

Abstract
| Draft Endorsed by the FEEDAP Panel | 17 May 2017 |
| Submitted for public consultation | 24 May 2017 |
| End of public consultation | 21 July 2017 |
| Adoption by the FEEDAP Panel | 27 September 2017 |
| Entry into force | 1 May 2018 |
Abstract
This guidance document is intended to assist the applicant in the preparation and the presentation of an application, as foreseen in Article 7.6 of Regulation (EC) No 1831/2003, for the authorisation of additives for use in animal nutrition. It specifically covers the assessment of the safety for the consumer.
Keywords: guidance, consumer safety, feed additives
Short abstract
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2017.EN-1305/full
Background and Terms of Reference
Regulation (EC) No 1831/20031 establishes the rules governing the Community authorisation of additives for use in animal nutrition. Moreover, Regulation (EC) No 429/20082 provides detailed rules for the implementation of Regulation (EC) No 1831/2003 as regards the preparation and the presentation of applications and the assessment and the authorisation of feed additives.
The Panel on Additives and Products or Substances used in Animal Feed (FEEDAP Panel) has adopted a series of guidance documents which aim at complementing Regulation (EC) No 429/2008 to support applicants in the preparation and submission of technical dossiers for the authorisation of additives for use in animal nutrition according to Regulation (EC) No 1831/2003.
The European Food Safety Authority (EFSA) asked its FEEDAP Panel to:
identify from the current guidance documents, those that need to be updated, taking into consideration the most recent scientific developments and the experience gained in the assessment of feed additives;
update the guidance documents in need of revision accordingly; this activity can be conducted in different rounds of activities on the basis of the priorities identified and on the feasibility of the revision according the resources available;
taking into account the sensitivity and the relevance of some of the guidance documents under revision and the entity of the revision itself (e.g. substantial or not), consider initiatives like preparatory info‐sessions or public consultations of the draft guidance documents. The relevant comments received in either step will have to be considered and addressed if appropriate in the final version of the guidance documents.
The first of the terms of reference was addressed by a statement of the EFSA FEEDAP Panel (2016), in which it was identified the need to update most of the guidance documents that it produced and set priorities for this update.
This output addresses the second and third terms of reference with regard to the update of the guidance documents dealing with the assessment of the safety of feed additives for the consumer. This guidance document underwent a public consultation (EFSA, 2017).
Scope of the guidance
This guidance document is intended to assist the applicant in the preparation and the presentation of its application for authorisation of a feed additive, as foreseen in Article 7.6 of Regulation (EC) No 1831/2003. This document does not substitute for the obligation of an applicant to comply with the requirements of Regulation (EC) No 1831/2003 and its implementing rules.
This document provides guidance on how to assess the safety for the consumer. This guidance is divided into five sections. The introduction provides the principles of the assessment of consumer safety. A catalogue of studies that may be needed is provided in Section 2 while the requirements for the different additives are listed in Section 3. Section 4 describes how to derive a highest safe intake for the consumer based on the outcome of the above studies. The safety for the consumer is assessed by comparing the exposure of the consumer to residues in food products to this safe intake. Finally, Section 5 describes how to calculate maximum residue levels (MRLs), when needed.
1. Introduction
The studies described in this guidance should be based on the additive for which authorisation is sought. As a principle, studies necessary to assess consumer safety should be carried out with the active substance. However, when the active substance is present in a fermentation product, the fermentation product should be tested. The fermentation product tested must be identical to that to be used in the commercial product. Where an additive has a number of active components, ideally each should be separately assessed and then consideration given to potential interactions. For complex mixtures, especially when the components cannot be fully identified/separated, the whole mixture should be assessed.
When data are provided in publications, evidence must be provided that the additive used in these studies is identical to that under application or, if not, would still allow conclusions on the additive under application to be made. For additives produced by fermentation, this includes the production strain.
Applicants should justify the omission from the dossier of any data or any deviations from the requirements detailed in this guidance.
Data submitted should allow an assessment of the safety for consumers exposed to food derived from animals given feed or water containing the additive. This should consider:
the metabolic fate and residues of the additive in the target species and laboratory animals,
the potential toxicity of the additive,
consumer exposure resulting from the consumption of food derived from animals exposed to the highest proposed use level of the additive.
2. Studies for the assessment of consumer safety
2.1. Absorption, distribution, metabolism and excretion (ADME) and residue studies
The aim of these studies is:
to establish the metabolic fate of the active substance(s) in laboratory animals and the target species as a basis for its toxicological evaluation,
to identify and quantify residues (parent compound and metabolites) of toxicological relevance in edible tissues and products and select the marker residue when required,
to establish the kinetics of total residues and the marker residue in tissues/products.
2.1.1. ADME studies
The studies should follow the principles described in the Organisation for Economic Co‐operation and Development (OECD) guideline 417 for the testing of chemicals as follows: ‘Studies examining the toxicokinetics […] of a chemical substance are conducted to obtain adequate information on its absorption, distribution, biotransformation (i.e. metabolism) and excretion […]. Basic [ADME] parameters determined from these studies will also provide information on the potential for accumulation of the test substance in tissues and/or products and the potential for induction/inhibition of biotransformation as a result of exposure to the test substance […] Adequate toxicokinetic data will be helpful to support the further acceptability and applicability of quantitative structure‐activity relationships, read‐across or grouping approaches in the safety evaluation of substances. […]
A radiolabelled test substance using 14C should be used for all mass balance and metabolite identification aspects of the study; however, if it can be demonstrated that […] mass balance and metabolite identification can be adequately evaluated using the unlabelled test substance, [and] the analytical specificity and sensitivity of the method used with non‐radioactive test substance is equal to or greater than that which could be obtained with the radiolabelled test substance,3 then, radiolabelled compound does not need to be used. Furthermore, other radioactive and stable isotopes may be used, particularly if the element is responsible for or is a part of the toxic portion of the compound. If possible, the radiolabel should be located in a core portion of the molecule which is metabolically stable (it is not exchangeable, is not removed metabolically as CO2, and does not become part of the one‐carbon pool of the organism). Labelling of multiple sites or specific regions of the molecule may be necessary to follow the metabolic fate of the compound'.
