revealed no behavioural abnormalities. Their weight gains, ERG, EEG, and plasma amino acids were similar to controls not consuming MSG. No evidence of gross obesity was developed’ (Wen et al., 1973 as referred to by JECFA, 1988).
‘Groups of 3 infant monkeys were dosed with a mixture of water and skimmed milk containing either added NaCl or MSG, on an equivalent mole/kg basis. Administration was via nasogastric tube. Other groups were injected subcutaneously with either a 25% aqueous solution of MSG or a 10% solution of NaCl. The doses ranged from 1 to 4 g/kg bw. All animals were sacrificed after dosing and the brains examined by combined light and electron microscopy. Infant animals given relatively low oral doses of MSG (1 and 2 g/kg) sustained small focal lesions confined primarily to the rostro‐ventral aspect of the infundibular nucleus. Animals treated with high subcutaneous doses developed lesions which spread throughout, and sometimes beyond, the infundibular nucleus. At all doses tested and by either route of administration, rapid necrosis of neurons (within 5 h) was observed. Measurements of blood glutamate levels suggested that the threshold for lesion formation in 1‐week old rhesus monkeys may be in the range of 200 mg/L’ (Olney et al., 1972 as referred to by JECFA, 1988).
‘Neonatal primates given 1‐4 g MSG/kg bw orally showed only elevated aspartate and glutamate blood levels; no hypothalamic lesions were noted’ (Boaz et al., 1974 as referred to by JECFA, 1988).
‘Three monkeys were given orally 2 mg MSG/kg bw at 3, 60, or 99 days of age; a fourth monkey aged 180 days served as a control. After 4 h, examination of the brains revealed no abnormalities. A further 16 monkeys were divided into five groups; four groups received orally 2 g MSG/kg bw and one group was given 4 g/kg bw Examination of the brains and of serum GOT and GPT showed no abnormalities’ (Newman et al., 1973 as referred to by JECFA, 1988).
In another experiment, ‘two monkeys aged 2 days were given 2 g MSG/kg bw and the brains were examined after 4 h. No abnormalities were seen’ (Newman et al., 1973 as referred to by JECFA, 1988).
‘No treatment‐related hypothalamic lesions were observed in neonatal rhesus monkeys killed 3 or 24 h or 8, 15, or 30 days following oral or subcutaneous administration of 0.25, 1 or 4 g MSG/kg bw The hypothalamus was examined by light and electron microscopy and the arcuate nuclei, median eminence, ependymal and glial cells were comparable to controls’ (Abraham et al., 1975 as referred to by JECFA, 1988).
‘Neonatal monkeys aged 1–14 days were given 1–4 g MSG/kg bw by gavage. No lesions were detected in the hypothalamus (Reynolds et al., 1976). No evidence of hypothalamic lesions was seen following in vitro exposure in 1 embryonic and 7 fetal brains’ (Reynolds et al., 1979 as referred to by JECFA, 1988).
‘Ten neonatal squirrel monkeys were divided into four groups of 2‐3 animals; 2 received infant formula, 3 received infant formula with 5% added MSG, 2 were given infant formula with 10% added MSG, and 3 were given infant formula with 20% added MSG from day 11 to day 21 post‐partum followed by observation for 9‐10 weeks. The animals were killed in the twelfth week. No abnormalities were observed in EEG or ERG scans and no retinal or hypothalamic lesions were detected’ (Wen et al., 1973 as referred to by JECFA, 1988).
‘One cynomolgus monkey and 1 bushbaby were given 10% MSG in their diets from the first to the eleventh month of age. No adverse effects were noted on growth or on EEG or ERG scans. In another experiment, a 3‐week old cynomolgus monkey was injected intramuscularly with 2.7 mg MSG/kg bw and observed for 2.5 years. No untoward effects were seen’ (Wen et al., 1973 as referred to by JECFA, 1988).
‘The administration of MSG to a 2‐day old rhesus monkey at a dose level of 4 g/kg bw in baby formula failed to induce any pathological changes in the hypothalamus’ (Heywood and James, 1979 as referred to by JECFA, 1988).
Overall, the Panel noted that many neurotoxicity studies have been performed in neonatal animals, weaning animals and animals after weaning. MSG was administered by the oral route (by gavage, in drinking water and in the diet).
In mice, rats and monkeys in the neonatal period, a specific sensitivity was noted. In neonatal mice, doses of 250 mg MSG/kg bw and higher induced neurotoxic effects. After the neonatal period, the doses which elicited neurotoxic effects were even higher. In adult mice, doses below 3,000 mg/kg bw were without effect and in one study even doses of up to 41 g/kg were tolerated without adverse effects. In neonatal rats, doses up to 500 mg/kg bw per day had no neurotoxic effect, whereas in most of the studies higher doses induced necrosis. In adult rats, no neurotoxic effects were noted at the tested doses. In dogs, 1,000 mg/kg bw MSG did not result in neurotoxic effects. In one study in infant monkeys (0.5–8 days old), subcutaneous administration as well as oral dosing led to focal lesions of the infundibular nucleus which were small and confined with oral dosing and wide spread with subcutaneous administration of 25% glutamate up to 4,000 mg/kg bw. Subcutaneous dosing did lead to remarkably higher glutamate levels in blood than oral dosing. Measurements of blood glutamate levels suggested that the threshold for lesion formation in 1‐week‐old rhesus monkeys may be in the range of 200 mg/L.
From the above reported studies, the Panel noted that:
Histopathological lesions of the CNS (mainly the arcuate nucleus in the hypothalamus) have been observed in mice (Olney and Ho, 1970; Abraham et al., 1971; Burde et al., 1971; Lemkey‐Johnston and Reynolds, 1974; Reynolds et al., 1976; Takasaki, 1978a, 1979; O'Hara and Takasaki, 1979; Takasaki and Yugari, 1980; Torii and Takasaki, 1983, 1987; Daabees et al., 1985; Yu et al., 2006; Shen et al., 2010), rats (Burde et al., 1971; Takasaki et al., 1979; Takasaki and Torii, 1983), guinea pigs (Heywood and Worden, 1979), and monkeys (Olney et al., 1972) following high doses of MSG or glutamic acid by gavage. In general, neonatal animals were more sensitive to the neuronal injury than adult animals, and mice were the most sensitive species. The lowest dose associated with lesions was 500 mg/kg bw as a single dose of MSG administered to 8‐day‐old mice, whereas no animal given glutamate at 250 mg/kg bw developed neuronal necrosis (Daabees et al., 1985). Also, in 6–12 day old mice (Reynolds et al., 1976) and in 10–12 day old mice (Olney and Ho, 1970), the dose of 500 mg/kg be elicited neuronal necrosis. At this dose, the peak plasma glutamate concentration was 880 μM (Daabees et al., 1985), and 3 out of 10 (Daabees et al., 1985) or 12 out of 23 (Olney and Ho, 1970) neonatal mice had hypothalamic neuronal necrosis. In none of the three studies were neuronal effects noted at a single dose of 250 mg/kg bw, where the plasma peak concentration of glutamate was 480 μM (which was 4–5 times the baseline level). FASEB (1995) concluded that neuronal lesions have been observed in rodents exposed to MSG orally but the data available are insufficient to allow for generalisation with regard to either dose or peak plasma glutamate concentrations associated with formation of neuronal lesions. The Panel agreed with this conclusion.
Histopathological lesions of the CNS were not observed in studies with rats (Wen et al., 1973; Semprini et al., 1974a; Huang et al., 1976; Takasaki and Torii, 1983), mice (Semprini et al., 1974a; Heywood et al., 1977; Takasaki, 1978b), hamsters (Takasaki and Torii, 1983) and monkeys (Wen et al., 1973) where glutamate was fed in the diet or drinking water, except in two studies where weanling animals were deprived of water overnight and then offered water containing 5–10% of MSG (Olney et al., 1980; Torii and Takasaki, 1983) or in newer studies where the relative volume of the arcuate nucleus was significantly decreased in the male offspring of rats fed diets containing about 20% of MSG (Fernandez‐Tresguerres, 2005; Hermanussen et al., 2006; this study is described in the section Other studies ‘Obesity’).
Only Olney et al. (1972) detected histopathological CNS lesions after oral doses of 1 and 2 g/kg bw in infant monkeys. In other studies, the authors did not observe histopathological lesions at the same or higher doses(Abraham et al., 1971; Golberg, 1973; HRC, 1971 confidential report, Oser et al., 1973; Heywood et al., 1972 confidential report, Heywood et al., 1971; Reynolds et al., 1971; Wen et al., 1973; Boaz et al., Abraham et al., 1975 Reynolds et al., 1979; Heywood and James, 1979 as referred to by JECFA, 1988)
Neurodevelopmental studies
Mice
In a two‐generation study including effects on the cellularity of the brain, Swiss albino mice were fed diets supplemented with 1% or 2% of vitamin mixtures and containing 0%, 1% or 2% (equal to 0, 1,300–2,500 and 2,700–6,100 mg/kg bw per day) of MSG (Semprini et al., 1974b). Females in the parent generation were fed MSG diets throughout pregnancy and lactation. Females in the F1 generation were fed the same diet as their mothers from weaning, were mated at 3 months to obtain the F2 generation and maintained on test diet until end of lactation. A number of animals from the F1 and F2 generation were sacrificed at 0, 15 or 30 days of age to study the cellularity of the CNS based on the amount of nucleic acids and protein in brain homogenate. Body weight and food intake were recorded daily during pregnancy and lactation in the parent and F1 generations. The percentage of pregnancies brought to term, the litter size, the average weight at birth and weaning, and the percentage reared was determined in the parent and F1 generations. The F1 control animals did not produce any litters. According to the authors, the only statistically significant difference was in increased mean body weight at weaning in the F1 generation fed the diet with 1% MSG and vitamins at 2% and increased rearing compare to controls. However, the authors also stated that diets with MSG increased the percentage of rearing and the average weight of the offspring at weaning, and attributed the effect to a probable but undefined lack in the basic diet. The Panel noted that the number of animals per group was not clearly indicated and statistical methods were not described.
‘Pregnant mice received MSG at levels of 0, 5, 10, or 15% in the diet or 5% in drinking water on the eighteenth day of gestation. The animals were killed and fetal brains were examined. Groups of lactating mice were put on the same regimes for 1‐4 days and the brains of the pups examined. Further groups of weanling mice were given similar diets for 1‐4 days and killed 3 h after feeding. No changes were seen in the arcuate nucleus of the fetuses, suckling pups, or weanlings’ (Takasaki, 1978b as referred to by JECFA, 1988).
In a three‐generation reproduction study, CD‐1 COBS mice were fed diets containing 0%, 1% or 4% of MSG (equal to a 0, 1,500 and 6,000 mg/kg bw per day for males and 0, 1,800 and 7,200 mg/kg bw per day for females (Anantharaman, 1979; study details described above). Histopathological examination of brain tissue was carried out in the F1 and F3 generations. The neural density, especially in the arcuate and other nuclei of the hypothalamus, in the basal ganglia, hippocampus, thalamus and cortex were observed in thousands of brain sections from hundreds of mice treated with MSG and no effects were found when compared to the control group. Special attention was given to the presence of ganglial cell degeneration and necrosis, phagocytosis of decaying ganglial cells, decreased density in ganglial cells, especially of the hypothalamic nuclei, disturbed bilateral symmetry of ganglial cell pattern, glial proliferation, altered glial cells or evidence of oedema and myelin changes were observed. The authors concluded that MSG did not induce brain lesions when administered via the oral route. The Panel agreed with this conclusion.
In a study on developmental neurobehaviour and histology of the hypothalamus, pregnant Kunming mice (number of animals not mentioned) were given orally a dose of 0, 2,500 or 4,000 mg/kg bw of MSG dissolved in distilled water at days 17–21 of pregnancy (Yu et al., 1997). Food intake of the offspring was recorded monthly. Offspring (10 animals/group) were weighed every 10 days for the first 60 days and at 90 days. The selection from the litters and the sex of the F1 pups for the behavioural tests and brain histology was not described. The hypothalamus of 10‐ and 90‐day‐old offspring mice was examined by light microscopy. Different behavioural studies (Y‐maze discrimination learning (10 animals/group), spatial learning, tail flick latency, rope crawling test were conducted in 60‐day‐old offspring. Mating of offspring was done from day 25 to 60. No significant differences were noted in food intake. The body weight was significantly increased from 30 to 90 days at both dose levels compared to controls. Histological changes were not detected in the hypothalamus. Y‐maze discrimination learning was significantly dose‐related impaired. Spatial learning and tail flick latency was not affected by treatment. The capacity for crawling along a rope was impaired at the beginning of training in both dose groups but no difference was observed after training. According to the authors, the reproductive capacity was not impaired. The mating resulted in pregnancies and normal offspring. The study was not well described and a section on the statistical treatment of the data is lacking. The figures which were presented to demonstrate histological changes were of poor quality. Considering all these limitations, the Panel decided not to use this study in hazard characterisation.
A study was performed to investigate the possible protection of ferulic acid against effects of MSG on developing mouse fetal brain (Yu et al., 2006). In this study, pregnant Kunming mice (n = unknown) were given doses of 0, 1,000, 2,000 or 4,000 mg MSG/kg bw dissolved in distilled water from GD 17–19 by gavage. The selection from the litters and the sex of the F1 pups for the behavioural tests and brain histology was not described. Y‐maze discrimination learning and open field test were conducted 3–5 times in 8–10 animals/group in 20‐ to 90–day‐old offspring. Offspring were mated to produce a second and a third generation. The same investigations concerning behaviour and histopathology as in the first generation were performed in the second and third generation. The expression of bcl‐2 and caspase‐3 in brain nerve cells was measured in 1‐day‐old first generation mice. In the hippocampi of 30‐day‐old offspring mice of the first generation, intracellular oedema, degeneration and necrosis of neurons, and hyperplasia were detected at maternal doses of 2,000 or 4,000 mg/kg bw (only microphotographs shown; no numbers of animals or lesions). Results for the 1,000 mg/kg group were not presented. Neuronal changes were not detected in the second and third generation of mice. Y‐maze discrimination learning was significantly impaired and locomotor activity was significantly increased in the first generation mice of mothers treated with 2,000 or 4,000 mg/kg bw. The behavioural changes were also apparent in the second generation. It is not clear whether these changes were statistically significant. Exposure to 2,000 mg/kg bw resulted in downregulation of bcl‐2 expression and upregulation of caspase‐3 expression in the brain nerve cells of offspring. Results are not shown for the other two doses. The statistical analysis is described only in general terms. It remains open whether adjustment for multiple testing was performed. The study as such could have a high relevance for risk assessment because of the multigenerational approach and the endpoints tested. However because of the mentioned flaws on the reporting and the statistical analysis applied the study has a low reliability and hence, the Panel assessed the findings as not suited for hazard characterisation.
Eight‐week‐old female Kunming mice (not further specified) were given 0 or 4,000 mg/kg of MSG by oral administration at days 17–21 of pregnancy (Xu et al., 2007). Pre‐protachykinin A mRNA was studied in the offspring mice at 10, 20, 30, 60 and 90 days of age. The authors reported that pre‐protachykinin A mRNA was significantly decreased in most brain regions. Pre‐protachykinin is a precursor for the two important neurotransmitter peptides, substance P and neurokinin A. The authors stated that these data provide insight into the vulnerability of the neurons in different brain regions to MSG. It is, however, unclear whether the findings have any functional meaning.
Pregnant Kunming mice (10 animals/group) were given MSG at single doses of 0, 1,000, 2,000 or 4,000 mg/kg bw by oral intubation at day 17 of pregnancy (Shen et al., 2010). The number of litters used for selection and the sex of the F1 pups for the behavioural tests and brain histology were not described. The expression of the N‐methyl‐d‐aspartic acid (NMDA) receptors of type 1 and neuropeptide Y in the hippocampi was visualised by immune histochemistry in 1‐day‐old offspring (4 animals/group). Different behavioural studies (Y‐maze discrimination learning, open field test, rope crawling test) were conducted 1–5 times in 10–22 animals/group in 20–105‐day‐old offspring. In the hippocampi intracellular oedema, degeneration and necrosis of neurons, and hyperplasia were detected in a dose‐dependent manner in the hippocampi of the offspring of mothers treated with all doses of MSG. Exposure to MSG resulted in dose‐dependent upregulation of expression of both NMDA receptor type 1 and neuropeptide Y in the hippocampi of the offspring of mothers treated with all doses of MSG. Y‐maze discrimination learning was significantly impaired and locomotor activity was significantly increased in a dose‐dependent manner in the offspring of mothers treated with 2,000 or 4,000 mg MSG/kg bw. It is not clear whether Y‐maze discrimination learning and locomotor activity was significantly changed at 1,000 mg/kg bw. The capacity for crawling along a rope was significantly impaired in the offspring of mothers treated with all doses of MSG in a non‐dose‐dependent manner.
The Panel noted that results for all the parameters studied were given only in figures as means and standard deviations of the means. The number of animals in the test was given as 14–19. The statistical analysis was described only in general terms. Adjustment for multiple testing was not performed. The study as such could have a high relevance for risk assessment because of the multigenerational approach and the endpoints tested. However, because of the mentioned flaws, the study has a low reliability and hence the Panel assessed the findings as not suited for hazard characterisation.
Rats
‘Groups of 10‐day old Charles‐River rats (10 male and 10 female) were dosed orally with 0.2 mL of either strained baby food containing no MSG, strained baby food containing MSG up to 0.4%, or strained baby food containing MSG equal to a dosage level of 0.5 g/kg, additional to that found in normal commercially distributed baby food (390 mg per jar). The rats were mated; half of the offspring were removed from parental females and sacrificed after 5 h. Histological studies were made of brains in the area of the hypothalamus at the roof and the floor of the third ventricle. The remaining rats were returned to parental females and allowed to grow to maturity (90 days post‐weaning), then sacrificed and histological studies made of the brain. No lesions were observed in the brains of animals sacrificed at either 5 h post‐treatment or after reaching maturity. Animals which were reared to maturity showed normal growth and food consumption (Geil, 1970 as referred to by JECFA, 1988).
In a two‐generation study in which also effects on the number and cell types of the hypothalamic arcuate region were investigated, SD rats were fed diets containing 0%, 1% or 2% of MSG (equal to 0, 530–980 or 1,040–1,835 mg/kg bw per day) and supplemented with 0.5%, 1% or 2% of vitamin mixtures the content of which was not further specified (Semprini, 1971). The number of females used for the parental generation was 54 and for the first generation 38. The numbers and sex for each measurement were not presented. Females in the parent generation were fed MSG diets throughout pregnancy (from GD 5) and lactation. Females in the F1 generation, fed the same diet as their mothers from weaning, were mated at 3–5 months to obtain the F2 generation and maintained on test diet until the end of lactation. It is not clear from the original publication whether the F1 male rats were dosed. A number of animals from the F1 and F2 generations were sacrificed at 0, 15 or 30 days of age to study the cellularity of the CNS based on the amount of nucleic acids and protein in brain homogenate. Body weight and food intake were recorded during pregnancy and lactation in the parent and F1 generations. The percentage of pregnancies brought to term, the litter size and the average weight at birth and weaning, and the breeding percentage was determined in the parent and F1 generations. The authors stated that diets with MSG increased the percentage of successful pregnancies and tended to increase the percentage of newborns reaching weaning age. In general, the brains of dosed F1 rats contained a smaller number of nuclei and larger cells than controls at birth. In contrast, the brains of dosed F2 rats contained a higher number of nuclei and smaller cells than controls at birth. The cellularity differences present at birth disappeared at weaning. It is unclear whether the study results have been statistically evaluated. Because of this methodological flaw, the Panel considered that the study does not contribute to the hazard characterisation of glutamate.
In a two‐generation study on effects on selected brain and liver metabolites, male and female Holtzman rats (n = unknown) were fed diets containing 0% or 10% of MSG (equivalent to 0 and 9,000 mg/kg bw per day) for 100 days before mating (Prosky and O'Dell, 1972). Administration of the test compound during mating, gestation and lactation was not described. After weaning, the F1 generation (n = 20) were continued on the same diet as their parents for 100 days and mated. From the F2 generation, 10 offspring from each group were sacrificed at days 1, 2, 3, 5, 10 and 21. The conception rate and pups per litter were noted; data not presented). Body, brain and liver weight of the offspring was recorded at the days of sacrifice. Brains were assayed for glutamic acid decarboxylase (GAD), GABA, glutamate, aspartate, protein and DNA. Livers were assayed for RNA, DNA, protein and glutamate. At day 5, the stomach content was analysed for glutamate. The relative brain weight was significantly higher at day 3, and the relative liver weight was significantly lower at days 3 and 5. GABA was significantly increased at day 1. These effects were not observed at later stages; the authors considered the effects as transient. Offspring of dosed dams had 20% more free glutamate than controls in their stomach. Treated offspring had rough, shaggy‐hair coats which became normal during the third week. No other significant differences were noted between dosed and control rats in any of the observed parameters. This limited study was not further considered for the hazard characterisation.
In a study on developmental neurotoxicity, SD rats (about 18 L/group) were given diets containing 0%, 1.7%, 3.4% or 5.1% of MSG (equal to 0, 1,900, 3,700 and 5,300 mg/kg bw per day in males, and 0, 1,600, 3,200 and 5,000 mg/kg bw per day in females in the prebreeding period; equal to 0, 1,800, 3,900 and 6,200 mg/kg bw per day in offspring males, and 0, 2,000, 4,300 and 6,600 mg/kg bw per day in offspring females) from 14 days prior to mating until conception for males and throughout gestation and lactation for the females (Vorhees et al., 1979). On postnatal day 1 (PND1), litters were reduced to 12 pups per litter. The offspring continued on the same diet until 90 days of age. Body weight and food intake for dams and offspring were recorded throughout the study. The length of gestation, litter size, sex distribution and number of dead newborns were noted. Before weaning, two animals/sex per litter were observed for physical and behavioural development (pinna detachment, incisor eruption, eye opening, forward locomotion, surface righting, cliff avoidance, swimming development, pivoting activity, auditory startle, visual placing). In the post‐weaning period, two animals/sex per litter went through different behavioural tests (residential activity wheel, open field, rotorod, appetite position discrimination, active avoidance, passive avoidance). Neurohistology (not including hypothalamus) and weighing of the brain were performed at weaning and at 90 days of age, respectively. No significant differences were found in body weights or food intake. The mortality of offspring from treated dams was increased although only significant in the middose group. The other measures of reproductive performance were unaffected. In the high‐dose group, delayed early swimming development, diminished rearing frequency in the open field, altered active avoidance acquisition and extinction, and prolonged day‐2 passive avoidance retention were observed. The high‐dose group also showed a trend towards decreased running wheel activity. In this study, the number of animals is sufficient, pup selection and reporting of the neurodevelopmental test by sex is correct. The statistical procedures are described in detail, adjustment for multiple testing was performed and the selected statistical test methods are adequate. Because of the effects on neurobehaviour at the highest dose, the Panel considered next lower dose of 3,200 mg/kg bw per day as the NOAEL.
