

Abstract
The European Commission asked EFSA for a scientific evaluation on the risk to human health of the presence of furan and methylfurans (2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran) in food. They are formed in foods during thermal processing and can co‐occur. Furans are produced from several precursors such as ascorbic acid, amino acids, carbohydrates, unsaturated fatty acids and carotenoids, and are found in a variety of foods including coffee and canned and jarred foods. Regarding furan occurrence, 17,056 analytical results were used in the evaluation. No occurrence data were received on methylfurans. The highest exposures to furan were estimated for infants, mainly from ready‐to‐eat meals. Grains and grain‐based products contribute most for toddlers, other children and adolescents. In adults, elderly and very elderly, coffee is the main contributor to dietary exposure. Furan is absorbed from the gastrointestinal tract and is found in highest amounts in the liver. It has a short half‐life and is metabolised by cytochrome P450 2E1 (CYP2E1) to the reactive metabolite, cis‐but‐2‐ene‐1,4‐dialdehyde (BDA). BDA can bind covalently to amino acids, proteins and DNA. Furan is hepatotoxic in rats and mice with cholangiofibrosis in rats and hepatocellular adenomas/carcinomas in mice being the most prominent effects. There is limited evidence of chromosomal damage in vivo and a lack of understanding of the underlying mechanism. Clear evidence for indirect mechanisms involved in carcinogenesis include oxidative stress, gene expression alterations, epigenetic changes, inflammation and increased cell proliferation. The CONTAM Panel used a margin of exposure (MOE) approach for the risk characterisation using as a reference point a benchmark dose lower confidence limit for a benchmark response of 10% of 0.064 mg/kg body weight (bw) per day for the incidence of cholangiofibrosis in the rat. The calculated MOEs indicate a health concern. This conclusion was supported by the calculated MOEs for the neoplastic effects.
Keywords: furan; 2‐methylfuran; 3‐methylfuran; 2,5‐dimethylfuran; food; risk assessment; BMD
Short abstract
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2017.EN-1066/full
Summary
The European Commission asked the European Food Safety Authority (EFSA) for a scientific evaluation on the risk to human health of the presence of furan and methylfurans (2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran) in food. To address this request, the EFSA Panel on Contaminants in the Food Chain (CONTAM) decided that the opinion should comprise the:
evaluation of the toxicity of furan, and 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran, collectively known as methylfurans, for humans considering all relevant toxicological endpoints;
estimation of the dietary exposure of the European Union (EU) population to furan, 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran from food, including the consumption patterns of specific groups of the population if appropriate;
assessment of the human health risks for the EU population, including specific groups of the population if appropriate, as the consequence of the estimated dietary exposure.
Furan, 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran are volatile compounds that are formed in foods during thermal processing. Furan can be formed in food from a variety of precursors including ascorbic acid, amino acids, carbohydrates, unsaturated fatty acids and carotenoids. Food characteristics, processing and cooking conditions, and the losses which mainly occur due to evaporation in the preparation of the food at the level of the consumer, determine the final concentration in the food as consumed.
Chronic dietary exposure to furan was estimated using a data set containing 9,663 samples provided by governmental organisations and 7,393 samples provided by commercial organisations. The data sets were comparable and were merged for the assessment. The highest concentrations of furan were found in whole roasted coffee beans, with a mean value of 4,579 μg/kg. High mean concentrations of furan were also found in ground roasted coffee (2,361 μg/kg), unspecified coffee solids (2,186 μg/kg), coffee imitates for brewing (1,922 μg/kg), instant coffee powder (310 μg/kg) and instant coffee imitates (127 μg/kg). Mean concentrations ranging from 20 to 57 μg/kg were found in composite foods (prepared salads, cereal‐based and vegetable‐based), ready‐to‐eat meals for infants and young children, soy sauce, bread and rolls, raw pasta, breakfast cereals, fine bakery wares and spirits. Occurrence data for 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran were not provided, and from the available literature data on the co‐occurrence of furan and methylfurans, a 2‐methylfuran/furan and 3‐methylfuran/furan ratio was calculated only for coffee, infant cereals, jarred baby‐food and cereal flakes. Co‐occurrence data on 2,5‐dimethylfuran were too limited to derive such a ratio.
The highest exposures to furan were estimated in the youngest population group, i.e. infants. The mean dietary exposures calculated for infants ranged from 0.14 to 0.99 μg/kg body weight (bw) per day (minimum lower bound (LB) to maximum upper bound (UB)). Regarding the 95th percentile exposures, which refer to highly exposed consumers within a given dietary survey and age class, the highest estimates were also observed for infants ranging from 0.27 to 1.8 μg/kg bw per day (minimum LB to maximum UB). Chronic dietary exposure of adults, elderly and very elderly was estimated to be on average between 0.11 and 0.75 μg/kg bw per day (minimum LB to maximum UB) and the 95th percentile was between 0.2 and 1.27 μg/kg bw per day (minimum LB to maximum UB) depending on the survey and age group. Ready‐to‐eat meals for infants and small children are the main contributor to the dietary exposure of infants. For adults, elderly and very elderly, the exposure is mainly driven by coffee. Grains and grain‐based products are the food group contributing most for toddlers, other children and adolescents, and it is also the second largest contributor in all other age classes. The influence of reheating commercially processed foods on furan concentrations is limited and depends on the consumer behaviour. Regarding the formation of furan during home cooking, a specific scenario based on toasting bread did not impact on the overall outcome of the exposure assessment. The inclusion of methylfurans, however, may significantly increase exposure. For the exposure scenario for the sum of furan, 2‐methylfuran and 3‐methylfuran, estimates for adults, elderly and very elderly showed the highest increase compared to the baseline scenario. This exposure was mainly driven by the high concentrations of 2‐methylfuran in coffee (four times higher than furan).
After oral exposure to rats, furan is rapidly and extensively absorbed from the gastrointestinal (GI) tract, distributed throughout the body and extensively metabolised. The major contributor to the metabolism of furan is cytochrome P450 2E1 (CYP2E1), resulting in opening of the furan ring and formation of cis‐but‐2‐ene‐1,4‐dialdehyde (BDA) which is very reactive. BDA has never been directly measured. However, results with trapping agents and the identification of urinary and biliary metabolites provide strong evidence that BDA is indeed a prime reactive intermediate in the metabolism of furan. BDA reacts readily with amino acids, glutathione (GSH) and biogenic amines and it has direct reactivity towards DNA. However, the data suggest that due to BDA reactivity with protein and non‐protein amino and thiol residues, access to DNA in vivo is restricted.
No oral studies have been performed with any of the three methylfurans considered in this opinion to address their kinetics in animals, but it is anticipated that they will be absorbed from the GI tract. Non‐extractable association of 2‐methylfuran with protein and DNA in the liver was reported after intraperitoneal (i.p.) administration. For 2‐methyl‐ and 3‐methylfuran, data from in vitro studies are available that show that they may become associated (non‐extractable) with lung and liver microsomal proteins. The reactive metabolic intermediates of 2‐methylfuran and 3‐methylfuran are the cis‐enedials 3‐acetylacrolein (4‐oxopent‐2‐enal) and 2‐methylbut‐2‐enedial, respectively. For 2,5‐dimethylfuran ring opening results in the formation of a reactive intermediate, the cis‐enedione 3(Z)‐hexene‐2,5‐dione. Similar to BDA, the reactive intermediate formed from 2,5‐dimethylfuran reacts readily with amino acids and with GSH and the corresponding structures for the adducts formed have been reported. For 2‐ and 3‐methylfuran metabolites, only very limited information is available, but from the structures of their primary metabolites and from studies with scavengers, it is anticipated that these will react with tissue components in a similar way to the primary metabolites of furan and 2,5‐dimethylfuran.
In experiments of a duration of up to 90 days, furan is strongly hepatotoxic and moderately nephrotoxic in rodents when applied by oral gavage. Rats seem to be more sensitive towards furan than mice. Furan leads to characteristic changes in serum markers related to hepatotoxicity as well as severe histopathological damage in the liver. After 90 days, significant increases in serum thyroid hormones were observed along with severe histopathological changes in the liver of male rats after a daily dose of 0.12 mg/kg bw, given 5 days a week.
In long‐term studies, furan was associated with toxicity in the liver. Cholangiofibrosis was observed in rats as an early and sensitive response with significant increases after 36 weeks at doses of 0.44 mg/kg bw and above. In mice, hepatocellular adenoma/carcinoma occurred at 104 weeks at doses of 4 mg/kg bw and above. In rats, cholangiocarcinomas were observed at the top dose of 8 mg/kg bw. No relevant malignancies were observed at doses at or below 2 mg/kg bw up to 104 weeks.
Furan did not induce gene mutations in bacteria. In the majority of the in vitro studies in mammalian cells, furan was able to induce chromosomal aberrations and sister chromatid exchanges; contrasting results were reported on the requirement for microsomal activation. BDA forms DNA adducts at the exocyclic N atom of the deoxycytosine, deoxyguanosine and deoxyadenosine nucleosides in in vitro model systems and in DNA from Salmonella Typhimurium TA104. BDA was able to directly induce mutations in bacteria and strand breaks and mutations in mammalian cells in vitro. In rodents, furan induced very low levels of DNA adducts in liver and kidney. The chemical structures of these adducts could not be defined but were not identical to those induced by BDA in in vitro model systems. Conflicting data were reported for the induction of DNA breaks in the liver. These breaks, likely reflecting oxidative stress‐induced DNA damage, occurred at doses showing mild liver toxicity. Chronic exposure to furan induced chromosomal damage in proliferating splenocytes from mice and rats. The DNA lesions responsible for these effects remain undefined. No clear induction of base substitutions was observed in transgenic rat models. A weak mutagenic activity was suggested by a single study in a transgenic mouse model. An excess of GC > TA transversions was observed at codon 61 of the Ha‐ras oncogene in furan‐induced liver tumours in mice. It is uncertain whether these mutations are the consequence of oxidative damage to DNA or reflect an expansion of pre‐existing spontaneous Ha‐ras mutations.
Histological changes in the testes, prostate gland, Leydig cells and seminal vesicles were observed in Wistar rats at 2 mg/kg bw (lowest dose tested) and above given during weaning and post‐puberty. No histological effects in reproductive organs were observed in adult rats and mice up to 8 mg/kg bw.
Furan induces oxidative stress. The binding of BDA to a range of target molecules leads ultimately to cell and tissue damage, mitochondrial dysfunction and fibrosis, primarily in the liver. There is clear evidence for the involvement of indirect mechanisms in the carcinogenic mode of action of furan. These include epigenetic changes, oxidative damage to DNA and regenerative hyperplasia, with all of these effects being accompanied by tissue damage. There is limited evidence of a direct mechanism (i.e. direct interaction with DNA) in the carcinogenic action of furan. The contributing factors in carcinogenesis are likely to vary according to dose, duration of exposure and degree of severity of liver cellular damage, inflammation and compensatory proliferation.
The liver is the primary target organ with respect to acute and short‐term (< 90 days) toxicity of 2‐ and 3‐methylfuran in rodents. There is also indication for kidney toxicity of 3‐methylfuran after 90 days of exposure. Liver toxicity in male rats given 3‐methylfuran (28 or 90 days) or 2‐methylfuran (28 days) by gavage was found to be the most sensitive adverse endpoint for both compounds. Their toxic potency was reported to be in the same order of magnitude as that for furan. No information on the genotoxic properties of 3‐methylfuran and limited information for 2‐methylfuran and 2,5‐dimethylfuran is available. Both 2‐methylfuran and 2,5‐dimethylfuran showed negative results in bacteria. There is some evidence that both compounds induce chromosomal damage in mammalian cells in vitro and there is little evidence that 2,5‐dimethylfuran can induce DNA breaks in vivo.
As there are few data available on the effect of furan in humans, the CONTAM Panel used data from experimental animals for the hazard characterisation. For non‐neoplastic effects, the CONTAM Panel selected the BMDL10 of 0.064 mg/kg bw per day (correcting for the applied dose regimen of 5 days per week) for the induction of cholangiofibrosis in male rats after 2 years as reference point. For neoplastic effects, the CONTAM Panel considered that the combined data set from two studies on the incidence of hepatocellular adenomas and carcinomas in female mice after 2 years is the most robust data set to derive a reference point and selected the BMDL10 of 1.31 mg/kg bw per day (correcting for the applied dose regimen of 5 days per week). In view of some indications for a direct genotoxic mechanism in the carcinogenic mode of action of furan, the CONTAM Panel decided that it was not appropriate to establish a tolerable daily intake (TDI) and used a margin of exposure (MOE) approach. The available information was insufficient to identify a reference point for the methylfurans. However, it was considered appropriate to assume dose additivity for hepatotoxicity of furan, 2‐methylfuran and 3‐methylfuran in the rat.
For non‐neoplastic effects, the calculated MOEs are below 100 in a number of dietary surveys, particularly for the high percentile exposure estimates for the younger age groups (infants and toddlers) and adults (including elderly). The CONTAM Panel concluded that these MOEs indicate a health concern. The CONTAM Panel noted that, with the exception of some surveys, the calculated MOEs for neoplastic effects of furan are smaller than 10,000, which, in accordance with the guidance given by the Scientific Committee, would indicate a health concern. However, there is uncertainty regarding the carcinogenic mode of action of furan. The CONTAM Panel considered the resulting MOEs for hepatocellular adenomas and carcinomas as supporting evidence for its conclusion, based on the hepatotoxicity of furan, that the current exposure to furan indicates a health concern. Based on a scenario for chronic dietary exposure to the sum of furan, 2‐methylfuran and 3‐methylfuran, MOE values for the incidence of cholangiofibrosis were calculated. From these MOE values, it becomes clear that methylfurans may add significantly to the overall exposure and therefore increase the concern for hepatotoxicity.
The CONTAM Panel considered that the impact of the uncertainties on the risk assessment of furan is moderate and that the assessment is likely to be conservative. For methylfurans, due to a lack of data, no full assessment could be performed and the uncertainties in the assessment are large.
The CONTAM Panel recommends producing additional data on the occurrence of methylfurans in food as well as data on the changes of furan and methylfurans concentrations during the distinct stages of coffee preparation for all coffee types. Furthermore, the CONTAM Panel recommends performing studies in vivo on the effect of furan on the genome to clarify the carcinogenic mode of action. Further information is also needed on the toxicity, including the genotoxic properties, of methylfurans.
1. Introduction
1.1. Background and Terms of Reference as provided by the requestor
BACKGROUND
Furan is formed in foods during thermal processing. It occurs in a variety of foods such as coffee, canned and jarred foods including baby food containing meat, and various vegetables. 2‐Methylfuran and 3‐methylfuran, have been found concurrently with furan, and apparently are also formed during thermal processing and are likely to undergo a similar metabolic fate to furan.
The Scientific Panel on Contaminants in the Food Chain adopted on 7 December 2004 a report on provisional findings on furan in food.1 They concluded from the available data that there is a relatively small difference between possible human exposures and the doses in experimental animals that produce carcinogenic effects, probably by a genotoxic mechanism. However, a reliable risk assessment would need further data on both toxicity and exposure.
In the meantime, EFSA published
on 2 December 2009, an external scientific report on furan in heat processed food products including home cooked food products and ready‐to‐eat products.2
on 18 December 2009, an external scientific/technical report on consumer exposure to furan from heat‐processed foods and kitchen air.3
on 22 August 2011, a scientific report with an update on furan levels in food from monitoring years 2004–2010 and exposure assessment.4
Furan was evaluated at the meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA) which was held in Rome, Italy, from 16 to 25 February 2010.5 The Committee concluded that the estimated margins of exposure (MOE) (480–960) indicate a human health concern for a carcinogenic compound that might act via a DNA‐reactive genotoxic metabolite.
Furthermore, the study report of the latest furan carcinogenicity bioassay, supported by an interagency agreement between the US National Institute of Environmental Health Sciences (NIEHS)/National Toxicology Program (NTP) and the US Food and Drug Administration (FDA) has been finalised and is now available (NCTR, 2015).
TERMS OF REFERENCE
In accordance with Art. 29 (1) (a) of Regulation (EC) No 178/2002, the Commission asks the European Food Safety Authority (EFSA) for a scientific opinion on the human health risks related to the presence of furan and methylfurans in food.
1.2. Interpretation of the Terms of Reference
The EFSA Panel on Contaminants in the Food Chain (CONTAM) concluded this opinion should comprise the:
evaluation of the toxicity of furan, and 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran, collectively known as methylfurans, for humans considering all relevant toxicological endpoints;
estimation of the dietary exposure of the European Union (EU) population to furan, and 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran, collectively known as methylfurans, from food, including the consumption patterns of specific groups of the population if appropriate;
assessment of the human health risks for the EU population, including specific groups of the population if appropriate, as the consequence of the estimated dietary exposure.
1.3. Supporting information for the assessment
1.3.1. Chemical and physical properties
1.3.1.1. Chemical properties
Furan [CAS‐number 110‐00‐9, C4H4O, molecular weight (MW) 68.07 g/mol] is the parent compound of five‐membered heterocycles containing one oxygen atom. Its structure is stabilised by resonance due to the delocalisation of the electrons of carbon and oxygen present in the p‐orbital in a 6π‐electron system. Its chemical structure is shown in Figure 1.
Figure 1.
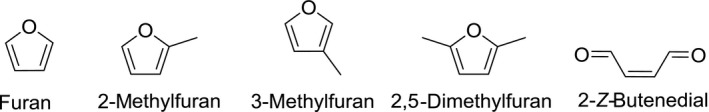
Chemical structures of furan, 2‐methylfuran, 3‐methylfuran, 2,5‐dimethylfuran and 2‐Z‐butenedial
Furan is highly susceptible to electrophilic aromatic substitution, mainly at the α‐position, yielding for instance 2‐alkylfurans (Hoydonckx et al., 2012). It is not clear to what extent such reactions are relevant in foods, as in general, furan is considered to be stable in foods. Hoydonckx et al. (2012) summarised the main chemical reactions applied to furan, but these are not relevant in a food context. The compound is highly flammable and may also form explosive peroxides. It is characterised by an ethereal odour.
2‐Methylfuran [CAS‐number 534‐22‐5, C5H6O, MW 82.1 g/mol] is also aromatic in nature, and its chemical properties are comparable to those of furan, although it should be noted it is more prone to electrophilic aromatic substitution (Hoydonckx et al., 2012). Its chemical structure is shown in Figure 1. It is reported to be characterised by a medium strength, chocolate like odour.
3‐Methylfuran [CAS‐number 930‐27‐8, C5H6O, MW 82.1 g/mol] (for structure see Figure 1) is the analogue of 2‐methylfuran. Like furan and 2‐methylfuran, 3‐methylfuran is also aromatic in nature. It can be supposed that its chemical properties are similar, but information is scarce.
2,5‐dimethylfuran [CAS‐number 625‐86‐5, C6H8O, MW 96.1 g/mol] is particularly studied because of its potential use as a liquid fuel as it can be produced from biomass‐derived fructose (Román‐Leshkov et al., 2007). This dimethylfuran is known as a potent scavenger of singlet oxygen (Noguchi et al., 1977; Mongin et al., 2016) and hydroxyl radicals (Okada and Okajima, 1998).
2‐Z‐Butenedial (commonly referred to as cis‐but‐2‐ene‐1,4‐dialdehyde (BDA)) [CAS‐number 3675‐13‐6, C4H4O2, MW 84.1 g/mol] has been identified as a key metabolite of furan (Chen et al., 1995). In view of its highly reactive character, it is synthesised in situ via the oxidative ring opening of furan using dimethyloxirane (Adger et al., 1991).
1.3.1.2. Physical properties
Furan is a colourless liquid with a density of 0.94 g/cm3 (20°C), a melting point of −85°C and a boiling point of 31.4°C at 101 kPa (760 mmHg). At 20°C, its vapour pressure is 66 kPa and at 25°C 80 kPa. Its solubility in water at 25°C is 10 g/L, and in acetone, benzene, ether and ethanol is greater than 100 g/L. The log Kow is 1.34 (Hoydonckx et al., 2012; NTP, 2014). It can darken upon storage.
2‐Methylfuran is a colourless liquid with a density of 0.92 g/cm3 (20°C), a melting point of −88.7°C and a boiling point of 63–64°C at 101 kPa. At 20°C, its vapour pressure amounts 16 kPa (Hoydonckx et al., 2012). Its solubility in water at 20°C is 3 g/L and the log Kow amounts to 1.85.6 It is readily soluble in organic solvents.
3‐Methylfuran is a colourless liquid with a density of 0.92 g/cm3 (18°C) and a boiling point of 65.5°C at 101 kPa (Asahina et al., 1924). On standing, it can turn yellow (Burness, 1956). The log Kow is 1.91 and the solubility in water is 3.03 g/L, as estimated using US‐EPA EPI‐suite.7 SciFinder8 reported an estimated solubility in water of 4.4 g/L and an estimated vapour pressure of 21 kPa (25°C). According to The Good Scents Company, 3‐methylfuran is soluble in ethanol, but further details are not provided.9 2,5‐Dimethylfuran is a colourless to yellow liquid with a density of 0.90 g/cm3 (20°C) and a boiling point of 94–96°C (Terent'ev and Kazitsyna, 1949) It has an estimated vapour pressure of 8 kPa at 25°C. Its (experimental) log Kow is 2.24 and from this value the solubility in water can be estimated to be approximately 1.5 g/L (US‐EPA‐EPI‐suite). However, SciFinder8 reported an estimated solubility in water of 4.3 g/L. According to The Good Scents Company, 2,5‐dimethylfuran is soluble in ethanol and propylene glycol, but further details are not provided.9
BDA is a major primary metabolite of furan (see Section 3.1.1.3), and for this reason, some information on this metabolite is presented here. Its boiling point was reported to be 54–61°C (1 kPa) (Hufford et al., 1952). In the absence of experimentally determined properties, the following estimations were obtained, using US‐EPA EPI‐suite: a vapour pressure of 0.5 kPa (25°C), log Kow 0.14 and a solubility in water of 9.8 g/L. However, different values were reported on SciFinder,8 namely a vapour pressure of 0.08 kPa (25°C), and a solubility in water of 124 g/L.
1.3.2. Analytical methods
Currently, no official standard methods are available for the analysis of furan and methylfurans in foods. Neither are there certified reference materials available. In 2008, the European Commission organised a proficiency test for furan using baby food as a sample in which 22 laboratories participated with 16 returning satisfactory results (Kubiak et al., 2008a,b). Commercial proficiency tests for furan using coffee or baby foods samples are organised by organisations such as Fapas.10 No proficiency tests were identified for methylfurans.
Laboratories use their in house‐validated analytical methods. Three analytical approaches are used most often for the determination of furan and methylfuran in foods. All of them are based on a mass‐spectrometric (MS) detection and quantification using stable isotope dilution assays with d4‐furan, d3‐2‐methylfuran and d3‐3‐methylfuran for, respectively, furan, 2‐methylfuran and 3‐methylfuran based on selective ion monitoring (SIM). For 2,5‐dimethylfuran, the use of deuterated analogues is not applied, probably because analytical standards are not available commercially. Separation is accomplished by capillary gas chromatography (GC).
The approaches differ in the isolation of the volatile analytes from the matrix. In the Goldmann et al. (2005) method, solid‐phase microextraction (SPME) (using typically a carboxen/polydimethylsiloxane fibre) is used in combination with cryofocusing. Bianchi et al. (2006) used a similar approach but omitted the cryofocusing step. Becalski et al. (2005, 2010) used a static headspace approach without the use of SPME, which is similar to the method proposed by FDA (2004b). The static head space approach and the use of SPME both proved to be fit for purpose as observed in a proficiency test organised by the Joint Research Centre of the European Commission in 2008 (Kubiak et al., 2008a,b). The limits of detection (LOD) of these methods typically range from 5 μg/kg to as low as 0.01 μg/kg, and limits of quantification (LOQ) from 25 μg/kg to 0.03 μg/kg (Kubiak et al., 2008a,b) for furan. For methylated furans, similar limits were reported (e.g. Becalski et al. (2010) reported an LOD for 2‐methylfuran amounting 0.05 μg/kg). These analytical limits are dependent upon the food matrix analysed (Becalski et al., 2016). Becalski et al. (2010) also reported the presence of trace amounts of furan present in blank samples and therefore considered only samples with concentrations exceeding 1 μg/kg as relevant.
Special care should be taken during the analysis because of the high volatility of the analyte and the internal standards. In addition, the possibility of artefactual furan formation should be considered. Adams et al. (2012) reported the artefactual furan formation on the SPME fibre from volatile precursors, especially at high fibre desorption temperatures. Also the additional formation of furan during the equilibration of the sample in the headspace vials has been reported (Senyuva and Gokmen, 2005).
Märk et al. (2006) used proton‐transfer reaction mass spectrometry (PTR‐MS) as an analytical tool to quantify furan and methylfuran. This technique could even be applied as an on‐line quality monitoring system as no chromatographic separation of the analytes is required.
1.3.3. Previous assessments
Furan was evaluated by the International Agency for Research on Cancer (IARC) (IARC, 1995). The IARC concluded that the evidence in humans for the carcinogenicity of furan was inadequate but there was sufficient evidence in experimental animals to classify furan as ‘possibly carcinogenic to humans’ (Group 2B).
In 2000, the US National Academy of Sciences (NAS) published a monograph on furan as an airborne contaminant in spacecraft and concluded that ‘…furan, or an active metabolite of furan, affects DNA indirectly through a mechanism involving cytotoxicity and does not react directly with the DNA in target cells’. In assessing the risk of furan‐induced cancers, the assumption was made that ‘an exposure concentration that adequately controls the risk of cholangiocarcinomas will also control leukemias and hepatocellular adenomas and carcinomas’. Biliary hyperplasia induced by furan in male rats in a 13 week NTP study (NTP, 1993) was used as a precursor lesion to the cholangiocarcinomas. From these data, a benchmark dose lower confidence limit for 1% increase in cancer incidence (BMDL01) of 0.09 mg/kg body weight (bw) per day was calculated (NAS, 2000).
During investigations relating to a review of a petition for certain uses of irradiation in food, the US Food and Drug Administration (FDA) identified furan in a number of foods that undergo heat treatment, such as canned and jarred foods. In May 2004, the FDA published a request for submission of data and information on furan (FDA, 2004a). The EFSA CONTAM Panel considered this as an emerging issue in food safety and decided to compile a scientific report comprising all the available data at the time on methods of analysis, occurrence, formation, exposure and toxicity (EFSA, 2004). From the limited data available at the time, the EFSA CONTAM Panel concluded that the weight of evidence indicates that furan‐induced carcinogenicity is probably attributable to a genotoxic mechanism. However, chronic toxicity leading to regenerative cell proliferation may indirectly amplify the tumour response. Furthermore, the CONTAM Panel concluded that there was a relatively small difference between possible human exposures and the doses in experimental animals that produce carcinogenic effects. However, for a reliable risk assessment, further data would be needed on both toxicity and exposure.
JECFA evaluated furan at its 72nd meeting in 2010 (FAO/WHO, 2011). The Committee concluded that furan is hepatotoxic and hepatocarcinogenic in rats and mice and considered carcinogenicity the critical endpoint for use in human health risk assessment. The Committee performed a benchmark dose (BMD) analysis of liver tumour incidence data from an oral bioassay in mice. The calculated benchmark dose lower confidence limits for 10% increase in cancer incidence (BMDL10) ranged from 1.34 to 1.89 mg/kg bw per day. The Committee used the value of 1.34 mg/kg bw per day, adjusted to 0.96 mg/kg bw per day to account for the dosing schedule used in the study, as the reference point to calculate MOEs. Dietary exposure estimates for mean exposure of the general population was 1 μg/kg bw per day and for high exposure 2 μg/kg bw per day, inclusive of children. The resulting MOEs were between 960 and 480 for the average and high dietary exposures, respectively. The Committee considered that these MOEs indicate a human health concern for furan which is a carcinogenic compound that might act via a DNA‐reactive genotoxic metabolite.
In 2012, the Norwegian Food Safety Authority published a risk assessment of furan exposure in the Norwegian population (VKM, 2012). On the basis of the available data, the VKM considered that the rat cholangiocarcinomas may be relevant for assessing human risk from furan and concluded that a genotoxic mechanism in furan‐induced carcinogenesis cannot be excluded. There was a high incidence (near 100%) of cholangiocarcinomas at all doses tested after a 2‐year treatment with furan (NTP, 1993). Since this endpoint showed a dose–response relationship after 9 months of exposure, the VKM chose this as the critical endpoint. They calculated a BMDL10 of 0.14 mg furan/kg bw per day. In applying a correction factor of 7 for shorter than full life‐time study duration (9 months instead of 2 years), a point of departure of 0.02 mg/kg bw per day was chosen. The resulting MOEs for 12‐month infants were between 29 and 13, and in adults, between 74 and 26 for average and high dietary exposures, respectively.
In addition, a number of risk assessments are available in which national exposure assessments were carried out (e.g. Lachenmeier et al., 2009; Bakhiya and Appel, 2010a,b; Minorczyk et al., 2012; Scholl et al., 2012a,b; Waizenegger et al., 2012; Health Canada, 2016).
1.3.4. European legislation
In order to protect public health, Article 2 of the Council Regulation (EEC) No 315/9311 stipulates that, where necessary, maximum tolerances for specific contaminants shall be established. Thus, a number of maximum tolerances for contaminants, natural plant toxicants as well as for process contaminants such as 3‐monochloropropane‐1,2‐diol are currently laid down in Commission Regulation (EC) No 1881/200612. Furan in food is currently not covered by this EU Regulation.
Commission Recommendation 2007/196/EC13 recommended that Member States should perform monitoring of the presence of furan in foodstuffs that have undergone heat treatment during the years 2007 and 2008. Results of this monitoring were published in an EFSA report (EFSA, 2011d), and included data for commercial foodstuffs as purchased without any further preparation14 and commercial foodstuffs analysed as consumed after further preparation in the laboratory.15
Both 2‐methylfuran and 2, 5‐dimethylfuran were on the Union list of flavourings and source materials for use in food and their conditions of use. However, they were removed from the Union list by Commission Regulation (EU) 246/201416, since their applications had been withdrawn by the industry. ‘The Good Scents Company’ (a website maintained by the flavourings industry) reported that 3‐methylfuran is not for use in flavourings.9
1.3.5. Precursors of furan and methylfurans in food
Furan can be formed in food from a variety of precursors including ascorbic acid, amino acids, carbohydrates, unsaturated fatty acids and carotenoids. The pathways described below have been investigated in model systems and have been confirmed for foods. These pathways have been simplified for clarity in Figures 2–6. The conditions for the formation of furan and methylfurans have been described in Section 3.2.3.
Figure 2.

Suggested formation pathway of furan from polyunsaturated lipids (adapted from Perez Locas and Yaylayan, 2004; Owczarek‐Fendor et al., 2010b)
Figure 6.
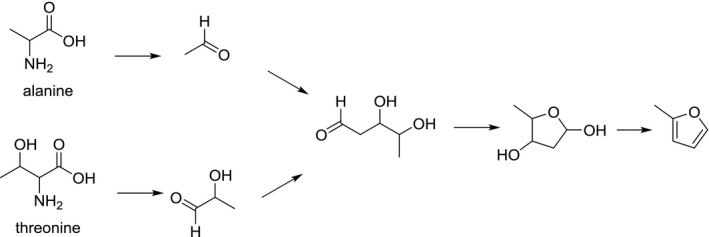
Suggested formation pathway of 2‐methylfuran (adapted from Märk et al., 2006)
The formation from unsaturated fatty acids and probably carotenoids originated from the formation of 4‐hydroxyalk‐2‐enals, particularly 4‐hydroxy‐but‐2‐enal, which is produced from the oxidation of but‐2‐enal which in turn can be produced from the oxidation of polyunsaturated omega‐3 fatty acids (Owczarek‐Fendor et al., 2010b) (Figure 2). 4‐Hydroxyalk‐2‐enals and other lipid peroxidation products are known to occur in palatable foods (Frankel, 2012).
Furan formation was reported during the thermal degradation of serine and cysteine (Perez Locas and Yaylayan, 2004). The formation pathway from serine involves the formation of acetaldehyde and glycolaldehyde which are both prone to aldol‐condensation giving rise to the crucial intermediate 2‐deoxyaldotetrose, which upon cyclisation and dehydration yields furan (Figure 3). Cysteine is supposed to react with a similar pathway (Perez Locas and Yaylayan, 2004).
Figure 3.
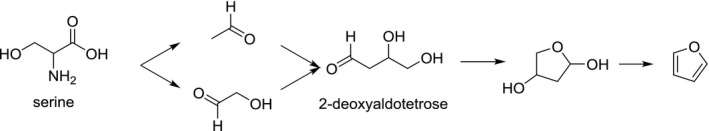
Suggested formation pathway of furan from serine (adapted from Perez Locas and Yaylayan, 2004; Limacher et al., 2008)
Other amino acids can also give rise to the formation of furan. Aspartic acid, alanine and threonine can degrade to acetaldehyde, but require an external source of glycolaldehyde (e.g. sugars) to form the 2‐deoxyaldotetrose intermediate (Perez Locas and Yaylayan, 2004).
Although amino compounds will favour the formation of furan from sugars (via the Maillard reaction), sugars on their own are able to generate furan. Various pathways have been suggested involving both the recombination of sugar fragmentation products from the intact sugar skeleton (Limacher et al., 2008) and the degradation of the intact sugar skeleton (Limacher et al., 2008; Van Lancker et al., 2011). The fragmentation pathway involves mainly the condensation of acetaldehyde and glycolaldehyde (Limacher et al., 2008). The formation from the intact sugar skeleton involves various parallel pathways, which depend on the type of sugar (Limacher et al., 2008). These reactions involve the formation of deoxy‐intermediates via dehydration reaction followed by cyclisation reactions and further dehydration. Finally, losses of mainly the C1 and C2 carbons of the sugar chain result in the formation of furan. The formation of furan in dry heating conditions is typically much higher than in pressure sterilisation conditions (Figure 4).
Figure 4.
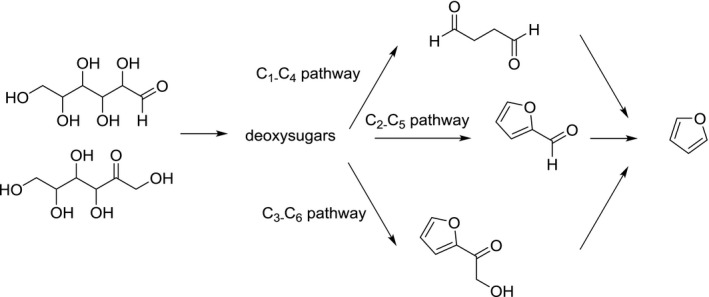
Suggested formation pathway of furan from hexoses (adapted from Limacher et al., 2008)
Furan formation from ascorbic acid is believed to start with the hydrolytic ring opening of ascorbic acid and elimination of water to form 4‐deoxyascorbic acid. This intermediate can generate 2‐deoxyaldotetrose. Alternatively, 4‐deoxyascorbic acid can result in the formation of 2‐furfural which in turn is a furan precursor (Limacher et al., 2007) (Figure 5).
Figure 5.
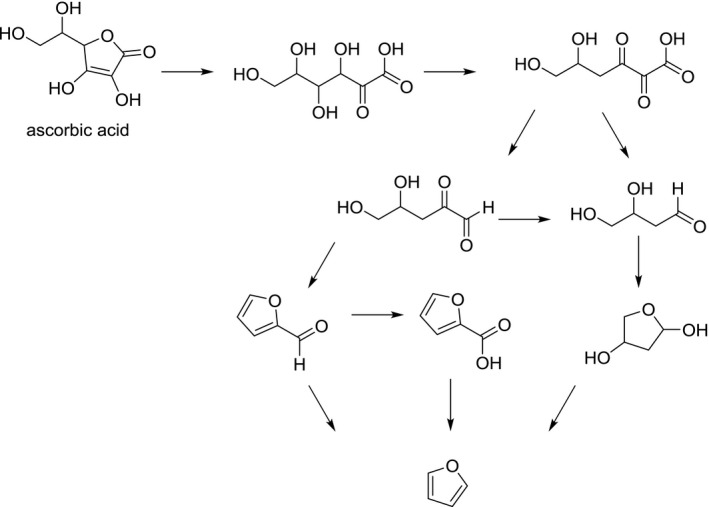
Suggested formation pathway of furan from ascorbic acid (adapted from Limacher et al., 2007)
The formation of methylfurans in food has received considerably less attention, although it was recently shown that 2‐methylfuran was the dominant furan in coffee (Becalski et al., 2016). Accordingly, the formation of methylfurans is less well understood. For 2‐methylfuran, a pathway based on the aldol condensation of the Strecker aldehydes lactaldehyde (originating from threonine) and acetaldehyde – similar to the glycoaldehyde/acetaldehyde pathway for furan – has been suggested (Märk et al., 2006) (Figure 6). Indeed, as model systems devoid of amino acids produced little or no methylfurans (Märk et al., 2006; Limacher et al., 2008), the presence of amino acids seems to play a key role in their formation. Limacher et al. (2007) reported the formation of 2‐methylfuran from the degradation of ascorbic acid, but acknowledged it was a minor degradation pathway. Märk et al. (2006) reported the formation of methylfuran from linolenic acid in the presence of α‐tocopherol or ferric ions. On basis of the findings of Adams et al. (2011), showing in model systems that α,β‐unsaturated aldehydes are potential precursors of the corresponding alkylated furans under roasting conditions, it could be suggested that 2‐pentenal may be a relevant precursor formed via the oxidation of linolenic acid. The formation pathway from 2‐pentenal proceeds similarly to the formation of furan outlined in Figure 2, requiring oxidative conditions. Adams et al. (2011) moreover showed that amino acids, such as phenylalanine catalyse the formation of alkylfurans from their corresponding α,β‐unsaturated aldehydes, although the impact for 2‐methylfuran from 2‐pentenal was not significant. No methylfuran formation was observed from linoleic acid in the presence of ferric ions or butylated hydroxytoluene (Märk et al., 2006).
1.3.6. Production, use and environmental fate
1.3.6.1. Industrial production and use
Furan is produced by decarbonylation of furfural using Pd catalysis. It can also be manufactured by partial oxidation of 1,3‐butadiene. It is mainly used for the production of α‐acetylfuran, 2,5‐dimethoxy‐2,5‐dihydrofuran, 2,2‐difurylpropane, 2,2‐di(tetrahydrofurfuryl)propane, pyrrole derivatives and thiophene. It is used as an intermediate in the production of pharmaceuticals, agricultural chemicals, lacquers and stabilisers or as a solvent for resins (IARC, 1995; Hoydonckx et al., 2012).
2‐Methylfuran is used as a solvent and as a raw material in the production of antimalarial drugs (chloroquine), methylfurfural, nitrogen and sulfur heterocycles, and functionally substituted aliphatic compounds. It is produced as a side product of furfuryl alcohol production.
2,5‐Dimethylfuran is a potential biofuel that can be obtained from bio‐based sources, such as fructose, via consecutive dehydration and hydrogenolysis reactions (Román‐Leshkov et al., 2007). Furan and 2‐methylfuran are also considered as potential biofuels (Liu et al., 2014).
1.3.6.2. Environmental fate
Furan and methylfurans are released into the atmosphere mainly from anthropogenic sources. Important sources are exhaust gas from diesel and gasoline engines and combustion of waste and biomass (e.g. wood) (IARC, 1995; Villanueva et al., 2009; Tapia et al., 2011). In addition to air, furan has been detected in river water and industrial effluents (IARC, 1995).
2. Data and methodologies
The CONTAM Panel applied the general principles for the assessment of chemicals in food as described by WHO/IPCS (2009) and any EFSA guidance documents pertaining to risk assessment and relevant for the present assessment (see Appendix A.3).
2.1. Supporting information for the assessment and information to be used in the dietary exposure assessment
2.1.1. Collection, selection of evidence
A search for recent reviews was conducted to identify scientific publications dealing with methods of analysis, chemistry, formation in food, exposure and occurrence of furan in food. Since it was expected to receive only limited information on the occurrence of methylfurans in food during the EFSA call for data (see Section 2.3.1), a search for original studies was also carried out. It was noted, that the information on dietary exposure of furan and methylfurans and on formation and methods of analysis of methylfurans was not sufficiently covered by the reviews. Therefore, specific searches were also conducted for these topics (see Appendix A, Section A.1). During the development of the exposure assessment, the Working Group (WG) identified the need for data on the influence of coffee preparation on furan levels in liquid coffee. An additional search was conducted to retrieve this information (see Appendix A, Section A.1).
The literature search was performed in May 2016 (March 2017 for information on coffee). Web of Science17 and PubMed18 were identified as databases appropriate for retrieving literature for the present evaluation. The references resulting from the literature search were imported and saved using a software package (EndNote19), which allows effective management of references and citations. Additionally, reviews and relevant scientific evaluations by national or international bodies were considered for the current risk assessment, i.e. IARC (1995), NAS, 2000, FDA (2004a), FAO/WHO (2011) and VKM, 2012. When relevant papers were identified during the risk assessment process (e.g. from other studies or reviews), they were also considered.
The references obtained were screened using title and abstract to identify relevant literature.
The VCF database20 on volatile compounds in food was consulted to identify papers on the occurrence of methylfurans.
EFSA also organised an Info Session with invited stakeholders to support the work of the CONTAM Panel and its working group in drafting this scientific opinion. The aim of this meeting was to collect information and data which are not readily available in the public domain regarding dietary exposure, home‐cooking and toxicity. Further information is available in EFSA's Register of Questions.21
2.1.2. Appraisal of evidence
The information retrieved was screened and evaluated by relevant domain experts from the CONTAM WG on furan in food and was used for the present assessment.
2.2. Hazard identification and characterisation
2.2.1. Collection and selection of evidence
EFSA outsourced an extensive literature search related to the toxicity of furan and its methyl analogues (contract: RC/EFSA/BIOCONTAM/2016/02). The aim of the assignment was to identify and collect all relevant literature regarding furan, 2‐methylfuran and 3‐methylfuran covering the following areas:
Area 1: Data on toxicokinetics (absorption, distribution, metabolism and excretion (ADME)) in experimental animals and humans and from in vitro studies.
Area 2: Data on oral toxicity in experimental animals.
Area 3: Data on in vitro and in vivo genotoxicity and mode of action (MoA).
Area 4: Data on observations in humans (including epidemiological studies, case reports and biomarkers of exposure).
The used methodology and the results are detailed in NFI (2017).
In addition to the literature searches outsourced by EFSA, a search for studies regarding the toxicity of 2,5‐dimethylfuran and the genotoxicity of BDA was conducted (see Appendix A, Section A.2). The literature search was performed in June 2016 using Web of Science17 and PubMed.18 The references resulting from the literature search were imported and saved using a software package (EndNote19), which allows effective management of references and citations.
It was regularly checked (last check May 2017) whether new papers had become available during the development of this scientific opinion.
Reviews, relevant scientific evaluations and toxicity studies by national or international bodies were also considered for the current risk assessment (see Section 2.1.1). When relevant papers were identified during the risk assessment process (e.g. from other studies or reviews), they were also considered.
In addition, an Info Session with invited stakeholders was organised (see Section 2.1.1).
2.2.2. Appraisal of evidence
The information retrieved has been screened and evaluated by relevant domain experts from the CONTAM WG on furan in food and has been used for the present assessment. Any limitations in the information used are documented in this scientific opinion.
Selection of the scientific papers for inclusion or exclusion was based on consideration of the extent to which the study was relevant to the assessment or on general study quality considerations (e.g. sufficient details on the methodology, performance and outcome of the study, on dosing, substance studied and route of administration and on statistical description of the results), irrespective of the results. Appendix A, Section A.2.3, provides an overview of the scientific papers (excluding abstracts and reviews) that emerged from the literature searches and seemed to be related to the toxicity and toxicokinetics of furan and methylfurans but upon examination were not included in the opinion as they did not meet the criteria stated above.
2.2.3. Benchmark dose analysis
Benchmark dose analysis was done according to EFSA guidance (EFSA Scientific Committee, 2017) and using the R package bmdModeling (see Appendix C).
2.3. Occurrence data submitted to EFSA
2.3.1. Data collection and validation
At the time of receiving the request for the scientific opinion from the European Commission, data on 2‐methylfuran and 3‐methylfuran were not available in the EFSA Chemical Occurrence database, and available data on furan were limited. The EFSA Evidence Management Unit (DATA Unit) initiated an ad hoc collection of data to compile occurrence data on furan, 2‐methylfuran and 3‐methylfuran levels in food.22 The European national food authorities and similar bodies, research institutions, academia and food business operators were invited to submit data. As 2,5‐dimethylfuran was not included in the initial request from the European Commission, the submission of occurrence data for 2,5‐dimethylfuran was not requested by EFSA.
The data submission to EFSA followed the requirements of the EFSA Guidance on Standard Sample Description for Food and Feed (EFSA, 2010a); occurrence data were managed following the EFSA standard operational procedures (SOPs) on ‘Data collection and validation’ and on ‘Data analysis of food consumption and occurrence data’. In accordance with Commission Recommendation 2007/196/EC13 on the monitoring of the presence of furan in foodstuffs, data providers were also requested to indicate whether commercial foodstuffs were analysed as purchased, disregarding any further preparation, or whether they were analysed as consumed after further preparation in the laboratory.
All data on furan and its methyl analogues in food available in the EFSA database by the end of November 2016 were used for the present assessment. Data received after this date were not included.
2.3.2. Data analysis
The data received were carefully evaluated by EFSA in view of cleaning and validating the data. Special attention was paid to the identification of duplicates and to the accuracy of different parameters such as ‘Analytical methods’, ‘Reporting unit’ and the coding of the different samples under FoodEx classification. The available information was also carefully analysed in order to identify those samples that were analysed either as purchased or as consumed. Upon identification of potential inconsistencies, data providers were contacted to provide further clarification. The outcome of the data analysis is shown in Section 3.2.1.
The left‐censored data (analytical data below the LOD/LOQ) were treated by the substitution method as recommended in the ‘Principles and Methods for the Risk Assessment of Chemicals in Food’ (WHO/IPCS, 2009). The same method is described in the EFSA scientific report ‘Management of left‐censored data in dietary exposure assessment of chemical substances’ (EFSA, 2010b), as an option in the treatment of left‐censored data. The guidance suggests that the lower bound (LB) and upper bound (UB) approach should be used for chemicals likely to be present in the food (e.g. naturally occurring contaminants, nutrients and mycotoxins). At the LB, results below the LOQ or LOD were replaced by zero; at the UB, the results below the LOD were replaced by the LOD and those below the LOQ were replaced by the value reported as LOQ. Additionally, a middle bound (MB) approach was used by assigning a value of LOD/2 or LOQ/2 to the left‐censored data. The use of different cut‐off values on the reported LOQs was also evaluated in order to reduce the uncertainty associated to the exposure estimations.
2.4. Food consumption data
The EFSA Comprehensive European Food Consumption Database (Comprehensive Database) provides a compilation of existing national information on food consumption at individual level. It was first built in 2010 (EFSA, 2011a; Huybrechts et al., 2011; Merten et al., 2011). Details on how the Comprehensive Database is used are published in the Guidance of EFSA (EFSA, 2011a). The latest version of the Comprehensive Database23 was used with subjects classified in different age classes as follows:
Infants: < 12 months old
Toddlers: ≥ 12 months to < 36 months old
Other children: ≥ 36 months to < 10 years old
Adolescents: ≥ 10 years to < 18 years old
Adults: ≥ 18 years to < 65 years old
Elderly: ≥ 65 years to < 75 years old
Very elderly: ≥ 75 years old.
Two additional surveys provided information on specific population groups: ‘Pregnant women’ (≥ 15 years to ≤ 45 years old; Latvia) and ‘Lactating women’ (≥ 28 years to ≤ 39 years old; Greece).
Overall, the food consumption data gathered by EFSA in the Comprehensive Database are the most complete and detailed data currently available in the EU. Consumption data were collected using single or repeated 24‐ or 48‐h dietary recalls or dietary records covering from 3 to 7 days per subject. As a result of the differences in the methods used for data collection, direct country‐to‐country comparisons can be misleading.
2.5. Food classification
Consumption data were classified according to the FoodEx classification system (EFSA, 2011b). FoodEx is a food classification system developed by EFSA in 2009 with the objective of simplifying the linkage between occurrence and food consumption data when assessing the exposure to hazardous substances. It contains 20 main food groups (first level), which are further divided into subgroups having 140 items at the second level, 1,261 items at the third level and reaching about 1,800 items (food names or generic food names) at the fourth level.
In 2011, a new version of FoodEx, named FoodEx2, was developed and is described in the scientific document ‘Report on the development of a Food Classification and Description System for exposure assessment and guidance on its implementation and use’ (EFSA, 2011c). The last release of FoodEx2 complements the previous hierarchical classification system of basic codes with more detailed food levels and gives the possibility of reporting additional information through the use of facets and facet descriptors such as processing or packaging (EFSA, 2015).
As the occurrence data were submitted to EFSA according to the FoodEx classification and only part of the food consumption data are currently coded according to the FoodEx2 classification, the occurrence data and the food consumption data were grouped at the most relevant level of the FoodEx classification. Where available, the FoodEx2 classification and its facet descriptors were also used to better identify consumption data referring to very specific foods such as commercially processed foods (e.g. canned and jarred foods) or toasted bread.
2.6. Exposure assessment
The CONTAM Panel considered that only chronic dietary exposure had to be assessed. As suggested by the EFSA WG on Food Consumption and Exposure (EFSA, 2011a), dietary surveys with only 1 day per subject were excluded from the current assessment because they are not adequate to assess repeated exposure. Similarly, subjects who participated only 1 day in the dietary studies, when the protocol prescribed more reporting days per individual, were also excluded from the chronic exposure assessment. When, for one particular country and age class, two different dietary surveys were available only the most recent one was used.
For calculating the chronic dietary exposure, food consumption and body weight data at the individual level were accessed in the Comprehensive Database. Occurrence data and consumption data were linked at the relevant FoodEx level (see also Section 2.5). For each individual of the selected surveys, the mean occurrence values of the different food samples collected (pooled European occurrence data) were combined with the average daily consumption of the corresponding food items, and the resulting exposures per food were summed in order to obtain the total chronic exposure at individual level (standardised by using the individual body weight). The mean and the 95th percentile of the individual exposures were subsequently calculated for each dietary survey and each age class separately.
Before linking the consumption data to the corresponding occurrence data, the following adjustments to the consumption data were made to reduce uncertainty and reach more accurate exposure estimates:
Consumption events for cereal‐based food for infants and young children were adjusted by a factor of 0.25 (when reconstituted with water) or 0.15 (when reconstituted with milk) when the eating occasions were reported as consumed (liquid) since the occurrence data also referred to the analysis of the food as purchased.
A number of consumption events reported for unspecified potatoes and potatoes products, unspecified alcoholic beverages or unspecified non‐alcoholic beverages was reclassified to a food category that was considered more appropriate for the assessment of furan.
A number of consumption events for boiled potatoes, main‐crop potatoes and new potatoes referred to powders, flakes, dried products or instant products. In view of the analysis on commercially processed products, classification as mashed potato powder was considered more appropriate for the assessment of furan.
Where a consumption event for a specific coffee beverage (e.g. cappuccino) was prepared from an instant powder, this consumption event was reclassified to the instant coffee beverages as this is considered more appropriate in the framework of the current assessment.
Legume vegetables, which are normally part of the vegetables and vegetable products, were reclassified to legumes (dried or green) in order to better match the available occurrence data for furan.
Human milk was not considered for the assessment due to lack of data available.
All analyses were made using the SAS Statistical Software (SAS enterprise guide 5.1).
2.7. Risk characterisation
The CONTAM Panel applied the general principles of the risk characterisation process for chemicals in food as described by WHO/IPCS (2009) and the relevant EFSA guidance documents (see Appendix A.3).
3. Assessment
The CONTAM Panel noted that the available information on 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran is limited and these compounds are therefore only described when information is available.
3.1. Hazard identification and characterisation
The CONTAM Panel noted that due to the high volatility of furan at room temperature, the handling of any furan preparation for treatment or sampling for analysis with/of furan is considered as critical. In some studies, it cannot be excluded that the outcome may have been influenced by losses of furan due to this effect. This issue is addressed in the uncertainty Section 3.5.
3.1.1. Toxicokinetics
The kinetics of furan and to a lesser extent also 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran have been studied in rodents, in hepatocyte cultures, in subcellular fractions and in various chemical model systems.
3.1.1.1. Absorption
A. Furan
Oral exposure
After administration of [2,5‐14C] furan (chemical and radiochemical purity both ≥ 99%) to male F344 rats at a dose level of 8 mg/kg bw (~ 0.37 MBq/kg bw), furan was quickly absorbed from the gastrointestinal (GI) tract to an extent of at least 80%. This estimate is based on excretion of radioactivity in exhaled air and urine and retention of radioactivity in tissues at 24 h post‐dosing. Radioactivity in faeces (22% of the dose) was not characterised and may have contained some unabsorbed furan (Burka et al., 1991).
Inhalation exposure
In dogs, approximately 95% of the inhaled furan is retained in the body and this percentage decreased to 91% when ventilation rate was increased. An inverse relationship was observed between retention and concentration in inhaled air (Egle and Gochberg, 1979a). Since absorption is virtually complete, the CONTAM Panel considered that the differences related to inhalation rate or concentration as reported by the study authors were of no biological significance.
B. Methylfurans
Inhalation exposure
In dogs, approximately 89% of the inhaled 2‐methylfuran is retained in the body and this percentage decreased to 83% when ventilation rate was increased (Egle and Gochberg, 1979b). For 2,5‐dimethylfuran, retention values of 63% decreasing to 57% were recorded with increasing ventilation rates. However, the limited set‐up of the experiments in this report and the high absorption of the substances do not allow firm conclusions on dependency on inhalation rate. The Panel considered that the slight differences related to inhalation rate were of no biological significance.
For 2‐methylfuran and 2,5‐dimethylfuran, the retention was not dependent on the concentration applied (Egle and Gochberg, 1979b).
3.1.1.2. Distribution
A. Furan
In vivo kinetics of furan in blood and liver were studied by Churchwell et al. (2015) in rats for up to 8 h after a single oral exposure to 0.92 mg (= 13.5 μmol) unlabelled furan/kg bw. The highest blood concentration (63 pmol/mL) was observed at 15 min post‐dosing (the 1st time point). In the liver, Cmax (547 pmol/g tissue) was observed 30 min post‐dosing. Over the observation period, liver concentrations were on average approximately six times higher than concentrations in blood, but concentrations in liver varied strongly between animals and between lobes. After 8 h, no furan could be detected in either blood or liver (LOD: ~ 0.75 pmol/mL in blood and ~ 1 pmol/g tissue in liver)
The distribution of furan‐related radioactivity retained after 24 h in various tissues in rats was studied after gavage dosing with 14C‐labelled furan (8 mg/kg bw (118 μmol/kg bw; 0.37 MBq/kg bw). The tissue distribution of total radioactivity (in percentage of the dose) was: liver 13%, kidney 0.45%, large intestine 0.13%, small intestine 0.15%, stomach (forestomach + glandular stomach) 0.09%, blood 0.42% and lung 0.02%. In total, the radioactivity in these tissues comprised 15 % of the dose after 24 h. Remaining tissues contained additional radioactivity approximating to 4% of the dose. No furan could be extracted from the blood, and from the liver, only 20% of the radioactivity could be extracted, but similar to blood this fraction did not contain furan. Seven days after treatment, the radioactivity had almost returned to the LOD. In a repeated dosing study (daily single dose for 8 days), 24 h after the last dose, concentrations in the liver were up by a factor of 4, and in blood and kidney up by a factor of 7 as compared to the concentrations after one dose (Burka et al., 1991).
After intraperitoneal (i.p.) administration of 4.1 mmol/kg bw furan to mice (ca. 280 mg/kg bw in sesame oil), furan levels were approximately 120, 490 and 50 nmol/g tissue in the kidney and approximately 190, 120 and 40 nmol/g tissue in the liver at 1, 2 or 5 h post‐dosing, respectively. No metabolites or other tissues were examined (Wiley et al., 1984).
B. Methylfurans
After i.p. administration of 4.1 mmol/kg bw 3‐methylfuran to mice (ca. 336 mg/kg bw in sesame oil), methylfuran concentrations were approximately 150, 380 and 50 nmol/g tissue in the kidney and approximately 240, 410 and 60 nmol/g tissue in the liver at 1, 2 or 5 h post‐dosing, respectively. No metabolites or other tissues were examined (Wiley et al., 1984).
Rats were dosed with 14C‐labelled 2‐methylfuran via i.p. injection at dose levels of 50–200 mg/kg bw (Ravindranath et al., 1986). At 12 h post‐dosing, total radioactivity was approx. four times higher in liver than in kidneys, and about 7 or 10 times higher in liver than in blood and lungs, respectively, at all dose levels. Peak levels of total radioactivity after a dose of 100 mg labelled 2‐methylfuran were reached after 2 h in kidneys, blood and lungs, but only after 8 h in the liver. Protein‐associated radioactivity reached a maximum value in all tissues after 8 h. Again, the liver had higher levels of total and non‐extractable radioactivity than the other three tissues. At 24 h post‐dosing, the concentration of total 14C had fallen to 58%, 67% or 83% of the peak levels in liver, kidneys and lungs.
The liver had the highest levels of 14C‐associated with DNA (equivalent to approximately 35 nmol 2‐methylfuran/mg DNA at an i.p. dose of 200 mg/kg). Minimal association of 14C with DNA was observed in the lungs. The peak of DNA associated 14C in the liver (32 nmol/mg DNA after 100 mg/kg bw) occurred at 1 h post‐dosing and in the kidneys at 4 h post‐dosing. At 24 h post‐dosing, the DNA‐associated 14C‐levels had dropped to 51% or 63% of the peak levels in liver and kidney, respectively (Ravindranath et al., 1986).
3.1.1.3. Metabolism
A. Furan
Figure 7 shows a graphical representation of the metabolites and adducts of furan.
Figure 7.
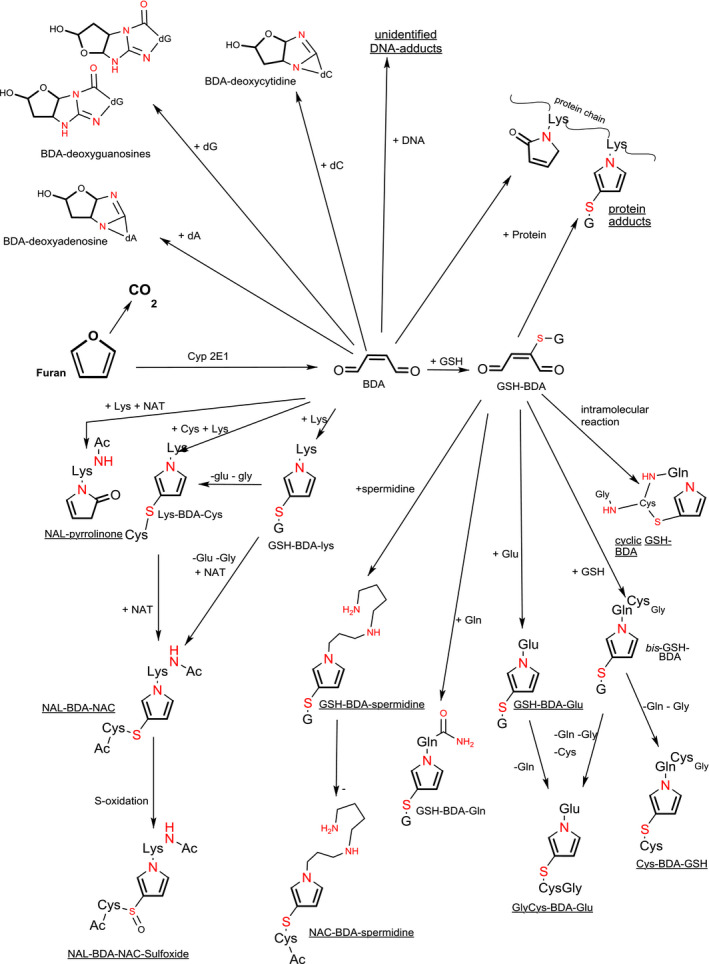
Metabolites and adducts of furan (compiled after Moro et al. (2012a,b), Peterson et al. (2011), Byrns et al. (2002), Neuwirth et al. (2012) and Phillips et al. (2014))
- Structural elements related to furan or BDA are in bold. Red‐coloured atoms belong to the abbreviated part of the molecules. Molecules with underlined names have been reported to occur in urine or bile after in vivo administration of furan to animals. Cys, Glu, Gln and Lys: cysteine, glutamate, glutamine and lysine; NAC: N‐acetylcysteine, NAL: N‐acetyllysine; NAT: N‐acetyltransferase, BDA: cis‐but‐2‐enedial; GSH or G: glutathione; dA deoxyadenosine; dG deoxyguanosine; dC deoxycytidine. Note that the Figure contains some representative structures especially from substances observed in vivo. Several structures that were only observed in vitro or in chemical systems have not been included in the presentation.