A justification of the choice and the molecular position of the label should be given, its specific (radio)activity, (radio)chemical purity and stability described.
2.1.1.1. ADME study in target animals
A study should be performed on a limited number of animals (e.g. at least three per sex for chickens for fattening, three for dairy cows) administered orally a single dose of the active substance. The dose should correspond to the highest proposed use level in feed. Mass balance, absorption, distribution (plasma/blood, tissues), excretion (urine, bile, faeces, milk or eggs, expired air (i.e. 14CO2), excretion via gills, where appropriate) and bioavailability if appropriate, should be measured.
Excreta (and plasma, if appropriate) should be collected for identification and quantification of unchanged test substance and metabolites. Efforts should be made to identify all metabolites present at 5% or greater of the administered dose or 10% of total radioactivity in the animal (whichever allows the greatest number of metabolites to be detected) and to provide a metabolic scheme for the test substance. Identification refers to the exact structural determination of components.
Metabolic pathways are assumed to be similar within a species. Therefore, not each category within a species needs to be examined. For example:
if data for chickens for fattening are provided, no data are necessary for chickens reared for laying, and in case of laying hens, only additional data for egg are required.
if data for calves for rearing are provided, no additional data for cattle for fattening is required (and vice versa), and in case of dairy cows, only additional data for milk are required.
if data for pigs for fattening are provided, no studies for piglets or sows are required (and vice versa).
Metabolic pathways are also assumed to be similar within physiologically similar species (Table 1). If metabolism data are available for the species/categories of column 1 in Table 1, then no specific studies need to be performed with the physiologically related species indicated in column 2. Otherwise, an indication of the metabolic pathway in the species under application is required. A comparison of metabolic profiles can be obtained through in vitro or in vivo studies.
Table 1.
Extrapolation of metabolism data from certain species/categories to physiologically related species
| From | To physiologically related species |
|---|---|
| Chickens for fattening | Other poultry for fattening (e.g. turkeys, ducks, geese, pheasants, quail, guinea fowl, ostrich) |
| Laying hens | Other birds kept for egg production (e.g. ducks, geese, pheasants, quail, ostrich) |
| Pigs | Other Suidae |
| Calves or cattle | Other growing ruminants (e.g. sheep, goat, buffalo) at the corresponding developmental stage |
| Dairy cows | Other dairy ruminants (e.g. goat, sheep, buffalo) |
| Salmon or trout | Other fish |
2.1.1.2. ADME study in laboratory animals
The purpose of these studies is to determine whether the metabolites to which the consumer will be exposed by consuming food products from animals fed with the additive are also produced by metabolism in the laboratory animals used in toxicological testing.
The proximity of metabolism in the target animals and laboratory animals may be demonstrated by comparison of metabolic profiles established by in vitro or in vivo studies.
If proximity cannot be established in vitro, then a metabolism study made with the laboratory animal is required. The laboratory animal species (and strain) used for the in vitro or in vivo comparative metabolism studies should be the same as used in toxicological studies to define the lowest no observed adverse effect level (NOAEL).
If metabolic proximity is not given, the applicant should address by other means the relevance to consumer safety of the specific metabolite(s) produced in the target species. If metabolic studies are available in humans, these data should be taken into consideration.
2.1.1.3. In vitro studies
Several questions concerning the metabolism of the substance may be addressed in in vitro studies using appropriate test systems. Fresh liver tissue slices, primary cell cultures from liver cells, freshly isolated hepatocytes and subcellular fractions (e.g. microsomes and cytosol or S9 fraction) from liver may be used to study possible metabolites.
2.1.2. Residue studies
The aim of residue studies is: (i) to determine total residues and (ii) to measure the marker residue of the active substance, in edible tissues and products at metabolic equilibrium and during withdrawal time. The marker residue is the residue selected for assay whose concentration is in a known relationship to total residues in tissues and products. Edible tissues are muscle/flesh, liver, kidney and fat (skin plus fat for poultry); products are milk, eggs and honey. The measurement of the active substance and its major metabolite(s) in excreta (urine, faeces, bile), edible tissues and products should be performed (see Section 2.1.1.1). Depending on structural alerts or toxicological considerations, the measurement of minor metabolites in tissues and products could be necessary. Consideration should also be given to the amount and nature of non‐extractable residues in edible tissues/products (covalently bound residues or incorporation into physiological body constituents such as fatty acids/lipids, carbohydrates or amino acids/proteins).
The marker residue should be selected and the ratios marker to total residues should be given for edible tissues and products, if appropriate. Where appropriate, metabolites of toxicological significance should also be measured.
The dose applied should correspond to the highest proposed use level and should preferably be administered via feed or water for drinking to animals until metabolic equilibrium (by default, until plasma steady state) is reached.
For those additives in which the consequences of the rate of depletion on residue concentration are needed (e.g. when MRLs are considered necessary), residues in tissues should be measured at additional sampling points after withdrawal (preferably three), spaced according to the rate of depletion from tissues. The same number of animals as listed in Sections 2.1.2.1 and 2.1.2.2 applies for each time point, respectively.