In a study on developmental neurobehaviour, pregnant Wistar rats (2–3 animals/group) were given drinking water containing 0% or about 5% of MSG (the amount of MSG in the water was adjusted to a daily dose of 10,000 mg/kg bw) at days 7–20 of pregnancy (Frieder and Grimm, 1984). The F1 pups were only obtained from 2 to 3 L and the observations were not divided by sex. Offspring (20–30 animals/group) were weighed at birth, at 28 days and at 6 months. At 20 and 35 days of age, the offspring (16–18 animals/group) were tested for general activity in an open field. Different behavioural studies (successive discrimination, simultaneous black‐white discrimination maze, acquisition of shock avoidance) were conducted in 6‐month‐old offspring (9–10 animals/group). The treated offspring had significantly lower birth weights than the controls. However, at 28 days and 6 months of age, the body weight was higher in the treated offspring than in the controls (only statistically significant at 28 days). At 35 days of age, the general activity of treated offspring was significantly reduced (more pronounced in males than females) compared to the controls. No significant differences were noted in the tests of successive discrimination and of acquisition of shock avoidance. However, in the simultaneous black–white discrimination maze, the treated offspring made significantly more errors than controls (more pronounced in males than females). The Panel noted that no correction was made for multiple testing and doing so would result in non‐statistically significant difference between treated and non‐treated animals.
In a study on developmental neurochemical effects, pregnant Wistar rats (10 animals/group) were given drinking water containing 0% or about 5% of MSG (the amount of MSG in the water was adjusted to a daily dose of 10,000 mg/kg bw) at days 7–20 of pregnancy (Frieder and Grimm, 1987). At 15, 21, 28 and 60 days of age the offspring (6 animals/sex per group) were killed and tested for various neurochemical parameters (choline uptake into synaptosomes from frontal cortex and hippocampus, choline acetyltransferase (ChAT) activity in both regions, uptake of Norepinephrine (NE) in the cortex and hypothalamus, uptake of γ‐aminobutyric acid (GABA) into synaptosomes from cortex and hippocampus (only at 60 days), ChAT activity in the striatum (only at 60 days)).The results were not separated by sex of the F1 animals. Choline uptake and ChAT activity in the frontal cortex of the treated offspring were significantly reduced at 15 days of age compared to the controls. Choline uptake in the frontal cortex gradually increased during development until it became significantly higher in treated offspring at 60 days of age compared to the controls. In male rats, the ChAT activity in the frontal cortex also increased and became significantly higher in treated offspring at 21 days of age compared to the controls. In male rats aged 60 days, both the choline uptake and ChAT activity in the hippocampus were significantly enhanced and the uptake of NE in the frontal cortex was significantly reduced in treated offspring compared to the controls. NE uptake in the hypothalamus was significantly increased in 21 day old and decreased in 28 day old male rats in treated offspring compared to the controls. The authors suggested that the neurochemical changes observed in this study may be related to the behavioural deficits seen in their previous study (Frieder and Grimm, 1984). The Panel noted that no correction was made for multiple testing and doing so would result in non‐statistically significant difference between treated and non‐treated animals.
In another study, pregnant Wistar rats (not further specified) were given l‐glutamate at a concentration of 0 or 1,000 mg/L (equal to 0 or 110 mg/kg bw per day) in the drinking water from gestational day 2 and during the whole gestational period (León et al., 2008). Brains of both mothers and their offspring were then isolated. Alpha Gi3 protein was significantly decreased in maternal brains. AlphaGi1 and alphaGi2 isoforms were increased in fetal brains. Adenosine 1 receptor functionality was significantly decreased in maternal brains. The authors stated that the results suggest that glutamate administered to pregnant rats modulates adenosine 1 receptor signalling in brains from mothers as well as offspring. It is unclear whether the findings have any functional meaning.
Pregnant Wistar rats (11–13 animals/group) were given drinking water containing 0 or 1,000 mg/L (equivalent to 0 and 120 mg/kg bw per day) of l‐glutamate from GD 2 onwards during the whole gestational period and lactation (López‐Zapata et al., 2011). Half of the dams and their offspring changed treatment for the lactational period; that is glutamate solution was replaced by water or vice versa. The effect on adenosine receptors in the brain was measured at day 15 after parturition in dams and offspring. Adenosine receptors were investigated because the adenosine pathway is one of the signalling pathways which could be modulated by l‐glutamate exposure. Adenosine 1 receptor was significantly downregulated in whole brain from both mothers and neonates exposed to l‐glutamate during gestation, lactation or both. Moreover, the adenosine 2A receptor was upregulated in neonates exposed to l‐glutamate during lactation. The authors stated that because of the important role played by adenosine in brain physiology special care should be taken when l‐glutamate is consumed in high doses during gestation and/or lactation.
Monkeys
Pregnant rhesus monkeys (4–6 animals/group) consumed 500 mL of water containing 0 or 4,000 mg MSG/kg bw per day for the last trimester of pregnancy (dosing period was 45–64 days) (Newman et al., 1973). After birth, each offspring was removed from its mother, observed for 4 h and sacrificed for histopathology of parts of the brain including the hypothalamic region. According to the authors, the duration of pregnancies and the infant birth weight were all within the accepted ranges and there were not any cases of delayed parturition or dystocia. The authors also stated that nursing, suckling and other behavioural patterns of the mother, as well as the behaviour of the infants were normal except for one dosed animal which killed its infant at birth. The study did not include a systematic protocol for the evaluation of behaviour. No histopathological abnormalities of the brain were detected.
Overall, studies on neurodevelopmental toxicity were performed in mice, rats and monkeys. Several studies showed flaws such as the low number of animals, selection procedure of the pups not described and no reporting of the sex of the F1 animals used for the behavioural tests, neurochemistry of brain histology and of the statistics used. The dietary study of Anantharaman (1979) in mice for the effects on neural density in brains of the F1 and F3 generation was adequate and showed no effects up to 7,200 mg MSG/kg bw per day. Other oral studies (Semprini et al., 1974b; Yu et al., 1997, 2006; Shen et al., 2010) in mice showed neurodevelopmental effects at dose levels from 2,000 mg/kg bw per day. However, in these studies, the statistical evaluation of the data and the reporting were considered of limited quality. Further oral studies (Semprini, 1971; Prosky and O'Dell, 1972 and Frieder and Grimm, 1984, 1987) in rats showed neurodevelopmental effects at 1,000–10,000 mg/kg bw per day. However, these studies and the reporting were considered limited by the Panel. The only study in monkeys showed no neurodevelopmental effects (behaviour and brain histopathology) at a dose of 4,000 mg/kg bw per day. The neurodevelopmental study was considered by the Panel of limited quality. From all these studies, the Panel considered that dietary neurodevelopmental toxicity study of Vorhees et al. (1979) in rats was adequate and a NOAEL of 3,200 mg MSG/kg bw per day was identified based on the absence of neurobehaviour effects which occurred at a higher dose.
3.5.8. Hypersensitivity, allergenicity and food intolerance
Mice
Male BALB/c mice with experimentally induced asthma were fed diets containing 0%, 0.5% or 5% of MSG monohydrate (equivalent to 0, 1,000 and 10,000 mg/kg bw per day) during the week before the first immunisation exposure and during the subsequent 3‐week period or once by gavage on the last day of the study (Yoneda et al., 2011). MSG feeding of asthmatic mice did not affect pulmonary eosinophil infiltration, production of Th2 cytokines, circulating IgE concentrations, or airway hyper‐responsiveness induced by methacholine. The oral gavage challenge with MSG solution did not exert any acute effects on pulmonary eosinophil infiltration or airway hyper‐responsiveness in the asthmatic mice.
Overall, the results of this study with a mouse model of asthma suggested that MSG was not involved in the development of asthma or in acute asthmatic responses.
Humans
Asthma
A possible association between intake of MSG and triggering of asthma attacks was first suggested in a case report in 1981 (Allen and Baker, 1981). Since then a small number of experimental studies have produced conflicting results. Three studies were only reported in abstract form with few experimental details available (Hosen, 1988; Germano et al., 1991; Altman et al., 1994) and are not considered in this risk assessment. Three of these studies have concluded that MSG may trigger asthma attacks in some people with asthma (Allen et al., 1987; Moneret‐Vautrin, 1987; Hodge et al., 1996), whereas three studies did not find such an association (Schwartzstein et al., 1987; Woods et al., 1998; Woessner et al., 1999).
Schwartzstein et al. (1987) concluded that it is highly unlikely that MSG is a contributing factor to bouts of bronchospasms in asthmatic patients because none of the 12 study patients responded. The study of Hodge et al., 1996 is a methodological study. Their results of non‐responsiveness in 11 out of 12 studied patients cannot be taken to assess the association between MSG exposure and asthmatic attacks in asthmatic patients. In the study of Woods et al. (1998) 12 asthmatic subjects with a perceived sensitivity towards MSG did not respond in a placebo‐controlled cross over challenge trial. In the study of Woessner et al. (1999) with 100 patients with asthma including 30 with a reported reaction of a MSG history no significant response was noted after 2,500 mg MSG in comparison to placebo treatment (Schwartzstein et al., 1987; Altman et al., 1994; Hodge et al., 1996; Woods et al., 1998). In five studies, the MSG history of the test subjects were either not reported, or 2 or less of the test subject were reported to have a history of asthmatic attacks after a Chinese meal (Moneret‐Vautrin, 1987; Schwartzstein et al., 1987; Hosen, 1988; Germano et al., 1991; Hodge et al., 1996). In the study by Allen et al. (1987) studied 32 asthmatic patients. Thirteen patients reacted to a dose of 2,500 mg MSG, half of them did so within 2 h following challenge but in the other half the reaction was delayed for up to 12 h. In the study of Moneret‐Vautrin (1987), two of 30 asthmatic patients responded with a mild bronchospasm 6–10 h after the ingestion of 2,500 mg MSG. The study by Schwartzstein et al. (1987) has been criticised for using a too short period of 4 h for lung measurements and it might also be a critical point that the largest challenge dose of 1,500 mg may has been too low (FSANZ, 2003). In the study by Hodge et al. (1996) one out of the 11 tested subjects was reported to have an asthma attack following MSG challenge in a double‐blind placebo‐controlled protocol. However, as the main purpose of the study was to compare two different methods of testing asthma reactions with different substances it is difficult to interpret the MSG result, as indicated by FSANZ (2003).
Based on the study by Allen et al. (1987), FASEB (1995) concluded that there appears to be a small subset of people with severe unstable asthma who may respond to doses of 1,500–2,500 mg of MSG given in capsules without food, but also that there is a pressing need to develop a more rigorous experimental protocol to test more critically the causal relationship. Others have suggested that when patients with unstable asthma are included and their medication is discontinued as in the studies by Allen et al. (Allen et al., 1987) and Moneret‐Vautrin (Moneret‐Vautrin, 1987), the positive responses are merely spontaneous asthma attacks (Stevenson, 2000). In the study by Allen et al. (Allen et al., 1987), 13 out of 32 subjects had a positive response, and in the study by Moneret‐Vautrin (Moneret‐Vautrin, 1987), 2 out of 30 subjects had a positive response. Both studies were conducted as single‐blinded placebo‐controlled studies where declines of peak expiratory flow (PEF) were used as an indicator of a positive response. The use of PEF as an indicator for a positive response has been criticised as the measurement can be influenced by subject bias (Stevenson, 2000). In most of the other studies, declines in forced expiratory volume in one‐second (FEV1) were used as an indicator of a positive response (Schwartzstein et al., 1987; Germano et al., 1991; Hodge et al., 1996; Woods et al., 1998; Woessner et al., 1999). One subject in each of the studies by Germano et al. (1991) and Woessner et al. (1999) had a positive reaction to MSG on single blind challenge but subsequent double blind challenge failed to reproduce the reaction. In the newest study by Woessner et al. (1999) subjects with unstable asthma were excluded from the study. Stevenson (2000) and also FSANZ (2003) concluded that the evidence for MSG‐induced asthma attacks is inconclusive, and that the newer studies suggest that MSG may not be a significant trigger of asthma attacks.
Overall, a small number of human challenge studies with different methodological flaws have produced conflicting results on a possible association between intake of MSG and triggering of asthma attacks. People with severe unstable asthma who had their medication withdrawn during the studies have had positive reactions in single‐blinded placebo‐controlled challenges with 1,500–2,500 mg of MSG given in capsules without food. However, it has been suggested that these positive responses may merely be spontaneous asthma attacks because of the withdrawal of the medicine. In the newest study where subjects with unstable asthma were excluded, one subject had a positive reaction to MSG on single blind challenge but subsequent double blind challenge failed to reproduce the reaction. The Panel considered that the studies did not provide conclusive evidence that MSG exposure is causally linked with elicitation of asthma attacks in the general population.
Urticaria and/or angio‐oedema
A review has evaluated the evidence for a relationship between MSG exposure and cutaneous reactions in the form of urticaria and/or angio‐oedema (Williams and Woessner, 2009). The six studies evaluated in the article review are summarised briefly below.
In four of the studies, the patients with a diagnosis of urticaria went through oral challenge with a number of food additives including MSG (Genton et al., 1985; Supramaniam and Warner, 1986; Botey et al., 1988; Simon, 2000). The three earliest of these studies had a number of design flaws that preclude drawing conclusions about an association. In Genton et al. (1985), four out of 19 adults were included in a single‐blind placebo‐controlled study. Four of them reacted to MSG after single‐blind placebo‐controlled an oral challenge where no attempt was made to disguise the taste of MSG, and where antihistamines were withdrawn fourteen days before the challenge. Supramaniam and Warner (1986) performed a placebo‐controlled multiple cross over study to several substances, three out of 36 children reacted to MSG after double‐blind placebo‐controlled challenge with capsules. The 4‐h interval used in this study between challenges with different the tested substances may have been too short to exclude the possibility that positive reactions may have been to other substances tested earlier. Botey et al. (1988) described four children who reacted to MSG in an uncontrolled challenge. In the studies by Supramaniam and Warner (1986) and by Botey et al. (1988) it is not mentioned whether medication was withdrawn before challenge.
In a newer and more well‐conducted study in 65 patients with active chronic idiopathic urticaria of which four reported a history of MSG induced urticaria, two patients reacted to MSG in a single‐blind placebo‐controlled challenge with 2,500 mg of MSG but subsequent double blind challenge failed to reproduce the reaction (Simon, 2000). In this study, the patients continued to take antihistamines at minimum effective dosage, and the challenge response was evaluated using objective reaction criteria compared with a baseline observation period in the same patient.
Two case reports on MSG intake and cutaneous reactions have been published (Squire, 1987; Asero, 2002b). In the newest study, a woman with a 12‐year history of chronic urticaria had decreased severity of symptoms and less need for antihistamines when placed on an additive free diet (Asero, 2002b). An open challenge consisting of a 3‐week unrestricted diet resulted in increased severity of urticaria which was again decreased when the additive free diet was resumed. Two separate double‐blind placebo‐controlled challenges with 100 mg of MSG resulted in both instances in severe urticaria within 45 min after challenge with MSG. In the older study, a man with recurrent attacks of angio‐oedema of the face and extremities reacted with angio‐oedema 16 h after ingestion of at least 250 mg of MSG in a single‐blinded placebo challenge (Squire, 1987). Dietary avoidance of MSG resulted in extended remission of attacks in this patient.
Overall, most of the studies suggesting a relationship between MSG and urticaria have design flaws that preclude drawing conclusions about an association. The Panel considered that, however, a case report (Asero, 2002b) with two positive separate double blind placebo controlled challenges in a patient provided some evidence to suggest that MSG may be a rare cause of urticaria.
Rhinitis
In two separate reports, three patients with chronic rhinitis symptoms attributed to MSG intake have been described (Asero, 2002a; Asero and Bottazzi, 2007). All three patients had marked improvement of nasal symptoms when placed on an additive free diet. An open challenge consisting of a 3‐week unrestricted diet resulted in relapse of nasal symptoms within a few days. When the additive free diet was resumed symptoms again improved. Double‐blind placebo‐controlled challenges with 100 mg of MSG resulted in all three patients in severe rhinitis within 4–24 h after challenge with MSG, and the symptoms lasted for a few days. Two of the patient went through a second double blind placebo controlled challenge which confirmed the results.
Williams and Woessner (2009) also reviewed the evidence for a relationship between MSG exposure and rhinitis. The two studies publications evaluated in the review are summarised briefly below.
Overall, the Panel concluded that case reports with two positive separate double blind placebo controlled challenges in two patients provide some evidence to suggest that MSG may be a cause of rhinitis.
3.5.9. Other studies
Obesity
An obesity syndrome was referred in Swiss albino mice treated in infancy with either single or multiple subcutaneous injections of MSG (Olney, 1969). The mice had a lower body weight when the treatment was stopped but became heavier than the control mice despite eating the same amount of feed or less (animals had free access to feed). The mice also had acute neuronal necrosis in several regions of the brain including the hypothalamus. It is the hypothesis of the author that the neonatal disruption of neuronal development might be the cause of adiposity. In 1976, MSG was administered by various methods to mice and rats of various ages and the incidence of obesity was determined by analysis of lipid content in the carcasses (Bunyan et al., 1976). At 20–30 weeks of age, the mean carcass lipid content was increased by about 120% in both sexes of mice exposed in the neonatal period to subcutaneous injections of MSG. The males but not the females had a significantly increased body weight (increased about 10%). Since then, the MSG‐mice model with s.c. injection of MSG has been repeatedly used as a model for obesity (Lutz and Woods, 2012).
FASEB reviewed in 1995 the studies available and assessed them. In many animal studies in which obesity and stunted growth were observed after neonatal parenteral dosing with MSG, lesions were observed in the arcuate nucleus of the hypothalamus and a decreased level of some of the hormones in the hypothalamic‐pituitary axis was found. In addition, the concentration of serum lipids, phospholipids, insulin and glucose was often increased.
Mice
Female C57BL/6 mice (number not stated) were given drinking water containing 0 or 640 mg MSG/L (equal to 0 and 97.2 mg/kg bw per day) for 3 weeks before mating, during pregnancy and lactation (Collison et al., 2010b). Male offspring (20 animals/group) continued their exposure to MSG (dose not exactly given, most probably 97 mg/kg bw) via drinking water until the age of 16 or 32 weeks. Food and water intake was recorded at 6 and at 28–30 weeks. Body weights and abdominal girth (marker of visceral adiposity) were recorded at 6 and 32 weeks. Markers in blood, indicating metabolic status (leptin, insulin, glucose, total cholesterol, high‐density lipoprotein cholesterol (HDL‐C), triglyceride, free fatty acids, adiponectin and retinol‐binding protein 4), were measured at 32 weeks. Liver and visceral fat were excised from four mice from each group for histology. The level of triglycerides in the liver was also determined. At 16‐week, RNA was isolated from liver and visceral fat from four mice from each group for gene expression analysis. No significant differences were noted in food intake and in initial or 32‐week body weight compared to control. However, the weight change from 6 to 32 weeks was significantly decreased while the change in abdominal girth was significantly increased. The water intake was significantly increased. Of the metabolic markers, triglycerides, free fatty acids, HDL‐C and insulin were significantly increased, but hepatic triglycerides remained unchanged. The histology of livers showed increased content of microvesicular vacuoles compared to controls. The histology of the visceral fat showed a significant increase in the mean adipocyte area. The expression of genes in the liver involved in lipid metabolism and bile synthesis was significantly elevated. In visceral adipose tissue, the expression of several genes involved in adipocytes differentiation and a transcription factor that controls lipid and cholesterol homeostasis was significantly increased, while the expression of mitochondrial respiratory chain components and a mitochondrial oxidative transcription factor was significantly decreased. As 28 different expression profiles were compared and no statistical adjustment done for multiple testing, the findings need to be confirmed in independent studies.
In a second study, C57BL/6 mice (number of animals not stated) were given drinking water containing 0 or 640 mg MSG/L (equal to 0 and 91.2 mg/kg bw per day) for 3 weeks before mating, during pregnancy and lactation (Collison et al., 2010a). Offspring (20 animals/sex per group) continued their exposure to MSG via drinking water until the age of 32 weeks. Food and water intake was recorded at 6 and at 28–30 weeks. Body weights and abdominal girth (marker of visceral adiposity) were recorded at 6 and 32 weeks. Markers in blood of metabolic status (leptin, insulin, glucose, total cholesterol, HDL‐C, low‐density lipoprotein cholesterol (LDL‐C), triglyceride, free fatty acids, adiponectin and retinol‐binding protein 4) were measured at 32 weeks. The spatial learning ability was assessed at 6, 16 and 32 weeks using the Morris Water Maze testing. No significant differences were noted in food and water intake and in initial and 32‐week body weight compared to control. However, the weight change from 6 to 32 weeks was significantly decreased in males and the change in abdominal girth was significantly decreased in females. The only metabolic disease marker that was significantly increased in both male and females was retinol‐binding protein 4. Insulin, triglycerides and free fatty acids were significantly increased and LDL‐C was significantly decreased in males. However, in this publication the abdominal girth change, the water intake, and the HDL‐C is not marked as significantly increased as they were in the former study by Collison et al. (2010b). The spatial learning ability was not significantly affected.
Adult male C57BL/6 mice (9 animals/group) were fed either a low‐fat (LF) diet (10 kcal% fat) or a very high‐fat (HF) diet (60 kcal% fat) with either two bottles of plain water or one bottle containing 1% of MSG in water (equivalent to 0 and 1,500 mg/kg bw per day) (Ren et al., 2011). Body weights and water and food intake were recorded weekly. At 15 weeks, the level of glycogen in liver and the level in blood of metabolic disease markers (leptin, insulin, glucose, total protein, total cholesterol, triglyceride, BUN, and non‐esterified fatty acids) were measured. In general, animals given a choice had a strong preference for the MSG solution. This preference was higher in the LF compared to the HF group. Intake of MSG neither influenced body weight nor the metabolic markers measured.