Oral administration of 14C‐labelled furan to rats resulted in an elimination of 26% of the dose as exhaled carbon dioxide. In addition, approximately 10% of the dose could not be extracted from liver tissue components (protein; DNA) and 20% of the dose was eliminated as urinary metabolites (Burka et al., 1991). The authors speculated that the carbon dioxide was formed after oxidative ring opening of furan. Although no direct evidence was available at that time, the authors suggested that similar to 2‐ and 3‐methylfuran (Ravindranath et al., 1984), furan itself could be converted into a reactive intermediate BDA and the formation of this dialdehyde was later confirmed in studies with rat liver microsomes using a trapping agent. No evidence for the formation of an epoxide was obtained, thus if such an intermediate was to play a role, it has to be very short‐lived (Chen et al., 1995).
B. Methylfurans
Ravindranath et al. (1984) showed that rat lung and liver microsomes can bioactivate 2‐ and 3‐methylfuran into reactive species. Using semicarbazide as a trapping agent, the reactive metabolites were identified as 3‐acetylacrolein (= 4‐oxopent‐2‐enal) for 2‐methylfuran and 2‐methylbut‐2‐enedial for 3‐methylfuran. Similar studies with 2‐methylfuran (Ravindranath and Boyd, 1985) showed that kidney microsomes can also bioactivate 2‐methylfuran. 3‐Acetylacrolein is very reactive towards microsomal protein and strongly inhibits bioactivation of 2‐methylfuran leading to the conclusion that 2‐methylfuran is a suicide substrate for cytochrome P450 (CYP). 2‐Methylfuran bioactivation by liver microsomes was strongly induced after pretreatment with phenobarbital, but not 3‐methylcholanthrene, and inhibited by piperonyl butoxide and even more by N‐octylimidazole (Ravindranath and Boyd, 1985).
Palmen and Evelo (1996) demonstrated that in aqueous incubates, 2‐methylfuran has no direct reactivity with glutathione (GSH). 2‐Methylfuran triggered partial depletion of GSH in haemolysates and also in intact human erythrocytes; in the latter case, only when co‐incubated with a (rat) microsomal bioactivating system. The authors concluded that 2‐methylfuran has to be bioactivated in order to deplete GSH, and that reactive metabolites can be generated in the erythrocyte cytosol, or in microsomal systems.
From general knowledge on biotransformation, it can be predicted that the alkylfurans may also be oxidised at the side‐chain resulting in the formation of furyl alcohols, which can be further oxidised to give the corresponding aldehydes and acids. However, these additional routes of metabolism and their consequences for the toxicity of the substances have not been studied.
C. Enzymes involved in furan biotransformation
As demonstrated using inducers or inhibitors of CYPs, the major contributor to the metabolism of furan is CYP2E1 (previously known as CYP‐J) (Garle and Fry, 1990; Carfagna et al., 1993; Parmar and Burka, 1993). A fair correlation was observed between the conversion of p‐nitrophenol, a standard substrate for CYP2E1 and the metabolism of furan in human microsomal incubations. Using various human recombinant CYPs, it was demonstrated that CYP2E1 was at least 5–10 times more active than CYP2D6, 3A4, 2J2, 1A2 and 2B6 forms. Minimal catalytic activity was observed for various other CYP forms (Gates et al., 2012). Parmar and Burka (1993) demonstrated that CYP‐catalysed biotransformation of furan results in inactivation of CYPs and that CYP2E1 is more heavily affected than other CYPs. A mechanistic explanation for CYP inactivation, based on quantum chemical methods involving the generation of furan‐epoxide and covalent binding of this intermediate to the haem centre and nucleophilic amino acid residues has been suggested by Taxak et al. (2013). From metabolism studies with 2‐methylfuran with inducers and inhibitors of CYP2E1 (Ravindranath and Boyd, 1985), it may be anticipated that also this alkylated furan and possibly also 3‐methylfuran are metabolised predominantly by CYP2E1, but contrary to furan, studies with purified CYP enzymes are not available for these two alkylfurans. Therefore, there is no direct evidence for an involvement of CYP2E1 in the biotransformation of 2‐ and 3‐methylfuran.
3.1.1.4. Binding of furan and methylfuran metabolites to tissue components
A. Binding to amino acids, glutathione and biogenic amines
Furan metabolites
BDA is a reactive α,β‐unsaturated dialdehyde, which has reactivity towards tissue components such as free amino acids and proteins. Garle and Fry (1990) demonstrated that rat liver microsomes can metabolise furan to a GSH‐reactive substance, which they did not further identify. Also, in hepatocytes in vitro, GSH can be depleted after exposure to furan (Carfagna et al., 1993), which is also indicative (but not definitive proof) of interaction of furan metabolites with this scavenger molecule. In chemical model systems, BDA reacted with GSH to form several complex (cyclic) structures, but in microsomal systems, only two bis‐GSH‐BDA adducts (an N‐alkyl‐3‐ and an N‐alkyl‐2‐thiopyrrole adduct) were observed (Chen et al., 1997). Additionally, a mono‐GSH‐BDA adduct has been observed in microsomal incubations with furan (Peterson et al., 2005). This mono‐GSH‐BDA adduct was observed in urine of rats treated with furan, indicating that also in vivo furan is converted to BDA. No urinary elimination of bis‐GSH‐BDA adducts was observed, but since these have a MW of around 660 g/mol, these could be preferentially excreted via the bile (Peterson et al., 2006). Metabolites found in rat bile, however, do not seem to include the bis‐GSH‐BDA adducts that were reported by Chen et al. (1997) and Peterson et al. (2006). Possible breakdown products of these have been reported such as cysteinylglycinyl‐BDA‐GSH, cysteinyl‐BDA‐GSH or cysteinylglycinyl‐BDA‐glutamate (Hamberger et al., 2010). Among the observed mono‐GSH adducts, cyclic reaction products were observed in which the thiol group had reacted with the double bond in BDA and a primary nitrogen had reacted with the two aldehydes, resulting in the formation of an N‐alkylated pyrrole or pyrrolinone moiety in microsomal incubates (Peterson et al., 2005) and in rat urine or bile (Peterson et al., 2006; Hamberger et al., 2010).
Apart from reactions with GSH, BDA has also been demonstrated to be reactive towards amino acids with primary amino groups in the side chain or/and to cysteine. As with the cyclic GSH conjugates these adducts contain N‐alkylated pyrrole or pyrrolinone moieties resulting e.g. from interaction with the amino groups from lysine or glutamine with the two aldehyde functions. Lysine‐BDA‐cysteine adducts as well as adducts of the N‐acetylated forms of these amino acids (N‐acetyllysinyl‐BDA‐N‐acetylcysteinyl (NAL‐BDA‐NAC)) have been demonstrated to be formed in chemical model systems (Chen et al., 1997). In concordance with this, BDA adducts of N‐acetylated lysine and cysteine were formed in human microsomal incubates (Gates et al., 2012) and could be detected in rat bile (Hamberger et al., 2010) after exposure in vivo. In chemical model systems, BDA together with GSH, can also react with other amines, such as asparagine, ornithine, putrescine, cadaverine, spermine or spermidine (Peterson et al., 2011). In contrast, in incubates of furan with rat hepatocytes next to mono‐GSH‐BDA, adducts of GSH‐BDA with ornithine (both at the Nδ‐ amino and the Nα‐amino; ~ 50% and ~ 25% of the total amount of amine adducts measured, respectively), lysine (Nε‐amino ~ 20%; Nα‐amino ~ 2%), spermidine (N1‐amino ~ 8%) and putrescine (< 1%) but no adducts to asparagine, cadaverine or spermine were found (Peterson et al., 2011). Lu et al. (2009) reported occurrence of a GSH‐BDA‐glutamine adduct in media of furan‐treated rat hepatocytes. Human hepatocytes (freshly isolated or cryopreserved) have higher or similar capacity to metabolise furan than rat and mouse hepatocytes (Kedderis and Held, 1996; Gates et al., 2014). In media of cryopreserved human and cryopreserved rat and mouse hepatocytes incubated with furan, the BDA adducts observed are similar as those reported by Peterson et al. (2011), but they vary in the amounts formed (Gates et al., 2014). Also, between hepatocytes from different human donors, there was a great difference in rate of formation of BDA adducts formation, despite similar levels of hepatocellular CYP2E1 activity (Gates et al., 2014). Therefore, these authors speculated that in humans alternative metabolic routes may be active to scavenge BDA (e.g. scavenging of the α,β‐unsaturated dialdehyde BDA by various CYPs) before it can react with e.g. GSH or amino acids. Of the amino‐adducts in urine from rats exposed to furan, next to NAC‐BDA‐lys, lower levels of NAC‐BDA‐spermidine, but no ornithine adducts could be found (Peterson et al., 2011). For the cysteine‐containing adducts also the corresponding sulfoxides have been reported in bile of rats and in urine of rats, mice and humans (Kellert et al., 2008b; Lu et al., 2009; Hamberger et al., 2010; Peterson et al., 2011; Grill et al., 2015). Lu and Peterson (2010) provided evidence that the cys‐BDA‐lys adduct is an important intermediate in the formation of the N‐acetylated, de‐aminated or sulfoxidated urinary metabolites of furan in vivo.
Urinary and biliary metabolites of furan could have arisen from reaction of BDA with GSH and free amino acids (cysteine, lysine and ornithine) in the cytosol (Kellert et al., 2008b; Hamberger et al., 2010). Based on the ratio of Nε‐lysine‐adducts over Nα‐lysine‐adducts, the lysine adducts are mainly formed with the Nε‐amino group, rather than with the Nα‐amino group. Based on changes in this ratio over time after furan exposure, Lu et al. (2009) argued that these lysine adducts arise from direct adduct formation with free lysine, especially shortly after exposure, but that at later points in time the observed adduct arises from degradation of BDA‐damaged proteins.
Methylfurans metabolites
Ravindranath et al. (1984) showed that reactive metabolites from 2‐ and 3‐methylfuran can bind (non‐extractable) to microsomal proteins. Cysteine was a very efficient scavenger of the reactive metabolite of 2‐methylfuran and to a lesser extent also GSH and semicarbazide (as a model compound). N‐acetylcysteine was less effective than GSH and lysine was a less effective scavenger than semicarbazide. Effective scavenging of acetylacrolein by cysteine was also observed in in vivo studies in rats exposed to 100 mg 2‐methylfuran /kg bw i.p. after treatment with various modulators of liver GSH synthesis (diethylmaleate (DEM), buthionine sulfoximine (BSO) and/or oxothiazolidinecarboxylate (OTZ)) (Ravindranath and Boyd, 1991).
Bile of rats dosed i.p. with 60 mg/kg bw of 2,5‐dimethylfuran contained a NAL‐dimethylpyrrole‐GSH conjugate after addition (ex vivo) of NAL to the bile sample. No GSH‐containing precursor for this metabolite could be detected, and also no other GSH‐containing conjugates with a putative reactive intermediate moiety were found in the bile, using liquid chromatography with tandem mass spectrometry (LC–MS/MS). The same pyrrole‐containing conjugate was also observed in microsomal incubates fortified with NAL and GSH and in a system in which 2,5‐dimethylfuran was chemically oxidised with subsequent treatment of the reaction products with NAL and GSH. However, no information was obtained as to the chemical nature of the reactive primary intermediate (Li et al., 2015). From similar in vitro studies but using 4‐bromobenzylamine instead of NAL, Wang et al. (2014b) concluded that the reactive intermediate formed from 2,5‐dimethylfuran by chemical or enzymatic oxidation could be (3Z)‐hex‐3‐ene‐2,5‐dione.
B. Binding to proteins
Furan
Parmar and Burka (1993) reported non‐extractable binding of 14C‐furan metabolites to proteins in microsomal incubates, with concomitant loss of enzymatic activities. The radioactivity became associated with CYP apoproteins as well as to the haem moieties. The level of binding depended on pretreatment of rats with a range of CYP‐activity modifiers before microsome isolation and on the presence of scavenging agents (e.g. primary amines and GSH).
Burka et al. (1991) observed that approximately 13% of the dose of 14C was still present in the liver 24 h after an oral dose of 8 mg/kg bw to rats. About 80% of this was non‐extractable and assumed to be covalently bound to tissue macromolecules, in particular to liver proteins.
Using immunoblotting on hepatocyte protein extracts, Lu et al. (2009) also demonstrated that the GSH‐BDA is sufficiently stable and reactive to become bound to proteins. The GSH‐BDA adduct may also migrate outside the cell and react with lysine or glutamine in cell culture media. Therefore, the authors further argued that BDA‐GSH adducts could also react with nucleophiles elsewhere in the tissue or in the body.
Phillips et al. (2014) studied the reactivity of BDA in the presence or absence of GSH against cytochrome c in aqueous solutions. With BDA alone, the predominant addition was 66 g/mol, which corresponds to a pyrrolinone‐modification of lysine (lys‐BDA) that may occur at various locations in the protein. To a much lower extent also additions of 48 (e.g. lys‐BDA‐lys cross‐link) or 84 g/mol (e.g. linear lys‐BDA‐mono‐adduct) were observed. Other tentatively identified adducts were with glutamine, histidine and arginine residues. Multiple adducts were also observed, (i.e. several BDA molecules reacted with different amino acid residues within one cytochrome c molecule) and adducts to adjacent lysine residues condensed further into bicyclic pyrrole‐pyrrolinone adducts. The addition of GSH to the incubates decreased the overall extent of adduct formation, and increased the mass of the protein with an addition of 355 g/mol. In the latter incubates, still some non‐GSH containing adducts were observed, but these were far less abundant than GSH‐BDA adducts. The 355 g/mol adducts were identified as GSH‐BDA‐lys adducts, again at several positions in the protein. The sites of lysine adduct formation were more selective for GSH‐BDA than for BDA itself (either in the absence or in presence of GSH). In the presence of GSH, double adducts (two times a 66 g/mol addition) and an addition of 84 g/mol were much less abundant than in the absence of GSH. The authors noted that cytochrome c has only two cysteine residues and both are already associated with the haem group of cytochrome c. Therefore, they considered that for cytochrome c the formation of a cys‐BDA‐lys crosslink is unlikely.
Nunes et al. (2016) treated rats daily with 0, 0.92, 2 or 4.4 mg furan/kg bw by gavage for 90, 180 or 360 days. In tryptic digests of liver histone H2B, a GSH‐BDA‐modified lysine 107 residue was observed, which was absent in the livers of the non‐exposed animals. The amount of GSH‐BDA‐modified peptide was dose‐related. No BDA‐modified (i.e. without the GSH moiety) peptides were seen. In recombinant human histone H2B homologue, incubated in vitro with BDA in the absence of GSH, no BDA‐modified lysine residue at position 108 (homologous to the rat lysine 107 residue) was observed. However, in incubates of histone octamer or free human histone H2B with BDA + GSH, the GSH‐BDA‐modified lysine residue 108 was found in a concentration‐dependent way. The authors suggested that GSH‐BDA has a higher reactivity towards proteins than BDA itself, and speculated that the higher reactivity of GSH‐BDA might also result in reactivity towards DNA bases. However, no data to support this are available and the results regarding reactivity towards proteins of free BDA as compared to GSH‐BDA are also opposite to those of Phillips et al. (2014; see above).
Methylfurans
2,5‐Dimethylfuran (0, 20, 35 or 70 mg/kg bw) was given to male mice by i.p. injection. Adduct formation was studied in proteins in liver, kidneys, heart, serum and lungs at different time‐points up to 36 h post‐dosing. 2,5‐Dimethylfuran adducts with cysteine‐ (at the SH group) and lysine‐ (at the Nε position) residues were found in all tissues examined but in liver proteins they were the most abundant. The occurrence of the adducts in liver proteins reached a peak at 2 h post dosing and decreased gradually thereafter. Adduct formation was dose‐related and also corresponded with serum alanine aminotransaminase (ALT) activity. The same lysine or cysteine adducts could be detected in supernatants of liver microsomes from non‐exposed mice, treated with 2,5‐dimethylfuran in the presence of lysine or cysteine. No adducts were found in the absence of reduced nicotinamide adenine dinucleotide phosphate, demonstrating that enzymatic activity is necessary for this bioactivation of 2,5‐dimethylfuran (Wang et al., 2015).
C. Binding to DNA
Formation of BDA‐nucleoside adducts has been extensively characterised. In chemical model systems, BDA reacts with the purified nucleosides deoxycytosine (dC), deoxyguanosine (dG) and deoxyadenosine (dA), but not deoxythymidine (dT), to form pairs of diastereomeric adducts. At pH = 6.5, BDA reacted only with dC, and this reaction was not pH dependent (Byrns et al., 2002). Reaction of BDA with dG and dA was pH‐dependent, in particular for dG, but at pH 7.0–7.5 there still was a preference of adduct formation of dC over the purines. At pH 8.0, BDA reaction rates with these nucleosides are in the order dG > dC >> dA (Byrns et al., 2002).
A general mechanism for the nucleoside adduct formation is proposed involving reaction of the C1 atom of BDA with the exocyclic N atom of the nucleoside (N4 of dC, N2 of dG and N6 of dA) followed by addition of the adjacent endocyclic nitrogen atoms (N3 of dC, N1 of dG and dA) at the BDA double bond and a subsequent ring closure of the added element to result in formation of an oxadiazabicyclo(3.3.0)‐octaimine ring system (Gingipalli and Dedon, 2001; Byrns et al., 2002). While adducts with dC are stable (half‐lives of 96 h at pH 2.8, 275 h at pH 7.4 and 13 h at pH 12), adducts with dG and dA rearrange quickly (half‐lives of 2.2–3.4 h for dG and 6 h for dA at pH 7.4; Gingipalli and Dedon, 2001; Byrns et al., 2002) and the initial ethano‐adducts of dG and dA decompose to form substituted etheno‐acetaldehyde adducts. These secondary adducts retain a reactive aldehyde with the potential to form crosslinks. The etheno‐acetaldehyde adducts also retain the BDA‐carbon atoms (Byrns et al., 2004, 2006).
Bohnert et al. (2004) provided additional kinetic and spectroscopic data of the reactions of cis‐ and trans‐BDA. Several products were observed after reaction of BDA (both isomers) with dA and dC at pH 7.4 in aqueous buffer systems. Apart from the mono‐adducts that were also found by Byrns et al. (2002), these included an additional pair of fluorescent compounds with similar high‐performance liquid chromatography (HPLC) retention times and with identical mass, each of which contains two molecules of BDA. The fluorescent adducts of dA contained the oxadiazabicyclo(3.3.0)‐octaimine ring system (see above), which was also found in the dC adducts. The reaction of trans‐BDA with dC produced two interchanging diastereomers at reaction rates that were nearly 10‐fold faster than the reaction with the cis‐isomer. However, it was noted that trans‐BDA is an endogenous oxidation product of deoxyribose and not a metabolite of furan.
The preferred reactivity of BDA with dC rather than dG and dA, as reported by Byrns et al. (2002) was also observed by Pluskota‐Karwatka et al. (2015). Similar oxadiazabiclyclooctaimine adducts were reported as before. Pluskota‐Karwatka et al. (2015) also mentioned that the BDA‐dC adduct can be converted into deoxyuridine by loss of the adducted BDA and subsequent deamination.
Sviatenko et al. (2012) applied quantum chemical computer assisted analysis to study the chemical interaction between BDA and dA. A similar technique was applied to study the interactions between BDA and dG (Sviatenko et al., 2014). For the reactions of BDA with dA and dG, the authors calculated that the primary reaction products would consist of four diastereoisomeric adducts for each nucleoside (for an example of these structure see Figure 7). Upon acidic or basic hydrolysis, the primary adducts of BDA‐dA and BDA dG, respectively, are converted into more stable secondary adducts that contain free aldehyde groups. Calculated UV and NMR spectra of the primary and secondary adducts as well as the predicted structures match well with experimental observations from published literature.
BDA‐dC and ‐dA adducts were also found in purified DNA treated with BDA and in DNA of Salmonella Typhimurium TA104 after exposure to BDA (Byrns et al., 2006).
Upon extensive extraction of rat liver homogenates and removal of the proteins, no radioactivity was associated with the residual liver DNA, not even after a (massive) i.p. dose of 243.5 MBq/kg bw (8 mg furan/kg bw; 2.07 GBq/mmol) of radioactive furan. This would indicate either absence or a low extent of binding of furan to DNA or an unstable nature of possible furan adducts (Burka et al., 1991).
Following administration by gavage of a single dose (0.1 or 2 mg/kg bw; 1.11 or 22.2 MBq/kg bw; 0.74 GBq/mmol) of [3,4‐14C]‐furan to F344 rats, a dose‐dependent increase of 14C was found in liver DNA at levels of 1.6 or 32.5 adducts/108 nucleotides at 2 h post‐dosing. The authors suggested that furan radioactivity was covalently bound to DNA. Metabolic incorporation into DNA bases during DNA synthesis occurred only to a minor extent. From differences in MS peaks, the authors concluded that the DNA adducts induced by furan in vivo were not the BDA‐dC, ‐dG or ‐dA adducts that have been reported in in vitro studies, but their identity could not further be established. Binding of furan to DNA was also reported in the kidneys at half the levels observed in the liver. Screening studies for the presence of DNA‐BDA cross‐links did not provide information that such cross‐links were formed, either in vivo or in BDA‐calf thymus incubates (Neuwirth et al., 2012).
The ability of BDA to form DNA adducts in the liver was investigated by Churchwell et al. (2015) in Fisher 344 rats administered furan by gavage (single doses ranging from 0.92 to 9.2 mg/kg). In addition, rats were gavaged (5 days per week) with 4.4 mg/kg bw for different lengths of time (45–360 days). Following both single dose or repeated exposure regimens, at 24 h after the last dose, livers were collected and analysed for the presence of BDA‐dC adducts by LC–MS/MS. The BDA‐dC levels did not increase (LOD: 0.4 adducts per 108 nucleotides) compared to the level present in liver DNA from control animals (1–2 adducts per 108 nucleotides).
In chemically defined systems, oligonucleotides containing a single‐furan‐modified nucleoside were selectively oxidised at the furan moiety to a reactive BDA‐like aldehyde. Fast and efficient formation of an interstrand DNA cross‐link was observed in the presence of a complementary DNA strand (Halila et al., 2005; Stevens and Madder, 2009). However, the BDA‐derived nucleoside adducts observed in vitro by Gingipalli and Dedon (2001) and Byrns et al. (2002, 2004) in single DNA strands have chemical structures that differ from the reactive BDA moieties that ultimately are formed upon oxidation of the furan‐modified oligonucleotides as described by Halila et al. (2005) and Stevens and Madder (2009). Therefore, the relevance for the in vivo toxicity of furan of the observed interstrand cross‐linking in oligonucleotides in the experimental designs from Halila et al. (2005) and Stevens and Madder (2009) is not clear. On the other hand, according to Stevens and Madder (2009), the adducts in the complementary oligonucleotide strands resemble the adducts formed by direct reaction of BDA with nucleotides.
3.1.1.5. Elimination
A. Furan
After a single oral dose of 8 mg [14C]‐furan/kg bw (0.37 MBq/kg bw) in rats, 11% of the dose was exhaled as unchanged furan within the first hour post‐dosing and an additional 3% was exhaled during the next 23 h (Burka et al., 1991). Exhaled carbon dioxide represented 26% of the dose. Urine contained 20% of the dose and comprised more than 10 peaks representing mixtures of metabolites, but these could not be further identified. The faeces contained 22% of the dose (not characterised). After 8 days of repeated dosing, the fraction of the dose eliminated via urine, increased from 20% to 33%. No change was observed in the extent of faecal elimination. Radioactivity from the liver was eliminated with a half‐life of 1.8 days. For blood and kidney, no half‐lives could be estimated. Hamberger et al. (2010) reported that in rats orally dosed with 5 mg furan/kg bw, several metabolites could be detected in bile. Some of these reached a peak within 1 h of dosing; others continued to increase in the bile during the 4 h observation period. The presence of parent furan was not studied. No indication was given or could be obtained as to the fraction of the dose that was eliminated via this route.
In a study by Churchwell et al. (2015), over the 8 h period of observation, the concentration in blood decreased log‐linearly with a half‐life of 1.3 h. The elimination in liver showed 2 log‐linear phases with half‐lives of 0.55 and 0.62 h, respectively. However, it is debatable whether this small difference has any biological meaning, given the high variability in furan concentrations in the liver.
A rapid elimination of furan from blood was also reported by Becalski et al. (2013), who gavage‐dosed rats with 2.9 mg/kg bw deuterated furan in corn oil. At 0.5 h post‐dosing, furan blood levels were approximately 13–42 ng/g and at 1.5 h post‐dosing, these levels had dropped to approximately 5 ng/g. Over the next 4.5 h, the furan blood concentration declined gradually to background level.
3.1.1.6. Biomarkers/biomonitoring
Kellert et al. (2008b) argued that estimation of human exposure to furan from concentration data in food is not reliable, because of the volatility of furan. They advocated a biomarker approach for furan exposure estimation. They treated rats with furan (0 or 40 mg/kg bw; n = 5) and analysed the urinary metabolite profile by principal component analysis. They observed a treatment‐related increase in mass spectrometer peaks for 13 compounds. For seven of these, the increase was statistically significant. Five urinary metabolites could be identified, all of them contained BDA in the form of a pyrrole nucleus. The pyrrole was formed by condensation of BDA with the Nε‐amino‐group of lysine or the Nα‐amino‐group of the glutamic acid moiety in GSH. In four of these metabolites, the double bond in BDA had also reacted with GSH‐thiol. One of these four products was an S‐methylated β‐lyase cleavage product. Two of the metabolites found (NAL‐BDA and NAL‐BDA‐NAC) were also observed in urine from control animals. The authors could not explain this observation, but argued that presence of furan in the feed was not a likely source.
The adducts of BDA with GSH or the cysteine‐BDA‐lysine crosslink adducts and downstream reaction products thereof (N‐acetylated; S‐oxidated, β‐lyase cleaved followed by S‐methylation) have been proposed as possible biomarkers for furan exposure (Kellert et al., 2008b; Lu and Peterson, 2010). Kellert et al. (2008b) further argued that the pattern of lysine‐containing adducts as eliminated via urine indicated that these are formed from BDA interaction with proteins, rather than with free amino acids, thereby being indicative of markers of past exposure, because shortly after exposure only a decrease in urinary excretion rate was observed for several GSH‐BDA conjugates, while the elimination of the lysine adducts did not show a decrease between 24 and 48 h post‐exposure.
Becalski et al. (2013) demonstrated that furan in animal feed does not contribute to the furan body burden in rats. Administration of 120 ng/kg bw of deuterated furan to rats did not result in detectable levels of labelled furan in blood or liver, while native furan was found at 0.09–0.18 ng/g blood or 0.04–0.12 ng/g liver. The labelled dose corresponded to 20 ng/animal. This is an exposure similar to that resulting from the consumption of control feed with a background furan content of approximately 2 ng/g feed. Based on this, the authors speculated that furan was formed from an endogenous source. If substantiated, endogenous formation of furan is likely to have implications regarding biomonitoring.
Grill et al. (2015) analysed the urine of smoking and non‐smoking human subjects (one cohort from Minnesota, one from Shanghai and one from Singapore) for the presence of furan metabolites. A BDA‐Nε‐lysine adduct and its N‐acetylated conjugate were observed and also NAC‐BDA‐lys together with the corresponding sulfoxide. The BDA‐lys adducts were the most abundant metabolites and were clearly elevated in urine from smokers as compared to non‐smokers in the Shanghai and Singapore cohorts, but not in the Minnesota cohort. Interference with other components in the urine hampered its quantification in 50% of the urine samples, notably all from USA, while the samples where the metabolite could be properly quantified were from Shanghai or Singapore. A similar rise in NAC‐BDA‐lys adduct and its sulfoxide was observed in smokers’ urine. For the USA samples, the concentration of NAC‐BDA‐lys sulfoxide was much higher than for the Asian samples. The urinary level of NAC‐BDA‐lys dropped significantly during the first 20 days after cessation of smoking, but remained higher than the level in urine from non‐smokers during the following 60 days, which the authors considered to be an indication of slow release from furan‐damaged proteins. In urine from furan‐treated rats and mice, the metabolic pattern for furan metabolites was different from that in humans: in rats and mice also NAL‐BDA‐NAC and their sulfoxides were observed, which were not seen in human urine. In mice urine, the prime metabolite was NAC‐BDA, while in rats the prime metabolite was the NAL‐BDA adduct. The non‐acetylated BDA‐lys adduct was not detectable in either mice or rat urine.
Mochalski and Unterkofler (2016) determined 19 volatile substances among which furan, 2‐methylfuran and 3‐methylfuran in the urine of 10 male and 9 female volunteers. Urine samples were collected in glass vials, which were immediately closed after urine production. Volatiles were analysed using headspace microextraction and GC‐SRI‐TOF‐MS. The three furans were found in all samples. Analytical results are given in Table 1.
Table 1.
Levels of furan, 2‐methylfuran and 3‐methylfuran in urine as reported by Mochalski and Unterkofler (2016)
| Compound | Concentration in urine range (median) | Limit of detection (nmol/L) | |
|---|---|---|---|
| nmol/L | nmol/mmol creatinine | ||
| Furan | 1.06–28.8 (11.3) | 0.1–4.75 (0.87) | 1.0 |
| 2‐Methylfuran | 1.74–6.88 (4.06) | 0.2–0.93 (0.34) | 0.1 |
| 3‐Methylfuran | 1.03–5.59 (2.97) | 0.12–0.92 (0.24) | 0.1 |
For furan, the levels ranged from 1.06 to 28.8 nmol/L (median 11.3 nmol/L) or from 0.1 to 4.75 nmol/mmol creatinine (median 0.87 nmol/mmol). For 2‐methylfuran and 3‐methylfuran, the concentrations ranged from 1.74 to 6.88 nmol/L (median 4.06 nmol/L) and from 1.03 to 5.59 nmol/L (median 2.97 nmol/L), respectively. No information was provided as to the possible source of the furans found, apart from the statement that all volunteers were non‐smokers. It was not stated whether there was a gender difference in urinary excretion of furans.
Two limited studies measuring urinary and plasma levels of furan were identified (Jun et al., 2008; Lee et al., 2009); however, the reported levels were inconsistent and therefore of limited value for this opinion.
3.1.1.7. Physiologically based pharmacokinetic (PBPK) modelling
Based on data from inhalation absorption studies and data on rate of metabolism of furan in hepatocyte suspensions, Kedderis et al. (1993) developed a PBPK model for rats. The model predicted that for an oral bolus dose of 8 mg/kg bw in rats, 84% of the dose would be metabolised and 16% of the dose would be exhaled unchanged, which is similar to the extent of metabolism and exhalation of furan in rats as observed by Burka et al. (1991) after the same oral bolus dose. The model also predicted that in rats for furan blood concentrations of 1 μM, the rate‐limiting step in the metabolism is the hepatic blood flow, rather than the maximum metabolic capacity of the liver. From the data presented, it can be calculated that the metabolism becomes saturated (90% of Vmax) at a blood concentration of 3.6 μM using a Km of 0.4 μM as determined in rat hepatocyte suspensions.
This model was modified to predict liver dosimetry in mice, rats and humans (Kedderis and Held, 1996). Metabolic parameters used were for Vmax 48, 18 or 19–44 nmol/h per 106 isolated hepatocytes, and for Km values 1.0, 0.4 or 2.1–3.3 μM, for mice, rats and humans (3 samples), respectively. The modified models predicted absorption of furan from inhaled air containing 10 ppm (27.8 mg/m3) for 4 h to be 4.1, 1.4 or 0.4 mg/kg bw, resulting in a liver exposure to furan metabolites of 1,075, 480 or 168 μM for mice, rats and humans. According to the humanised model, for air concentrations up to 300 ppm (830 mg/m3) the exposure of the liver to furan metabolites is not dependent on Vmax but only on hepatic blood flow, that restricts the availability of substrate for biotransformation. The authors argue that induction status of CYP2E1 is therefore not relevant for individual differences in the rate of hepatic furan bioactivation in humans. Kedderis (1997) also applied the humanised model for the estimation of hepatic furan bioactivation for an oral furan bolus dose of 2 mg/kg bw. For this route, the extent of bioactivation of furan would be flow‐limited and independent on the CYP2E1 induction status of the liver. However, the modelling conditions for the latter prediction were not reported, especially not with respect to the furan absorption rate from the GI tract. Nevertheless, the conclusion that the metabolic clearance rate of furan depends on the delivery of furan to the liver, rather than on hepatic CYP2E1 activity, was supported by a generic PBPK model (Poulin and Krishnan, 1998).
3.1.1.8. Summary and discussion
After oral exposure of rats, furan is rapidly and extensively absorbed from the GI tract and distributed throughout the body. At 24 h after exposure, major tissues where furan‐related material can be found are liver and kidneys and to a lesser extent lungs and intestines. Furan is extensively metabolised in the body and from a dose of 8 mg/kg bw to the rat, only 11% was exhaled as unchanged furan. The remainder was eliminated as carbon dioxide or as polar metabolites in urine or bile/faeces. At 24 h post‐dosing, no unchanged furan could be detected in liver or blood of rats, and this is in line with the very short half‐life of the substance in these tissues: approximately 40 min in liver and approximately 1.3 h in blood. Although at 24 h post‐dosing some furan‐related material could be extracted from liver tissue, 80% of the residual material in the liver appeared to be non‐extractable. Extensive non‐extractable association of furan and 2‐methylfuran in the liver was also reported after i.p. administration. For 2‐methyl and 3‐methylfuran, data from in vitro studies are available that show that they may become associated (non‐extractable) with lung and liver microsomal proteins.
No oral studies have been performed with 2‐ or 3‐methylfuran or with 2,5‐dimethylfuran to address their kinetics in animals, but inhalation studies in dogs with furan and 2‐methylfuran and to a lesser extent also with 2,5‐dimethylfuran indicate that these three alkylated furans will be absorbed from the gastrointestinal tract but the extent of this is unclear.
As indicated above, the major route for elimination of furan is via metabolic conversion. By analogy with 2‐methylfuran and 3‐methylfuran, it was suggested that this metabolism would involve opening of the furan ring to result in the formation of a very reactive cis‐enedial intermediate, i.e. BDA. This metabolite has never been directly measured. However, results with trapping agents and the identification of urinary and biliary metabolites, in comparison to reaction products of BDA with various biomacromolecules or amino acids in purified chemical model systems, provide strong evidence that BDA is indeed a prime reactive intermediate in the metabolism of furan. The corresponding reactive metabolic intermediates of 2‐ and 3‐methylfuran are the cis‐enedials 3‐acetyl acrolein (4‐oxopent‐2‐enal) and 2‐methylbut‐2‐enedial, respectively. Also for 2,5‐dimethylfuran, ring opening results in the formation of a reactive intermediate, 3(Z)‐hexene‐2,5‐dione. Induction‐inhibition studies, but also studies with various recombinant CYPs indicate that CYP2E1 is by far the most important (but not exclusively) monooxygenase that catalyses the conversion of furan into BDA.
As a result of its reactivity, BDA reacts very quickly with amino acids and GSH. The same applies for the reactive intermediate formed from 2,5‐dimethylfuran. For the intermediates of 2‐methyl‐ and 3‐methylfuran, reactivity towards amino acids and GSH has not been studied in great depth. However, it may be anticipated based on structural similarity and similar reactivity towards scavengers such as semicarbazide, cysteine, NAC and microsomal proteins, that these will react with tissue components in a similar way to the primary metabolites of furan and 2,5‐dimethylfuran. A plethora of low molecular weight reaction products has been reported. Reactive (alkyl)furan metabolites may react with:
amino acids (in particular lysine): reacts with the two terminal aldehyde groups of BDA to give a cyclic pyrrole derivative. This reaction may involve the side chain terminal amino group of the amino acid or the amino acid in α‐position to the carboxylic acid. For these reaction products, the corresponding N‐acetylated forms have been reported. BDA can also react with free amino groups in proteins (in particular from lysine). The same reactions have been reported for the cis‐enedione formed from 2,5‐dimethylfuran.
GSH or cysteine: react directly with the double bond in these α,β‐unsaturated carbonyls at their thiol group. The latter reaction does not take away the reactivity of the two aldehydes, and consequentially, these GSH or cysteine adducts may react further, either with the amino groups from free amino acids or with amino groups protruding from protein amino acid residues. Since cysteine or GSH have several reactive functional groups, also bicyclic adducts have been reported, in which the thiol group is connected to the BDA double bond and the free amino group has reacted with the two BDA aldehydes, resulting in a pyrrole ring.
Reactivity of BDA towards biogenic amines has also been reported.
As a result of the reactivity towards amino acids, GSH, biogenic amines and proteins, the urinary excretion is biphasic: in first instance, the main excretion of urinary furan metabolites consists of metabolites that were formed from BDA and free amino acids or GSH within the cell, but later on the urinary excretion of furan‐related material is a result from protein turn‐over that liberates the furan‐damaged amino acid residues. Also, the elimination of furan (radioactivity) from, e.g. liver is biphasic with a rapid elimination of non‐bound radioactivity and a slower and more prolonged elimination of protein‐bound furan.
More intriguing is the possibility of furan metabolites reacting with DNA, which could be an important factor in the genotoxicity and formation of tumours. Studies in chemical model systems have demonstrated that BDA can readily react with dC and to a lesser extent dG and dA, but not dT. Again, a plethora of reaction products was obtained. While BDA‐dC is rather stable (with a half‐life of about 300 h at pH = 7.4), the adducts of dG and dA rearrange rather quickly with half‐lives of 2.2–3.4 h for dG and 6 h for dA at physiological pH. Nevertheless, these adducts retain the BDA‐carbon atoms and this should reveal their presence in studies in which radioactive furan is given to animals in vivo. When purified DNA or bacterial cultures are exposed to BDA, DNA adduct formation can be demonstrated.
Although it is clear that BDA has direct reactivity towards DNA, it is not clear from the available data that BDA‐DNA adducts will be formed after exposure of rats to furan in vivo. In earlier in vivo studies with rats, at 24 h post‐dosing, furan radioactivity in liver was associated with protein but not with DNA, not even when a dose as high as 243.5 MBq/kg bw (8 mg/kg; 2.07 GBq/mmol) was administered. In a study in which rats were treated with furan, using accelerator mass spectrometry, the presence of DNA adducts in the liver (1.6 or 32 adducts per 108 nucleotides) and kidneys (0.5 or 13 adducts per 108 nucleotides) at 2 h post‐dosing with 0.1 or 2 mg radiolabelled furan/kg bw (1.11 or 22.2 MBq/kg bw; 0.74 GBq/mmol), respectively, was demonstrated. However, the adducts observed could not be identified as either BDA‐dC, BDA‐dG or BDA‐dA adducts. The possibility was suggested that the observed adducts were related to DNA‐DNA or DNA‐protein cross‐links, but no definitive evidence for this was presented. In another study in rats, no increase in BDA‐dC levels (LOD: 0.4 adducts per 108 nucleotides) above the level present in commercial control DNA and in liver DNA from control animals (~ 1–2 adducts per 108 nucleotides) was observed in livers of rats following administration after a single dose of furan (ranging from 0.92 to 9.2 mg/kg), as well as after repeated dosing (5 days per week) with 4.4 mg/kg bw for 45–360 days, as determined using LC–MS/MS. In any case, the potency of covalent binding to DNA in vivo is not very high or the reaction products are very short‐lived. The data further suggest that BDA reacts readily with protein and non‐protein amino and thiol residues in liver, thereby restricting access to DNA.
Based on the studies in rats, and on furan metabolism rates measured in rodent and human hepatocytes, a PBPK model has been constructed for rats, mice and humans. Applying this model, the rate of exhalation of furan in rats after an oral bolus dose was well described. The model further predicts that after an oral bolus dose of 2 mg/kg bw the exposure of the liver to furan metabolites depends on the blood flow to the liver rather than the metabolic capacity of the liver since at the blood concentrations reached, the metabolic capacity of the liver is far from saturated. The efficient metabolism of furan will result in an effective first‐pass elimination and will therefore limit the systemic exposure to furan after oral intake. The bioactivation of furan depends less on the liver CYP2E1 activity (the main furan‐metabolising enzyme) than on liver blood flow. Activity of GSH conjugating enzymes may be of less relevance, because BDA has a high rate of spontaneous reactivity towards GSH.
It has been argued that estimation of furan exposure based on concentration in foods is not reliable, and biomarkers of exposure might be an alternative. However, from the available kinetics data, it is not possible to conclude on a methodology to reliably estimate exposure to furan in humans from the excretion of furan or furan metabolites. The main reason for this is that there is no information as to the quantity of metabolites in human excreta, not for total amounts of metabolites and also not for individual metabolites. Even for rats, this information is not available, since virtually all metabolism studies have focused only on qualitative determination and identification of furan metabolites in urine or bile and did not pay attention to quantitative aspects of metabolism and excretion. Nevertheless, several studies have suggested that three metabolites might be good candidates for biomonitoring furan exposure: NAL‐BDA‐NAC, its sulfoxidation product and the sulfoxidated α‐keto‐derivative of NAC‐BDA‐lys. The urinary metabolite pattern may also provide information on the level of past exposure, because several metabolites seem to emerge from protein breakdown rather than direct adduct formation to small biomolecules (amino acids; GSH), which are probably more rapidly eliminated. However, further work is needed to validate the use of these metabolites as biomarkers of exposure. A few studies have also tried to monitor unmetabolised furan in human blood and urine. In many samples furan was not detectable. In addition, the volatility of furan requires very stringent treatment of samples, because otherwise artificially low furan levels might result. Given the short half‐life of furan, at least in rats, but probably also in humans, looking at unchanged furan would only provide an indication for very recent exposure to furan. It is further noted that furan may also be formed endogenously, which may need to be taken into account.
Concluding remarks
After oral administration, furan is extensively oxidised in the liver to form a reactive intermediate metabolite BDA. BDA may be further metabolised to carbon dioxide. Alternatively, it can spontaneously react with free amino‐ or thiol groups in low molecular weight tissue components (e.g. GSH, cysteine, lysine and other amino acids and biogenic amines). The resulting reaction products can be eliminated from the body via urine or bile. A study with oral dosing indicated that approx. 55% of an oral gavage dose may be eliminated via these two routes, predominantly within the first 24 h after exposure. The rate of metabolism is determined by the delivery of furan to the liver, and not by its enzymatic capacity. From the data available, it may be assumed that the fraction of an oral dose that is converted to BDA will be dose dependent, but this has not been studied. The relative extent of formation and elimination of the BDA secondary reaction products has not been investigated, but several of these (e.g. NAL‐BDA‐NAC) have been suggested to be useful biomarkers of exposure. BDA can form adducts with protein (including histones) after exposure to furan in vivo, especially with lysine, and form adducts with free nucleosides (in particular with deoxyribose guanosine, ‐cytosine and –adenine) and DNA adducts in purified DNA and bacteria. In one in vivo study, binding of furan to DNA has been reported, but this result could not be reproduced in another study. The DNA adducts found were different from the BDA adducts formed with the free nucleosides and could not be identified. No DNA‐DNA cross‐links have been reported.
In addition, for 2‐methyl‐, 3‐methyl‐ and 2,5‐dimethylfuran, oxidative opening of the furan ring has been reported with similar reactivity towards amino acids and GSH. For these alkylfurans, also oxidation of the methyl‐ring substituent groups may be expected, but there are no quantitative or qualitative studies available that address these routes of biotransformation.
The relevance of these findings for the toxicity and genotoxicity of furan and the alkylfurans will be discussed in Sections 3.1.2.4 and 3.1.4.
3.1.2. Toxicity in experimental animals
3.1.2.1. Acute toxicity (single exposure)
In adult male Sprague–Dawley rats and male Swiss mice, furan was injected i.p., while another group of adult male Swiss mice inhaled air enriched with furan vapour (Egle and Gochberg, 1979a). The i.p. LD50 values were 5.2 mg/kg bw in rats and 7.0 mg/kg bw in mice. The LC50 value as a vapour was 0.12 mg/L in mice. In the lungs, fluid accumulation and inflammatory reactions were found after inhalation.
Wiley et al. (1984) injected furan i.p. in male ICR mice. Twenty‐four hours after a dose of 347 mg/kg bw, the animals were sacrificed. Histopathological inspection revealed slight proximal tubular necrosis of the kidneys and massive centrilobular necrosis of the liver. Dosage of 3‐methylfuran (254 mg/kg bw) claimed to be ‘equimolar’ was reported not to lead to significant damage in these organs. The CONTAM Panel noted that the dose levels mentioned in the abstract and in the text are inconsistent and not equimolar. The CONTAM Panel noted that the dose level for furan by far exceeded the LD50 value reported by Egle and Gochberg (1979a).
Bronchiolar necrosis was observed 8 h after i.p. application of 100–200 mg/kg bw of 3‐methylfuran to NIH Swiss mice (age and gender not specified) (Boyd et al., 1978). Gammal et al. (1984) administered 213 or 254 mg/kg bw of 3‐methylfuran i.p. to adult male ICR mice. After 24 h, severe bronchiolar necrosis was observed.
3.1.2.2. Subacute and subchronic toxicity (< 90 days duration)
A. Furan
Several short‐term toxicity studies with furan were identified, which are described below and summarised in Table 2.
Table 2.
Effects of orally administered furan (by gavage) in acute, subacute and subchronic (&le 90 days) studies in experimental animals
| Species (number of animals per group) | Dosage (mg/kg bw) | Duration/time of observation | Outcomea | NOAEL (LOAEL) in mg/kg bw per dayb | Reference |
|---|---|---|---|---|---|
|
B6C3F1/CrIBR male mouse (5) Fischer 344/CrIBR male rat (5) |
Single 30 (rats), 50 (mice) | 12, 24, 48 h, 4 and 8 days |
Increased plasma liver enzymes after 12 h and later LI increased after 48 h |
One dose level only | Wilson et al. (1992) |
|
B6C3F1/CrIBR male mouse (5) Fischer 344/CrIBR male rat (5) |
Single, 15, 27, 39. 50 (mice) 0, 8, 15, 22, 30 (rats) | 24 h | Increased plasma liver enzymes at 27 mg/kg bw (male mice) and 15 mg/kg bw (male rats) | (15; mice) (8; rats) | |
|
B6C3F1/CrIBR male mouse (5) Fischer 344/CrIBR male and female rat (5) |
30 (rats), 50 (mice) | 6 weeks, 5 days a week | Hepatic necrosis and inflammation, bile duct hyperplasia and metaplasia in rats | One dose level only | |
|
B6C3F1/CrIBR male mouse (5) Fischer 344/CrIBR male rat (5) |
30 (rats), 50 (mice) | 1, 3 and 6 weeks, 5 days a week | increased hepatocellular LI (proliferation) | One dose level only | |
|
B6C3F1 male and female mouse (5) Fischer 344 male and female rat (5) |
0, 5, 10, 20, 40 or 80 mg (male rats) 0, 10, 20, 40, 80 and 160 (mice and female rats) |
16 days, 5 days a week |
Rats (observation): mottled and enlarged livers Mice: no organ changes |
Range‐finding study | NTP (1993) |
|
B6C3F1 male and female mouse (10) Fischer 344 male and female rat (10) |
0, 4, 8, 15, 30 or 60 (rats and female mice) 0, 2, 4, 8, 15 or 30 (male mice) |
13 weeks, 5 days a week | Increase in relative and absolute liver weight, histopathological changes in the liver | (2; male mice) (4; rats and female mice) | NTP (1993) |
| Fischer 344 male rat (3–6) | 45 | 6 weeks, 5 days a week | Reduced liver weight, cholangiofibrosis, biliary metaplasia, biliary cirrhosis | One dose level only | Sirica et al. (1994b) |
| B6C3F1 female mouse (6–11) | 4, 8 and 15 | 3 weeks, 5 days a week | Increase in serum ALT, SDH, apoptotic index, subcapsular inflammation | 4 | Fransson‐Steen et al. (1997) |
| 15 | 3–5 days a week over 2 weeks – 2 days (15 days in total) | Increase in liver weight, serum ALT, SDH and bile acids | One dose level only | ||
| B6C3F1 female mouse (15) | 0.5, 1, 2 and 4 | 3 weeks, 5 days a week | Increased serum ALT, subcapsular inflammation | (0.5) | Moser et al. (2009) |
| B6C3F1 male mouse (3–11) | 2, 4, 8 and 15 | 28 days, 5 days a week | Hepatic necrosis, hepatocellular proliferation | 4 | Cordelli et al. (2010) |
| Sprague–Dawley Crl CD1 BR male rat (3–5) | 30 | 8 h, 1, 3, 7, 10, 12 and 20 days, and 1, 2 and 3 months, 5 days a week | Hepatic necrosis, hepatocellular and biliary proliferation | one dose level only | Hickling et al. (2010b) |
| Fischer 344 male rat (3–5) | 0.1, 0.5, 2 | 28 days, 5 days a week | Increased DNA synthesis, increased subcapsular mitosis, apoptosis and inflammation | (0.1) | Mally et al. (2010) |
| Wistar juvenile male rat (8) | 2, 4 and 8 | 90 days, 7 days a week | Increased hepatic TNF‐α | (2) | Selmanoglu et al. (2012) |
| Fischer 344 male and female rat (12) | 0.03, 0.12, 0.5, 2 and 8 | 90 days, 5 days a week | Increase in serum T4 in males c | 0.03 | Gill et al. (2010) |
| B6C3F1 male and female mouse (16) | 0.03, 0.12, 0.5, 2 and 8 | 90 days, 5 days a week | Distinct histopathological changes, changes in clinical blood parameters (BUN and phosphorous in serum) c | 0.12 | Gill et al. (2011) |
| Wistar male rat (8) | 2, 4 and 8 | 90 days, 7 days a week | Islet of Langerhans congestion | 4 | Karacaoglu et al. (2012) |
| Wistar juvenile male rat (8) | 2, 4 and 8 | 90 days, 7 days a week | Decrease in relative thymus weight, histological changes in the thymus | 2 | Kockaya et al. (2012) |
| Sprague–Dawley male (10) | 16 | 30 daysd | Changes in blood cells, blood cell phagocytic activity, serum immune parameters, oxidative stress in the spleen, changes in splenic lymphoid cells | One dose level only | Alam et al. (2017) |
ALP: alkaline phosphatase; ALT: alanine aminotransaminase; BUN: blood urea nitrogen; LDL: low‐density lipoprotein; LI: labelling index; LOAEL: lowest‐observed‐adverse‐effect level; NOAEL: no‐observed‐adverse‐effect level; SDH: sorbitol dehydrogenase; TNF: tumour necrosis factor.
Most sensitive endpoint given in bold.
LOAEL (in italics) or NOAEL (in bold) for the most sensitive endpoint affected schedule.
See comments under ‘Summary’.
Information on dose regimen not available.
Wilson et al. (1992) published results from treatment of male mice or male and female rats at the highest NTP bioassay dose in each species (15 mg/kg bw per day or 8 mg/kg bw per day, respectively) 5 days a week by gavage. In male and female rats, necrosis and inflammation were observed along the subcapsular visceral surface of the left or caudate liver lobes after one week. A similar picture was evident in mice after 3 weeks. After 6 weeks of furan treatment, male and female rats exhibited bile duct hyperplasia and metaplasia in the areas of fibrosis along the subcapsular visceral surface of the left or caudate liver lobes. This was not observed in mice. The fold increase in hepatocyte labelling index (DNA synthesis) in treated animals relative to the combined controls measured at weeks 1, 3 and 6 was 25‐, 12‐ and 3‐fold in male mice, 3‐, 9‐ and 7‐fold in male rats, and 13‐, 11‐ and 16‐fold in female rats, respectively.
In a study carried out for the NTP (1993), adult F344/N rats and B6C3F1 mice were treated with furan, dissolved in corn oil, by gavage. Groups of five male rats received doses of 0, 5, 10, 20, 40 or 80 mg of furan/kg bw per day and groups of five female rats and five mice of each sex received doses of 0, 10, 20, 40, 80 and 160 mg/kg bw per day over 16 days, 5 days a week. All male and female mice and female rats which had received 160 mg/kg bw, all male and female rats and all male and four out of five female mice which had received 80 mg/kg bw per day, and three out of five male mice which had received 40 mg/kg bw per day died by day 8. All furan‐treated animals showed motor inactivity or reduced activity. Final mean body weights of male rats which had received 20 mg/kg bw per day and of male and female rats which had received 40 mg/kg bw were significantly lower than in the controls. Final mean body weights of male mice which had received 10 or 20 mg/kg bw per day were significantly greater than controls. At necropsy, mottled and enlarged livers were observed in male rats after 20, 40 or 80 mg/kg bw and in female rats after 40, 80 or 160 mg/kg bw. No treatment‐related lesions were observed in mice.
In a second experiment carried out for the NTP (1993), groups of 10 rats of each sex and groups of 10 female mice received 0, 4, 8, 15, 30 or 60 mg of furan per kg bw in corn oil by gavage 5 days per week for 13 weeks. Groups of 10 male mice received 0, 2, 4, 8, 15 or 30 mg of furan per kg bw in corn oil by gavage 5 days per week for 13 weeks. Nine male and four female rats that received 60 mg/kg bw died before the end of the experiment. There were no treatment‐related deaths in mice. Significantly lower final mean body weights were observed in male rats that received 15 or 30 mg/kg bw, in female rats that received 60 mg/kg bw and in male mice that received 30 mg/kg bw compared to controls. Relative and absolute liver weights were increased in all furan‐treated groups in both species, as were relative and absolute kidney weights in all furan‐treated groups of female rats. Thymus weights were decreased in all furan‐treated groups of rats. In rats, furan administration resulted in bile duct hyperplasia, cholangiofibrosis, cytomegaly and degeneration of hepatocytes, and nodular hyperplasia of hepatocytes. Kidney tubular dilatation and necrosis were observed in rats that received 30 or 60 mg/kg bw. In rats, exposed to 60 mg/kg bw furan, thymic atrophy and testicular or ovarian atrophy were also observed. In mice, similar hepatic lesions were found in all furan‐treated dose groups, while bile duct hyperplasia and cholangiofibrosis were observed in groups of mice receiving 30 or 60 mg/kg bw.
The CONTAM Panel noted that even at the lowest dose levels (4 mg/kg bw in rats; 2 mg/kg bw in mice), significant hepatic lesions were found.
Sirica et al. (1994b) applied 45 mg furan/kg bw by gavage to young adult male Fischer 344 rats over 6 weeks, 5 days a week. Furan reduced liver weight and led to pronounced cholangiofibrosis with associated metaplasia mainly found in the right liver lobe together with biliary cirrhosis. In contrast to normal cholangiolar structures, the metaplastic cholangia‐like cells were surrounded by a basement membrane positive for both laminin and type IV collagen. The cells were stained positive for gamma‐glutamyl transpeptidase and cytokeratin 8, while cytokeratin 19 was strongly expressed in all of the biliary epithelial cells, but in just some of the metaplastic cells.
In a first experiment, 50‐day‐old female B6C3F1 mice received furan by daily gavage, 5 days per week over a period of 3 weeks (15 days of treatment) at doses of 4, 8 or 15 mg/kg bw per day (Fransson‐Steen et al., 1997). In a second experiment, 15 mg/kg bw furan was initially applied by gavage for three consecutive days followed by 2 weeks with five consecutive days per week and over a final week with two consecutive days. Thus, animals were exposed for five and two consecutive days immediately prior to necropsy in the first and second experiments, respectively. In the second experiment, a separate group received 100 mg/kg of the non‐specific CYP‐inhibitor, aminobenzotriazole (ABT), 7 days per week beginning 5 days before the start of furan dosing.
In the first experiment, ALT and sorbitol dehydrogenase24 (SDH) were measured in the serum and the apoptotic index in the liver, and all were significantly increased at 8 mg/kg bw and above. At the two highest dose levels, histopathological examination revealed minor subcapsular inflammation as well as sporadic necrosis. In the second experiment, 15 mg/kg bw furan resulted in a significant increase in liver weight, while serum levels of ALT, SDH and total bile acids were increased at this dose level. Co‐treatment with ABT attenuated these effects while it caused a mild lipid vacuolisation of the liver. The labelling index of hepatocellular nuclei was increased significantly at 15 mg/kg bw, the apoptotic index at 8 and 15 mg/kg bw. Co‐treatment with ABT abrogated these effects completely.
Moser et al. (2009) treated female B6C3F1 mice with 0.5 1, 2 and 4 mg furan/kg bw by gavage over 3 weeks, 5 days per week. There was a dose‐dependent and significant hepatotoxicity (measured as serum ALT) at and above 0.5 mg/kg bw and a mild subcapsular inflammation of the liver at the lowest dose. Cell proliferation as measured by 5‐bromo‐2‐deoxyuridine (BrdU) incorporation was enhanced at 8 mg/kg bw applied in a long‐term carcinogenicity study.
Cordelli et al. (2010) gave furan by daily gavage over 28 days, 5 days a week, to male B6C3F1 mice at dose levels of 2, 4, 8 and 15 mg/kg bw per day. The animals were sacrificed and the livers were removed for further investigation, 24 h after the last gavage. Histopathological inspection revealed some necrosis and regenerative hyperplasia (at 8 and 15 mg/kg bw) and an increase in apoptotic figures (at 15 mg/kg bw). Furthermore, at the highest dose level, an increase in nuclear ploidy was found. DNA methylation and the number of γ‐H2AX foci were unchanged. At the highest dose level, overexpression of DNA damage response‐related genes was noted. At a very high single dose level of 250 mg/kg bw diffuse alteration of liver parenchyma with areas of necrosis and steatosis in surrounding hepatocytes was found 3 h after application.
Hickling et al. (2010b) administered furan to adult male Sprague–Dawley Crl CD1 BR rats by gavage in corn oil at 30 mg/kg bw (five daily doses per week, per gavage in corn oil). After 8 h, 1, 3, 7, 10, 12 and 20 days, and 1, 2 and 3 months of treatment, animals were sacrificed and livers were examined. The administration of furan caused centrilobular and subcapsular injury (necrosis) and subsequent proliferation of hepatocytes. Later, a rapid proliferation of ductular cells expanding into the parenchymal area accompanied by a subtype of liver fibroblasts occurred. The ductular structures either differentiated into hepatocytes or transformed into ductular cholangiofibrotic structures.
Mally et al. (2010) treated male Fischer 344 rats orally (gavage) at doses of 0.1, 0.5 and 2 mg/kg bw for 28 days (5 days per week), with an interim sacrifice after 5 days of treatment. An off‐dose recovery group (0 and 2 mg/kg bw dose groups only) was kept for additional 2 weeks (recovery period) after the end of the 4‐week treatment period. Statistically significant proliferative changes were restricted to rats treated with furan at 2 mg/kg bw. An approximate twofold and threefold increase in BrdU labelling above background was observed in the low‐ (0.1 mg/kg bw) and mid‐ (0.5 mg/kg bw) dose group, respectively. Furthermore, the authors observed an increased mitotic activity in the subcapsular regions of the left and caudate lobe accompanied by an occasional apoptotic cell and inflammatory foci with accumulation of mononuclear cells.
Selmanoglu et al. (2012) treated juvenile male Wistar rats by gavage with 2, 4 or 8 mg/kg bw per day over 90 days. A 7 days per week dose schedule was applied (Karacaoğlu, 2016, E‐mail communication, 25 October 2016). Reduced liver weights and significant changes in serum ALT, alkaline phosphatase (ALP) and low‐density lipoprotein (LDL) were observed at 4 mg/kg bw and above. At the lowest dose level (2 mg/kg bw), hepatic tumour necrosis factor (TNF)‐α was significantly increased and numerous histopathological alterations of the liver as well as morphological changes of the glomerula (kidney) were found.
Gill et al. (2010) treated male and female Fischer 344 rats over 90 days by gavage with 0.03, 0.12, 0.5, 2.0 and 8.0 mg/kg bw per day, 5 days per week. The liver was found to be the major target organ with changes in serum enzymes, increased liver weight and various histological lesions (pattern and severity changing with the dose level). There was a significant increase in serum thyroxine (T4) and triiodothyronine (T3) in males. The CONTAM Panel noted that there was no clear dose response for changes in T3. The increase in thyroid hormones was not accompanied by histological thyroid changes. At a dose level above 0.5 mg/kg bw, changes in clinical biochemistry and haematological parameters were observed, while mild histopathological alterations in the liver were observed at a dose level of > 0.12 mg/kg bw per day and at 0.12 mg/kg bw per day and above in the caudate lobe. At this dose level, serum T4 was increased significantly in male rats, and further increased with increasing furan doses.