Two types of studies are in principle required:
2.1.2.1. Total residue study
A study of total residues should be made with the labelled active substance, administered until metabolic equilibrium in tissues is reached. The minimum number of healthy animals selected for tissue analysis is three for dairy cows, sows and laying hens, four for cattle, pigs, poultry and related minor species and rabbits, 10 for salmonids and other aquatic species. Equal sex distribution should be applied as appropriate. For poultry species, age/body weight of the birds used for this study should reflect approximately the middle of the production period. For residues in products, the following number of animals should be used: for milk, at least eight cows (24 h pooled milk); for eggs, the sufficient number of laying hens to collect 10 eggs; for honey, six bee hives. The parent compound and identified metabolites (see Section 2.1.1.1) should be determined in edible tissues and products. The marker residue should be selected from this study, and the ratios marker to total residues should be established.
2.1.2.2. Marker residue study
The minimum number of healthy animals selected for tissue analysis is four for dairy cows, cattle, pigs, sows and related minor species and rabbits, six for poultry, and 10 for salmonids and other aquatic species. Equal sex distribution should be applied as appropriate. For residues in products, the same number of animals as in Section 2.1.2.1 applies. The minimum administration period of the additive should be 28 days, for animals for fattening for the 28 days prior to slaughter. The samples should be collected at the end of the administration period. Measurements of the marker residue concentration (MRC) should use a validated analytical method with sufficient sensitivity.
2.1.3. Relevance of residues to safety assessment
As a first approach, total residues measured in edible tissues and products from target animals administered the (radio)labelled active substance are considered of toxicological relevance.
In a second step, toxicological relevant residues could be derived from total residues by discounting, on a quantitative basis:
-
1
– the labelled fraction incorporated into physiological endogenous compounds and
-
2
– metabolites with evidence of a reduced toxicological relevance with regard to the parent compound.
Identification of toxicologically relevant metabolites can make use of structural alerts.
2.2. Toxicological studies
The safety of the additive is assessed on the basis of the toxicological studies performed in vitro and in vivo on laboratory animals.
The basic set of toxicological studies includes studies on:
-
1
genotoxicity
-
2
subchronic oral toxicity
This may be augmented as necessary by studies on:
-
3
chronic oral toxicity
-
4
reproduction toxicity including prenatal developmental toxicity
-
5
carcinogenicity
Further studies providing additional information necessary for the assessment of the safety of the active substance and its residues should be conducted if there is any reason for concern. This could include in vitro and in silico studies (e.g. read‐across among substances with similar chemical structures) to improve understanding of toxicity mechanisms and to compare the toxicity of parent substance with its metabolites.
Toxicological studies on particular metabolites may be necessary if metabolic proximity cannot be demonstrated between the target species and the laboratory test species. If toxicological data are available from studies in humans, these should be taken into consideration.
2.2.1. Genotoxicity studies
To identify active substances and, if appropriate, their metabolites and degradation products with mutagenic and genotoxic properties, a selected combination of different genotoxicity tests should be carried out. The in vitro tests should be performed with and without mammalian metabolic activation system, and the compatibility of the test material with the test system should be taken into account.
The following two in vitro tests are recommended as the first step (EFSA Scientific Committee, 2011):
-
1
– a bacterial reverse mutation test (OECD guideline 471), and
-
2
– an in vitro mammalian cell micronucleus test (OECD guideline 487).
This combination of tests fulfils the basic requirements to cover the three genetic endpoints with the minimum number of tests; the bacterial reverse mutation assay covers gene mutations and the in vitro micronucleus test covers both structural and numerical chromosome aberrations.
Consideration should be given to whether specific features of the test substance might require substitution or integration of one or more of the recommended in vitro tests with other in vitro or in vivo tests in the basic battery. In the event of positive results from the in vitro battery, all the available relevant data on the test substance should be reviewed, and where necessary, an appropriate in vivo study (or studies) should be conducted to assess whether the genotoxic potential observed in vitro is expressed in vivo.
The following in vivo tests are recommended as follow‐up studies:
-
1
– a mammalian erythrocyte micronucleus test (OECD guideline 474),
-
2
– a transgenic rodent somatic and germ cell gene mutation assays (OECD guideline 488),
-
3
– an in vivo Comet assay (OECD guideline 489).
The in vivo micronucleus test covers the endpoints of structural and numerical chromosomal aberrations and is an appropriate follow‐up for in vitro clastogens and aneugens. There may be circumstances in which an in vivo mammalian bone marrow chromosome aberration test (OECD guideline 475) may be an alternative follow‐up test.
In the in vivo genotoxicity studies, in case of negative results, it is important that evidence of target cell exposure is obtained and it may be necessary to consider other relevant tissues (e.g. site of contact tissues for highly reactive substances which are not systemically available).
Transgenic rodent assays can detect point mutations and small deletions and are without tissue restrictions. The in vivo Comet assay is considered a useful indicator test in terms of its sensitivity to substances which cause gene mutations and/or structural chromosomal aberrations and can be applied to many target tissues. In order to reduce the number of animals used, a single rodent study combining the analysis of different endpoints (e.g. micronucleus and comet assay) may be considered.
In specific situations, the use of other studies (e.g. mechanistic studies) may be considered in order to define the in vivo genotoxic potential of a substance.
2.2.2. Subchronic oral toxicity studies
To investigate the subchronic toxic potential of the active substance, at least one study on a rodent species must be submitted with duration of at least 90 days. If the information from a rodent study is not a suitable basis for consumer risk assessment, a further study in a non‐rodent species may be required. The test item must be administered orally (preferably incorporated into the diet) with at least three levels in addition to a control group to obtain a dose response. The highest dose used should normally be associated with evidence of adverse effects. The lowest dose level should not produce any evidence of toxicity.