MSG has been administered by various methods including in the diet and drinking water to CFLP mice of various ages and the incidence of obesity was determined by analysis of lipid content in the carcasses (Bunyan et al., 1976). No obesity was detected at 18 weeks of age in the offspring (5–34 animals/sex per group) of female mice (4–11 animals/group) which were fed diets containing 0 or 100,000 mg/kg of MSG (equivalent to 0 and 20,000 mg/kg bw per day) either from 3 weeks before mating until weaning or from the 14th day of pregnancy until weaning. Neither was obesity detected at 23 weeks of age in female mice (20 animals/group) which were given drinking water containing 0 or 20,000 mg/L of MSG (equivalent to 0 and 3,000 mg/kg bw per day) from weaning onward.
Overall, conflicting results have been observed concerning influence on metabolic parameters and obesity when MSG was given on the oral route to mice.
Rats
Pregnant Wistar rats (8 females/group) were given 0, 2,500 or 5,000 mg/day of MSG with the diet (according to the publication ‘2,500 g, respectively, 5,000 mg MSG accounted for some 10%, respectively, 20% of dry weight of average daily food ration’) from day 14 of pregnancy to the end of weaning (Fernandez‐Tresguerres, 2005; Hermanussen et al., 2006). After weaning and gender separation, feeding with MSG was continued in the offspring (6–9 pups/sex per group) at the same doses for up to 90 days. For comparison, pregnant rats in a fourth group were not fed MSG but the offspring was injected s.c. with 4 g/kg bw every second day from day 1 to 10 of life. All litters were weighed weekly and food intake was recorded daily. Half of the pups were killed on day 30 and the other half on day 90. The brain was removed for histomorphometric analysis of the relative volume of the arcuate nucleus. Anterior pituitaries were removed for analysis of growth hormone (GH) content. Trunk blood was collected for analysis of plasma GH, insulin‐like growth factor‐1 (IGF‐1) and leptin. The birth weight was significantly decreased in the offspring of mothers fed 5 g of MSG. The weight at weaning was significantly dose‐related decreased in the offspring of mothers fed MSG. No changes were noted in the weight at weaning of pups injected s.c. The weights of pups were not specifically reported for any other times during the study. It was, however, mentioned that rats orally treated with MSG were smaller and had lower body weight than controls. It was also mentioned that the gravity index (specific weight, measured in air and water), however, indicated that animals fed MSG contained significantly more body fat both at day 30 and 90. The relative volume of the arcuate nucleus was significantly decreased at 90 days in offspring of both sexes injected s.c. as well as in male offspring fed the high dose. In general, the levels of plasma GH and IGF‐1 were significantly decreased in MSG treated rats (dose‐related in the orally fed rats) although no difference was noted on day 90 for the group fed 2,500 mg/day. The effect was in general more marked in males compared to females. The level of leptin was significantly increased in s.c. dosed rats. The animals fed MSG had significantly and dose‐related increased water (threefold at the highest dose) and food (almost twofold at the highest dose) intake. No difference was noted for food and water intake for rats dosed s.c. The amount of GH in the anterior pituitaries was not reported. In another study in pregnant rats a high dose of MSG (diet ad libitum contained 100,000 mg MSG/kg (equivalent to 7,000 mg/kg bw per day) induced increase in body fat at day 10 and day 20 of the pregnancy compare to controls without increase in body weight (Afifi and Abbas, 2011). In the offspring, the body weight was statistically increased. In the pregnant animals, serum insulin, leptin and glucose were elevated. The study was of limited quality as the only dose used was not given and could not be calculated because of lack of information. The authors hypothesised that the effect on fat content, insulin, leptin and glucose as well as the increased weight in the pups could be due to a metabolic imbalance as the leptin receptors gene expression was reduced in the hypothalamus.
In 10‐day‐old Wistar rats (7–14 animals/group, sex not specified) dosed once by gavage with 1,300, 1,400 or 1,500 mg/kg bw per day of MSG in a 10% aqueous solution, hypothalamic lesions (necrotic neurons in the arcuate nucleus) were induced from 1,400 mg/kg bw per day (Takasaki et al., 1979). Based on this result, 10‐day‐old Wistar rats (12 animals/sex per group) were dosed by gavage with 0 or 500 mg/kg bw per day of MSG in a 10% aqueous solution from day 10 to 19 of life. Another group of 10‐day old rats was also given 500 mg/kg bw per day by gavage for 10 days followed by a diet containing 5% of MSG (equal to 7,200 mg/kg bw per day in males and 7,400 mg/kg bw per day in females) for 10 days after weaning. Food intake, body weight, body length and tail length were recorded every third week for 11 months. Food intake was also recorded daily for the 10 days after weaning in all groups. Blood pressure, heart rate, temperature, haematology and serum glucose and triglyceride were measured at 11 months of age. At the same time, organ weights were determined and histology performed on the major organs. The only effect observed in treated rats was a slightly reduced food intake in female rats during the 10 days where the diet contained MSG. There were no brain lesions and no obesity (evaluated by Lee's index, which correlates well with carcass fat content) in treated rats.
Male SD rats (4–6 animals/group) had access to either two bottles of plain water or one bottle containing 1% of MSG (equivalent to 500 mg/kg bw per day) in water and another one containing plain water during lactation and for 15 weeks after weaning (Kondoh and Torii, 2008). After weaning, the rats were fed either a LF, HF, ultra‐high‐fat (UHF) or high‐sucrose (HS) diet. Body weights and water and food intake were recorded twice a week. At 15 weeks, the naso‐anal length, blood pressure, metabolic markers in blood (leptin, insulin, glucose, total cholesterol, triglyceride, and albumin) and abdominal fat were measured. Animals given a choice had a strong preference for the MSG solution irrespective of the diet. Rats with access to MSG weighed significantly less, had less abdominal fat, and had lower leptin levels than rats with access only to water (when the data from the four diet groups were combined). The food intake was similar in all groups. Intake of MSG did not influence the blood pressure, the naso‐anal length, and the rest of the metabolic disease markers measured. Comparable results were obtained when older male rats (9 animals/group, aged 8–23 weeks) were fed HF diet and either plain water or water/water with 1% of MSG. MSG ingestion did not influence the plasma glutamate concentration in male rats (8 animals/group) fed HF diet and either plain water or water/water with 1% of MSG from postnatal day 21 to postnatal day 24.
Male Wistar rats (8 animals/group) were fed either a standard diet or a fibre‐enriched diet with or without 100,000 mg MSG/kg bw per day for 45 days (Diniz et al., 2005). Food intake was recorded daily, and body weights were recorded once a week. At 45 days, markers in blood of metabolic disease and oxidative stress (leptin, insulin, glucose, triacylglycerol, lipid hydroperoxide, total antioxidant substances) were measured. No significant differences were noted in body weights. The food intake was higher in the group fed a standard diet supplemented with MSG, and the same group also had increased levels of insulin, leptin, glucose, triacylglycerol and lipid hydroperoxide, whereas the level of total antioxidant substances was decreased. No significant changes were noted between the group fed a fibre‐enriched diet and the group fed fibre‐enriched diet supplemented with MSG. The Panel noted that the oral LD50 values for MSG or glutamic acid are between 16,600 and 19,900 mg/kg bw in rats, the dose in the study seems to be not correct and the study results cannot be taken into consideration for hazard characterisation.
MSG has been administered by various methods including in the diet and drinking water to CFY rats of various ages and the incidence of obesity was determined by analysis of lipid content in the carcasses (Bunyan et al., 1976). No obesity was detected at 17 weeks of age in female rats (20 animals/group) which were given drinking water containing 0 or 20 g MSG/L (equivalent to 0 and 1,800 mg/kg bw per day) from weaning onward. Neither was obesity detected at 14 weeks of age in rats (6–8 animals/sex per group) which were fed diets containing 0 or 20 g MSG/kg (equivalent to 0 and 1,800 mg/kg bw per day) from weaning onward.
Overall, an influence of MSG on the regulation of appetite has been indicated in one study in which the male offspring of rats fed diets containing about 20% of MSG were smaller and had lower body weight but more body fat than controls although they had increased water and feed intake. In addition, the relative volume of the arcuate nucleus and the levels of plasma GH and IGF‐1 were significantly decreased. An association between MSG and increased body weight has not been detected in several studies in young and adult rats given MSG in the diet or drinking water. In one study, rats administered MSG in the drinking water weighed significantly less, had less abdominal fat, and had lower serum leptin levels than rats having access to drinking water without MSG although the food intake was similar in all groups and the plasma glutamate level was unaffected.
Cats
Domestic female cats (2 animals/group) were fed diets containing 0 or 1.125% of MSG (equal to 0 and 201.4 mg/kg bw per day) for 3 weeks before mating, during pregnancy and lactation (Collison et al., 2011). Male offspring continued on these diets for 9 months. Food intake was recorded twice daily. Body weights were recorded approximately every second week. At 3 and 9 months of age, total body scans were performed to determine body composition, percentage of total body fat, and bone mineral content. Blood chemistry including various hormones (insulin, leptin, adiponectin, cortisol, IGF‐1) was measured at 9 months. A glucose tolerance test was also performed at 9 months. At 3 months, no significant differences were determined in adiposity and body weight. However, between 3 and 9 months of age the weight gain and body fat gain was significantly higher (1.4‐fold) in the MSG treated group than in controls. Fasting insulin levels were significantly higher (2.4‐fold) than in controls and remained significantly elevated after 20 min of glucose challenge. No significant difference existed for fasting glucose levels but the clearance rate of glucose was significantly increased (1.6‐fold). Serum lipids did not differ significantly from control.
Overall, the weight gain and body fat gain in the male offspring was significantly higher between 3 and 9 months of age compared to the control group. Fasting insulin levels were significantly higher and remained significantly elevated after 20 min of glucose challenge. No significant difference was observed for fasting glucose levels but the clearance rate of glucose was significantly increased.
Humans
In a longitudinal study, the association between consumption of MSG and incidence of overweight was examined in the China Health and Nutrition Survey (CHNS) nationwide prospective open‐cohort including 10,095 apparently healthy adults aged 18–65 at entry between 1991 and 2006 (He et al., 2011). Surveys of the cohort were conducted in 1991, 1993, 1997, 2000, 2004 and 2006. Household food consumption was determined with a weighed food inventory for 3 consecutive days. Dietary intake at the individual level was assessed by using 24‐h recalls for the same 3 days. Individual reported consumption was adjusted by the amount weighed at the household level if large differences existed between the household and individual level. MSG consumption at the household level was measured by direct weighing of the MSG container and of soy sauce before and after the 3‐day periods. The accuracy of MSG intake at the individual level was validated in six households using riboflavin as an adherence marker. Pregnant women and those with an implausible daily MSG intake (> 10 g/day) were excluded from the study. Overweight was defined as a body mass index (BMI) ≥ 25 kg/m2 in the main analysis and as a BMI ≥ 23 kg/m2 in the sensitivity analysis. The mean follow‐up period was 5.5 years. The mean cumulative MSG intake was 2.2 g/day. Participants with a high MSG intake had a higher BMI, higher income, higher intakes of total energy and sodium, and lower physical activity levels. BMI showed a significant, dose dependent association with MSG consumption, after adjustment for potential confounders (age, sex, urban residence, region, smoking status, alcohol consumption, income, education, physical activity, and intake of total energy, sodium, potassium and calcium) and control for clustering of data at multiple levels (individual, household, and community). The adjusted hazard ratio of overweight was 1.33 (95% CI: 1.01, 1.75) for participants in the highest quintile (4,190 mg/day as median MSG intake) of MSG intake compared with those in the lowest quintile (630 mg/day as median MSG intake) after adjustment for potential confounders. The study has several weaknesses which were addressed by Bursey et al. (2011). The Panel noted that among the weaknesses the reliability of the measurements of MSG intake at the individual level and the physical activity were the main critical elements which render the findings questionable.
In a subset of the national study in nutrition and health in China, starting in 2002, which represents the Jiangsu Nutrition cohort, data from 1,282 Chinese men and women from the province of Jiangsu (rural and urban samples) were analysed to determine whether a greater MSG intake was associated with a clinically significant weight gain over 5 years (Shi et al., 2010b). MSG intake and body weight were quantitatively assessed in 2002 and the body weight followed up in 2007. Participants were excluded from the study if they had extreme values of weight change (> 20 kg) over the 5 years, or had known diabetes, stroke or cancer in 2002 (i.e. at the beginning of this study). Dietary intake patterns were assessed by a series of detailed questions about the usual frequency and quantity of intake of thirty‐three food groups and beverages. Each household was specifically asked about its usual monthly consumption of MSG and other seasonings. Individual consumption of MSG was calculated from the household consumption and was adjusted for the proportion of the household energy intake by each individual. By adding the glutamate content of all foods or seasonings eaten by an individual per day, the average total glutamate intake was calculated. Individual intake of nutrients and vegetable oil was assessed using a 3‐day weighed food diary. The mean MSG intake was 3,800 mg/day. Out of the 1,227 participants 72 reported no use of MSG. The median intakes of MSG across quartiles were 800, 2,000, 3,700 and 6,900 mg/day, respectively. Over the 5‐year period (2002–2007), MSG intake was not associated with significant weight gain (≥ 5%) after adjusting for age, sex, multiple lifestyle factors and energy intake. An inverse association between MSG intake and 5% weight gain was noted when also adjusting for total glutamate intake. However, this association disappeared when the model was adjusted for either rice intake or food patterns.
The association of MSG intake and overweight was examined in a cross‐sectional study with 752 healthy Chinese adults, aged 40–59 years, randomly sampled from three rural villages in north and south China in 1997 (He et al., 2008). Dietary intake was assessed by four in‐depth multipass 24‐h recalls within on average 3 weeks. MSG users were asked to demonstrate the amount added during food preparation and the amounts shaken out were weighed by trained interviewers. MSG in soy sauce, commercially processed foods and restaurant foods was also accounted for. Overweight was defined as a BMI ≥ 25 kg/m2 or BMI ≥ 23 kg/m2. The great majority of participants did not use commercially processed foods and prepared their foods at home. 82.4% of the participants used MSG. The average intake was 0.33 g/day. In general, MSG users had higher BMI, and higher intake of animal protein, fat, cholesterol and calories, and lower intake of vegetable protein, total carbohydrate, starch, fibre, and magnesium than non‐users. MSG intake was positively related to BMI after adjustment for potential confounders (age, sex, smoking status, physical activity, and intake of total energy, sodium, animal protein, saturated fat, monounsaturated fat, carbohydrate and fibre). In MSG users, the prevalence of overweight was significantly and dose‐related higher than in non‐users. The multivariable‐adjusted odds‐ratios of overweight were 2.10 (BMI ≥ 23 kg/m2) and 2.75 (BMI ≥ 25 kg/m2) for users in the highest tertile of MSG intake (700 mg/day as median MSG intake) compared to non‐users.
A total of 57 women (ethnic origin not mentioned – study performed in Pennsylvania, US, 23 obese (BMI > 29.9 kg/m2) and 34 normal weight (BMI < 25 kg/m2)) aged 21–40 years took part in a study intended to examine whether obese women exhibit altered unami taste perception compared to normal‐weight women (Pepino et al., 2010). Exclusion criteria included taking medication (except birth control pills), diabetes, pregnancy, lactation, chronic rhinitis, and food allergies. The women were asked to abstain from smoking and eating 12 h before testing and were given a standard breakfast to standardise the level of hunger or satiation. The women were tested at two occasions, separated by at least 5 days in a randomised cross‐over fashion. Treatment consisted of MSG or sucralose to test for taste detection thresholds, for preferences, for perceived intensity of suprathreshold concentrations, and for discrimination between 29 mmol/L MSG and 29 mmol/L NaCl. To detect the taste of MSG, obese women required a significantly higher concentration compared with the normal weight women. Obese women also preferred a significantly higher concentration of MSG in a soup‐like vehicle. No differences existed between obese and normal weight women in their ability to perceive savouriness and saltiness of different MSG concentrations. Neither were there any differences in their ability to discriminate MSG from salt. Based on these results, the authors suggested that an increased MSG intake may be a consequence rather than a cause of obesity when positive associations are reported in epidemiological studies.
In a single blind randomised intervention study, nursing home elderly in the Netherlands were given 0 or 300 mg of MSG added daily to the animal protein part of the cooked meal for 16 weeks (Essed et al., 2007). The control group consisted of 23 elderly and the MSG group of 19 elderly. No significant differences were found in energy intake and body weight between the two groups.
Overall, in some studies in animals, specifically in mice, MSG, mainly in extremely high doses did increase body weight and fat and influenced indices of the metabolic status. Similar results were obtained from studies in rats.
In summary, in humans, positive associations between consumption of MSG and an increased weight have been reported in one longitudinal and in one cross‐sectional study from China. Another Chinese cross‐sectional study did not find an association between MSG intake and weight gain (≥ 5%). Similarly, no significant differences were found between MSG intake and body weight gain in a blind intervention study of nursing home elderly Dutch. Overall, the Panel considered that equivocal results were found in 3 epidemiological studies and no effect was seen in an interventional study, making it unlikely that the observed effects in animals were relevant for human hazard characterisation of glutamate.
MSG symptom complex and headache
FASEB (1995) evaluated the possible role of MSG in eliciting or mediating the Chinese restaurant syndrome (CRS) and concluded that ‘With regard to a role of MSG in producing adverse effects in humans, there is sufficient evidence to support the existence of a subgroup of the general population of otherwise healthy individuals who may respond to large doses (> 3 g) under specific conditions of use. In addition, there may be a small subgroup of previously diagnosed unstable asthmatics who also may respond to large doses of MSG under specific conditions of use. The mechanisms of these reactions are unknown at this time; however, no evidence exists to support the ability of orally ingested glutamate to produce neurotoxic or lesioning effects in humans’.
The FSANZ evaluated the evidence for a relationship between MSG exposure and (a) the Chinese restaurant syndrome and (b) the induction of an asthmatic reaction in susceptible individuals (FSANZ, 2003). The FSANZ concluded that ‘There is no convincing evidence that MSG is a significant factor in causing systemic reactions resulting in severe illness or mortality. The studies conducted to date on CRS have largely failed to demonstrate a causal association with MSG. Symptoms resembling those of CRS may be provoked in a clinical setting in small numbers of individuals by the administration of large doses of MSG without food. However, such effects are neither persistent nor serious and are likely to be attenuated when MSG is consumed with food. In terms of more serious adverse effects such as the triggering of bronchospasm in asthmatic individuals, the evidence does not indicate that MSG is a significant trigger factor’.
A brief overview of the studies reported in these two evaluations is presented. A possible association between intake of MSG and clinical symptoms in humans was first implicated in 1968 where a recurring syndrome occurring after eating at Chinese restaurants was referred to (Kwok, 1968). The syndrome named ‘Chinese restaurant syndrome’ consisted of numbness at the back of the head that radiated to the arms and the back, weakness and palpitations. Shortly after similar and additional symptoms (facial numbness, lacrimation, periorbital fasciculations, syncope, headache) were described by others (Beron, 1968; Davies, 1968; Gordon, 1968; Kandall, 1968; McCaghren, 1968; Menken, 1968; Migden, 1968; Rath, 1968; Schaumburg, 1968). In one case report, 10 out of 100 people who ingested a soup at a hotel fell sick within ten min and the soup was analysed to contain an unusual high concentration (31 g/L) of MSG (Rudin et al., 1989). FASEB (1995) suggested to call the range of symptoms for ‘MSG symptom complex’ and established a list of symptoms that constitutes the syndrome: burning sensation in the back of neck, forearms, and chest; facial pressure/tightness; chest pain; headache; nausea; palpitations; numbness in back of neck radiating to arms and back; tingling, warmth, weakness in face, temples, upper back, neck and arms; bronchospasm (observed in asthmatics only); drowsiness, weakness. Affected individuals often only reported one or a few of the symptoms at any one time.
Two case reports have suggested that the more serious effect cardiac arrhythmia may be a feature of the MSG symptom complex in certain susceptible individuals (Gann, 1977; Goldberg, 1982) but no confirmatory evidence linking the reactions to the MSG content of foods is available.
A few food symptom surveys in the 1970s have examined effects associated with intake of MSG (Go et al., 1973; Kerr et al., 1977, 1979; Reif‐Lehrer, 1977). As none of the studies reported quantitative data on the intake of MSG, no conclusions can be made from these studies on a possible association between MSG intake and clinical symptoms.
Numerous challenge studies have been conducted in an attempt to confirm or reject the hypothesis that MSG intake may cause clinical symptoms. Some studies have reported significant and/or dose‐related increases in clinical symptoms after ingestion of MSG (Schaumburg et al., 1969; Rosenblum et al., 1971; Kenney and Tidball, 1972; Kenney, 1974, 1979; Gore and Salmon, 1980; Yang et al., 1997), whereas others reported no or an equivocal association between MSG intake and increases in clinical symptoms (Morselli and Garattini, 1970; Zanda et al., 1973; Kenney, 1986; Wilkin, 1986; Tarasoff and Kelly, 1993; Altman et al., 1994; Geha et al., 2000; Prawirohardjono et al., 2000).
According to the FSANZ (2003) and FASEB (1995), many of the studies conducted before 1995 had numerous methodological flaws. Many of the studies were unblinded or single blinded and did not use appropriate placebos. Not all of the double‐blinded studies took the necessary steps to disguise the taste of MSG. Many of the studies included very few subjects and thus there was not any statistical analysis of the results. In some studies, subjects that had no previous history of reactions to MSG were included and in others the criteria for inclusion or exclusion of subjects were omitted. It was not always clear whether the subjects were fed or fasted before challenge with MSG. These points are especially important since no objective clinical measures have been associated with the subjective clinical symptoms. With the exception of one study (Kenney, 1974) in which 5,000 mg of MSG was given with a small meal, there is no evidence for symptoms in humans when oral MSG exposure is concurrent with the intake of a meal.
Since the FASEB review (FASEB, 1995), only three further studies have been conducted of which two (Yang et al., 1997; Geha et al., 2000) have been reviewed by FSANZ (2003). The two other newer studies were better conducted than the older studies. Both studies were double‐blinded, used a liquid rather than capsule vehicle, controlled for the taste of MSG, used subjects self‐identified as reactors to MSG, and used an appropriate placebo. In addition, the study by Geha et al. (2000) used three separate double‐blind challenges as recommended by FASEB (1995) without accompanying food in fasted subjects and a fourth challenge with food. The last two challenges were with capsules. Both the Geha et al. (2000) and Yang et al. (1997) studies showed that the frequency of reported symptoms increased with increasing dose with a threshold of 2,500 mg. Only 1.8% of the subjects that had self‐reported symptoms to MSG in the study by Geha et al. (2000) consistently had reactions to MSG through the first three challenges but their symptoms differed through the challenges. No reproducible responses to MSG was noted when MSG in capsules was administered in the course of a light meal. According to FSANZ (2003) and the review of Williams and Woessner (2009), the results of the newer studies appear to be consistent with the conclusions from the FASEB review (FASEB, 1995).