Gill et al. (2011) published a study where male and female B6C3F1 mice were treated by gavage over 90 days with 0.03, 0.12, 0.5, 2.0 and 8.0 mg/kg bw per day, 5 days per week. Serum ALT was increased at the highest dose level and liver weights were increased at the highest dose in females only. At the two highest dose levels, histological changes in the liver were observed in both sexes and also at 0.5 mg/kg bw in males. For the kidney, clinical parameters were also altered, but they were not accompanied by histological changes. At 0.5 mg/kg bw, blood urea nitrogen (BUN) was decreased in both sexes. The CONTAM Panel noted that the levels reported for controls (10.96–11.45 g/L) are not in agreement with published data for BUN in mice (e.g. 80–330 mg/L; University of Minnesota Reference Values, https://www.ahc.umn.edu/rar/refvalues.html). In male mice, Gill et al. (2011) found an increase in blood phosphorous (controls: 0.8–0.9 mmol/L) at a dose of 0.12 mg/kg bw of furan. The CONTAM Panel noted that phosphorous measurement in serum is highly vulnerable to blood sample handling. Likewise, haemolysis can cause an up to 30% increase in phosphorous measurements (UK Association for Clinical Biochemistry, 201225). Upon request, the corresponding author stated that the dimension given in the paper was erroneous and should read mmol/L instead of g/L (Gill, 2017, e‐mail communication, 31 January 2017).
Karacaoglu et al. (2012) treated male Wistar rats with 2, 4 and 8 mg/kg bw by gavage over 90 days. A 7 days per week dose schedule was applied (Karacaoğlu, 2016, e‐mail communication, 25 October 2016). At the highest dose, there was significant congestion in the islets of Langerhans in the pancreas, while the organ weights of adrenal glands and pancreas were unchanged.
Kockaya et al. (2012) treated young male Wistar rats orally (gavage) with doses of 2, 4 and 8 mg/kg bw per day for 90 days. A 7 days per week dose schedule was applied (Karacaoğlu, 2016, E‐mail communication, 25 October 2016). At the end of the experiment, thymus of the rats were examined morphologically, histopathologically and immunohistochemically. At the two highest doses, relative thymus weights were decreased significantly. A histopathological examination revealed various changes in the thymus at these dose levels including enlargement of interstitial connective tissue, decrease in thymic lymphocyte counts and haemorrhage.
Alam et al. (2017) treated male Sprague–Dawley rats by gavage with 16 mg/kg bw over 30 days. Furan treatment resulted in significantly elevated red blood cell count, haemoglobin, white blood cell count, lymphocytes and granulocytes and reduced phagocytic parameters in white blood cells. It decreased serum total protein, albumin, globulin, immunoglobulin M (IgM), IgG and interleukin 4 (IL4), with a significant increase in serum TNFα and 8‐oxodeoxyguanosine. In the spleen, furan decreased the GSH content and GST activity and increased the malondialdehyde levels. Histopathologically, furan led to a moderate depletion in splenic lymphoid cells.
B. Methylfurans
The CONTAM Panel identified three short‐term toxicity studies on methylfurans, which are described below. All the no‐observed‐adverse‐effect levels (NOAEL) mentioned in this section are based on limited data and should therefore be considered as preliminary.
Gill et al. (2014) treated male Fischer 344 rats (n = 10/group) by gavage with 0.4, 1.5, 3, 6, 12 and 25 mg 2‐methylfuran/kg bw per day over 28 days, 5 days a week. Mild histological hepatic lesions such as apoptotic hepatocytes, abnormally pigment Kupffer cells and inflammatory infiltrations were observed at 1.5 mg/kg bw and increased in severity with increasing dose levels. Furthermore, serum cholesterol and T3, T4 were also increased at this dose level. In the thyroid gland, no histopathological changes were found. The CONTAM panel noted that the dose responses for T3 and cholesterol were rather flat, while there was a dose‐dependent statistically significant increase of T4 at lower doses which disappeared at higher doses (inconsistent dose response). Thus, a NOAEL of 0.4 mg/kg bw per day could be identified by the CONTAM Panel. A slight increase in peripheral eosinophils observed at this dose level could not be confirmed at higher dosage.
Gill et al. (2015) treated male Fischer 344 rats (n = 10/group) by gavage with 0.1, 0.3, 1.5, 3, 6, 12 and 25 mg 3‐methylfuran/kg bw per day over 28 days, 5 days a week. Statistically significant histological changes of the liver were noted at 1.5 mg/kg bw per day and gross changes were noted beginning at 3 mg/kg bw per day. At the highest dose, alterations in serum enzyme activities indicating hepatic toxicity were observed. There was also a significant increase in serum T3 and T4 without histopathological changes in the thyroid. From these data, a NOAEL of 0.3 mg/kg bw per day could be derived for hepatic histopathological changes in the rat.
Gill et al. (2017, in preparation) treated male and female Fischer 344 rats (n = 12/group) by gavage daily (5 days per week) for 90 days with 3‐methylfuran at final doses of 0.0 0.02, 0.075, 0.25, 1.0 and 4.0 mg/kg bw per day. Based on a severity score, statistically significant differences were found for some types of hepatic lesions (zone 1 hepatocytes with eosinophilic, i.e. altered cytoplasm and hyperchromasia) in the 1.0 mg/kg bw male rats while in females, statistical differences appeared at the 4.0 mg/kg bw dose. Furthermore, the authors reported a number of hepatic histopathological observations which were not statistically significant when based on a severity score. These included very mild lesions characterised by hyperchromasia of chromatin with enlarged nucleoli and increased cytoplasmic eosinophilia in the 0.25 mg/kg bw dose group, in the left and caudate lobes of the males. Small numbers of cells were pale or had mild microvesicular steatosis. The numbers of hepatic mitoses were slightly increased and slight proliferation of oval cells was noted in the left and caudate lobes. In females at the same dose level (0.25 mg/kg bw), similar mild changes and slight oval cell proliferation were observed. There was no statistical analysis or long‐term comparison with controls. The extent and severity of the alterations increased with increasing doses. In the 4.0 mg/kg bw group, massive nodular changes of the liver were observed. Serum ALP and bilirubin were significantly enhanced in males at 1.0 mg/kg bw and in females et 4.0 mg/kg bw. In the highest dose group, serum parameters indicative of diminished renal function, i.e. serum creatinine (at 4.0 mg/kg bw) and uric acid (at 1.0 mg/kg bw) were also increased in males. Based on these findings, including statistically non‐significant histopathological observations in the liver, the authors suggested a NOAEL of 0.075 mg/kg bw per day. Based on benchmark dose modelling, the authors also derived BMDL10 values of 0.08 mg/kg bw for liver focal necrosis and oval cell hyperplasia in the left lobe in males, and of 0.05–0.17 mg/kg bw for liver lesions (oval cell hyperplasia and karyocytomegaly in the left lobe) in females.
C. Observed effects for methylfurans compared to other alkylfurans
The CONTAM Panel reviewed the literature on the toxicity of several alkylated furans in order to obtain more information on possible structure–activity relationships with respect to the pattern of adverse effects. Appendix B gives an overview of the reported effects for different alkylfurans. Such information was expected to be possibly instrumental in addressing the question if certain alkylated furans could eventually be considered together with furan. For this purpose, also non‐oral routes of exposure were considered, since the number of oral studies with alkylated furans is very limited and it was expected that such studies might provide more insight into MoAs of the compounds of interest.
For 2‐ and 3‐methylfuran, pulmonary and hepatic toxicity was shown. Pulmonary toxicity was observed upon inhalation but also upon i.p. application. There is also indication for kidney toxicity of 3‐methylfuran after 90 days exposure. 2‐ and 3‐Ethylfuran exhibited renal, pulmonary and hepatic toxicity in different species. For 3‐pentylfuran, adverse renal and hepatic but not pulmonary effects were reported (no inhalation studies). Thus, no clear structure–effect relationship for the alkylated furans with respect to the pattern of adverse effects/the target organs could be derived.
D. Summary
Taken together, in experiments of a duration of up to 90 days, furan is strongly hepatotoxic and moderately nephrotoxic in rodents when applied by oral gavage. Rats seem to be more sensitive towards furan than mice. Furan leads to characteristic changes in serum markers related to hepatotoxicity as well as severe histopathological damage in the liver. These effects are particularly pronounced in centrilobular and subcapsular areas and can be attenuated by suppression of CYP activity. After 90 days, distinct histopathological changes and significant increases in serum thyroid hormones were seen in male rats even after a daily dose of 0.12 mg/kg bw, given 5 days a week. For effects of this type, a NOAEL of 0.03 mg/kg bw per day for furan was derived by the authors. The observations in B6C3F1 mice were not considered sufficient by the CONTAM Panel for dose–response analysis since they were not quantified (histopathology).
2‐ and 3‐Methylfuran exerted toxicity in the liver, kidney and lung in rodents. After oral application to rats, furan, 2‐methylfuran and 3‐methylfuran led to a similar overall picture of liver toxicity. Although the studies on methylfurans are not sufficiently robust to identify a specific reference point for the individual compounds, they show that the toxic potencies are in the same order of magnitude, whether expressed as dose or molarity.
3.1.2.3. Long‐term toxicity (including carcinogenicity)
There have been two long‐term toxicity studies carried out under the NTP, one in 1993 and a follow‐up study in 2015. The findings of these studies are described below and the details are shown in Tables 3–6. The first NTP study was carried out on male and female Fischer 344/N rats and B6C3F1 mice. Furan was given by gavage in corn oil 5 days per week for 104 weeks. The doses in rats were 0, 2, 4 and 8 and in mice were 0, 8 and 15 mg/kg bw. There was clear evidence of carcinogenicity and survival decreasing with increasing dose.
Table 3.
Incidence of neoplastic lesions and statistical analysis results derived from male and female F344/N rats and male F344/N Nctre rats from 2‐year carcinogenicity assays with furan
| Lesiona | Dosage (mg/kg bw per day) | Incidenceb | Reference | |
|---|---|---|---|---|
| Male | Female | |||
| Cholangiocarcinoma before re‐evaluationc |
0 2 4 8 |
0/50 43/50*** 48/50*** 49/50*** |
0/50 49/50*** 50/50*** 48/50*** |
NTP (1993), Maronpot et al. (1991) |
| Hepatocellular adenoma/carcinoma |
0 2 4 8 |
1/50 5/50 22/50*** 35/50*** |
0/50 2/50 4/50 8/50** |
NTP (1993) |
| Interstitial cell adenoma of testesd |
0 2 4 8 |
41/50 36/50 39/50 43/50 |
NTP (1993) | |
| Hepatocellular carcinoma |
0 2 4 8 |
0/50 1/50 6/50* 18/50*** |
0/50 0/50 0/50 1/50 |
Maronpot et al. (1991) |
| Hepatocellular adenoma |
0 2 4 8 |
1/50 4/50 18/50*** 27/50*** |
0/50 2/50 4/50 7/50** |
Maronpot et al. (1991) |
| Mononuclear cell leukaemia |
0 2 4 8 |
8/50 11/50 17/50* 25/50*** |
8/50 9/50 17/50* 21/50** |
NTP (1993) |
| Malignant mesothelioma |
0 2 4 8 |
1/50 1/50 3/50 3/50 |
– | NTP (1993) |
| Mononuclear cell leukaemia |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
47/150 56/150 36/100 44/100* 29/50*** 18/50 27/50*** 28/50*** |
– | NCTR (2015) |
| Malignant mesothelioma of the tunica vaginalis |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
6/150 8/150 1/100 2/100 0/50 2/50 2/50 6/50* |
– | NCTR (2015) |
| Follicular cell adenoma |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
0/150 2/150 5/99 0/100 1/50 1/49 0/50 0/50 |
– | NCTR (2015) |
| C‐cell adenoma |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
11/150 15/150 9/99 13/100 7/50 8/49 1/50 8/50* |
– | NCTR (2015) |
| C‐cell adenoma or carcinoma |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
12/150 21/150 11/99 14/100 8/50 9/49 1/50 9/50* |
– | NCTR (2015) |
bw: body weight.
Only the major tumour sites and/or those showing significant effects at the lower dose are listed.
*Equals significant at > 0.05; **equals significant at > 0.01; ***equals significant at > 0.001.
Re‐examination of 23 of these samples at a later date revealed that only in the highest dose cholangiocarcinomas were present.
This was the terminology used in the paper, however, this is equivalent to mesotheliomas of the tunica vaginalis
Von Tungeln et al. (2017) specified that the F344/N NCTR substrain differs from the Charles River substrain used in the previous furan bioassay (NTP, 1993). However, the NCTR F344/N sub‐strain has tumourigenic responses that are comparable to those of other F344/N substrains used in previously reported carcinogenicity bioassays such as acrylamide.
Table 6.
Summary of non‐neoplastic lesions in male rats taken from NCTR (2015)
| Lesion | Dosage (mg/kg bw per day) | Incidence/time of assessmenta | ||
|---|---|---|---|---|
| 36 weeks | 60 weeks | 104 weeks | ||
| Cholangiofibrosis |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
0/20 0/20 0/20 0/20 0/20 6/20*** 17/20*** 19/20*** |
0/20 0/10 0/10 0/10 0/10 10/10*** 10/10*** 8/10*** |
0/149 0/150 0/99 1/100 38/50*** 49/49*** 47/50*** 49/49*** |
| Oval cell hyperplasia |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
0/20 0/20 0/20 0/20 0/20 0/20 1/20 16/20*** |
0/20 0/10 0/10 0/10 0/10 0/10 3/10* 7/10*** |
14/149 15/150 10/99 8/100 7/50 6/49 14/50*** 33/49*** |
| Mixed cell foci |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
0/20 0/20 0/20 0/20 1/20 0/20 1/20 5/20* |
1/20 0/10 1/10 0/10 1/10 1/10 3/10 6/10** |
7/149 7/150 6/99 3/100 5/50 3/49 6/50 13/49*** |
| Biliary tract hyperplasia |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
1/20 1/20 0/20 1/20 0/20 0/20 6/20* 19/20*** |
7/20 3/10 1/10 2/10 1/10 2/10 3/10 8/10* |
89/149 86/150 5,999 56/100 29/50 25/49 32/50 43/49*** |
| Bile duct subcapsular hyperplasia |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
0/20 0/20 0/20 0/20 1/20 12/20*** 19/20*** 20/20*** |
0/20 0/10 0/10 0/10 1/10 9/10*** 10/10*** 7/10*** |
– |
| Hepatocyte hypertrophy |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
0/20 0/20 0/20 0/20 1/20 0/20 2/20 12/20*** |
0/20 0/10 0/10 0/10 0/10 5/10** 4/10** 8/10*** |
– |
| Periportal cytoplasmic alteration |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
0/20 0/20 0/20 0/20 0/20 0/20 7/20** 19/20*** |
0/20 0/10 0/10 1/10 0/10 1/10 7/10*** 8/10*** |
– |
| Subcapsular fibrosis |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
0/20 0/20 0/20 0/20 3/20 14/20*** 20/20*** 20/20*** |
0/20 0/10 0/10 0/10 4/10** 9/10** 10/10** 7/10*** |
– |
| Subcapsular chronic inflammation |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
0/20 0/20 0/20 0/20 2/20 14/20*** 20/20*** 20/20*** |
0/20 0/10 0/10 0/10 8/10*** 9/10** 10/10** 7/10*** |
– |
| Subcapsular pigmentation |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
0/20 0/20 0/20 0/20 2/20 14/20*** 20/20*** 20/20*** |
0/20 0/10 0/10 0/10 8/10*** 9/10** 10/10** 7/10*** |
– |
| Basophilic foci |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
– | – |
28/149 16/150 19/99 18/100 11/50 13/49 10/50 21/49*** |
| Regenerative hyperplasia |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
– | – |
0/149 1/150 1/99 2/100 1/50 1/49 7/50*** 12/49*** |
| Cytoplasmic vacuolisation |
0 0.02 0.044 0.092 0.2 0.44 0.92 2 |
– | – |
23/149 23/150 14/99 19/100 12/50 18/49*** 23/50*** 37/49*** |
bw: body weight.
*Equals significant at > 0.05; **equals significant at > 0.01; ***equals significant at > 0.001.
In the rat study, cholangiocarcinomas were found in both males and females at all doses of furan (NTP, 1993). Hepatocellular carcinomas were observed at 4 and 8 mg/kg bw in the males but none occurred in the female rats (Maronpot et al., 1991). Male rats developed hepatocellular adenomas at 4 and 8 mg/kg bw but these only occurred in the females at the highest dose (Maronpot et al., 1991). When combined (hepatocellular adenoma/carcinoma), the results were significant for males at the two higher doses and for females at the highest dose only (Table 3). Mononuclear cell leukaemia was observed in both males and females at the two higher doses. A high incidence of mesothelioma of the tunica vaginalis (including in the control) was also reported.26 Overall, males are more sensitive than female rats. Other non‐malignant effects were observed as shown in Tables 3 and 6.
Histologically, it is difficult to distinguish between cholangiocarcinoma and cholangiofibrosis and a consensus report by Thoolen et al. (2010) provided clear guidelines for a standardised nomenclature and differential diagnosis of microscopic lesions in the hepatobiliary system of commonly used laboratory rodents. Samples (23 from 2 mg/kg bw and 6 from 8 mg/kg bw) from the NTP study (1993) were re‐examined following the guidelines described by Thoolen et al. (2010) and it is now clear that cholangiocarcinomas were only observed at the highest dose (NCTR, 2015; Von Tungeln et al., 2017) while cholangiofibrosis was observed at the lower doses.
As the first study used exposure to furan that induced effects at all doses, a second study was undertaken to determine the dose–response relationship in male rats using a significantly lower dose range, 0, 0.02, 0.044, 0.092, 0.2, 0.44, 0.92 and 2 mg/kg bw per day, 5 days per week for 104 weeks. Interim analyses were carried out at 36 and 60 weeks. The doses were chosen to ensure a range of concentrations that would give a lower tumour incidence in order to establish the dose response relationship. The group sizes in this study were not equal with higher numbers of animals in the lower dose groups to maximise the chance of detecting tumours in these low exposed groups. There was a dose‐related decrease in survival but this did not reach significance.
In the NCTR study, mononuclear cell leukaemia was observed in all groups, including the control. There was a statistically significant increase at doses of 0.092 mg/kg bw and above. However, the incidence was within the historical control range for this endpoint and rat strain within the same laboratory (NCTR, 2015). The Fischer 344 rat is known to develop a high incidence of mononuclear cell leukaemia (Caldwell, 1999). This tumour type is unique to the rat and occurs in aged, untreated F‐344 rats at a high and variable rate. It is common only to the Fischer 344 strain. Significant increases in C‐cell adenoma or carcinoma, and malignant mesothelioma of the tunica vaginalis were noted at the top dose of 2 mg/kg bw. Bearing in mind the high spontaneous incidence of mesotheliomas, C‐cell adenoma and carcinoma and of the observed leukaemias in the Fischer 344 rat specifically the relevance of these tumours to human risk assessment is questionable. There were no treatment‐related neoplastic changes at 36 or 60 weeks. There were no significant increases in hepatocellular adenoma or carcinoma observed in the NCTR study (Von Tungeln et al., 2017). The cholangiocarcinomas, observed at the highest dose (8 mg/kg bw per day) in the NTP (1993) study after reclassification, were not seen in the NCTR study with dose levels up to 2 mg/kg bw per day (2015; Von Tungeln et al., 2017).
Of the non‐neoplastic effects, the most sensitive endpoint was cholangiofibrosis with increases observed from 0.2 mg/kg bw and above. The dose response was non‐linear showing a low incidence (1%) at 0.092 mg/kg bw and a high incidence (≥ 76%) from 0.2 mg/kg bw and above. Cholangiofibrosis was also noted at 36 and 60 weeks with the severity increasing with time from minimal (36 weeks) to minimal to mild (60 weeks). Other significant changes observed were biliary tract hyperplasia, cytoplasmic vacuolisation of oval cells and regenerative hypertrophy (Table 3). Interim analyses at 36 and 60 weeks showed similar responses with the significance at lower doses increasing with the time (NCTR, 2015; Von Tungeln et al., 2017).
In a separate study, male rats were exposed to 30 mg/kg bw furan by gavage (n = 50) in corn oil 5 days per week for 13 weeks and then followed up for 9, 15 and 24 months (NTP, 1993). Cholangiocarcinomas were observed in all rats at all time points. Hepatocellular carcinoma was first noted at 15 months.
In the equivalent mouse studies, both male and female mice developed hepatocellular adenomas and adenomas/carcinomas at both doses tested (8 and 15 mg/kg bw; Table 4). Male mice also showed hepatocellular carcinomas at both doses (Table 4; NTP, 1993). Carcinomas were only observed in female mice at the higher dose. In a later study, female mice were treated by gavage with 0, 0.5, 1.0, 2.0, 4.0 and 8 mg/kg bw per day furan in corn oil 5 days per week for 104 weeks. Hepatocellular adenomas were observed at the two highest doses and hepatocellular carcinomas only at 8 mg/kg bw. The combined hepatocellular adenoma/carcinoma incidence was significant at 4 and 8 mg/kg bw (Moser et al., 2009).
Table 4.
Tumour incidence and statistical analysis results derived from B6C3F1 mice from 2‐year carcinogenicity assays with furan
| Tumoura | Dosage (mg/kg bw per day) | Incidenceb | Reference | |
|---|---|---|---|---|
| Male | Female | |||
| Hepatocellular adenoma |
0 0.5 1.0 2.0 4.0 8.0 |
– |
3/36 4/72 4/53 4/41 11/36* 25/39*** |
Moser et al. (2009) |
| Hepatocellular carcinoma |
0 0.5 1.0 2.0 4.0 8.0 |
– |
0/36 4/72 2/53 1/41 2/36 11/39*** |
Moser et al. (2009) |
| Hepatocellular adenoma or carcinoma |
0 0.5 1.0 2.0 4.0 8.0 |
– |
3/36 8/72 6/53 5/41 12/36* 29/39*** |
Moser et al. (2009) |
| Hepatocellular carcinoma |
0 8 15 |
7/50 32/50*** 34/50*** |
2/50 7/50 27/50*** |
NTP (1993) |
| Hepatocellular adenoma |
0 8 15 |
20/50 33/50** 42/50*** |
5/50 31/50*** 48/50*** |
NTP (1993) |
| Benign Pheochromocytoma |
0 8 15 |
1/49 6/50 10/50*** |
2/50 1/50 6/50 |
NTP (1993) |
| Hepatocellular adenoma/carcinoma |
0 8 15 |
26/50 44/50*** 50/50*** |
7/50 34/50*** 50/50*** |
NTP (1993) |
bw: body weight.
Only the major tumour sites and/or those showing significant effects at the lower dose are listed.
*Equals significant at > 0.05; **equals significant at > 0.01; ***equals significant at > 0.001.
Johansson et al. (1997) undertook a study in preweaning B6C3F1 mice to investigate the dose–response relationship to furan over 95 weeks. In this study, they compared single (400 mg/kg bw on post‐natal day (PND) 15) vs multiple (6 doses of 200 mg/kg bw administered on PND 3, 6, 9, 12, 15 and 18) exposures to furan. The single exposure group showed an overall increase in multiplicity of liver tumours but not in prevalence. Neither of these observations was significant. In the multiple dosed group, a statistically significant increase in both multiplicity and prevalence of liver tumours was noted. PCR analysis of the tumours revealed mutations on codon 61 of the Ha‐ras1 gene in 82% of the single dose group while only 32% of the multiple does group had the Ha‐ras1 mutation. Their findings confirm the hepatocarcinogenicity of furan in mice and suggest that the dose of carcinogen may influence the activation of Ha‐ras1 that results in liver tumours.
Morton et al. (2002) used furan as an exemplar chemical to evaluate the Eker rat as a model for short‐term exposure to nephrotoxins and carcinogens. Rats were exposed to 8 mg/kg bw furan in olive oil for 5 days/week for 4 or 6 months. Furan treatment did not result in any increase in the number or incidence of preneoplastic or neoplastic lesions at all sites and at either time.
Høie et al. (2016) investigated the tumourigenic potential of 2,5‐dimethylfuran administered orally to the C57BL/6J‐Apc Min/+ mouse (Min). The study included Min mouse and Min mouse expressing the human sulfotransferase 1A1 and 1A2 (Min/hSULT mice). In untreated Min/hSULT mice, the spontaneous tumours in the small intestine were smaller, but not fewer, compared to the Min mouse. Colonic tumour incidence and tumour load were also decreased in the untreated Min/hSULT mice. Mice were exposed to 5, 25 and 50 mg/kg bw by gavage in corn oil three times per week for 6 weeks (a total of 18 doses), no consistent significant changes in intestinal or colon tumours or in aberrant crypt foci were seen in male or female Min mice exposed to 2,5‐dimethylfuran. There was a significant increase in the incidence of colonic tumours in the female, but not the male, mice expressing hSULT at the medium dose but this was not significant at either the low or high doses. Exposure to 2,5‐dimethylfuran increased the tumour number in the middle to distal part of the small intestine and colon, the area where the majority of the spontaneous tumours are found. These effects of 2,5‐dimethylfuran in Min/hSULT females were suggested to be due to the low background level of tumours in the untreated females. There were no effects of 2,5‐dimethylfuran in the Min mice. Overall the data for the tumourigenicity of 2,5‐dimethylfuran in metabolically competent mice were not convincing.
In summary, most of the cholangiocarcinomas in rats reported in the NTP study were later reclassified as cholangiofibrosis and only at the top dose of 8 mg/kg bw, cholangiocarcinomas were confirmed. In the NCTR study where a lower dose range was used, cholangiofibrosis was observed at doses of 0.2 mg/kg bw and above and no neoplastic findings of relevance to human risk assessment were observed. In the mouse studies, hepatocellular adenomas and carcinomas were observed. The male rats and mice appear more sensitive to furan exposure for hepatocellular adenoma and carcinoma development. In a short‐term tumour model system, 2,5‐dimethylfuran did not enhance colorectal tumour formation.
Table 5.
Summary of non‐neoplastic lesions in rats and mice taken from NTP (1993)
| Lesion | Dosage (mg/kg bw per day) | Incidencea | |
|---|---|---|---|
| Rats | Male | Female | |
| Biliary tract cysts |
0 2 4 8 |
0/50 44/50** 47/50** 49/50** |
0/50 49/50** 50/50** 46/50** |
| Biliary tract fibrosis, multifocal |
0 2 4 8 |
0/50 44/50** 48/50** 49/50** |
0/50 49/50** 50/50** 49/50** |
| Biliary tract hyperplasia |
0 2 4 8 |
0/50 44/50** 48/50** 49/50** |
0/50 49/50** 48/50** 49/50** |
| Biliary tract inflammation chronic multifocal |
0 2 4 8 |
0/50 44/50** 48/50** 49/50** |
0/50 49/50** 50/50** 49/50** |
| Biliary tract metaplasia |
0 2 4 8 |
0/50 44/50*** 48/50*** 49/50*** |
0/50 49/50*** 50/50*** 49/50*** |
| Hepatocyte cytomegaly |
0 2 4 8 |
0/50 35/50** 46/50** 49/50** |
0/50 44/50** 50/50** 49/50** |
| Hepatocyte degeneration |
0 2 4 8 |
0/50 33/50** 46/50** 49/50** |
0/50 35/50** 49/50** 47/50** |
| Hepatocyte hyperplasia, multifocal |
0 2 4 8 |
0/50 33/50 46/50** 49/50** |
0/50 32/50 47/50** 46/50** |
| Hepatocyte necrosis, multifocal |
0 2 4 8 |
0/50 32/50** 46/50** 49/50** |
0/50 18/50** 46/50** 47/50** |
| Hepatocyte vacuolisation, cytoplasmic |
0 2 4 8 |
0/50 39/50** 45/50** 49/50** |
0/50 43/50** 49/50** 47/50** |
| Kupffer cell pigmentation, multifocal |
0 2 4 8 |
0/50 44/50** 48/50** 49/50** |
0/50 49/50** 50/50** 48/50** |
| Mice | |||
| Multifocal hyperplasia liver |
0 8 15 |
1/50 44/50** 49/50** |
0/50 7/50 11/50 |
| Biliary tract fibrosis |
0 8 15 |
0/50 45/50** 49/50** |
0/50 47/50** 50/50** |
| Biliary tract hyperplasia |
0 8 15 |
0/50 46/50** 49/50** |
0/50 47/50** 50/50** |
| Biliary tract inflammation |
0 8 15 |
0/50 44/50** 49/50** |
2/50 48/50** 50/50** |
| Hepatocyte hepatomegaly |
0 8 15 |
8/50 45/50** 50/50** |
0/50 48/50** 50/50** |
| Hepatocyte degeneration |
0 8 15 |
0/50 43/50** 43/50** |
0/50 47/50** 48/50** |
| Hepatocyte necrosis |
0 8 15 |
2/50 39/50** 41/50** |
0/50 44/50** 47/50** |
| Kupffer cell pigmentation |
0 8 15 |
2/50 43/50** 50/50** |
5/50 48/50** 50/50** |
bw: body weight.
**equals significant at > 0.01; ***equals significant at > 0.001.
3.1.2.4. Genotoxicity
A. Furan
The genotoxicity of furan has been studied in bacteria, and mammalian cells in vitro, and in vivo. The available studies are summarised below and further details are given in Table 7.
Table 7.
Genotoxicity of furan
| Test system | Cells/animals | Concentration/treatment | Result | Comments | Reference |
|---|---|---|---|---|---|
| In vitro (bacteria) | |||||
| Bacterial reverse mutation assay (Ames test) | S. Typhimurium TA98, TA100, TA1535, TA1537 | 33–10,000 μg/plate | Negative | Aroclor 1,254‐induced male Sprague–Dawley rat and Syrian hamster liver S9 | NTP (1993); Mortelmans et al. (1986) |
| Ames test | S. Typhimurium TA98, TA100 |
56–225,000 μg/plate +/− S9 |
Positive: only in TA98 (+S9) |
Positive only at the lowest dose in TA98 Clear effect only + S9 |
Shinohara et al. (1986) |
| Ames test | S. Typhimurium TA98, TA100 | 54, 272, 1,361, 6,807 μg/ plate | Positive: only in TA100 (+/− S9) | Weak effects | Lee et al. (1994) |
| In vitro (mammalian cells) | |||||
| Forward mutation assay | L5178Y tk+/tk− mouse lymphoma cell line | 125–3,800 μg/mL (−S9) | Positive: ≥ 1,139 μg/mL |
Relative total growth was about 77% Not tested: + S9 |
McGregor et al. (1988) |
| SCEs | CHO cells | 1.6, 5, 16, 50, 160, 500 μg/mL |
Positive –S9: 1.6–160 μg/mL +S9: only at 500 μg/mL |
S9 from Aroclor 1,254‐induced Sprague–Dawley rat liver | NTP (1993) |
| CA | CHO cells | 100, 160, 300, 500, 1,000 μg/mL |
Positive −S9: 100–500 μg/mL +S9: ≥ 500 μg/mL |
S9 from Aroclor 1,254‐induced Sprague–Dawley rat liver | NTP (1993) |
| CA | CHO cells | 3 h exposures, up to 13,614 μg/mL +/− S9 from Aroclor 1,254‐induced rat liver | Positive: Chromatid breaks and chromatid exchanges only in the presence of S9 | No information on toxicity | Stich et al. (1981) |
| SCEs | V79‐Mz‐hCYP2E1‐SULT1 cell line | 0.2–1,089 μg/mL | Positive: both V79‐hCYP2E1‐SULT1 and parental V79‐Mz cell line | Unusual constant increase over the whole dose range. Marginal increase in comparison to the parental V79‐Mz cell line | Glatt et al. (2005) |
| Micronucleus assay | Lymphocytes from two non‐smoking women | 136, 340, 511, 681, 1021, 1,361, 6,807 μg/mL +/− rat liver homogenate. | Negative | Aroclor 1,254‐induced rat liver might contain low CYP2E1 activity. Cytotoxicity: 1,361 and 6,807 μg/mL | Durling et al. (2007) |
| Micronucleus assay, tk +/ tk − mutation assay and DNA breaks by Comet assays | L5178Y tk + /tk − mouse lymphoma cell line | 15–211 μg/mL (~ 225–3,100 μM) | Negative | Only assayed in the absence of S9. No cytotoxicity was observed at any of the tested doses | Kellert et al. (2008a) |
| In vivo | |||||
| Sex‐linked recessive lethal assay | Drosophila melanogaster | Feeding (10,000 mg/kg) and injection (25,000 mg/kg) | Negative | – | NTP (1993), Foureman et al. (1994) |
| Unscheduled DNA synthesis in hepatocytes | Hepatocytes from furan‐treated male F344 rats or B6C3F1 mice | Single gavage treatment: 5, 30, 100 mg/kg bw (rats) and 10, 50, 100, 200 mg/kg bw (mice) | Negative | – | Wilson et al. (1992) |
| SCEs in bone marrow | Male B6C3F1 mice | i.p.: 87.5, 175, 350 mg/kg bw (23 h sampling) and 25, 50,100 mg/kg bw (42 h sampling) | Negative | – | NTP (1993) |
| CA in bone marrow | Male B6C3F1 mice | i.p.: 87.5, 175, 350 mg/kg bw (17 h harvest); 62.5, 125, 250 mg/kg bw (36 h harvest) | Positive: only at 250 mg/kg furan with 36 h sampling time (2 experiments) | – | NTP (1993) |
| Micronucleus assay in peripheral erythrocytes (flow cytometer‐based) | BALB/c and CBA mice |
BALB/c mice: i.p. (0, 50, 75, 90, 110, 125, 150, 175, 200, 250, 300 mg/kg bw) and s.c. (0, 150 and 275 mg/kg bw) CBA mice: i.p. (0 and 225 mg/kg bw) |
Negative | No significant depression of cell proliferation in any of the experiments. A single sampling time (42 h) was used | Durling et al. (2007) |
| Measurements of DNA 8‐oxodeoxyguanosine in liver by immune fluorescence | Sprague–Dawley rats |
Gavage (30 mg/kg bw per day, 5 daily doses per week). Time points: 1, 3, 7, 10, 12, 20 days and 1, 2 and 3 months In addition, a 3‐month treatment + 1‐month off was included |
Positive: Increased DNA 8‐oxodeoxyguanosine levels in areas of centrilobular necrosis. Persistence of DNA oxidation after recovery time in areas affected by cholangiofibrosis | – | Hickling et al. (2010a) |
| Micronuclei, SSBs and DNA cross‐links by Comet assays, DSBs by γ‐H2AX foci in the spleen. | B6C3F1 mice | Gavage for 4 weeks with 2, 4, 8 and 15 mg/kg bw per day |
Positive: micronuclei in mitogen‐stimulated splenocytes (4–15 mg/kg) Positive: γ‐H2AX foci (8 and 15 mg/kg) Negative: Comet assays |
All assays gave negative results in quiescent spleen lymphocytes | Leopardi et al. (2010) |
| DSBs by γ‐H2AX foci; SSBs and DNA crosslinks by Comet assays in the liver. | B6C3F1 mice |
Gavage for 28 days (2, 4, 8, 15 mg/kg bw per day, 5 days per week) Single oral dose (15, 100, 250 mg/kg bw) |
Negative: γ‐H2AX foci and Comet assays (28‐day exposure) Positive: DNA breaks and crosslinks (acute exposure at 250 mg/kg bw) |
28‐day exposure: Increased polyploidy in liver cells Increased expression of several DNA repair genes |
Cordelli et al. (2010) |
| DNA adducts, micronuclei, CAs, SCEs and SSBs and DNA crosslinks by Comet assays | F344 rats | Adducts determination: [3,4 14C]‐furan (0.1 and 2 mg/kg bw) for 2 h by gavage. Genotoxicity: Oral administration of 0.1, 0.5 and 2 mg/kg bw per day for 5 and 28 days |
Positive: some evidence of DNA adducts in the liver and kidney Positive: CAs in proliferating splenocytes Negative: micronuclei, CAs, SCEs and Comet assays in bone marrow and peripheral blood |
DNA adducts apparent in 14 C‐furan treated rats are not the same of those induced by BDA (MS peaks are not identical). Some indication of DNA damage in the liver by Comet assay (a reduction of tail moment and tail intensity after 28 days of furan treatment and increased DNA strand breaks after two weeks recovery) | Neuwirth et al. (2012) |
| DNA‐protein crosslinks by Comet assays | Turkey eggs | Furan injection in eggs at 23 days incubation (136–1,360 μg). Hepatocytes preparation 24 h later | Positive: Formation of DNA protein crosslinks | – | Jeffrey et al. (2012) |
| Micronucleus and Comet assay +/‐Fpg and EndoIII in liver and bone marrow. | F344 rats | Gavage for 4 days (2, 4, 8, 12, 16 mg/kg bw per day) |
Positive: Comet assays in the liver (8–16 mg/kg bw per day) Negative: Comet assays in the bone marrow and micronuclei in peripheral blood |
Increased proliferation in the liver. Up‐ and down‐regulation of several DNA repair, apoptotic and cell cycle genes | Ding et al. (2012) |
| Micronuclei in normochromatic erythrocytes and reticulocytes, mutation assays at Pig‐A and Hprt, liver cII transgene mutation assay, liver Comet assay. | Transgenic Big Blue rats | Gavage for 1 and 8 weeks (2, 8, 16, 30 mg/kg bw per day; 5 days per week) and sampling time 24 h after the end of the treatment |
Positive: Comet assay in the liver (16 and 30 mg/kg bw per day) Negative: micronuclei and mutation assays |
Measurements of mutations at Hprt, Pig‐A and cII transgene and micronuclei were performed after 1 or 8 weeks of treatment | McDaniel et al. (2012) |
| Mutations in the cII transgene in the liver | Transgenic Big Blue B6C3F1 mice | Gavage for 6 weeks (15 mg/kg bw per day; 5 days per week, 24 h sampling time) and for 3 weeks (once weekly, 1 week sampling time) |
Negative: no increase in mutation frequency under either treatment condition Positive: changes in mutational spectra |
Small increase in GC>CG transversions (6‐week treatment); a larger shift with increases in GC>TA, GC>CG, AT>TA transversions in the 3‐week protocol | Terrell et al. (2014) |
| Mutations in the gpt transgene and Spi‐ in the liver | Transgenic gpt F344 male and female rats | Gavage for 13 weeks (0, 2, 8 mg/kg bw per day) | Negative: no increase in mutation frequencies. No changes in mutational spectra at the gpt gene | Increased number of GST‐P foci, PCNA+ hepatocytes and cyclin d1 and cyclin e1 at 8 mg/kg bw per day. Cholangiofibrosis observed in the caudate lobe | Hibi et al. (2017) |
| Analysis of Ha‐ras mutations | Liver of furan‐treated B6C3F1 mice | Samples from NTP study (1993) | Positive: 9 Ha‐ras mutations: 4/5 GC>TA transversions at codon 61 and 2 GC>CG + 2 GC>TA transversions at codon 117 | The localisation of Ha‐ras mutations in furan‐treated livers differs from that of untreated animals (100% vs 60% mutations at codon 61 in untreated and furan‐treated animals, respectively). | Reynolds et al. (1987) |
| Analysis of Ha‐ras mutations | Liver tumours induced in infant B6C3F1 mice (adenomas + carcinomas) | Treatment in preweaning mice (day 15): i.p. 400 mg/kg bw (single dose) or 6 doses of 200 mg/kg bw (at day 15). | Positive: only in single dose exp: codon 61 mutations were 23/28 (82%) (vs 1/3 (33%) in parallel controls) (mostly C>A transversions) | Statistically significant increased incidence and multiplicity of hepatocellular neoplasms in the multiple treatment group and only increased multiplicity in the single dose group | Johansson et al. (1997) |
| Analysis of Ha‐ras mutations by ACB‐PCR | Liver of furan‐treated B6C3F1 mice | Gavage: 0, 1, 2, 4 and 8 mg/kg bw per day (5 days per week) over a 3‐week period | Negative: No increase in mutations frequency at codon 61 (CAA>AAA and CAA>CTA) | Relatively insensitive assay | Banda et al. (2013) |
| DNA adduct formation (BDA‐dC adduct) | Serum and liver of furan‐treated Fischer 344 rats | Gavage: 0.92–9.2 mg/kg bw (single dose) and 4.4 mg/kg bw per day (multiple doses, 45–360 days) | Negative: No increase over background levels | Background levels in untreated rats: 1.2–2.4 adducts ×108 nucleotides | Churchwell et al. (2015) |
| 8‐Oxodeoxyguanosine | Serum of furan‐treated Sprague–Dawley rats | Gavage. 16 mg/kg bw per day over 30 days | Positive: large increases in 8‐oxodeoxyguanosine levels | Method: ELISA kit | Alam et al. (2017) |
| ROS and 8‐oxodeoxyguanosine | Serum of furan‐treated BALB/c mice | i.p. 8 mg/kg bw per day over 7 days | Positive: increased ROS levels; threefold increase in 8‐oxodeoxyguanosine | Method: ELISA kit | Wang et al. (2014a) |
A: adenosine; ACB‐PCR: Allele‐specific competitive blocker‐polymerase chain reaction; BDA: cis‐but‐2‐ene‐1,4‐dialdehyde; bw: body weight; C: cytidine; CA: chromosomal aberrations; CHO: Chinese hamster ovary; dC: 2’‐deoxycytidine; DSB: double strand break; ELISA: enzyme‐linked immunosorbent assay; G: guanosine; i.p.: intraperitoneal; ROS: reactive oxygen species; s.c.: subcutaneous; SCE: sister chromatid exchange; SSB: single strand break; T: thymidine.
The CONTAM Panel noted that in vitro studies with furan are often hampered by the properties of the compound. First, the high volatility of the furan exhibiting a boiling temperature of 31.4°C leads to its rapid evaporation from the test vessels under open conditions. In closed systems that used to prevent evaporation of furan, however, an equilibrium between the liquid and the gas phase is rapidly achieved, thus markedly decreasing the initial concentration of furan in the liquid phase. Finally, reducing the headspace in order to avoid this effect may cause a lack of oxygen which is required for CYP‐catalysed metabolic activation of furan. Thus, quantitative considerations based on in vitro experiments with furan must be made with caution.
Bacteria
Furan did not induce gene mutations in S. Typhimurium strains TA100, TA1535, TA1537 and TA98 in the presence or absence of exogenous metabolic activation (S9) (Mortelmans et al., 1986; NTP, 1993). In another report, positive results were reported only in strain TA98 (but not in strain TA100) in the presence of S9 (Shinohara et al., 1986). Finally, a weak positive effect was reported only in strain TA100 both in the presence and absence of S9 fraction (Lee et al., 1994).
In vitro genotoxicity in mammalian cells
In vitro tests for genotoxicity in mammalian cells were generally positive. In the NTP study (1993), furan induced trifluorothymidine resistance in mouse L5178Y lymphoma cells in the absence of S9 (McGregor et al., 1988), sister chromatid exchanges (SCEs) and chromosomal aberrations (CA) in Chinese hamster ovary (CHO) cells, with and without S9 (NTP, 1993).
In another report, an increase in CA (chromatid breaks and chromatid exchanges) was observed in CHO cells exposed to furan (up to 200 mM) only in the presence of S9 (Stich et al., 1981).
Furan induced SCEs in a modified V79 cell line that stably expressed human CYP2E1 and SULT1A1. No information on the activity of CYP2E1 was reported. The increment in the number of SCEs was small and showed no clear dose response, with only a marginal increase in the hCYP2E1‐SULT1A1‐expressing cells in comparison to the parental cell line (Glatt et al., 2005).
In the presence or absence of metabolic activation, no significant increase in the frequency of micronuclei was observed in human lymphocytes derived from two non‐smoking women (Durling et al., 2007).
The genotoxicity of furan was also investigated in L5178Y tk +/‐ mouse lymphoma cells. Furan did not induce mutations at the tk locus, micronuclei, and DNA strand breaks as measured by Comet assays (Kellert et al., 2008a). The CONTAM Panel noted that all the assays were performed at furan concentrations devoid of any cytotoxic effects.
In vivo studies
Furan did not induce sex‐linked recessive lethal mutations in germ cells of male Drosophila melanogaster when administered either by feeding or by injection (NTP, 1993; Foureman et al., 1994).
Furan did not induce unscheduled DNA synthesis (UDS) in hepatocytes isolated after single gavage treatment of male F344 rats or male B6C3F1 mice (Wilson et al., 1992).
In male B6C3F1 mice, furan administered by i.p. induced CA but not SCEs in bone marrow cells. The CONTAM Panel noted that the significant increase in the frequency of CA was observed only at the highest tested dose and required the use of an extended harvesting protocol to maximise the detection of these effects (NTP, 1993).
Furan administered by i.p or subcutaneous injection to BALB/c and CBA mice did not induce micronuclei in peripheral blood (a single sampling time of 42 h was used; Durling et al., 2007).
Furan was administered by gavage to Sprague–Dawley rats from 8 h up to 20 days and for 1, 2 and 3 months. Increased DNA 8‐oxodeoxyguanosine levels as measured by immunofluorescence were observed in the liver. This increase was associated with inflammation in areas adjacent to, as well as within, necrotic regions. Persistence of DNA oxidation was also observed in cholangiofibrotic lesions, biliary epithelia, metaplastic ducts and hepatocytes in proximity of inflamed areas in animals sampled at the end of 3‐month treatment or after 1‐month recovery (Hickling et al., 2010a).
Furan induced micronuclei and double‐strand breaks (DSBs) as measured by γ‐H2AX foci (a known biomarker for DNA‐strand breaks) in mitogen‐stimulated splenocytes from B6C3F1 mice treated by gavage for 4 weeks. Negative results were obtained in quiescent spleen lymphocytes. No induction of single strand breaks (SSBs) or DNA crosslinks as measured by Comet assays were observed (Leopardi et al., 2010). The CONTAM Panel noted that this study represents evidence of genotoxicity of orally administered furan in mice. It also indicates that for primary DNA damage in the spleen to be converted into frank DSBs and chromosomal damage requires DNA replication.
Furan‐induced DNA damage was investigated in the liver of B6C3F1 mice following a 28‐day repeated exposure by gavage. No induction of DSBs (as measured by γ‐H2AX induction) or SSBs and DNA crosslinks was observed in liver cells following the 28‐day treatment. In contrast, an increase of SSBs and DNA crosslinks, as measured by Comet assays, was observed in the liver of mice receiving a single very high oral dose of 250 mg/kg bw. Increased expression of DNA repair genes following the 28‐day treatment was also observed (Cordelli et al., 2010).
Following a 2 h administration by gavage of a single dose of [3,4‐14C]‐furan to F344 rats, a dose‐dependent increase of 14C was found by mass spectrometry in liver and kidney DNA, suggesting that furan metabolites were able to bind covalently to DNA (Neuwirth et al., 2012). The number of DNA adducts induced in the liver were 1.6 and 32.5 adducts/108 nucleotides at 0.1 and 2.0 mg/kg bw of furan. However, individual DNA adducts could not be identified and it was demonstrated that these adducts are not identical to those induced in chemical model systems by the reactive BDA metabolite. Metabolic incorporation of furan into DNA bases during DNA synthesis occurred only to a minor extent. In a parallel oral 28‐day study, furan induced an increase in CA in proliferating splenocytes. No increase in micronuclei, CA, SCEs or strand breaks by Comet assays were observed in bone marrow and peripheral blood (Neuwirth et al., 2012). At 2 mg/kg bw, furan produced an increase in DNA strand breaks in the liver as measured in the Comet assay. This occurred, however, after a 14‐day recovery period following a 28‐day exposure (Neuwirth et al., 2012). The CONTAM Panel recognises that this study provides some evidence of the ability of furan to covalently bind to DNA in vivo. However, in view of the low number of furan‐induced DNA adducts their biological significance remains to be clarified. Finally, this study confirms in splenocytes from furan‐treated rats the previously observed increase in chromosomal damage in proliferating mouse splenocytes (Leopardi et al., 2010).
DNA‐protein crosslinks were studied by injecting furan into turkey eggs at 23 days of incubation. Hepatocytes were prepared from the embryos 24 h later and analysed by alkaline Comet assays (tail length and moment) for the presence of strand breaks and DNA protein crosslinks. Furan treatment produced a reduction in DNA migration suggesting the formation of DNA crosslinks. Comet tail length was increased following treatment with proteinase K indicating that these were due to DNA‐protein crosslinks (Jeffrey et al., 2012).
Furan administered to F344 rats by gavage for 4 days increased DNA strand breaks (as measured by Comet assays) in the liver but not in the bone marrow (Ding et al., 2012). Maximal DNA damage was observed 1 h after the last furan treatment and decreased with time (3, 6 and 8 h) to reach control levels by 16 h. Comet assays were performed also in the presence of Fpg and EndoIII (enzymes involved in the repair of oxidised bases). In both cases, the number of SSBs increased (in the range 12–16 and 4–16 mg/kg bw for Fpg and EndoIII, respectively) indicating the presence of oxidised purines and pyrimidines. No induction of micronuclei was observed in the peripheral blood (although signs of toxicity were present in the bone marrow). The CONTAM Panel noted that furan‐induced DNA strand breaks, identified by Comet assays, in the liver show a relatively short half‐life.
The potential of furan to produce systemic genotoxicity was evaluated by treating female Big Blue transgenic F344 rats by gavage for 1 and 8 weeks (five times a week) with doses of furan used in the cancer bioassay (2 and 8 mg/kg bw) and two higher doses (16 and 30 mg/kg bw). No increases of micronuclei in peripheral blood, Hprt mutations in lymphocytes, Pig‐A mutations in spleen lymphocytes and peripheral red blood cells, and transgene cII mutations in the liver were observed. In line with the Ding et al. (2012) study, liver DNA damage as measured by Comet assay was increased at the two highest doses (McDaniel et al., 2012).
Furan was administered to female Big Blue cII transgenic B6C3F1 mice by gavage either for 6 weeks with 5 daily doses or for 3 weeks with a single weekly dose (15 mg/kg bw). Furan did not increase mutation frequency under either treatment regimen. However, in the 6‐week protocol, there was a change in the liver cII mutational spectrum (a significant reduction in GC>AT transitions and an increase in CG>GC transversions). A much larger furan‐dependent shift in mutational spectra was observed in the liver of mice receiving three weekly doses of furan (a significant increase in transversions including GC>TA, GC>CG and AT>TA). The mutagenic potential of BDA in the same target gene was also investigated by in vitro treatment of mouse embryo fibroblasts derived from this transgenic mouse strain. Again, no increase in mutation frequency was observed, but BDA treatment induced a shift in mutational spectrum with a significant increase in AT>CG transversions (Terrell et al., 2014).
Gpt and Spi‐ mutations were measured in the liver of gpt delta rats treated with furan at doses of 2 and 8 mg/kg bw for 13 weeks. No increase in mutation frequencies was observed and mutational spectra did not identify furan‐specific gpt mutations. In the high dose group, the number and areas of glutathione S‐transferase placental form (GST‐P)‐positive foci, the number of PCNA‐positive hepatocytes and the mRNA levels of cyclin d1 and cyclin e1 were increased. Cholangiofibrosis was only observed in the caudate lobe. The authors conclude that cell proliferation, but not genotoxic mechanisms, contribute to the early stages of furan‐induced hepatocarcinogenesis (Hibi et al., 2017).
Analysis of liver neoplasms (hepatocellular adenomas/carcinomas) induced by furan in B6C3F1 mice (from the NTP 1993 study) showed the presence of dominant transforming oncogenes (13/29, 45%). Activating Ha‐ras mutations were found both in codon 61 and in codon 117, the majority being GC>TA transversions. The authors concluded that the spectrum of furan‐induced mutations in the Ha‐ras oncogene differs from that of untreated animals, mostly because the localisation of mutations in untreated animals occurs only at codon 61 (Reynolds et al., 1987).
The presence of Ha‐ras mutations was also analysed in DNA from liver tumours induced in preweaning B6C3F1 mice administered a single or multiple (6x) doses of furan. Mutations in codon 61 of the Ha‐ras gene were present in 82% of liver neoplasms (adenoma + carcinoma) occurring in mice treated with a single dose, while in the concomitant controls these occurred in 33% of the animals and in the historical ones in 54% of the animals. In contrast, in mice treated with multiple doses only 32% of the tumours had codon 61 Ha‐ras1 mutations. The majority of mutations associated with furan exposure in codon 61 were G>T transversions (74%) followed by A>G transitions (22%). A single G>T transversion in codon 117 was also identified in an adenoma induced by this multiple treatment. In historical controls, the majority of codon 61 Ha‐ras1 mutations were transversions followed by transitions (58% and 30%, respectively) (Johansson et al., 1997).
To verify whether furan treatment caused an early expansion of pre‐existing spontaneous Ha‐ras mutations, allele‐specific competitive blocker polymerase chain reaction (ACB‐PCR) was used to quantify CAA>CTA and CAA>AAA mutations in codon 61 of the Ha‐ras gene. Mutation analyses were performed in liver DNA from female B6C3F1 mice treated by gavage with furan over a 3‐week period. No difference in mutation frequencies at codon 61 was identified between controls and furan‐treated groups (Banda et al., 2013). The CONTAM Panel noted the limitations of the study including the low sensitivity of the technique.
Treatment of Sprague–Dawley rats by gavage with furan for 30 days resulted in large increases (10‐fold) in serum levels of 8‐oxodeoxyguanosine as measured by enzyme‐linked immunosorbent assay (ELISA) (Alam et al., 2017).
Treatment (i.p.) of BALB/c mice with furan for 7 days resulted in increased serum levels of reactive oxygen species (ROS) and 8‐oxodeoxyguanosine as measured by ELISA (Wang et al., 2014a).
B. cis ‐2‐Butene‐1,4‐dial (BDA)
The genotoxicity of BDA has been studied in bacteria and mammalian cells in vitro and the available studies are summarised below and further details are given in Table 8. The reactivity of BDA with DNA is discussed in Section 3.1.1.4.
Table 8.
Genotoxicity of cis‐2‐Butene‐1,4‐dial (BDA) in vitro
| Test system | Experimental system | Concentration/treatment | Result | Comments | Reference |
|---|---|---|---|---|---|
| Formation of ICLs | In situ oxidation of furan containing oligonucleotide | Positive: formation of DNA interstrand cross‐link in duplex oligonucleotides | – | Halila et al. (2005) | |
| Formation of dC and dA adducts | Calf thymus DNA and DNA from BDA‐treated S. Typhimurium TA104 | Positive: formation of DNA adducts in both systems | – | Byrns et al. (2006) | |
| Bacterial reverse mutation assay (Ames test) | S. Typhimurium TA104 | 118, 143, 177, 244, 362 μg/plate | Positive |
Toxic at doses > 177 μg/plate BDA mutagenic potential revealed in a specific strain sensitive to aldehydes |
Peterson et al. (2000) |
| SSBs and crosslinks by alkaline elution | CHO‐K1 | 14, 42, 126 μg/mL | Positive: both end‐points | High concentrations (in the mM range) but no apparent toxicity | Marinari et al. (1984) |
|
Forward mutation Comet and micronucleus assays |
L5178Y tk+/− mouse lymphoma cells | 0.5, 1.1, 2.1, 4.2 μg/mL |
Positive: Comet and mutation (at ≥ 70% survival) Negative: micronucleus assays and crosslinks |
– | Kellert et al. (2008a) |
| Mutation induction at the cII transgene | In vitro treatment of mouse embryo fibroblasts from Big Blue B6C3F1 mice | 0.2 and 0.4 μg/mL | Negative: no increase in mutation frequency Positive::change in mutational spectrum | Increase in AT > CG transversions | Terrell et al. (2014) |
BDA: cis‐but‐2‐ene‐1,4‐dialdehyde; CHO: Chinese hamster ovary; dA: 2’‐deoxyadenosine; dC: 2’‐deoxycytidine; dG: 2’‐deoxyguanosine; SSB: single strand break; ICL: inter‐strand crosslink.
In the absence of exogenous metabolic activation, BDA induced a dose‐dependent increase of mutations in S. Typhimurium TA104. Pre‐incubation of BDA with GSH inhibited the toxic and mutagenic effects of the compound. In contrast, BDA was not mutagenic in S. Typhimurium strains TA97, TA98, TA100 and TA102 (Peterson et al., 2000).
dC and dA adducts were detected in calf thymus DNA treated in vitro with BDA. These adducts were also present in DNA isolated from S. Typhimurium strain TA104 treated with BDA (Byrns et al., 2006).
The DNA damaging capacity of BDA was investigated by alkaline elution in CHO‐K1 cells. Both SSBs and DNA crosslinks were observed at non‐toxic doses (Marinari et al., 1984). The ability to form DNA‐protein crosslinks observed in CHO cells was not confirmed by Kellert et al. (2008a) in a Comet assay.
Genotoxicity of BDA was investigated in L5178Y tk +/− mouse lymphoma cells. A concentration‐dependent increase in mutation frequency at the tk locus and in the number of DNA breaks as measured by Comet assays was observed. No concentration related increases were observed for micronuclei (Kellert et al., 2008a).
No increase in mutation frequency at the cII locus was observed in BDA‐treated mouse embryo fibroblasts derived from Big Blue B6C3F1 mice. However, BDA treatment induced a shift in mutational spectrum with a concentration‐dependent increase in AT > CG transversions only (Terrell et al., 2014).
C. 2‐Methylfuran
The genotoxicity of 2‐methylfuran has been studied in bacteria and mammalian cells in vitro and the available studies are summarised below and further details are given in Table 9.
Table 9.
Genotoxicity of 2‐methylfuran and, 2,5‐dimethylfuran
| Compound | Test system | Cells/animals | Concentration/treatment | Result | Comments | Reference |
|---|---|---|---|---|---|---|
| 2‐Methylfuran | Bacterial reverse mutation assay (Ames test) | S. Typhimurium TA98, TA100, TA102 | 0.9–90,000 μg (+/−S9) | Negative |
Use of a pre‐incubation assay to maximise the reaction of volatile compounds with bacteria Pre‐incubation assay |
Aeschbacher et al. (1989) |
| 2‐Methylfuran | Ames test | S. Typhimurium TA98, TA100, TA1535, TA102 TA104 | 33–6,666 μg/plate (+/−S9) | Negative: TA98, TA100, TA1535, TA102 |
Questionable response in strain TA104 Pre‐incubation assay |
Zeiger et al. (1992) |
| 2‐Methylfuran | Ames test | S. Typhimurium TA98, TA100 | 14–55 μg (+/−S9) | Negative | – | Shinohara et al. (1986) |
| 2‐Methylfuran | Chromosome aberrations | CHO cells | 4,105–12,315 μg/mL | Positive (+/−S9): | Increased chromatid breaks and chromatid exchanges. Decreased clastogenicity in the presence of S9. No information on toxicity | Stich et al. (1981) |
| 2,5‐Dimethylfuran | Ames test | S. Typhimurium TA97, TA98, TA100, TA1535 | 10–3,333 μg/plate (+/−S9) | Negative | Pre‐incubation assay | Zeiger et al. (1992) |
| 2,5‐Methylfuran | Ames test | S. Typhimurium TA98, TA100, | 16–64 μg (+/−S9) | Negative | – | Shinohara et al. (1986) |
| 2,5‐Dimethylfuran | Chromosome aberrations | CHO cells | 2,5‐Dimethylfuran (up to 1,922 μg/mL) | Positive (+/−S9): | Increased chromatid breaks and chromatid exchanges. Decreased clastogenicity in the presence of S9. No information on toxicity | Stich et al. (1981) |
| 2,5‐Dimethylfuran | In vitro micronucleus assay | BM cells prepared from C57BL/6J mice | 48 and 96 μg/mL (1 h exposure) | Positive (+/−S9) | Same increase in micronuclei both in the absence and presence of S9 | Fromowitz et al. (2012) |
| 2,5‐Dimethylfuran | Single strand breaks by Comet assays | Wild‐type and human hSULT /CYP2E1‐co‐expressing V79 cells | 48, 96, 144 and 192 μg/mL (30 min exposure) | Positive: both wild‐type and hSULT/CYP2E1‐co‐expressing V79 | Increase in breaks at > 50% survival | Huffman et al. (2016) |
| 2,5‐Dimethylfuran | Comet assays in kidney, colon and liver | hSULT and wild‐type FVBN mice | Gavage: 75, 150, 300 mg/kg bw |
Negative: wild‐type mice Positive: hSULT mice |
Positive at a single dose in kidney and colon but not in the liver No evidence of interstrand crosslinks formation |
Høie et al. (2015) |
CHO: Chinese hamster ovary.