Protocols for these studies should comply with the OECD guidelines 408 (rodents) or 409 (non‐rodents).
2.2.3. Chronic oral toxicity studies
Based on the results of the basic set of studies, there could be an indication to perform a chronic oral study, which should be performed in at least one species and should be of at least 12 months' duration. The species chosen should be the most appropriate on the basis of all available scientific data, including the results of the 90‐day studies. The default species is the rat. If a second study is necessary, another rodent or a non‐rodent mammalian species should be used. The test item must be administered orally (preferably incorporated into the diet) with at least three dose levels in addition to a control group to obtain a dose response.
Protocols for these studies should comply with the OECD guideline 452.
2.2.4. Carcinogenicity studies
If there are indications, from previous tests and/or read‐across, of a potential tumorigenic effect of the test substance, either from genotoxic or non‐genotoxic mechanisms, a carcinogenicity study should be performed in order to derive a relevant point of departure. Investigations can be made by a combined chronic toxicity/carcinogenicity study according to OECD guideline 453 or by a carcinogenicity study according to OECD guideline 451. Other methods could be used if validated.
2.2.5. Reproduction toxicity studies
If there are indications, from previous toxicity studies and/or read‐across, of a potential effect of the test substance on the reproductive system, studies of reproductive function must be carried out. These studies comprise:
a reproductive toxicity study in one species; and
prenatal developmental toxicity studies in two species.
The test item must be administered orally (preferably incorporated into the diet) at least at three levels in addition to a control group to obtain a dose response.
2.2.5.1. Reproduction toxicity study
Protocols for the reproduction toxicity studies should comply with OECD guideline 416 (two‐generation reproduction toxicity) or 443 (extended one‐generation reproductive toxicity study).
2.2.5.2. Prenatal developmental toxicity studies
The objective is to detect any adverse effects on the pregnant female and the development of the embryo and foetus as a result of exposure through the gestation period. The preferred species are rats and rabbits. If a study on one of the species shows adverse effects on the progeny in the absence of maternal toxicity, there is no requirement to perform a study in a second species.
Protocols should be in line with OECD guideline 414.
2.2.6. Other studies
Further studies should be conducted if there are reasons for concern not covered by the studies listed above (e.g. if the pharmacodynamic properties of the active substance are such that there is a potential for effects on particular organs or functions of the organism). Such studies may include examination of pharmacological effects, effects in juvenile (prepubertal) animals, immunotoxicity, neurotoxicity (including developmental neurotoxicity) and/or endocrine‐mediated effects.
3. Required studies for different additives
For some additives (Section 3.1), safety for the consumer can be presumed without the need for specific information. For all other additives, the number and type of studies needed to establish the safety of the additive for the consumer will depend on the nature and use of the additive.
3.1. Additives for which no safety studies are required
No studies concerning the safety of use of the additive for the consumers (ADME, residue and toxicological studies) are required for:
additives for which no exposure of the target animals to the active substance (or related substances, including contaminants) will occur. Appropriate analytical data to determine that the active substance (or related substances) is not detectable in feed at the time of feeding should be provided;
additives for which evidence exists that the substance (and potential metabolites/degradation products) is not significantly absorbed in the target animal except additives with a measurable fraction of nanoparticles;
microorganisms considered by EFSA to qualify for the qualified presumption of safety (QPS) approach to safety assessment or when its biology is sufficiently well known to allow pathogenic/toxigenic strains to be excluded by direct testing4;
enzymes and amino acids produced by microorganisms considered by EFSA to qualify for the QPS approach to safety assessment;
enzymes and amino acids produced by genetically modified microorganisms for which the recipient strain is considered by EFSA to qualify for the QPS approach to safety assessment and for which the molecular characterisation of the event does not give rise to concern;
enzymes, not excluded above, for which there is evidence that low molecular weight material (below 10,000 Da) has been removed from the product;
silage additives where it can be demonstrated that the active substance(s) and agent(s) occur as normal constituents of silage and use of the additive does not substantially increase their concentration compared to silage prepared without use of the additive (i.e. where there is no substantial change in exposure);
food additives for which an acceptable daily intake (ADI) is not specified or which are authorised or approved as components of foodstuffs in the European Union (EU) without any restriction provided that the use as feed additive would not lead to a pattern of metabolites different from that of the species (laboratory animals, humans) used to assess the safety of the additive in food.
3.2. Additives for which a restricted set of safety studies is required
3.2.1. ADME studies
No ADME studies are required:
if the active component(s) of the additive consists only of microorganisms, enzymes; or
if the active substance(s) is naturally present in significant amounts in food or feedingstuffs or the substance as absorbed is a normal constituent of body fluids or tissues; or
for nutritional additives; or
in the species covered in column 2 of Table 1 when these data are available for the relevant species in column 1; or
in minor terrestrial species if data are available from three major terrestrial species (cattle, pigs and chickens).
ADME studies are required for all substances not exempted above.
3.2.2. Residue studies
No residue studies are required if:
the active component(s) of the additive consists only of microorganisms, enzymes, amino acids, ‘vitamins pro‐vitamins and chemically well‐defined substances, having similar effect’ which do not accumulate in tissues/products or ‘compounds of trace elements’ already authorised; or
-
the additive is intended to be used in:
-
1
– the species covered in column 2 of Table 1 if metabolic similarity with the relevant species in column 1 of Table 1 is given or demonstrated,
-
2
– horses, if the residue pattern and distribution in a major ruminant and pigs are similar,
-
3
– all other food‐producing animals (including rabbits but excluding fish and bees) if the residue patterns and distribution in cattle, pigs and chicken are similar,
-
1
Provided that the concentrations of the additive in feed among the species are similar.