After the review of FSANZ (2003), Williams and Woessner (2009) reviewed the data until this time. The authors underscored the findings, already mentioned in the former reviews, that no symptoms have been observed with doses up to 3,000 mg per meal. Effects were noted in studies where more than 3,000 mg up to 5,000 mg were given, specifically under the condition when MSG was given on an empty stomach without food.
In 2016, Obayashi and Nagamura performed a systematic review on the relationship between MSG intake and headache which is one of the symptoms of the MSG symptom complex (Obayashi and Nagamura, 2016). The authors identified two experimental studies performed after the previous reviews (Baad‐Hansen et al. (2010) and Shimada et al. (2013)). In both placebo‐controlled blinded studies, high MSG doses were used (6,000 and 9,000 mg/day). The Panel noted that in the study by Shimada et al. (2013) from 14 healthy subjects (9 females, 5 males), eight reported headache within the 1 week of treatment with 9,000 mg MSG/day, whereas two had headache when on placebo. In the study of Baad‐Hansen et al. (2010) with 14 healthy men, two doses (6,000 and 9,000 mg/day) were tested. The Panel noted that whereas there was a statistically significant higher number of subjects reporting headache with the 6,000 mg/day dose, this was not found with the higher dose of 9,000 mg/day.
Overall, in some individuals there was evidence for a dose‐related response to an oral MSG challenge as a bolus dose of 3,000 mg or more. The mechanism behind the MSG symptom complex is not known.
Blood pressure
Humans
In a double‐blinded placebo‐controlled three‐armed crossover study, 14 healthy men with no history of adverse effects to MSG drank sugar‐free soda that contained either MSG (75 or 150 mg/kg bw) or NaCl as placebo (24 mg/kg bw) after an overnight fast (Baad‐Hansen et al., 2010). Plasma glutamate level, blood pressure, heart rate, different measures of pericranial muscle pain and reported adverse effects were assessed for 2 h. At 30 min after exposure, the plasma glutamate level increased by 395% and 556% in the low‐ and high‐dose MSG groups, respectively. Systolic blood pressure, subjectively reported pericranial muscle tenderness and stomach ache were significantly increased after the high dose of MSG. As a control experiment to rule out sodium loading as an explanation for any change in blood pressure, five healthy men drank sugar‐free soda that contained sodium in the form of sodium chloride at an equimolar dose to the high‐dose MSG and the blood pressure and heart rate were followed over 2 h. The blood pressure and heart rate was not significantly affected. The Panel noted that the reported increase in blood pressure was thus likely due to glutamate and not to the sodium intake.
Data from 1,227 Chinese men and women from the province of Jiangsu (rural and urban areas) were analysed to determine whether a greater MSG intake was associated with a significant change in blood pressure over 5 years (Shi et al., 2011). MSG intake and blood pressure were quantitatively assessed in 2002 and followed up in 2007. Participants were excluded from the study if they had extreme values of weight change (> 20 kg) over the 5 years, or had known diabetes, stroke or cancer in 2002. Dietary intake patterns were assessed by a series of detailed questions about the usual frequency and quantity of intake of thirty‐three food groups and beverages. Each household was specifically asked about its usual monthly consumption of MSG and other seasonings. Individual consumption of MSG was calculated from the household consumption and was adjusted for the proportion of the household energy intake by each individual. By adding the glutamate content of all foods or seasonings eaten by an individual per day, the average total glutamate intake was also calculated. Individual intakes of nutrients and vegetable oil were also assessed using a 3‐day weighed food diary. The mean MSG intake was 3,800 mg/day. 72 out of the 1,227 participants reported no use of MSG. The median intakes of MSG across quartiles were 800, 2,000, 3,700 and 6,900 mg/day, respectively. Longitudinally, MSG intake was associated with a significant increase in both systolic and diastolic blood pressure after adjusting for age, sex, multiple lifestyle factors, overweight, family history of hypertension, hypertension medication, and the intake of energy, sodium, fibre and potassium in the group of participants with a median intake of 3,700 mg/day (third quartile) and 6,900 mg/day (forth quartile). Among women and those taking hypertension medications the positive association between MSG intake and increased blood pressure (only diastolic for medication) was highly significant.
The association of intake of glutamic acid with blood pressure was examined in a cross‐sectional study with 4,680 healthy adults, aged 40–59 years, randomly sampled from 17 populations in China, Japan, the United Kingdom and the United States in 1996–1999 (Stamler et al., 2009). Dietary intake was assessed by four in‐depth multipass 24‐h recalls and by two 24‐h urine collections within on average 3 weeks. At the same visits, the blood pressure was measured. The use of MSG for participants from Japan and the United Kingdom was negligible. For participants from China and the United states, the use of MSG was quantitated. Glutamic acid was the predominant dietary amino acid averaging 15.7 g glutamic acid contained in protein per day. Only 2% of the participants reported use of dietary supplements containing glutamic acid averaging 500 mg/day. Blood pressure decreased with increasing glutamic acid consumption (as a percentage of total protein), an inverse relationship being also present after control for multiple non‐dietary and dietary confounders.
Overall, in a double‐blinded placebo controlled crossover study with 14 healthy men, the systolic blood pressure was significantly increased after single oral exposure to MSG at 150 mg/kg bw (corresponding to 10,500 mg with a body weight of 70 kg). In an epidemiological study with 1,227 Chinese people, MSG intake was associated with a significant increase in systolic and diastolic blood pressure over 5 years in the group with median intake of 3,700 mg/day and 6,900 mg/day after adjusting for various confounding factors. The Panel noted that in this study the method used to estimate the daily intake was prone to errors (no individual estimation of consumption). In a cross‐sectional study with 4,680 healthy adults from China, Japan, the United Kingdom and the United States, where only 2% of the participants reported use of dietary supplements containing glutamic acid averaging 500 mg/day, glutamic acid intake (averaging 15,700 mg glutamic acid contained in protein per day) was inversely related to blood pressure. Therefore, the Panel considered that there is some evidence that high intake of MSG (more than 3,000 mg/day) may increase both systolic and diastolic blood pressure.
3.5.9.1. Changes in hormone levels
Rats
Wistar rats (12–24 animals/sex per group) were dosed orally with 0 or 500 mg/kg bw per day of MSG in an aqueous solution from day 10 to 20 of life (Matsuzawa et al., 1979), and in another group (12–24 animals/sex per group), this was followed by a diet containing 5% of MSG (equivalent to 6,000 mg/kg bw per day) for 10 days after weaning. Females were checked for vaginal opening and vaginal smears were taken daily for longer periods within the first year of life to determine oestrous cycles. At 3 and 12 months, 12 females from each group were sacrificed and the weight of the anterior pituitary, ovaries, uterus and adrenals were determined. The levels of luteinising hormone (LH), follicle stimulating hormone (FSH) and oestradiol in serum, and of LH and FSH in anterior pituitaries were also measured. At 5 and 12 months, six males from each group were sacrificed and the weight of the anterior pituitary, testes, seminal vesicles and adrenals were determined. The levels of LH, FSH and testosterone in serum, and of LH and FSH in anterior pituitaries were also measured. Histological examination was performed on ovaries and testes. The Panel noted that no significant differences were reported between dosed and control offspring in any of the parameters examined.
Humans
In an open trial, 10,000 mg of glutamic acid dissolved in 700 mL of saline was given orally to 11 healthy volunteers of both sexes aged 23–32 years (Carlson et al., 1989). The study started two and a half hours after the breakfast. Blood samples were taken at 15 min intervals for 30 min before and 2.5 h after the administration of test substances (besides glutamate intake of capsules with taurine, cysteine, and aspartic acid was studied). Serum glutamate, LH, PRL, GH, cortisol, and TSH were measured in the blood samples. Serum glutamate levels increased markedly over baseline values after intake of glutamic acid. Glutamic acid significantly stimulated the secretion of PRL and cortisol to approximately twice baseline values. The other amino acids examined did not have any effects on the hormone levels.
In a placebo‐controlled crossover study, 12,700 mg of MSG was given orally in a cold flavoured drink (300 mL) to eight healthy men with a mean age of 25.6 years after an overnight fast (Fernstrom et al., 1996; Fernstrom, 2000). The placebo drink contained 3,000 mg of NaCl instead of MSG. The volunteers were also treated with a high protein meal and intravenous (i.v.) injection of thyrotropin‐releasing hormone (TRH) for comparison of prolactin (PRL) release. Blood samples were taken via an indwelling venous line at 20 minute intervals for the next 4 h. Plasma glutamate, glucose, LH, FSH, PRL, GH, testosterone, cortisol, thyroid‐stimulating hormone (TSH), total triiodothyronine (T3), total thyroxine (T4) and insulin were measured in the blood samples. The maximum level of plasma glutamate was reached after 60 min and was approximately 11 times the baseline value after drinking of the MSG‐containing solution. The plasma glutamate level was not significantly increased after the intake of the protein meal. Both the MSG treatment and the high protein meal significantly increased the level of insulin. The level of glucose was unaffected by MSG exposure. Prolactin was significantly increased after treatment with a high protein meal (twofold) and after injection of TRH (10‐fold), but not after MSG treatment. Plasma levels of LH, FSH, testosterone, GH, and cortisol were unaffected by any of the treatments. TSH and thyroid hormones were only affected by TRH treatment.
In a double‐blind placebo‐controlled crossover study, 10,000 mg of MSG were given orally in capsules to 18 healthy volunteers aged 19–28 years together with 75,000 mg of glucose after an overnight fast (Chevassus et al., 2002). Blood samples were taken at 15 min intervals for the next 2 h. Plasma glutamate, glucose, insulin and glucagon were measured in the blood samples. The maximum level of plasma glutamate was reached after 75 min and was highly variable between individuals. MSG significantly enhanced the glucose‐induced insulin secretion in a plasma glutamate concentration dependent manner. The level of glucose and glucagon in plasma was unaffected by MSG exposure.
Overall, in two studies in fasted humans, the level of insulin was increased following a single oral dose of 10,000 mg or more of MSG but the level of glucose was unaffected. Glutamic acid significantly stimulated the secretion of PRL and cortisol to approximately twice baseline values in an open trial with 10,000 mg glutamic acid. Both effects were not observed in another study with 12,700 mg of MSG. The Panel considered that high doses (10,000 mg and more) of MSG increased plasma insulin level whereas equivocal results were obtained concerning the influence of MSG on PRL and cortisol levels.
3.6. Discussion
MSG is one of the substances possessing the property to evoke the so‐called umami taste which can result in a plethora of physiological effects. Glutamate is also an important excitatory amino acid in the central nervous system. The function of umami receptors which are found throughout the body is not well understood. However, there are some indications that they could play a role in the regulation of the immune response.
Glutamate can be produced by different manufacturing processes. The reports of older studies did not characterise the glutamate used, whereas in the studies performed after 2007 the MSG used was well characterised with a high purity (> 98%). However, the studies were performed with a MSG produced by a single company which also sponsored the studies and differences in the production process were present. The Panel noted that the manufacturing process for the MSG used in the newer studies performed in 2006 and 2007 were different. In particular, the studies which were used in this assessment were partially performed with MSG produced by a manufacturing process different than the process evaluated by the Panel in 2015, but that no details on the new manufacturing process were submitted to EFSA. The Panel noted that the assessment of this different manufacturing process is out of the remit of the current re‐evaluation of glutamic acid–glutamates (E 620–625) as food additives. However, the Panel decided that given the high purity a read‐across could be performed and the study results obtained with the specific MSG could be used for MSG in general
Glutamate is absorbed in the intestine and it is presystemically metabolised in the gut wall. There was evidence that concomitant ingestion of nutrients along with MSG limits the plasma concentration rise in glutamate. Evidence was limited for increased brain glutamate following increased plasma glutamate by even high dose MSG ingestion (10 g) via the oral route.
Glutamic acid and its salts had a low acute toxicity.
Short‐term and subchronic studies showed no effects of MSG treatment up to doses of roughly 5,000 mg/kg bw per day (short‐term studies) and 5,250 mg/kg bw per day in one limit dose test and 2,700 mg/kg bw per day in a study performed following the OECD Guideline 408. In studies with protocols according to OECD, performed to fulfil regulatory requirements, increased kidney weight and increased spleen weight was found in rats in chronic studies and studies to investigate reproductive toxicity at doses of 939 mg/kg bw per day in males and 1,039 mg/kg bw per day in females and above. However, the increase in organ weight was not accompanied by histopathological changes. Hence, the Panel considered the extent of increased kidney and spleen weight as not adverse. In these studies, the NOAEL was the highest dose tested.
The available genotoxicity data indicated that MSG did not induce point mutation in bacteria, chromosomal aberrations, UDS and DNA single‐strand breaks in mammalian cells in vitro. Similarly, in vivo, MSG did not induce SCE, micronuclei and chromosomal aberrations both in somatic and germ cells. The Panel considered this data set sufficiently robust to evaluate its genotoxicity and cover by read‐across the limited or missing data for glutamic acid, monopotassium glutamate, calcium diglutamate, monoammonium glutamate and magnesium diglutamate. On this basis, the Panel considered that glutamic acid (E 620), monosodium glutamate (E 621), monopotassium glutamate (E 622), calcium diglutamate (E 623), monoammonium glutamate (E 624) and magnesium diglutamate (E 625) did not raise concern with regards to genotoxicity when used as food additives.
In three 2‐year studies in rats, no increased tumour rate was observed up to the highest doses tested (1,800 mg/kg bw per day, 133 mg/kg bw per day, 1,982 mg/kg bw per day in males and 2,320 mg/kg bw per day, 73 mg/kg bw per day and 2,311 mg/kg bw per day in females). The only observed effect was a significant increase in relative kidney weights in both sexes in the 5% group (12.7% increase in males and 11.9% increase in females) in the study by Shibata et al. (1995) which, however, was not considered as adverse effect. Thus, the NOAELs identified by the Panel from the three 2‐year studies were the highest doses tested. There was no indication of carcinogenicity.
No adverse effects were observed in the following reproductive and developmental toxicity studies: a three‐generation reproduction toxicity study in mice with a NOAEL of 4% MSG in the diet (equal to 6,000 and 7,200 mg/kg bw per day in males and females, the highest dose tested) (Anantharaman, 1979), a two‐generation reproduction toxicity study in rats with a NOAEL of 5% MSG in the diet (equal to 3,131 and 3,496 mg/kg bw per day for males and females, the highest dose tested) for parental, reproductive and developmental toxicity (Biosafety Research Center, 2008 [Documentation provided to EFSA n. 6]); and a dietary prenatal developmental toxicity study in rats with a NOAEL of 5% MSG (equal to 3,019 mg/kg bw per day), the highest dose tested, was identified for maternal and developmental toxicity (Biosafety Research Center, 2007c [Documentation provided to EFSA n. 5]).
Effects were seen in some neurodevelopmental studies which had substantial flaws. The Panel considered them as unsuitable to be used for hazard characterisation.
Only the neurodevelopmental toxicity study of Vorhees et al. (1979) had an adequate quality. In this study in the high‐dose group delayed early swimming development, diminished rearing frequency in the open field, altered active avoidance acquisition and extinction, and prolonged day‐2 passive avoidance retention were observed. The Panel considered the effects as adverse and determined the NOAEL from this study as 3.4% of MSG (equal to 3,700 mg/kg bw per day in males and 3,200 mg/kg bw per day in females) based on the absence of neurobehaviour effects which occurred at a higher dose.
Overall, histopathological lesions of the CNS (mainly the arcuate nucleus in the hypothalamus) were observed in rats, mice, guinea pigs, and monkeys following high doses of MSG or glutamic acid by subcutaneous injection or gavage. In general, neonatal animals were most sensitive to the neuronal injury, and mice were the most sensitive species. At a single dose of 250 mg/kg bw (peak plasma concentration of 480 μM), no lesions were observed and the minimum dose associated with lesions was 500 mg/kg bw (peak plasma glutamate concentration of 880 μM. Lesions of the CNS were not observed in studies with rats, mice, hamsters and monkeys where glutamate was fed in the diet or drinking water, except in two studies where weanling mice were deprived of water overnight and then offered water containing 5–10% of MSG or in a newer study where the relative volume of the arcuate nucleus was significantly decreased in the male offspring of rats fed diets containing about 20% of MSG.
The Panel noted that neonatal mice in which the neurotoxic effects have been observed have biological and physiological specificities which make them different from adult mice and other species. The capacity of glutamate metabolism in the gastrointestinal wall is lower than in the adult resulting in elevated blood concentrations even after low doses of glutamate (e.g. 250–500 mg/kg per day). Further, the BBB is less developed leading to elevated glutamate levels in the brain. Therefore, the findings in neonatal mice cannot be extrapolated to the human situation, with the possible exception of the neonatal period. In addition, the Panel noted that direct exposure to neonates (young infants) is not in the scope of the re‐evaluation of glutamates as food additives and, therefore, the histopathological lesions of the CNS and behavioural changes observed in neonatal animals are not relevant for the assessment of glutamic acid–glutamates (E 620–625) when used as food additives.
In 1969, obesity was reported in mice treated in infancy with either single or multiple subcutaneous injections of MSG (Olney, 1969). In further studies, obesity and stunted growth were observed after neonatal parenteral dosing with MSG, and in some of the studies lesions were observed in the arcuate nucleus in hypothalamus and a decreased level of some of the hormones in the hypothalamic–pituitary axis was found. In addition, the concentration of serum lipids, phospholipids, insulin and glucose (metabolic disease markers) was often increased. In humans, equivocal results were found in three epidemiological studies and no effect was seen in an interventional study, making it unlikely that the observed effects in animals are relevant for human hazard characterisation of glutamate.
In reviewing the existing studies, an expert panel of FASEB (1995) and the FSANZ (2003) concluded that some individuals may experience the MSG symptom complex to an oral MSG challenge of 3,000 mg or more when given without food. With the exception of one study in which 5,000 mg of MSG was given with a small meal, there was no evidence for symptoms in humans when oral MSG exposure is concurrent with the intake of a meal. A recent review on the relationship between MSG intake and headache, one of the symptoms of the MSG symptom complex, identified two studies not included in former reviews. In one of the blinded, placebo‐controlled studies in healthy subjects 9,000 mg MSG elicited headache in a higher number of subjects than placebo. The result was in agreement with the findings in the former reviews.
Case reports and human studies have addressed a possible association between intake of MSG and urticaria, rhinitis, blood pressure or hormonal changes. In newer case reports, positive separate double‐blind placebo‐controlled challenges in patients give some evidence to suggest that MSG may be a rare cause of urticaria and of rhinitis.
Glutamic acid significantly stimulated the secretion of the hormones prolactin and cortisol to approximately twice the baseline values in an open trial with 11 healthy volunteers given 10,000 mg of glutamic acid dissolved in saline. However, in a newer placebo‐controlled crossover study with eight healthy men, prolactin was significantly increased (twofold) after intake of a high protein meal but not after intake of 12,700 mg of MSG in a cold flavoured drink.
In a double‐blinded placebo‐controlled crossover study with 14 healthy men, the systolic blood pressure was significantly increased after single oral exposure to MSG at 150 mg/kg bw. In an epidemiological study with 1,282 Chinese participants, MSG intake was associated with a significant but clinically not relevant increase in systolic and diastolic blood pressure in participants of the third and fourth quartile of intake (mean MSG intake of 3,700 mg/day and 6,900 mg/day, respectively) over 5 years after adjusting for various confounding factors. In a cross‐sectional study with 4,680 healthy adults from China, Japan, the United Kingdom and the United States, with low glutamic acid intake in the whole study population no increase in blood pressure was noted.
The Panel assessed the suitability of human data to be used for the derivation of a HBGV. The MSG symptom complex was rarely seen when doses below 3,000 mg (corresponding to 42.9 mg/kg bw per day using a body weight of 70 kg) were ingested but a clear dose‐response relationship could not be established. Blood pressure increases were seen in the only dose of 150 mg/kg tested in an experimental study. From epidemiological studies, no dose response could be derived for this endpoint. Insulin increase was seen with the only dose tested of 10,000 mg MSG in healthy subjects. Hence, the human studies and information on effects were not suitable to derive an HBGV.
Considering the available animal studies, it can be stated that no adverse effects were observed in the repeated dose studies with oral dosing. Also, the appropriate reproductive and developmental studies showed no effect for the endpoints reproduction and development at the highest doses tested. The Panel considered the statistically significant increase in relative kidney weight observed in several studies being not an adverse effect because the effect size was small (below 20%) and not accompanied by histopathological findings. From other animal studies in which adverse effects were mentioned the majority had methodological flaws which renders the studies not suitable for deriving a reference point. Only in the neurodevelopmental toxicity study of Vorhees et al. (1979) a NOAEL could be identified based on the absence of neurobehaviour effects which were seen at a higher dose. In addition, the study had an adequate quality. Hence, the Panel decided to use the NOAEL of this study (3,200 mg MSG/kg bw per day) as reference point to derive an ADI.
Applying the default uncertainty factor of 100 to the NOAEL of 3,200 mg/kg bw per day, the Panel derived an ADI of 32 mg MSG/kg bw per day or 27.8 mg glutamate/kg bw per day. The value of 27.8 mg glutamate/kg bw per day was rounded to derive a group ADI of 30 mg/kg bw per day, expressed as glutamic acid.
The Panel noted that the ADI is below the doses which have been associated with the MSG symptom complex (> 42.9 mg/kg bw per day), headache (85.8 mg/kg bw per day), blood pressure increase (150 mg/kg bw per day (Baad‐Hansen et al., 2010) and also insulin increase (> 143 mg/kg bw per day) (Chevassus et al., 2002) in humans.
To assess the dietary exposure to glutamic acid–glutamates (E 620–625) from their use as food additives, the exposure was calculated based on different exposure scenarios as described in Section 3.4.1. As glutamic acid–glutamates (E 620 and E 625) are authorised to be used in a wide range of foods, the Panel did not identify brand loyalty to specific food categories and therefore selected the refined non‐brand loyal scenario (Section 3.4.1) as the most relevant exposure scenario for the safety evaluation of these food additives; it is assumed that the population would probably be exposed long‐term to the food additive present at the mean reported use in processed food.