2‐Methylfuran was negative for induction of gene mutations in S. Typhimurium strains TA100, TA102 and TA98 in the presence or absence of exogenous metabolic activation. A pre‐incubation assay in closed vials was used to prevent the loss of 2‐methylfuran (Aeschbacher et al., 1989). Negative results were also reported by Shinohara et al. (1986) in strains TA100 and TA98 and by Zeiger et al. (1992) in strains TA100, TA98, TA97 and TA1535. Equivocal results were reported for strain TA104 (Zeiger et al., 1992).
CHO cells exposed to 2‐methylfuran showed an increased number of CA (chromatid breaks and chromatid exchanges) in the absence of S9 (Stich et al., 1981). The CONTAM Panel noticed the lack of requirement of metabolic activation of 2‐methylfuran clastogenicity which is remarkable because of the lack of chemical reactivity of unmetabolised 2‐methylfuran.
D. 2,5‐Dimethylfuran
The genotoxicity of 2,5‐dimethylfuran has been studied in bacteria, mammalian cells in vitro, and in vivo. The available studies are summarised below and further details are given in Table 9.
2,5‐Dimethylfuran was negative for induction of gene mutations in S. Typhimurium strains TA100, TA1535, TA97 and TA98 in the presence or absence of exogenous metabolic activation. A pre‐incubation assay in closed vials was used to prevent the loss of 2,5‐dimethylfuran (Zeiger et al., 1992). These negative results were confirmed in another study with S. Typhimurium strains TA100 and TA98 (Shinohara et al., 1986).
CHO cells exposed to 2,5‐dimethylfuran (up to 1,922 μg/mL) showed an increased number of CA (chromatid breaks and chromatid exchanges). The clastogenic activity was significantly decreased in the presence of S9 (Stich et al., 1981). The CONTAM Panel noticed that, similarly to 2‐methylfuran, 2,5‐dimethylfuran clastogenicity does not seem to require S9‐mediated metabolic activation.
The ability of 2,5‐dimethylfuran to induce micronuclei was investigated in in vitro cultures of murine erythropoietic cells. Exposure to 2,5‐dimethylfuran induced an increase in micronuclei frequency both in the absence and presence of S9 (Fromowitz et al., 2012).
2,5‐Dimethylfuran induced a concentration‐dependent increase in DNA breaks as measured by Comet assays in V79 cells, irrespective of the co‐expression in these cells of the human CYP2E1 and SULT1A1 sulfotransferase (Huffman et al., 2016).
A single oral dose of 2,5‐dimethylfuran was administered by gavage to wild‐type and FVBN mice expressing the human SULT1A1/A2 (hSULT mice). Induction of DNA damage was evaluated in vivo by alkaline single cell gel electrophoresis (Comet assays) 2 h after the end of treatment. No genotoxicity is associated with 2,5‐dimethylfuran exposure of wild‐type mice, while increased levels of DNA damage were observed in the kidney and colon, but not in the liver, of 2,5‐dimethylfuran treated hSULT mice. These increases were modest and occurred at a single 2,5‐dimethylfuran dose (150 mg/kg bw). No evidence of interstrand crosslinks formation was observed (Høie et al., 2015). The authors anticipated that 2,5‐dimethylfuran can be hydroxylated at the methyl groups and subsequently conjugated with sulfate involving SULT1A1 which may lead to genotoxic breakdown products.
2,5‐Dimethylfuran has been shown to act as a scavenger of singlet oxygen (Noguchi et al., 1977; Mongin et al., 2016) and hydroxyl radicals (Okada and Okajima, 1998). In a limited in vitro study, DNA adducts induced by benzo [A] pyrene were decreased in the presence of 2,5 dimethylfuran used as scavenger of singlet oxygen (Bryla and Weyand, 1991). The CONTAM Panel noted that the possible role of 2,5‐dimethylfuran as a scavenger of singlet oxygen in vivo remains to be determined.
Summary of genotoxicity studies on furan and methylated derivatives.
Furan was unable to induce gene mutations in bacteria (with the exception of a single report showing a weak mutagenic effect). When furan genotoxic potential was investigated in mammalian cells in vitro, both negative and positive results were reported. In the majority of the studies, furan was able to induce chromosomal aberrations and sister chromatid exchanges. However, the role of metabolic activation in these studies is unclear, with one study showing positive results only in the presence of S9 and several others where furan genotoxicity was decreased by the presence of S9. In view of the volatility, quantitative considerations based on in vitro experiments with furan must be made with caution.
The primary microsomal metabolite of furan, via CYP2E1, is BDA. This can form DNA adducts at the exocyclic N atom of the dC, dG and dA nucleosides. In vitro studies indicate that BDA‐induced adducts on dG and dA are unstable, with the initial ethano adduct decomposing into substituted etheno‐acetaldehyde adducts. These secondary adducts retain a reactive aldehyde with the potential to form DNA inter‐strand crosslinks (ICLs). BDA is directly mutagenic in S. Typhimurium TA104, in which BDA‐induced DNA adducts were also identified. It also induces strand breaks and mutations in mammalian cells in vitro. One in vitro study suggests that BDA can induce ICLs in mammalian cells.
In vivo studies suggested that furan was able to bind covalently to DNA in the liver and kidney of repeatedly exposed rats. Although in one study a specific DNA adduct was not detected, in a separate study DNA adducts were detected but their molecular nature was not identified. It was demonstrated that these adducts are not identical to those induced in chemical model systems by the reactive BDA metabolite. In addition, in view of the low number of furan‐induced DNA adducts their biological significance remains to be clarified.
In several in vivo studies, no increase in chromosomal damage was observed either in the bone marrow or in peripheral blood of mice and rats exposed to furan. The single exception was the NTP study (1993) in which a dose‐related increase in chromosome aberrations in the bone marrow was found in mice following an acute treatment with unusually high doses of furan.
In rodent liver, the genotoxic potential of furan was mainly investigated by measuring strand breaks by Comet assays. This assay measures strand breaks and alkali‐labile sites, including apurinic/apyrimidinic (AP) sites, which might arise from the loss of a damaged base or as intermediates during base excision repair. In general, negative results have been reported. Two studies showed that DNA breaks/AP sites induced by repeated exposure to furan appear rapidly and disappear within a few hours post‐treatment. This observation might explain the negative results observed in several studies in which liver cells were analysed at late time points post‐treatment. Thus, observations based on Comet assays at late post‐treatment times are likely to be of limited relevance. The CONTAM Panel recognises that disappearance of strand breaks measured by the Comet assay does not necessarily indicate error‐free, non‐mutagenic DNA repair.
Evidence of chromosomal damage (micronuclei, chromosome aberrations and DSBs) was observed in proliferating, but not in resting, splenocytes of mice and rats repeatedly exposed to furan at the same doses used in the NTP carcinogenicity assay reported in 1993. These data indicate that cell proliferation is required to convert the primary furan‐induced DNA damage to detectable DNA lesions. This premutagenic DNA damage could be due to ICLs that require replication to be converted into frank lesions or secondary events associated with oxidative stress‐induced DNA damage.
Studies investigating the formation of ICLs following in vivo furan exposure were generally negative. Limited evidence comes from Comet assays in the liver of mice exposed to unusual high furan doses. DNA‐protein crosslinks were induced in hepatocytes from embryos following furan injection in turkey eggs.
It is possible that some of the genotoxic effects of furan are secondary to oxidative stress caused by repeated furan administration. Increased levels of ROS and 8‐oxodeoxyguanosine were indeed observed in sera of furan‐treated rats and mice. The sensitivity of DNA from the liver of treated animals to digestion by EndoIII and Fpg, enzymes that, respectively, act on oxidised purines and pyrimidines, confirms the presence of oxidatively damaged DNA after relatively short exposures to furan. Finally, DNA 8‐oxodeoxyguanosine persists in areas of furan‐induced rat cholangiofibrosis long after furan exposure had been discontinued.
However, furan‐induced oxidative stress is not associated with a clear induction of base substitutions in transgenic models. Furan is weakly mutagenic in the liver of a mouse transgenic model, with the mutational spectrum differing from that induced by BDA in cultured mouse cells. In contrast, consistent negative results, either in reporter or endogenous genes, were observed in transgenic rats. Moreover (GST‐P)‐positive foci, a marker of a preneoplastic phenotype in hepatocellular tumours, were increased in the liver of rats treated with carcinogenic doses of furan in the absence of any furan‐specific mutation. These data indicate that mutagenicity is not likely to contribute to the early stages of furan‐induced hepatocarcinogenesis.
Analysis of activating mutations in the Ha‐ras oncogene in liver tumours from furan‐treated B6C3F1 mice show a strong selective bias, with the vast majority being GC > TA transversions at codon 61. Since this type of transversion is also the major mutational class in control animals, it cannot be excluded that this result simply reflects an expansion of pre‐existing spontaneous Ha‐ras mutations.
Information on the genotoxic properties of methylated furans is limited. Both 2‐methylfuran and 2,5‐dimethylfuran showed negative results in bacteria. There is some evidence that both compounds can induce chromosomal damage in mammalian cells in vitro and a limited evidence that 2,5‐dimethylfuran can induce DNA breaks, but not ICLs, in vivo.
Concluding remarks
In vitro tests have provided contradictory indications as to the genotoxicity of furan. It is possible that these studies may be hampered by technical difficulties arising from the specific physicochemical properties of this compound.
In contrast, the CYP2E1‐mediated metabolite of furan, i.e. BDA, can form DNA adducts, and possibly crosslinks, in in vitro assays and is able to directly induce mutations in bacteria and mammalian cells in vitro.
In vivo, furan can induce the formation of low levels of covalent DNA adducts in liver and kidney. However, the precise nature of these DNA adducts has not been established. Conflicting data were reported for the induction of DNA breaks in the liver. These discrepancies may be related to the timing of measurements, dosing and assays sensitivities. These breaks, likely reflecting oxidative stress‐induced DNA damage, occurred at doses showing limited liver cytotoxicity. A role for oxidative stress in furan‐induced liver carcinogenesis is suggested by molecular analysis of Ha‐ras mutations in furan‐induced liver tumours indicating an excess of GC > TA transversions at codon 61 in mice. This is a mutational class commonly associated with oxidatively damaged DNA which is also present in spontaneous liver tumours. However, furan‐induced oxidative stress is not associated with a clear induction of base substitutions in transgenic rodent models. A single study suggests a weak mutagenic activity in a transgenic mouse model, while negative results were consistently reported in transgenic rat models. Finally, there is convincing evidence that chronic exposure to furan induces chromosomal damage in proliferating splenocytes from mice and rats, but the DNA lesions responsible for these effects remain undefined. In particular, it is unclear whether the observed chromosomal instability is due to direct damage to DNA (formation of ICLs or DNA adducts) or is the consequence of secondary events associated with oxidative stress‐induced DNA damage.
3.1.2.5. Developmental and reproductive toxicity
In F344 rats treated up to 8 mg/kg bw per day by gavage, no effect was observed in testes, epididymis, prostate, seminal vesicles, ovaries, uterus or vagina (Gill et al., 2010).
In contrast, Wistar rats with similar exposure but during weaning and post‐puberty (3–4 weeks old treated for 90 days) exhibited a significant increase in the weight of prostate glands and decreased weight of seminal vesicles at 8 but not at 4 mg/kg bw per day or below. Dose‐dependent histological changes (including some at the lowest dose tested of 2 mg/kg bw per day) were recorded in the testes, prostate gland, seminal vesicles and Leydig cells with an increase in apoptotic cells in the testes (Karacaoglu and Selmanoglu, 2010). No effects on sperm counts or sperm morphology were observed.
Steroidogenic acute regulating protein (stAR) mRNA was increased at all dose levels studied in F344 rats (0.03 mg/kg bw and above given daily for 90 days) while the mRNA for the cholesterol side‐chain cleavage enzyme (CYP11A1) was increased by furan at 8 mg/kg bw per day along with an increase in intratesticular testosterone suggesting an ability of furan to interfere with testicular steroidogenesis (Cooke et al., 2014).
Gill et al. (2011) found no effect of furan on testes or ovaries in B6C3F1 mice when examined microscopically following doses up to 8 mg/kg bw per day by gavage, 5 days per week for 90 days.
Furan was studied in embryos of Xenopus laevis and found to be non‐teratogenic having 96 h LC50 and EC50 values that were very similar (approximately 44 and 40 mM, respectively Williams et al., 2014).
Kara et al. (2016) used male Wistar rats, made diabetic through treatment with streptozotoxin, and exposed them to 40 mg/kg bw furan by gavage daily for 28 days. Testicular malondialdehyde levels, and the activities of CAT, GPx, SOD and GST were found to be higher than in the control and diabetic control animals and the effects were inhibited by treatment of animals with the antioxidant lycopene suggesting oxidative stress in testes. This was associated with a lowering of plasma FSH, LH and testosterone levels in animals treated with furan.
Male Sprague–Dawley rats (3–4 weeks old) were administered furan by gavage in corn oil at 4 mg/kg per day for 5 days per week for 90 days. This treatment also produced oxidative stress in testes as evidenced by an increase in malondialdehyde and decreased activity of antioxidant enzymes. These effects were associated with decreased plasma testosterone concentrations (El‐Akabawy and El‐Sherif, 2016).
In conclusion, histological changes in the testes, prostate gland, Leydig cells and seminal vesicles were observed in Wistar rats at 2 mg/kg bw (lowest dose of furan tested) and above given during weaning and post‐puberty. No histological effects in reproductive organs were observed in adult rats and mice up to 8 mg/kg bw.
3.1.3. Observations in humans
In a study in Korea, blood (Lee et al., 2009) and urine (Jun et al., 2008) samples were collected from 100 volunteers (49% males, 51% males age ranging from 30 to 70 years; see also Section 3.1.1.6). The authors in the two study reports stated that the participants consumed a regular Korean diet and avoided large amounts of furan‐rich foods. However, they also stated that: ‘Since a database on amounts of furan in the typical foods is not available so far, an actual intake of furan per individual was not shown in this study. Also, other factors such as smoking were not considered for actual intake of furan’. Furan could be detected in 56 urine samples out of 100 samples collected with average values of 0.5–1.3 μg/L, with a concentration above the LOQ (1.0 μg/L) in 15 volunteers. The highest concentration found was 3.14 μg/L. In 21 volunteers, furan could be detected in the plasma (LOD: 0.3 μg/L) with 17 samples above the LOQ (1.0 μg/L) ranging from 1.2 to 17.9 μg/L. From the data provided, it is not possible to correlate the outcome of plasma and urine analysis for the individual volunteers. No information is provided on the handling of non‐detects or on the handling of levels < LOQ. It was not stated how much time elapsed between the last consumption of food and the collection of urine, and whether 24 h urines were collected or spot samples. However, in the paper Jun et al. (2008), it was stated that that blood samples were collected after a 12 h fasting period.
Jun et al. (2008) further investigated correlations between average urinary furan levels (above LOD) and plasma ALT, aspartate amino transferase (AST) and γ‐glutamyl transpeptidase (γ‐GT) as markers of liver toxicity. There were good correlations between ALT, AST and γ‐GT, but for urinary furan only a significant correlation with plasma γ‐GT activity was found, which remained significant when covariates like age, sex, body weight, triglyceride and diastolic blood pressure were taken into account. The study authors further mentioned that mean urinary furan levels in males were higher than in females and that a similar trend was observed for plasma furan levels. However, elsewhere in the paper, they mention that quantifiable levels of furan in plasma were not found in any of the volunteers, which invalidates this comparison. The study did not investigate correlations between plasma furan levels and plasma indicators of liver toxicity. Residual confounding cannot be excluded and no graphical representation was given of the regression observed. In addition, for only 15 individuals, quantifiable urinary furan levels were observed, which puts doubts to the validity of the observed correlation. From these two studies (Lee et al., 2009; Jun et al., 2008), it is impossible to conclude that at the reported levels of blood and urinary furan, there is an effect of furan on the liver.
These two studies (Jun et al. 2008; Lee et al., 2009) show many inconsistencies and are therefore of limited value for this opinion.
3.1.4. Mode of action
Mode of action in hepatocellular toxicity
Furan depletes GSH and is cytotoxic to rodent hepatocytes both in vitro and in vivo evidently through CYP2E1‐mediated conversion to BDA (see Section 3.1.1.3). Likewise, the corresponding unsaturated aldehydes, acetylacrolein and methylbutenedial, were found to be reactive intermediates of 2‐ and 3‐methylfurans (Ravindranath et al., 1984).
Chen et al. (1997) also concluded that reactions between BDA and amino acids may be involved in the toxicity of furan. Nunes et al. (2016) provided evidence that a reactive BDA‐GSH adduct can bind to lysine 107 of histone H2B in male F344 rats treated with 0.92 mg/kg bw per day for 90 days.
In vitro, the EC50 has been estimated at 27.2 μg/mL in rat hepatocytes using an oxygen controlled closed system to avoid loss of furan by evaporation (Brueck et al., 2009). Hepatic toxicity associated with oxidative stress produced in mice by daily i.p. injection of furan (8 mg/kg bw) was inhibited by treatment with the antioxidant, salidroside, suggesting a role of ROS in hepatotoxicity (Yuan et al., 2013). Following a single dose of radiolabelled furan (2 mg/kg bw) in rats, protein binding analyses suggested particular disturbance of pathways related to mitochondrial energy production, redox regulation and protein folding that are likely to combine to alter cellular homeostasis leading to hepatotoxicity (Hamberger et al., 2010; Moro et al., 2012a). This is in accordance with the ability of furan to uncouple oxidative phosphorylation (Mugford et al., 1997). Twenty‐four hours after a single oral dose of 30 mg/kg bw to rats, a significant inhibition of activities of glyceraldehyde‐3‐phosphate dehydrogenase and succinate dehydrogenase, increased activity of enoyl‐CoA hydratase and enhanced production of ketone bodies, was indicative of inhibited glycolysis, enhanced fatty acid oxidation and mitochondrial dysfunction (Ramm et al., 2016).
Furan was found to significantly alter the level of thyroxine‐T4 and triiodothyronine‐T3 in rats at 0.12 mg/kg bw and above (Section 3.1.2.2) leading Gill et al. (2010) to conclude on a NOAEL of 0.03 mg/kg bw in rats (90 day study). The elevation of triiodothyronine‐T3 and thyroxine‐T4 reported at 0.12 mg/kg bw and above may relate to altered hepatic metabolism relating to these hormones. However, no direct evidence is available.
Furan induced cholangiofibrosis which was extensive following 2 mg/kg bw per day but no significant incidence was found at 0.02, 0.04 or 0.092 mg/kg bw per day in rats (NCTR, 2015; Von Tungeln et al., 2017). Cholangiofibrosis is only observed in rodents. However, it is an adverse outcome, which has to be taken seriously with regard to risk assessment (Weber, 2017).
In conclusion, furan induces oxidative stress. BDA binds covalently to a range of target molecules, including proteins and GSH. These features lead ultimately to cell and tissue damage, mitochondrial dysfunction and fibrosis, primarily in the liver.
Mode of action in carcinogenesis
The observation that furan is also occurring in food has generated a number of new studies that try to explain the MoA underlying the observed carcinogenicity in animals (see Section 3.1.2.3). These studies have demonstrated the production of hepatocellular carcinomas, adenomas and cholangiocarcinomas in rodents. The formation of diagnosed cholangiocarcinoma appears to be a feature of a high dose (8 mg/kg bw) and secondary to hepatotoxicity.
Findings suggest that both genotoxic and non‐genotoxic modes contribute to furan carcinogenesis depending upon the level of exposure (dose and route), the tumour type, species and gender.
Furan can be metabolised by CYP2E1 to BDA and this represents a mechanism leading to hepatic cytotoxicity and possibly to genotoxicity in mammalian cells. The mechanism and potency of genotoxicity of furan in vivo is, however, uncertain (see Section 3.1.2.4). The dial metabolite (BDA) has been found to produce substituted 1,N4‐etheno‐2′‐dC; 1,N6‐etheno‐2′‐dA and 1,N2‐etheno‐2′‐dG adducts in chemical reactions (Byrns et al., 2002, 2004, 2006). The dC and dA adducts were also detected in S. Typhimurium strain TA104 treated with a mutagenic concentration of the dial metabolite (Byrns et al., 2006). Churchwell et al. (2015) could not detect an elevation of the adduct BDA‐dC in furan‐treated rat liver DNA using LC–MS/MS after single doses up to 9.2 mg/kg bw or repeated dosing with 4.4 mg/kg bw (5 days per week) for up to 360 days. However, furan has been found to produce DNA adducts in rat tissues albeit at very low levels, suggesting a low potency (Neuwirth et al., 2012).
There is some evidence of chromosomal damage observed in vivo, but the underlying mechanism(s) has not been clarified at the molecular level (see Section 3.1.2.4). Since DNA crosslinking, DNA adducts or DNA oxidation are each potential mechanisms, it is not possible to determine if the chromosomal damage is direct or indirect.
Furan can also induce a range of cellular responses potentially relevant to cancer in rat and mouse liver. In rats, the most sensitive pathological endpoint following 2‐year exposure by gavage was cholangiofibrosis seen markedly at 0.2 mg/kg and above (NCTR, 2015; Von Tungeln et al., 2017). Increased BrdU‐labelling of hepatocytes, indicative of cell proliferation, also showed a lowest‐observed‐effect‐level (LOAEL) (statistically significant) of 2 mg/kg bw in rats administered furan by gavage 5 days per week for 5 and 28 days (Mally et al., 2010).
In mice, there was an increase in BrdU‐labelling in hepatocytes following treatment with 8 mg/kg bw but not 4 mg/kg bw furan for 28 days, 5 days per week (Cordelli et al., 2010). Subcapsular inflammation was seen in the mouse liver at the lowest dose level employed (0.5 mg/kg bw) when given 5 days per week for 3 weeks (Moser et al., 2009). In addition, after 90 days dosing (5 days per week) of mice hepatocyte apoptosis was observed with a NOAEL of 0.12 mg/kg bw (Gill et al., 2011). From their short‐ and long‐term toxicity studies in mice, Moser et al. (2009) concluded that there was an association between furan‐induced hepatotoxicity, compensatory cell replication and liver tumour formation at 4 mg/kg bw furan.
Gene expression changes in the liver following furan doses of 4 and 40 mg/kg bw for up to 14 days in Sprague–Dawley rats were in accord with fibrosis (Hamadeh et al., 2004). Statistically significant changes in the expression of a number of genes involved in oxidative stress, apoptosis and cell‐cycle were seen in rats after 2 mg/kg bw per day furan over 90 days (Curran et al., 2014; Yauk et al., 2014; Dong et al., 2016) or following 4 weeks of treatment with 0.1 and 2 mg/kg bw furan (Chen et al., 2010). Male rats responded more than female rats. Genes associated with DNA repair, cell cycle and apoptosis were also altered in expression at 2 mg/kg and above in rats exposed by gavage daily up to 3 days (Ding et al., 2012). Following a single gavage dose of 30 mg/kg bw to male rats, a wave of compensatory hepatocyte proliferation following necrosis was associated with a transient increase in the expression of c‐myc, c‐fos and Ha‐ras genes. However, a later, sustained increase in expression of c‐myc was seen and suggested to be independent of cell proliferation (Butterworth et al., 1994).
Gpt delta rats were given furan at 0, 2 or 8 mg/kg bw per day by gavage for 13 weeks (assumed daily) and the frequency of hepatic mutations in gpt or Spi were not altered despite an increase in PCNA‐positive hepatocytes, cyclin d1 and cyclin e1 mRNA levels and in the number and area of GST‐P‐positive foci at 8 mg/kg bw. These results do not support a role of genotoxicity in the production of the observed GST‐P‐ positive foci (Hibi et al., 2017).
Furan doses of 0.92, 2.0 or 4.4 mg/kg bw per day for 360 days in F344 rats also caused a dose‐dependent hepatic global DNA demethylation and a sustained decrease in the levels of histone methylation (de Conti et al., 2014). Reduction of histone lysine acetylation following treatment of F344 rats with 8 mg/kg bw of furan 5 days per week up to 90 days persisted after removal of the treatment (de Conti et al., 2015). The p53 target microRNA, miR‐34a, was significantly increased at 2 mg/kg bw per day (Dong et al., 2016). de Conti et al. (2016) showed that treatment of rats with furan at 0.92 or 2.0 mg/ kg bw per day for 90, 180 and 360 days led to increased expression of various microRNAs (miR‐34a, miR‐93, miR‐200a, miR‐200b and miR‐224) which was reversible following cessation of the dosing. In contrast, there was a sustained, reduced expression of miR‐375 in morphologically normal and in cholangiofibrotic liver tissue. This is a tumour suppressing microRNA involved in the regulation of Yes‐associated protein 1 which is a feature of hepatic carcinogenesis and the effects were seen in conjunction with increased methylation, both of cytosine in DNA and of lysine in histone H3K9 and H3K27 at the miR‐375 gene. BDA‐GSH binding to lysine 107 of histone H2B (see above, Nunes et al., 2016) may contribute to epigenetic interference.
In liver lesions of F344 male rats treated with furan (0.92 or 2 mg/kg bw daily for 104 weeks), 1,336 and 1,541 genes were found to be differentially expressed compared to control rat liver at the two doses, respectively. Of 1,001 transcripts mutually altered in expression at both doses, the expression level of a total of 42 of these was found to be negatively correlated with the methylation of their associated CpG Islands (Tryndyak et al., 2017). Thus, changes in DNA methylation appear to be involved in some of the gene expression changes induced by furan.
With regard to the induction of cholangiocarcinoma in rats, which is only seen at the top dose studied (8 mg/kg bw), evidence has been provided for an apparent sustained biliary cell and hepatocellular proliferation that persisted without recovery provided that the dosage is sufficient to cause extensive centrilobular hepatocellular toxicity (see Table 2; Hickling et al., 2010a,b). Indeed a biliary epithelial cell origin has been proposed for cholangiocarcinomas produced by furan (Sirica, 1996; Sirica et al., 1994a,b, 1997). Aberrant expression of HGF/SF expression in both furan–induced cholangiocarcinoma epithelium and in intestinal metaplastic epithelium, suggested that the latter are putative precancerous lesions (Lai et al., 2000). At a high dose of 30 mg/kg bw, this is associated with metaplasia in the liver and an inflammatory response involving excessive production of ROS, epigenetic changes and irreversible enhanced expression of DNA‐damage responsive genes, cytokines and microRNAs (Elmore and Sirica, 1991, 1992, 1993; Hickling et al., 2010a,b; Chen et al., 2012).
Changes in hepatic gene expression were also observed in mouse liver following furan exposure. Thus, in mice treated with a carcinogenic dose of 4 and 8 mg/kg bw per day, similar changes in hepatic gene expression were observed and the profile of expression changes differed from that at 2 mg/kg bw in which a more adaptive response was considered by the authors. Changes in the expression of NF‐kappaB and c‐Jun were suggestive of liver regeneration and oxidative stress‐mediated NRF2 activity. Chronic inflammation was suggested as a critical event in the transition between the adaptive changes and adversity (Jackson et al., 2014). Moreover, in mice treated for 3 weeks, long‐non‐coding RNA was differentially expressed at carcinogenic doses of 4 and 8 mg/kg bw but not at 1 or 2 mg/kg bw (Recio et al., 2013; Webster et al., 2014a,b). Cordelli et al. (2010) found altered expression of genes involved in the DNA damage response in mice treated with 15 mg/kg bw per day furan for 28 days although this was not associated with a change in DNA methylation.
Yauk et al. (2016) assessed the profile of gene expression produced by furan (68, 136 and 390 μg/mL, 4 h exposure) in human TK6 cells with rat liver S9 (induced by 5% ethanol to elevate CYP2E1). By comparing gene expression profiles for both genotoxic and non‐genotoxic chemicals, they concluded that the profile of furan matched that of a genotoxic chemical for 28 selected gene (TGx‐28.65) biomarkers. However, principal component analysis and dendrogram indicated that the profile of gene expression, albeit with a limited set of reporter genes for oxidative stress, was similar to acetaminophen that induces oxidative stress (Jaeschke et al., 2012) under the conditions of the study. Their analysis of micronuclei induction supported their conclusion that furan is clastogenic at high concentrations associated with oxidative stress. Furihata et al. (2016) were able to discriminate furan from 8 genotoxic carcinogens based on the hepatic gene expression profile of 100 candidate marker genes at 4 and 48 h after an i.p. dosage (30 mg/kg bw) in B6C3F1 male mice. These two studies do not support a direct genotoxicity of furan.
A computational analysis sought significant disease associations with either furan or 2,5‐dimethylfuran using data from the Comparative Toxicogenomics Database. There were inferred associations between furan and lung neoplasms, liver injury and cirrhosis based on 21 gene interactions. There were no detected direct associations between 2,5‐dimethylfuran and disease (Phuong et al., 2012).
In human lung alveolar epithelial cells, 2,5‐dimethylfuran at concentrations up to 2 mM showed no effect on viability or changes in gene expression as assessed using a human microarray platform (Cheah et al., 2013).
In conclusion, when considering MoAs, the contributing factors are likely to vary according to dose, length of exposure and degree of severity of liver cellular damage, inflammation and regenerative hyperplasia.
It is possible that, based on the potential for genotoxicity (evidenced in some but not all studies in vitro, and limited evidence in vivo of some chromosomal damage of unknown origin), the mode of carcinogenic action of furan, in rats and mice, may involve genotoxicity. However, there is clear evidence for the involvement of indirect mechanisms in the carcinogenic MoA of furan. These include epigenetic changes, oxidative damage to DNA and regenerative hyperplasia, with all of these effects being accompanied by tissue damage. Extensive protein binding of the reactive metabolite BDA restricts access to DNA. On the other hand, protein binding may contribute to the epigenetic changes (histone binding). Theoretically inactivation of proteins involved in DNA processing may also contribute to genomic instability, however, no studies were conducted with furan.
In particular, oxidative stress with associated production of DNA 8‐oxodeoxyguanosine in liver and 8‐oxodeoxyguanosine in blood, gene expression alteration and epigenetic changes are evidently involved along with an inflammatory and cell proliferative component to the MoA. This is particularly evident in the liver of rodents at 2 mg/kg bw and above accompanied by toxicity.
3.1.5. Considerations of critical effects and dose–response analysis
3.1.5.1. Considerations of critical effects
In rodents, furan was shown to be hepatotoxic and nephrotoxic when applied by oral gavage. The liver is the most sensitive organ. Furan leads to characteristic changes in markers related to hepatotoxicity as well as severe histopathological damage in the liver. The observed changes in blood parameters, including the effect on thyroid hormones, are considered secondary to changes in liver function. Hepatic toxicity is thought to be due to conversion of furan into the highly reactive metabolite, BDA. In long‐term experiments hepatic necrosis, cholangiofibrosis, regenerative hyperplasia, metaplasia and adenomas/carcinomas are observed. The CONTAM Panel noted that non‐neoplastic effects have been observed at low doses, i.e. cholangiofibrosis in a 2‐year study in male rats at doses as low as 0.2 mg/kg bw per day (NCTR, 2015) and an increase in serum T4 levels in a 90‐day study in male rats at doses of 0.12 mg/kg bw per day (Gill et al., 2010).
Neoplastic lesions comprise hepatocellular adenomas/carcinoma, mononuclear cell leukaemia, C‐cell adenoma/carcinoma and mesothelioma of the tunica vaginalis. The mononuclear cell leukaemia, C‐cell adenoma/carcinoma and mesothelioma of the tunica vaginalis were not considered critical for the risk assessment for reasons discussed in Section 3.1.2.3. The CONTAM Panel selected the incidence of hepatocellular adenomas/carcinoma as critical effect for the neoplastic changes.
There is clear evidence for the involvement of indirect mechanisms in the carcinogenic MoA of furan. These include epigenetic changes, oxidative damage to DNA and regenerative hyperplasia, with all of these effects being accompanied by tissue damage. In addition, extensive protein binding of the reactive furan‐metabolite BDA restricts access to DNA, and binding to histones may also contribute to the reported epigenetic changes.
There is limited evidence of a direct mechanism (i.e. direct interaction with DNA) in the carcinogenic action of furan. A very low level (see Section 3.1.1.4) of altered DNA bases was observed in vivo but their chemical structures could not be identified. These altered DNA bases are not identical to the adducts formed by reaction of the furan metabolite, BDA, with isolated DNA and DNA in bacteria. Nevertheless, there is evidence of chromosomal damage in vivo, but it is unknown whether this is caused by a direct or indirect mechanism.
The CONTAM Panel selected the incidence of cholangiofibrosis in male rats, increase in serum T4 in male and female rats and the incidence of hepatocellular adenoma and carcinoma in mice and male rats for the dose–response analysis of furan, but considered that there was insufficient information for the methylfurans.
3.1.5.2. Dose–response analysis (including benchmark dose analysis)
The BMD analysis performed followed the updated guidance of the Scientific Committee on BMD modelling (EFSA Scientific Committee, 2017) and a detailed description of the BMD analysis performed by the Panel can be found in Appendix C. The reported BMD10, BMDL10 and BMDU10 have not been corrected for the applied dose regimen (i.e. 5 days per week), unless indicated.
As described in Section 3.1.5.1, the CONTAM Panel considered the induction of cholangiofibrosis in rats as a critical effect following oral exposure to furan. The CONTAM Panel selected the most recent chronic study with male rats for dose–response modelling of the incidence of cholangiofibrosis after 36 weeks and after 2 years (NCTR, 2015; Von Tungeln et al., 2017). This study is particularly suitable for dose–response modelling as 8 dose levels (0, 0.02, 0.044, 0.092, 0.2, 0.44, 0.92 and 2 mg/kg bw per day, 5 days per week for 104 weeks) were tested with a higher number of animals per dose group in the lower dose range (see Table 6). A benchmark dose analysis was performed using the EFSA web‐tool, which is based on the R‐package PROAST 61.3 (see Appendix C). The default benchmark response (BMR) for quantal data was selected, i.e. an extra risk of 10% compared with the background risk. Using model averaging, the resulting BMDL10 for cholangiofibrosis after 36 weeks was 0.22 mg/kg bw and after 2 years 0.09 mg/kg bw (see Table 10).
Table 10.
Summary of the BMD analysis, using model averaging
| Response variable | Species and sex | BMD10 (mg/kg bw per day)a | BMDL10 (mg/kg bw per day)a | BMDU10 (mg/kg bw per day)a |
|---|---|---|---|---|
| Cholangiofibrosis after 36 weeks | Male rats | 0.3 | 0.22 | 0.41 |
| Cholangiofibrosis after 2 years | Male rats | 0.11 | 0.09 | 0.12 |
| Hepatocellular adenoma or carcinoma; combined data set from Moser et al. (2009) and NTP (1993) | Female mice | 2.6 | 1.84 | 4.00 |
| Hepatocellular carcinoma; combined data set from Moser et al. (2009) and NTP (1993) | ||||
|
Female mice Female mice |
5.72 7.21 |
4.18 4.82 |
7.24 9.10 |
|
| Hepatocellular adenoma or carcinoma; data set from Moser et al. (2009) only | Female mice | 2.4 | 1.45 | 3.59 |
| Hepatocellular carcinoma; data set from Moser et al. (2009) only | Female mice | 5.34 | 3.45 | 7.55 |
For a 5 days per week dosing regimen.
Although the effects on thyroid hormones are considered as secondary to changes in liver function, the CONTAM Panel performed a dose–response analysis of the increase of the serum T4 levels in a 90‐day study in male and female rats (Gill et al., 2010). The BMD analysis showed that there was insufficient information in these data on the dose–response to calculate a BMDL (i.e. the BMDL–BMDU interval ranged from 0 to infinite). Since the effects are observed in the same dose range as cholangiofibrosis, the changes in thyroid hormones were not used further in the risk assessment.
The induction of hepatocellular adenomas or carcinomas following furan exposure has been reported in male rats and male and female mice. The CONTAM Panel selected the incidence of hepatocellular adenomas or carcinomas (combined) and hepatocellular carcinomas (only) in female mice after 2 years reported in two independent studies (NTP, 1993; Moser et al., 2009) for BMD analysis, since this was a more robust data set covering the low dose‐range compared to the male mice and rats. A BMD analysis of the combined data from both studies (Moser et al. (2009) and NTP (1993); see Appendix C, Table C.8), using the studies as a covariate, was performed. Model averaging was used. The resulting BMDL10 for hepatocellular adenomas or carcinomas was 1.84 mg/kg bw per day for both studies combined. For hepatocellular carcinomas alone, the shape of the dose–response curves of the two studies differed and the resulting BMDL10 were 4.18 and 4.82 mg/kg bw for the Moser et al. and the NTP study, respectively. When analysing the Moser et al. study alone, the BMDL10 values were 1.45 and 3.45 mg/kg bw per day for hepatocellular adenomas or carcinomas and hepatocellular carcinomas, respectively.
Table C.8.
Data on the incidence of hepatocellular adenomas/carcinomas in female mice used for BMD analysis
| Dose (mg/kg bw per day)a | n hepatocellular adenomas and carcinomas | n hepatocellular carcinomas | n total | Reference |
|---|---|---|---|---|
| 0.0 | 3 | 0 | 36 | Moser et al. (2009) |
| 0.0 | 7 | 2 | 50 | NTP (1993) |
| 0.5 | 8 | 4 | 72 | Moser et al. (2009) |
| 1.0 | 6 | 2 | 53 | Moser et al. (2009) |
| 2.0 | 5 | 1 | 41 | Moser et al. (2009) |
| 4.0 | 12 | 2 | 36 | Moser et al. (2009) |
| 8.0 | 29 | 11 | 39 | Moser et al. (2009) |
| 8.0 | 34 | 7 | 50 | NTP (1993) |
| 15.0 | 50 | 27 | 50 | NTP (1993) |
N: number of animals; bw: body weight.
For a 5 days per week dosing regimen.
From this analysis, the CONTAM Panel selected the BMDL10 of 0.09 mg/kg bw per day (5 days per week dosing regimen) for the induction of cholangiofibrosis in male rats after 2 years as a reference point for non‐neoplastic effects. This corresponds to 0.064 mg/kg bw per day (correcting for the applied dose regimen of 5 days per week). For neoplastic effects, the CONTAM Panel considered that the combined data set from NTP (1993) and Moser et al. (2009) on the incidence of hepatocellular adenomas and carcinomas in female mice after 2 years is the most robust data set to derive a reference point and selected the BMDL10 of 1.84 mg/kg bw per day (5 days per week dosing regimen). This corresponds to 1.31 mg/kg bw per day (correcting for the applied dose regimen of 5 days per week).
3.1.6. Possibilities for derivation of a health‐based guidance value (HBGV)
Based on the available information, the CONTAM Panel concluded that there is clear evidence for the involvement of indirect mechanisms in the carcinogenic MoA of furan. However, there are also some indications for a direct genotoxic mechanism. Therefore, the CONTAM Panel considered it inappropriate to establish a tolerable daily intake (TDI). The CONTAM Panel selected a BMDL10 of 0.064 mg/kg bw per day for the incidence of cholangiofibrosis and a BMDL10 of 1.31 mg/kg bw per day for the incidence of hepatocellular adenomas or carcinomas as reference points and to use an MOE approach for the risk characterisation.
3.2. Occurrence data
3.2.1. Factors influencing furan and methylfurans levels in food
Both the food matrix and the process variables will have an impact on the formation and loss of furan and methylfurans in foods. Currently, most of the information is related to furan. In view of its chemical properties, it is supposed that the formed furan is chemically stable during storage of foods, although limited studies examining stability are available (Guenther et al., 2010). Losses for furan are typically linked to its high volatility (evaporation). First, the factors impacting furan formation in foods and model systems will be summarised, followed by reviewing the factors influencing the evaporative losses of furan. Similarly, the available information on methylfurans will be discussed.
Factors influencing furan formation in food and model systems
As outlined in Section 1.3.5, several precursors and formation mechanisms have been suggested. Most of these precursors have been identified in simplified model systems in which the complexity of a real food matrix cannot be mimicked. Indeed, it should be realised that in foods, multiple precursors can be present, and that their formation pathways may interfere with each other. It also seems that interaction with non‐precursor matrix components (e.g. starch) may impact the furan formation (e.g. Owczarek‐Fendor et al., 2010a).
Only a limited number of studies have tried to corroborate the specific role of particular precursors in real food systems for which stable isotope labelled precursors are necessary. Such studies revealed that the role of some precursors can be largely overestimated. Limacher et al. (2008) reported that only 21% of the formed furan originated from the available sugars in the pumpkin puree studied, while the remaining 79% was suggested to result from lipids, although this reaction pathway was not considered or investigated.
In an acidic environment, it has been shown that sugars are less efficient precursors in model systems compared to neutral or alkaline conditions (e.g. Limacher et al., 2008; Owczarek‐Fendor et al., 2012). For ascorbic acid, an acidic pH favours the conversion to furan (e.g. Limacher et al., 2007; Owczarek‐Fendor et al., 2010a). Care needs to be taken in the interpretation of these observations as the pH of a model system is sometimes difficult to control, it varies as a function of the temperature, and some buffers, like phosphate, are supposed to enhance furan formation as well (e.g. Fan et al., 2008).
The redox potential is another matrix‐relevant parameter. The use of antioxidants in model systems reduces lipid peroxidation and furan formation (Becalski and Seaman, 2005). Metal ions, as pro‐oxidants have the opposite effect (Becalski and Seaman, 2005). It should be noticed that the different reports are not always consistent (Märk et al., 2006). Reduction of the available amount of oxygen in food products (in potato purées supplemented with, respectively, ascorbic acid, fructose and poly‐unsaturated fatty acids; Palmers et al., 2016) or model systems (Märk et al., 2006) resulted in a variable decrease in the furan formation.
The process variables time and temperature have an impact on furan levels in food. Sterilisation treatments typically result in higher furan levels compared to pasteurisation treatments (Fan et al., 2008). Dry heating, generally more intense than sterilisation or pasteurisation treatments, typically results in higher furan levels (Limacher et al., 2007, 2008). This is consistent with the observation that the highest concentrations of furan are reported in roasted coffee (EFSA, 2010c). Furan levels in coffee increase with the degree of roasting (Guenther et al., 2010; Altaki et al., 2011; Arisseto et al., 2011) and this effect is attributed to the increase in roasting temperature. Coffees with the same degree of roasting but roasted using different time–temperature conditions show different levels, with the highest levels observed after quick roasting at high temperature (Altaki et al., 2011). Also, during deep‐frying the applied time‐temperature combination influences the furan level. DTU (2009) studied the levels of furan in chips and crisps during deep‐frying as a function of different time‐temperature combinations. No difference was observed in the level of furan in chips fried at 160°C for 6 mins or 175°C for 4 mins. However, higher levels were found in chips fried for 3 mins at 190°C. For crisps, the authors observed an increase in furan level with increased frying temperature and decreased frying time (7 mins at 150°C vs 3.5 mins at 190°C). In toast, on the other hand, the influence of increasing toasting time, without changing the toasting temperature, was studied. The level of furan increased with toasting time and consequently with the degree of browning.
In aqueous solutions, it was initially accepted that temperatures of 90–100°C resulted in a limited amount of furan formation and that typically higher temperatures were necessary (e.g. Fan et al., 2008; Owczarek‐Fendor et al., 2010a). Subsequently, it was shown that during storage at 35°C, thermally treated fruit juices were able to generate furan, while the same authors observed limited additional furan formation during storage of vegetable purées at 4°C (Palmers et al., 2015a,c). Presumably, precursors formed during thermal treatment or storage are further converted to furan during long‐term storage, without the need for additional thermal input. In comparison, conventional sterilisation techniques, high‐pressure high‐temperature sterilisation or alternative heat treatments (e.g. dielectric heating) resulted in a lower furan formation due to a lower time‐temperature impact (Palmers et al., 2015b).
In addition, the influence of ionising and UV‐C radiation on furan formation has been studied (Fan, 2005, 2015; Fan and Sokorai, 2008; Hu et al., 2016), but given the limited application of these techniques at EU level the Panel did not consider this for this opinion.
Factors influencing furan losses in food
Van Lancker et al. (2009) showed that the evaporation of furan is highly influenced by the composition of foods (e.g. lipids, starch). In foods such as coffee, although coffee is roasted at temperatures above 200°C, furan is still present. The retention of furan in this matrix can be explained by the interaction of furan with matrix components, such as the lipids and the low molecular weight extractables (Van Lancker et al., 2009).
Given the volatility of furan, various studies considered the evaporation of furan from samples before consumption, typically during reheating and open storage conditions (e.g. Kim et al., 2009; Fromberg et al., 2014; Palmers et al., 2015a). Appendix D, Table D.1 gives an overview of furan concentrations in commercially processed foods, before and after heating by the consumer, reported in the scientific literature. As shown in Appendix D, Table D.1, limited evaporation occurs and variable results are obtained, depending upon the particular experimental conditions (e.g. time, temperature, stirring, type of container).
Table D.1.
Influence of heating by the consumer on furan levels in commercially heat‐processed foods as reported in scientific literature
| Matrix | n | Concentration (μg/kg)e | Reduction factord | Reference | |
|---|---|---|---|---|---|
| As purchased | As consumed | ||||
| Foods for infants and small children | |||||
| Varied vegetables | 3 | 7.3 ± 0.6 |
6.0 ± 0.3a 5.2 ± 0.2a 4.0 ± 0.2c |
1.2 1.4 1.8 |
Altaki et al. (2017) |
| Vegetables cream with pasta | 3 | 29 ± 1 |
25 ± 1a 22 ± 1a 17 ± 1c |
1.2 1.3 1.7 |
Altaki et al. (2017) |
| Chicken stew with vegetables | 3 | 38 ± 2 |
32 ± 2a 25 ± 2a 18 ± 1c |
1.2 1.5 2.1 |
Altaki et al. (2017) |
| Chicken with rice | 3 | 16 ± 1 |
15 ± 1a 13 ± 1a 10 ± 1c |
1.1 1.2 1.6 |
Altaki et al. (2017) |
| Pea with pork meat | 3 | 53 ± 3 |
60 ± 3a 41 ± 2a 32 ± 1c |
0.9 1.3 1.7 |
Altaki et al. (2017) |
| Pork meat with pasta and vegetables | 3 | 49 ± 2 |
46 ± 2a 32 ± 3a 23 ± 2c |
1.1 1.5 2.1 |
Altaki et al. (2017) |
| Beef meat with rice | 3 | 44 ± 3 |
46 ± 2a 34 ± 2a 25 ± 2c |
1.0 1.3 1.8 |
Altaki et al. (2017) |
| Jardinière beef meat | 3 | 51 ± 3 |
56 ± 2a 41 ± 2a 35 ± 2c |
0.9 1.2 1.5 |
Altaki et al. (2017) |
| Lamb meat stew with potato | 3 | 34 ± 2 |
37 ± 2a 25 ± 2a 22 ± 2c |
0.9 1.4 1.5 |
Altaki et al. (2017) |
| Lamb meat stew with green bean | 3 | 43 ± 2 |
36 ± 3a 30 ± 1a 23 ± 1c |
1.2 1.4 1.9 |
Altaki et al. (2017) |
| Monkfish with potato | 3 | 84 ± 4 |
85 ± 1a 65 ± 3a 45 ± 3c |
1.0 1.3 1.9 |
Altaki et al. (2017) |
| Hake with rice | 3 | 69 ± 3 |
76 ± 2a 55 ± 2a 45 ± 2c |
0.9 1.3 1.5 |
Altaki et al. (2017) |
| Chicken | 1m | 23 ± 6f |
17 ± 19g 18 ± 17c |
1.4 1.3 |
Hasnip et al. (2006) |
| Vegetable | 1m | 32 ± 8f |
31 ± 35g 20 ± 19c |
1.0 1.6 |
Hasnip et al. (2006) |
| Retort packaged baby food 1 | n.r. | 28.3 ± 0.5 |
19.0 ± 1.2a 20.8 ± 1.1b |
1.5 1.4 |
Kim et al. (2009) |
| Retort packaged baby food 2 | n.r. | 10.8 ± 0.6 |
9.1 ± 0.5a 9.6 ± 0.2b |
1.2 1.1 |
Kim et al. (2009) |
| Soybean milk 1 | n.r. | 4.3 ± 0.3 |
4.3 ± 0.2a 3.6 ± 0.2b |
1.0 1.2 |
Kim et al. (2009) |
| Soybean milk 2 | n.r. | 14.9 ± 0.8 |
12.0 ± 0.4a 12.3 ± 0.2b |
1.2 1.2 |
Kim et al. (2009) |
| Infants vegetable meal | 1 | 45n | 23l | 2 | DTU (2009) |
| Herbs, spices and condiments | |||||
| Tomato‐based sauce | 1m | 12 ± 3f |
6 ± 6h 9 ± 10b 7 ± 8a 6 ± 7b |
2.0 1.3 1.7 2.0 |
Hasnip et al. (2006) |
| Sweet and sour vegetable sauce | 1 | 13n | 7l | 1.8 | DTU (2009) |
| Composite food | |||||
| Baked beans | 1m | 24 ± 6f |
31 ± 35g 24 ± 23h |
0.8 1.0 |
Hasnip et al. (2006) |
| Baked beans | 1 | 102n | 42l | 2.4 | DTU (2009) |
| Stew | 1m | 25 ± 6f |
17 ± 16h 26 ± 30b 17 ± 19a 16 ± 19b |
1.5 1.0 1.5 1.6 |
Hasnip et al. (2006) |
| Korean seasoned pork canned | n.r. | 45.2 ± 1.5 |
31.6 ± 4.7i 24.3 ± 8.5j |
1.4 1.9 |
Kim et al. (2009) |
| Luncheon meat canned | n.r. | 3.9 ± 0.5 |
2.9 ± 0.4i 2.7 ± 0.9j |
1.3 1.4 |
Kim et al. (2009) |
| Meat sauce | 1 | 40n | 16l | 2.5 | DTU (2009) |
| Tomato soup | 1 | 25n |
32g 26h |
0.8 1.0 |
Crews (2009) |
| Vegetable soup | 1m | 17 ± 4f |
19 ± 11h 21 ± 15g |
0.9 0.8 |
Hasnip et al. (2006) |
| Soup | 10 | 7–47 | 1–16l | 1.5–12.5 | DTU (2009) |
| Vegetables canned | 1m | 4 ± 2f |
4 ± 5h 3 ± 5b 3 ± 4a 3 ± 4b |
1.0 1.3 1.3 1.3 |
Hasnip et al. (2006) |
| Tuna canned | n.r. | 12.6 ± 0.1 | 2.5 ± 0.2k | 5.0 | Kim et al. (2009) |
| Mackerel pike canned | n.r. | 13.7 ± 0.9 | 3.1 ± 0.3k | 4.4 | Kim et al. (2009) |
N: number of samples.
Heating experiments that were not considered relevant for heating carried out by consumers were not added to this table (e.g. heating baby food for 1 h at 80°C). Experiments for which the furan concentrations were below the LOD/LOQ were not included in this table.
Microwave; without lid (uncapped jar/dish/open casserole).
Microwave; with lid (glass/plastic lid).
Hot water bath; without lid (uncapped jars).
Calculated from (mean) concentrations reported by the authors by dividing the furan concentration in the food as purchased by the furan concentration in the food as consumed.
Mean ± standard deviation unless stated otherwise.
Mean ± measurement uncertainty (including uncertainty due to analysis and heating).
Microwave; without specification regarding lid.
Saucepan heating with stirring.
Microwave; heating to 50°C; without specification regarding lid.
Microwave; heating to 70°C; without specification regarding lid.
Heating without a lid to 100°C.
Calculated based on % of furan left after heating as reported by the authors; heating unspecified.
Heating experiments were carried out at least in duplicate.
Single concentration.
From the information available in scientific literature, the CONTAM Panel concluded that the influence of reheating commercially processed foods on furan levels is limited and highly dependent on the consumer behaviour which is not predictable. Moreover, it should be noted that some consumers might consume these products without heating.
During beverage preparation however, furan levels decrease substantially due to a combination of dilution, evaporation and partial extraction. Guenther et al. (2010) estimated losses regardless of the dilution for each process/handling step of roasted coffee: grinding (40%), degassing (20%), shelf life (0% as long as original package is closed, 25% when original package is opened), brewing (55%) and standing time (10–35%). Several other studies investigated the effect of the brewing method in more detail. An overview of furan levels in coffee before and after brewing retrieved from those studies is provided in Appendix D, Table D.2. In this table, only the studies that provided sufficient information on any of the following parameters were retained: brewing method, amount of solid coffee used for brewing, volume of brew obtained and concentrations of furan before and after brewing. These data allowed the CONTAM Panel to calculate for each sample an overall reduction factor by dividing the furan concentration in the food as purchased by the furan concentration in the food as consumed. This reduction factor comprises a dilution and a loss factor. The loss factor is considered as an indicator for the amount of furan lost during the brewing process, regardless of the dilution effects. The higher the loss factor, the higher the amount of furan lost during the brewing process.
Table D.2.
Influence of beverage preparation by the consumer on furan levels as reported in scientific literature
| Coffee type | N | Amount of solid (g)a | Volume of brew (mL)a | Concentration (μg/kg)a | Dilution factora | Loss factora | Overall reduction factora , b | Reference | |
|---|---|---|---|---|---|---|---|---|---|
| Solid | Brew | ||||||||
| Ground coffee | |||||||||
| Boiled/Turkish coffee | 16 | 50 | 500 | 3,457.5 (1,373–5,580) | 29 (12–81) | 10 | 11.6 (3.6–45.6) | 115.9 (35.9–455.7) | Arisseto et al. (2011) |
| Espresso | 7 | 5 | 75 | 1,770 (1,490–2,020) | 43 (28–60) | 15 | 2.5 (1.7–4.3) | 37.3 (26.1–64.3) | Kuballa et al. (2005) |
| Espresso | 5 | 8 | 64 (62–67) | 3,493 (2,254–5,938) | 98 (72–199) | 8 (7.8–8.4) | 4.6 (2.1–6.4) | 37 (17.6–49.6) | Zoller et al. (2007) |
| Filter coffee | 20 | 50 | 500 | 3,457.5 (985–5,697) | 97.5 (19–279) | 10 | 4 (2–7.8) | 39.6 (20–78.4) | Arisseto et al. (2011) |
| Filter coffee | 1 | 32 | 740 | 2,080 | 46.7 | 23.1 | 1.9 | 44.5 | Becalski et al. (2016) |
| Filter coffee | 3 | 40 | 1,000 | 1,892 (1,681–1,966) | 56.7 (51.5–64.3) | 25 | 1.4 (1–1.5) | 34.7 (26.1–36.7) | DTU (2009) |
| Filter coffee | 15 | 40 | 850 | 1,790 (790–3,390) | 15 (8–66) | 21.3 | 6.4 (1.1–8.9) | 135.6 (23.3–190) | Kuballa et al. (2005) |
| Filter coffee | 1 | 8 | 130 | 1,438 | 40 | 16.3 | 2.2 | 36 | Zoller et al. (2007) |
| French press/infusion | 1 | 51 | 1,000 | 2,080 | 77.6 | 19.6 | 1.4 | 26.8 | Becalski et al. (2016) |
| Coffee beans | |||||||||
| Espresso | 6 | 10 | 150 | 4,725 (3,510–6,100) | 91.5 (57–115) | 15 | 3.5 (2.1–6.2) | 52.5 (30.8–93.7) | Kuballa et al. (2005) |
| Filter coffee (machine) | 5 | 40 | 850 | 4,910 (3,600–6,100) | 16 (9–33) | 21.3 | 15.7 (5.1–23.9) | 333.8 (109.1–508.3) | Kuballa et al. (2005) |
| Filter coffee (manual) | 5 | 40 | 850 | 4,910 (3,600–6,100) | 20 (17–24) | 21.3 | 12 (8.1–14) | 254.2 (171.4–296.7) | Kuballa et al. (2005) |
| French press/infusion | 5 | 40 | 850 | 4,910 (3,600–6,100) | 51 (33–66) | 21.3 | 4.5 (3.5–6.5) | 95.4 (74.4–137.6) | Kuballa et al. (2005) |
| Cartridge coffee | |||||||||
| Not specified | 3 | 8 (7–9) | 210 | 2,300 (2,110–2,660) | 59.1 (57.6–59.2) | 26.3 (23.3–30) | 1.5 (1.5–1.5) | 38.9 (35.7–46.2) | Becalski et al. (2016) |
| Instant coffee | |||||||||
| Instant coffee | 2 | 2 | 60 | 960 (820–1,100) | 31.5 (28–35) | 30 | 1 | 30.4 (29.3–31.4) | Altaki et al. (2009) |
| Instant coffee | 2 | 1.4 (1.2–1.5) | 112.5 (75–150) | 977.5 (625–1,330) | 5.5 (4.3–6.6) | 81.3 (62.5–100) | 2.2 (2–2.3) | 173.4 (145.3–201.5) | DTU (2009) |
| Instant coffee | 7 | 10 | 500 | 530 (240–2,200) | 11 (3–25) | 50 | 1.5 (0.8–3.1) | 75.7 (41.8–157.1) | Kuballa et al. (2005) |
| Instant coffee | 14 | 2 (1.7–2) | 150 (65–150) | 1,702 (44–2,150) | 19 (1–51.3) | 75 (32.5–89.8) | 1.1 (0.6–1.4) | 82.4 (41.9–97.9) | Zoller et al. (2007) |
Median (range).
The overall reduction factor is calculated by dividing the furan concentration in the food as purchased by the furan concentration in the food as consumed. This reduction factor comprises a dilution and a loss factor.
Although highly variable results are obtained, some trends were identified when merging the individual loss factors obtained throughout the different studies (Appendix D, Table D.2). The highest losses of furan were observed when whole coffee beans are used as a starting material, indicating that loss of furan does not only occur during the brewing process itself, but also during the grinding. When ground coffee was used as a starting material, the highest loss factors were obtained for boiled/Turkish coffee (Median = 11.6; P25 = 5.9; P75 = 18.5). Loss factors for filter coffees (Median = 4.2; P25 = 2.3; P75 = 5.9) were also found to be higher compared to espresso coffees (Median = 2.8; P25 = 2.3; P75 = 4.4). Altaki et al. (2011) explained the higher transfer of furan to espresso brews by the higher pressure applied in the espresso machine which increases the furan extraction compared to a filter coffee maker. Also, the time difference between both brewing methods to prepare a cup of coffee may explain the higher levels of furan in espresso (± 30 s for espresso vs ± 100 s for filter coffee). For instant coffees, the loss of furan during beverage preparation is considered negligible as the median loss factor amounted to 1.1 (P25 = 1.0; P75 = 1.4).
Several researchers studied the impact of stirring and the standing‐time (time between preparation and consumption) on furan levels. In general, stirring the sample results in a decrease of furan, while variable effects have been observed during standing. Mesias and Morales (2014) studied the effect of standing and stirring on the furan level in brewed coffee: Standing at room temperature for 5 min resulted in a loss of 74%, while standing at room temperature for 5 min in combination with 30 s stirring resulted in a loss of 64%. Stirring on the other hand for 5 min resulted in losses of 94% and coffee that was kept for 8 h in a sealed thermo had furan levels that were 98% lower. Also, Kim et al. (2009) and Becalski et al. (2016) observed a decrease of the furan in brewed coffee during standing. Roberts et al. (2008) on the other hand demonstrated that furan levels in composite foods that were left to stand without stirring did not change significantly, while stirring the foods resulted in lower furan levels. Altaki et al. (2017) on the other hand observed losses between 22% and 47% when baby food was standing for 5 min. For methylfurans, few studies are available. Palmers et al. (2014) confirmed the susceptibility of heat‐treated tomato based products for 2‐methylfuran and 3‐methylfuran formation as reported in a survey of Becalski et al. (2010), while for other vegetable products (broccoli, carrot, pumpkin, potato) formation was restricted (up to 127 μg/kg in tomato product for the sum of 2‐ and 3‐methylfuran, others < 20 μg/kg). It is, however, not clear which factors are responsible for these remarkable differences.
Factors influencing methylfurans levels in food
Although amino acid degradation is considered as an important pathway in the 2‐methylfuran formation, addition of supplementary glucose to a pumpkin puree (stimulating the Maillard reaction) did not result in a significant increase of 2‐methylfuran (Limacher et al., 2008). From the methylfuran survey data, however, it is obvious that various factors should have an impact on the formation of methylfurans in foods (Becalski et al., 2010).
In contrast to furan, methylfurans seem to be prone to degradation or loss in some foods during storage. Palmers et al. (2015a) observed that during the storage of pasteurised tomato purée at 35°C, a reduction was seen from 127 μg/kg to 13 μg/kg, while in other vegetable purée or fruit juices, this was not observed or at least not to this extent. The decrease observed at 4°C was lower (24%). The cause of the decrease is not known.
Summary
The food characteristics, processing and cooking conditions, and the losses which occur mainly due to evaporation in the preparation of the food at the level of the consumer, affect the final concentration in the food as consumed.