For the following substances, the requirement for residue data is limited to marker residue (Section 2.1.2.2) concentrations comparing the tissue/products levels in an untreated group and in the group supplemented with the highest proposed concentration without a withdrawal time:
substances which are a natural constituent of body fluids or tissues or are naturally present in food or feedingstuffs if the use of the additive substantially increases the intake or tissue retention;
for colourants which add colour to food of animal origin;
‘vitamins, pro‐vitamins and chemically well‐defined substances, having similar effect’ that have a potential for accumulation in the tissues/products which are not already authorised;
‘compounds of trace elements’ not already authorised;
additives already authorised in food for which a health‐based guidance value is established.
A complete set of residue studies are required for all substances not exempted above.
3.2.3. Toxicological studies
Toxicological studies are not required for:
non‐xenobiotic chemicals highly purified (as a guide < 1% of unidentified material on a dry matter basis).
nutritional additives already authorised.
Toxicological studies are required for all additives not exempted above.
For microorganisms and fermentation products, a basic set of toxicity studies should be provided consisting of genotoxicity/mutagenicity tests and a subchronic (90‐day) oral toxicity study.
For xenobiotic substances (defined as chemicals which are not a natural component of the organism exposed to them), the basic set of toxicological studies augmented as appropriate by other studies (see Section 2.2).
In addition to the above, for substances for reduction of the contamination of feed by mycotoxins that modify the chemical structure of mycotoxins the following should be considered:
The mycotoxin metabolites/degradation products derived from the mycotoxin should be identified.
Any major metabolite(s)/degradation products(s) of the mycotoxin should be examined for genotoxicity and for toxicity after oral administration and then compared to that of the parent mycotoxin. The endpoints selected should include mycotoxin‐specific effects if appropriate.
3.3. Additives for which a complete set of safety studies are required
For all additives not exempted above, a complete set of studies listed under Section 2 should be provided.
For coccidiostats and histomonostats (and potentially other additives, see Section 5), the data set submitted should contain the elements necessary for establishing risk management tools as MRLs and withdrawal periods.
4. Assessment of consumer safety
4.1. Determination of a safe dose
The safe dose is derived from the dose response of toxicological or pharmacological effects and usually expressed as a NOAEL5 as mg per kg body weight (bw) per day. Where only a lowest observed adverse effect level (LOAEL) is available or the distance between the dose levels tested is large, a benchmark dose (BMD) (e.g. BMDL5 for continuous data, BMDL10 for quantal data) should be calculated instead.
In general, the BMD approach can be used to derive a value which can substitute for a NOAEL. It makes extended use of dose–response data and provides a quantification of the uncertainty and variability in the dose–response data. For details on how to apply the BMD approach, see the EFSA guidance on ‘Use of the benchmark dose approach in risk assessment’ (EFSA Scientific Committee,2017).
The overall NOAEL used for the risk characterisation of the substance is selected from the results of all the studies available. In principle, the lowest NOAEL should be taken. Any deviation from this principle should be fully justified. All findings from previous sections together with all other relevant published data (including any relevant information on the effects of the active substance on human) and, where appropriate, information on chemicals having a closely related chemical structure should be taken into consideration.
4.2. Highest safe intake
The highest safe intake is given by health‐based guidance value (e.g. ADI, tolerable upper intake level (UL)) based on the outcome of toxicological studies and applying an appropriate uncertainty factor (UF). The health‐based guidance value (mg of active substance per kg bw per day) is derived by dividing the selected reference point (e.g. NOAEL, BMDL, LOAEL) by an appropriate UF.
The UF used to determine the ADI of an active substance should take into consideration the nature of the biological effects and the quality of the data used to identify the NOAEL. The overall default UF of 100 (10x10 for inter‐ and intraspecies extrapolation) is normally used in calculating the ADI from laboratory animal data provided that the toxicological package consists at least of genotoxicity studies, a chronic study and reproduction studies. If the NOAEL of a subchronic study has to be used in place of that of a chronic study, an additional UF of 2 should be applied (EFSA Scientific Committee, 2012). Higher factors might be applied in order to account for additional sources of uncertainty in the data or when the NOAEL is set on the basis of a particularly critical endpoint, such as prenatal development toxicity or endocrine disruption. When data on the active substance are available for humans a lower UF (≤ 10) may be used.
The use of an ADI for assessing consumer safety of feed additives normally requires that the consumer of food derived from treated animals is exposed to a pattern of residues comparable to that formed in the laboratory animal used for deriving that ADI. If metabolic proximity between the laboratory animal used for deriving the ADI and the target species cannot be demonstrated, additional toxicological studies with the metabolites specific to the target species should be done.
For some additives (e.g. nutritional additives), it may be more appropriate to base the safety assessment on the UL. This is defined as the maximum level of total chronic daily intake of a nutrient (from all sources) judged (by national or international scientific bodies) to be unlikely to pose a risk of adverse health effects to consumers or specific groups of consumers.
In the case of an additive which is not genotoxic or genotoxic and carcinogenic and for which a health‐based guidance value cannot be established, the margin of safety (MOS) approach may be used to conclude whether or not there would be a risk at the proposed maximum use level (EFSA ANS Panel, 2012). The MOS is calculated as the ratio between the lowest NOAEL usually from a 90‐day subchronic toxicity study and the dietary exposure of consumers to the toxicologically relevant residue.
4.3. Consumer exposure
4.3.1. Consumption data
Consumption of edible tissues and products as derived from the EFSA Comprehensive European Food Consumption Database (Comprehensive Database) will be used to assess exposure to residues from the use of feed additives in different EU countries, age classes and special population groups.