The Panel noted that in the non‐brand‐loyal scenario, the mean exposure exceeded the dose which has been associated with the MSG symptom complex (> 42.9 mg/kg bw per day) in toddlers and children and the high exposure (95th percentile) in infants, toddlers, children and adolescents. The main food contributing to the mean exposure in the non‐brand‐loyal scenario were fine bakery wares for toddlers, children, adolescents, adults and for elderly, and soups and broths for infants. The main contributing food categories for which industry reported use data were fine bakery wares (FC 07.2) for toddlers (up to 75%), children (up to 80%), adolescents (up to 78%) and adults (up to 70%), and for infant ‘soups and broths’ (FC 12.5) (up to 90%). For the elderly, soups and broths and fine bakery wares were important sources of exposure. Other relevant food contributors for which use levels were available were sauces, meat and meat products and seasoning and condiments.
Considering additional exposure from food categories which may contain glutamic acid–glutamate due to natural presence and for which analytical data were available, the ADI was exceeded at the mean and high (95th percentile) level by all population groups, except for the mean exposure in elderly. In addition, the Panel further noted that in this exposure assessment scenario exposure in infants, toddlers and children at the high level (95th percentile) was higher than the doses which have been associated with adverse effects in humans such as the MSG symptom complex (> 42.9 mg/kg bw per day), headache (85.8 mg/kg bw per day), blood pressure increase (150 mg/kg bw per day) and also insulin increase (> 143 mg/kg bw per day).
Considering the food supplements consumers only scenario, the ADI was exceeded at the mean level in children, and at the high level (95th percentile) for all population groups.
Overall, the Panel considered that the uncertainties identified would, in general, result in an overestimation of the exposure to glutamic acid–glutamates (E 620–625) from their use as food additives according to Annex II in both the maximum level and refined exposure scenarios. For more details, see Section 3.4.4.
4. Conclusions
Considering the toxicological database and based on the NOAEL of 3,200 mg MSG/kg bw per day identified in a neurodevelopmental toxicity study and applying an uncertainty factor of 100, the Panel derived a group ADI of 30 mg/kg bw per day, expressed as glutamic acid, for glutamic acid and glutamates (E 620–625).
The Panel noted that the ADI is below the doses which have been associated with the MSG symptom complex (> 42.9 mg/kg bw per day), headache (85.8 mg/kg bw per day), blood pressure increase (150 mg/kg bw per day) and also insulin increase (> 143 mg/kg bw per day) in humans.
The Panel noted that considering only food categories authorised according Annex II, in the refined non‐brand‐loyal exposure assessment scenario, exposure estimates exceeded not only the proposed ADI but also doses which were associated with adverse effects in humans such as the MSG symptom complex for all population groups at the high level (95th percentile) but not at the mean. The high exposure in infants deserves special attention and further evaluation of the sources.
The Panel also noted that in the refined non‐brand‐loyal exposure assessment scenario, when considering all the available data for glutamic acid–glutamate, which include all their identified sources in the diet – food additive use, natural presence and addition as nutrient – exposure estimates largely exceeded the proposed ADI for all population groups at the high level (95th percentile) and at the mean (except for the elderly) as well as the doses, which in humans, are associated with adverse effects such as the MSG symptom complex.
5. Recommendations
The Panel recommended that:
the European Commission considers revising the maximum permitted levels, in particular, in food categories contributing the most to the overall exposure to glutamic acid and its salts:, fine bakery wares, soups and broths, sauces, meat and meat products, seasoning and condiments and food supplements.
the European Commission considers revising the current limits for toxic elements ‐arsenic and lead‐ in the EU specifications for glutamic acid (E 620) and lead in EU specifications for monosodium glutamate (E 621), monopotassium glutamate (E 622), calcium diglutamate (E 623), monoammonium glutamate (E 624) and magnesium diglutamate (E 625) in order to ensure that they will not be a significant source of exposure to those toxic elements in food.
Documentation provided to EFSA
Asociación Española de Exportadores e Industriales de Aceitunas de Mesa (ASEMESA), 2015. Data on usage levels of glutamic acid‐glutamates (E 620–625) in foods in response to the EFSA call for food additives usage level and/or concentration data in food and beverages intended for human consumption (Batch 4). Submitted to EFSA on 25 November 2015.
AVIKO, 2016. Data on usage levels of glutamic acid‐glutamates (E 620–625) in foods in response to the EFSA call for food additives usage level and/or concentration data in food and beverages intended for human consumption (Batch 4). Submitted to EFSA on 10 May 2016.
Biosafety Research Center, 2007a. Monosodium L‐glutamate monohydrate produced by a new method: 90‐day repeated dose toxicity study by dietary administration in rats. Experiment No. 9957 (258‐057). Submitted by Ajinomoto, 22 August 2016.
Biosafety Research Center, 2007b. Monosodium L‐glutamate monohydrate produced by a new method: 90‐day repeated oral dose toxicity study in dogs. Experiment No. 9959 (258‐059). Submitted by Ajinomoto, 22 August 2016.
Biosafety Research Center, 2007c. Monosodium L‐glutamate monohydrate produced by a new method: teratogenicity study in rats. Experiment No. 9958 (258‐058). Submitted by Ajinomoto, 22 August 2016.
Biosafety Research Center, 2008. Monosodium L‐glutamate monohydrate produced by a new method: reproduction study in rats. Experiment No. 9976 (258‐060). Submitted by Ajinomoto, 22 August 2016.
Center International de Toxicologie (CIT), 1997. 4‐week toxicity study by oral administration (dietary admixture) in rats. CIT/Study No 14458 TSR/MSG (MSG)/Société Orsan. Submitted by Ajinomoto, 22 August 2016.
Center International de Toxicologie (CIT), 1997. Complementary 4‐week toxicity study by oral administration (dietary admixture) in rats. CIT/Study No 14716 TSR/MSG (RC035/01)/Société Orsan. Submitted by Ajinomoto, 22 August 2016.
Degussa AG, 1883. Über die toxikologische Prüfung von L‐glutaminsaüre nach einmaliger oraler Gabe an der Ratte. Bericht‐Nr Ind‐Tox‐277‐82/83. Submitted by Ajinomoto, 22 August 2016.
ECU (European Committee for Umani), 2010. Response to the call for scientific data on miscellaneous food additives permitted in the EU and belonging to several functional classes, 2010. Submitted by ECU, 9 December 2010.
FDE (FoodDrinkEurope), 2016. Data on usage levels of glutamic acid‐glutamates (E 620–625) in foods in response to the EFSA call for food additives usage level and/or concentration data in food and beverages intended for human consumption (Batch 4). Submitted to EFSA on 31 May 2016.
Hatano Research Institute, 2006a. Chromosomal aberration test of monosodium L‐glutamate monohydrate produced by a new method using cultured Chinese hamster lung cells. Contract No. 06‐K‐078. Submitted by Ajinomoto, 22 August 2016.
Hatano Research Institute, 2006a. Micronucleus test of monosodium L‐glutamate monohydrate produced by a new method. Contract No. 06‐K‐077. Submitted by Ajinomoto, 22 August 2016.
Hatano Research Institute, 2007. Chromosomal aberration test of monosodium L‐glutamate monohydrate produced by a new method (Lot No. 20061222BLD3) using cultured Chinese hamster lung cells. Contract No. G‐06‐090. Submitted by Ajinomoto, 22 August 2016.
IGTC (International Glutamate Technical Committee), 2010. Response to the call for scientific data on miscellaneous food additives permitted in the EU and belonging to several functional classes, 2010. Literature search on glutamate. Submitted by IGTC, 25 November 2010.
Notox, 2010a. Toxicokinetic assessment of L‐glutamic acid. Notox Project 493748. Submitted by Ajinomoto, 22 August 2016.
Notox, 2010b. Evaluation of the mutagenicity activity of L‐glutamic acid in the salmonella typhimurium reserve mutation assay and the Escherichia coli reserved mutation assay (with independent repeat). Notox Project 493744. Submitted by Ajinomoto, 22 August 2016
Pre‐evaluation document prepared by DTU, February 2013.
Specialised Nutrition Europe (SNE), 2016. Data on usage levels of glutamic acid‐glutamates (E 620–625) in foods in response to the EFSA call for food additives usage level and/or concentration data in food and beverages intended for human consumption (Batch 4). Submitted to EFSA on 30 May 2016.
TNO (Netherlands Organisation for Applied Scientific Research), 2013a. Bacterial reverse mutation test with Monosodium L‐Glutamate, monohydrate. TNO project number 093.25061/01.41. Submitted by Ajinomoto, 4 November 2016.
TNO (Netherlands Organisation for Applied Scientific Research), 2013b. In vitro micronucleus test with Monosodium L‐Glutamate, monohydrate in cultured human lymphocytes. TNO project number 093.25061/01.47. Submitted by Ajinomoto, 4 November 2016.
TNO (Netherlands Organisation for Applied Scientific Research), 2014. Repeated‐dose (13‐week) oral toxicity study in rats with Monosodium L‐Glutamate monohydrate produced by a GMM production strain. TNO project number 093.25059. Submitted by Ajinomoto, 4 November 2016.
Abbreviations
- ADI
acceptable daily intake
- ANS
EFSA Scientific Panel on Food Additives and Nutrient Sources added to Food
- AOAC
Association of Official Analytical Chemists
- ASEMESA
Asociación Española de Exportadores e Industriales de Aceitunas de Mesa
- BBB
blood–brain barrier
- BMI
body mass index
- BUN
blood urea nitrogen
- bw
body weight
- CAS
Chemical Abstracts Service
- ChAT
choline acetyltransferase
- CHL
Chinese hamster lung
- CHNS
China Health and Nutrition Survey
- CIT
Center International de Toxicologie
- CNS
central nervous system
- CONTAM
EFSA Panel on Contaminants in Food Chain
- CRS
Chinese restaurant syndrome
- DMF
1,1′‐dimethylferrocene
- ECF
extracellular fluid
- ECHA
European Chemicals Agency
- ECG
electrocardiograph
- EEG
electroencephalogram
- EINECS
European Inventory of Existing Chemical Substances
- ERG
electroretinogram
- FAO
Food and Agriculture Organization of the United Nations
- FASEB
Federation of American Societies for Experimental Biology
- FCS
food categorisation system
- FDA
Food and drug administration
- FDE
FoodDrinkEurope
- FEV1
Forced expiratory volume in 1 second
- FID
flame ionisation detector
- FSANZ
Food Standard Australia New Zealand
- FSH
Follicle stimulating hormone
- GABA
γ‐aminobutyric acid
- GAD
glutamic acid decarboxylase
- GC
gas chromatography
- GD
gestation day
- GH
growth hormone
- GLP
Good Laboratory Practice
- GNPD
Global New Products Database
- GOT
glutamate Oxaloacetate Transferase
- GPT
glutamate pyruvate transaminase
- HBGV
health‐based guidance value
- HDL‐C
high‐density lipoprotein cholesterol
- HF
high fat
- HPLC
high‐performance liquid chromatography
- HPTLC
high‐performance thin layer chromatography
- HS
high sucrose
- IEC
ion‐exchange chromatography
- IgE
immunoglobulin E
- IGF‐1
insulin‐like growth factor‐1
- i.p.
intraperitoneal
- i.v.
intravenous
- JECFA
Joint FAO/WHO Expert Committee on Food Additives
- LD50
median lethal dose
- LDL‐C
low‐density lipoprotein cholesterol
- LF
low fat
- LH
luteinising hormone
- LOD
limit of detection
- log Po/w
octanol/water partition coefficient
- LOQ
limit of quantification
- MB
medium Bound
- mGluR
metabotropic glutamate receptors
- MNPCE
micronucleated polychromatic erythrocytes
- mRNA
messenger RNA
- MS
mass spectrometry
- MSG
monosodium glutamate
- NaCl
sodium chloride
- NAD
nicotinamide adenine dinucleotide
- NADP
nicotinamide adenine dinucleotide phosphate
- NCE
normochromatic erythrocytes
- NDA
EFSA Panel on Dietetic Products, Nutrition and Allergies
- NMDA
N‐methyl‐d‐aspartate
- NOAEL
no observed adverse effect level
- MPL
maximum permitted level
- ODC
ornithine decarboxylase
- OECD
Organisation for Economic Co‐operation and Development
- PEF
peak expiratory flow
- PCE
polychromatic erythrocytes
- PRL
prolactin
- QS
quantum satis
- RBC
red blood cell
- REACH
Registration, Evaluation, Authorisation and Restriction of Chemicals
- s.c.
subcutaneously
- SCE
sister chromatid exchanges
- SCF
Scientific Committee on Food
- SD rats
Sprague–Dawley rats
- SGOT
serum glutamate oxaloacetate transferase
- SGPT
serum glutamate pyruvate transaminase
- SNE
Specialised Nutrition Europe
- T1R1
taste receptor type 1 member 1
- T1R3
taste receptor type 1 member 3
- T3
triiodothyronine
- T4
total thyroxine
- TemaNord
is a publishing series for results of the often research‐based work that working groups or projects under Nordic Council of Ministers have put in motion
- TG
test guideline
- TLC
thin‐layer chromatography
- TNO
Netherlands Organisation for Applied Scientific Research
- TRH
thyrotropin‐releasing hormone
- TSH
thyroid stimulating hormone
- UDS
unscheduled DNA synthesis
- UHF
ultra‐high‐fat
- UV
ultraviolet
- WBC
white blood cell
- WHO
World Health Organization
Appendix A – Summary of the reported use levels in food (mg/kg or mg/L) of glutamic acid–glutamates (E 620–625) provided by industry
Appendix A can be found in the online version of this output (‘Supporting information’ section): https://doi.org/10.2903/j.efsa.2017.4910
Appendix B – Summary of analytical results (mg/kg or mg/L) of glutamic acid–glutamates (E 620–625) provided by Members States for food categories authorised according to Annex II
Appendix B can be found in the online version of this output (‘Supporting information’ section): https://doi.org/10.2903/j.efsa.2017.4910
Appendix C – Summary of all analytical results (mg/kg or mg/L) of glutamic acid–glutamates (E 620–625) provided by Members States
Appendix C can be found in the online version of this output (‘Supporting information’ section): https://doi.org/10.2903/j.efsa.2017.4910
Appendix D – Number and percentage of food products labelled with glutamic acid–glutamates (E 620–625) out of the total number of food products present in the Mintel GNPD per food sub‐category between 2012 and 2017
Appendix D can be found in the online version of this output (‘Supporting information’ section): https://doi.org/10.2903/j.efsa.2017.4910
Appendix E – Concentration levels of glutamic acid–glutamates (E 620–625) used in the exposure scenarios (mg/kg or mg/L as appropriate)
Appendix E can be found in the online version of this output (‘Supporting information’ section): https://doi.org/10.2903/j.efsa.2017.4910
Appendix F – Summary of total estimated exposure of glutamic acid–glutamates (E 620–625) per population group and survey: mean and 95th percentile (mg/kg bw per day)
Appendix F can be found in the online version of this output (‘Supporting information’ section): https://doi.org/10.2903/j.efsa.2017.4910
Appendix G – Main food categories contributing to exposure to glutamic acid–glutamates (E 620–625) (> 5% to the total mean exposure)
Appendix G can be found in the online version of this output (‘Supporting information’ section): https://doi.org/10.2903/j.efsa.2017.4910
Supporting information
Summary of the reported use levels in food (mg/kg or mg/L) of glutamic acid–glutamates (E 620–625) provided by industry
Summary of analytical results (mg/kg or mg/L) of glutamic acid–glutamates (E 620–625) provided by Members States for food categories authorised according to Annex II
Summary of all analytical results (mg/kg or mg/L) of glutamic acid–glutamates (E 620–625) provided by Members States
Number and percentage of food products labelled with glutamic acid–glutamates (E 620–625) out of the total number of food products present in the Mintel GNPD per food sub‐category between 2012 and 2017
Concentration levels of glutamic acid–glutamates (E 620–625) used in the exposure scenarios (mg/kg or mg/L as appropriate)
Summary of total estimated exposure of glutamic acid–glutamates (E 620–625) per population group and survey: mean and 95th percentile (mg/kg bw per day)
Main food categories contributing to exposure to glutamic acid–glutamates (E 620–625) (> 5% to the total mean exposure)
Suggested citation: EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food) , Mortensen A, Aguilar F, Crebelli R, Di Domenico A, Dusemund B, Frutos MJ, Galtier P, Gott D, Gundert‐Remy U, Leblanc J‐C, Lindtner O, Moldeus P, Mosesso P, Parent‐Massin D, Oskarsson A, Stankovic I, Waalkens‐Berendsen I, Woutersen RA, Wright M, Younes M, Boon P, Chrysafidis D, Gürtler R, Tobback P, Altieri A, Rincon AM and Lambré C, 2017. Scientific Opinion on the re‐evaluation of glutamic acid (E 620), sodium glutamate (E 621), potassium glutamate (E 622), calcium glutamate (E 623), ammonium glutamate (E 624) and magnesium glutamate (E 625) as food additives. EFSA Journal 2017;15(7):4910, 90 pp. 10.2903/j.efsa.2017.4910
Requestor: European Commission
Question numbers: EFSA‐Q‐2011‐00577; EFSA‐Q‐2011‐00578; EFSA‐Q‐2011‐00579; EFSA‐Q‐2011‐00580; EFSA‐Q‐2011‐00581; EFSA‐Q‐2011‐00582
Panel members: Fernando Aguilar, Riccardo Crebelli, Alessandro Di Domenico, Birgit Dusemund, Maria Jose Frutos, Pierre Galtier, David Gott, Ursula Gundert‐Remy, Claude Lambré, Jean‐Charles Leblanc, Oliver Lindtner, Peter Moldeus, Alicja Mortensen, Pasquale Mosesso, Dominique Parent‐Massin, Agneta Oskarsson, Ivan Stankovic, Ine Waalkens‐Berendsen, Rudolf Antonius Woutersen, Matthew Wright and Maged Younes.
Acknowledgements: The Panel wishes to thank Davide Arcella and Adamantia Papaioannou for the support provided to this scientific output. The ANS Panel wishes to acknowledge all European competent institutions, Member State bodies and other organisations that provided data for this scientific output.
Adopted: 21 June 2017
Notes
Regulation (EC) No 1333/2008 of the European Parliament and of the Council of 16 December 2008 on food additives. OJ L 354, 31.12.2008, p. 16–33.
Commission Regulation (EU) No 257/2010 of 25 March 2010 setting up a programme for the re‐evaluation of approved food additives in accordance with Regulation (EC) No 1333/2008 of the European Parliament and of the Council on food additives. OJ L 80, 26.3.2010, p. 19–27.
COM(2001) 542 final.
Food Additives in Europe 2000, Status of safety assessments of food additives presently permitted in the EU, Nordic Council of Ministers, TemaNord 2002, 560.
Commission Regulation (EU) No 231/2012 of 9 March 2012 laying down specifications for food additives listed in Annexes II and III to Regulation (EC) No 1333/2008 of the European Parliament and of the Council. OJ L 83, 22.3.2012, p. 1–295.
Regulation (EC) No 609/2013 of the European Parliament and of the Council of 12 June 2013 on food intended for infants and young children, food for special medical purposes, and total diet replacement for weight control and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulation (EC) No 41/2009 and (EC) No 953/2009. OJ L 181, 29.6.2013, p. 35–56.
Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH). OJ L 396, 30.12.2006, p. 1–520.
Available online: http://ec.europa.eu/consumers/cosmetics/cosing/index.cfm?fuseaction=search.simple
Call for scientific data on miscellaneous food additives permitted in the EU and belonging to several functional classes Published: 9 June 2010 and modified on 16 June 2010. Available online: https://www.efsa.europa.eu/en/dataclosed/call/ans100609
Call for food additives usages level and/or concentration data in food and beverages intended to human consumption. Deadline for submission: 31 May 2016. Available online: https://www.efsa.europa.eu/en/data/call/151012
Available online: http://www.efsa.europa.eu/en/datexfoodcdb/datexfooddb.htm
SciFinder® the choice for chemistry researchTM.
Call for food additives usage level and/or concentration data in food and beverages intended for human consumption. Published 27 March 2013. Available online: http://www.efsa.europa.eu/en/dataclosed/call/130327
Available online: http://www.frida.fooddata.dk/ShowList.php?compid=150&lang=en
Missing Bulgaria, Cyprus, Estonia, Latvia, Lithuania, Luxembourg, Malta and Slovenia.
http://www.gnpd.com/sinatra/home/ accessed on 3/3/2017.
Available online: http://www.efsa.europa.eu/en/press/news/150428.htm
Available online: http://www.efsa.europa.eu/en/datexfoodcdb/datexfooddb.htm
Directive 2002/46/EC of the European Parliament and of the Council of 10 June 2002 on the approximation of the laws of the Member States relating to food supplements. OJ L 183, 12.7.2002, p. 51–57.
Total number of food categories is 67 because FCS 12.1.2 is authorised at quantum satis as E 620–625 and at 10,000 mg/kg as Group I.
MSG monohydrate produced by a genetically modified microorganism (GMM) production strain already evaluated by EFSA (EFSA ANS Panel, 2015).
References
- Abraham R, Dougherty W, Golberg L and Coulston F, 1971. The response of the hypothalamus to high doses of MSG in mice and monkeys ‐ cytochemistry and ultrastructural study of lysosomal changes. Experimental and Molecular Pathology, 15, 43–60. [DOI] [PubMed] [Google Scholar]
- Abraham R, Swart J, Golberg L and Coulston F, 1975. Electron microscopic observations of hypothalami in neonatal rhesus monkeys (Macaca mulatta) after administration of monosodium‐L‐glutamate. Experimental and Molecular Pathology, 23, 203–213. [DOI] [PubMed] [Google Scholar]
- Afifi MM and Abbas AM, 2011. Monosodium glutamate versus diet induced obesity in pregnant rats and their offspring. Acta Physiologica Hungarica, 98, 177–188. [DOI] [PubMed] [Google Scholar]
- Afraa A, Mounir A and Zaid A, 2013. Colorimetric determination of monosodium glutamate in food samples using L‐glutamate oxidase. Clinical Journal of Applied and Environmental Biology 19, 1069–1072. [Google Scholar]
- Airoldi L, Bonfanti M, Ghezzi P, Salmona M and Garattini S, 1980. Effect of oral MSG on glutamic acid levels in the nucleus arcuatus of the hypothalamus and on serum osmolality of adult and infant mice. Toxicology Letters, 7, 107–111. [DOI] [PubMed] [Google Scholar]
- Albin RL and Greenamyre JT, 1992. Alternative excitotoxic hypotheses. Journal of Neurology, 42, 733–738. [DOI] [PubMed] [Google Scholar]
- Ali MM, Bawari M, Misra UK and Babu GN, 2000. Locomotor and learning deficits in adult rats exposed to monosodium‐L‐glutamate during early life. Neuroscience Letters, 284, 57–60. [DOI] [PubMed] [Google Scholar]
- Allen DH and Baker GJ, 1981. Chinese‐restaurant asthma. New England Journal of Medicine, 305, 1154–1155. [DOI] [PubMed] [Google Scholar]
- Allen DH, Delohery J and Baker G, 1987. Monosodium L‐glutamate‐induced asthma. The Journal of Allergy and Clinical Immunology, 80, 530–537. [DOI] [PubMed] [Google Scholar]
- Altman DR, Fitzgerald T and Chiaramonte LT, 1994. Double‐blind placebo‐controlled challenge (DBPCC) of persons reporting adverse reactions to MSG (MSG). Journal of Allergy and Clinical Immunology, 93, 303 (abstract 844). [Google Scholar]
- Amorim M, Pereira JO, Gomes D, Pereira CD, Pinheiro H and Pintado M, 2016. Nutritional ingredients from spent brewer's yeast obtained by hydrolysis and selective membrane filtration integrated in a pilot phase. Journal of Food Engineering, 185, 42–47. [Google Scholar]
- Anantharaman K, 1979. In utero and dietary administration of monosodium L‐glutamate to mice: reproductive performance and development in a multigeneration study. In: Filer LJ Jr, Garattini S, Kare MR, Reynolds WA and Wurtman RJ (eds.). Glutamic Acid: Advances in Biochemistry and Physiology. Raven Press, New York. pp. 231–253. [Google Scholar]
- AOAC (Association of Official Analytical Chemists), 2000. AOAC Official Methods 970.37. Monosodium glutamate in food. Potentiometric titration method. First Action 1970. Chapter 47 – Food Additives: Direct, pp. 50−51.