3.2.2. Occurrence data submitted to EFSA
Although the call for data requested the submission of chemical occurrence data on furan, 2‐methylfuran and 3‐methylfuran, only data for furan were made available within this framework. The data for the present assessment were provided by national authorities from Austria, Belgium, China, Cyprus, the Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Ireland, Italy, Lithuania, Luxembourg, the Netherlands, Norway, Poland, Slovakia, Slovenia, Spain and United Kingdom, and by the European Breakfast Cereal Association (CEEREAL), the European Coffee Federation (ECF) and Specialised Nutrition Europe (SNE).
An initial number of 18,141 analytical results on furan in food were available. This data set also included the 5,500 records included in the latest report on furan in food (EFSA, 2011d). These 5,500 records were previously validated by EFSA and incorporated in the current assessment without further involvement of the data providers. The remaining data were subject to the validation processes described in Section 2.3.2, and data providers were invited to clarify possible inconsistencies identified by EFSA's DATA Unit. Where such a clarification was not received from the data provider, the CONTAM Panel was required to apply certain assumptions. These assumptions, and the number of records impacted are summarised in Annex A, Table A.1.
Furthermore, the overall data set was carefully analysed before being used to estimate dietary exposure and a total of 378 records were excluded from the final data set. Criteria for exclusion of these analytical results are also detailed in Annex A, Table A.1.
3.2.2.1. Governmental data
The data set retained for assessment included 10,370 analytical results from governmental organisations. As requested through the call for data, Member States reported analytical results for commercial foodstuffs as purchased, disregarding any further preparation, and commercial foodstuffs analysed as consumed after further preparation in the laboratory. Since the data set also included samples analysed both as purchased and as consumed, the number of analytical results reported above actually corresponds to a total of 9,663 samples. Samples were taken in 21 Member States (Figure 8) throughout the years 2004–2016 (Figure 9).
Figure 8.
Number of governmental samples reported by Member State
Figure 9.
Number of governmental samples reported by year
Apart from the above‐mentioned samples that were analysed both as purchased and as consumed (n = 707), available samples were analysed only once, either as purchased (n = 8,531) or as consumed (n = 237). For some remaining samples (n = 188), data providers did not clearly specify whether the samples were analysed as purchased or as consumed, but on the basis of the observed levels these samples were assumed to be analysed as purchased.
In view of the exposure assessment, the data were grouped at different FoodEx levels taking into consideration several factors including the similarities between food categories, the number of samples and the concentrations observed (Annex A, Table A.2). The CONTAM Panel agreed that where a sample was analysed both as purchased and as consumed, only the analytical result for the sample analysed as consumed would be retained for assessment because this is the most representative for the consumer exposure. Furthermore, in order to assess the impact of food and beverage preparation on the concentration levels of furan, reduction factors were calculated for each sample that was analysed both as purchased and as consumed (samples with left‐censored results were excluded). For those food categories where no dilution occurs during food preparation (i.e. heating of commercially processed foods), the median reduction factors are reported in Table 11.
Table 11.
Reduction of furan levels during heating of commercially processed foods for which no dilution occurs
| Food group | Number of samples | Median reduction factora |
|---|---|---|
| Grains and grain‐based products | 3 | 1.1 |
| Vegetables and vegetable products (including fungi) | 13 | 1.1 |
| Legumes, nuts and oilseeds | 4 | 1.3 |
| Fruit and fruit products | 1 | 3.2 |
| Meat and meat products (including edible offal) | 1 | 1.6 |
| Herbs, spices and condiments | 18 | 1.1 |
| Food for infants and small children | 157 | 1.1 |
| Composite food (including frozen products) | 130 | 1.2 |
| Snacks, desserts and other foods | 2 | 1 |
Note that in this specific case the reduction factor is equal to the loss factor.
These data indicate that heating of commercially available foods (where no dilution occurs) has a limited impact on the levels of furan with median reduction factors generally ranging from 1 to 1.2. Median reduction factors in legumes, fruit and fruit products, and meat and meat products were higher (amounting to 1.3, 3.2 and 1.6, respectively), but the number of samples in these food groups was not considered sufficient to demonstrate that a significant loss of furan is expected during preparation of such food items. These findings are consistent with findings in scientific literature (see Section 3.2.1) and it was agreed that for these food categories further distinction between foods analysed as purchased or consumed would not be meaningful.
A similar analysis was attempted for foods and beverages where furan concentrations are expected to decline, not only due to the possible loss of furan during preparation, but also through the effect of dilution (i.e. cereal‐based products for infants and young children, infant and follow‐on formulae, and cocoa, tea, coffee and coffee imitate beverages). However, in most of these samples, the preparation method applied by the laboratories was not reported and it could not be assessed to what extent the decline of furan concentrations in these samples was driven by the effect of dilution or by the loss of furan during preparation. For these reasons, the information on coffee retrieved from scientific literature was considered more useful (Appendix D, Table D.2). Meanwhile, it was agreed that for these food categories furan concentrations in the food as purchased are not comparable to the furan concentrations in the food as consumed. Samples analysed as purchased were reported under the corresponding solid or powder, while the samples analysed as consumed were reported under the corresponding beverage or food.
The Panel also noted that the food category ‘Coffee beans, roasted’ may have been used by some Member States to report any unspecified roasted coffee sample (regardless whether it was ground or not). In order to allow for a better comparison between ground and unground roasted coffee, only the samples where the product description explicitly refers to grains or beans were retained in this food category. All other samples were reported as unspecified coffee beans and coffee products. A similar analysis was made for coffee imitates, in order to make a distinction between instant coffee imitates and coffee imitates for brewing.
Based on this classification, the highest number of samples was reported for ‘Foods for infants and small children’ with a particular attention to ready‐to‐eat meals for infants and young children (n = 2,090), and ‘Vegetables and vegetable products (including fungi)’, which includes the coffee beans and solid coffee products (n = 1,547) as well as the solid coffee imitates (n = 136). Other well‐represented food groups are composite foods (n = 1,292) and grains and grain‐based products (n = 1,207). These also correspond to the most relevant food categories in terms of furan levels.
The highest concentrations of furan were found in whole roasted coffee beans, with a mean value of 3,956 μg/kg and the highest individual value of 12,810 μg/kg. The mean values in ground roasted coffee (2,446 μg/kg) and unspecified coffee solids (2,186 μg/kg) were significantly lower, suggesting a loss of furan during the process of grinding, while the mean concentration in instant coffee powder was found to be even lower (429 μg/kg). A similar observation was made for coffee imitates where concentrations in solids for brewing (1,922 μg/kg) were higher compared to the instant powders (127 μg/kg). Due to the low number of left‐censored data within these food categories, the difference between lower bound and higher bound estimations was negligible.
In prepared salads and cereal‐ and vegetable‐based composite foods, mean furan concentrations ranged from 10 to 20 μg/kg, while in other composite foods (including the unspecified ones) mean levels ranged between 20 and 34 μg/kg. On average, levels in ready‐to‐eat meals for infants and young children were similar (31 μg/kg), but in this case, the quality of the data was not sufficient to allow for further disaggregation on the basis of the meal content. Comparable mean levels were also found in bread and rolls, raw pasta, breakfast cereals and fine bakery wares (16–30 μg/kg).
All remaining samples included a wide range of other foods, most of which are also expected to be commercially prepared foods (including canned and jarred products). Nevertheless, mean levels of furan in these food categories were generally found to be below 20 μg/kg, with the exception of soy sauce (32 μg/kg) and spirits (57 μg/kg).
Almost 90% of the samples were taken through objective, selective or convenient sampling while for the majority of the remaining samples the sampling strategy was not specified. Suspect sampling was reported for only 0.4% of the samples. However, the CONTAM Panel noted that there are no legal limits or indicative values for furan, and that the compound is present due to processing and not due to intentional use or environmental contamination. Therefore, the Panel considered it would be difficult in practice to identify suspect samples and that it cannot be excluded that the product was placed on the market. It was therefore decided to retain those samples in the data set.
The majority of the samples were analysed by means of different GC–MS techniques, except for approximately 27% of the samples where Member States did not provide a detailed classification of the method. LOQs were reported for more than 97% of the samples (Figure 10). Samples where the LOQ value was omitted either referred to a sample with quantifiable levels or to a sample with residues below the LOD.
Figure 10.
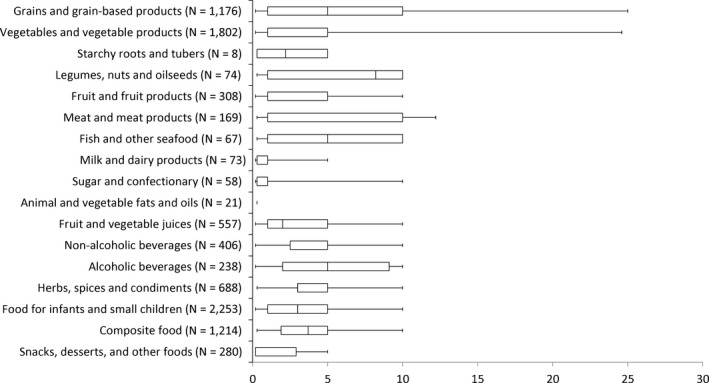
Distribution of reported limits of quantification in μg/kg, presented by food group
- Vertical line represent the median value, box outline represent 25th and 75th percentiles and whiskers represent 5th and 95th percentiles.
Furan may also be a constituent of the food additive caramel colours and the food flavouring rum ether. Caramel colours were re‐evaluated by the ANS Panel (EFSA ANS Panel, 2011) and according to a refined exposure assessment combining all caramel colours (EFSA, 2012), the main contributors to the mean exposure of toddlers and children to caramel colours were identified as ‘Dairy products and analogues’, ‘Salts, spices, soups, sauces, salads and protein products’, ‘Non‐alcoholic beverages’, ‘Edible ices’ and ‘Bakery wares’. For adults, elderly and very elderly, the main contributors to the mean exposure were ‘Salts, spices, soups, sauces, salads and protein products’, ‘Non‐alcoholic beverages’ and ‘Alcoholic beverages, including alcohol‐free and low‐alcohol counterparts’. Rum ether was recently evaluated by the CEF Panel (EFSA CEF Panel, 2017). For this flavouring, a refined exposure assessment was not carried out, but use levels were reported for ‘Edible ices’, ‘Confectionery’, ‘Chewing gum’, Cereal and cereal products derived from cereal grains, roots and tubers, and pulses and legumes, excluding bakery wares of food category 7.0’, ‘Bakery wares’, ‘Meat and meat products’, ‘Salts, spices, soups, sauces, salads, protein products (including soya bean protein products) and fermented soya bean products’, ‘Non‐alcoholic beverages’ and ‘Alcoholic beverages’.
Although slightly different food classifications are applied in the area of food additives and flavourings, it can be concluded that the above reported food categories are covered by the available occurrence data on furan. It is therefore expected that most of the furan levels resulting from the use of caramel colours and rum ether have been captured in the final data set, possibly contributing to the relatively high levels of furan observed in certain food items (e.g. fine bakery wares, soy sauce and spirits).
Occurrence of 2‐methylfuran and 2,5‐dimethylfuran resulting from their use as a food flavouring is no longer expected since both substances were removed from the Union list of flavourings (see Section 1.3.4).
3.2.2.2. Commercial data
A total number of 7,393 analytical results reported by commercial organisations were retained. Samples analysed both as purchased and as consumed were in this case not reported; hence the available data set refers to the same number of samples. The highest number of samples was reported by ECF, mainly covering the years 2010–2016. From 2012 onwards, samples were also reported by CEEREAL and SNE (Figure 11).
Figure 11.
Number of samples reported by commercial organisations, presented by year
- CEEREAL: European Breakfast Cereal Association, ECF: European Coffee Federation, SNE: Specialised Nutrition Europe.
In view of a possible comparison, the commercial data were grouped in the same way as for the governmental data (Annex A, Table A.2). Reported samples mainly referred to coffee beans and coffee products (n = 6,796), coffee beverages (n = 131), ready‐to‐eat meals for infants and young children (n = 348), and breakfast cereals (n = 67). All samples were analysed as purchased, except for the samples in coffee beverage which were assumed to be solid coffee samples analysed as consumed. However, as the corresponding solid samples could not be identified, a detailed impact assessment of coffee preparation on the levels of furan was not possible.
For most commercial data (over 99%), the sampling strategy remained unspecified but, as for the governmental data, this is not expected to have a major impact on the assessment.
Most samples were analysed using a GC–MS method, while for 114 samples in coffee beverage the data provider was not able to retrieve the adequate classification for the method. LOQs were reported for approximately 17% of the samples, ranging from 3 to 10 μg/kg. For the remaining samples LOQs were not reported, but all these samples referred to quantifiable levels of furan in roasted coffee beans (incl. ground coffee), instant coffee and coffee beverages.
In view of comparing the governmental and commercial data, several statistical tests were considered but they were not adequate, in particular, for those food categories with a high percentage of left‐censored data. Nevertheless, looking at the most predominant food categories (coffee beans and coffee products, coffee beverages, ready‐to‐eat meals for infants and young children, and breakfast cereals), commercial data were generally found to be higher or within the same range as the governmental data. One exception was identified for instant coffee powder where the mean of the governmental data is approximately 50% higher compared to the commercial data. Due to the high number of commercial samples, inclusion of the commercial data in the assessment would significantly lower the estimated mean for instant coffee powder. On the other hand, the CONTAM Panel also noted that furan levels in instant coffee liquid are relatively low and that contribution of instant coffee to the overall exposure is expected to be low, as already indicated in a previous assessment by EFSA (2011d). The Panel therefore concluded that it would be appropriate to merge both data sets in view of the exposure assessment.
3.2.3. Previously reported occurrence data in the open literature
EFSA and JECFA reviewed occurrence data on furan in foods up to 2010 (EFSA, 2011d; FAO/WHO, 2011). A literature search for recent reviews was conducted. However, since the occurrence of methylfurans was not addressed in the identified reviews, a separate search to identify papers regarding the occurrence of 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran was conducted. In addition, the VCF database20 on volatile compounds in food was consulted to identify papers on methylfurans.
There are numerous citations reporting the occurrence of furan and methylfurans in the aroma profiles of foods, of which some examples are presented below. These studies refer mainly to commercially heat‐processed foods, however, some report the natural occurrence of furan and methylfurans in unprocessed foods.
It should be noted that some of the reviews described in this Section considered data that are included in the EFSA occurrence data set described in Section 3.3.2.
A. Furan
Several papers reviewing the occurrence of furan in food were published after 2010 (Moro et al., 2012b; Stadler, 2012; Mariotti et al., 2013; Seok et al., 2015; Santonicola and Mercogliano, 2016). These reviews conclude that the highest levels of furan occur in roasted coffee (powder and beans). High furan levels are also observed in foods that received a heat treatment in sealed containers such as baby foods, soups, baked beans and meat products. However, furan also occurs in low moisture foods that were not heated in a closed container, such as cereals, crackers, cookies (biscuits).
These conclusions are in accordance with JECFA review of furan including over 3,300 food samples from 14 EU countries and Switzerland. Additionally, data from 6 other countries were considered. The report of the review stated, ‘The range of national mean levels of furan for foods with the highest contamination levels were as follows: roasted coffee (powder) (814–4,590 μg/kg), instant coffee (powder) (90–783 μg/kg), brewed roasted coffee (34–113 μg/kg), baby food (19–96 μg/kg), soya sauce (16–52 μg/kg), canned fish (6–76 μg/kg) and baked beans (27–581 μg/kg)’ (FAO/WHO, 2011).
Besides the presence in commercially heat‐processed foods, furan has also been reported to occur in home‐cooked foods. Appendix E, Table E.1 gives an overview of furan concentrations in home‐cooked foods reported in scientific literature. The reported levels are in general low (< 10 μg/kg) except for home‐made toast, pizza, chips, candied potatoes, apple cake, caramel and crisps.
Table E.1.
Levels of furan in home‐cooked foods reported in scientific literature
| Processing/matrix | Concentration (μg/kg) | Reference | |
|---|---|---|---|
| Raw/as purchased | As consumed | ||
| Toasting | |||
| White bread | < 2 | 39 ± 48a (n = 5) | Hasnip et al. (2006) |
| White bread | < 3–3 (n = 3) | 260 (187–385)b (n = 5) | Crews (2009) |
| Wholemeal bread | < 3–3 (n = 3) | 150 (32–229)b (n = 7) | Crews (2009) |
| Bread | < 2.4 (n = 2) | < 2.4–179c , k (n = 11) | DTU (2009) |
| Toast bread | < 2 (n = 2) | < 2–18 (n = 4) | Zoller et al. (2007) |
| Baking | |||
| Part baked bread | < 3 (n = 1) | 7 (3–11)d (n = 4) | Crews (2009) |
| Cookies | < 3 (n = 1) | 7 (5–9)d (n = 4) | Crews (2009) |
| Pizza (chilled) | < 3 (n = 1) | 4 (< 3–6)d (n = 4) | Crews (2009) |
| Pizza (frozen) | 3 (n = 1) | 19 (10–27)d (n = 4) | Crews (2009) |
| Breaded chicken pieces | < 3 (n = 1) | 7 (6–8)d (n = 4) | Crews (2009) |
| Breaded vegetables | < 3 (n = 1) | 6 (4–9)d (n = 5) | Crews (2009) |
| Breaded fish | < 3 (n = 1) | 5 (4–5)d (n = 5) | Crews (2009) |
| Cream potatoes | n.r. | < 2.4–2.4h (n = 2) | DTU (2009) |
| Chips (oven) | 3 (n = 1) | (< 3–3)d (n = 4) | Crews (2009) |
| Chips made from industrially made products (oven) | 3–15g (n = 9) | DTU (2009) | |
| Apple pie | 4.4 (n = 1) | DTU (2009) | |
| Apple cake | 23.4 (n = 1) | DTU (2009) | |
| Frying | |||
| Fish meat balls | < 2.4 (n = 1) | < 2.4–3.1c (n = 4) | DTU (2009) |
| Candied potatoes | 3.3–15.5h (n = 3) | DTU (2009) | |
| Pancake | < 2.4–2.6c (n = 5) | DTU (2009) | |
| Caramel | < 2 (n = 2) | 51–570g , j (n = 4) | Zoller et al. (2007) |
| Deep‐frying | |||
| Chips made from fresh potatoes | < 3 (n = 1) | 12 (8–16)e (n = 4) | Crews (2009) |
| Chips made from industrially made products | 11–21f (n = 9) | DTU (2009) | |
| Crisps made from fresh potatoes | 12–51f (n = 10) | DTU (2009) | |
| Dried slices of white, purple and sweet potato | 6–43f , i | Yuan et al. (2016) | |
| Roasting | |||
| Fresh onion | < 2 (n = 1) | < 3–5 (n = 2) | Zoller et al. (2007) |
N: number of samples.
Mean ± measurement uncertainty; toasting for 4 min in a preheated domestic toaster.
Mean (range); until fully browned.
Range; different toasting/roasting/frying degrees.
Mean (range); baked in a pre‐heated oven set at 190–220°C.
Mean (range); fried at 180°C in rapeseed oil for 6 min, followed by 3 min at 190°C.
Range; different time‐temperature combinations.
Range; different heating temperatures.
Range; completely home‐made or made from an industrially made product.
Numbers read from a graph.
For one sample, a concentration of 1,956 μg/kg was reported; however, the authors indicated that this was unedible due to the bitter taste.
The level of 179 μg/kg was reported for black toast.
No furan (concentration < LOQ of 2.4 μg/kg) was quantified in home‐made oatmeal porridge, home‐made soups, sweet and sour pork, meat sauce (Bolognese), omelette, mashed potatoes, marinated potato salad, syrup cake, cookies, caramel sauce, lemon cream, fruit compote, white bread and tea buns. Also, in meat balls, no furan was detected; however, in the crust of heavily fried meat balls, a level of 2.4 μg/kg was quantified (DTU, 2009; Fromberg et al., 2014). Arisseto et al. (2012, 2013) studied the formation of furan during pressure cooking of beans, whole rice, soy beans, beef, pork, potato and cassava but no quantifiable results were measured (LOQ = 2.4 μg/kg). Furan was also not detected (LOD = 3 μg/kg) in microwaved chips (Crews, 2009) and freshly home‐cooked vegetable, cereal, potato and fruit‐based baby foods (LOD = 0.15 μg/kg) (Lachenmeier et al., 2009).
Juaniz et al. (2016) studied the formation of furan in frozen precooked bread‐coated foods during frying at 190°C in fresh or reheated oil. The data are reported in a figure and therefore not included in Appendix E, Table E.1. Using fresh oil, the furan levels ranged from 16 μg/kg (fish fingers) to 115 μg/kg (onion rings). With the exception of ham croquettes and tuna pasties, furan levels were higher when reheated oil was used (ranging from 12 μg/kg in tuna pasties to 172 μg/kg in onion rings). Furan was not quantified (< LOQ of 2.3 μg/kg) in chopped vegetables (yellow onion, green peper, cardoon, cabbage and chicory) that were fried (115/108°C) or griddled (150/110°C) with the exception of griddled onion (3.5 ± 0.3 μg/kg; mean ± standard deviation; n = 3).
B. Co‐occurrence of furan with methylfurans
Several studies have investigated the co‐occurrence of furan, 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran. Appendix E Table E.2 gives an overview of the concentrations reported in studies in which furan and at least one methylfuran was analysed.
Table E.2.
Co‐Occurrence of furan and methylfurans in commercially processed foods
| Matrix | n | Concentration (μg/kg)a | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Furan | 2‐Methylfuran | 3‐Methylfuran | 2,5‐Dimethylfuran | |||||||
| Range | Meanb | Range | Mean | Range | Mean | Range | Mean | |||
| Grains and grain‐based products | ||||||||||
| Cereal flakes | 18 | 9–63 | 22.3 | 12–86 | 23.5 | < RL (5)–6 | 3.3 | n.a. | n.a. | DPEc |
| Vegetables and vegetable products (including fungi) | ||||||||||
| Artichoke hearts | 1 | 8.5 | 8.5 | 4.6 | 4.6 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Asparagus | 4 | 2.8–11.4 | 5.5 | < LOD (1)–3.2 | 2.3 | < LOD (1)–4.4 | 2.6 | n.a. | n.a. | Becalski et al. (2010) |
| Beets | 4 | 11.7–338 | 100.4 | 1.6–30.0 | 9.0 | < LOD (1)–4.3 | 3.2 | n.a. | n.a. | Becalski et al. (2010) |
| Mushrooms | 5 | 11.3–26.0 | 17.2 | 8.4–12.4 | 10.2 | < LOD (1)–1.2 | 0.6 | n.a. | n.a. | Becalski et al. (2010) |
| Tomatoes | 5 | 6.3–19.6 | 14.3 | 16.5–42.0 | 24.8 | 86.9–144 | 111.8 | n.a. | n.a. | Becalski et al. (2010) |
| Carrots | 5 | 26.9–70.9 | 43.9 | 4.1–22.0 | 8.9 | < LOD (1)–11.8 | 4.3 | n.a. | n.a. | Becalski et al. (2010) |
| Corn | 5 | 28.1–52.1 | 36.1 | 5.9–14.2 | 8.9 | 57.9–151 | 116 | n.a. | n.a. | Becalski et al. (2010) |
| Hearts of palm | 2 | 1.1–1.6 | 1.4 | < LOD (1) | 0.5 | 1.4–2.7 | 2.1 | n.a. | n.a. | Becalski et al. (2010) |
| Ground roasted coffee | 1 | 4,590 | 4,590 | 8,680 | 8,680 | 450 | 450 | n.a. | n.a. | Becalski et al. (2010) |
| Ground roasted coffee | 117 | 762–5434 | 3,145 | 4,724–34,378 | 18,338 | 190–2,597 | 694 | n.a. | n.a. | DPEc |
| Regular ground coffee | 15 | 715–2800 | 2,200 | 4,890–13,100 | 9,470 | 311–615 | 447 | n.a. | n.a. | Becalski et al. (2016) |
| Cartridge ground coffee | 3 | 2,110–2,660 | 2,360 | 9,440–13,100 | 10,700 | 427–555 | 508 | n.a. | n.a. | Becalski et al. (2016) |
| Decaffeinated ground coffee | 7 | 1,640–3,450 | 2,450 | 6,570–13,500 | 10,400 | 274–628 | 463 | n.a. | n.a. | Becalski et al. (2016) |
| Decaffeinated instant coffee powder | 7 | 32.0–896 | 329 | 129–6,150 | 1,800 | 9.01–213 | 75.2 | n.a. | n.a. | Becalski et al. (2016) |
| Instant coffee | 2 | 279–547 | 413 | 759–764 | 761.5 | 40.4–48.6 | 44.5 | n.a. | n.a. | Becalski et al. (2010) |
| Regular instant coffee powder | 7 | 46.8–742 | 233 | 200–6,200 | 1,595 | 11.9–270 | 72.9 | n.a. | n.a. | Becalski et al., 2016) |
| Instant coffee | 64 | 17–3,394 | 627 | 135–15,932 | 2,472 | 10–1,853 | 161 | n.a. | n.a. | DPEc |
| Starchy roots and tubers | ||||||||||
| Potatoes | 4 | 20.2–114 | 65.2 | < LOD (1)–2.7 | 1.7 | < LOD (1)–1.5 | 1.0 | n.a. | n.a. | Becalski et al. (2010) |
| Legumes, nuts and oilseeds | ||||||||||
| Green and waxy beans | 5 | 48.5–69.7 | 59.8 | 9.1–29.3 | 17.9 | 7.1–20.9 | 13.2 | n.a. | n.a. | Becalski et al. (2010) |
| Beans | 5 | 29.6–83.8 | 54.7 | 3.0–15.5 | 8.6 | 3.9–7.6 | 5.8 | n.a. | n.a. | Becalski et al. (2010) |
| Chick peas | 5 | 26.8–73.5 | 40.3 | 13.9–21.6 | 18.2 | 5.5–8.8 | 7.2 | n.a. | n.a. | Becalski et al. (2010) |
| Kidney beans | 5 | 18.2–195 | 75.5 | 3.4–15.6 | 6.9 | 4.7–10.9 | 6.8 | n.a. | n.a. | Becalski et al. (2010) |
| Fruit and fruit products | ||||||||||
| Applesauce | 5 | 6.3–19.2 | 11.1 | 3.7–14.3 | 7.7 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Mixed fruit | 4 | 4.8–51.2 | 27.2 | 1.6–7.1 | 5.0 | < LOD (1)–1.1 | 0.7 | n.a. | n.a. | Becalski et al. (2010) |
| Peaches | 2 | 13.4–21.9 | 17.7 | 4.6–9.6 | 7.1 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Pineapple | 3 | 3.4–6.7 | 4.8 | 1.9–3.8 | 2.9 | 1.2–2.5 | 2.0 | n.a. | n.a. | Becalski et al. (2010) |
| Meat and meat products (including edible offal) | ||||||||||
| Flakes of chicken | 1 | 29.8 | 29.8 | 152 | 152 | 2.9 | 2.9 | n.a. | n.a. | Becalski et al. (2010) |
| Flakes of ham | 3 | 13.9–14.6 | 14.2 | 58.1–85.7 | 76.1 | < LOD (1)–1.6 | 1.1 | n.a. | n.a. | Becalski et al. (2010) |
| Luncheon meat | 1 | 39.0 | 39.0 | 24.4 | 24.4 | 1.7 | 1.7 | n.a. | n.a. | Becalski et al. (2010) |
| Meat products | 11 | < LOQ (1.0)–1.3 | 0.6 | < LOQ (1.0)–2.6 | 0.7 | n.a. | n.a. | < LOQ (1.0) | 0.5 | Shen et al. (2016) |
| Meat spreads | 5 | 51.0–172 | 100.7 | 26.2–54.8 | 36.9 | 10.4–28.8 | 17.9 | n.a. | n.a. | Becalski et al. (2010) |
| Fish and other seafood (including amphibians, reptiles, snails and insects) | ||||||||||
| Oysters | 1 | 171 | 171 | 149 | 149 | 28.3 | 28.3 | n.a. | n.a. | Becalski et al. (2010) |
| Salmon | 5 | 9.3–18.1 | 13.4 | 38.8–109 | 65.3 | 7.9–16.1 | 12.2 | n.a. | n.a. | Becalski et al. (2010) |
| Sardines in water, oil or sauce | 4 | 17.5–65.5 | 33.5 | 15.9–21.6 | 18.0 | 1.3–5.2 | 2.5 | n.a. | n.a. | Becalski et al. (2010) |
| Tuna | 5 | 15.4–27.9 | 21.8 | 14.4–39.6 | 24.8 | 1.2–1.7 | 1.5 | n.a. | n.a. | Becalski et al. (2010) |
| Milk and dairy products | ||||||||||
| Milk and milk beverages | 15 | < LOQ (1.0)–8.0 | 2.2 | < LOQ (1.0)–7.9 | 2.1 | n.a. | n.a. | < LOQ (1.0)–88.8 | 8.7 | Shen et al. (2016) |
| Sugar and confectionary | ||||||||||
| Chocolates | 6 | < LOQ (1.0) | 0.5 | < LOQ (1.0)–1.4 | 0.7 | n.a. | n.a. | < LOQ (1.0) | 0.5 | Shen et al. (2016) |
| Fruit and vegetable juices | ||||||||||
| Juice and drinks | 3 | 6.7–16.7 | 13.1 | 1.3–2.0 | 1.6 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Clamato | 3 | 6.7–9.5 | 8.3 | < LOD (1) | 0.5 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Cocktail | 1 | 10.8 | 10.8 | 3.7 | 3.7 | 2.6 | 2.6 | n.a. | n.a. | Becalski et al. (2010) |
| Fruit juices | 15 | < LOQ (0.5)–1.9 | 0.5 | < LOQ (0.5)–2.0 | 0.8 | n.a. | n.a. | < LOQ (0.5)–0.5 | 0.3 | Shen et al. (2016) |
| Tomato drinks | 4 | 4.3–14.1 | 8.0 | 9.5–20.6 | 13.5 | 22.9–55.7 | 42.0 | n.a. | n.a. | Becalski et al. (2010) |
| Non‐alcoholic beverages (excepting milk‐based beverages) | ||||||||||
| Brewed coffee | 1 | 820 | 820 | 3,900 | 3,900 | n.a. | n.a. | n.a. | n.a. | Ochiai et al. (2014) |
| Brewed medium roast or bold roast coffee | 16 | 25.1–69.5 | 38.7 | 109–287 | 172 | 5.0–9.5 | 6.4 | n.a. | n.a. | Becalski et al. (2016) |
| Coffee | 15 | 47–2,821 | 885 | 117–5,982 | 1,328 | n.a. | n.a. | 32–466 | 217 | Fromberg et al. (2014) |
| Decaffeinated coffee | 12 | 6.63–121 | 53.1 | 24.4–365 | 184 | 1.1–12.0 | 6.7 | n.a. | n.a. | Becalski et al. (2016) |
| Espresso coffee | 12 | 34.9–352 | 157 | 135–1,360 | 583 | 6.5–39.3 | 19.0 | n.a. | n.a. | Becalski et al. (2016) |
| Alcoholic beverages | ||||||||||
| Alcoholic drinks | 3 | < LOQ (0.5) | 0.3 | < LOQ (0.5)–2.0 | 0.9 | n.a. | n.a. | < LOQ (0.5)–0.5 | 0.3 | Shen et al. (2016) |
| Beer | 13 | < LOQ (0.5)–0.8 | 0.4 | < LOQ (0.5)–2.4 | 0.9 | n.a. | n.a. | < LOQ (0.5) –3.4 | 0.6 | Shen et al. (2016) |
| Herbs, spices and condiments | ||||||||||
| Ketchup | 4 | 28.7–286 | 101.6 | 2.5–18.1 | 8.9 | < LOD (1)–11.3 | 6.7 | n.a. | n.a. | Becalski et al. (2010) |
| Tomato ketchup | 2 | 1.8–2.1 | 2.0 | < LOQ (1.0)–2.7 | 1.6 | n.a. | n.a. | < LOQ (1.0) | 0.5 | Shen et al. (2016) |
| Chilli sauce | 2 | 72.8–89.2 | 81.0 | 5.4–13.5 | 9.5 | 3.8–17.3 | 10.6 | n.a. | n.a. | Becalski et al. (2010) |
| Seafood sauce | 2 | 7.7–77.3 | 42.5 | 1.2–20.6 | 10.9 | < LOD (1)–7.3 | 3.9 | n.a. | n.a. | Becalski et al. (2010) |
| Soy sauce | 14 | 7.3–63.8 | 33.1 | 1.7–43.6 | 15.2 | n.a. | n.a. | < LOQ (0.5)–10.8 | 2.9 | Shen et al. (2016) |
| Worcestershire sauce | 1 | 17.4 | 17.4 | 4.2 | 4.2 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Vinegar | 15 | < LOQ (0.5)–34.3 | 10.3 | < LOQ (0.5)–34.5 | 8.5 | n.a. | n.a. | < LOQ (0.5)–2.0 | 0.5 | Shen et al. (2016) |
| Food for infants and small children | ||||||||||
| Baby‐food purees | 11 | < LOQ (1.0)–16.2 | 5.1 | < LOQ (1.0)–13.5 | 3.6 | n.a. | n.a. | < LOQ (1.0) | 0.5 | Shen et al. (2016) |
| Banana graham pie | 1 | 43.9 | 43.9 | 4.9 | 4.9 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Chicken cacciatore | 1 | 331 | 331 | 35.5 | 35.5 | 22.9 | 22.9 | n.a. | n.a. | Becalski et al. (2010) |
| Junior oatmeal with mixed fruit | 1 | 120 | 120 | 9.5 | 9.5 | 5.1 | 5.1 | n.a. | n.a. | Becalski et al. (2010) |
| Infant formulas | 12 | < LOQ (1.0)–27.8 | 10.5 | < LOQ (1.0)–4.4 | 1.3 | n.a. | n.a. | < LOQ (1.0)–17.1 | 3.9 | Shen et al. (2016) |
| Infant formula with iron (add water) | 1 | 26.7 | 26.7 | 8.7 | 8.7 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Squash | 1 | 66.2 | 66.2 | 7.5 | 7.5 | 1.6 | 1.6 | n.a. | n.a. | Becalski et al. (2010) |
| Strained blueberry | 1 | 32.1 | 32.1 | 2.6 | 2.6 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Strained carrots | 1 | 27.8 | 27.8 | 2.6 | 2.6 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Strained creamed corn | 1 | 51.5 | 51.5 | 16.6 | 16.6 | 4.3 | 4.3 | n.a. | n.a. | Becalski et al. (2010) |
| Strained custard | 1 | 199 | 199 | 50.2 | 50.2 | 1.0 | 1.0 | n.a. | n.a. | Becalski et al. (2010) |
| Strained green beans | 1 | 78.3 | 78.3 | 7.4 | 7.4 | 3.2 | 3.2 | n.a. | n.a. | Becalski et al. (2010) |
| Strained strawberries | 1 | 8.5 | 8.5 | 1.18 | 1.18 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Strained mixed vegetables | 1 | 54.0 | 54.0 | 4.2 | 4.2 | 1.6 | 1.6 | n.a. | n.a. | Becalski et al. (2010) |
| Strained peas | 1 | 82.4 | 82.4 | 10.5 | 10.5 | 3.1 | 3.1 | n.a. | n.a. | Becalski et al. (2010) |
| Strawberries | 1 | 27.4 | 27.4 | 1.41 | 1.41 | < LOD (1) | 0.5 | n.a. | n.a. | Becalski et al. (2010) |
| Sweet potatoes | 1 | 239 | 239 | 15.7 | 15.7 | 17.5 | 17.5 | n.a. | n.a. | Becalski et al. (2010) |
| Commercial baby foods based on flour or cereals | 6 | 22.7–88.2 | 38.6 | 30.5–88.8 | 68.3 | n.a. | n.a. | 69.4–230.3 | 153.1 | Habibi et al. (2013) |
| Vegetable and beef jars | 1 | 128 | 128 | 18.8 | 18.8 | 6.6 | 6.6 | n.a. | n.a. | Becalski et al. (2010) |
| Vegetable/chicken jars | 1 | 121 | 121 | 14.3 | 14.3 | 9.5 | 9.5 | n.a. | n.a. | Becalski et al. (2010) |
| Baby food in jar | 35 | 20–67 | 35.6 | < RL (5)–12 | 6.8 | < RL (5)–12 | 5.2 | n.a. | n.a. | DPEc |
| Infant cereals | 28 | < RL (5)–32 | 8.3 | 7–118 | 15.4 | < RL (5)–9 | 3.4 | n.a. | n.a. | DPEc |
| Composite food (including frozen products) | ||||||||||
| Baked beans | 5 | 368–824 | 580.6 | 61.4–152 | 91.3 | 9.3–20.4 | 13.5 | n.a. | n.a. | Becalski et al. (2010) |
| Pasta sauce | 3 | 15.2–42.7 | 32.8 | 4.1–13.2 | 9.1 | 3.6–8.6 | 5.8 | n.a. | n.a. | Becalski et al. (2010) |
| Peanut butter | 1 | 10.6 | 10.6 | 9.2 | 9.2 | 3.2 | 3.2 | n.a. | n.a. | Becalski et al. (2010) |
| Pizza sauce | 4 | 24.2–111 | 59.6 | 10.7–25.6 | 16.1 | 7.9–11.9 | 9.8 | n.a. | n.a. | Becalski et al. (2010) |
| Salsa | 3 | 10.8–25.8 | 19.3 | 4.8–7.9 | 6.5 | 7.4–12.5 | 9.1 | n.a. | n.a. | Becalski et al. (2010) |
| Canned food | 12 | < LOD (2.4)–47 | 20.5 | < LOD (2.4)–8.0 | 4.7 | n.a. | n.a | < LOD (2.4) – 105 | 67d | Fromberg et al. (2014) |
| Chilli con carne | 5 | 241–863 | 386.4 | 35.9–73.0 | 51.8 | 25.2–55.7 | 38.8 | n.a. | n.a. | Becalski et al. (2010) |
| Gourmet antipasto | 1 | 27.0 | 27.0 | 46.4 | 46.4 | 9.1 | 9.1 | n.a. | n.a. | Becalski et al. (2010) |
| Pasta meals | 5 | 151–1,230 | 396.4 | 22.9–105 | 41.9 | 10.2–49.5 | 18.8 | n.a. | n.a. | Becalski et al. (2010) |
| Stew and meatball entrees | 4 | 34.6–1,030 | 308.6 | 6.6–132 | 46.5 | 2.0–58.9 | 17.6 | n.a. | n.a. | Becalski et al. (2010) |
| Cream of mushroom (soup) | 1 | 93.5 | 93.5 | 11.5 | 11.5 | 1.7 | 1.7 | n.a. | n.a. | Becalski et al. (2010) |
| Tomato paste | 5 | 59.7–200 | 123.0 | 24.2–76.8 | 50.4 | 14.9–98.2 | 51.6 | n.a. | n.a. | Becalski et al. (2010) |
| Tomato sauce | 5 | 17.0–35.5 | 23.6 | 3.1–45.7 | 15.8 | 5.8–86.8 | 24.5 | n.a. | n.a. | Becalski et al. (2010) |
| Tomato sauce | 3 | < RL (5) | 0.3 | 6–10 | 8.3 | 5–7 | 6.3 | n.a. | n.a. | DPEc |
DPE: documentation provided to EFSA by FoodDrinkEurope; LOD: limit of detection; LOQ: limit of quantification; n.a. not analysed n: number of samples; RL: reporting limit.
Applied method of analysis was headspace sampling followed by analysis using GC–MS unless stated otherwise.
The mean values were calculated by EFSA using the middle bound approach if they were not provided in the publication.
Data on the co‐occurrence of furan, 2‐methylfuran and 3‐methylfuran in jarred baby foods, cereal flakes, roasted and grounded coffee, soluble coffee, infant cereals and tomato sauce. December 2016. Submitted by FoodDrinkEurope to EFSA, outside the ad hoc call for occurrence data
Calculated using mean values reported by the authors.
Health Canada published levels of furan, 2‐methylfuran and 3‐methylfuran for a wide range of different foods mainly packaged in cans or jars (n = 176; fruit products, vegetable products, mixed products, meat products, coffee and baby foods). The highest concentrations of 2‐methylfuran were reported for coffee. In all coffee samples (n = 3), 2‐methylfuran occurred at higher concentrations compared to furan, while 3‐methylfuran concentrations were well below the concentrations of furan. In all 17 samples of baby food, the levels of 2‐methylfuran and 3‐methylfuran were well below the levels of furan. For the other foods, the highest 2‐methylfuran concentrations (± 150 μg/kg) were found in a sample of baked beans, flakes of chicken and whole smoked oysters. In general, the 2‐methylfuran concentration was below the concentration of furan, but the CONTAM Panel noted that, particularly for canned/jarred tomatoes (including sauces and drinks), some meat products, salmon and tuna, higher or similar concentrations of 2‐methylfuran compared to furan were reported. As for 2‐methylfuran, the 3‐methylfuran concentration was generally below the concentration of furan, but the CONTAM Panel noted that, particularly for canned/jarred corn and tomatoes (including drinks), the concentration of 3‐methylfuran was higher than the concentrations reported for furan and 2‐methylfuran (Becalski et al., 2010).
The conclusions regarding the co‐occurrence of furan, 2‐methylfuran and 3‐methylfuran in coffee observed by Becalski et al. (2010) were confirmed by the same authors in 2016. Commercially brewed coffee (n = 40) and non‐brewed coffee (n = 48) samples were collected in Canada and analysed for the presence of furan, 2‐methylfuran and 3‐methylfuran. In all samples analysed, 2‐methylfuran was present at the highest concentrations and 3‐methylfuran at the lowest concentrations. In non‐brewed coffee, the highest 2‐methylfuran concentrations were reported for regular ground coffee, decaffeinated ground coffee and cartridge ground coffee (mean: 9,470, 10,400, 10,700 μg/kg, respectively). In comparison, the mean levels of furan in these coffees were 2,200, 2,450 and 2,360 μg/kg, respectively. Lower levels of furan, 2‐methylfuran and 3‐methylfuran were reported for instant coffee powder; the mean concentrations of 2‐methylfuran were 1,595 and 1,800 μg/kg in regular and decaffeinated instant coffee power, respectively, while the mean concentrations of furan were 233 and 329 μg/kg, respectively. For non‐brewed coffee, the authors reported mean 2‐methylfuran/furan proportions ranging from 4.24 to 6.84 and mean 3‐methylfuran/furan proportions ranging from 0.19 to 0.31. For commercially brewed coffee, the highest mean 2‐methylfuran concentration was reported for espresso (583 μg/kg), while lower mean concentrations were reported for medium roast or bold roast coffee (172 μg/kg) and decaffeinated coffee (184 μg/kg). The mean concentrations of furan in these coffees were 157, 38.7 and 53.1 μg/kg, respectively. For commercially brewed coffee, the authors reported mean 2‐methylfuran/furan proportions ranging from 3.5 to 4.4 and mean 3‐methylfuran/furan proportions ranging from 0.12 to 0.17 (Becalski et al., 2016).
Shen et al. (2016) analysed heat‐processed Chinese foods samples for the presence of furan, 2‐methylfuran, 2,5‐dimethylfuran and other alkylfurans not in the scope of this Scientific Opinion. The study included soy sauce, vinegars, fruit juices, beer and other alcoholic beverages, milk and milk beverages, infant formula, coffee, salad sauce, tomato ketchup, baby‐food purees, chocolates and meat products. The information on the coffee samples was not included in this Scientific Opinion due to uncertainty regarding the coffee type and the representiveness for coffee on the EU market. 2,5‐Dimethylfuran was detected less frequently and at lower concentrations than furan and 2‐methylfuran in soy sauce and vinegar. In fruit juices, beer and other alcoholic drinks, 2‐methylfuran was most frequently detected; however, all three furans (i.e. furan, 2‐methylfuran, 2,5‐dimethylfuran) only occurred at low concentrations (≤ 2.4 μg/kg). 2,5‐Dimethylfuran was detected in 2 samples of raw milk at relatively high concentrations (34.5 and 88.8 μg/kg), while furan and 2‐methylfuran were not detected in these samples. Both compounds on the other hand occurred in one condensed milk sample and five milk powder samples at low but similar concentrations (≤ 8 μg/kg), while 2,5‐dimethylfuran was not detected. In infant formula, 2,5‐dimethylfuran was detected more frequently compared to 2‐methylfuran but in general at concentrations below the furan concentrations. 2,5‐Dimethylfuran was not detected in any of the samples of meat products (n = 11), chocolates (n = 6), tomato ketchup (n = 2), baby food purees (n = 11) and salad sauce (n = 1). In these food groups, furan and 2‐methylfuran were also not detected or at low levels (< 3 μg/kg), with the exception of baby food purees, in which furan was detected at concentrations up to 16.2 μg/kg and 2‐methylfuran up to 13.5 μg/kg.
Chaichi et al. (2013) analysed three coffee samples (ground, instant and a coffee‐mix) to evaluate the reliability of a newly developed a headspace liquid‐phase microextraction GC/MS method for the analysis of furan, 2‐methylfuran and 2,5‐dimethylfuran. 2‐methylfuran was in all three samples present at the highest concentration. Chaichi et al. (2015) used the same method to analyse 67 coffee samples collected in supermarkets and hypermarkets in Tehran, Iran and the results confirm the occurrence of 2‐methylfuran at high levels in coffee. However, the CONTAM Panel noted that only for furan a labelled standard is used in this method and only to correct for the variation in injection volume. Therefore, these studies are not further considered in the assessment.
The co‐occurrence of furan, 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran in the headspace of brewed coffee has also been reported in several other studies (e.g. Kallio et al., 1990; Shimoda and Shibamoto, 1990; Leino et al., 1992).
Fromberg et al. (2014) analysed furan, 2‐methylfuran and 2,5‐dimethylfuran in coffee samples (n = 15) and canned food (n = 12). The mean concentration of 2‐methylfuran in coffee (1,328 μg/kg) was higher than the concentration of furan (885 μg/kg) and 2,5‐dimethylfuran (217 μg/kg). In canned food, the highest mean concentration was reported for 2,5‐dimethylfuran (67 μg/kg), while the levels of furan and 2‐methylfuran were 20.5 and 4.7 μg/kg, respectively.
Habibi et al. (2013) analysed six commercial baby food samples based on flour or cereals for the presence of furan, 2‐methylfuran and 2,5‐dimethylfuran. 2,5‐Dimethylfuran was the dominant component and furan was present at the lowest concentrations in all except one sample. Levels of 2,5‐dimethylfuran ranged from 69.42 to 230.25 μg/kg. These results are in contradiction with Shen et al. (2016) that reported that 2,5‐dimethylfuran was below the LOQ (1 μg/kg) in all analysed commercial baby food purees (n = 11). However, it should be noted that these samples were fruit or vegetable based.
Bouseta et al. (1992) analysed 84 honey samples to determine their aroma profiles using dynamic headspace GC–MS. Furan was reported to occur at concentrations up to 256 μg/kg (% of left‐censored data not reported). 2‐Methylfuran was detected in 15 samples (18%) at concentrations up to 29 μg/kg.
The co‐occurrence of furan and the methylfurans has also been reported in cooked meat. Galt and MacLeod (1984) cooked beef for 1 or 4 h in an oven at 205°C and analysed the volatiles in the aroma. 3‐Methylfuran and traces of furan and 2‐methylfuran were present after 4 h but none of the compounds was present after 1 h. Also, in the volatiles from cooked mutton, the co‐occurrence of furan and 2‐methylfuran was reported (Nixon et al., 1979). Macleod and Ames (1986b) studied the effect of heat on beef aroma in steak cooked at 104°C or 171°C for 1, 2 or 6 min. Traces of furan and 2‐methylfuran were detected but not in the same aroma isolates. The same authors reported the occurrence of 2‐methylfuran in ground beef that had been cooked at 104°C for 2 min followed by 171°C for 6 min (Macleod and Ames, 1986a). The occurrence of 2,5‐dimethylfuran was reported in roasted chicken at a concentration of 1 μg/kg (250°C for 75 min) (Noleau and Toulemonde, 1986) and in roast beef heated to internal temperatures of 85°C and 95°C at a concentration of 6.9 and 1.2 μg/kg, respectively (Vercellotti et al., 1987), while the other furan compounds studied in this opinion were not reported to be present.
In addition, EFSA received data from FoodDrinkEurope, representing the European food and drink industry, on the occurrence of furan, 2‐methylfuran and 3‐methylfuran in jarred baby foods, cereal flakes, roasted and grounded coffee, soluble coffee, infant cereals and tomato sauce. A few datapoints on 2,5‐dimethylfuran were also received; however, since no furan levels were reported for these samples, the data could not be used for the evaluation of the co‐occurrence of furan and the methylfurans. In all baby food samples (n = 35), the highest concentration was observed for furan. In infant cereals, on the other hand, the highest concentration was reported in general for 2‐methylfuran (mean: 15.4 μg/kg vs 7.3 and 1.6 μg/kg for furan and 3‐methylfuran, respectively). In cereal flakes, similar levels were reported for furan and 2‐methylfuran (mean: 22 and 24 μg/kg, respectively) while 3‐methylfuran was present at concentrations below or close to the reporting limit (5 μg/kg). A large data set on roasted and grounded coffee (n = 117) and soluble coffee (n = 64) confirmed the conclusion from the scientific literature that 2‐methylfuran occurs in coffee at concentrations higher than furan. In tomato sauce (n = 3), furan occurred at concentrations below the reporting limit, while both 2‐methylfuran and 3‐methylfuran were quantified although at low levels (≤ 10 μg/kg).
From the available data on the co‐occurrence of furan and methylfurans, the CONTAM Panel concluded that only for coffee, infant cereals, jarred baby‐food and cereal flakes a 2‐methylfuran/furan and 3‐methylfuran/furan ratio can be calculated (Appendix E, Table E.3). The reported ratios are calculated from the samples for which both furans were quantified. These food groups are also important contributors to furan exposure as shown in previous exposure assessments (see Section 3.3.3). For the other food groups, only limited data were available that does not allow drawing firm conclusions.
Table E.3.
2‐Methylfuran/furan and 3‐methylfuran/furan ratios in commercially processed foods
| Food | n | Concentration (μg/kg)a | Mean ratiob | Median ratio | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Furan | 2‐Methylfuran | 3‐Methylfuran | 2MF/F | 3MF/F | 2MF/F | 3MF/F | ||||||
| Range | Meanc | Range | Mean | Range | Mean | |||||||
| Grains and grain‐based products | ||||||||||||
| Cereal flakes | 18 | 9–63 | 22.3 | 12–86 | 23.5 | < RL (5)–6 | 3.3 | 1.10 | 0.17 (n = 5) | 1.12 | 0.15 (n = 5) | DPEd |
| Vegetables and vegetable products (including fungi) | ||||||||||||
| Ground roasted coffee | 1 | 4,590 | 4,590 | 8,680 | 8,680 | 450 | 450 | 1.89 | 0.10 | – | – | Becalski et al. (2010) |
| Ground roasted coffee | 117 | 762–5,434 | 3,145 | 4,724–34,378 | 18,338 | 190–2,597 | 694 | 5.9 | 0.2 | 5.7 | 0.2 | DPEd |
| Regular ground coffee | 15 | 715–2,800 | 2,200 | 4,890–13,100 | 9,470 | 311–615 | 447 | 4.30 | 0.20 | n.r. | n.r. | Becalski et al. (2016) |
| Cartridge ground coffee | 3 | 2,110–2,660 | 2,360 | 9,440–13,100 | 10,700 | 427–555 | 508 | 4.53 | 0.22 | n.r. | n.r. | Becalski et al. (2016) |
| Decaffeinated ground coffee | 7 | 1,640–3,450 | 2,450 | 6,570–13,500 | 10,400 | 274–628 | 463 | 4.24 | 0.19 | n.r. | n.r. | Becalski et al. (2016) |
| Decaffeinated instant coffee powder | 7 | 32.0–896 | 329 | 129–6,150 | 1,800 | 9.01–213 | 75.2 | 5.47 | 0.23 | n.r. | n.r. | Becalski et al. (2016) |
| Instant coffee | 2 | 279–547 | 413 | 759–764 | 761.5 | 40.4–48.6 | 44.5 | 2.06 | 0.12 | n.r. | n.r. | Becalski et al. (2010) |
| Regular instant coffee powder | 7 | 46.8–742 | 233 | 200–6,200 | 1,595 | 11.9–270 | 72.9 | 6.84 | 0.31 | n.r. | n.r. | Becalski et al. (2016) |
| Instant coffee | 64 | 17–3,394 | 627 | 135–15,932 | 2,472 | 10–1,853 | 161 | 4.26 | 0.39 | 3.85 | 0.22 | DPEd |
| Non‐alcoholic beverages (excepting milk‐based beverages) | ||||||||||||
| Brewed coffee | 1 | 820 | 820 | 3,900 | 3,900 | n.a. | n.a. | 4.76 | – | – | – | Ochiai et al. (2014) |
| Brewed medium roast or bold roast coffee | 16 | 25.1–69.5 | 38.7 | 109–287 | 172 | 5.0–9.5 | 6.4 | 4.4 | 0.17 | 4.2 | 0.17 | Becalski et al. (2016) |
| Coffee | 15 | 47–2,821 | 885 | 117–5,982 | 1,328 | n.a. | n.a. | 1.50e | – | n.r. | – | Fromberg et al. (2014) |
| Decaffeinated coffee | 12 | 6.63–121 | 53.1 | 24.4–365 | 184 | 1.1–12.0 | 6.7 | 3.5 | 0.13 | 3.7 | 0.15 | Becalski et al. (2016) |
| Espresso coffee | 12 | 34.9–352 | 157 | 135–1,360 | 583 | 6.5–39.3 | 19.0 | 3.7 | 0.12 | 3.9 | 0.14 | Becalski et al. (2016) |
| Food for infants and small children | ||||||||||||
| Baby food in jar | 35 | 20–67 | 35.6 | < RL (5)–12 | 6.8 | < RL (5)–12 | 5.2 | 0.24 (n = 22) | 0.19 (n = 19) | 0.23 (n = 22) | 0.17 (n = 19) | DPEd |
| Infant cereals | 28 | < RL (5)–32 | 8.3 | 7–118 | 15.4 | < RL (5)–9 | 3.4 | 1.71 (n = 15) | 0.33 (n = 5) | 1.4 (n = 15) | 0.26 (n = 5) | DPEd |
2MF/F: 2‐methylfuran/furan ratio; 3MF/F: 3‐methylfuran/furan ratio; DPE: Documentation provided to EFSA by FoodDrinkEurope; n.a.: not analysed; n.c.: not calculated; n.r. not reported; n: number of samples; RL: reporting limit.
Applied method of analysis was Headspace sampling followed by analysis using GC–MS unless otherwise stated.
The ratios were estimated by EFSA, with the exception of those food products reported in Becalski et al. (2016) where the ratios were provided and are reproduced in this table. Ratios were only calculated when both furan and the methylfuran was quantified.
The mean values were calculated by EFSA using the middle bound approach if they were not provided in the publication.
Data on the co‐occurrence of furan, 2‐methylfuran and 3‐methylfuran in jarred baby foods, cereal flakes, roasted and grounded coffee, soluble coffee, infant cereals and tomato sauce. December 2016. Submitted by FoodDrinkEurope to EFSA, outside the ad hoc call for occurrence data.
Calculated using mean values reported by the authors.
The CONTAM Panel noted the higher ratios for solid coffee compared to brewed coffee. This difference might be explained by the fact that 2‐ and 3‐methylfuran are more apolar compared to furan and consequently the extraction to the liquid phase might be lower. Since coffee is consumed in the liquid form, only data on brewed coffee were considered. From the available data, a median 2‐methylfuran/furan ratio of 4.0 was calculated (average 2‐methylfuran/furan ratio = 4.0) (Table 12). The overall median 3‐methylfuran/furan ratio for brewed coffee was 0.15 (average 3‐methylfuran/furan ratio = 0.15).
Table 12.
Overview of calculated median methylfuran/furan ratios
| Food | Corresponding FoodEx categories | Median 2‐methylfuran/furan ratio | Median 3‐methylfuran/furan ratio |
|---|---|---|---|
| Coffee | Coffee beverage | 4.0 | 0.15 |
| Cereal flakes | Breakfast cereals | 1.1 | 0.15 |
| Infant cereals | Cereal‐based food for infants and young children | 1.4 | 0.26 |
| Jarred baby foods | Ready‐to‐eat meals for infants and young children | 0.23 | 0.17 |
Based on a small data set (n = 18), the CONTAM Panel noted that similar concentrations of furan and 2‐methylfuran occur in cereal flakes and calculated a median 2‐methylfuran/furan ratio of 1.1. 3‐Methylfuran on the other hand occurred at lower concentrations and was only quantified in 5 out of the 18 samples with a median 3‐methylfuran/furan ratio of 0.15. Also, in infant cereals (n = 28), 2‐methylfuran was an important contributor to the total concentration of furans with a median 2‐methylfuran/furan ratio of 1.4 (average = 1.7) (ratio calculated from 15 samples). 3‐Methylfuran was only a minor contributor and the % of left‐censored data was high. The median 3‐methylfuran/furan ratio was 0.26 (ratio calculated from 5 samples).
Several data sources were identified reporting the co‐occurrence of furan and methylfurans in jarred baby foods (see Appendix E, Table E.2). The CONTAM Panel noted the differences in furan levels among different data sets. For example, Shen et al. (2016) reported a mean furan concentration for Chinese baby food purees of 5.1 μg/kg (range: < 1.0–16.2 μg/kg; n = 11) while higher levels were reported by FoodDrinkEurope (mean: 35.6 μg/kg; range: 20–67 μg/kg; n = 35). The CONTAM Panel noted that this mean value is close to the mean MB furan level of 31 μg/kg calculated from the data set used for dietary exposure (see Section 3.2.2). In addition, the profile of the furans was different, showing higher methylfuran/furan ratios for the Chinese data compared to the European data. Considering these differences and the uncertainty regarding the representativeness of samples collected outside Europe for the jarred baby foods available on the European market, the CONTAM Panel decided to use only the data on co‐occurrence submitted by FoodDrinkEurope. From these data a median 2‐methylfuran/furan ratio of 0.23 (ratio calculated from 22 samples) and a 3‐methylfuran/furan ratio of 0.17 (ratio calculated from 19 samples) were calculated.
The CONTAM Panel noted that in canned/jarred tomatoes, 2‐methylfuran and 3‐methylfuran are important contributors to the total level of furans. This has also been observed in other canned/jarred tomato products such as tomato juices and tomato sauce. However, this observation is made on a small data set and the data are too limited to calculate ratios.
C. 2‐methylfuran
In addition to the studies on co‐occurrence described above, the occurrence of 2‐methylfuran has been reported by several authors. However, it should be noted that in most studies the volatile constituents were analysed.
2‐Methylfuran has been reported to occur in the volatile constituents of milk (Coppa et al., 2011), creams used as fillers of bakery products (Ramos et al., 2009), okra (Ames and Macleod, 1990) and roasted cocoa beans (Gill et al., 1984). Traces of 2‐methylfuran have been detected during analysis of volatile constituents in nectarines (Takeoka et al., 1988), acerola fruit (Pino and Marbot, 2001) and peppers (Rodríguez‐Burruezo et al., 2010). Also, in a study on tomato volatiles, 2‐methylfuran was found in one of 10 samples of tomato juice, albeit at a low concentration (9% of the standard, not quantified in the overall extractive). No 2‐methylfuran was reported for fresh tomatoes, tomato paste, tomato puree and canned diced tomatoes. Its presence was attributed to the heat processing of the cooked tomato product analysed (Marković et al., 2007).
Goldberg et al. (1999) analysed 2‐methylfuran in 12 types of distilled spirits by HPLC‐UV. The highest concentrations were measured in bourbon (mean = 3,000 μg/L); however, it was also detected in Armagnac, cognac, rye whiskey, blended scotch and rum (concentrations not reported). The CONTAM Panel noted that the analysis was carried out using HPLC‐UV which is not commonly used for furan and methylfuran analysis (see Section 1.3.2). The occurrence of 2‐methylfuran in distilled spirits should therefore be confirmed by another technique.
D. 3‐methylfuran
The co‐occurrence of furan and 3‐methylfuran has been demonstrated in some studies as described above. 3‐methylfuran has also been reported to be present in the volatile compounds from wild rocket (Diplotaxis tenuifolia L.) during storage (Luca et al., 2016).
E. 2,5‐dimethylfuran
In addition to the studies on co‐occurrence described above, the occurrence of 2,5‐dimethylfuran has been reported by some authors. However, it should be noted that in most studies the volatile constituents were analysed.
Alasalvar et al. (2003, 2004) published two reports from studies examining volatiles in hazelnut varieties. The average 2,5‐dimethylfuran levels found in roasted nuts was 431 μg/kg and ranged from 14 to 75 μg/kg in different varieties of unroasted nuts. Traces of 2,5‐dimethylfuran have been reported to occur in Rooibos tea (Habu et al., 1985) and in the volatile components of okra (Ames and Macleod, 1990) and cooked eggs (Macleod and Cave, 1975).