The Comprehensive Database provides a compilation of existing national information on food consumption at individual level. Details on how the Comprehensive Database is used are published in the Guidance of EFSA (EFSA, 2011). For each EU country and age class, only the latest survey available in the Comprehensive Database will be used. Within the dietary studies, subjects are classified in different age classes as follows:
Infants: < 12 months old
Toddlers: ≥ 12 months to < 36 months old
Other children: ≥ 36 months to < 10 years old
Adolescents: ≥ 10 years to < 18 years old
Adults: ≥ 18 years to < 65 years old
Elderly: ≥ 65 years to < 75 years old
Very elderly: ≥ 75 years old
While the residue data reported for feed additives refer to the raw agricultural commodities (RAC), the Comprehensive Database includes consumption data for foods as consumed, such as composite foods (e.g. pizza) and other single foods or ingredients (e.g. cheese). In order to match those consumption data with the available residue data for feed additives, the consumption data reported in the Comprehensive Database have been converted into RAC equivalents. This involved identification and quantification of the single ingredients present in each composite food, and a subsequent conversion of these single ingredients into their RACs by means of conversion factors, where relevant. For assessing the exposure to feed additives, the following list of commodities will be considered relevant: meat, fat, liver, other offals (including kidney), milk, eggs, honey, fish and seafood. Consumption data for meat, fat, liver and other offals will be considered for mammals and poultry separately.
4.3.2. Occurrence and residue data
The dossier should contain all relevant occurrence/residue data which allow the assessor to estimate the total relevant occurrence/residues in the food commodities listed above (i.e. the arithmetic mean plus two standard deviations or the highest single value in case of less than six animals) resulting from the use of the additive as described by the applicant.
When assessing additives intended for multispecies use, the value for the species with the highest concentration of residues in a given tissue of poultry, mammals and fish will be taken as representative for that specific food commodity in all poultry, mammals and fish, respectively.
The residue concentration in muscle and fat (skin + fat for poultry) will be applied to the intake of meat according to the following proportions: mammals 80% muscle and 20% fat, poultry 90% muscle and 10% fat (skin + fat).
The residue concentration in kidney will be applied to the intake of other offals, while in the case of poultry, the concentration of skin fat will be applied to the fat intake.
4.3.3. Exposure methodology
Depending on the nature of the health‐based guidance derived, either a chronic or acute exposure assessment may be required.
For chronic exposure assessments, dietary surveys with only one reporting day per individual will not be considered as they are not adequate to assess repeated exposure. For each individual, the total relevant residues will be combined with the average daily consumptions of the corresponding food commodities, and the resulting exposures per food will be summed in order to obtain total chronic exposure at individual level (standardised by using the individual body weight). The mean and the higher percentile (usually the 95th percentile) of the individual exposures will be subsequently calculated for each dietary survey and each age class separately.
As opposed to the chronic exposure assessments, acute exposure will be assessed for all dietary surveys available and will be carried out for each RAC value separately. In this case, the total relevant residue for the commodity under assessment will be combined with the total corresponding consumption within each single day. The higher percentile (usually the 95th percentile) exposures based on the consuming days only will be calculated for each food commodity, dietary survey and age class separately. In case the exposure assessment reveals more than one food commodity approaching the health‐based guidance value alternative scenarios including addition of acute exposures within a single day may additionally be envisaged.
A web‐based tool will be made available by EFSA, supporting assessors in the calculation of chronic and acute exposure estimates according to the above methodology.
4.3.4. Outcome of the assessment
If the highest proposed feed concentration of an additive would result in a consumer exposure exceeding the highest safe level, measures to reduce consumer exposure should be taken. These measures could consist of a reduction of the proposed feed concentration of the additive and/or the introduction of a withdrawal period and maximum residue limits, if necessary.
5. MRLs and withdrawal period
Where the levels of residues of an additive in food from animals fed with that additive might have an adverse effect on human health, setting of MRLs for the active substance or for its metabolites in the relevant foodstuffs of animal origin and a withdrawal period may be needed. MRLs are generally not considered necessary if a withdrawal time is not required.
Maximum residue limit means the maximum concentration of residue (expressed as μg marker residue per kg of edible wet tissue or product) which may be accepted by the EU to be legally permitted or recognised as acceptable in food. It is based on the type and amount of residue considered to be without any toxicological risk for human health as expressed by a health‐based guidance value. In principle, the MRL cannot be set in the absence of such a value.
The validity of the MRLs with respect to the exposure of consumers to total residues will be evaluated using the approach as described in Section 5.1 (Figure 1).
Figure 1.
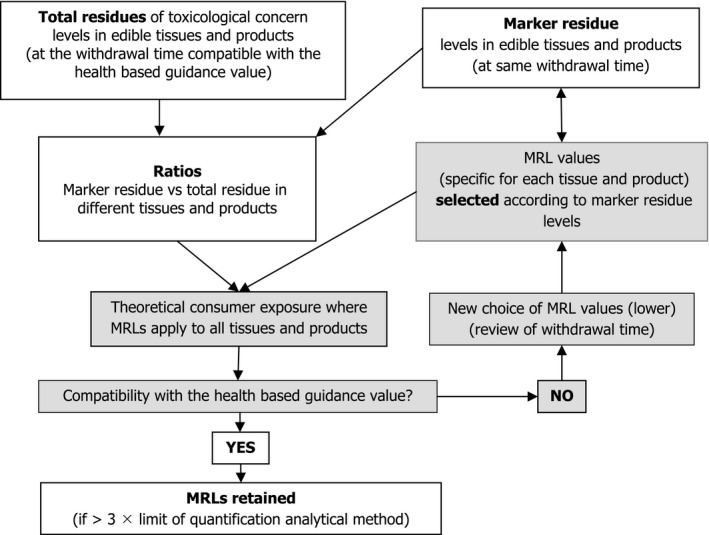
Rationale for setting MRLs
For certain additives, residues could arise below the MRL values in milk, eggs or meat which could nonetheless interfere with food quality in particular food‐processing procedures. For such additives, it may be appropriate to consider a ‘maximum (food product) processing compatible residue’ (MPCR) in addition to establishing MRL values.