- Asero R, 2002a. Food additives intolerance: A possible cause of perennial rhinitis. The Journal of Allergy and Clinical Immunology, 110, 937–938. [DOI] [PubMed] [Google Scholar]
- Asero R, 2002b. Multiple intolerance to food additives. The Journal of Allergy and Clinical Immunology, 110, 531–532. [DOI] [PubMed] [Google Scholar]
- Asero R and Bottazzi G, 2007. Chronic rhinitis with nasal polyposis associated with sodium glutamate intolerance. International Archives of Allergy and Immunology, 144, 159–161. [DOI] [PubMed] [Google Scholar]
- Ault A, 2004. The MSG story: The commercial production of MSG and other amino acids. Journal of Chemical Education, 81, 347–355. [Google Scholar]
- Baad‐Hansen L, Cairns B, Ernberg M and Svensson P, 2010. Effect of systemic MSG (MSG) on headache and pericranial muscle sensitivity. Cephalalgia, 30, 68–76. [DOI] [PubMed] [Google Scholar]
- Balazs R, 1965. Control of glutamate oxidation in brain and liver mitochondrial systems. The Biochemical Journal, 95, 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu AK, Chattopadhyay P, Roychudhuri U and Chakraborty R, 2006. A biosensor based on co‐immobilized l‐glutamate oxidase and l‐glutamate dehydrogenase for analysis of monosodium glutamate in food. Biosensors and Bioelectronics, 21, 1968–1972. [DOI] [PubMed] [Google Scholar]
- Battaglia FC, 2000. Glutamine and glutamate exchange between the fetal liver and the placenta. Journal of Nutrition, 130, 974S–977S. [DOI] [PubMed] [Google Scholar]
- Bellisle F, 1999. Glutamate and the UMAMI taste: sensory, metabolic, nutritional and behavioural considerations. A review of the literature published in the last 10 years. Neuroscience and Biobehavioral Reviews, 23, 423–438. [DOI] [PubMed] [Google Scholar]
- Benjamin AM and Quastel JH, 1974. Fate of L‐glutamate in the brain. Journal of Neurochemistry, 23, 457–464. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Drejer J, Schousboe A and Diemer NH, 1984. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. Journal of Neurochemistry, 43, 1369–1374. [DOI] [PubMed] [Google Scholar]
- Beron E, 1968. Chinese‐restaurant syndrome. The New England Journal of Medicine, 278, 1123. [PubMed] [Google Scholar]
- Berry HK, Butcher RE, Elliot LA and Brunner RL, 1974. The effect of monosodium glutamate on the early biochemical and behavioural development of the rat. Developmental Psychobiology, 7, 165–173, as referred to by JECFA, 1988. [DOI] [PubMed] [Google Scholar]
- Boaz DP, Stegink LD, Reynolds WA, Filer LJ Jr, Pitkin RM and Brummel MC, 1974. Monosodium glutamate metabolism in the neonatal primate. Federation Proceedings, 33, 651, as referred to by JECFA, 1988. [Google Scholar]
- Bodor R, Žúborová M, Ölvecká E, Madajová V, Masár M, Kaniansky D and Stanislawski B, 2001. Isotachophoresis and isotachophorsis‐zone electrophoresis of food additives on a chip with column‐coupling separation channels. Journal of Separation Science, 24, 801–809. [Google Scholar]
- Bogdanov MB, Tjurmina OA and Wurtman RJ, 1996. Consumption of a high dietary dose of MSG fails to affect extracellular glutamate levels in the hypothalamic arcuate nucleus of adult rats. Brain Research, 736, 76–81. [DOI] [PubMed] [Google Scholar]
- Botey J, Cozzo M, Marin A and Eseverri JL, 1988. MSG and skin pathology in pediatric allergology. Allergologia et Immunopathologia (Madr), 16, 425–428. [PubMed] [Google Scholar]
- Bravenboer J, Putten AVD, Verwaal W and van Bevel‐Salm M, 1995. Validation of methods. Food Law Enforcement Practitioners. Rep. No. 95‐001, January 1995.
- Bunyan J, Murrell EA and Shah PP, 1976. The induction of obesity in rodents by means of MSG. British Journal of Nutrition, 35, 25–39. [DOI] [PubMed] [Google Scholar]
- Burde RM, Schainker B and Kayes J, 1971. Acute effect of oral and subcutaneous administration of MSG on the arcuate nucleus of the hypothalamus in mice and rats. Nature, 233, 58–60. [DOI] [PubMed] [Google Scholar]
- Burrin DG and Stoll B, 2009. Metabolic fate and function of dietary glutamate in the gut. American Journal of Clinical Nutrition, 90, 850S–856S. [DOI] [PubMed] [Google Scholar]
- Bursey RG, Watson L and Smriga M, 2011. A lack of epidemiologic evidence to link consumption of monosodium L‐glutamate and obesity in China, Comments. The American Journal of Clinical Nutrition, 94, 958–960. [DOI] [PubMed] [Google Scholar]
- Caccia S, Garattini S, Ghezzi P and Zanini MG, 1982. Plasma and brain levels of glutamate and pyroglutamate after oral MSG to rats. Toxicology Letters, 10, 169–175. [DOI] [PubMed] [Google Scholar]
- Carlson HE, Miglietta JT, Roginsky MS and Stegink LD, 1989. Stimulation of pituitary hormone secretion by neurotransmitter amino acids in humans. Metabolism, 38, 1179–1182. [DOI] [PubMed] [Google Scholar]
- Castillo J, Dávalos A, Naveiro J and Noya M, 1996. Neuroexcitatory amino acids and their relation to infarct size and neurological deficit in ischemic stroke. Stroke, 27, 1060–1065. [DOI] [PubMed] [Google Scholar]
- Chaparro‐Huerta V, Rivera‐Cervantes MC, Torres‐Mendoza BM and Beas‐Zarate C, 2002. Neuronal death and tumor necrosis factor alpha response to glutamate‐induced excitotoxicity in the cerebral cortex of neonatal rats. Neuroscience Letters, 333, 95–98. [DOI] [PubMed] [Google Scholar]
- Chapman J and Zhou M, 1999. Microplate‐based fluorometric methods for the enzymatic determination of L‐glutamate: application in measuring L‐glutamate in food samples. Analytica Chimica Acta, 402, 47–52. [Google Scholar]
- Chevassus H, Renard E, Bertrand G, Mourand I, Puech R, Molinier N, Bockaert J, Petit P and Bringer J, 2002. Effects of oral monosodium (L)‐glutamate on insulin secretion and glucose tolerance in healthy volunteers. British Journal of Clinical Pharmacology, 53, 641–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Maulucci‐Gedde M and Kriegstein AR, 1987. Glutamate neurotoxicity in cortical cell culture. Journal of Neuroscience, 7, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM, Garland EM, Cano M, St John MK, Khachab M, Wehner JM and Arnold LL, 1995. Effects of sodium ascorbate, sodium saccharin and ammonium chloride on the male rat urinary bladder. Carcinogenesis, 16, 2743–2750. [DOI] [PubMed] [Google Scholar]
- Collison KS, Makhoul NJ, Inglis A, Al‐Johi M, Zaidi MZ, Maqbool Z, Saleh SM, Bakheet R, Mondreal R, Al‐Rabiah R, Shoukri M, Milgram NW and Al‐Mohanna FA, 2010a. Dietary trans‐fat combined with MSG induces dyslipidemia and impairs spatial memory. Physiology and Behavior, 99, 334–342. [DOI] [PubMed] [Google Scholar]
- Collison KS, Maqbool ZM, Inglis AL, Makhoul NJ, Saleh SM, Bakheet RH, Al‐Johi MA, Al‐Rabiah RK, Zaidi MZ and Al‐Mohanna FA, 2010b. Effect of dietary MSG on HFCS‐induced hepatic steatosis: expression profiles in the liver and visceral fat. Obesity, 18, 1122–1134. [DOI] [PubMed] [Google Scholar]
- Collison KS, Zaidi MZ, Saleh SM, Inglis A, Mondreal R, Makhoul NJ, Bakheet R, Burrows J, Milgram NW and Al‐Mohanna FA, 2011. Effect of trans‐fat, fructose and MSG feeding on feline weight gain, adiposity, insulin sensitivity, adipokine and lipid profile. British Journal of Nutrition, 106, 218–226. [DOI] [PubMed] [Google Scholar]
- Conacher HBS, Iyengar JR, Milles WF and Botting HG, 1979. Gas‐liquid chromatographic determination of monosodium glutamate in soups and soup bases. Journal Association of Official Analytical Chemists, 62, 604–609. [PubMed] [Google Scholar]
- Coppola ED, Christie SN and Gordon JA, 1975. Fast, short‐column separation and fluorimetric determination of monosodium glutamate in foods. Journal of the AOAC, 58, 58–61. [PubMed] [Google Scholar]
- Daabees TT, Finkelstein MW, Stegink LD and Applebaum AE, 1985. Correlation of glutamate plus aspartate dose, plasma amino acid concentration and neuronal necrosis in infant mice. Food and Chemical Toxicology, 23, 887–893. [DOI] [PubMed] [Google Scholar]
- Davies NE, 1968. Chinese‐restaurant syndrome. The New England Journal of Medicine, 278, 1124. [DOI] [PubMed] [Google Scholar]
- De Castro A, Sáncheza AH, Beatoa VM, Casadob FJ and Montaño A, 2014. Stability of monosodium glutamate in green table olives and pickled cucumbers as a function of packing conditions and storage time. Food Additives and Contaminants: Part A, 31, 1158–1164. [DOI] [PubMed] [Google Scholar]
- De Flora S, Zanacchi P, Camoirano A, Bennicelli C and Badolati GS, 1984. Genotoxic activity and potency of 135 compounds in the Ames reversion test and in a bacterial DNA‐repair test. Mutation Research ‐ Genetic Toxicology, 133, 161–198. [DOI] [PubMed] [Google Scholar]
- De Groot AP, Feron VJ and Immel HR, 1988. Induction of hyperplasia in the bladder epithelium of rats by a dietary excess of acid or base: implications for toxicity/carcinogenicity testing. Food and Chemical Toxicology, 26, 425–434. [DOI] [PubMed] [Google Scholar]
- Deckmann K and Kummer W, 2016. Chemosensory epithelial cells in the urethra: sentinels of the urinary tract. Histochemistry and Cell Biology, 146, 673–683. [DOI] [PubMed] [Google Scholar]
- Dennis SC, Land JM and Clark JB, 1976. Glutamate metabolism and transport in rat brain mitochondria. Biochemical Journal, 156, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz YS, Faine LA, Galhardi CM, Rodrigues HG, Ebaid GX, Burneiko RC, Cicogna AC and Novelli ELB, 2005. MSG in standard and high‐fiber diets: metabolic syndrome and oxidative stress in rats. Nutrition, 21, 749–755. [DOI] [PubMed] [Google Scholar]
- Drauz K, Grayson I, Kleemann A, Krimmer H‐P, Leuchtenberger W and Weckbecker C, 2000. Amino Acids. In: Ullmann's Encyclopedia of Industrial Chemistry. Wiley‐VCH Verlag GmbH & Co. KGaA, p. 1–58, 10.1002/14356007.a02_057.pub2 [DOI] [Google Scholar]
- Ebert AG, 1979a. The dietary administration of L‐MSG, DL‐MSG and L‐glutamic acid to rats. Toxicology Letters, 3, 71–78. [Google Scholar]
- Ebert AG, 1979b. The dietary administration of MSG or glutamic acid to C‐57 black mice for two years. Toxicology Letters, 3, 65–70. [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2009. Scientific Opinion on arsenic in food. EFSA Journal 2009;7(10):1351, 199 pp. 10.2903/j.efsa.2009.1351 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2010. Scientific Opinion on lead in food. EFSA Journal 2010;8(4):1570, 151 pp. 10.2903/j.efsa.2010.1570 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2012. Scientific Opinion on lead dietary exposure in the European population. EFSA Journal 2012;10(7):2831, 59 pp. 10.2903/j.efsa.2012.2831 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2014. Scientific Opinion on dietary exposure to inorganic arsenic in the European population. EFSA Journal 2014;12(3):3597, 68 pp. 10.2903/j.efsa.2014.3597 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2007. Scientific opinion of the Scientific Committee related to uncertainties in dietary exposure assessment. EFSA Journal 2007;5(1):438, 54 pp. 10.2903/j.efsa.2007.438 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2010. Management of left‐censored data in dietary exposure assessment of chemical substances. EFSA Journal 2010;8(3):1557, 96 pp. 10.2903/j.efsa.2010.1557 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011a. Use of the EFSA Comprehensive European Food Consumption Database in Exposure Assessment. EFSA Journal 2011;9(3):2097, 34 pp. 10.2903/j.efsa.2011.2097 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011b. Evaluation of the FoodEx, the food classification system applied to the development of the EFSA Comprehensive European Food Consumption Database. EFSA Journal 2011;9(3):1970, 27 pp. 10.2903/j.efsa.2011.1970 [DOI] [Google Scholar]
- EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources Added to Food), 2015. Scientific opinion on the safety of the change in the production method of L‐glutamic acid (E 620), monosodium L‐glutamate (E 621), monopotassium L‐glutamate (E 622), calcium di‐L‐glutamate (E 623), monoammonium glutamate (E 624) and magnesium di‐L‐glutamate (E 625). EFSA Journal 2015;13(1):3981, 20 pp. 10.2903/j.efsa.2015.3981 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2009. Guidance of the Scientific Committee on Transparency in the Scientific Aspects of Risk Assessments carried out by EFSA. Part 2: general principles. EFSA Journal 2009;7(7):1051, 22 pp. 10.2903/j.efsa.2009.1051 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2012. Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data. EFSA Journal 2012;10(3):2579, 32 pp. 10.2903/j.efsa.2012.2579 [DOI] [Google Scholar]
- Erecińska M and Silver IA, 1990. Metabolism and role of glutamate in mammalian brain. Progress in Neurobiology, 35, 245–296. [DOI] [PubMed] [Google Scholar]
- Eriksson LS, Law DH, Hagenfeldt L and Wahren J, 1983. Nitrogen metabolism of the human brain. Journal of Neurochemistry, 41, 1324–1328. [DOI] [PubMed] [Google Scholar]
- Essed NH, van SWA, kok FJ and de GC, 2007. No effect of 16 weeks flavor enhancement on dietary intake and nutritional status of nursing home elderly. Appetite, 48, 29–36. [DOI] [PubMed] [Google Scholar]
- Fahmy MA and Donya SM, 2001. MSG genotoxicity in mice. Bulletin of the National Research Centre (Egypt), 26, 191–204. [Google Scholar]
- FASEB (Federation of American Societies for Experimental Biology), 1995. Analysis of adverse reactions to MSG (MSG). In: Raitan DJ, Talbot JM, Fisher KD (eds.). American Institute of Nutrition, Bethesda, MD. pp. 1–324. [Google Scholar]
- FDA (Food and Drug Administration), online. US. Department of Health and Human Services. Questions and answers on monosodium glutamate (MSG). Available online [Access on 21 June 2017]: https://www.fda.gov/food/ingredientspackaginglabeling/foodadditivesingredients/ucm328728.htm
- Fernandez‐Tresguerres HJA, 2005. Effect of MSG given orally on appetite control (a new theory for the obesity epidemic). Anales de la Real Academia Nacional de Medicina, 122, 341–355; discussion 355–360. [PubMed] [Google Scholar]
- Fernstrom JD, 2000. Pituitary hormone secretion in normal male humans: acute responses to a large, oral dose of MSG. Journal of Nutrition, 130, 1053S–1057S. [DOI] [PubMed] [Google Scholar]
- Fernstrom JD, Cameron JL, Fernstrom MH, McConaha C, Weltzin TE and Kaye WH, 1996. Short‐term neuroendocrine effects of a large oral dose of MSG in fasting male subjects. Journal of Clinical Endocrinology and Metabolism, 81, 184–191. [DOI] [PubMed] [Google Scholar]
- Foran L, Blackburs K and Klesza RJ, 2017. Auditory hindbrain atrophy and anomalous calcium binding protein expression after neonatal exposure to MSG. Neuroscience, 344, 406–417. [DOI] [PubMed] [Google Scholar]
- Frieder B and Grimm VE, 1984. Prenatal MSG (MSG) treatment given through the mother's diet causes behavioral deficits in rat offspring. International Journal of Neuroscience, 23, 117–126. [DOI] [PubMed] [Google Scholar]
- Frieder B and Grimm VE, 1987. Prenatal MSG causes long‐lasting cholinergic and adrenergic changes in various brain regions. Journal of Neurochemistry, 48, 1359–1365. [DOI] [PubMed] [Google Scholar]
- FSANZ (Food Standards Australia New Zealand), 2003. MSG. A Safety Assessment. Technical Report Series, 20, Available online: http://www.foodstandards.gov.au/_srcfiles/MSG%20Technical%20Report.pdf
- Fujita H, Aoki N and Sasaki M, 1994. Mutagenicity of food additives with Salmonella typhimurium TA97 and TA102. IX. Annual Report of Tokyo Metropolitan Research Laboratory of Public Health, 45, 191–199. [Google Scholar]
- Furihata C, Yamakoshi A, Takezawa R and Matsushima T, 1989. Various sodium salts, potassium salts, a calcium salt and an ammonium salt induced ornithine decarboxylase and stimulated DNA synthesis in rat stomach mucosa. Japanese Journal of Cancer Research, 80, 424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Gabriel AM, 2015. AMTaste receptors in the gastrointestinal system. Flavour, 4, 14–17. [Google Scholar]
- Gann D, 1977. Ventricular tachycardia in a patient with Chinese restaurant syndrome. Southern Medical Journal, 70, 879–881. [DOI] [PubMed] [Google Scholar]
- Gayte‐Sorbier A, Airaudo CB and Armand P, 1985. Stability of glutamic acid and MSG under model system conditions: influence of physical and technological factors. Journal of Food Science, 50, 350–352, 360. [Google Scholar]
- Geha RS, Beiser A, Ren C, Patterson R, Greenberger PA, Grammer LC, Ditto AM, Harris KE, Shaughnessy MA, Yarnold PR, Corren J and Saxon A, 2000. Multicenter, double‐blind, placebo‐controlled, multiple‐challenge evaluation of reported reactions to MSG. The Journal of Allergy and Clinical Immunology, 106, 973–980. [DOI] [PubMed] [Google Scholar]
- Geil RG, 1970. Prelim. Comm. Gerber Products Co. Submitted to WHO in 1973, as referred to by JECFA, 1988.
- Genton C, Frei PC and Pécoud A, 1985. Value of oral provocation tests to aspirin and food additives in the routine investigation of asthma and chronic urticaria. Journal of Allergy and Clinical Immunology, 76, 40–45. [DOI] [PubMed] [Google Scholar]
- Germano P, Cohen SG, Hahn B and Metcalfe DD, 1991. An evaluation of clinical reactions to MSG (MSG) in asthmatics using a blinded, placebo‐controlled challenge. Journal of Allergy and Clinical Immunology, 87, 177 (abstract 155). [Google Scholar]
- Go G, Nakamura FH, Rhoads GG and Dickinson LE, 1973. Long‐term health effects of dietary MSG. Hawaii Medical Journal, 32, 13–17. [PubMed] [Google Scholar]
- Golberg L, 1973. Unpublished report from the Institute of Experimental Pathology and Toxicology, Albany Medical College. Submitted to WHO in 1973, as referred to by JECFA, 1988.