3.3. Dietary exposure assessment for humans
3.3.1. Mean and high dietary exposure
Chronic dietary exposure was estimated across Europe following the methodology described in Section 2.6. Hence a total of 35 dietary surveys, carried out in 19 different Member States, were selected for this assessment. These dietary surveys and the number of subjects available per age class are described in Annex A, Table A.3.
While the occurrence data for coffee and coffee imitates mainly referred to solid samples and a limited number of samples was available for coffee and coffee imitate beverages, most of the selected surveys reported consumption for coffee and coffee imitate beverages. Occurrence data from solid coffee and coffee imitate samples were therefore converted to the corresponding beverages by applying specific factors for dilution and loss of furan during brewing (see also Section 3.2.1). Concentrations from all samples (i.e. liquid samples and converted solid samples) were subsequently pooled to obtain a mean furan concentration for each beverage. Possible losses of furan during standing of coffee and coffee imitate beverages after brewing were not considered because this standing time is highly dependent on consumer behaviour and therefore not predictable. The adjusted concentrations for coffee and coffee imitate beverages obtained through this conversion are summarised in Table 13. On average, estimations for the major coffee types were consistent with the mean furan concentrations observed in the limited number of samples reported for those beverages.
Table 13.
Summary of the adjusted occurrence data for coffee and coffee imitate beverages
| Coffee beverage | DFa | LFb | Liquid samples | Solid samplesc | Pooled samplesd | |||
|---|---|---|---|---|---|---|---|---|
| N | Mean (μg/kg) | N | Mean (μg/kg) | N | Mean (μg/kg) | |||
| Coffee, Americano | 18 | 2.3 | 164 | 51.2 | 4,605 | 2,361.2 | 4,769 | 56.8 |
| Coffee, macchiato | 7 | 2.3 | 0 | – | 4,605 | 2,361.2 | 4,605 | 146.7 |
| Coffee, cappuccino | 18 | 2.3 | 1 | 31 | 4,605 | 2,361.2 | 4,606 | 57 |
| Coffee, espresso | 7 | 2.3 | 17 | 81.6 | 4,605 | 2,361.2 | 4,622 | 146.4 |
| Coffee, unspecified | 18 | 2.3 | 179 | 59.7 | 4,605 | 2,361.2 | 4,784 | 57.1 |
| Coffee, with milk | 18 | 2.3 | 0 | – | 4,605 | 2,361.2 | 4,605 | 57 |
| Imitates, brewed | 18 | 2.3 | 0 | – | 28 | 1,921.9 | 28 | 46.4 |
| Imitates, instant | 50 | 1 | 18 | 1 | 108 | 126.5 | 126 | 2.3 |
| Coffee, instant | 63 | 1 | 54 | 4.9 | 1,457 | 309.5 | 1,511 | 4.9 |
DF: dilution factor; LF: loss factor; N: number of samples.
The dilution factors usually applied by EFSA for Americano, cappuccino, espresso and instant coffee were extrapolated to the other coffee (imitate) beverages, except for instant coffee imitates where (compared to the dilution factor for instant coffee) a more conservative dilution factor was suggested during the consultation of stakeholders.
For each brewing method, the loss factor selected by EFSA corresponds to the lowest reliable percentile (25th percentile) of the loss factors reported in Section 3.2.1.
All coffee drinks were assumed to be prepared from roasted and ground coffee beans, except for instant coffee and coffee imitates where a specific category of solid samples is available.
Prior to the pooling of liquid and solid samples, occurrence values for the solid samples were divided by the corresponding dilution and loss factors to obtain an estimate for the corresponding beverages as consumed.
For those consumption events referring to the solid coffee or coffee imitate rather than the prepared beverage, only a loss factor was applied assuming that all these solids were consumed as a beverage and loss of furan occurred during the preparation of those beverages. An overview of all furan concentrations used for the chronic dietary exposure calculations (including the adjusted concentrations for coffee and coffee imitates) is provided in Annex A, Table A.4.
Furthermore, EFSA identified a number of specific food categories where a consumption event may refer either to fresh produce or commercially processed products (Annex A, Table A.5). As the occurrence data for furan mainly refer to commercially processed products and furan is not expected to occur in fresh produce, exposure through these food categories would be overestimated. In view of reducing the uncertainty, consumption events reported for these food categories were analysed, identifying those that referred to a canned, jarred, brined, pickled, smoked or cured product. This analysis was performed by applying a text search to the original food description or by searching for the corresponding facet descriptor when a dietary survey was already coded according to the FoodEx2 classification. The overall contribution of commercially processed products was then calculated for each food category and each dietary survey. Considering that such products have been under‐reported in a number of surveys, only the highest contribution of commercially processed products among dietary surveys was retained for each food category (expressed in % in Annex A, Table A.5), and subsequently applied to all consumers, regardless of the dietary survey. Any food item that is not listed in Annex A, Table A.5 was assumed to be commercially processed.
Table 14 summarises the chronic dietary exposure estimates for furan across the 35 dietary surveys. Detailed summary statistics on the exposure estimates calculated for each dietary survey are presented in Annex A, Table A.6.
Table 14.
Summary statistics of chronic dietary exposure assessments to furan across European dietary surveys (baseline scenario)
| Age classa | N | Mean dietary exposure (μg/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants | 6 | 0.14 | 0.21 | 0.40 | 0.49 | 0.87 | 0.99 |
| Toddlers | 10 | 0.22 | 0.31 | 0.36 | 0.46 | 0.52 | 0.65 |
| Other children | 18 | 0.19 | 0.27 | 0.28 | 0.36 | 0.45 | 0.52 |
| Adolescents | 17 | 0.11 | 0.14 | 0.17 | 0.22 | 0.26 | 0.31 |
| Adults | 17 | 0.11 | 0.14 | 0.32 | 0.35 | 0.50 | 0.54 |
| Elderly | 14 | 0.12 | 0.15 | 0.32 | 0.36 | 0.58 | 0.61 |
| Very elderly | 12 | 0.13 | 0.16 | 0.28 | 0.32 | 0.71 | 0.75 |
| Age class a | N | 95th percentile dietary exposure c (μg/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimum b | Median b | Maximum b | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants | 5 | 0.27 | 0.42 | 1.11 | 1.27 | 1.60 | 1.82 |
| Toddlers | 7 | 0.34 | 0.46 | 0.59 | 0.75 | 0.96 | 1.08 |
| Other children | 18 | 0.29 | 0.40 | 0.47 | 0.60 | 0.75 | 0.86 |
| Adolescents | 17 | 0.19 | 0.25 | 0.31 | 0.39 | 0.50 | 0.58 |
| Adults | 17 | 0.20 | 0.25 | 0.63 | 0.69 | 1.18 | 1.22 |
| Elderly | 14 | 0.24 | 0.28 | 0.66 | 0.70 | 1.25 | 1.27 |
| Very elderly | 9 | 0.27 | 0.32 | 0.52 | 0.56 | 0.92 | 0.96 |
bw: body weight; LB: lower bound; N: number of surveys; UB: upper bound.
Section 2.4 describes the age range within each age class.
Estimates were rounded to two decimal places.
The 95th percentile estimates obtained on dietary surveys/age classes with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
The difference between LB and UB estimates was found to be small, indicating that the uncertainty due to left‐censored data is limited. The highest chronic exposures were estimated in the youngest population group, i.e. infants. Among the mean dietary exposures calculated for infants, the highest LB estimate amounted to 0.87 μg/kg bw per day while the highest UB estimated amounted to 0.99 μg/kg bw per day. Regarding the 95th percentile exposures, which refer to highly exposed consumers within a given dietary survey and age class, the highest estimates were also observed for infants ranging from 1.60 to 1.82 μg/kg bw per day (LB/UB). Dietary exposure in specific groups of the population, namely pregnant and lactating women, were within the range of exposure estimates in the adult population.
3.3.2. Contributions of different food groups
Contribution of the different food groups across dietary surveys is summarised by age class in Annex A, Table A.7. These contributions were calculated on the basis of the middle bound (MB) exposure estimates.
Food for infants and small children is the main contributor to the dietary exposure of infants to furan. At the MB exposure, ready‐to‐eat meals for infants and small children accounted for up to 70% of the exposure (median = 53%). One dietary survey did not report consumption of ready‐to‐eat meals for infants and small children, which resulted in the lowest exposure estimate among infant dietary surveys (see also Table 14). In this specific survey, where exposure is most likely underestimated, the main contributors to the exposure were infant and follow‐on formula liquid (48%). The contribution of food for infants and small children in toddlers is still important, but much lower compared to infants (max = 31%; median = 7%). In other age classes, contribution of this food group to the exposure is negligible.
The group of non‐alcoholic beverages was found to be the main contributor in adults, elderly and very elderly. For all three age classes, the median contribution among surveys exceeds 50% and contributions up to 85% were observed for specific surveys. Depending on the survey, these high contributions are driven by either Americano coffee, espresso coffee or unspecified coffee, which is consistent with the high concentrations of furan observed in coffee (see Section 3.2.2). Although high concentrations of furan were estimated for other coffee beverages, consumption of these beverages is less frequently reported and their contribution to the exposure is much lower. While non‐alcoholic beverages may still contribute significantly to the exposure of adolescents (max = 24%; median = 12%, also driven by coffee beverages), their contribution to younger age classes is low. It is also worth mentioning that in some surveys consumption of coffee was not reported as a beverage but as a solid, which is included in the FoodEx category of vegetables and vegetable products. This explains the high contribution of vegetables and vegetable products observed for certain surveys (21–47%). Excluding these surveys, however, contributions to furan exposure from vegetables and vegetable products are all below 10%.
Grains and grain‐based products is another important contributor throughout all age classes, in particular, for toddlers, adolescents and other children where it is the main contributor. The median contribution among surveys ranges from 14% for infants to 49% for adolescents, with contributions reaching up to 65% for certain surveys. A specific subcategory driving the contributions of this food group cannot be identified.
A last important contributor to the dietary exposure is the group of composite foods, accounting for more than 25% in several surveys. The main subcategories driving the contribution of this food group are ready‐to‐eat soups and, to a smaller extent, cereal‐based dishes.
Apart from a few exceptions, food groups other than those mentioned above generally accounted for less than 10% of the dietary exposure.
3.3.3. Additional dietary exposure scenarios
In addition to the exposure assessment for furan reported under Section 3.3.1, hereafter referred to as the baseline scenario, specific scenarios were considered in order to assess the influence of several parameters, such as the impact of household preparation of food and the contribution of 2‐ and 3‐methylfuran to the dietary exposure. Each of these scenarios was based on the baseline scenario, including some additional assumptions which are further detailed in the following sections.
Reheating of ready‐to‐eat meals for infants and small children
The influence of reheating commercially processed foods on furan concentrations is limited and highly dependent on the consumer behaviour which is not predictable (see Section 3.2.1). It was therefore decided in the baseline scenario not to consider losses during re‐heating of ready‐to‐eat meals for infants and small children. However, ready‐to‐eat meals being the main contributor to the dietary exposure of infants, an additional scenario was considered where all ready‐to‐eat meals for infants and small children were assumed to be reheated in a hot‐water bath without lid. Among the heating techniques summarised in Appendix D, Table D.1, this heating technique leads to the highest losses of furan with a median reduction factor of 1.7. Hence, in view of this scenario, furan concentrations in ready‐to‐eat meals for infants and small children were divided by the aforementioned reduction factor.
Chronic dietary exposure estimates assuming that all ready‐to‐eat meals for infants and small children are re‐heated in a hot‐water bath (without lid) are summarised in Table 15, across all surveys. Detailed summary statistics on the exposure estimates calculated for each dietary survey are presented in Annex A, Table A.8. Infants and toddlers being the only age classes affected by the current scenario, other age classes are not presented in Table 15.
Table 15.
Summary statistics of the specific scenario assessing chronic dietary exposure to furan across European dietary surveys, assuming that all ready‐to‐eat meals for infants and small children are re‐heated in a hot‐water bath (without lid)
| Age classa | N | Mean dietary exposure (μg/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants | 6 | 0.14 | 0.21 | 0.29 | 0.37 | 0.61 | 0.72 |
| Toddlers | 10 | 0.22 | 0.31 | 0.34 | 0.44 | 0.52 | 0.65 |
| Age class a | N | 95th percentile dietary exposure c (μg/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimum b | Median b | Maximum b | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants | 5 | 0.27 | 0.42 | 0.70 | 0.84 | 1.15 | 1.32 |
| Toddlers | 7 | 0.34 | 0.46 | 0.57 | 0.74 | 0.77 | 0.91 |
bw: body weight; LB: lower bound; N: number of surveys; UB: upper bound.
Section 2.4 describes the age range within each age class.
Estimates were rounded to two decimal places.
The 95th percentile estimates obtained on dietary surveys/age classes with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Results indicate that heating of ready‐to‐eat meals for infants and small children is mainly expected to impact on the dietary exposure of infants. According to this scenario, the exposure estimates for infants are now in the same range as the exposure estimates for the adult population groups. Compared to the outcome of the baseline scenario for infants, the present scenario is expected to reduce the MB dietary exposure by 16–33% depending on the dietary survey. Only one dietary survey for infants was not impacted by the present scenario. However, as for the baseline scenario, this survey did not report consumption of ready‐to‐eat meals for infants and small children, and resulted in the lowest exposure estimate among all infant dietary surveys.
For toddlers, the impact on the mean dietary exposure is limited but for highly exposed consumers (95th percentile exposure) a reduction by up to 18% may be expected (median = 2%).
Home‐cooking: toasting bread
Furan has been reported in home‐cooked foods. The highest furan concentrations for these foods were reported for toasted bread and caramel (see Appendix E, Table E.1). Considering the importance of toasted bread in some diets, a scenario assessing the impact of toasting bread on the dietary exposure was considered by the CONTAM Panel.
For this scenario, a furan concentration of 260 μg/kg was assumed for all toasted bread, which corresponds to the highest mean concentration reported for fully browned toast in Appendix E, Table E.1. To identify consumption records referring to toasted bread, any consumption record containing ‘toast’ in its original food description was selected, and where the consumption data were already coded according to FoodEx2, consumption records containing the processing facet for toasting (A07HC) were also considered. Furthermore, any consumption record reported as toast bread was assumed to be toasted.
Chronic dietary exposure estimates for this scenario are summarised in Table 16, across all surveys. Detailed summary statistics on the exposure estimates calculated for each dietary survey are presented in Annex A, Table A.9.
Table 16.
Summary statistics of the specific scenario assessing chronic dietary exposure to furan across European dietary surveys, assuming that all toasted bread is fully browned (home‐cooking)
| Age classa | N | Mean dietary exposure (μg/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants | 6 | 0.14 | 0.21 | 0.41 | 0.49 | 0.90 | 1.01 |
| Toddlers | 10 | 0.22 | 0.31 | 0.40 | 0.49 | 0.68 | 0.77 |
| Other children | 18 | 0.19 | 0.27 | 0.33 | 0.41 | 0.47 | 0.55 |
| Adolescents | 17 | 0.11 | 0.14 | 0.19 | 0.25 | 0.28 | 0.34 |
| Adults | 17 | 0.13 | 0.16 | 0.32 | 0.36 | 0.57 | 0.61 |
| Elderly | 14 | 0.14 | 0.17 | 0.33 | 0.37 | 0.63 | 0.66 |
| Very elderly | 12 | 0.13 | 0.16 | 0.31 | 0.35 | 0.76 | 0.80 |
| Age class a | N | 95th percentile dietary exposure c (μg/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimum b | Median b | Maximum b | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants | 5 | 0.27 | 0.42 | 1.11 | 1.27 | 1.75 | 1.94 |
| Toddlers | 7 | 0.34 | 0.46 | 0.70 | 0.82 | 1.35 | 1.47 |
| Other children | 18 | 0.29 | 0.40 | 0.60 | 0.71 | 0.91 | 1.00 |
| Adolescents | 17 | 0.22 | 0.28 | 0.39 | 0.47 | 0.61 | 0.66 |
| Adults | 17 | 0.24 | 0.29 | 0.67 | 0.72 | 1.27 | 1.31 |
| Elderly | 14 | 0.30 | 0.33 | 0.73 | 0.76 | 1.32 | 1.36 |
| Very elderly | 9 | 0.28 | 0.32 | 0.56 | 0.59 | 0.92 | 0.96 |
bw: body weight; LB: lower bound; N: number of surveys; UB: upper bound.
Section 2.4 describes the age range within each age class.
Estimates were rounded to two decimal places.
The 95th percentile estimates obtained on dietary surveys/age classes with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Based on these results, toasted bread is expected to have a limited impact on the exposure of infants where the mean and the 95th percentile MB exposure estimates increased by up to 16% and 9%, respectively.
For all other age classes, the impact on the dietary exposure is very variable depending on the dietary survey. While a high number of exposure estimates remained unchanged compared to the baseline scenario, the highest increases were observed for the 95th percentile exposures (MB), ranging from 65% to 116% depending on the age class. These differences may partially be explained by the fact that some surveys did not clearly report consumption of toasted bread, but it is also expected that dietary habits may be different among Member States.
Nevertheless, the high increases reported above did not necessarily refer to the surveys giving the highest exposure estimates under the baseline scenario and, when comparing the maximum exposure estimates from the current scenario (Table 16) with the maximum exposure estimates from the baseline scenario (Table 14), the overall impact of toasting bread on the outcome of the exposure assessment is limited.
Contribution of 2‐ and 3‐methylfuran to the exposure
Due to the lack of occurrence data for 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran in food, these compounds could not be considered in the baseline exposure assessment. While for coffee beverages, breakfast cereals, cereal‐based food for infants and young children, and ready‐to‐eat meals for infants and young children, indicative ratios for 2‐ and 3‐methylfuran over furan were reported (see Section 3.2.3, Table 12), literature data were too limited to derive such ratios for 2,5‐dimethylfuran. Hence, in order to assess the possible contribution of 2‐ and 3‐methylfuran to the exposure, occurrence values for the food categories reported in Table 12 were adjusted according to the following equation:
MeanTF = MeanF × (1 + Ratio2MF/F + Ratio3MF/F)
Where:
MeanTF: mean concentration for the sum of furan, 2‐methylfuran and 3‐methylfuran
MeanF: mean concentration for furan
Ratio2MF/F: ratio of 2‐methylfuran over furan
Ratio3MF/F: ratio of 3‐methylfuran over furan
For those consumption events referring to a solid coffee product, the ratios derived for coffee beverage were applied assuming that these solids were consumed as a beverage.
The food categories adjusted for this scenario are expected to provide a good indication for the contribution of 2‐ and 3‐methylfuran to the overall exposure because these food categories were also identified as the main contributors to the exposure calculated under the baseline scenario.
Chronic dietary exposure estimates for the sum of furan, 2‐methylfuran and 3‐methylfuran are summarised in Table 17, across all surveys. Detailed summary statistics on the exposure estimates calculated for each dietary survey are presented in Annex A, Table A.10.
Table 17.
Summary statistics of the specific scenario assessing chronic dietary exposure to the sum of furan, 2‐methylfuran and 3‐methylfuran across European dietary surveys
| Age classa | N | Mean dietary exposure (μg/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants | 6 | 0.17 | 0.25 | 0.52 | 0.61 | 1.21 | 1.35 |
| Toddlers | 10 | 0.28 | 0.36 | 0.40 | 0.50 | 0.63 | 0.77 |
| Other children | 18 | 0.23 | 0.31 | 0.31 | 0.39 | 0.49 | 0.60 |
| Adolescents | 17 | 0.15 | 0.18 | 0.21 | 0.27 | 0.44 | 0.50 |
| Adults | 17 | 0.21 | 0.24 | 1.14 | 1.18 | 2.25 | 2.29 |
| Elderly | 14 | 0.27 | 0.30 | 1.15 | 1.19 | 2.64 | 2.68 |
| Very elderly | 12 | 0.26 | 0.30 | 1.02 | 1.05 | 3.27 | 3.31 |
| Age class a | N | 95th percentile dietary exposure c (μg/kg bw per day) | |||||
| Minimum b | Median b | Maximum b | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants | 5 | 0.34 | 0.49 | 1.54 | 1.70 | 2.21 | 2.36 |
| Toddlers | 7 | 0.51 | 0.64 | 0.63 | 0.81 | 1.25 | 1.40 |
| Other children | 18 | 0.37 | 0.49 | 0.55 | 0.67 | 1.31 | 1.40 |
| Adolescents | 17 | 0.29 | 0.33 | 0.51 | 0.58 | 1.31 | 1.36 |
| Adults | 17 | 0.46 | 0.50 | 2.66 | 2.70 | 5.73 | 5.78 |
| Elderly | 14 | 0.96 | 0.98 | 2.92 | 2.96 | 6.11 | 6.14 |
| Very elderly | 9 | 0.78 | 0.84 | 2.34 | 2.37 | 4.00 | 4.02 |
bw: body weight; LB: lower bound; N: number of surveys; UB: upper bound.
Section 2.4 describes the age range within each age class.
Estimates were rounded to two decimal places.
The 95th percentile estimates obtained on dietary surveys/age classes with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
When considering the sum of furan, 2‐methylfuran and 3‐methylfuran, the highest chronic exposures are estimated for adults, the elderly and the very elderly. In these cases, the highest mean exposure was calculated for the very elderly, ranging from 3.27 to 3.31 μg/kg bw per day (LB and UB estimates, respectively). Compared to the exposures calculated under the baseline scenario, the median contribution of 2‐ and 3‐methylfuran to the overall exposure amounted to 245%, going up to 317% for one survey. Looking at the highly exposed consumers (95th percentile exposure), the highest exposure is calculated for the elderly ranging from 6.11 to 6.14 μg/kg bw per day (LB and UB estimates, respectively). In this case, the median contribution of 2‐ and 3‐methylfuran corresponded to 351%, going up to 387% for one survey.
The high exposures calculated under this scenario for adults, elderly and very elderly are mainly driven by the high occurrence of 2‐methylfuran in coffee (four times higher than furan), while coffee was already the major contributor to the exposure of furan in these age classes (see also Section 3.3.2). For the younger age groups (infants, toddlers, adolescents and other children), the major contributors to the exposure were food for infants and small children, and grains and grain‐based products. The concentrations of 2‐ and 3‐methylfuran were much lower in these food groups. Therefore, in the younger age groups, the contribution of these two compounds to the overall exposure is estimated to be much lower than in the adult age groups.
3.3.4. Previously reported dietary exposure
The occurrence of furan in food was reported annually by the EFSA (2009, 2010c, 2011d). The report published in 2011 includes all data collected between 2004 and 2010. In addition, it presents exposure estimates for different populations. The analysis included a total of 5,050 analytical results for furan content in food submitted by 20 countries. Mean furan exposure across surveys was estimated to range between 0.03 and 0.59 μg/kg bw per day for adults, between 0.02 and 0.13 μg/kg bw per day for adolescents, between 0.04 and 0.22 μg/kg bw per day for other children, between 0.05 and 0.31 μg/kg bw per day for toddlers, and between 0.09 and 0.22 μg/kg bw per day for infants. A major contributor to exposure for adults was brewed coffee with an average of 85% of total furan exposure. Major contributors to furan exposure in toddlers and other children were fruit juice, milk‐based products and cereal‐based products, whereas in addition for toddlers jarred baby foods were major contributors. Table 18 gives an overview of the reported exposure levels.
Table 18.
Overview of previously reported dietary exposure levels
| Age groupa | Location | Mean dietary exposure (μg/kg bw per day | High dietary exposure (μg/kg bw per day | Reference |
|---|---|---|---|---|
| Infants | EU | 0.09–0.22 | 0.89–0.97 | EFSA (2011d) |
| Infants | USA, EU and Brazil | 0.27–1.01 | 0.99–1.34 | FAO/WHO (2011) |
| Infants | France | 0.14–0.84 | 0.28–1.52 | Anses (2016) |
| Infants | Norway | 0.62–0.65 | 1.43 | VKM (2012) |
| Infants | Canada | 0.65 | 1.76 | Health Canada (2016) |
| Toddlers | EU | 0.05–0.31 | 0.15–1.38 | EFSA (2011d) |
| Toddlers | France | 0.37 | 0.78 | Anses (2016) |
| Toddlers | Canada | 0.32 | 0.70 | Health Canada (2016) |
| Children 1–6 years | USA, EU and Brazil | 0.08–0.23 | n.a. | FAO/WHO (2011) |
| Other children | EU | 0.04–0.22 | 0.06–0.46 | EFSA (2011d) |
| Adolescents | EU | 0.02–0.13 | 0.05–0.31 | EFSA (2011d) |
| Adults (> 18 years) | EU | 0.03–0.59 | 0.08–1.29 | EFSA (2011d) |
| Adults | USA, EU and Brazil | 0.25–1.17 | 0.6–2.22 | FAO/WHO (2011) |
| Adults (18–70 years) | Norway | 0.27 | 0.77 | VKM (2012) |
| Adults (> 18 years) | Canada | 0.14–0.21 | 0.33–0.48 | Health Canada (2016) |
n.a.: not available; bw: body weight.
Data are organised according to age classes specified in Section 2.4 unless otherwise specified.
JECFA evaluated furan at its 72nd meeting in 2010 (FAO/WHO, 2011) and considered dietary exposure estimates for furan submitted by the USA, EU and Brazil. In general, the mean dietary exposure was between 0.25 and 1.17 μg/kg bw per day for adults, between 0.08 and 0.23 μg/kg bw per day for children 1–6 years of age, and between 0.27 and 1.01 μg/kg bw per day for infants. The high dietary exposure was between 0.6 and 2.22 μg/kg bw per day for adults and between 0.99 and 1.34 μg/kg bw per day for infants. Coffee was the main contributor to dietary furan exposure for adults, and breakfast cereals was the main contributor for children. Based on these data, JECFA decided to use for the risk characterisation a value of 1 μg/kg bw per day to represent mean dietary exposure and 2 μg/kg bw per day to represent high dietary exposure (see also Section 1.3.3).
In 2012, the Norwegian Food Safety Authority assessed the dietary exposure to furan as part of a full risk assessment (VKM, 2012; see also Section 1.3.3). The mean dietary exposure was the highest in 12‐month old infants; being 0.62 μg/kg bw per day (0.65 μg/kg bw per day in non‐breastfed infants). For adults (18–70 years old), the mean dietary furan exposure was 0.27 μg/kg bw per day. The 95th percentile dietary exposure was 1.43 μg/kg bw per day for 12‐month‐old infants and 0.77 μg/kg bw per day for adults. Coffee was the most important contributor to dietary furan exposure in adults. For the younger age groups, jarred baby food and breakfast cereals (especially sweet breakfast cereals) were the highest contributors.
The French Agency for Food, Environmental and Occupational Health & Safety (anses; Agence nationale de sécurité sanitaire alimentation, environment, travail) published in 2016 the outcome of a total diet study (TDS) in children younger than 3 years of age. The mean dietary exposure to furan was 0.14, 0.60, 0.84 and 0.37 μg/kg bw per day (UB) for 1–4 months, 5–6 months, 7–12 months and 13–36 months old infants, respectively. High dietary exposure (90th percentile) was 0.28, 1.29, 1.52 and 0.78 μg/kg bw per day for these age groups, respectively. For the youngest age group (1–4 months), jarred baby food (vegetables) and infant formula27 were the main contributors and for the older age groups jarred baby food (vegetables/vegetables with meat/vegetables with fish) (anses, 2016).
Health Canada carried out a deterministic exposure assessment for furan and the sum of furan and 2‐methylfuran in 2010 using occurrence data reported by Becalski et al. (2005, 2010). This exposure assessment was updated in 2016 using a probabilistic approach and the same occurrence data but more recent consumption data. The mean dietary exposure to furan and the sum of furan and 2‐methylfuran was the highest in infants (0.65 and 0.69 μg/kg bw per day, respectively) and toddlers (0.32 and 0.40 μg/kg bw per day, respectively). For adults (age groups greater than 18 years of age), the mean dietary exposures to furan and the sum of furan and 2‐methylfuran were 0.18 and 0.20 μg/kg bw per day, respectively. However, it should be noted that the presence of 2‐methylfuran in brewed coffee and rehydrated instant coffee was not taken into account in this assessment. Consequently, the exposure to furan and 2‐methylfuran of the age groups that consume coffee are expected to be higher than the estimated intakes. It should also be noted that foods such as toast, breakfast cereals and milk were not included in the exposure assessment. The major contributors to the dietary exposure of children up to 13 years of age were canned baked beans, fruit juices and sauces and condiments, while coffee was the most important contributor in all age classes greater than 18 years (Health Canada, 2016).
3.3.5. Non‐dietary sources of exposure
In addition to dietary exposure, exposure through inhalation during cooking occurs. Crews (2009) measured furan in the air during cooking. Low and variable concentrations were detected when the cooking activity was less than 10 mins, while higher levels were observed when the cooking took longer than 10 mins. The authors calculated the amount of furan inhaled during cooking which varied between 11 and 523 ng furan depending on the food, the duration of the processing and type of processing technique. However, the authors indicated that these values should be considered as rough estimates only.
There are numerous reports of furan exposure through cigarette smoke. Furan is listed as one of the ‘Harmful and Potentially Harmful Constituents in Tobacco Products and Tobacco Smoke’ on the US Food and Drug Administration's website.28 The exposure to furan per ‘puff’ (40 mL inhalation) was reported to be about 8.4 μg (Egle and Gochberg, 1979a). Pouli et al. (2003) reported a higher level of furan in cigarette smoke than 2,5‐dimethylfuran (37.3 vs 19.4 μg/cigarette). The same observation was made by Hatzinikolaou et al. (2006), although lower levels were reported (20.2 vs 10.7 μg/cigarette). Baek and Jenkins (2004) reported besides the presence of furan and 2,5‐dimethylfuran in cigarette smoke also the presence of 2‐methylfuran at similar levels as furan.
Furan29 and methylfurans are also used in industrial settings (see also Section 1.3.6).
3.4. Risk characterisation
3.4.1. Furan
Non‐neoplastic effects
The CONTAM Panel selected the BMDL10 of 0.064 mg/kg bw per day (corrected for the applied dose regimen of 5 days per week) for the induction of cholangiofibrosis in male rats after 2 years as reference point for the risk characterisation of the non‐neoplastic effects.
Comparison of the chronic dietary exposure to furan across dietary surveys and age groups reported above (Table 14) to this BMDL10 of 0.064 mg/kg bw per day, results in MOE values (Table 19) that range from 584 (minimum LB) to 65 (maximum UB) for the mean exposure estimates, and from 338 (minimum LB) to 35 (maximum UB) for the 95th percentile exposure estimates across dietary surveys and age groups.
Table 19.
Margins of exposure (MOE) values for the incidence of cholangiofibrosis across dietary surveys and age groups
| Age group | MOE calculated from mean dietary exposure | MOE calculated from P95 dietary exposure | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Minimum | Median | Maximum | Minimum | Median | Maximum | |||||||
| LB | UB | LB | UB | LB | UB | LB | UB | LB | UB | LB | UB | |
| Infants | 459 | 306 | 161 | 131 | 74 | 65 | 238 | 153 | 58 | 51 | 40 | 35 |
| Toddlers | 292 | 207 | 179 | 140 | 124 | 99 | 189 | 140 | 109 | 86 | 67 | 60 |
| Other children | 338 | 238 | 230 | 179 | 143 | 124 | 222 | 161 | 137 | 107 | 86 | 75 |
| Adolescents | 584 | 459 | 378 | 292 | 247 | 207 | 338 | 257 | 207 | 165 | 129 | 111 |
| Adults | 584 | 459 | 201 | 184 | 129 | 119 | 321 | 257 | 102 | 93 | 54 | 53 |
| Elderly | 536 | 429 | 201 | 179 | 111 | 105 | 268 | 230 | 97 | 92 | 51 | 51 |
| Very elderly | 495 | 402 | 230 | 201 | 91 | 86 | 238 | 201 | 124 | 115 | 70 | 67 |
N: number of dietary surveys; LB: lower bound; UB: upper bound; P95: 95th percentile.
The CONTAM Panel considered that based on the available toxicity data and taking inter‐ and intra‐ species variations into account, a MOE of 100 or higher would be sufficient to conclude a low health concern for the non‐neoplastic effects. The calculated MOEs for the incidence of cholangiofibrosis are below 100 in a number of dietary surveys, particularly for the high percentile exposure estimates for the younger age groups (infants and toddlers) and adults (including elderly). The CONTAM Panel concluded that these MOEs indicate a health concern.
Neoplastic effects
The CONTAM Panel selected the BMDL10 of 1.31 mg/kg bw per day (corrected for the applied dose regimen of 5 days per week) for the induction of hepatocellular adenomas and carcinomas in female mice after 2 years as a reference point for the risk characterisation of the neoplastic effects.
Comparison of the chronic dietary exposure to furan across dietary surveys and age groups reported above (Table 14) to this BMDL10 of 1.31 mg/kg bw per day, results in MOE values (Table 20) that range from 11,948 (minimum LB) to 1,328 (maximum UB) for the mean exposure estimates, and from 6,917 (minimum LB) to 722 (maximum UB) for the 95th percentile exposure estimates across dietary surveys and age groups.
Table 20.
Margins of exposure (MOE) values for the incidence of hepatocellular adenomas and carcinomas across dietary surveys and age groups
| Age group | MOE calculated from mean dietary exposure | MOE calculated from P95 dietary exposure | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Minimum | Median | Maximum | Minimum | Median | Maximum | |||||||
| LB | UB | LB | UB | LB | UB | LB | UB | LB | UB | LB | UB | |
| Infants | 9,388 | 6,259 | 3,286 | 2,682 | 1,511 | 1,328 | 4,868 | 3,129 | 1,184 | 1,035 | 821 | 722 |
| Toddlers | 5,974 | 4,240 | 3,651 | 2,857 | 2,527 | 2,022 | 3,866 | 2,857 | 2,228 | 1,752 | 1,369 | 1,217 |
| Other children | 6,917 | 4,868 | 4,694 | 3,651 | 2,921 | 2,527 | 4,532 | 3,286 | 2,796 | 2,190 | 1,752 | 1,528 |
| Adolescents | 11,948 | 9,388 | 7,731 | 5,974 | 5,055 | 4,240 | 6,917 | 5,257 | 4,240 | 3,370 | 2,629 | 2,266 |
| Adults | 11,948 | 9,388 | 4,107 | 3,755 | 2,629 | 2,434 | 6,571 | 5,257 | 2,086 | 1,905 | 1,114 | 1,077 |
| Elderly | 10,952 | 8,762 | 4,107 | 3,651 | 2,266 | 2,155 | 5,476 | 4,694 | 1,991 | 1,878 | 1,051 | 1,035 |
| Very elderly | 10,110 | 8,214 | 4,694 | 4,107 | 1,851 | 1,752 | 4,868 | 4,107 | 2,527 | 2,347 | 1,429 | 1,369 |
N: number of dietary surveys; LB: lower bound; UB: upper bound; P95: 95th percentile.
For substances that are both genotoxic and carcinogenic, the EFSA Scientific Committee stated that an MOE of 10,000 or higher, if based on the BMDL10 from an animal carcinogenicity study, would be of low concern from a public health point of view (EFSA, 2005). The CONTAM Panel noted that, with the exception of some dietary surveys, the calculated MOEs are smaller than 10,000, which would indicate a health concern for substances that are both genotoxic and carcinogenic. However, there is uncertainty regarding the carcinogenic MoA of furan.
As discussed in Section 3.1.5 on critical effects, there is clear evidence for the involvement of indirect mechanisms in the carcinogenic MoA of furan. These include epigenetic changes, oxidative damage to DNA and regenerative hyperplasia, with all these effects being accompanied by tissue damage. In addition, extensive protein binding of BDA restricts access to DNA, and binding to histones may also contribute to the reported epigenetic changes. There is limited evidence of a direct mechanism (i.e. direct interaction with DNA) in the carcinogenic action of furan. A very low level of altered DNA bases was observed in vivo but their chemical structures could not be identified. These altered DNA bases are not identical to the adducts formed by reaction of BDA with DNA. Nevertheless, there is evidence of chromosomal damage in vivo, but it is unknown whether this is caused by a direct or indirect mechanism.
Although there is uncertainty regarding the role of a direct genotoxic mechanism in the carcinogenic action of furan, the CONTAM Panel considered the resulting MOEs for hepatocellular adenomas and carcinomas as supporting evidence for its conclusion, based on the hepatotoxicity of furan, that the current exposure to furan indicates a health concern.
3.4.2. Additivity of furan and methylfurans
For the methylfurans, insufficient information is available to identify a reference point. 2‐Methylfuran, 3‐methylfuran and 2,5‐dimethylfuran have all been shown to convert to reactive species via ring opening evidently via CYP2E1 similar to the metabolic pathway of furan. Also, as with furan, these methylfurans have been found to give rise to non‐extractable binding to protein (and, with 2‐methylfuran, to DNA) in the liver. Only limited information is available on 2‐methylfuran and 2,5‐dimethylfuran genotoxicity. There is possible involvement of alternative routes of metabolic activation for 2,5‐dimethylfuran. Finally, the hepatic toxicity of both 2‐ and 3‐methylfuran in rodents exhibited a potency of the same order of magnitude to that of furan. Based on the above comparisons, it was considered appropriate for hepatotoxicity to assume dose additivity for furan, 2‐methylfuran, 3‐methylfuran. However, since there is no information on hepatotoxicity of 2,5‐dimethylfuran in vivo the CONTAM Panel is unable to include this in the additivity assumption.
Due to the lack of occurrence data on methylfurans in the available data set, a scenario was designed to estimate the chronic dietary exposure to the sum of furan, 2‐methylfuran and 3‐methylfuran based on 2‐methylfuran/furan and 3‐methylfuran/furan ratios for some selected foods (see Section 3.3.3). Comparison of the chronic dietary exposure calculated for this scenario (Table 17) to the BMDL10 of 0.064 mg/kg bw per day for the incidence of cholangiofibrosis, results in MOE values (Table 21) that range from approximately 430 (minimum LB) to 20 (maximum UB) for the mean exposure estimates, and from 220 (minimum LB) to 10 (maximum UB) for the 95th percentile exposure estimates across dietary surveys and age groups.
Table 21.
Margins of exposure (MOE) values for the incidence of cholangiofibrosis based on a scenario for chronic dietary exposure to the sum of furan, 2‐methylfuran and 3‐methylfuran across dietary surveys and age groups
| Age group | MOE calculated from mean dietary exposure | MOE calculated from P95 dietary exposure | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Minimum | Median | Maximum | Minimum | Median | Maximum | |||||||
| LB | UB | LB | UB | LB | UB | LB | UB | LB | UB | LB | UB | |
| Infants | 378 | 257 | 124 | 105 | 53 | 48 | 189 | 131 | 42 | 38 | 29 | 27 |
| Toddlers | 230 | 179 | 161 | 129 | 102 | 83 | 126 | 100 | 102 | 79 | 51 | 46 |
| Other children | 280 | 207 | 207 | 165 | 131 | 107 | 174 | 131 | 117 | 96 | 49 | 46 |
| Adolescents | 429 | 357 | 306 | 238 | 146 | 129 | 222 | 195 | 126 | 111 | 49 | 47 |
| Adults | 306 | 268 | 56 | 54 | 29 | 28 | 140 | 129 | 24 | 24 | 11 | 11 |
| Elderly | 238 | 214 | 56 | 54 | 24 | 24 | 67 | 66 | 22 | 22 | 11 | 10 |
| Very elderly | 247 | 214 | 63 | 61 | 20 | 19 | 82 | 77 | 27 | 27 | 16 | 16 |
N: number of dietary surveys; LB: lower bound; UB: upper bound; P95: 95th percentile
Note: the additive exposure has been calculated as mg/kg bw per day and not on a molar basis.
The CONTAM Panel noted that the calculated MOEs for the incidence of cholangiofibrosis are below 100 for more than 50% of the dietary surveys for the adult age groups (including elderly and very elderly) for both mean and high percentile exposure estimates. Also, for the younger age groups, MOEs below 100 were calculated for some dietary surveys. From these figures, it becomes clear that methylfurans may add significantly to the overall exposure and therefore increase the concern for hepatotoxicity.
3.5. Uncertainty analysis
The evaluation of the inherent uncertainties in the assessment of exposure to furan and methylfurans has been performed following the guidance of the Opinion of the Scientific Committee related to Uncertainties in Dietary Exposure Assessment (EFSA, 2007). In addition, the report on ‘Characterizing and Communicating Uncertainty in Exposure Assessment’ has been considered (WHO/IPCS, 2008).
3.5.1. Assessment objectives
The objectives of the assessment were clearly specified in the terms of reference.
3.5.2. Exposure scenario/exposure model
To estimate chronic dietary exposure to furan, EFSA used a final data set containing 9,663 samples provided by governmental organisations and 7,393 samples provided by commercial organisations. The amount of occurrence data submitted differs considerably depending on food products and reporting data provider, with most of the samples (~ 80%) collected in only five Member States (and ~ 60% originating from one single Member State). There is therefore uncertainty on whether possible country‐based differences in the levels of furan in diverse food commodities are well represented. Likewise, the lack of information on the analytical method used to analyse some food samples (~ 30% of the governmental data) adds some uncertainty to the concentrations of furan reported for some food commodities.
Another uncertainty regarding the occurrence data refers to the roasting degree of coffee, as the available data did not allow differentiating furan concentrations in light, medium and dark roast coffee. While a consumer's preference for dark roast coffee is expected to increase the dietary exposure to furan, the exposure estimate for light roast coffee consumers will most likely be lower. Furthermore, in order to estimate the occurrence of furan in prepared coffee and coffee imitate beverages, certain assumptions were made on the dilution and loss of furan during beverage preparation. On average, estimations for the major coffee types were consistent with the furan concentrations measured in a limited number of beverage samples. Considering the wide variety of coffee preparation methods, uncertainty remains regarding other very specific preparation methods that may occur in practice. In addition, possible losses of furan during standing of beverages after brewing could not be considered in the present assessment because the standing time is highly dependent on consumer behaviour and therefore not predictable. Overall, these uncertainties may lead to an overestimation or underestimation of exposure.
Furan may also occur in food additives and flavourings, which may be used in a wide range of food products. While it is expected that most of the furan concentrations resulting from those uses have been captured in the final data set, some minor food products may not be covered by the available data hereby resulting in a minor underestimation of the exposure estimates.
Uncertainties and limitations related to the use of the EFSA Comprehensive Food Consumption Database have already been described by EFSA (EFSA, 2011a) and are not further detailed in this opinion. However, among those with a particular implication for the dietary exposure to furan, the uncertainty associated to commercially processed foods should be mentioned because for certain food groups (e.g. vegetables and vegetables products) consumption data have been mostly reported at the level of raw commodities (e.g. tomatoes), without systematic information on whether these were commercially processed products (e.g. canned tomatoes). Following a more detailed analysis of the Comprehensive Database, the highest contribution of commercially processed products among dietary surveys was derived for each food category and applied to all consumers, regardless of the dietary survey. Assuming the same contribution for all consumers may underestimate the upper tail exposures for high consumers of commercially processed products. However, as this contribution refers to the maximum contribution observed among dietary surveys, mean exposures are expected to be overestimated. Furthermore, eating occasions reported for composite food were assumed to be commercially processed as no information is available on whether they referred to homemade dishes or ready‐to‐eat foods. Also in this case, an overestimation of the exposure is likely.
The Comprehensive Database also contains limited information on consumer's preferences regarding coffee consumption (e.g. roasting degree, coffee type). In particular, unspecified coffee beverage is the most frequently reported coffee beverage in the Comprehensive Database. While these coffee beverages were handled as the most frequently consumed coffee within Europe (i.e. Americano coffee or filtered coffee), these consumption events might in fact refer to other coffee types with higher or lower furan concentrations. For several surveys, consumption events for coffee and coffee imitates were reported as the solid. Options for refinement of the exposure through solid coffee were limited and conservative assumptions were applied. Hence, an overestimation of the exposure is expected in these surveys.
Additional uncertainty in the exposure assessment to furan is related to the reheating of commercially processed foods, which may be particularly important for infants where ready‐to‐eat meals are the most important contributor to the exposure. While some reheating techniques have a very limited impact on the concentrations of furan (therefore not considered in the main exposure scenario), heating of a meal in a hot water bath without lid may decrease the exposure by approximately 15–30%. Due to the limited information, home‐cooking habits were also not considered in the main exposure scenario but the example of toasting bread was assessed in a separate scenario. While fully browned toasting of bread was found to increase significantly the exposure estimates for some population groups (up to 116%), the overall impact on the outcome of the assessment was limited.
The highest level of uncertainty is associated to the lack of occurrence data on 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran. While data for these compounds were too limited for consideration in the main exposure scenario, an additional scenario was elaborated where EFSA considered indicative occurrence values for 2‐ and 3‐methylfuran in the food categories contributing the most to the exposure. This scenario indicated that 2‐ and 3‐methylfuran may increase the exposure for adults, elderly and very elderly by more than 300%, and it is expected that exposure estimates would increase even more in case occurrence data for 2,5‐dimethylfuran would be available.
In addition to dietary exposure to furan and methylfurans, exposure via inhalation may occur in humans but this was not taken into account in this risk assessment.
3.5.3. Model input (parameters)
Currently, no official standard methods are available for the analysis of furan and methylfurans in foods. Analytical results used for the exposure assessment were therefore obtained using different analytical methods and varying LOQ/LODs. Although a large proportion of left‐censored results was found in certain food groups (i.e. grains and grain‐based products, foods for infants and young children and composite foods), the difference between LB and UB estimates was found to be small, particularly for adults, elderly and very elderly where coffee is the main contributor to the dietary exposure. This indicates that the uncertainty due to left‐censored data is limited.
Furthermore, analytical methods for furan mostly rely on the use of headspace analysis, while a limited additional formation of furan during the equilibration of the sample in the headspace vials has been reported (see Section 1.3.2). This may result in a minor overestimation of furan concentrations in food.
3.5.4. Other uncertainties
Although many studies on the toxicity of furan have been published, several sources of uncertainty have been identified. The greatest degree of uncertainty concerns the carcinogenic MoA of furan i.e. whether or not furan is directly genotoxic and the extent to which this may contribute to carcinogenicity at levels to which humans are exposed.
For studies that attempt to estimate concentrations of furan or methylfurans in cell cultures or in tissues, there is uncertainty regarding the loss via evaporation. This potentially could lead to an overestimate of exposure both in vitro and in vivo. In vitro studies that used a reduced headspace to avoid evaporation may have incurred oxygen depletion leading to compromised metabolic capacity, introducing uncertainty in the interpretation of the results. In tissues of dosed animals, the evaporation may affect the measured residual concentrations.
Regarding methylfurans, there are limited studies on absorption, metabolism and toxicity. Carcinogenicity data are limited to one study on 2,5‐dimethylfuran. In addition to the demonstrated ring opening of methylfurans, there is a theoretical possibility of oxidation of the side‐chain leading to furyl alcohols which can subsequently be activated by sulfonation, aldehydes and acids but this possibility has not been assessed. Therefore, a conclusion regarding the potential for the methylfurans to be carcinogenic in a similar way to furan cannot be made. Based on evidence of reactivity of metabolites and similarity in hepatotoxic potency, it was considered reasonable, despite uncertainty, to combine furan, 2‐methylfuran and 3‐methylfuran as an additive in assessment of hepatotoxicity.
3.5.5. Summary of uncertainties
In Table 22, a summary of the uncertainty evaluation is presented, highlighting the main sources of uncertainty and indicating an estimate of whether the respective source of uncertainty might have led to an over‐ or underestimation of the exposure or the resulting risk.
Table 22.
Summary of qualitative evaluation of the impact of uncertainties on the risk assessment of the dietary exposure of furan and methylfurans in food
| Sources of uncertainty | Direction |
|---|---|
| Extrapolation of the occurrence data to the whole of Europe | +/−a |
| Lack of occurrence data in coffee in relation to its roasting degree | +/− |
| Assumptions on the preparation of coffee and coffee imitate beverages | +/− |
| Assumptions on the contribution of commercially processed foods to the consumption | +/− |
| Assumptions on the consumption of unspecified coffee beverages | +/− |
| Exclusion of reheating commercially prepared foods in the baseline exposure scenario | + |
| Exclusion of home‐cooking practices in the baseline exposure scenario | − |
| Exclusion of methylfurans in the baseline exposure scenario | − |
| Evaporation of furan in tissues and cell cultures | − |
| Potential for a direct genotoxic mechanism in the carcinogenic mode of action of furan | − |
| Potential genotoxicity and carcinogenicity of methylfurans | − |
| Using an additive approach for furan and methylfurans regarding hepatotoxicity | +/− |
+ = uncertainty with potential to cause over‐estimation of exposure/risk; − = uncertainty with potential to cause under‐estimation of exposure/risk.
The CONTAM Panel considered that the impact of the uncertainties on the risk assessment of furan is moderate and that the assessment is likely to be conservative. For methylfurans, due to a lack of data, no full assessment could be performed and the uncertainties in the assessment are large.
4. Conclusions
Furan, 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran are volatile compounds that are formed in foods during thermal processing. Furan can be formed in food from a variety of precursors including ascorbic acid, amino acids, carbohydrates, unsaturated fatty acids and carotenoids.
4.1. Occurrence/Exposure
To estimate chronic dietary exposure to furan, EFSA used a final data set containing 9,663 samples provided by governmental organisations and 7,393 samples provided by commercial organisations. Governmental data and commercial data were found to be comparable and it was considered appropriate to merge both data sets.
Food characteristics, processing and cooking conditions, and the losses which occur mainly due to evaporation in the preparation of the food at the level of the consumer, determine the final concentration in the food as consumed.
The highest concentrations of furan were found in whole roasted coffee beans, with a mean value of 4,579 μg/kg. Mean concentrations in ground roasted coffee (2,361 μg/kg) and unspecified coffee solids (2,186 μg/kg) were lower, suggesting a loss of furan during the process of grinding. High concentrations of furan were also found in coffee imitates for brewing (1,922 μg/kg), instant coffee powder (310 μg/kg) and instant coffee imitates (127 μg/kg).
During coffee beverage preparation furan concentrations decrease substantially due to a combination of dilution, evaporation and partial extraction. Highest losses of furan during beverage preparation were observed for boiled/Turkish coffee (median loss factor of 11.6), and losses for filter coffees (median loss factor of 4.2) were also found to be higher compared to espresso coffees (median loss factor of 2.8). For instant coffees, the loss of furan during beverage preparation is considered negligible.
Mean concentrations ranging from 20 to 57 μg/kg were found in composite foods (prepared salads, cereal‐based and vegetable‐based), ready‐to‐eat meals for infants and young children, soy sauce, bread and rolls, raw pasta, breakfast cereals, fine bakery wares and spirits. In all remaining samples, including a wide range of mainly commercially prepared foods, mean furan concentrations were below 20 μg/kg.
Occurrence data for 2‐methylfuran, 3‐methylfuran and 2,5‐dimethylfuran were not provided, and from the available literature data on the co‐occurrence of furan and methylfurans, a 2‐methylfuran/furan and 3‐methylfuran/furan ratio was calculated only for coffee, infant cereals, jarred baby‐food and cereal flakes. Co‐occurrence data on 2,5‐dimethylfuran were too limited to derive such a ratio.
The highest exposures to furan were estimated in the youngest population group, i.e. infants. The mean dietary exposures calculated for infants ranged from 0.14 to 0.99 μg/kg bw per day (minimum LB to maximum UB). Regarding the 95th percentile exposures, which refer to highly exposed consumers within a given dietary survey and age class, the highest estimates were also observed for infants ranging from 0.27 to 1.8 μg/kg bw per day (minimum LB to maximum UB).
Ready‐to‐eat meals for infants and small children are the main contributor to the dietary exposure of infants. For adults, elderly and very elderly, the exposure is mainly driven by coffee. Grains and grain‐based products is the most contributing food group for toddlers, other children and adolescents, and it is also the second largest contributor in all other age classes. Other important contributors to the exposure of most age classes are ready‐to‐eat soups, and to a smaller extent, cereal‐based composite foods.
Overall, the influence of reheating commercially processed foods on furan concentrations is limited and depends on the consumer behaviour. Regarding ready‐to‐eat meals for infants and young children, reheating the meals in a hot water bath (without lid) may reduce the dietary exposure of infants by approximately 15–30% depending on the dietary pattern. This results in an exposure comparable to the exposures in the adult age classes.
In addition, furan can also be formed during home cooking. A scenario based on toasting bread, the process that caused the highest increase in the foods studied, showed that although for some specific subpopulations a significant increase of the exposure was observed the overall outcome of the exposure assessment (including all subpopulations) was not impacted.
In the exposure scenario for the sum of furan, 2‐methylfuran and 3‐methylfuran, estimates for adults, elderly and very elderly showed the highest increase compared to the baseline scenario. The highest mean exposure, calculated for the very elderly, was approximately 3.3 μg/kg bw per day (LB = UB), while the highest 95th percentile exposure was approximately 6.1 μg/kg bw per day (LB = UB) for the elderly. This exposure was mainly driven by the high concentrations of 2‐methylfuran in coffee (four times higher than furan).
4.2. Hazard identification and characterisation
Toxicokinetics
Furan
After oral exposure to rats, furan is rapidly and extensively absorbed from the GI tract, distributed throughout the body and extensively metabolised to give carbon dioxide and polar metabolites in urine or bile/faeces.
Furan has a very short half–life in liver and blood: approximately 40 min in liver and approximately 1.3 h in blood. Although at 24 h post‐dosing some furan‐related material could be extracted from liver tissue, 80% of the residual material in the liver appeared to be non‐extractable.
The major route for elimination of furan is via metabolic conversion, resulting in opening of the furan ring and formation of BDA which is very reactive. BDA has never been directly measured. However, results with trapping agents and the identification of urinary and biliary metabolites provide strong evidence that BDA is indeed a prime reactive intermediate in the metabolism of furan.
CYP2E1 is by far the most important monooxygenase that catalyses the conversion of furan into BDA.
BDA reacts readily with amino acids, GSH and biogenic amines. A plethora of adducts has been reported. Elimination of these adducts via urine is biphasic: in first instance, the main excretion consists of BDA coupled to free amino acids or GSH. Later, the urinary excretion of furan‐related material is a result from protein turn‐over that liberates the furan‐damaged amino acid residues.
BDA has direct reactivity towards DNA. However, the data suggest that BDA reacts readily with protein and non‐protein amino and thiol residues, thereby restricting access to DNA in vivo.
It is currently impossible to select any furan metabolite as a quantitative biomarker of exposure. Furan may also be formed endogenously, which may need to be taken into account.
Methylfurans
No oral studies have been performed with any of the three methylfurans considered in this opinion to address their kinetics in animals, but it is anticipated that the three methylfurans will be absorbed from the GI tract.
Non‐extractable association of 2‐methylfuran with protein and DNA in the liver was reported after i.p. administration. For 2‐ and 3‐methylfuran data from in vitro studies are available that show that they may become associated (non‐extractable) with lung and liver microsomal proteins.
The reactive metabolic intermediates of 2‐ and 3‐methylfuran are the cis‐enedials 3‐acetylacrolein (4‐oxopent‐2‐enal) and 2‐methylbut‐2‐enedial, respectively. Also, for 2,5‐dimethylfuran, ring opening results in the formation of a reactive intermediate, the cis‐enedione 3(Z)‐hexene‐2,5‐dione.
Similar to BDA, the reactive intermediate formed from 2,5‐dimethylfuran reacts readily with amino acids and with GSH and corresponding structures for the adducts formed have been reported. For 2‐ and 3‐methylfuran metabolites, only very limited information is available, but from the structures of their primary metabolites and from studies with scavengers, it is anticipated that these will react with tissue components in a similar way to the primary metabolites of furan and 2,5‐dimethylfuran. For the methylfurans or their metabolites, there is no direct information on reactivity towards DNA. However, limited genotoxicity studies provide an indication that such interactions may occur.
Toxicity
Furan
In experiments of a duration of up to 90 days, furan is strongly hepatotoxic and moderately nephrotoxic in rodents when applied by oral gavage. Rats seem to be more sensitive towards furan than mice. Furan leads to characteristic changes in serum markers related to hepatotoxicity as well as severe histopathological damage in the liver.
The hepatotoxic effects are particularly pronounced in centrilobular and subcapsular areas and can be attenuated by suppression of CYP activity.
After 90 days, significant increases in serum thyroid hormones were observed along with severe histopathological changes in the liver of male rats after a daily dose of 0.12 mg/kg bw, given 5 days a week.
In long‐term studies, furan was associated with toxicity in the liver. Cholangiofibrosis was observed in rats as an early and sensitive response with significant increases after 36 weeks at doses of 0.44 mg/kg bw and above.
In mice, hepatocellular adenoma/carcinoma occurred at 104 weeks at doses of 4 mg/kg bw and above. In rats, cholangiocarcinomas were observed at the top dose of 8 mg/kg bw. No relevant malignancies were observed at doses at or below 2 mg/kg bw up to 104 weeks.
Furan did not induce gene mutations in bacteria.
In the majority of the in vitro studies in mammalian cells, furan was able to induce chromosomal aberrations and sister chromatid exchanges; contrasting results were reported on the requirement for microsomal activation.
BDA, the CYP2E1‐mediated metabolite of furan, formed DNA adducts at the exocyclic N atom of the dC, dG and dA nucleosides in in vitro model systems and in DNA from S. Typhimurium TA104. BDA was able to directly induce mutations in bacteria and strand breaks and mutations in mammalian cells in vitro.
In rodents, furan induced very low levels of DNA adducts in liver and kidney. The chemical structures of these adducts could not be defined but were not identical to those induced by BDA in in vitro model systems.
Conflicting data were reported for the induction of DNA breaks in the liver. These breaks, likely reflecting oxidative stress‐induced DNA damage, occurred at doses showing mild liver toxicity. Chronic exposure to furan‐induced chromosomal damage in proliferating splenocytes from mice and rats. The DNA lesions responsible for these effects remain undefined.
No clear induction of base substitutions was observed in transgenic rat models. A weak mutagenic activity was suggested by a single study in a transgenic mouse model.
An excess of GC > TA transversions was observed at codon 61 of the Ha‐ras oncogene in furan‐induced liver tumours in mice. It is uncertain whether these mutations are the consequence of oxidative damage to DNA or reflect an expansion of pre‐existing spontaneous Ha‐ras mutations.
Histological changes in the testes, prostate gland, Leydig cells and seminal vesicles were observed in Wistar rats at 2 mg/kg bw (lowest dose tested) and above given during weaning and post‐puberty. No histological effects in reproductive organs were observed in adult rats and mice up to 8 mg/kg bw.
Methylfurans
The liver is the primary target organ with respect to acute and short‐term (< 90 days) toxicity of 2‐ and 3‐methylfuran in rodents. Pulmonary toxicity was observed upon inhalation but also upon i.p. application. There is also indication for kidney toxicity of 3‐methylfuran after 90 days of exposure.
No clear differences in structure–effect relationship for the investigated alkylated furans with respect to the pattern of adverse effects/the target organs were identified.
Liver toxicity in male rats given 3‐methylfuran (28 or 90 days) or 2‐methylfuran (28 days) by gavage was found to be the most sensitive adverse endpoint for both compounds. Their toxic potency was reported to be in the same order of magnitude as that for furan.
No information on the genotoxic properties of 3‐methylfuran and limited information for 2‐methylfuran and 2,5‐dimethylfuran is available. Both 2‐methylfuran and 2,5‐dimethylfuran showed negative results in bacteria. There is some evidence that both compounds induce chromosomal damage in mammalian cells in vitro and there is little evidence that 2,5‐dimethylfuran can induce DNA breaks in vivo.
Mode of action
Furan induces oxidative stress. BDA binds covalently to a range of target molecules, including proteins and GSH. These features lead ultimately to cell and tissue damage, mitochondrial dysfunction and fibrosis, primarily in the liver.
There is clear evidence for the involvement of indirect mechanisms in the carcinogenic MoA of furan. These include epigenetic changes, oxidative damage to DNA and regenerative hyperplasia, with all of these effects being accompanied by tissue damage.
There is limited evidence of a direct mechanism (i.e. direct interaction with DNA) in the carcinogenic action of furan.
The contributing factors in carcinogenesis are likely to vary according to dose, duration of exposure and degree of severity of liver cellular damage, inflammation and compensatory proliferation.
Considerations of critical effects, dose–response modelling and possibilities for derivation of a health based guidance value
Since only few data are available on the effect of furan in humans, the CONTAM Panel used data in experimental animals for the hazard characterisation.