The withdrawal time comprises the period after cessation of the administration of the additive which is necessary to enable the residue levels to fall below the MRLs. An experimental withdrawal up to 12 h is considered a practical zero‐day withdrawal.
5.1. Calculation of MRLs
Individual MRLs will ideally be set for the different tissues or products of target animal species. The individual MRLs in different tissues/products will reflect the depletion kinetics and variability of the residue levels within those tissues/products in the animal species. Variability is normally reflected by using the 95% confidence limit of the mean. If the confidence limit cannot be calculated due to low number of samples, the highest individual value should be taken instead. The definitions used in deriving an MRL are listed in Table 2.
Table 2.
Definitions used in deriving an MRL
| i‐j | Individual tissues/products (liver, kidney, muscle, skin + fat, milk, eggs) at different times |
| MRLi‐j | Maximum residue limit in tissues/products (μg marker substance/kg) |
| TRCi‐j | Total residue concentration in individual tissues/products (μg/kg) |
| MRCi‐j | Marker residue concentration for individual tissues/products (μg/kg) |
| RMTRi‐j | Ratio MRCi‐j to TRCi‐j for individual tissues/products |
| DITR |
Dietary intake of total residues (μg/kg body weight per day) Calculation follows Section 4.3 (consumer exposure) by the use of a web‐based tool provided by EFSA |
| DITRM |
Dietary intake of total residues (μg/kg body weight per day) calculated from MRLs of individual tissues/products divided by RMTR Calculation follows Section 4.3 (consumer exposure) by the use of a web‐based tool provided by EFSA |
Deriving a MRL is an iterative process which is summarised in Figure 1.
The different steps are the following:
select the minimum withdrawal time for which the exposure (dietary intake of total residues (DITR)) is below the health‐based guidance value. If that value is exceeded at each withdrawal time for which data are available, a new data set should be generated using a longer withdrawal time or lower dietary concentrations of the additive.
for all tissues/products, calculate the ratios marker vs. total toxicologically relevant metabolites corresponding to the withdrawal selected in step (i). Ideally, the ratio values will be in the same range (similar) for all withdrawal times. In cases where a full data set is not available because values fall below the limit of detection an extrapolation of ratio marker to total residues (RMTR) may be acceptable.
where the marker residue represents a major fraction of total residues and as total residue concentrations (TRCs) and MRCs are determined in separate experiments, TRCs may appear to be lower than MRCs which leads to ratios > 1. In that case, MRCs will be considered as the reference and TRCs back calculated using the RMTR from another experiment, in which TRCs and MRCs have been measured.
considering the MRCs measured in the different tissues/products (including two standard deviations or the highest values where a reduced number of animals is available as a guide), and taking into consideration the limit of quantification of the analytical method of the marker residue in the different tissues/products, select a first set of MRL values.
check whether the DITR MRL (DITRM) obtained from the proposed MRLs is below the health‐based guidance value and close to the DITR. If the health‐based guidance is exceeded, then a set of lower MRLs should be selected and the comparison repeated.
In all cases, an analytical method of sufficient sensitivity must be available before MRLs can be set. The limit of quantification of the method should be at least three times lower than the MRL.
5.2. Existing MRLs
Where MRLs for an active substance are in force within EU (e.g. Regulation (EC) No 37/2010)6, the assessment of the safety of the additive for the consumer is based on:
a verification that the toxicological data set submitted or retrieved from the updated scientific literature would not modify the health‐based guidance value on which the MRL is based,
a marker residue study performed with the additive at the highest proposed use level.
If the concentration of residues in tissues and products is above the existing MRLs, the conditions of use of the additive (e.g. withdrawal time, use level) should be reconsidered.
5.3. Extrapolation of MRLs
Provided that the levels of the additive in the feed of the different species considered are essentially similar, MRLs for the different edible tissues and products can be extrapolated:
within physiologically similar species (see Table 1). This would not exclude the setting of lower MRLs for these species based on the outcome of residue studies.
to horses when MRLs for a major ruminant and pigs exist.
to all other minor species of food‐producing animals except fish if identical MRLs were derived in tissues of cattle, pigs and chicken.
Glossary
- Active agent
Any viable microorganism intended to be used as/in a feed additive and that provides the intended effect.
- Active substance
Any substance or mixture of substances intended to be used as/in a feed additive and that provides the intended effect.
- Excretion
Process(es) by which an administered substance and/or its metabolites are removed from the body (OECD guideline 417 (2010)).
- Feed additive
Substances, microorganisms or preparations other than feed materials and premixtures which are intentionally added to feed or water in order to perform one or more functions mentioned in Article 5.3 of Regulation (EC) No 1831/2003.
- Marker residue
The residue selected for assay whose concentration is in a known relationship to total residues in tissues and products, ideally constant during depletion.
- Target tissue(s)
The edible tissue(s) selected to monitor for residues in the target animals, including, where appropriate, milk or eggs.