- Goldberg LH, 1982. Supraventricular tachyarrhythmia in association with the Chinese restaurant syndrome. Annals of Emergency Medicine, 11, 333. [DOI] [PubMed] [Google Scholar]
- Gordon ME, 1968. Chinese‐restaurant syndrome. The New England Journal of Medicine, 278, 1123–1124. [PubMed] [Google Scholar]
- Gore ME and Salmon PR, 1980. Chinese restaurant syndrome: fact or fiction? Lancet, 1, 251–252. [DOI] [PubMed] [Google Scholar]
- Graham TE, Sgro V, Friars D and Gibala MJ, 2000. Glutamate ingestion: the plasma and muscle free amino acid pools of resting humans. American Journal of Physiology‐Endocrinology and Metabolism, 278, E83–E89. [DOI] [PubMed] [Google Scholar]
- Hawkins RA, 2009. The blood‐brain barrier and glutamate. American Journal of Clinical Nutrition, 90, 867S–874S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Sanada K, Sagawa N, Yamada N and Kido K, 1978. Umbilical vein‐artery differences of plasma amino acids in the last trimester of human pregnancy. Biology of the Neonate, 34, 11–18. [DOI] [PubMed] [Google Scholar]
- Haynes WM, 2012. CRC Handbook of Chemistry and Physics on DVD. CRC Press. Taylor & Francis Group, Boca Raton, 7‐1 and 5‐96 pp. [Google Scholar]
- Hays SP, Ordonez JM, Burrin DG and Sunehag AL, 2007. Dietary glutamate is almost entirely removed in its first pass through the splanchnic bed in premature infants. Pediatric Research, 62, 353–356. [DOI] [PubMed] [Google Scholar]
- He K, Zhao L, Daviglus ML, Dyer A, Horn LV, Garside D, Shu L, Guo D, Wu Y, Zhou B and Stamler J, 2008. Association of monosodium glutamate intake with overweight in Chinese adults: the INTERMAP study. Obesity, 16, 1875–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Du S, Xun P, Sharma S, Wang H, Zhai F and Popkin B, 2011. Consumption of MSG in relation to incidence of overweight in Chinese adults: China Health and Nutrition Survey (CHNS). American Journal of Clinical Nutrition, 93, 1328–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanussen M, Garcia AP, Sunder M, Voigt M, Salazar V and Tresguerres JAF, 2006. Obesity, voracity, and short stature: the impact of glutamate on the regulation of appetite. European Journal of Clinical Nutrition, 60, 25–31. [DOI] [PubMed] [Google Scholar]
- Hertz L, 1979. Functional interactions between neurons and astrocytes I. Turnover and metabolism of putative amino acid transmitters. Progress in Neurobiology, 13, 277–323. [DOI] [PubMed] [Google Scholar]
- Heywood R and James RW, 1979. An attempt to induce neurotoxicity in an infant rhesus monkey with MSG. Toxicology Letters, 4, 285–286. [Google Scholar]
- Heywood R and Worden AN, 1979. Glutamate toxicity in laboratory animals. In: Filer LJ Jr, Garattini S, Kare MR, Reynolds WA and Wurtman RJ (eds.). Glutamic Acid: Advances in Biochemistry and Physiology. Raven Press, New York. pp. 203–215. [Google Scholar]
- Heywood R, Palmer AK and Hague PH, 1971. Unpublished report submitted to Central Research Laboratories, Ajinomoto Co., Inc. Submitted to WHO in 1973, as referred to by JECFA, 1988.
- Heywood R, Palmer AK, Newman AJ, Barry DH and Edwards FP, 1972. Unpublished report submitted to Central Research Laboratories, Ajinomoto Co., Inc. Submitted to WHO in 1973, as referred to by JECFA, 1988.
- Heywood R, James RW and Worden AN, 1977. The ad libitum feeding of MSG to weanling mice. Toxicology Letters, 1, 151–155. [Google Scholar]
- Hodge L, Yan KY and Loblay RL, 1996. Assessment of food chemical intolerance in adult asthmatic subjects. Thorax, 51, 805–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosen H, 1988. Re: airway effects of MSG. Journal of Asthma, 25, 111–112. [PubMed] [Google Scholar]
- Huang PC, Lee NY, Wu TJ, Yu SL and Tung TC, 1976. Effect of MSG supplementation to low protein diets on rats. Nutrition Reports International, 13, 477–486. [Google Scholar]
- HRC (Huntington Research Centre), 1971. Unpublished report dated 18 October. Submitted to WHO in 1973, as referred to by JECFA, 1988.
- Industrial‐Bio‐test‐Laboratories , 1973. Mutagenic study with accent brand monosodium L‐glutamate in albino mice. Northbrook, IL, USA, 1‐12, as referred to by JECFA, 1988.
- Isa IM and Ghani SA, 2009. A non‐plasticized chitosan based solid state electrode for flow injection analysis of glutamate in food samples. Food Chemistry, 112, 756–759. [Google Scholar]
- Ishidate M Jr, Sofuni T, Yoshikawa K, Hayashi M, Nohmi T, Sawada M and Matsuoka A, 1984. Primary mutagenicity screening of food additives currently used in Japan. Food and Chemical Toxicology, 22, 623–636. [DOI] [PubMed] [Google Scholar]
- Iwata S, Ichimura M, Matsuzawa Y, Takasaki Y and Sasaoka M, 1979. Behavioural studies in rats treated with monosodium l‐glutamate during the early stages of life. Toxicology Letters, 4, 345–357. [Google Scholar]
- Janarthanan C and Mottola HA, 1998. Enzymatic determinations with rotating bioreactors: determination of glutamate in food products. Analytica Chimica Acta, 369, 147–155. [Google Scholar]
- Janeczko MJ, Stoll B, Chang X, Guan X and Burrin DG, 2007. Extensive gut metabolism limits the intestinal absorption of excessive supplemental dietary glutamate loads in infant pigs. Journal of Nutrition, 137, 2384–2390. [DOI] [PubMed] [Google Scholar]
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 1971. Evaluation of food additives: specifications for the identity and purity of food additives and their toxicological evaluation: some extraction solvents and certain other substances; and a review of the technological efficacy of some antimicrobial agents. Fourteenth Report of the Joint FAO/WHO Expert Committee on Food Additives. WHO Technical Report Series, 462, Available online: http://whqlibdoc.who.int/trs/WHO_TRS_462.pdf [PubMed]
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 1974. Toxicological evaluation of certain food additives with a review of general principles and of specifications. Seventeenth Report of the Joint FAO/WHO Expert Committee on Food Additives. WHO Technical Report Series, 539, and corrigendum, Available online: http://whqlibdoc.who.int/trs/WHO_TRS_539.pdf [PubMed]
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 1987. Evaluation of certain food additives and contaminants. Thirty‐first Report of the Joint FAO/WHO Expert Committee on Food Additives. WHO Technical Report Series, 759, and corrigendum, Available online: http://whqlibdoc.who.int/trs/WHO_TRS_759.pdf [PubMed]
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 1988. L‐Glutamic acid and its ammonium, calcium, monosodium and potassium salts. Toxicological evaluation of certain food additives. Prepared by the thirty‐first meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA) at the meeting in Geneva 16–25 February 1987. Food Additives Series, 22, Available online: http://www.inchem.org/documents/jecfa/jecmono/v22je12.htm
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2000. Guidelines for the preparation of toxicological working papers for the Joint FAO/WHO Expert Committee on Food Additives. Geneva, Switzerland.
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2005. Evaluation of certain food additives. Sixty‐third Report of the Joint FAO/WHO Expert Committee on Food Additives. WHO Technical Report Series, 928, Available online: http://whqlibdoc.who.int/trs/WHO_TRS_928.pdf
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2006a. Amino acids and related substances. Safety evaluation of certain food additives. Prepared by the sixty‐third meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA) at the meeting in Geneva 8–17 June 2004. Food Additives Series, 54, Available online: http://www.inchem.org/documents/jecfa/jecmono/v54je01.pdf
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2006b. Monograph 1. Combined compendium of food additive specifications. All specifications monographs from the 1st to the 65th meeting (1956–2005). Volume 4, Available online: http://www.fao.org/ag/agn/jecfa-additives/search.html
- Kandall SR, 1968. Chinese‐restaurant syndrome. The New England Journal of Medicine, 278, 1123. [PubMed] [Google Scholar]
- Kampha W, Meevootisom V and Wiyakrutta S, 2004. Spectrophotometric enzymatic cycling method using L‐glutamate dehydrogenase and D‐phenylglycine aminotransferase for determination of L‐glutamate in foods. Analytica Chimica Acta, 520, 133–139. [Google Scholar]
- Kenndler E and Huber JFK, 1980. Simultaneous identification and quantification of the food additives glutamate, guanosine 5′‐monophosphate and inosine 5′‐monophosphate by isotachophoresis. Z Lebensm Unters Forsch, 171, 292–296. [Google Scholar]
- Kenney RA, 1974. Effect of concurrent caloric intake on the response to oral monosodium l‐glutamate in susceptible subjects. Journal of Food Science, 39, 414–415. [Google Scholar]
- Kenney RA, 1979. Placebo controlled studies of human reaction to oral monosodium L‐glutamate. In: Filer LJ Jr, Garattini S, Kare MR, Reynolds WA and Wurtman RJ (eds.). Glutamic Acid: Advances in Biochemistry and Physiology. Raven Press, New York. pp. 363–373. [Google Scholar]
- Kenney RA, 1986. The Chinese restaurant syndrome: an anecdote revisited. Food and Chemical Toxicology, 24, 351–354. [DOI] [PubMed] [Google Scholar]
- Kenney RA and Tidball CS, 1972. Human susceptibility to oral monosodium L‐glutamate. American Journal of Clinical Nutrition, 25, 140–146. [DOI] [PubMed] [Google Scholar]
- Kerr GR, Wu‐Lee M, El‐Lozy M, McGandy R and Stare FJ, 1977. Objectivity of food‐symptomatology surveys. Questionnaire on the “Chinese restaurant syndrome”. Journal of the American Dietetic Association, 71, 263–268. [PubMed] [Google Scholar]
- Kerr GR, Wu‐Lee M, McGandy R and Stare FJ, 1979. Prevalence of the ‘Chinese restaurant syndrome’. Journal of the American Dietetic Association, 75, 29–33. [PubMed] [Google Scholar]
- Khera KS, Whitta LL and Nera EA, 1970. Unpublished results of Research Laboratories, Food and Drug Directorate, Ottawa, Canada. Submitted to WHO in 1970, as referred to by JECFA, 1988.
- Kim DH and Yang KH, 1987. Effects of MSG on unscheduled DNA synthesis and DNA single‐strand breaks in primary cultures of rat hepatocytes. Environmental Mutagens and Carcinogens, 7, 65–71. [Google Scholar]
- Kondoh T and Torii K, 2008. MSG intake suppresses weight gain, fat deposition, and plasma leptin levels in male Sprague‐Dawley rats. Physiology and Behavior, 95, 135–144. [DOI] [PubMed] [Google Scholar]
- Krebs HA and Bellamy D, 1960. The interconversion of glutamic acid and aspartic acid in respiring tissues. The Biochemical Journal, 75, 523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs HA and Veech RL, 1969. Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Advances in Enzyme Regulation, 7, 397–413. [DOI] [PubMed] [Google Scholar]
- Krishna Veni N, Karthika D, Surya Devi M, Rubini MF, Vishalini M and Pradeepa YJ, 2010. Analysis of monosodium L‐glutamate in food products by high‐performance thin layer chromatography. Journal of Young Pharmacists, 2(3), 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok RHM, 1968. Chinese‐restaurant syndrome. The New England Journal of Medicine, 278, 796. [DOI] [PubMed] [Google Scholar]
- Lau OW and Mok CS, 1995. Indirect conductometric detection of amino acids after liquid chromatographic separation Part II. determination of monosodium glutamate in foods. Analytica Chimica Acta, 302, 45–52. [Google Scholar]
- Lee N, Foustoukos DI, Sverjenskya DA, Hazen RM and Cody GD, 2014a. Hydrogen enhances the stability of glutamic acid in hydrothermal environments. Chemical Geology, 386, 184–189. [Google Scholar]
- Lee N, Foustoukos DI, Sverjenskya DA, Hazen RM and Cody GD, 2014b. The effects of temperature, pH and redox state on the stability of glutamic acid in hydrothermal fluids. Geochimica et Cosmochimica Acta, 135, 66–86. [Google Scholar]
- Lemkey‐Johnston N and Reynolds WA, 1972. Incidence and extent of brain lesions in mice following ingestion of monosodium glutamate (MSG). The Anatomical Record, 172, 354, as referred to by JECFA, 1988. [Google Scholar]
- Lemkey‐Johnston N and Reynolds WA, 1974. Nature and extent of brain lesions in mice related to ingestion of MSG. Light and electron microscope study. Journal of Neuropathology and Experimental Neurology, 33, 74–97. [DOI] [PubMed] [Google Scholar]
- Lemkey‐Johnston N, Butler V and Reynolds WA, 1975. Brain damage in neonatal mice following MSG administration: possible involvement of hypernatremia and hyperosmolality. Experimental Neurology, 48, 292–309. [DOI] [PubMed] [Google Scholar]
- León D, Albasanz JL, Castillo CA and Martín M, 2008. Effect of glutamate intake during gestation on adenosine A1 receptor/adenylyl cyclase pathway in both maternal and fetal rat brain. Journal of Neurochemistry, 104, 435–445. [DOI] [PubMed] [Google Scholar]
- Liang JC and Pacchierotti F, 1988. Cytogenetic investigation of chemically‐induced aneuploidy in mouse spermatocytes. Mutation Research, 201, 325–335. [DOI] [PubMed] [Google Scholar]
- Liming W, Jinhui Z, Xiaofeng X, Yi L and Jing Z, 2009. Fast determination of 26 amino acids and their content changes in royal jelly during storage using ultra‐performance liquid chromatography. Journal of Food Composition and Analysis, 22, 242–249. [Google Scholar]
- Litton‐Bionetics , 1975a. Mutagenic evaluation of compound FDA 73‐58, 000997‐42‐2, monopotassium glutamate. US Department of Commerce, National Technical Information Service PB‐254.511, as referred to by JECFA, 1988.
- Litton‐Bionetics , 1975b. Mutagenic evaluation of compound FDA 75‐11, 007558‐63‐6, monoammonium glutamate, FCC. US Department of Commerce, National Technical Information Service PB‐254.512, as referred to by JECFA, 1988.
- Litton‐Bionetics , 1977. Mutagenic evaluation of compound FDA 75‐65, L‐glutamic acid, FCC. US Department of Commerce, National Technical Information Service PB‐266.889, as referred to by JECFA, 1988.
- López‐Zapata A, León D, Castillo CA, Albasanz JL and Martín M, 2011. Maternal glutamate intake during gestation and lactation regulates adenosine A₁ and A(2A) receptors in rat brain from mothers and neonates. Neuroscience, 199, 133–142. [DOI] [PubMed] [Google Scholar]
- Lukondeh T, Ashbolt NJ and Rogers PL, 2003. Evaluation of Kluyveromyces marxianus as a source of yeast autolysates. Journal of Industrial Microbiology and Biotechnology, 30, 52–56. [DOI] [PubMed] [Google Scholar]
- Lutz TA and Woods SC, 2012. Overview of animal models of obesity. Current Protocols in Pharmacology CHAPTER: Unit5.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maga JA, 1983. Flavor potentiators. Critical Reviews in Food Science and Nutrition, 18, 231–312. [DOI] [PubMed] [Google Scholar]
- Malki A, Fiedler J, Fricke K, Ballweg I, Pfaffl MW and Krautwurst D, 2015. Class I odorant receptors, TAS1R and TAS2R taste receptors, are markers for subpopulations of circulating leukocytes. Journal of Leukocyte Biology, 97, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RL, Lloyd HG and Cowan AI, 1994. The early events of oxygen and glucose deprivation: setting the scene for neuronal death? Trends in Neurosciences, 17, 251–257. [DOI] [PubMed] [Google Scholar]
- Matsuzawa Y, Yonetani S, Takasaki Y, Iwata S and Sekine S, 1979. Studies on reproductive endocrine function in rats treated with monosodium l‐glutamate early in life. Toxicology Letters, 4, 359–371. [Google Scholar]
- McCaghren TJ, 1968. Chinese‐restaurant syndrome. The New England Journal of Medicine, 278, 1123. [PubMed] [Google Scholar]
- McCall A, Glaeser BS, Millington W and Wurtman RJ, 1979. MSG neurotoxicity, hyperosmolarity, and blood‐brain barrier dysfunction. Neurobehavioral Toxicology, 1, 279–283. [PubMed] [Google Scholar]
- McLaughlan JM, Noel FJ, Botting HG and Knipfel JE, 1970. Blood and brain levels of glutamic acid in young rats given MSG. Nutrition Reports International, 1, 131–138. [Google Scholar]
- Menken M, 1968. Chinese‐restaurant syndrome. The New England Journal of Medicine, 278, 1123. [PubMed] [Google Scholar]
- Migden W, 1968. Chinese‐restaurant syndrome. The New England Journal of Medicine, 278, 1123. [PubMed] [Google Scholar]
- Miller AL, Hawkins RA and Veech RL, 1975. Decreased rate of glucose utilization by rat brain in vivo after exposure to atmospheres containing high concentrations of CO2. Journal of Neurochemistry, 25, 553–558. [DOI] [PubMed] [Google Scholar]
- Mizutani F, Sato Y, Hirata Y and Yabuki S, 1998. High‐throughput flow‐injection analysis of glucose and glutamate in food and biological samples by using enzyme/polyion complex bilayer membrane‐based electrodes as the detectors. Biosensors and Bioelectronics, 13, 809–815. [DOI] [PubMed] [Google Scholar]
- Moneret‐Vautrin DA, 1987. MSG‐induced asthma: study of the potential risk of 30 asthmatics and review of the literature. Allergie et Immunologie, 19, 29–35. [PubMed] [Google Scholar]
- Monno A, Vezzani A, Bastone A, Salmona M and Garattini S, 1995. Extracellular glutamate levels in the hypothalamus and hippocampus of rats after acute or chronic oral intake of MSG. Neuroscience Letters, 193, 45–48. [DOI] [PubMed] [Google Scholar]
- Montaño A, Sanchez AH and De Castro A, 2000. Changes in the amino acid composition of green olive brine due to fermentation by pure culture of bacteria. Journal of Food Science, 65, 1022–1027. [Google Scholar]
- Montevecchi G, Masino F and Antonelli A, 2011. Pyroglutamic acid development during grape must cooking. European Food Research and Technology, 232, 375–379. [Google Scholar]
- Morselli PL and Garattini S, 1970. MSG and the Chinese restaurant syndrome. Nature, 227, 611–612. [DOI] [PubMed] [Google Scholar]
- Murphy‐Royal C, Dupuis J, Groc L and Oliet SHR, 2017. Astroglial glutamate transporters in the brain: regulating neurotransmitter homeostasis and synaptic transmission. Journal of Neuroscience Research. 10.1002/jnr.24029 [DOI] [PubMed] [Google Scholar]
- Mushahwar IK and Koeppe RE, 1971. The toxicity of monosodium glutamate in young rats. Biochimica et Biophysica Acta, 244, 318–321. [DOI] [PubMed] [Google Scholar]
- Muslim NZ, Ahmad M, Heng LY and Sad B, 2012. Optical biosensor test strip for the screening and direct determination of L‐glutamate in food samples. Sensors and Acutuators B, 161, 493–497. [Google Scholar]
- Neame KD and Wiseman G, 1958. The alanine and oxo acid concentrations in mesenteric blood during the absorption of L‐glutamic acid by the small intestine of the dog, cat and rabbit in vivo. Journal of Physiology, 140, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AJ, Heywood R, Palmer AK, Barry DH, Edwards FP and Worden AN, 1973. Administration of monosodium L‐glutamate to neonatal and pregnant rhesus monkeys. Toxicology, 1, 197–204. [DOI] [PubMed] [Google Scholar]
- Nguyen TT and Sporns P, 1985. Decomposition of the flavor enhancers, MSG, inosine‐5′‐monophosphate and guanosine‐5′‐monophosphate during canning. Journal of Food Science, 50, 812–814, 822. [Google Scholar]
- Nikolelis DP, 1987. Kinetic ‐ potentiometric determination of monosodium glutamate in soups and soup bases and of glutamic dehydrogenase. Analyst, 112, 763–765. [DOI] [PubMed] [Google Scholar]
- Nunes RS and Cavalheiro ETG, 2007. Thermal Behavior of glutamic acid and its sodium, lithium and ammonium salts. Journal of Thermal Analysis and Colorimetry, 87, 627–630. [Google Scholar]
- Obayashi Y and Nagamura Y, 2016. Does monosodium glutamate really cause headache? A systematic review of human studies. The Journal of Headache and Pain, 17, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara Y and Takasaki Y, 1979. Relationship between plasma glutamate levels and hypothalamic lesions in rodents. Toxicology Letters, 4, 499–505. [Google Scholar]
- O'Hara Y, Fukimoto T, Ichimura M and Kirimura J, 1970. Unpublished report from Central Research Laboratories Ajinomoto Co. Inc. Submitted to WHO in 1970, as referred to by JECFA, 1988.
- Olney JW and Sharpe LG, 1969. Brain lesions in an infant Rheus monkey treated with monosodium glutamate. Science, 166, 385–388. [DOI] [PubMed] [Google Scholar]
- Olney JW and Ho O‐L, 1970. Brain damage in infant mice following oral intake of glutamate, aspartate or cysteine. Nature, 227, 609–611. [DOI] [PubMed] [Google Scholar]
- Olney JW, Sharpe LG and Feigin RD, 1972. Glutamate‐induced brain damage in infant primates. Journal of Neuropathology and Experimental Neurology, 31, 464–488. [DOI] [PubMed] [Google Scholar]
- Olney JW, Labruyere J and de Gubareff T, 1980. Brain damage in mice from voluntary ingestion of glutamate and aspartate. Neurobehavioral Toxicology, 2, 125–129. [PubMed] [Google Scholar]
- Oser BL, Carson S, Vogin EE and Cox GE, 1971. Oral and subacutaneous administration of monosodium glutamate to infant rodents and dogs. Nature, 229, 411–413. [DOI] [PubMed] [Google Scholar]
- Oser BL, Bailey DE, Morgareidge K, Carson S and Vogin EE, 1973. Unpublished report submitted to International Glutamate Technical Committee. Submitted to WHO in 1973, as referred to by JECFA, 1988.