For non‐neoplastic effects, the CONTAM Panel selected the BMDL10 of 0.09 mg/kg bw per day (5 days per week dosing regimen) for the induction of cholangiofibrosis in male rats after 2 years as reference point. This corresponds to 0.064 mg/kg bw per day (correcting for the applied dose regimen of 5 days per week).
For neoplastic effects, the CONTAM Panel considered that the combined data set from NTP (1993) and Moser et al. (2009) on the incidence of hepatocellular adenomas and carcinomas in female mice after 2 years is the most robust data set to derive a reference point and selected the BMDL10 of 1.84 mg/kg bw per day (5 days per week dosing regimen). This corresponds to 1.31 mg/kg bw per day (correcting for the applied dose regimen of 5 days per week).
In view of some indications for a direct genotoxic mechanism in the carcinogenic MoA of furan, the CONTAM Panel decided that it was not appropriate to establish a TDI and used an MOE approach.
The available information was insufficient to identify a reference point for the methylfurans. However, it was considered appropriate to assume dose additivity for hepatotoxicity of furan, 2‐methylfuran and 3‐methylfuran in the rat.
4.3. Risk characterisation
For non‐neoplastic effects, MOE values that range from 584 (minimum LB) to 65 (maximum UB) for the mean exposure estimates, and from 338 (minimum LB) to 35 (maximum UB) for the 95th percentile exposure estimates across dietary surveys and age groups have been calculated. The calculated MOEs are below 100 in a number of dietary surveys, particularly for the high percentile exposure estimates for the younger age groups (infants and toddlers) and adults (including elderly). The CONTAM Panel concluded that these MOEs indicate a health concern.
For neoplastic effects, MOE values that range from 11,948 (minimum LB) to 1,328 (maximum UB) for the mean exposure estimates, and from 6,917 (minimum LB) to 722 (maximum UB) for the 95th percentile exposure estimates across dietary surveys and age groups have been calculated.
The CONTAM Panel noted that, with the exception of some surveys, the calculated MOEs for neoplastic effects of furan are smaller than 10,000, which, in accordance with the guidance given by the Scientific Committee, would indicate a health concern. However, there is uncertainty regarding the carcinogenic MoA of furan. The CONTAM Panel considered the resulting MOEs for hepatocellular adenomas and carcinomas as supporting evidence for its conclusion, based on the hepatotoxicity of furan, that the current exposure to furan indicates a health concern.
Based on a scenario for chronic dietary exposure to the sum of furan, 2‐methylfuran and 3‐methylfuran, MOE values for the incidence of cholangiofibrosis range from approximately 430 (minimum LB) to 20 (maximum UB) for the mean exposure estimates, and from 220 (minimum LB) to 10 (maximum UB) for the 95th percentile exposure estimates across surveys and age groups have been calculated. From these figures, it becomes clear that methylfurans may add significantly to the overall exposure and therefore increase the concern for hepatotoxicity.
5. Recommendations
There is a need for additional data on the occurrence of methylfurans in food.
There is a need for additional data on the changes of furan and methylfurans concentrations during the distinct stages of coffee preparation for all coffee types.
Studies in vivo on the effect of furan on the genome to clarify the carcinogenic MoA are needed.
Further information is needed on the toxicity, including the genotoxic properties, of methylfurans.
Documentation provided to EFSA
Data on the co‐occurrence of furan, 2‐methylfuran and 3‐methylfuran in jarred baby foods, cereal flakes, roasted and grounded coffee, soluble coffee, infant cereals and tomato sauce. December 2016. Submitted by FoodDrinkEurope, outside the ad‐hoc call for occurrence data (see Section 2.3.1).
Gill S, Kavanagh M, Cherry W, Bourque C, Caldwell D, Wang G and Bondy G. A 90‐Day Subchronic Gavage Toxicity Study in Fischer 344 Rats with 3‐methylfuran. August 2017. Submitted by Health Canada.
Abbreviations
- ABT
aminobenzotriazole
- ACB‐PCR
allele‐specific competitive blocker‐polymerase chain reaction
- ADME
absorption, distribution, metabolism and excretion
- AIC
Akaike information criterion
- ALP
alkaline phosphatase
- ALT
alanine aminotransaminase
- anses
Agence nationale de sécurité sanitaire alimentation, environnement, travail
- AP
apurinic/apyrimidinic
- BDA
cis‐but‐2‐ene‐1,4‐dialdehyde
- BMD
benchmark dose
- BMDL
benchmark dose lower confidence limit
- BMDU
benchmark dose upper confidence limit
- BrdU
5‐bromo‐2‐deoxyuridine
- BSO
buthionine sulfoximine
- BUN
blood urea nitrogen
- bw
body weight
- CA
chromosomal aberrations
- CEEREAL
European Breakfast Cereal Association (),
- CHO
Chinese hamster ovary
- CONTAM
EFSA Panel on Contaminants in the Food Chain
- Cys
cysteine
- CYP
cytochrome P450
- dA
2’‐deoxyadenosine
- DATA
EFSA Evidence Management Unit
- dC
2’‐deoxycytidine
- DEM
diethylmaleate
- dG
2’‐deoxyguanosine
- DPE
Documentation provided to EFSA by FoodDrinkEurope
- DSB
double strand break
- dT
2’‐deoxythymidine
- EC50
half maximal effective concentration
- ECF
European Coffee Federation
- ELISA
enzyme‐linked immunosorbent assay
- FDA
US Food and Drug Administration
- γ‐GT
γ‐glutamyl transpeptidase
- GC
gas chromatography
- GC‐SRI‐TOF‐MS
gas chromatography‐selective reagent ionization‐time of flight‐mass spectrometry
- GI
gastrointestinal
- Gln
glutamine
- Glu
glutamate
- GSH
glutathione
- GST‐P
glutathione S‐transferase placental form
- HBGV
health‐based guidance value
- HPLC
high‐performance liquid chromatography
- IARC
International Agency for Research on Cancer
- ICL
inter‐strand crosslink
- Ig
immunoglobulin
- IL
interleukin
- i.p.
intraperitoneal
- IPCS
International Programme on Chemical Safety
- JECFA
Joint FAO/WHO Expert Committee on Food Additives
- Kow
octanol/water partition coefficient
- LB
lower bound
- LC
left‐censored
- LC–MS/MS
liquid chromatography/tandem mass spectrometry
- LC50
lethal concentration, median
- LD50
lethal dose, median
- LDL
low‐density lipoprotein
- LOAEL
lowest‐observed‐adverse‐effect level
- LOD
limit of detection
- LOQ
limit of quantification
- Lys
lysine
- Max
maximum
- MB
middle‐bound
- Min
minimum
- ML
maximum level
- MoA
mode of action
- MOE
margin of exposure
- MS
mass spectrometry
- MW
molecular weight
- N
Number
- n.r.
not reported
- NADH
nicotinamide adenine dinucleotide
- NAC
N‐acetylcysteinyl
- NAL
N‐acetyllysinyl
- NAS
US National Academy of Sciences
- NAT
N‐acetyltransferase
- NIEHS
US National Institute of Environmental Health Sciences
- NOAEL
no‐observed‐adverse‐effect level
- NTP
National Toxicology Program
- OTZ
oxothiazolidinecarboxylate
- PBPK
physiologically based pharmacokinetic
- PND
post‐natal day
- PTR‐MS
proton‐transfer reaction mass spectrometry
- ROS
reactive oxygen species
- SCE
sister chromatid exchange
- SDH
sorbitol dehydrogenase
- SIM
selective ion monitoring
- SNE
Specialised Nutrition Europe
- SPME
solid‐phase microextraction
- SOP
standard operational procedure
- SSB
single strand break
- stAR
steroidogenic acute regulating protein
- SULT
sulfotransferase
- TDI
tolerable daily intake
- TDS
total diet study
- TNF
tumour necrosis factor
- UB
upper bound
- UDS
unscheduled DNA synthesis
- UV
ultraviolet
- VKM
Norwegian Scientific Committee for Food Safety
- WG
Working group
- WHO
World Health Organization
Appendix A – Identification and selection of evidence relevant for the risk assessment of furan and methylfurans in food
A.1. Literature search for supporting information for the assessment
A literature search for recent reviews, in combination with a limited literature search for topics not sufficiently covered by reviews, was used to identify the scientific literature. The used search strings and the number of results in web of science and PubMed are given below.
A.1.1. Furan, 2‐methylfuran and 3‐methylfuran
Search for reviews
A. Web of Science
Used search string: TOPIC: (Furan OR 2‐methylfuran OR 3‐methylfuran) AND TOPIC: (food) Refined by: DOCUMENT TYPES: (REVIEW); Timespan=2006–2016; Search language=Auto
Results in Web of Science: 78
B. PubMed
Used search string: ((((Furan OR 2‐methylfuran OR 3‐methylfuran)) AND food) AND review) AND (“2006/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in PubMed: 26
Search for information regarding the occurrence, formation and analysis of 2‐methylfuran and 3‐methylfuran
A. Web of Science
Used search string: TOPIC: (2‐methylfuran OR 3‐methylfuran) AND TOPIC: (food OR formation OR analys*) Timespan=2006–2016; Search language=Auto
Results in Web of Science: 187
B. PubMed
Used search string: (((2‐methylfuran OR 3‐methylfuran)) AND (food OR formation OR analys*)) AND (“2006/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in PubMed: 60
Search for previous exposure assessments
A. Web of Science
Used search string: TOPIC: (furan OR 2‐methylfuran OR 3‐methylfuran) AND TOPIC: (exposure and food) NOT TOPIC: (PCDD OR PCDF); Timespan=2006–2016; Search language=Auto
Results in Web of Science: 228
B. PubMed
Used search string ((((furan OR 2‐methylfuran OR 3‐methylfuran)) AND (exposure and food)) NOT (PCDD OR PCDF)) AND (“2006/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in PubMed: 49
Overall results
From these searches, 628 references were identified and after removal of the duplicates 466 references were screened for relevance.
A.1.2. 2,5‐dimethylfuran
Search for reviews
C. Web of Science
Used search string: TOPIC: (2,5‐dimethylfuran) AND TOPIC: (food) Refined by: DOCUMENT TYPES: (REVIEW); Timespan=2006–2016; Search language=Auto
Results in Web of Science: 0
D. PubMed
Used search string: ((((2,5‐dimethylfuran)) AND food) AND review) AND (“2006/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in PubMed: 0
Search for information regarding the occurrence, formation and analysis of 2,5‐dimethylfuran
C. Web of Science
Used search string: TOPIC: (2,5‐dimethylfuran) AND TOPIC: (food OR formation OR analys*) Timespan=2006–2016; Search language=Auto
Results in Web of Science: 123
D. PubMed
Used search string: (((2,5‐dimethylfuran)) AND (food OR formation OR analys*)) AND (“2006/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in PubMed: 66
Search for previous exposure assessments
C. Web of Science
Used search string: TOPIC: (2,5‐dimethylfuran) AND TOPIC: (exposure and food) NOT TOPIC: (PCDD OR PCDF); Timespan=2006‐2016; Search language=Auto
Results in Web of Science: 5
D. PubMed
Used search string ((((2,5‐dimethylfuran)) AND (exposure and food)) NOT (PCDD OR PCDF)) AND (“2006/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in PubMed: 5
Overall results
From these searches, 199 references were identified and after removal of the duplicates 159 references were screened for relevance.
A.1.3. Coffee
Since most of the occurrence data on coffee were reported for the solid, while most coffee consumption data were reported as liquid, the WG carried out an additional literature search to identify papers regarding furan and methylfurans in coffee.
A. Web of Science
Used search string: TOPIC: (coffee) AND TOPIC: (furan or 2‐methylfuran or 3‐methylfuran or 2,5‐dimethylfuran) Indexes=SCI‐EXPANDED, SSCI, A&HCI, CPCI‐S, CPCI‐SSH, BKCI‐S, BKCI‐SSH, ESCI, CCR‐EXPANDED, IC Timespan=2006–2017
Results in Web of Science: 114
B. PubMed
Used search string: (((“Coffee”[Mesh] or coffee)) AND (furan or 2‐methylfuran or 3‐methylfuran or 2,5‐dimethylfuran)) AND (“2006/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in PubMed: 53
From these searches, 167 references were identified and after removal of the duplicates 119 references were screened for relevance.
A.2. Literature search for hazard identification and characterisation
In addition to the literature search outsourced by EFSA (see NFI, 2017 for further details), some specific searches were performed to identify the scientific literature. The used search strings and the number of results in web of science and PubMed are given below.
A.2.1. Search for papers regarding the genotoxicity caused by cis‐2‐butene‐1,4‐dial
A. Web of Science
Used search string: TOPIC: (“cis‐2‐butene‐1,4‐dial” OR “maleic dialdehyde”) AND TOPIC: (genotox* OR muta* OR DNA OR damage OR repair OR clastogen* OR aneugen* OR chromosom*); Timespan=1990–2016; Search language=Auto
Results in Web of Science: 152
B. PubMed
Used search string: (((“cis‐2‐butene‐1,4‐dial” OR “maleic dialdehyde”)) AND (genotox* OR muta* OR DNA OR damage OR repair OR clastogen* OR aneugen* OR chromosom*)) AND (“1990/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in Web of Science: 57
From these searches, 209 references were identified and after removal of the duplicates 141 references were screened for relevance.
A.2.2. Search for papers regarding the toxicity of 2,5‐dimethylfuran
A.2.2.1. Toxicokinetics
A. Web of Science
Used search string: TOPIC: (2,5‐dimethylfuran) AND TOPIC: (absor* OR tissue* OR metaboli* OR excret* OR kinetic* OR toxicokinetic* OR pharmacokinetic* OR degrad* OR biotrans*); Timespan=1990–2016; Search language=Auto
Results in Web of Science: 110
B. PubMed
Used search string: ((2,5‐dimethylfuran) AND ((absor* OR tissue* OR metaboli* OR excret* OR kinetic* OR toxicokinetic* OR pharmacokinetic* OR degrad* OR biotrans*))) AND (“1990/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in Web of Science: 47
A.2.2.2. Toxicity in experimental animals
A. Web of Science
Used search string: TOPIC: (2,5‐dimethylfuran) AND TOPIC: ((oral OR diet* OR gavage OR feed OR food OR organ* OR tissue* OR cancer* OR carcino* OR tumor* OR tumour* OR tox* OR immun* OR teratogen* OR rat OR mouse OR mice OR rabbit*)); Timespan=1990–2016; Search language=Auto
Results in Web of Science: 98
B. PubMed
Used search string: ((2,5‐dimethylfuran) AND (oral OR diet* OR gavage OR feed OR food OR organ* OR tissue* OR cancer* OR carcino* OR tumor* OR tumour* OR tox* OR immun* OR teratogen* OR rat OR mouse OR mice OR rabbit*)) AND (“1990/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in Web of Science: 68
A.2.2.3. Genotoxicity and mode of action
A. Web of Science
Used search string: TOPIC: ((2,5‐dimethylfuran) AND TOPIC: (genotox* OR mode OR action OR mechanism* OR muta* OR DNA OR damage OR repair OR clastogen* OR aneugen* OR chromosom*)); Timespan=1990–2016; Search language=Auto
Results in Web of Science: 102
B. PubMed
Used search string: ((2,5‐dimethylfuran) AND (genotox* OR mode OR action OR mechanism* OR muta* OR DNA OR damage OR repair OR clastogen* OR aneugen* OR chromosom*)) AND (“1990/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in Web of Science: 36
A.2.2.4. Observations in humans
A. Web of Science
Used search string: TOPIC: (2,5‐dimethylfuran) AND TOPIC: (epidemio* OR biomarker* OR exposure* OR case* OR poison* OR cohort* OR cross‐sectional OR random* OR work*); Timespan=2004–2016; Search language=Auto
Results in Web of Science: 80
B. PubMed
Used search string: ((2,5‐dimethylfuran) AND (epidemio* OR biomarker* OR exposure* OR case* OR poison* OR cohort* OR cross‐sectional OR random* OR work*)) AND (“2004/01/01”[Date ‐ Publication] : “3000”[Date ‐ Publication])
Results in Web of Science: 35
From these searches, 576 references were identified and after removal of the duplicates 298 references were screened for relevance.
A.2.3. Papers not included in this scientific opinion
The list below gives an overview of the scientific papers (excluding abstracts and reviews) related to the toxicity and toxicokinetics of furan and methylfurans that were identified in the scientific literature (see Section 2.2) but not included in the opinion.
Bas H, Pandir D and Kalender S, 2016. Furan‐induced hepatotoxic and hematologic changes in diabetic rats: the protective role of lycopene. Arhiv za Higijenu Rada i Toksikologiju, 67, 194–203. https://doi.org/10.1515/aiht-2016-67-2762
Bluhm K, Seiler TB, Anders N, Klankermayer J, Schaeffer A and Hollert H, 2016. Acute embryo toxicity and teratogenicity of three potential biofuels also used as flavor or solvent. The Science of the Total Environment, 566–567, 786‐795. https://doi.org/10.1016/j.scitotenv.2016.05.055
Carrette LL, Gyssels E, De Laet N and Madder A, 2016. Furan oxidation based cross‐linking: a new approach for the study and targeting of nucleic acid and protein interactions. Chemical Communications, 52, 1539–1554. https://doi.org/10.1039/c5cc08766j
Carrette LL, Morii T and Madder A, 2013. Toxicity inspired cross‐linking for probing DNA‐peptide interactions. Bioconjugate Chemistry, 24, 2008–2014. https://doi.org/10.1021/bc400327q
Dang NL, Hughes TB, Miller GP and Swamidass SJ, 2017. Computational approach to structural alerts: furans, phenols, nitroaromatics, and thiophenes. Chemical Research in Toxicology, 30, 1046–1059. https://doi.org/10.1021/acs.chemrestox.6b00336
Deferme L, Wolters J, Claessen S, Briede J and Kleinjans J, 2015. Oxidative stress mechanisms do not discriminate between genotoxic and nongenotoxic liver carcinogens. Chemical Research in Toxicology, 28, 1636–1646. https://doi.org/10.1021/acs.chemrestox.5b00222
Doyle M, Sexton KG, Jeffries H and Jaspers I, 2007. Atmospheric photochemical transformations enhance 1,3‐butadiene‐induced inflammatory responses in human epithelial cells: the role of ozone and other photochemical degradation products. Chemico‐Biological Interactions, 166, 163–169. https://doi.org/10.1016/j.cbi.2006.05.016
Fry JR, Hammond AH, Garle MJ and Lal K, 1993. Comparison of xenobiotic‐mediated cytotoxicity in rat cultured‐hepatocytes and the V79 Chinese‐Hamster lung fibroblast cell‐line ‐ can metabolically‐activated hepatotoxins be identified by selective cytotoxicity to hepatocytes. Atla‐Alternatives to Laboratory Animals, 21, 8–12.
Garcia‐Gomez D, Bregy L, Barrios‐Collado C, Vidal‐de‐Miguel G and Zenobi R, 2015. Real‐time high‐resolution tandem mass spectrometry identifies furan derivatives in exhaled breath. Analytical Chemistry, 87, 6919–6924. https://doi.org/10.1021/acs.analchem.5b01509
Hamadeh HK, Jayadev S, Gaillard ET, Huang Q, Stoll R, Blanchard K, Chou J, Tucker CJ, Collins J, Maronpot R, Bushel P and Afshari CA, 2004. Integration of clinical and gene expression endpoints to explore furan‐mediated hepatotoxicity. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 549, 169–183. https://doi.org/10.1016/j.mrfmmm.2003.12.021
Higgins LG, Cavin C, Itoh K, Yamamoto M and Hayes JD, 2008. Induction of cancer chemopreventive enzymes by coffee is mediated by transcription factor Nrf2. Evidence that the coffee‐specific diterpenes cafestol and kahweol confer protection against acrolein. Toxicology and Applied Pharmacology, 226, 328–337. https://doi.org/10.1016/j.taap.2007.09.018
Huang Q, Jin X, Gaillard ET, Knight BL, Pack FD, Stoltz JH, Jayadev S and Blanchard KT, 2004. Gene expression profiling reveals multiple toxicity endpoints induced by hepatotoxicants. Mutation Research, 549, 147–167. https://doi.org/10.1016/j.mrfmmm.2003.12.020
Inamdar AA, Masurekar P and Bennett JW, 2010. Neurotoxicity of fungal volatile organic compounds in Drosophila melanogaster. Toxicological Sciences, 117, 418–426. https://doi.org/10.1093/toxsci/kfq222
Kamijima M, Sobue G, Ichihara G, Shibata E, Ono Y, Kondo H, Villanueva MB, Itoh T, Mitsuma T and Takeuchi Y, 1996. Toxic effects of hexane derivatives on cultured rat Schwann cells. Toxicology, 108, 25–31.
Lai GH and Sirica AE, 1999. Establishment of a novel rat cholangiocarcinoma cell culture model. Carcinogenesis, 20, 2335–2340.
McMurtry RJ and Mitchell JR, 1977. Renal and hepatic necrosis after metabolic activation of 2‐substituted furans and thiophenes, including furosemide and cephaloridine. Toxicology and Applied Pharmacology, 42, 285–300. https://doi.org/10.1016/0041-008x(77)90005-9
Muller A, Briviba K, Graf V, Greiner R, Herrmann C, Kuballa T and Stahl MR, 2013. UV‐C treatment using a Dean vortex technology ‐ impact on apple juice enzymes and toxicological potential. Innovative Food Science & Emerging Technologies, 20, 238–243. https://doi.org/10.1016/j.ifset.2013.07.010
Ochi T and Ohsawa M, 1985. Participation of active oxygen species in the induction of chromosomal aberrations by cadmium chloride in cultured Chinese hamster cells. Mutation Research, 143, 137–142.
Pandir D, 2015. Assessment of the DNA damage in human sperm and lymphocytes exposed to the carcinogen food contaminant furan with comet assay. Brazilian Archives of Biology and Technology, 58, 773–780. https://doi.org/10.1590/s1516-89132015050269
Radaeva S, Ferreira‐Gonzalez A and Sirica AE, 1999. Overexpression of C‐NEU and C‐MET during rat liver cholangiocarcinogenesis: a link between biliary intestinal metaplasia and mucin‐producing cholangiocarcinoma. Hepatology, 29, 1453–1462. https://doi.org/10.1002/hep.510290524
Ramos‐Marquez ME, Grijalva G and Armendariz‐Borunda J, 2002. Ductular hyperplasia is characterized by an over expression of c‐Myc in bile duct ligation plus furan injured rats: possible role of interleukin‐6. Hepatology Research, 22, 127–138. https://doi.org/10.1016/s1386-6346(01)00121-8
Sahlberg B, Gunnbjornsdottir M, Soon A, Jogi R, Gislason T, Wieslander G, Janson C and Norback D, 2013. Airborne molds and bacteria, microbial volatile organic compounds (MVOC), plasticizers and formaldehyde in dwellings in three North European cities in relation to sick building syndrome (SBS). The Science of the Total Environment, 444, 433–440. https://doi.org/10.1016/j.scitotenv.2012.10.114
Sprankle CS, Goldsworthy TL, Goldsworthy SM, Wilson DM and Butterworth BE, 1994. Expression of the hepatocyte growth factor and c‐MET genes during furan‐induced regenerative cell proliferation in the livers of B6C3F1 mice and F‐344 rats. Cell Proliferation, 27, 529–539. https://doi.org/10.1111/j.1365-2184.1994.tb01490.x
Stanley LA, Blackburn DR, Devereaux S, Foley J, Lord PG, Maronpot RR, Orton TC and Anderson MW, 1994. Ras mutations in methylclofenapate‐induced B6C3F1 and C57BL/10J mouse liver tumours. Carcinogenesis, 15, 1125–1131.
Upreti KK, Das M and Khanna SK, 1991. Role of antioxidants and scavengers on argemone oil‐induced toxicity in rats. Archives of Environmental Contamination and Toxicology, 20, 531–537.
Vu CC and Peterson LA, 2005. Synthesis of a 2’‐deoxyguanosine adduct of cis‐2‐butene‐1,4‐dial, a reactive metabolite of furan. Chemical Research in Toxicology, 18, 1012–1017. https://doi.org/10.1021/tx049647d
Walinder R, Ernstgard L, Johanson G, Norback D, Venge P and Wieslander G, 2005. Acute effects of a fungal volatile compound. Environmental Health Perspectives, 113, 1775–1778.
Wang J, Yang Z, Lin L, Zhao Z, Liu Z and Liu X, 2012. Protective effect of naringenin against lead‐induced oxidative stress in rats. Biological Trace Element Research, 146, 354–359. https://doi.org/10.1007/s12011-011-9268-6
Zaidi SI, Agarwal R, Eichler G, Rihter BD, Kenney ME and Mukhtar H, 1993. Photodynamic effects of new silicon phthalocyanines: in vitro studies utilizing rat hepatic microsomes and human erythrocyte ghosts as model membrane sources. Photochemistry and Photobiology, 58, 204–210.
A.3. EFSA guidance documents applied for the risk assessment
The following EFSA guidance pertaining to risk assessment were followed for the development of the risk assessment:
EFSA (European Food Safety Authority), 2005. Opinion of the Scientific Committee on a request from EFSA related to a harmonised approach for risk assessment of substances which are both genotoxic and carcinogenic. EFSA Journal 2005;282, 1–31.
EFSA (European Food Safety Authority), 2007. Guidance of the Scientific Committee on a request from EFSA related to Uncertainties in Dietary Exposure Assessment. EFSA Journal 2007;4(12):438, 54 pp. https://doi.org/10.2903/j.efsa.2007.438
EFSA (European Food Safety Authority), 2009. Guidance of the Scientific Committee on transparency in the scientific aspects of risk assessments carried out by EFSA. Part 2: general principles. EFSA Journal 2009;1051, 1–22.
EFSA (European Food Safety Authority), 2010. Standard sample description for food and feed. EFSA Journal 2010;8(1):1457, 54 pp. https://doi.org/10.2903/j.efsa.2010.1457
EFSA (European Food Safety Authority), 2010. Management of left‐censored data in dietary exposure assessment of chemical substances. EFSA Journal 2010;8(3):1557, 96 pp. https://doi.org/10.2903/j.efsa.2010.1557
EFSA (European Food Safety Authority), 2011. Use of the EFSA Comprehensive European Food Consumption Database in Intakes Assessment. EFSA Journal 2011;9(3):2097, 34 pp. https://doi.org/10.2903/j.efsa.2011.2097
EFSA Scientific Committee 2011. Scientific Opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379, 69 pp. DOI: https://doi.org/10.2903/j.efsa.2011.2379
EFSA Scientific Committee 2012. Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data. EFSA Journal 2012;10(3):2579, 32 pp. https://doi.org/10.2903/j.efsa.2012.2579
EFSA Scientific Committee, 2012. Scientific Opinion on Risk Assessment Terminology. EFSA Journal 2012;10(5):2664, 43 pp. DOI: https://doi.org/10.2903/j.efsa.2012.2664
EFSA Scientific Committee, Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen KH, More S, Mortensen A, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Silano V, Solecki R, Turck D, Aerts M, Bodin L, Davis A, Edler L, Gundert‐Remy U, Sand S, Slob W, Bottex B, Abrahantes JC, Marques DC, Kass G and Schlatter JR, 2017. Update: use of the benchmark dose approach in risk assessment. EFSA Journal, 15, n/a‐n/a. https://doi.org/10.2903/j.efsa.2017.465
Appendix B – Effects of different alkylfurans in experimental animals
| Compound/species | Treatment | Outcome | Reference |
|---|---|---|---|
| 2‐methylfuran | |||
| Rat | ≥ 1,218 μmol/kg bw, i.p. (single exposure) | Liver necrosis, pulmonary bronchiolar lesions | Ravindranath et al. (1986) |
| Rat, Fischer 344, male | 5, 18, 37, 73, 146, 305 μmol/kg bw per day by gavage over 28 days | Mild histological lesions and inflammatory infiltrations in the liver, Changes in serum cholesterol and T3, T4 | Gill et al. (2014) |
| 3‐methylfuran | |||
| Mouse, NIH Swiss, male | 100–200 mg (radiolabelled)/kg bw i.p. single exposure or 200 μmol (radiolabelled)/L, inhalation, 1 h | Bronchiolar necrosis, binding of radioactivity in the lung, 8 h after i.p. injection or 24 h after inhalation | Boyd et al. (1978) |
| Rat, CD/CR, female Syrian hamster, female | 148 and 322 μmol/L, inhalation, 1 h |
Hamster: Bronchiolar necrosis after 1 day Rat: Bronchiolar necrosis after 1 day |
Haschek et al. (1983) |
| Mouse, BALB/c, male | 14 and 37 μmol/L, inhalation, 1 h | Bronchiolar necrosis after 1 day | Haschek et al. (1984) |
| Mouse, ICR, male | 3,094 μmol/kg bw i.p., (single exposure) | No effects in liver and kidney after 24 h | Wiley et al. (1984) |
| Mouse, ICR, male | 2,600 and 3,100 μmol/kg bw i.p. (single exposure) | Severe bronchiolar necrosis after 24 h | Gammal et al. (1984) |
| Syrian hamster, male and female | 344 μmol/L, inhalation, 2 h once a week over 10 weeks | No pulmonary effects | Witschi et al. (1985) |
| Rat, Fischer 344, male | 1, 4, 18, 37, 73, 146 and 305 μmol/kg bw per day by gavage over 28 days | Histological and gross changes of the liver, changes in serum liver enzymes, increase in serum T3 and T4 | Gill et al. (2015) |
| Rat, Fischer 344, male and female | 0, 0.2, 1, 3, 12 and 49 μmol/kg bw by gavage over 90 days (5 days per week) | Liver lesions, kidney lesions, increase in markers for liver and kidney injury in serum | Gill et al. (2017, in press) |
| 2‐ethylfuran | |||
| Mouse, Swiss, male | 2,081 μmol/kg bw, i.p. (single exposure) | Hepatic centrilobular and renal proximal tubular necrosis after 36 h | McMurtry and Mitchell (1977) |
| Mouse, ICR, male | 2,600 μmol/kg bw, i.p. |
Kidney: proximal tubular necrosis liver: focal hydropic degeneration after 24 h |
Wiley et al. (1984) |
| Mouse, ICR, male | 2,600 μmol/kg bw, i.p. (single exposure) | Severe bronchiolar necrosis after 24 h | Gammal et al. (1984) |
| 3‐ethylfuran | |||
| Mouse, ICR, male | 3,100 μmol/kg bw, i.p. (single exposure) |
Kidney: proximal tubular necrosis liver: focal hydropic degeneration after 24 h |
Wiley et al. (1984) |
| Mouse, ICR, male | 3,100 μmol/kg bw, i.p. (single exposure) | Severe bronchiolar necrosis, slight perivascular oedema after 24 h | Gammal et al. (1984) |
| 3‐pentylfuran | |||
| Mouse, ICR, male | 1,800 μmol/kg bw, i.p. (single exposure) | Kidney: proximal tubular necrosis, liver: centrilobular necrosis after 24 h | Wiley et al. (1984) |
| Mouse, ICR, male | 1,800 and 2,600 μmol/kg bw, i.p. (single exposure) | No pulmonary effects after 24 h | Gammal et al. (1984) |
bw: body weight; i.p.: intraperitoneal.
Appendix C – Benchmark dose analysis
The text below describes the benchmark dose (BMD) analysis of the incidence of cholangiofibrosis in male rats and the incidence of hepatocellular adenomas/carcinomas in female mice. BMD analysis was done according to the EFSA guidance (EFSA Scientific Committee, 2017). The following was applied in all analyses.
Software used
Results are obtained using the R‐package bmdModeling.
Fitting benchmark dose models is based on the R‐package proast61.3.
Averaging results from multiple fitted benchmark dose models is based on the methodology in Wheeler and Bailer (2008).
Model averaging
Model averaging was used for all tested endpoints.
Dose–response model sets
No deviation from the recommended defaults. Default set of fitted models:
| Model | Number of parameters | Formula | |
|---|---|---|---|
| Null | 1 | y = a | |
| Full | No of groups | y = groupmean | |
| Logistic | 2 |
|
|
| Probit | 2 |
|
|
| Log‐logistic | 3 |
|
|
| Log‐probit | 3 |
|
|
| Weibull | 3 |
|
|
| Gamma | 3 | y = pgamma(bx; c) | |
| Two‐stage | 3 |
|
Procedure for selection of BMDL
There was no deviation from the procedure described in the flow chart to obtain the final BMD confidence interval.
Figure C.1.
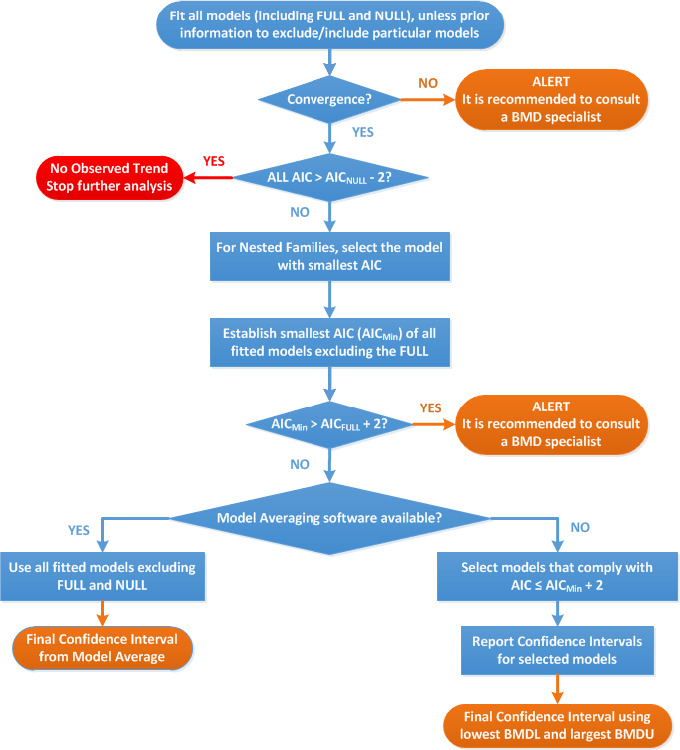
Flow chart for selection of BMDL
C.1. Incidence of cholangiofibrosis
C.1.1. Data description
Data from male F344/N NCTR rats treated for 36 weeks or 2 years with furan in corn oil by gavage 5 times per week (NCTR, 2015). Doses used in this BMD analysis were not adjusted for the dose regimen and the reported results consequently correspond to a 5 days per week dosing regimen.
The analysed endpoints are:
cholangiofibrosis after 36 weeks of treatment (Cholfib_36Wk);
cholangiofibrosis after 2 years of treatment (Cholfib_2y).
Table C.1.
Data on the incidence of cholangiofibrosis used for BMD analysis
| Dose (mg/kg bw per day)a | After 36 weeks of treatment | After 2 years of treatment | ||
|---|---|---|---|---|
| N | N total | N | N total | |
| 0.000 | 0 | 20 | 0 | 149 |
| 0.020 | 0 | 20 | 0 | 150 |
| 0.044 | 0 | 20 | 0 | 99 |
| 0.092 | 0 | 20 | 1 | 100 |
| 0.200 | 0 | 20 | 38 | 50 |
| 0.440 | 6 | 20 | 49 | 49 |
| 0.920 | 17 | 20 | 47 | 50 |
| 2.000 | 19 | 20 | 49 | 49 |
N: number of animals; bw: body weight.
For a 5 days per week dosing regimen.
C.1.2. Selection of the BMR
A default benchmark response (BMR) of 10% (extra risk compared with the background risk) and a 90% interval around the BMD were selected as recommended by EFSA Scientific Committee (2017).
C.1.3. Results for the incidence of cholangiofibrosis after 36 weeks
Table C.2.
Result for the incidence of cholangiofibrosis after 36 weeks using model averaging
| Model | Number of parameters | Log‐likelihood | AIC | BMD | BMDL | BMDU | Converged | Accepted AIC |
|---|---|---|---|---|---|---|---|---|
| Null | 1 | −92.10 | 186.20 | NA | NA | NA | Yes | |
| Full | 8 | −24.64 | 65.28 | NA | NA | NA | Yes | |
| Logistic | 2 | −32.17 | 68.34 | 0.35 | 0.27 | 0.44 | Yes | No |
| Log‐logistic | 3 | −25.78 | 57.56 | 0.31 | 0.22 | 0.39 | Yes | Yes |
| Log‐probit | 3 | −25.97 | 57.94 | 0.30 | 0.21 | 0.38 | Yes | Yes |
| Weibull | 3 | −28.99 | 63.98 | 0.24 | 0.16 | 0.33 | Yes | No |
| Gamma | 3 | −27.38 | 60.76 | 0.28 | 0.19 | 0.37 | Yes | No |
| Two‐stage | 3 | −29.27 | 64.54 | 0.27 | 0.23 | 0.33 | No | No |
AIC: Akaike information criterion; BMD: benchmark dose; BMDL: benchmark dose lower confidence limit; BMDU: benchmark dose upper confidence limit.
It was noted that the Probit model of the PROAST version 61.3 was not fitting the data.
Table C.3.
Model weights used in model averaging
| Logistic | Log‐logistic | Log‐probit | Weibull | Gamma | |
|---|---|---|---|---|---|
| Estimated model weights | 0 | 0.48 | 0.4 | 0.02 | 0.1 |
Given 1,000 generated data sets, the BMDL is the 5th percentile of all parametric bootstrap BMD values and the BMDU is the 95th percentile.
Estimated the BMD based on the averaged response model which is a weighted average of the accepted models’ response values.
Table C.4.
Calculated BMD, BMDL and BMDU values (mg/kg bw per day for a 5 days per week dosing regimen) for the incidence of cholangiofibrosis after 36 weeks using model averaging
| BMD | BMDL | BMDU |
|---|---|---|
| 0.3 | 0.22 | 0.41 |
Figure C.2.
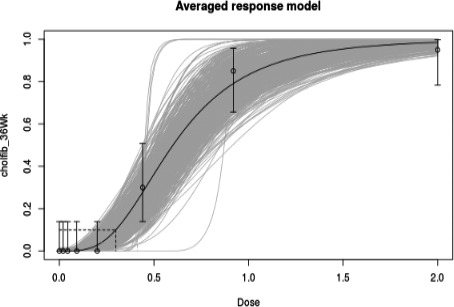
Averaged dose–response model for the incidence of cholangiofibrosis after 36 weeks
C.1.4. Results for the incidence of cholangiofibrosis after 2 years
Table C.5.
Result for the incidence of cholangiofibrosis after 2 years using model averaging
| Model | Included covariate(s) | Number of parameters | Log‐likelihood | AIC | BMD | BMDL | BMDU | Converged | Accepted AIC |
|---|---|---|---|---|---|---|---|---|---|
| Null | 1 | −401.99 | 805.98 | NA | NA | NA | Yes | ||
| Full | 8 | −44.50 | 105.00 | NA | NA | NA | Yes | ||
| Logistic | 2 | −100.79 | 205.58 | 0.11 | 0.10 | 0.13 | Yes | No | |
| Log‐logistic | 3 | −60.70 | 127.40 | 0.11 | 0.09 | 0.12 | Yes | No | |
| Log‐probit | 3 | −66.76 | 139.52 | 0.10 | 0.09 | 0.11 | Yes | No | |
| Weibull | 3 | −89.97 | 185.94 | 0.08 | 0.07 | 0.09 | Yes | No | |
| Gamma | 3 | −78.53 | 163.06 | 0.10 | 0.08 | 0.11 | Yes | No | |
| Two‐stage | 3 | −93.07 | 192.14 | 0.10 | 0.09 | 0.11 | No | No |
AIC: Akaike information criterion; BMD: benchmark dose; BMDL: benchmark dose lower confidence limit; BMDU: benchmark dose upper confidence limit.
It was noted that none of the fitted models is at least as good as the full model; all fitted models’ AIC values are larger than full model's AIC + 2. However, the confidence band contains the observed values (see Figure C.3), and therefore, it was decided that model averaging can be used. Nevertheless, it is recommended to generate more data on the incidence of cholangiofibrosis in the dose range 0.092–0.2 mg/kg bw per day to better characterise the dose–response.
Figure C.3.
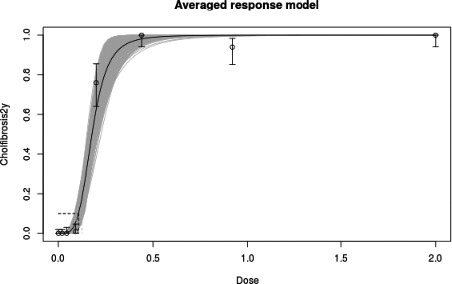
Averaged dose–response model for the incidence of cholangiofibrosis after 2 years
It was noted that the Probit model of the PROAST version 61.3 was not fitting the data.
Table C.6.
Model weights used in model averaging
| Logistic | Log‐logistic | Log‐probit | Weibull | Gamma | |
|---|---|---|---|---|---|
| Estimated model weights | 0.000 | 0.998 | 0.002 | 0.000 | 0.000 |
Given 1,000 generated data sets, the BMDL is the 5th percentile of all parametric bootstrap BMD values and the BMDU is the 95th percentile.
Estimated the BMD based on the averaged response model which is a weighted average of the accepted models’ response values.
Table C.7.
Calculated BMD, BMDL and BMDU values (mg/kg bw per day for a 5‐day per week dosing regimen) for the incidence of cholangiofibrosis after 2 years using model averaging
| BMD | BMDL | BMDU |
|---|---|---|
| 0.11 | 0.09 | 0.12 |
C.2. Incidence of hepatocellular adenomas/carcinomas in female mice
C.2.1. Data description
Hepatocellular adenomas and carcinomas in female B6C3F1 mice have been reported in two separate 2‐year carcinogenicity assays with furan (NTP, 1993 and Moser et al., 2009). A combined analysis of the Moser et al. (2009) and NTP (1993) data was performed (see Table 4 of the opinion), as well as an analysis for the Moser et al. (2009) data alone. Doses used in this BMD analysis were not adjusted for the dose regimen and the reported results consequently correspond to a 5‐day per week dosing regimen.
C.2.2. Selection of the BMR
A default benchmark response (BMR) of 10% (extra risk compared with the background risk) and a 90% interval around the BMD were selected as recommended by EFSA Scientific Committee (2017).
C.2.3. Results for the incidence of hepatocellular adenomas and carcinomas as reported by Moser et al. (2009) and NTP (1993)
A combined analysis of the incidence of hepatocellular adenomas and carcinomas reported in two separate studies (NTP, 1993; Moser et al., 2009) was performed, using study as a covariate. The effect of study as covariate was not significant and therefore the data from both studies were merged.
Table C.9.
Result for the incidence of hepatocellular adenomas and carcinomas reported by Moser et al. (2009) and NTP (1993) using model averaging
| Model | Number of parameters | Log‐likelihood | AIC | BMD | BMDL | BMDU | Converged | Accepted AIC |
|---|---|---|---|---|---|---|---|---|
| Null | 1 | −279.17 | 560.34 | NA | NA | NA | Yes | |
| Full | 7 | −166.62 | 347.24 | NA | NA | NA | Yes | |
| Logistic | 2 | −168.70 | 341.40 | 2.00 | 1.73 | 2.30 | Yes | Yes |
| Probit | 2 | −168.44 | 340.88 | 1.83 | 1.61 | 2.09 | Yes | Yes |
| Log‐logistic | 3 | −169.19 | 344.38 | 3.42 | 2.45 | 4.75 | Yes | No |
| Log‐probit | 3 | −168.54 | 343.08 | 3.41 | 2.50 | 4.46 | Yes | No |
| Weibull | 3 | −167.20 | 340.40 | 3.02 | 2.08 | 4.20 | Yes | Yes |
| Gamma | 3 | −167.79 | 341.58 | 3.22 | 2.23 | 4.32 | Yes | Yes |
| Two‐stage | 3 | −167.81 | 341.62 | 2.39 | 2.16 | 2.66 | Yes | Yes |
AIC: Akaike information criterion; BMD: benchmark dose; BMDL: benchmark dose lower confidence limit; BMDU: benchmark dose upper confidence limit.
Table C.10.
Model weights used in model averaging
| Logistic | Probit | Log‐logistic | Log‐probit | Weibull | Gamma | Two‐stage | |
|---|---|---|---|---|---|---|---|
| Estimated model weights | 0.16 | 0.2 | 0.04 | 0.07 | 0.26 | 0.14 | 0.14 |
Given 1,000 generated data sets, the BMDL is the 5th percentile of all parametric bootstrap BMD values and the BMDU is the 95th percentile.
Estimated the BMD based on the averaged response model which is a weighted average of the accepted models’ response values.
Table C.11.
Calculated BMD, BMDL and BMDU values (mg/kg bw per day for a 5 days per week dosing regimen) for the incidence of hepatocellular adenomas/carcinomas reported by Moser et al. (2009) and NTP (1993) using model averaging
| BMD | BMDL | BMDU |
|---|---|---|
| 2.6 | 1.84 | 4 |
Figure C.4.
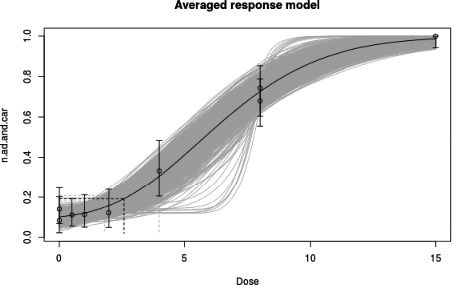
Averaged dose–response model for the incidence of hepatocellular adenomas/carcinomas after 2 years reported by Moser et al. (2009) and NTP (1993)
C.2.4. Results for the incidence of hepatocellular carcinomas as reported by Moser et al. (2009) and NTP (1993)
A combined analysis of the incidence of hepatocellular carcinomas reported in two separate studies (NTP, 1993; Moser et al., 2009) was performed, using study as a covariate. The effect of study as covariate was significant and the studies were therefore not combined.
Table C.12.
Result for the incidence of hepatocellular carcinomas reported by Moser et al. (2009) and NTP (1993), using study as a covariate and using model averaging
| Model | Included covariate(s) | Number of parameters | Log‐likelihood | AIC | BMD – study.Moser | BMDL – study.Moser | BMDU – study.Moser | BMD – study.NTP | BMDL – study.NTP | BMDU – study.NTP | Converged | Accepted AIC | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Null | 1 | −165.92 | 333.84 | NA | NA | NA | NA | NA | NA | Yes | ||
| 2 | Full | 7 | −125.20 | 264.40 | NA | NA | NA | NA | NA | NA | Yes | ||
| 3 | Logistic | 2 | −126.28 | 256.56 | 6.26 | 5.46 | 7.14 | 6.26 | 5.46 | 7.14 | Yes | Yes | |
| 4 | Logistic | a | 3 | −126.05 | 258.10 | 5.91 | 5.00 | 7.19 | 5.91 | 5.00 | 7.19 | Yes | Yes |
| 5 | Logistic | b | 3 | −125.22 | 256.44 | 5.13 | 4.22 | 6.66 | 6.73 | 5.75 | 7.80 | Yes | Yes |
| 6 | Logistic | a, b | 4 | −125.20 | 258.40 | 5.14 | 4.22 | 6.65 | 6.61 | 5.30 | 8.08 | Yes | Yes |
| 7 | Probit | 2 | −126.30 | 256.60 | 5.74 | 5.00 | 6.59 | 5.74 | 5.00 | 6.59 | Yes | Yes | |
| 11 | Log‐logistic | 3 | −125.95 | 257.90 | 6.44 | 4.81 | 7.95 | 6.44 | 4.81 | 7.95 | Yes | Yes | |
| 12 | Log‐logistic | a | 4 | −125.93 | 259.86 | 6.45 | 4.83 | 7.94 | 6.45 | 4.83 | 7.94 | Yes | No |
| 13 | Log‐logistic | b | 4 | −124.55 | 257.10 | 5.87 | 4.62 | 7.19 | 7.90 | 5.85 | 9.91 | Yes | Yes |
| 14 | Log‐logistic | a, b | 5 | −124.52 | 259.04 | 5.88 | 4.62 | 7.18 | 8.01 | 5.88 | 10.94 | Yes | No |
| 15 | Log‐probit | 3 | −125.87 | 257.74 | 6.49 | 4.94 | 7.91 | 6.49 | 4.94 | 7.91 | Yes | Yes | |
| 16 | Log‐probit | a | 4 | −125.84 | 259.68 | 6.49 | 4.95 | 7.90 | 6.49 | 4.95 | 7.90 | Yes | No |
| 17 | Log‐probit | b | 4 | −124.50 | 257.00 | 5.90 | 4.66 | 7.22 | 7.82 | 5.91 | 9.59 | Yes | Yes |
| 18 | Log‐probit | a, b | 5 | −124.49 | 258.98 | 5.90 | 4.66 | 7.22 | 7.88 | 5.91 | 10.38 | Yes | No |
| 19 | Weibull | 3 | −126.04 | 258.08 | 6.36 | 4.62 | 7.99 | 6.36 | 4.62 | 7.99 | Yes | Yes | |
| 20 | Weibull | a | 4 | −126.03 | 260.06 | 6.36 | 4.62 | 7.99 | 6.36 | 4.62 | 7.99 | Yes | No |
| 21 | Weibull | b | 4 | −124.60 | 257.20 | 5.80 | 4.53 | 7.15 | 7.99 | 5.76 | 10.14 | Yes | Yes |
| 22 | Weibull | a, b | 5 | −124.56 | 259.12 | 5.82 | 4.53 | 7.13 | 8.13 | 5.80 | 11.29 | Yes | No |
| 23 | Gamma | 3 | −125.93 | 257.86 | 6.44 | 4.77 | 7.94 | 6.44 | 4.77 | 7.94 | Yes | Yes | |
| 24 | Gamma | a | 4 | −125.91 | 259.82 | 6.45 | 4.78 | 7.93 | 6.45 | 4.78 | 7.93 | Yes | No |
| 25 | Gamma | b | 4 | −124.52 | 257.04 | 5.86 | 4.61 | 7.19 | 7.88 | 5.85 | 9.73 | Yes | Yes |
| 26 | Gamma | a, b | 5 | −124.50 | 259.00 | 5.87 | 4.61 | 7.18 | 7.97 | 5.87 | 10.57 | Yes | No |
| 27 | Two‐stage | 3 | −126.23 | 258.46 | 5.80 | 5.09 | 6.71 | 5.80 | 5.09 | 6.71 | Yes | No | |
| 28 | Two‐stage | a | 4 | −126.23 | 260.46 | 5.79 | 5.08 | 6.72 | 5.79 | 5.08 | 6.72 | Yes | No |
| 29 | Two‐stage | b | 4 | −125.88 | 259.76 | 5.09 | 3.99 | 6.96 | 5.99 | 5.17 | 7.06 | Yes | No |
| 30 | Two‐stage | a, b | 5 | −125.87 | 261.74 | 5.07 | 3.98 | 6.96 | 6.01 | 5.17 | 7.16 | Yes | No |
AIC: Akaike information criterion; BMD: benchmark dose; BMDL: benchmark dose lower confidence limit; BMDU: benchmark dose upper confidence limit.
It was noted that the Probit implementation in PROAST version 61.3 does not support covariate adjustment.
Table C.13.
Model weights used in model averaging
| Logistic | Probit | Log‐logistic | Log‐probit | Weibull | Gamma | Two‐stage | |
|---|---|---|---|---|---|---|---|
| Estimated model weights | 0.19 | 0.18 | 0.14 | 0.15 | 0.13 | 0.14 | 0.07 |
Given 200 generated data sets, the BMDL is the 5th percentile of all parametric bootstrap BMD values and the BMDU is the 95th percentile.
Estimated the BMD based on the averaged response model which is a weighted average of the accepted models’ response values.
Table C.14.
Calculated BMD, BMDL and BMDU values (mg/kg bw per day for a 5 days per week dosing regimen) for the incidence of hepatocellular carcinomas reported by Moser et al. (2009) and NTP (1993) using model averaging
| BMD | BMDL | BMDU | |
|---|---|---|---|
| study.Moser | 5.72 | 4.18 | 7.24 |
| study.NTP | 7.21 | 4.82 | 9.10 |
Figure C.5.
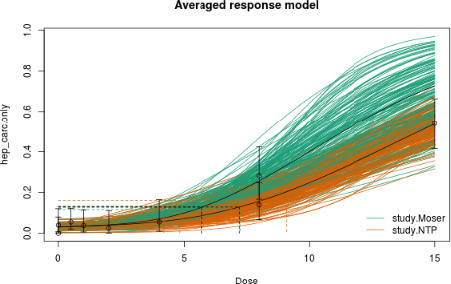
Averaged dose–response model for the incidence of hepatocellular carcinomas after 2 years reported by Moser et al. (2009) and NTP (1993)
C.2.5. Results for the incidence of hepatocellular adenomas and carcinomas as reported by Moser et al. (2009)
Table C.15.
Result for the incidence of hepatocellular adenomas and carcinomas reported by Moser et al. (2009) using model averaging
| Model | Number of parameters | Log‐likelihood | AIC | BMD | BMDL | BMDU | Converged | Accepted AIC |
|---|---|---|---|---|---|---|---|---|
| Null | 1 | −148.52 | 299.04 | NA | NA | NA | Yes | |
| Full | 6 | −114.48 | 240.96 | NA | NA | NA | Yes | |
| Logistic | 2 | −114.94 | 233.88 | 2.03 | 1.71 | 2.41 | Yes | Yes |
| Probit | 2 | −115.09 | 234.18 | 1.87 | 1.59 | 2.21 | Yes | Yes |
| Log‐logistic | 3 | −114.65 | 235.30 | 2.78 | 1.76 | 3.87 | Yes | Yes |
| Log‐probit | 3 | −114.62 | 235.24 | 2.85 | 1.89 | 3.88 | Yes | Yes |
| Weibull | 3 | −114.73 | 235.46 | 2.62 | 1.53 | 3.88 | Yes | Yes |
| Gamma | 3 | −114.66 | 235.32 | 2.76 | 1.65 | 3.88 | Yes | Yes |
| Two‐stage | 3 | −114.82 | 235.64 | 2.34 | 2.02 | 2.77 | No | Yes |
AIC: Akaike information criterion; BMD: benchmark dose; BMDL: benchmark dose lower confidence limit; BMDU: benchmark dose upper confidence limit.
Table C.16.
Model weights used in model averaging
| Logistic | Probit | Log‐logistic | Log‐probit | Weibull | Gamma | |
|---|---|---|---|---|---|---|
| Estimated model weights | 0.26 | 0.23 | 0.13 | 0.13 | 0.12 | 0.13 |
Given 200 generated data sets, the BMDL is the 5th percentile of all parametric bootstrap BMD values and the BMDU is the 95th percentile.
Estimated the BMD based on the averaged response model which is a weighted average of the accepted models’ response values.
Table C.17.
Calculated BMD, BMDL and BMDU values (mg/kg bw per day for a 5 days per week dosing regimen) for the incidence of hepatocellular adenomas and carcinomas reported by Moser et al. (2009) using model averaging
| BMD | BMDL | BMDU |
|---|---|---|
| 2.4 | 1.45 | 3.59 |
Figure C.6.
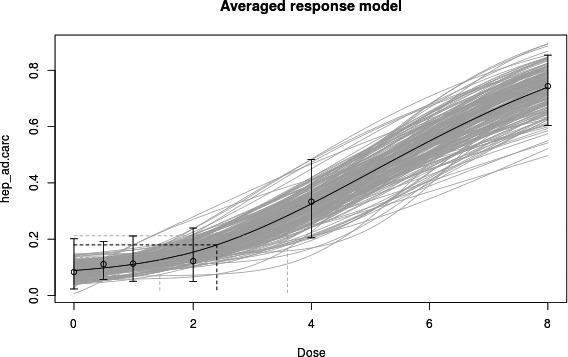
Averaged dose–response model for the incidence of hepatocellular adenomas and carcinomas reported by Moser et al. (2009)
C.2.6. Results for the incidence of hepatocellular carcinomas as reported by Moser et al. (2009)
Table C.18.
Result for the incidence of hepatocellular carcinomas reported by Moser et al. (2009), using model averaging
| Model | Number of parameters | Log‐likelihood | AIC | BMD | BMDL | BMDU | Converged | Accepted AIC |
|---|---|---|---|---|---|---|---|---|
| Null | 1 | −71.83 | 145.66 | NA | NA | NA | Yes | |
| Full | 6 | −59.59 | 131.18 | NA | NA | NA | Yes | |
| Logistic | 2 | −61.78 | 127.56 | 5.14 | 4.22 | 6.66 | Yes | Yes |
| Probit | 2 | −61.94 | 127.88 | 4.89 | 3.92 | 6.60 | Yes | Yes |
| Log‐logistic | 3 | −63.31 | 132.62 | 2.85 | 1.66 | 5.21 | Yes | No |
| Log‐probit | 3 | −61.34 | 128.68 | 5.84 | 4.06 | 8.24 | Yes | Yes |
| Weibull | 3 | −63.19 | 132.38 | 2.88 | 1.69 | 5.11 | Yes | No |
| Gamma | 3 | −61.35 | 128.70 | 5.97 | 4.02 | 7.95 | Yes | Yes |
| Two‐stage | 3 | −61.95 | 129.90 | 5.07 | 3.98 | 6.95 | Yes | No |
AIC: Akaike information criterion; BMD: benchmark dose; BMDL: benchmark dose lower confidence limit; BMDU: benchmark dose upper confidence limit.
Table C.19.
Model weights used in model averaging
| Logistic | Probit | Log‐logistic | Log‐probit | Weibull | Gamma | Two‐stage | |
|---|---|---|---|---|---|---|---|
| Estimated model weights | 0.29 | 0.25 | 0.02 | 0.16 | 0.03 | 0.16 | 0.09 |
Given 200 generated data sets, the BMDL is the 5th percentile of all parametric bootstrap BMD values and the BMDU is the 95th percentile.
Estimated the BMD based on the averaged response model which is a weighted average of the accepted models’ response values.
Table C.20.
Calculated BMD, BMDL and BMDU values (mg/kg bw per day for a 5 days per week dosing regimen) for the incidence of hepatocellular carcinomas reported by Moser et al. (2009) using model averaging
| BMD | BMDL | BMDU |
|---|---|---|
| 5.34 | 3.45 | 7.55 |
Figure C.7.
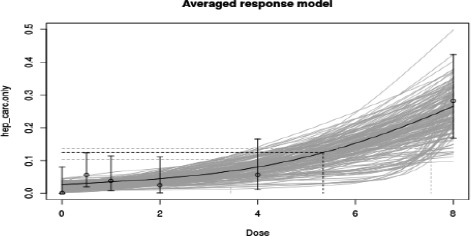
Averaged dose–response model for the incidence of hepatocellular carcinomas reported by Moser et al. (2009)
Appendix D – Factors influencing furan and methylfuran levels in food
Appendix E – Previously reported occurrence data
Annex A – Occurrence data submitted to EFSA and dietary exposure assessment for humans
Annex A can be found in the online version of this output, under the section ‘Supporting information’, at: http://onlinelibrary.wiley.com/doi/10.2903/j.efsa.2017.5005/full
Description: The annex is an excel file which presents tables from Tables A1–A10 on furan occurrence and dietary exposure assessment for humans.
Supporting information
Occurrence data submitted to EFSA and dietary exposure assessment for humans
Suggested citation: EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) , Knutsen HK, Alexander J, Barregård L, Bignami M, Brüschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl‐Kraupp B, Hogstrand C, Hoogenboom LR, Nebbia CS, Oswald IP, Petersen A, Rose M, Roudot A‐C, Schwerdtle T, Vleminckx C, Vollmer G, Chipman K, De Meulenaer B, Dinovi M, Mennes W, Schlatter J, Schrenk D, Baert K, Dujardin B and Wallace H, 2017. Scientific opinion on the risks for public health related to the presence of furan and methylfurans in food. EFSA Journal 2017;15(10):5005, 142 pp. 10.2903/j.efsa.2017.5005
Requestor: European Commission
Question number: EFSA‐Q‐2016‐00025
Panel members: Jan Alexander, Lars Barregård, Margherita Bignami, Beat Brüschweiler, Sandra Ceccatelli, Bruce Cottrill, Michael Dinovi, Lutz Edler, Bettina Grasl‐Kraupp, Christer Hogstrand, Laurentius (Ron) Hoogenboom, Helle Katrine Knutsen, Carlo Stefano Nebbia, Isabelle P. Oswald, Annette Petersen, Martin Rose, Alain‐Claude Roudot, Tanja Schwerdtle, Christiane Vleminckx, Günter Vollmer and Heather Wallace.
Acknowledgements: EFSA wishes to thank the hearing experts: Diana Doell and Ruud Woutersen and EFSA staff member: José Cortinas Abrahantes for the support provided to this scientific output. The CONTAM Panel acknowledges all European competent institutions and other stakeholders that provided occurrence data on furan and methylfurans in food, and supported the data collection for the Comprehensive European Food Consumption Database.