- Tolerable upper intake level (UL)
Maximum level of total chronic daily intake of a nutrient (from all sources) judged (by national or international scientific bodies) to be unlikely to pose a risk of adverse health effects to consumers or specific groups of consumers
- Withdrawal period
The withdrawal time comprises the period after cessation of the administration of the additive which is necessary to enable the residue levels to fall below a level which does not pose a risk to consumers.
- Xenobiotic
Chemicals which are not a natural component of the organism exposed to them. Physiological substances whose use results in much higher concentrations than usual in the organism exposed to them may be treated as xenobiotics.
Abbreviations
- ADI
acceptable daily intake
- ADME
absorption, distribution, metabolism and excretion
- BMD
benchmark dose
- BMDL
benchmark dose level
- DITR
dietary intake of total residues
- DITRM
dietary intake of total residues (calculated from proposed) MRLs
- FEEDAP Panel
EFSA Panel on Additives and Products or Substances used in Animal Feed
- LOAEL
lowest observed adverse effect level
- MOS
margin of safety
- MPCR
maximum (food product) processing compatible residue
- MRC
marker residue concentration
- MRL
maximum residue level
- NOAEL
no observed adverse effect level
- NOEL
no observed effect level
- OECD
Organisation for Economic Cooperation and Development
- QPS
qualified presumption of safety
- RAC
raw agricultural commodities
- RMTR
ratio marker to total residues
- TRC
total residue concentration
- UF
uncertainty factor
- UL
tolerable upper intake level
Suggested citation: EFSA FEEDAP Panel (EFSA Panel on Products or Substances used in Animal Feed) , Rychen G, Aquilina G, Azimonti G, Bampidis V, Bastos ML, Bories G, Chesson A, Cocconcelli PS, Flachowsky G, Gropp J, Kolar B, Kouba M, López‐Alonso M, López Puente S, Mantovani A, Mayo B, Ramos F, Saarela M, Villa RE, Wallace RJ, Wester P, Anguita M, Dujardin B, Galobart J and Innocenti ML, 2017. Guidance on the assessment of the safety of feed additives for the consumer. EFSA Journal 2017;15(10):5022, 17 pp. 10.2903/j.efsa.2017.5022
Requestor: EFSA
Question number: EFSA‐Q‐2016‐00822
Panel members: Gabriele Aquilina, Giovanna Azimonti, Vasileios Bampidis, Maria de Lourdes Bastos, Georges Bories, Andrew Chesson, Pier Sandro Cocconcelli, Gerhard Flachowsky, Jürgen Gropp, Boris Kolar, Maryline Kouba, Marta López‐Alonso, Secundino López Puente, Alberto Mantovani, Baltasar Mayo, Fernando Ramos, Guido Rychen, Maria Saarela, Roberto Edoardo Villa, Robert John Wallace and Pieter Wester.
Acknowledgements: The Panel wishes to thank the members of the Working Group on guidance update including Paul Brantom for the preparatory work on this scientific output and Davide Arcella and Rita Sousa for the support provided to this scientific output.
Adopted: 27 September 2017
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2017.EN-1305/full
Notes
Regulation (EC) No 1831/2003 of the European Parliament and of the Council of 22 September 2003 on additives for use in animal nutrition. OJ L 268, 18.10.2003, p. 29.
Commission Regulation (EC) No 429/2008 of 25 April 2008 on detailed rules for the implementation of Regulation (EC) No 1831/2003 of the European Parliament and of the Council as regards the preparation and the presentation of applications and the assessment and the authorisation of feed additives. OJ L 133, 22.5.2008, p. 1.
Chromatographic techniques couple to high‐resolution mass spectrometric methods.
For substances for reduction of the contamination of feed with mycotoxins based on QPS microorganisms, the effects of the metabolites/degradation products on consumer safety should be studied.
For the purpose of determining consumer safety, the NOAEL from toxicological studies and the no observed effect level (NOEL) from pharmacological studies are used in an equivalent manner.
Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin.
References
- EFSA (European Food Safety Authority), 2011. Guidance of EFSA on the use of the EFSA comprehensive European Food Consumption Database in Exposure Assessment. EFSA Journal 2011;9(3):2097, 34 pp. 10.2903/j.efsa.2011.2097 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2017. Outcome of the public consultation on the draft Guidance on the assessment of the safety of feed additives for the consumer. EFSA supporting publication 2017;EN‐1305. 30 pp. 10.2903/sp.efsa.2017.EN-1305 [DOI] [Google Scholar]
- EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food), 2012. Guidance for submission for food additive evaluations. EFSA Journal 2012;10(7):2760, 60 pp. 10.2903/j.efsa.2012.2760 [DOI] [Google Scholar]
- EFSA FEEDAP Panel (EFSA Panel on Additives and Products or Substances used in Animal Feed), 2016. Statement of the panel: Analysis of the need for an update of the guidance documents. EFSA Journal 2016;14(5):4473, 8 pp. 10.2903/j.efsa.2016.4473 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2011. Scientific Opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379, 69 pp. 10.2903/j.efsa.2011.2379 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2012. Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data. EFSA Journal 2012;10(3):2579, 32 pp. 10.2903/j.efsa.2012.2579 [DOI] [Google Scholar]
- EFSA Scientific Committee , Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen KH, More S, Mortensen A, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Silano V, Solecki R, Turck D, Aerts M, Bodin L, Davis A, Edler L, Gundert‐Remy U, Sand S, Slob W, Bottex B, Abrahantes JC, Marques DC, Kass G and Schlatter JR, 2017. Update: Guidance on the use of the benchmark dose approach in risk assessment. EFSA Journal 2017;15(1):4658, 41 pp. 10.2903/j.efsa.2017.4658 [DOI] [PMC free article] [PubMed] [Google Scholar]