- Owen G, Cherry CP, Prentice DE and Worden AN, 1978a. The feeding of diets containing up to 4% MSG to rats for 2 years. Toxicology Letters, 1, 221–226. [Google Scholar]
- Owen G, Cherry CP, Prentice DE and Worden AN, 1978b. The feeding of diets containing up to 10% MSG to beagle dogs for 2 years. Toxicology Letters, 1, 217–219. [Google Scholar]
- Pardridge WM, 1979. Regulation of amino acid availability to brain: selective control mechanisms for glutamate. In: Filer LJ Jr, Garattini S, Kare MR, Reynolds WA and Wurtman RJ (eds.). Glutamic Acid: Advances in Biochemistry and Physiology. Raven Press, New York. pp. 125–137. [Google Scholar]
- Pepino MY, Finkbeiner S, Beauchamp GK and Mennella JA, 2010. Obese women have lower MSG taste sensitivity and prefer higher concentrations than do normal‐weight women. Obesity, 18, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkin RM, Reynolds WA, Stegink LD and Filer LJ Jr, 1979. Glutamate metabolism and placental transfer in pregnancy. In: Filer LJ Jr, Garattini S, Kare MR, Reynolds WA and Wurtman RJ (eds.). Glutamic Acid: Advances in Biochemistry and Physiology. Raven Press, New York. pp. 103–110. [Google Scholar]
- Prawirohardjono W, Dwiprahasto I, Astuti I, Hadiwandowo S, Kristin E, Muhammad M and Kelly MF, 2000. The administration to Indonesians of monosodium L‐glutamate in Indonesian foods: an assessment of adverse reactions in a randomized double‐blind, crossover, placebo‐controlled study. Journal of Nutrition, 130, 1074S–1076S. [DOI] [PubMed] [Google Scholar]
- Price MT, Olney JW, Lowry OH and Buchsbaum S, 1981. Uptake of exogenous glutamate and aspartate by circumventricular organs but not other regions of brain. Journal of Neurochemistry, 36, 1774–1780. [DOI] [PubMed] [Google Scholar]
- Prosky L and O'Dell RG, 1972. Biochemical changes of brain and liver in neonatal offspring of rats fed monosodium‐l‐glutamate. Experientia, 28, 260–263. [DOI] [PubMed] [Google Scholar]
- Ragginer C, Lechner A, Bernecker C, Horejsi R, Möller R, Wallner‐Blazek M, Weiss S, Fazekas F, Schmidt R, Truschnig‐Wilders M and Gruber HJ, 2012. Reduced urinary glutamate levels are associated with the frequency of migraine attacks in females. European Journal of Neurology, 19, 1146–1150. [DOI] [PubMed] [Google Scholar]
- Rath J, 1968. Chinese‐restaurant syndrome. The New England Journal of Medicine, 278, 1123. [PubMed] [Google Scholar]
- Raugel PJ, 1999. Rapid Food Analysis and Hygiene Monitoring. Kits, instruments and systems. Volume I. Springer‐Verlag V, Berlin, Heidelberg, 109–110. [Google Scholar]
- Reeds PJ, Burrin DG, Jahoor F, Wykes L, Henry J and Frazer EM, 1996. Enteral glutamate is almost completely metabolized in first pass by the gastrointestinal tract of infant pigs. American Journal of Physiology‐Endocrinology and Metabolism, 270, E413–E418. [DOI] [PubMed] [Google Scholar]
- Reif‐Lehrer L, 1977. A questionnaire study of the prevalence of Chinese restaurant syndrome. Federation Proceedings, 36, 1617–1623. [PubMed] [Google Scholar]
- Ren X, Ferreira JG, Yeckel CW, Kondoh T and de AIE, 2011. Effects of ad libitum Ingestion of MSG on Weight Gain in C57BL6/J Mice. Digestion, 83, 32–36. [DOI] [PubMed] [Google Scholar]
- Reynolds WA, Lemkey‐Johnston N, Filer LJ Jr and Pitkin RM, 1971. MSG: absence of hypothalamic lesions after ingestion by newborn primates. Science, 172, 1342–1344. [DOI] [PubMed] [Google Scholar]
- Reynolds WA, Butler V and Lemkey‐Johnston N, 1976. Hypothalamic morphology following ingestion of aspartame or MSG in the neonatal rodent and primate: a preliminary report. Journal of Toxicology and Environment Health, 2, 471–480. [DOI] [PubMed] [Google Scholar]
- Reynolds WA, Lemkey‐Johnston N and Stegink LD, 1979. Morphology of the fetal monkey hypothalamus after in utero exposure to monosodium L‐glutamate. In: Filer LJ (ed.). Glutamic Acid. Raven Press, New York, USA. pp. 217–229, as referred to by JECFA, 1988. [Google Scholar]
- Rosenblum I, Bradley JD and Coulston F, 1971. Single and double blind studies with oral MSG in man. Toxicology and Applied Pharmacology, 18, 367–373. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, 1996. Excitotoxicity hypothesis. Journal of Neurology, 47(4 Suppl. 2), S19–S25. [DOI] [PubMed] [Google Scholar]
- Rudin O, Stauffer E, Cramer Y and Kramer M, 1989. Glutamic acid group poisoning. So‐called Chinese restaurant syndrome. Beitrage zur Gerichtlichen Medizin, 47, 69–71. [PubMed] [Google Scholar]
- Sano C, 2009. History of glutamate production. American Journal of Clinical Nutrition, 90, 728S–732S. [DOI] [PubMed] [Google Scholar]
- SCF (Scientific Committee for Food), 1991. Reports of the Scientific Committee for Food, 25th Series. First series of food additives of various technological functions. Opinion expressed on 18 May 1990.
- SCF (Scientific Committee for Food), 2001. Guidance on submissions for food additive evaluations by the Scientific Committee on Food. SCF/CS/ADD/GEN/26 Final. 12 July 2001.
- Schaumburg H, 1968. Chinese‐restaurant syndrome. The New England Journal of Medicine, 278, 1122. [DOI] [PubMed] [Google Scholar]
- Schaumburg HH, Byck R, Gerstl R and Mashman JH, 1969. Monosodium L‐glutamate: its pharmacology and role in the Chinese restaurant syndrome. Science, 163, 826–828. [DOI] [PubMed] [Google Scholar]
- Schultz SG, Yu‐Tu L, Alvarez OO and Curran PF, 1970. Dicarboxylic amino acid influx across brush border of rabbit ileum: effects of amino acid charge on the sodium‐amino acid interaction. Journal of General Physiology, 56, 621–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Foster AC, French ED, Whetsell WO Jr and Köhler C, 1984. Excitotoxic models for neurodegenerative disorders. Life Sciences, 35, 19–32. [DOI] [PubMed] [Google Scholar]
- Schwartzstein RM, Kelleher M, Weinberger SE, Weiss JW and Drazen JM, 1987. Airway effects of MSG in subjects with chronic stable asthma. Journal of Asthma, 24, 167–172. [DOI] [PubMed] [Google Scholar]
- Schwerin P, Bessman SP and Waelsch H, 1950. The uptake of glutamic acid and glutamine by brain and other tissues of the rat and mouse. Journal of Biological Chemistry, 184, 37–44. [PubMed] [Google Scholar]
- Seidenberg JM and Becker RA, 1987. A summary of the results of 55 chemicals screened for developmental toxicity in mice. . Teratogenesis, Carcinogenesis, and Mutagenesis, 7, 17–28. [DOI] [PubMed] [Google Scholar]
- Semprini ME, 1971. Effects of MSG (MSG) administration on rats during the intrauterine life and the neonatal period. Quaderni Della Nutrizione, 31, 85–100. [Google Scholar]
- Semprini ME, Conti L, Ciofi‐Luzzatto A and Mariani A, 1974a. Effect of oral administration of MSG (MSG) on the hypothalamic arcuate region of rat and mouse: a histological assay. Biomedicine, 21, 398–403. [PubMed] [Google Scholar]
- Semprini ME, D'Amicis A and Mariani A, 1974b. Effect of MSG on fetus and newborn mouse. Nutrition and Metabolism, 16, 276–284. [DOI] [PubMed] [Google Scholar]
- Seo HJ, Sham H‐D, Jin HY, Lee WH, Hwang HS, Park S‐A, Kim YS, Choi SC, Lee S, Oh KJ, Kim BS, Park BR and Lee MY, 2010. Chronic administration of MSG under chronic variables stress impaired hypothalamic‐pituitary‐adrenal axis function in rats. The Korean Journal of Physiology and Pharmacology, 14, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SA, Yoon GH, Kim H and Kim MO, 2015. Vitamin C neuroprotection against dose‐dependent glutamate induced neurodegeneration in the postnatal brain. Neurochemical Research, 40, 875–884. [DOI] [PubMed] [Google Scholar]
- Shen J, Yu L, Ma R, Zhang Y, Zhang X, Fang J and Yu T, 2010. Potent protection of Danshensu (β‐3, 4‐dihydroxyphenyl‐lactic acid) against excitotoxic effects of maternal intragastric administration of MSG at a late stage of pregnancy on developing mouse fetal brain. Neural Regeneration Research, 5, 1559–1567. [Google Scholar]
- Shi Z, Luscombe‐Marsh ND, Wittert GA, Yuan B, Dai Y, Pan X and Taylor AW, 2010b. MSG is not associated with obesity or a greater prevalence of weight gain over 5 years: findings from the Jiangsu Nutrition Study of Chinese adults. British Journal of Nutrition, 104, 457–463. [DOI] [PubMed] [Google Scholar]
- Shi Z, Yuan B, Taylor AW, Dai Y, Pan X, Gill TK and Wittert GA, 2011. MSG is related to a higher increase in blood pressure over 5 years: findings from the Jiangsu Nutrition Study of Chinese adults. Journal of Hypertension, 29, 846–853. [DOI] [PubMed] [Google Scholar]
- Shibata MA, Tanaka H, Kawabe M, Sano M, Hagiwara A and Shirai T, 1995. Lack of carcinogenicity of monosodium L‐glutamat in Fischer 344 rats. Food and Chemical Toxicology, 33, 383–391. [DOI] [PubMed] [Google Scholar]
- Shimada A, Cairns BE, Vad N, Ulriksen K, Lynge Pedersen AM, Svensson P and Baad‐Hansen L, 2013. Headache and mechanical sensitization of human pericranial muscles after repeated intake of monosodium glutamate (MSG). The Journal of Headache and Pain, 14, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon RA, 2000. Additive‐induced urticaria: experience with MSG (MSG). Journal of Nutrition, 130, 1063S–1066S. [DOI] [PubMed] [Google Scholar]
- Smith QR, 2000. Transport of glutamate and other amino acids at the blood‐brain barrier. Journal of Nutrition, 130, 1016S–1022S. [DOI] [PubMed] [Google Scholar]
- Sporns P, 1982. Rapid high performance liquid chromatographic determination of monosodium glutamate in food. Association of Official Analytical Chemists, 65, 567–571. [PubMed] [Google Scholar]
- Squire ENJ, 1987. Angio‐oedema and MSG. The Lancet, 329, 988. [DOI] [PubMed] [Google Scholar]
- Stamler J, Brown IJ, Daviglus ML, Chan Q, Kesteloot H, Ueshima H, Zhao L, Elliott P and Grp IR, 2009. Glutamic acid, the main dietary amino acid, and blood pressure the INTERMAP study (international collaborative study of macronutrients, micronutrients and blood pressure). Circulation, 120, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stańska K and Krzeski A, 2016. The umami taste: from discovery to clinical use. The Polish Otolaryngology, 70, 10–15. [DOI] [PubMed] [Google Scholar]
- Stegink LD, Filer LJ Jr and Baker GL, 1972. MSG. Effect on plasma and breast milk amino acid levels in lactating women. Proceedings of the Society for Experimental Biology and Medicine, 140, 836–841. [DOI] [PubMed] [Google Scholar]
- Stegink LD, Brummel MC, Boaz DP and Filer LJ Jr, 1973a. MSG metabolism in the neonatal pig. Conversion of administered glutamate into other metabolites in vivo. Journal of Nutrition, 103, 1146–1154. [DOI] [PubMed] [Google Scholar]
- Stegink LD, Filer LJ Jr and Baker GL, 1973b. MSG metabolism in the neonatal pig. Effect of load on plasma, brain, muscle, and spinal fluid free amino acid levels. Journal of Nutrition, 103, 1138–1145. [DOI] [PubMed] [Google Scholar]
- Stegink LD, Shepherd JA, Brummel MC and Murray LM, 1974. Toxicity of protein hydrolysate solutions; correlation of glutamate dose and neuronal necrosis to plasma amino acid levels in young mice. Toxicology, 2, 285–299, as referred to by JECFA, 1988. [DOI] [PubMed] [Google Scholar]
- Stegink LD, Filer LJJ and Baker GL, 1983. Plasma amino acid concentrations in normal adults fed meals with added monosodium L‐glutamate and aspartame. Journal of Nutrition, 113, 1851–1860. [DOI] [PubMed] [Google Scholar]
- Stevenson DD, 2000. MSG and asthma. Journal of Nutrition, 130, 1067S–1073S. [DOI] [PubMed] [Google Scholar]
- Supramaniam G and Warner JO, 1986. Artificial food additive intolerance in patients with angio‐oedema and urticaria. Lancet, 328, 907–909. [DOI] [PubMed] [Google Scholar]
- Takasaki Y, 1978a. Studies on brain lesion by administration of monosodium L‐glutamate to mice. I. Brain lesions in infant mice caused by administration of monosodium L‐glutamae. Toxicology, 9, 293–305. [DOI] [PubMed] [Google Scholar]
- Takasaki Y, 1978b. Studies on brain lesions after administration of monosodium L‐glutamate to mice. II. Absence of brain damage following administration of monosodium L‐glutamate in the diet. Toxicology, 9, 307–318. [DOI] [PubMed] [Google Scholar]
- Takasaki Y, 1979. Protective effect of mono‐ and disaccharides on glutamate‐induced brain damage in mice. Toxicology Letters, 4, 205–210. [Google Scholar]
- Takasaki Y and Torii K, 1983. Effect of water restriction on the development of hypothalamic lesions in weanling rodents given MSG. II. Drinking behaviour and physiological parameters in rats (Rattus norvegicus) and golden hamsters (Mesocricetus auratus). Toxicology Letters, 16, 195–210. [DOI] [PubMed] [Google Scholar]
- Takasaki Y and Yugari Y, 1980. Protective effect of arginine, leucine and preinjection of insulin on glutamate neurotoxicity in mice. Toxicology Letters, 5, 39–44. [DOI] [PubMed] [Google Scholar]
- Takasaki Y, Sekine S, Matsuzawa Y, Iwata S and Sasaoka M, 1979. Effects of parenteral and oral administration of monosodium l‐glutamate (MSG) on somatic growth in rats. Toxicology Letters, 4, 327–343. [Google Scholar]
- Takasaki Y, Narui K and Shioya S, 1990. Toxicity of salts of L‐glutamate. Acute toxicity of four salts of L‐glutamate in mice and rats, and mutagenicity test. Iyakuhin Kenkyu, 21, 257–264. [Google Scholar]
- Tamano S, Tanaka H, Kawabe M, Asakawa E, Sano M, Shioya S, Shirai T and Fukushima S, 1993. No enhancing effects of calcium/magnesium salts of L‐glutamate and L‐ascorbate on tumor development in a rat medium‐term multiorgan carcinogenesis bioassay. Journal of Toxicology and Environment Health, 39, 43–58. [DOI] [PubMed] [Google Scholar]
- Tarasoff L and Kelly MF, 1993. Monosodium L‐glutamate: a double‐blind study and review. Food and Chemical Toxicology, 31, 1019–1035. [DOI] [PubMed] [Google Scholar]
- Torii K and Takasaki Y, 1983. Effect of water restriction on the development of hypothalamic lesions in weanling rodents given MSG. I. Drinking behavior and physiological parameters in mice (Mus musculus). Toxicology Letters, 16, 175–194. [DOI] [PubMed] [Google Scholar]
- Torii K and Takasaki Y, 1987. Mealing and related hormone release suppress hypothalamic lesions of neonatal mice by L‐glutamate. Brain Research Bulletin, 18, 547–554. [DOI] [PubMed] [Google Scholar]
- Udomsopagit S, Suphantharika M, Ènnecke WK, Bilitewski U and Bhumiratana A, 1998. Determination of L‐glutamate in various commercial soy sauce products using flow injection analysis with a modified electrode. World Journal of Microbiology and Biotechnology, 14, 543–549. [Google Scholar]
- Urena‐Guerrero ME, Lopez‐Pe rez SJ and Beas‐Zarate C, 2003. Neonatal MSG treatment modifies glutamic acid decarboxylase activity during rat brain postnatal development. Neurochemistry International, 42, 269–276. [DOI] [PubMed] [Google Scholar]
- Vahjen W, Bradley J, Bilitewski U and Schmid RD, 1991. Mediated enzyme electrode for the determination of L‐glutamate. Analytical Letters, 24, 1445–1452. [Google Scholar]
- Vorhees CV, Butcher RE, Brunner RL and Sobotka TJ, 1979. A developmental test battery for neurobehavioral toxicity in rats: a preliminary analysis using MSG calcium carrageenan, and hydroxyurea. Toxicology and Applied Pharmacology, 50, 267–282. [DOI] [PubMed] [Google Scholar]
- Walaas I and Fonnum F, 1978. The effect of parenteral glutamate treatment on the localization of neurotransmitters in the mediobasal hypothalamus. Brain Research, 153, 549–562. [DOI] [PubMed] [Google Scholar]
- Welcome MO, Mastorakis NE and Pereverzev VA, 2015. Sweet taste receptor signaling network: possible implication for cognitive functioning. Neurology Research International, 2015, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen C‐P, Hayes KC and Gershoff SN, 1973. Effects of dietary supplementation of MSG on infant monkeys, weanling rats, and suckling mice. The American Journal of Clinical Nutrition, 26, 803–813. [DOI] [PubMed] [Google Scholar]
- WHO (World Health Organization), 2009. Principles and methods for the risk assessment of chemicals in food, International Programme on Chemical Safety, Environmental Health Criteria 240. Chapter 2: risk assessment and its role in risk analysis.
- Wilkin JK, 1986. Does MSG cause flushing (or merely “glutamania”)? Journal of the American Academy of Dermatology, 15, 225–230. [DOI] [PubMed] [Google Scholar]
- Williams AN and Woessner KM, 2009. MSG ‘allergy’: menace or myth? Clinical and Experimental Allergy, 39, 640–646. [DOI] [PubMed] [Google Scholar]
- Windmueller HG, 1982. Glutamine utilization by the small intestine. Advances in Enzymology and Related Areas of Molecular Biology, 53, 201–237. [DOI] [PubMed] [Google Scholar]
- Windmueller HG and Spaeth AE, 1974. Uptake and metabolism of plasma glutamine by the small intestine. Journal of Biological Chemistry, 249, 5070–5079. [PubMed] [Google Scholar]
- Windmueller HG and Spaeth AE, 1975. Intestinal metabolism of glutamine and glutamate from the lumen as compared to glutamine from blood. Archives of Biochemistry and Biophysics, 171, 662–672. [DOI] [PubMed] [Google Scholar]
- Woessner KM, Simon RA and Stevenson DD, 1999. MSG sensitivity in asthma. The Journal of Allergy and Clinical Immunology, 104, 305–310. [DOI] [PubMed] [Google Scholar]
- Woods RK, Weiner JM, Thien F, Abramson M and Walters EH, 1998. The effects of MSG in adults with asthma who perceive themselves to be MSG‐intolerant. The Journal of Allergy and Clinical Immunology, 101, 762–771. [DOI] [PubMed] [Google Scholar]
- Workman AD, Palmer JN, Adappa ND and Cohen NA, 2015. The Role of Bitter and Sweet Taste Receptors in Upper Airway Immunity. Current Allergy and Asthma Reports, 15, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CM, Wang Z and Wu QH, 2000. Volatile compounds produced from monosodium glutamate in common food cooking. Journal of Agricultural and Food Chemistry, 48, 2438–2442. [DOI] [PubMed] [Google Scholar]
- Xing W and Na R, 1996. Amino acids excess increase SCEs in human lymphocytes. Mutation Research, 372, 75–78. [DOI] [PubMed] [Google Scholar]
- Xu L, Zhao Y, Zhan SQ, Tang XD, Guo Y, Wang HS and Yang C, 2007. Temporal and spatial expression of preprotachykinin A mRNA in the developing filial mice brain after maternal administration of MSG at a late stage of pregnancy. Neuroscience (San Diego, CA, U. S.), 145, 974–980. [DOI] [PubMed] [Google Scholar]
- Yang WH, Drouin MA, Herbert M, Mao Y and Karsh J, 1997. The MSG symptom complex: assessment in a double‐blind, placebo‐controlled, randomized study. The Journal of Allergy and Clinical Immunology, 99, 757–762. [DOI] [PubMed] [Google Scholar]
- Yılmaz D and Karakuş E, 2011. Construction of a potentiometric glutamate biosensor for determination of glutamate in some real samples. Artificial Cells, Blood Substitutes, and Biotechnology, 39, 385–391. [DOI] [PubMed] [Google Scholar]
- Yoneda J, Chin K, Torii K and Sakai R, 2011. Effects of oral MSG in mouse models of asthma. Food and Chemical Toxicology, 49, 299–304. [DOI] [PubMed] [Google Scholar]
- Yonetani S, 1967. Unpublished report from Central Research Laboratories Ajinomoto Co. Inc. Submitted to WHO in 1970, as referred to by JECFA, 1988.
- Yonetani S, Ishii H and Kirimura J, 1979. Effect of dietary administration of monosodium L‐glutamate on growth and reproductive functions in mice. Oyo Yakuri, 17, 143–152. [Google Scholar]
- Yoshida T, 1978. L‐MSG (MSG). Kirk‐Othmer Encyclopedia of Chemical Technology. 3rd Edition. Vol. 2. John Wiley and Sons, New York, pp. 410–421. As cited in FASEB 1995 by “as referred to by FASEB, 1995”. [Google Scholar]
- Yu T, Zhao Y, Shi W, Ma R and Yu L, 1997. Effects of maternal oral administration of MSG at a late stage of pregnancy on developing mouse fetal brain. Brain Research, 747, 195–206. [DOI] [PubMed] [Google Scholar]
- Yu L, Zhang Y, Ma R, Bao L, Fang J and Yu T, 2006. Potent protection of ferulic acid against excitotoxic effects of maternal intragastric administration of MSG at a late stage of pregnancy on developing mouse fetal brain. European Neuropsychopharmacology, 16, 170–177. [DOI] [PubMed] [Google Scholar]
- Zanda G, Francios P, Tognoni G, Rizzo M, Standen SM, Morselli PL and Garattin S, 1973. Double‐blind study on effects of MSG in man. Biomedicine, 19, 202–204. [PubMed] [Google Scholar]
- Zeiger E, Anderson B, Haworth S, Lawlor T and Mortelmans K, 1992. Salmonella mutagenicity tests: V. Results from the testing of 311 chemicals. Environmental and Molecular Mutagenesis, 19, 2–141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of the reported use levels in food (mg/kg or mg/L) of glutamic acid–glutamates (E 620–625) provided by industry
Summary of analytical results (mg/kg or mg/L) of glutamic acid–glutamates (E 620–625) provided by Members States for food categories authorised according to Annex II
Summary of all analytical results (mg/kg or mg/L) of glutamic acid–glutamates (E 620–625) provided by Members States
Number and percentage of food products labelled with glutamic acid–glutamates (E 620–625) out of the total number of food products present in the Mintel GNPD per food sub‐category between 2012 and 2017
Concentration levels of glutamic acid–glutamates (E 620–625) used in the exposure scenarios (mg/kg or mg/L as appropriate)
Summary of total estimated exposure of glutamic acid–glutamates (E 620–625) per population group and survey: mean and 95th percentile (mg/kg bw per day)
Main food categories contributing to exposure to glutamic acid–glutamates (E 620–625) (> 5% to the total mean exposure)