Adopted: 20 September 2017
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2017.EN-1066/full
Notes
Report of the Scientific Panel on Contaminants in the Food Chain on provisional findings on furan in food, EFSA Journal 2004; 137:1–20, Available online: http://www.efsa.europa.eu/sites/default/files/scientific_output/files/main_documents/137.pdf
EFSA Journal 2011;9(9):2347 http://www.efsa.europa.eu/sites/default/files/scientific_output/files/main_documents/2347.pdf
WHO Technical Report Series, No. 959, 2011, Evaluation of certain contaminants in food (Seventy‐second report of the Joint FAO/WHO Expert Committee on Food Additives). Available online: http://whqlibdoc.who.int/trs/WHO_TRS_959_eng.pdf?ua=1
http://www.thegoodscentscompany.com/data/rw1023171.html (Accessed: 30 June 2016)
US EPA. [2012]. Estimation Programs Interface Suite™ for Microsoft® Windows, v 4.11. United States Environmental Protection Agency, Washington, DC, USA. Available online: https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface#what (Accessed: 30 June 2016)
Predicted with Advanced Chemistry Development (ACD/Labs) Software V11.02 (© 1994‐2017 ACD/Labs)
http://www.thegoodscentscompany.com/data/rw1435541.html (Accessed: 30 June 2016)
Council Regulation (EEC) No 315/93 of 8 February 1993 laying down Community procedures for contaminants in food. OJ L 37, 13.2.1993, p. 1–5.
Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. OJ L 364, 20.12.2006, p. 5–24.
Commission Recommendation (EC) No 2007/196 of 28 March 2007 on the monitoring of the presence of furan in foodstuffs. OJ L 88, 29.3.2007, p. 56–57.
Commercial foodstuffs as purchased disregarding any further preparation, e.g. coffee powder, juices, jars and cans not heated before consumption.
Commercial foodstuffs analysed as consumed after further preparation in the laboratory,: e.g. brewed coffee, canned and jarred products heated before consumption. If available, preparation should follow label instructions. Food prepared at home on basis of fresh ingredients (e.g. vegetable soup with fresh vegetables, home‐made Irish stew) is not subject of this monitoring programme.
Commission Recommendation (EU) No 246/2014 of 13 March 2014 amending Annex I to Regulation (EC) No 1334/2008 of the European Parliament and of the Council as regards removal from the Union list of certain flavouring substances. OJ L 74, 14.3.2014, p. 58–60.
Web of Science (WoS), formerly ISI Web of Knowledge, Thomson Reuters. Available online: http://thomsonreuters.com/thomson-reuters-web-of-science/
PubMed, Entrez Global Query Cross‐Database Search System, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), Department of the National Institutes of Health (NIH), United States Department of Health and Human Services. Available online: http://www.ncbi.nlm.nih.gov/pubmed/
EndNote X5, Thomson Reuters. Available online: http://endnote.com/
See mandate documents at: http://registerofquestions.efsa.europa.eu/roqFrontend/questionsListLoader?mandate=M-2016-0012
The CONTAM Panel noted that the enzyme is called ‘sorbital dehydrogenase’ in the paper, and assumed it is sorbitol dehydrogenase.
In the paper, this was noted as interstitial cell adenoma of testes but is in fact mesothelioma of the tunica vaginalis.
Reported by the authors as ‘Préparations 1er âge’.
References
- Adams A, Bouckaert C, Van Lancker F, De Meulenaer B and De Kimpe N, 2011. Amino acid catalysis of 2‐alkylfuran formation from lipid oxidation‐derived alpha, beta‐unsaturated aldehydes. Journal of Agricultural and Food Chemistry, 59, 11058–11062. 10.1021/jf202448v [DOI] [PubMed] [Google Scholar]
- Adams A, Van Lancker F, De Meulenaer B, Owczarek‐Fendor A and De Kimpe N, 2012. On‐fiber furan formation from volatile precursors: a critical example of artefact formation during solid‐phase microextraction. Journal of Chromatography B, 897, 37–41. 10.1016/j.jchromb.2012.04.005 [DOI] [PubMed] [Google Scholar]
- Adger BM, Barrett C, Brennan J, McKervey MA and Murray RW, 1991. Oxidation of furans with dimethyldioxirane. Journal of the Chemical Society‐Chemical Communications, 1553–1554. 10.1039/c39910001553 [DOI] [Google Scholar]
- Aeschbacher H, Wolleb U, Löliger J, Spadone J and Liardon R, 1989. Contribution of coffee aroma constituents to the mutagenicity of coffee. Food and Chemical Toxicology, 27, 227–232. [DOI] [PubMed] [Google Scholar]
- Alam RT, Abu Zeid EH and Imam TS, 2017. Protective role of quercetin against hematotoxic and immunotoxic effects of furan in rats. Environmental Science and Pollution Research, 24, 3780–3789. 10.1007/s11356-016-8108-9 [DOI] [PubMed] [Google Scholar]
- Alasalvar C, Shahidi F and Cadwallader KR, 2003. Comparison of natural and roasted Turkish tombul hazelnut (Corylus avellana L.) volatiles and flavor by DHA/GC/MS and descriptive sensory analysis. Journal of Agricultural and Food Chemistry, 51, 5067–5072. 10.1021/jf0300846 [DOI] [PubMed] [Google Scholar]
- Alasalvar C, Odabasi AZ, Demir N, Balaban MÖ, Shahidi F and Cadwallader KR, 2004. Volatiles and flavor of five Turkish hazelnut varieties as evaluated by descriptive sensory analysis, electronic nose, and dynamic headspace analysis/gas chromatography‐mass spectrometry. Journal of Food Science, 69, SNQ99–SNQ106. 10.1111/j.1365-2621.2004.tb13382.x [DOI] [Google Scholar]
- Altaki MS, Santos FJ and Galceran MT, 2009. Automated headspace solid‐phase microextraction versus headspace for the analysis of furan in foods by gas chromatography–mass spectrometry. Talanta, 78, 1315–1320. 10.1016/j.talanta.2009.02.003 [DOI] [PubMed] [Google Scholar]
- Altaki MS, Santos FJ and Galceran MT, 2011. Occurrence of furan in coffee from Spanish market: contribution of brewing and roasting. Food Chemistry, 126, 1527–1532. 10.1016/j.foodchem.2010.11.134 [DOI] [PubMed] [Google Scholar]
- Altaki MS, Santos FJ, Puignou L and Galceran MT, 2017. Furan in commercial baby foods from the Spanish market: estimation of daily intake and risk assessment. Food Additives and Contaminants. Part A, 1–12. 10.1080/19440049.2016.1278080 [DOI] [PubMed] [Google Scholar]
- Ames JM and Macleod G, 1990. Volatile components of okra. Phytochemistry, 29, 1201–1207. 10.1016/0031-9422(90)85429-J [DOI] [Google Scholar]
- Anses (Agence nationale de sécurité sanitaire alimentation, environnement, travail), 2016. Étude de l'alimentation totale infantile. 372.
- Arisseto AP, Vicente E, Ueno MS, Amelia S, Tfouni V and Toledo MCD, 2011. Furan levels in coffee as influenced by species, roast degree, and brewing procedures. Journal of Agricultural and Food Chemistry, 59, 3118–3124. 10.1021/jf104868g [DOI] [PubMed] [Google Scholar]
- Arisseto AP, Vicente E, Furlani RPZ, Ueno MS, Pereira ALD and Toledo MCF, 2012. Occurrence of furan in commercial processed foods in Brazil. Food Additives and Contaminants: Part A, 29, 1832–1839. 10.1080/19440049.2012.713030 [DOI] [PubMed] [Google Scholar]
- Arisseto AP, Vicente E and Toledo MCF, 2013. Investigation on furan levels in pressure‐cooked foods. International Journal of Food Science, 2013, 4. 10.1155/2013/904349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina Y, Murayama Y, Shibata B, Kariyone T, Kuwada S and Asano M, 1924. Elsholtzia ketone, a contribution to furan chemistry. Acta Phytochimica, 2, 1–23. [Google Scholar]
- Baek S‐O and Jenkins RA, 2004. Characterization of trace organic compounds associated with aged and diluted sidestream tobacco smoke in a controlled atmosphere — volatile organic compounds and polycyclic aromatic hydrocarbons. Atmospheric Environment, 38, 6583–6599. 10.1016/j.atmosenv.2004.08.016 [DOI] [Google Scholar]
- Bakhiya N and Appel KE, 2010. Toxicity and carcinogenicity of furan in human diet. Archives of Toxicology, 84, 563–578. 10.1007/s00204-010-0531-y [DOI] [PubMed] [Google Scholar]
- Banda M, Recio L and Parsons BL, 2013. ACB‐PCR measurement of spontaneous and furan‐induced H‐ras codon 61 CAA to CTA and CAA to AAA mutation in B6C3F1 mouse liver. Environmental and Molecular Mutagenesis, 54, 659–667. 10.1002/em.21808 [DOI] [PubMed] [Google Scholar]
- Becalski A and Seaman S, 2005. Furan precursors in food: a model study and development of a simple headspace method for determination of furan. Journal of AOAC International, 88, 102–106. [PubMed] [Google Scholar]
- Becalski A, Forsyth D, Casey V, Lau BPY, Pepper K and Seaman S, 2005. Development and validation of a headspace method for determination of furan in food. Food Additives and Contaminants, 22, 535–540. 10.1080/02652030500129170 [DOI] [PubMed] [Google Scholar]
- Becalski A, Hayward S, Krakalovich T, Pelletier L, Roscoe V and Vavasour E, 2010. Development of an analytical method and survey of foods for furan, 2‐methylfuran and 3‐methylfuran with estimated exposure. Food Additives and Contaminants: Part A, 27, 764–775. 10.1080/19440040903473332 [DOI] [PubMed] [Google Scholar]
- Becalski A, Turcotte A‐M, Cooke GM and Gill SS, 2013. Investigation of possible endogenous formation of furan in Fischer‐344 rat. Toxicological and Environmental Chemistry, 95, 814–822. 10.1080/02772248.2013.796785 [DOI] [Google Scholar]
- Becalski A, Halldorson T, Hayward S and Roscoe V, 2016. Furan, 2‐methylfuran and 3‐methylfuran in coffee on the Canadian market. Journal of Food Composition and Analysis, 47, 113–119. 10.1016/j.jfca.2016.01.006 [DOI] [Google Scholar]
- Bianchi F, Careri M, Mangia A and Musci M, 2006. Development and validation of a solid phase micro‐extraction–gas chromatography–mass spectrometry method for the determination of furan in baby‐food. Journal of Chromatography A, 1102, 268–272. 10.1016/j.chroma.2005.10.056 [DOI] [PubMed] [Google Scholar]
- Bohnert T, Gingipalli L and Dedon PC, 2004. Reaction of 2′‐deoxyribonucleosides with cis‐ and trans‐1,4‐dioxo‐2‐butene. Biochemical and Biophysical Research Communications, 323, 838–844. 10.1016/j.bbrc.2004.08.164 [DOI] [PubMed] [Google Scholar]
- Bouseta A, Collin S and Dufour JP, 1992. Characteristic aroma profiles of unifloral honeys obtained with a dynamic headspace GC‐MS system. Journal of Apicultural Research, 31, 96–109. [Google Scholar]
- Boyd MR, Statham CN, Franklin RB and Mitchell JR, 1978. Pulmonary bronchiolar alkylation and necrosis by 3‐methylfuran, a naturally occurring potential atmospheric contaminant. Nature, 272, 270–271. [DOI] [PubMed] [Google Scholar]
- Brueck J, Schneider T and Schrenk D, 2009. In vitro toxicity of furan and its metabolite(s) in liver cells. Toxicology Letters, 189, S231–S232. 10.1016/j.toxlet.2009.06.496 [DOI] [Google Scholar]
- Bryla P and Weyand EH, 1991. Role of activated oxygen species in benzo[a]pyrene: DNA adduct formation in vitro . Free Radical Biology and Medicine, 11, 17–24. [DOI] [PubMed] [Google Scholar]
- Burka LT, Washburn KD and Irwin RD, 1991. Disposition of [14C]furan in the male F344 rat. Journal of Toxicology and Environmental Health, 34, 245–257. 10.1080/15287399109531564 [DOI] [PubMed] [Google Scholar]
- Burness DM, 1956. β‐Keto acetals. II. Synthesis of 3‐methyl‐ and 3‐phenyl‐furans. The Journal of Organic Chemistry, 21, 102–104. 10.1021/jo01107a021 [DOI] [Google Scholar]
- Butterworth BE, Sprankle CS, Goldsworthy SM, Wilson DM, Goldsworthy TL, Sprankle CS, Goldsworthy SM, Wilson DM and Goldsworthy TL, 1994. Expression of myc, fos, and Ha‐ras in the livers of furan‐treated F344 rats and B6C3F1 mice. Molecular Carcinogenesis, 9, 24–32. [DOI] [PubMed] [Google Scholar]
- Byrns MC, Predecki DP and Peterson LA, 2002. Characterization of nucleoside adducts of cis‐2‐butene‐1,4‐dial, a reactive metabolite of furan. Chemical Research in Toxicology, 15, 373–379. [DOI] [PubMed] [Google Scholar]
- Byrns MC, Vu CC and Peterson LA, 2004. The formation of substituted 1, N‐6‐etheno‐2 ‘‐deoxyadenosine and 1, N‐2‐etheno‐2 ‘‐deoxyguanosine adducts by cis‐2‐butene‐1,4‐dial, a reactive metabolite of furan. Chemical Research in Toxicology, 17, 1607–1613. 10.1021/tx049866z [DOI] [PubMed] [Google Scholar]
- Byrns MC, Vu CC, Neidigh JW, Abad JL, Jones RA and Peterson LA, 2006. Detection of DNA adducts derived from the reactive metabolite of furan, cis‐2‐butene‐1,4‐dial. Chemical Research in Toxicology, 19, 414–420. 10.1021/tx050302k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell DJ, 1999. Review of mononuclear cell leukemia in F‐344 rat bioassays and its significance to human cancer risk: a case study using alkyl phthalates. Regulatory Toxicology and Pharmacology, 30, 45–53. 10.1006/rtph.1999.1305 [DOI] [PubMed] [Google Scholar]
- Carfagna MA, Held SD and Kedderis GL, 1993. Furan‐induced cytolethality in isolated rat hepatocytes: correspondence with in vivo dosimetry. Toxicology and Applied Pharmacology, 123, 265–273. 10.1006/taap.1993.1245 [DOI] [PubMed] [Google Scholar]
- Chaichi M, Mohammadi A and Hashemi M, 2013. Optimization and application of headspace liquid‐phase microextraction coupled with gas chromatography‐mass spectrometry for determination of furanic compounds in coffee using response surface methodology. Microchemical Journal, 108, 46–52. 10.1016/j.microc.2012.12.009 [DOI] [Google Scholar]
- Chaichi M, Ghasemzadeh‐Mohammadi V, Hashemi M and Mohammadi A, 2015. Furanic compounds and furfural in different coffee products by headspace liquid‐phase micro‐extraction followed by gas chromatography‐mass spectrometry: survey and effect of brewing procedures. Food Additives and Contaminants: Part B, 8, 73–80. 10.1080/19393210.2014.981601 [DOI] [PubMed] [Google Scholar]
- Cheah NP, Pennings JLA, Vermeulen JP, van Schooten FJ and Opperhuizen A, 2013. In vitro effects of aldehydes present in tobacco smoke on gene expression in human lung alveolar epithelial cells. Toxicology in Vitro, 27, 1072–1081. 10.1016/j.tiv.2013.02.003 [DOI] [PubMed] [Google Scholar]
- Chen LJ, Hecht SS and Peterson LA, 1995. Identification of cis‐2‐butene‐1,4‐dial as a microsomal metabolite of furan. Chemical Research in Toxicology, 8, 903–906. [DOI] [PubMed] [Google Scholar]
- Chen LJ, Hecht SS and Peterson LA, 1997. Characterization of amino acid and glutathione adducts of cis‐2‐butene‐1,4‐dial, a reactive metabolite of furan. Chemical Research in Toxicology, 10, 866–874. 10.1021/tx9700174 [DOI] [PubMed] [Google Scholar]
- Chen T, Mally A, Ozden S and Chipman JK, 2010. Low doses of the carcinogen furan alter cell cycle and apoptosis gene expression in rat liver independent of DNA methylation. Environmental Health Perspectives, 118, 1597–1602. 10.1289/ehp.1002153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Williams TD, Mally A, Hamberger C, Mirbahai L, Hickling K and Chipman JK, 2012. Gene expression and epigenetic changes by furan in rat liver. Toxicology, 292, 63–70. 10.1016/j.tox.2011.10.020 [DOI] [PubMed] [Google Scholar]
- Churchwell MI, Scheri RC, Von Tungeln LS, da Costa GG, Beland FA and Doerge DR, 2015. Evaluation of serum and liver toxicokinetics for furan and liver DNA adduct formation in male Fischer 344 rats. Food and Chemical Toxicology, 86, 1–8. 10.1016/j.fct.2015.08.029 [DOI] [PubMed] [Google Scholar]
- Crews C, 2009. Consumer exposure to furan from heat‐processed foods and kitchen air. EFSA Supporting Publication 2009;6(9):EN‐30, 65 pp. 10.2903/sp.efsa.2009.en-30 [DOI] [Google Scholar]
- Cooke GM, Taylor M, Bourque C, Curran I, Gurofsky S and Gill S, 2014. Effects of furan on male rat reproduction parameters in a 90‐day gavage study. Reproductive Toxicology, 46, 85–90. 10.1016/j.reprotox.2014.02.003 [DOI] [PubMed] [Google Scholar]
- Coppa M, Martin B, Pradel P, Leotta B, Priolo A and Vasta V, 2011. Effect of a hay‐based diet or different upland grazing systems on milk volatile compounds. Journal of Agricultural and Food Chemistry, 59, 4947–4954. 10.1021/jf2005782 [DOI] [PubMed] [Google Scholar]
- Cordelli E, Leopardi P, Villani P, Marcon F, Macri C, Caiola S, Siniscalchi E, Conti L, Eleuteri P, Malchiodi‐Albedi F and Crebelli R, 2010. Toxic and genotoxic effects of oral administration of furan in mouse liver. Mutagenesis, 25, 305–314. 10.1093/mutage/geq007 [DOI] [PubMed] [Google Scholar]
- Curran IH, Williams A, Dong H, Gill S, Wade MG, Yauk CL and Kuo B, 2016. Toxicogenomic assessment of liver responses following subchronic exposure to furan in fischer F344 rats miRNA. Envirnopmental and Molecular Mutagensis, 57, 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Conti A, Kobets T, Escudero‐Lourdes C, Montgomery B, Tryndyak V, Beland FA, Doerge DR and Pogribny IP, 2014. Dose‐ and time‐dependent epigenetic changes in the livers of fisher 344 rats exposed to furan. Toxicological Sciences, 139, 371–380. 10.1093/toxsci/kfu044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Conti A, Kobets T, Tryndyak V, Burnett SD, Han T, Fuscoe JC, Beland FA, Doerge DR and Pogribny IP, 2015. Persistence of furan‐induced epigenetic aberrations in the livers of F344 rats. Toxicological Sciences, 144, 217–226. 10.1093/toxsci/kfu313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Conti A, Tryndyak V, Doerge DR, Beland FA and Pogribny IP, 2016. Irreversible down‐regulation of miR‐375 in the livers of Fischer 344 rats after chronic furan exposure. Food and Chemical Toxicology, 98, 2–10. 10.1016/j.fct.2016.06.027 [DOI] [PubMed] [Google Scholar]
- Ding W, Petibone DM, Latendresse JR, Pearce MG, Muskhelishvili L, White GA, Chang CW, Mittelstaedt RA, Shaddock JG, McDaniel LP, Doerge DR, Morris SM, Bishop ME, Manjanatha MG, Aidoo A and Heflich RH, 2012. In vivo genotoxicity of furan in F344 rats at cancer bioassay doses. Toxicology and Applied Pharmacology, 261, 164–171. 10.1016/j.taap.2012.03.021 [DOI] [PubMed] [Google Scholar]
- Dong HY, Gill S, Curran IH, Williams A, Kuo B, Wade MG and Yauk CL, 2016. Toxicogenomic assessment of liver responses following subchronic exposure to furan in Fischer F344 rats. Archives of Toxicology, 90, 1351–1367. 10.1007/s00204-015-1561-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DTU (Technical University of Denmark), 2009. Furan in heat processed food products including home cooked food products and ready‐to‐eat products. Scientific report submitted to EFSA. EFSA Supporting Publication 2009;7(9):EN‐1, 49 pp. 10.2903/sp.efsa.2009.en-1 [DOI] [Google Scholar]
- Durling L, Svensson K and Abramssonzetterberg L, 2007. Furan is not genotoxic in the micronucleus assay in vivo or in vitro . Toxicology Letters, 169, 43–50. 10.1016/j.toxlet.2006.08.020 [DOI] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2004. Report of the Scientific Panel on Contaminants in the Food Chain on provisional findings on furan in food. EFSA Journal 2004;2(12):137, 20 pp. 10.2903/j.efsa.2004.137 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2007. Guidance of the Scientific Committee on a request from EFSA related to uncertainties in dietary exposure assessment. EFSA Journal 2007;4(12):438, 54 pp. 10.2903/j.efsa.2007.438 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2009. Technical report of EFSA prepared by Data Collection and Exposure Unit (DATEX) on “Monitoring of furan levels in food”. EFSA Journal 2009;7(6):304r, 23 pp. 10.2903/j.efsa.2009.304r [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2010a. Standard sample description for food and feed. EFSA Journal 2010;8(1):1457, 54 pp. 10.2903/j.efsa.2010.1457 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2010b. Management of left‐censored data in dietary exposure assessment of chemical substances. EFSA Journal 2010;8(3):1557, 96 pp. 10.2903/j.efsa.2010.1557 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2010c. Update of results on the monitoring of furan levels in food. EFSA Journal 2010;8(7):1702, 18 pp. 10.2903/j.efsa.2010.1702 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011a. Use of the EFSA Comprehensive European Food Consumption Database in Intakes Assessment. EFSA Journal 2011;9(3):2097, 34 pp. 10.2903/j.efsa.2011.2097 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011b. Evaluation of the FoodEx, the food classification system applied to the development of the EFSA Comprehensive European Food Consumption Database. EFSA Journal 2011;9(3):1970, 27 pp. 10.2903/j.efsa.2011.1970 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011c. Report on the development of a Food Classification and Description System for exposure assessment and guidance on its implementation and use. EFSA Journal 2011;9(12):2489, 84 pp. 10.2903/j.efsa.2011.2489 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011d. Update on furan levels in food from monitoring years 2004‐2010 and exposure assessment. EFSA Journal, 2011;9(9):2347, 33 pp. 10.2903/j.efsa.2011.2347 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2012. Refined exposure assessment for caramel colours (E 150a, c, d). EFSA Journal 2012;10(12):3030, 39 pp. 10.2903/j.efsa.2012.3030 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2015. The food classification and description system FoodEx2 (revision 2). EFSA supporting publication 2015:EN‐804, 90 pp. 10.2903/sp.efsa.2015.en-804 [DOI] [Google Scholar]
- EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food), 2011. Scientific Opinion on the re‐evaluation of caramel colours (E 150 a,b,c,d) as food additives. EFSA Journal 2011;9(3):2004, 103 pp. 10.2903/j.efsa.2011.2004 [DOI] [Google Scholar]
- EFSA CEF Panel (EFSA Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids), Silano V, Bolognesi C, Castle L, Cravedi J‐P, Engel K‐H, Fowler P, Franz R, Grob K, Gurtler R, Husøy T, Kärenlampi S, Milana MR, Penninks A, Tavares Poças MF, Smith A, Tlustos C, Wölfle D, Zorn H, Zugravu C‐A, Beckman Sundh U, Benigni R, Brimer L, Mulder G, Oskarsson A, Svendsen C, Martino C and Mennes W, 2017. Scientific Opinion of Flavouring Group Evaluation 500 (FGE.500): rum ether. EFSA Journal 2017;15(8):4897, 53 pp. 10.2903/j.efsa.2017.4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2012. Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data. EFSA Journal 2012;10(3):2579, 32 pp. 10.2903/j.efsa.2012.2579 [DOI] [Google Scholar]
- EFSA Scientific Committee , Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen KH, More S, Mortensen A, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Silano V, Solecki R, Turck D, Aerts M, Bodin L, Davis A, Edler L, Gundert‐Remy U, Sand S, Slob W, Bottex B, Abrahantes JC, Marques DC, Kass G and Schlatter JR, 2017. Update: use of the benchmark dose approach in risk assessment. EFSA Journal 2017;15(1):4658, 41 pp. 10.2903/j.efsa.2017.4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egle JL and Gochberg BJ, 1979a. Respiratory retention and acute toxicity of furan. American Industrial Hygiene Association Journal, 40, 310–314. 10.1080/15298667991429633 [DOI] [PubMed] [Google Scholar]
- Egle JL and Gochberg BJ, 1979b. Retention of inhaled 2‐methylfuran and 2,5‐dimethylfuran. American Industrial Hygiene Association Journal, 40, 866–869. 10.1080/15298667991430415 [DOI] [PubMed] [Google Scholar]
- El‐Akabawy G and El‐Sherif NM, 2016. Protective role of garlic oil against oxidative damage induced by furan exposure from weaning through adulthood in adult rat testis. Acta Histochemica, 118, 456–463. 10.1016/j.acthis.2016.04.008 [DOI] [PubMed] [Google Scholar]
- Elmore LW and Sirica AE, 1991. Phenotypic characterization of metaplastic intestinal glands and ductular hepatocytes in cholangiofibrotic lesions rapidly induced in the caudate liver lobe of rats treated with furan. Cancer Research, 51, 5752–5759. [PubMed] [Google Scholar]
- Elmore LW and Sirica AE, 1992. Sequential appearance of intestinal mucosal cell types in the right and caudate liver lobes of furan‐treated rats. Hepatology, 16, 1220–1226. [PubMed] [Google Scholar]
- Elmore LW and Sirica AE, 1993. “Intestinal‐type” of adenocarcinoma preferentially induced in right/caudate liver lobes of rats treated with furan. Cancer Research, 53, 254–259. [PubMed] [Google Scholar]
- Fan X, 2005. Formation of furan from carbohydrates and ascorbic acid following exposure to ionizing radiation and thermal processing. Journal of Agricultural and Food Chemistry, 53, 7826–7831. 10.1021/jf051135x [DOI] [PubMed] [Google Scholar]
- Fan XT, 2015. Furan formation from fatty acids as a result of storage, gamma irradiation, UV‐C and heat treatments. Food Chemistry, 175, 439–444. 10.1016/j.foodchem.2014.12.002 [DOI] [PubMed] [Google Scholar]
- Fan X and Sokorai KJB, 2008. Effect of ionizing radiation on furan formation in fresh‐cut fruits and vegetables. Journal of Food Science, 73, C79–C83. 10.1111/j.1750-3841.2007.00622.x [DOI] [PubMed] [Google Scholar]
- Fan X, Huang L and Sokorai KJB, 2008. Factors affecting thermally induced furan formation. Journal of Agricultural and Food Chemistry, 56, 9490–9494. 10.1021/jf801612c [DOI] [PubMed] [Google Scholar]
- FAO/WHO (Food and Agriculture Organization of the United Nations/World Health Organization), 2011. Joint FAO/WHO Expert Committee on Food Additives (JECFA) Rome Feb 2010: Furan. Technical Report Series 959/Food Additives Series 63 (2011). Available online: http://apps.who.int/food-additives-contaminants-jecfa-database/chemical.aspx?chemID=5884
- FDA (US Food and Drug Administration), 2004a. Furan in Food, Thermal Treatment; Request for Data and Information. Federal Register Vol. 69 No 90 (2004) 25911. Available online: https://www.gpo.gov/fdsys/pkg/FR-2004-05-10/pdf/04-10588.pdf
- FDA (US Food and Drug Administration), 2004b. Determination of furan in foods. Available online: http://www.fda.gov/ohrms/dockets/ac/04/briefing/4045b2_10_furan%20method.pdf
- Foureman P, Mason JM, Valencia R and Zimmering S, 1994. Chemical mutagenesis testing in Drosophila. IX. Results of 50 coded compounds tested for the national toxicology program. Environmental and Molecular Mutagenesis, 23, 51–63. 10.1002/em.2850230109 [DOI] [PubMed] [Google Scholar]
- Frankel EN, 2012. Oily Press Lipid Library Series. Vol 18. Lipid oxidation. 2nd Edition. Woodhead Publishing, Cambridge, UK. 488 pp. [Google Scholar]
- Fransson‐Steen R, Goldsworthy TL, Kedderis GL and Maronpot RR, 1997. Furan‐induced liver cell proliferation and apoptosis in female B6C3F1 mice. Toxicology, 118, 195–204. 10.1016/s0300-483x(97)03618-4 [DOI] [PubMed] [Google Scholar]
- Fromberg A, Mariotti MS, Pedreschi F, Fagt S and Granby K, 2014. Furan and alkylated furans in heat processed food, including home cooked products. Czech Journal of Food Sciences, 32, 443–448. [Google Scholar]
- Fromowitz M, Shuga J, Wlassowsky AY, Ji Z, North M, Vulpe CD, Smith MT and Zhang L, 2012. Bone marrow genotoxicity of 2,5‐dimethylfuran, a green biofuel candidate. Environmental and Molecular Mutagenesis, 53, 488–491. 10.1002/em.21707 [DOI] [PubMed] [Google Scholar]
- Furihata C, Watanabe T, Suzuki T, Hamada S and Nakajima M, 2016. Collaborative studies in toxicogenomics in rodent liver in JEMS.MMS; a useful application of principal component analysis on toxicogenomics. Genes and Environment, 38, 15. 10.1186/s41021-016-0041-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galt AM and MacLeod G, 1984. Headspace sampling of cooked beef aroma using Tenax GC. Journal of Agricultural and Food Chemistry, 32, 59–64. 10.1021/jf00121a016 [DOI] [Google Scholar]
- Gammal LM, Wiley RA, Traiger G, Haschek WM and Baraban S, 1984. Toxicity‐distribution relationships among 3‐alkylfurans in the mouse lung. Toxicology, 30, 177–184. [DOI] [PubMed] [Google Scholar]
- Garle MJ and Fry JR, 1990. Reactive metabolite formation catalysed by cytochrome P‐450j. Toxicology in Vitro, 4, 493–496. [DOI] [PubMed] [Google Scholar]
- Gates LA, Lu D and Peterson LA, 2012. Trapping of cis‐2‐butene‐1,4‐dial to measure furan metabolism in human liver microsomes by cytochrome P450 enzymes. Drug Metabolism and Disposition, 40, 596–601. 10.1124/dmd.111.043679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates LA, Phillips MB, Matter BA and Peterson LA, 2014. Comparative metabolism of furan in rodent and human cryopreserved hepatocytes. Drug Metabolism and Disposition, 42, 1132–1136. 10.1124/dmd.114.057794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MS, Macleod AJ and Moreau M, 1984. Volatile components of cocoa with particular reference to glucosinolate products. Phytochemistry, 23, 1937–1942. 10.1016/s0031-9422(00)84945-6 [DOI] [Google Scholar]
- Gill S, Bondy G, Lefebvre DE, Becalski A, Kavanagh M, Hou Y, Turcotte AM, Barker M, Weld M, Vavasour E and Cooke GM, 2010. Subchronic oral toxicity study of furan in Fischer‐344 rats. Toxicologic Pathology, 38, 619–630. 10.1177/0192623310368978 [DOI] [PubMed] [Google Scholar]
- Gill S, Kavanagh M, Barker M, Weld M, Vavasour E, Hou Y and Cooke GM, 2011. Subchronic oral toxicity study of furan in B6C3F1 Mice. Toxicologic Pathology, 39, 787–794. 10.1177/0192623311412980 [DOI] [PubMed] [Google Scholar]
- Gill SS, Kavanagh M, Cherry W, Barker M, Weld M and Cooke GM, 2014. A 28‐day gavage toxicity study in male Fischer 344 rats with 2‐methylfuran. Toxicologic Pathology, 42, 352–360. 10.1177/0192623313482526 [DOI] [PubMed] [Google Scholar]
- Gill S, Kavanagh M, Cherry W, Barker M, Weld M and Cooke GM, 2015. A 28‐day gavage toxicity study in Fischer 344 rats with 3‐methylfuran. Toxicologic Pathology, 43, 221–232. 10.1177/0192623314534537 [DOI] [PubMed] [Google Scholar]
- Gill S, Kavanagh M, Cherry W, Bourque C, Caldwell D, Wang G and Bondy G, 2017, in preparation. A 90‐Day Subchronic Gavage Toxicity Study in Fischer 344 Rats with 3‐methylfuran. [DOI] [PubMed]
- Gingipalli L and Dedon PC, 2001. Reaction of cis ‐ and trans ‐2‐butene‐1,4‐dial with 2’‐deoxycytidine to form stable oxadiazabicyclooctaimine adducts. Journal of American Chemical Society, 123, 2664–2665. 10.1021/ja0056421 [DOI] [PubMed] [Google Scholar]
- Glatt H, Schneider H and Liu Y, 2005. V79‐hCYP2E1‐hSULT1A1, a cell line for the sensitive detection of genotoxic effects induced by carbohydrate pyrolysis products and other food‐borne chemicals. Mutation Research, 580, 41–52. 10.1016/j.mrgentox.2004.11.005 [DOI] [PubMed] [Google Scholar]
- Goldberg DM, Hoffman B, Yang J and Soleas GJ, 1999. Phenolic constituents, furans, and total antioxidant status of distilled spirits. Journal of Agricultural and Food Chemistry, 47, 3978–3985. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Périsset A, Scanlan F and Stadler RH, 2005. Rapid determination of furan in heated foodstuffs by isotope dilution solid phase micro‐extraction‐gas chromatography – mass spectrometry (SPME‐GC‐MS). The Analyst, 130, 878–883. 10.1039/b419270b [DOI] [PubMed] [Google Scholar]
- Grill AE, Schmitt T, Gates LA, Lu D, Bandyopadhyay D, Yuan JM, Murphy SE and Peterson LA, 2015. Abundant rodent furan‐derived urinary metabolites are associated with tobacco smoke exposure in humans. Chemical Research in Toxicology, 28, 1508–1516. 10.1021/acs.chemrestox.5b00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther H, Hoenicke K, Biesterveld S, Gerhard‐Rieben E and Lantz I, 2010. Furan in coffee: pilot studies on formation during roasting and losses during production steps and consumer handling. Food Additives and Contaminants: Part A, 27, 283–290. 10.1080/19440040903317505 [DOI] [PubMed] [Google Scholar]
- Habibi H, Mohammadi A, Hoseini H, Mohammadi M and Azadniya E, 2013. Headspace liquid‐phase microextraction followed by gas chromatography‐mass spectrometry for determination of furanic compounds in baby foods and method optimization using response surface methodology. Food Analytical Methods, 6, 1056–1064. 10.1007/s12161-012-9510-7 [DOI] [Google Scholar]
- Habu T, Flath RA, Mon TR and Morton JF, 1985. Volatile components of Rooibos tea (Aspalathus linearis). Journal of Agricultural and Food Chemistry, 33, 249–254. 10.1021/jf00062a024 [DOI] [Google Scholar]
- Halila S, Velasco T, Clercq PD and Madder A, 2005. Fine‐tuning furan toxicity: fast and quantitative DNA interchain cross‐link formation upon selective oxidation of a furan containing oligonucleotide. Chemical Communications, 936, 10.1039/b415092a [DOI] [PubMed] [Google Scholar]
- Hamadeh HK, Jayadev S, Gaillard ET, Huang Q, Stoll R, Blanchard K, Chou J, Tucker CJ, Collins J, Maronpot R, Bushel P and Afshari CA, 2004. Integration of clinical and gene expression endpoints to explore furan‐mediated hepatotoxicity. Mutation Research, 549, 169–183. 10.1016/j.mrfmmm.2003.12.021 [DOI] [PubMed] [Google Scholar]
- Hamberger C, Kellert M, Schauer UM, Dekant W and Mally A, 2010. Hepatobiliary toxicity of furan: identification of furan metabolites in bile of male F344/N rats. Drug Metabolism and Disposition, 38, 1698–1706. 10.1124/dmd.109.031781 [DOI] [PubMed] [Google Scholar]
- Haschek WM, Boyd MR, Hakkinen PJ, Owenby CS and Witschi H, 1984. Acute inhalation toxicity of 3‐methylfuran in the mouse ‐ pathology, cell‐kinetics, and respiratory rate effects. Toxicology and Applied Pharmacology, 72, 124–133. 10.1016/0041-008x(84)90256-4 [DOI] [PubMed] [Google Scholar]
- Haschek WM, Morse CC, Boyd MR, Hakkinen PJ and Witschi HP, 1983. Pathology of acute inhalation exposure to 3‐methylfuran in the rat and hamster. Experimental and Molecular Pathology, 39, 342–354. 10.1016/0014-4800(83)90063-1 [DOI] [PubMed] [Google Scholar]
- Hasnip S, Crews C and Castle L, 2006. Some factors affecting the formation of furan in heated foods. Food Addit Contam, 23, 219–227. 10.1080/02652030500539766 [DOI] [PubMed] [Google Scholar]
- Hatzinikolaou DG, Lagesson V, Stavridou AJ, Pouli AE, Lagesson‐Andrasko L and Stavrides JC, 2006. Analysis of the gas phase of cigarette smoke by gas chromatography coupled with UV‐diode array detection. Analytical Chemistry, 78, 4509–4516. 10.1021/ac052004y [DOI] [PubMed] [Google Scholar]
- Health Canada , 2016. Update on the assessment of exposure to furan from the Canadian retail food supply. 13 pp.
- Hibi D, Yokoo Y, Suzuki Y, Ishii Y, Jin M, Kijima A, Nohmi T, Nishikawa A and Umemura T, 2017. Lack of genotoxic mechanisms in early‐stage furan‐induced hepatocellular tumorigenesis in gpt delta rats. Journal of Applied Toxicology, 37, 142–149. 10.1002/jat.3331 [DOI] [PubMed] [Google Scholar]
- Hickling KC, Hitchcock JM, Oreffo V, Mally A, Hammond TG, Evans JG and Chipman JK, 2010a. Evidence of oxidative stress and associated DNA damage, increased proliferative drive, and altered gene expression in rat liver produced by the cholangiocarcinogenic agent furan. Toxicologic Pathology, 38, 230–243. 10.1177/0192623309357946 [DOI] [PubMed] [Google Scholar]
- Hickling KC, Hitchcock JM, Chipman JK, Hammond TG and Evans JG, 2010b. Induction and progression of cholangiofibrosis in rat liver injured by oral administration of furan. Toxicologic Pathology, 38, 213–229. 10.1177/0192623309357945 [DOI] [PubMed] [Google Scholar]
- Høie AH, Svendsen C, Brunborg G, Glatt H, Alexander J, Meinl W and Husøy T, 2015. Genotoxicity of three food processing contaminants in transgenic mice expressing human sulfotransferases 1A1 and 1A2 as assessed by the in vivo alkaline single cell gel electrophoresis assay. Environmental and Molecular Mutagenesis, 56, 709–714. 10.1002/em.21963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høie AH, Svendsen C, Rasmussen T, Alexander J and Husoy T, 2016. Intestinal tumor development in C57BL/6J‐Apc Min /+ mice expressing human sulphotransferases 1A1 and 1A2 after oral exposure to 2,5‐dimethylfuran. Anticancer Research, 36, 545–553. [PubMed] [Google Scholar]
- Hoydonckx HE, Van Rhijn WM, Van Rhijn W, De Vos DE and Jacobs PA, 2012. Furfural and derivatives. In: Elvers B (ed.). Ullmann's Encyclopedia of Industrial Chemistry, John Wiley and Sons, NEW York. pp. 285–313. [Google Scholar]
- Hu G, Zhu Y, Hernandez M, Koutchma T and Shao S, 2016. An efficient method for the simultaneous determination of furan, 2‐methylfuran and 2‐pentylfuran in fruit juices by headspace solid phase microextraction and gas chromatography‐flame ionisation detector. Food Chemistry, 192, 9–14. 10.1016/j.foodchem.2015.06.100 [DOI] [PubMed] [Google Scholar]
- Huffman MP, Høie AH, Svendsen C, Brunborg G, Murkovic M, Glatt H and Husoy T, 2016. An in vitro study on the genotoxic effect of substituted furans in cells transfected with human metabolizing enzymes: 2,5‐dimethylfuran and furfuryl alcohol. Mutagenesis, 31, 597–602. 10.1093/mutage/gew025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufford DL, Tarbell DS and Koszalka TR, 1952. Maleic and fumaric dialdehydes, Δ4‐tetrahydrophthalaldehyde and related compounds. Journal of the American Chemical Society, 74, 3014–3018. 10.1021/ja01132a018 [DOI] [Google Scholar]
- Huybrechts I, Sioen I, Boon PE, Ruprich J, Lafay L, Turrini A, Amiano P, Hirvonen T, De Neve M, Arcella D, Moschandreas J, Westerlund A, Ribas‐Barba L, Hilbig A, Papoutsou S, Christensen T, Oltarzewski M, Virtanen S, Rehurkova I, Azpiri M, Sette S, Kersting M, Walkiewicz A, Serra‐Majem L, Volatier J‐L, Trolle E, Tornaritis M, Busk L, Kafatos A, Fabiansson S, De Henauw S and Van Klaveren JD, 2011. Dietary exposure assessments for children in Europe (the EXPOCHI project): rationale, methods and design. Archives of Public Health, 69, 4. 10.1186/0778-7367-69-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC (International Agency for Research on Cancer), 1995. Furan. IARC monographs. Volume 63, 393–407. Available online: http://monographs.iarc.fr/ENG/Monographs/vol63/mono63-16.pdf
- Jackson AF, Williams A, Recio L, Waters MD, Lambert IB and Yauk CL, 2014. Case study on the utility of hepatic global gene expression profiling in the risk assessment of the carcinogen furan. Toxicology and Applied Pharmacology, 274, 63–77. 10.1016/j.taap.2013.10.019 [DOI] [PubMed] [Google Scholar]
- Jaeschke H, McGill MR and Ramachandran A, 2012. Oxidant stress, mitochondria, and cell death mechanisms in drug‐induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metabolism Reviews, 44, 88–106. 10.3109/03602532.2011.602688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey AM, Brunnemann KD, Duan JD, Schlatter J and Williams GM, 2012. Furan induction of DNA cross‐linking and strand breaks in turkey fetal liver in comparison to 1,3‐propanediol. Food and Chemical Toxicology, 50, 675–678. 10.1016/j.fct.2011.11.011 [DOI] [PubMed] [Google Scholar]
- Johansson E, Reynolds S, Anderson M and Maronpot R, 1997. Frequency of Ha‐ras‐1 gene mutations inversely correlated with furan dose in mouse liver tumors. Molecular Carcinogenesis, 18, 199–205. [DOI] [PubMed] [Google Scholar]
- Juaniz I, Zocco C, Mouro V, Cid C and De Pena MP, 2016. Effect of frying process on furan content in foods and assessment of furan exposure of Spanish population. LWT‐Food Science and Technology, 68, 549–555. 10.1016/j.lwt.2015.12.061 [DOI] [Google Scholar]
- Jun H‐J, Lee K‐G, Lee Y‐K, Woo G‐J, Park YS and Lee S‐J, 2008. Correlation of urinary furan with plasma γ‐glutamyltranspeptidase levels in healthy men and women. Food and Chemical Toxicology, 46, 1753–1759. 10.1016/j.fct.2008.01.013 [DOI] [PubMed] [Google Scholar]
- Kallio H, Leino M, Koullias K, Kallio S and Kaitaranta J, 1990. Headspace of roasted ground coffee as an indicator of storage time. Food Chemistry, 36, 135–148. 10.1016/0308-8146(90)90097-N [DOI] [Google Scholar]
- Kara O, Bas H and Pandir D, 2016. Furan toxicity on testes and protective role of lycopene in diabetic rats. Journal of the Turkish‐German Gynecological Association, 17, 191–196. 10.5152/jtgga.2016.16144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karacaoğlu E, 2016, Re: About Furan dose regiment. Message to Dieter Schrenk. 25 October 2016. Email.
- Karacaoglu E and Selmanoglu G, 2010. Effects of heat‐induced food contaminant furan on reproductive system of male rats from weaning through postpuberty. Food and Chemical Toxicology, 48, 1293–1301. 10.1016/j.fct.2010.02.026 [DOI] [PubMed] [Google Scholar]
- Karacaoglu E, Selmanoglu G and Kilic A, 2012. Histopathological effects of the food contaminant furan on some endocrine glands of prepubertal male rats. Turkish Journal of Medical Sciences, 42, 1207–1213. 10.3906/sag-1112-17 [DOI] [Google Scholar]
- Kedderis GL, 1997. Extrapolation of in vitro enzyme induction data to humans in vivo . Chemico‐Biological Interactions, 107, 109–121. [DOI] [PubMed] [Google Scholar]
- Kedderis GL and Held SD, 1996. Prediction of furan pharmacokinetics from hepatocyte studies: comparison of bioactivation and hepatic dosimetry in rats, mice, and humans. Toxicology and Applied Pharmacology, 140, 124–130. 10.1006/taap.1996.0205 [DOI] [PubMed] [Google Scholar]
- Kedderis GL, Carfagna MA, Held SD, Batra R, Murphy JE and Gargas ML, 1993. Kinetic analysis of furan biotransformation by F‐344 rats in vivo and in vitro . Toxicology and Applied Pharmacology, 123, 274–282. 10.1006/taap.1993.1246 [DOI] [PubMed] [Google Scholar]
- Kellert M, Brink A, Richter I, Schlatter J and Lutz WK, 2008a. Tests for genotoxicity and mutagenicity of furan and its metabolite cis‐2‐butene‐1,4‐dial in L5178Y tk+/− mouse lymphoma cells. Mutation Research/Genetic Toxicology and Environmental Mutagenesis, 657, 127–132. 10.1016/j.mrgentox.2008.08.014 [DOI] [PubMed] [Google Scholar]
- Kellert M, Wagner S, Lutz U and Lutz WK, 2008b. Biomarkers of furan exposure by metabolic profiling of rat urine with liquid chromatography‐tandem mass spectrometry and principal component analysis. Chemical Research in Toxicology, 21, 761–768. 10.1021/tx7004212 [DOI] [PubMed] [Google Scholar]
- Kim TK, Lee YK, Park YS and Lee KG, 2009. Effect of cooking or handling conditions on the furan levels of processed foods. Food Additives and Contaminants: Part A, 26, 767–775. 10.1080/02652030902774656 [DOI] [PubMed] [Google Scholar]
- Kockaya EA, Kilic A, Karacaoglu E and Selmanoglu G, 2012. Does furan affect the thymus in growing male rats? Drug and Chemical Toxicology, 35, 316–323. 10.3109/01480545.2011.619191 [DOI] [PubMed] [Google Scholar]
- Kuballa T, Stier S and Strichow N, 2005. Furan in kaffee und kaffeegetränken. Deutsche Lebensmittel‐Rundschau, 101, 229–235. [Google Scholar]
- Kubiak A, Karasek L and Wenzl T, 2008a. Proficiency test on the determination of furan in baby food. JRC Scientific and Technical Reports. Available online: http://publications.jrc.ec.europa.eu/repository/bitstream/JRC48419/jrc48419%20%20eur%2023544.pdf
- Lachenmeier DW, Reusch H and Kuballa T, 2009. Risk assessment of furan in commercially jarred baby foods, including insights into its occurrence and formation in freshly home‐cooked foods for infants and young children. Food Additives and Contaminants: Part A, 26, 776–785. 10.1080/02652030802714018 [DOI] [PubMed] [Google Scholar]
- Lai GH, Radaeva S, Nakamura T and Sirica AE, 2000. Unique epithelial cell production of hepatocyte growth factor/scatter factor by putative precancerous intestinal metaplasias and associated “intestinal‐type” biliary cancer chemically induced in rat liver. Hepatology, 31, 1257–1265. 10.1053/jhep.2000.8108 [DOI] [PubMed] [Google Scholar]
- Lee H, Bian SS and Chen YL, 1994. Genotoxicity of 1,3‐dithiane and 1,4‐dithiane in the CHO/SCE assay and the Salmonella/microsomal test. Mutation Research, 321, 213–218. [DOI] [PubMed] [Google Scholar]
- Lee YK, Jung SW, Lee SJ and Lee KG, 2009. Analysis of residual furan in human blood using solid phase microextraction‐gas chromatography/mass spectrometry (SPME‐GC/MS). Food Science and Biotechnology, 18, 379–383. [Google Scholar]
- Leino M, Kaitaranta J and Kallio H, 1992. Comparison of changes in headspace volatiles of some coffee blends during storage. Food Chemistry, 43, 35–40. 10.1016/0308-8146(92)90238-W [DOI] [Google Scholar]
- Leopardi P, Cordelli E, Villani P, Cremona TP, Conti L, De Luca G and Crebelli R, 2010. Assessment of in vivo genotoxicity of the rodent carcinogen furan: evaluation of DNA damage and induction of micronuclei in mouse splenocytes. Mutagenesis, 25, 57–62. 10.1093/mutage/gep043 [DOI] [PubMed] [Google Scholar]
- Li C, Lin D, Gao H, Hua H, Peng Y and Zheng J, 2015. N‐acetyl lysine/glutathione‐derived pyrroles as potential ex vivo biomarkers of bioactivated furan‐containing compounds. Chemical Research in Toxicology, 28, 384–393. 10.1021/tx500334m [DOI] [PubMed] [Google Scholar]
- Limacher A, Kerler J, Conde‐Petit B and Blank I, 2007. Formation of furan and methylfuran from ascorbic acid in model systems and food. Food Additives and Contaminants, 24, 122–135. 10.1080/02652030701393112 [DOI] [PubMed] [Google Scholar]
- Limacher A, Kerler J, Davidek T, Schmalzried F and Blank I, 2008. Formation of furan and methylfuran by Maillard‐type reactions in model systems and food. Journal of Agricultural and Food Chemistry, 56, 3639–3647. 10.1021/jf800268t [DOI] [PubMed] [Google Scholar]
- Liu D, Togbe C, Tran L‐S, Felsmann D, Osswald P, Nau P, Koppmann J, Lackner A, Glaude P‐A, Sirjean B, Fournet R, Battin‐Leclerc F and Kohse‐Hoeinghaus K, 2014. Combustion chemistry and flame structure of furan group biofuels using molecular‐beam mass spectrometry and gas chromatography ‐ Part I: Furan. Combustion and Flame, 161, 748–765. 10.1016/j.combustflame.2013.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D and Peterson LA, 2010. Identification of furan metabolites derived from cysteine‐cis‐2‐butene‐1,4‐dial‐lysine cross‐links. Chemical Research in Toxicology, 23, 142–151. 10.1021/tx9003215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Sullivan MA, Phillips MB and Peterson LA, 2009. Degraded protein adducts of cis‐2‐butene‐1,4‐dial are urinary and hepatocyte metabolites of furan. Chemical Research in Toxicology, 22, 997–1007. 10.1021/tx800377v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca A, Mahajan PV and Edelenbos M, 2016. Changes in volatile organic compounds from wild rocket (Diplotaxis tenuifolia L.) during modified atmosphere storage. Postharvest Biology and Technology, 114, 1–9. 10.1016/j.postharvbio.2015.11.018 [DOI] [Google Scholar]
- Macleod G and Ames JM, 1986a. Capillary gas‐chromatography ‐ mass‐spectrometric analysis of cooked ground‐beef aroma. Journal of Food Science, 51, 1427–1434. [Google Scholar]
- Macleod G and Ames JM, 1986b. The effect of heat on beef aroma: comparisons of chemical composition and sensory properties. Flavour and Fragrance Journal, 1, 91–104. 10.1002/ffj.2730010302 [DOI] [Google Scholar]
- Macleod AJ and Cave SJ, 1975. Volatile flavor components of eggs. Journal of the Science of Food and Agriculture, 26, 351–360. 10.1002/jsfa.2740260316 [DOI] [Google Scholar]
- Mally A, Graff C, Schmal O, Moro S, Hamberger C, Schauer UM, Bruck J, Ozden S, Sieber M, Steger U, Schrenk D, Hard GC, Chipman JK and Dekant W, 2010. Functional and proliferative effects of repeated low‐dose oral administration of furan in rat liver. Molecular Nutrition and Food Research, 54, 1556–1567. 10.1002/mnfr.201000064 [DOI] [PubMed] [Google Scholar]
- Marinari UM, Ferro M, Bassi AM, Sciaba L, Finollo R and Brambilla G, 1984. DNA‐damaging activity of biotic and xenobiotic aldehydes in Chinese hamster ovary cells. Cell Biochemistry and Function, 2, 243–248. 10.1002/cbf.290020411 [DOI] [PubMed] [Google Scholar]
- Mariotti MS, Granby K, Rozowski J and Pedreschi F, 2013. Furan: a critical heat induced dietary contaminant. Food and Function, 4, 1001–1015. 10.1039/c3fo30375f [DOI] [PubMed] [Google Scholar]
- Märk J, Pollien P, Lindinger C, Blank I and Märk T, 2006. Quantitation of furan and methylfuran formed in different precursor systems by proton transfer reaction mass spectrometry. Journal of Agricultural and Food Chemistry, 54, 2786–2793. 10.1021/jf052937v [DOI] [PubMed] [Google Scholar]
- Marković K, Vahčić N, Ganić KK and Banović M, 2007. Aroma volatiles of tomatoes and tomato products evaluated by solid‐phase microextraction. Flavour and Fragrance Journal, 22, 395–400. 10.1002/ffj.1811 [DOI] [Google Scholar]
- Maronpot RR, Giles HD, Dykes DJ and Irwin RD, 1991. Furan‐induced hepatic cholangiocarcinomas in Fischer 344 rats. Toxicologic Pathology, 19, 561–570. [DOI] [PubMed] [Google Scholar]
- McDaniel LP, Wei D, Dobrovolsky VN, Shaddock JGJ, Mittelstaedt RA, Doerge DR and Heflich RH, 2012. Genotoxicity of furan in Big Blue rats. Mutation Research/Genetic Toxicology and Environmental Mutagenesis, 742, 72–78. [DOI] [PubMed] [Google Scholar]
- McGregor DB, Brown A, Cattanach P, Edwards I, McBride D, Riach C and Caspary WJ, 1988. Responses of the l5178y tk+/tk− mouse lymphoma cell forward mutation assay: III. 72 Coded chemicals. Environmental Mutagenesis, 12, 85–154. 10.1002/em.2860120111 [DOI] [PubMed] [Google Scholar]
- Merten C, Ferrari P, Bakker M, Boss A, Hearty A, Leclercq C, Lindtner O, Tlustos C, Verger P, Volatier JL and Arcella D, 2011. Methodological characteristics of the national dietary surveys carried out in the European Union as included in the European Food Safety Authority (EFSA) Comprehensive European Food Consumption Database. Food Additives and Contaminants: Part A, 28, 975–995. 10.1080/19440049.2011.576440 [DOI] [PubMed] [Google Scholar]
- Mesias M and Morales FJ, 2014. Reliable estimation of dietary exposure to furan from coffee: an automatic vending machine as a case study. Food Research International, 61, 257–263. 10.1016/j.foodres.2013.07.054 [DOI] [PubMed] [Google Scholar]
- Minorczyk M, Góralczyk K, Struciński P, Hernik A, Czaja K, Lyczewska M, Korcz W, Starski A and Ludwicki JK, 2012. Risk assessment for infants exposed to furan from ready‐to‐eat thermally processed food products in Poland. Roczniki Panstwowego Zakladu Higieny, 63, 403–410. [PubMed] [Google Scholar]
- Mochalski P and Unterkofler K, 2016. Quantification of selected volatile organic compounds in human urine by gas chromatography selective reagent ionization time of flight mass spectrometry (GC‐SRI‐TOF‐MS) coupled with head‐space solid‐phase microextraction (HS‐SPME). Analyst, 141, 4796–4803. 10.1039/c6an00825a [DOI] [PubMed] [Google Scholar]
- Mongin C, Golden JH and Castellano FN, 2016. Liquid PEG polymers containing antioxidants: a versatile platform for studying oxygen‐sensitive photochemical processes. ACS Applied Materials and Interfaces, 8, 24038–24048. 10.1021/acsami.6b05697 [DOI] [PubMed] [Google Scholar]
- Moro S, Chipman JK, Antczak P, Turan N, Dekant W, Falciani F and Mally A, 2012a. Identification and pathway mapping of furan target proteins reveal mitochondrial energy production and redox regulation as critical targets of furan toxicity. Toxicological Sciences, 126, 336–352. 10.1093/toxsci/kfs005 [DOI] [PubMed] [Google Scholar]
- Moro S, Chipman JK, Wegener J‐W, Hamberger C, Dekant W and Mally A, 2012b. Furan in heat‐treated foods: formation, exposure, toxicity, and aspects of risk assessment. Molecular Nutrition and Food Research, 56, 1197–1211. 10.1002/mnfr.201200093 [DOI] [PubMed] [Google Scholar]
- Mortelmans K, Haworth S, Lawlor T, Speck W, Tainer B and Zeiger E, 1986. Salmonella mutagenicity tests: II. Results from the testing of 270 chemicals. Environmental Mutagenesis, 8, 56–119. 10.1002/em.2860080803 [DOI] [PubMed] [Google Scholar]
- Morton LD, Youssef AF, Lloyd E, Kiorpes AL, Goldsworthy TL and Fort FL, 2002. Evaluation of carcinogenic responses in the Eker rat following short‐term exposure to selected nephrotoxins and carcinogens. Toxicology Pathology, 30, 559–564. [DOI] [PubMed] [Google Scholar]
- Moser GJ, Foley J, Burnett M, Goldsworthy TL and Maronpot R, 2009. Furan‐induced dose‐response relationships for liver cytotoxicity, cell proliferation, and tumorigenicity (furan‐induced liver tumorigenicity). Experimental and Toxicologic Pathology, 61, 101–111. 10.1016/j.etp.2008.06.006 [DOI] [PubMed] [Google Scholar]
- Mugford CA, Carfagna MA and Kedderis GL, 1997. Furan‐mediated uncoupling of hepatic oxidative phosphorylation in fischer‐344 rats: an early event in cell death. Toxicology and Applied Pharmacology, 144, 1–11. 10.1006/taap.1997.8121 [DOI] [PubMed] [Google Scholar]
- NAS (National Academy of Sciences), 2000. Spacecraft maximum allowable concentrations for selected airborne contaminants: Volume 4, 386 pp. National Academies Press, Washington, DC. Available online: http://www.nap.edu/catalog/9786.html
- NCTR (National Center for Toxicological Research), 2015. Two‐year carcinogenicity bioassay of furan in F344 rats. Technical report for NCTR experiment No E2168.01 (Test No. E2168.02). 102 pp.
- Neuwirth C, Mosesso P, Pepe G, Fiore M, Malfatti M, Turteltaub K, Dekant W and Mally A, 2012. Furan carcinogenicity: DNA binding and genotoxicity of furan in rats in vivo . Molecular Nutrition and Food Research, 56, 1363–1374. 10.1002/mnfr.201200226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NFI (National Food Institute, Technical University of Denmark), 2017. Identifying and collecting relevant literature related to the oral toxicity of furan and its methyl analogues, 2‐methylfuran and 3‐methylfuran. EFSA supporting publication 2017:EN‐1066. 28 pp. 10.2903/sp.efsa.2017.en-1066 [DOI] [Google Scholar]
- Nixon LN, Wong E, Johnson CB and Birch EJ, 1979. Nonacidic constituents of volatiles from cooked mutton. Journal of Agricultural and Food Chemistry, 27, 355–359. 10.1021/jf60222a044 [DOI] [Google Scholar]
- Noguchi T, Takayama K and Nakano M, 1977. Conversion of 2,5‐dimethylfuran to 2‐hydroxy‐5‐hydroperoxy‐2,5‐dimethyldihydrofuran, a true o‐1(2) derived reaction in aqueous o‐1(2) generating systems. Biochemical and Biophysical Research Communications, 78, 418–423. 10.1016/0006-291x(77)91270-0 [DOI] [PubMed] [Google Scholar]
- Noleau I and Toulemonde B, 1986. Quantitative study of roasted chicken flavor. Lebensmittel‐Wissenschaft and Technologie, 19, 122–125. [Google Scholar]
- NTP (National Toxicology Program), 1993. Toxicology and carcinogenesis studies of furan (CAS No. 110‐00‐9) in F344 Rats and B6C3F1 Mice (Gavage Studies). National Toxicology Program Technical Reports Series, 402, 286 pp. [PubMed]
- NTP (National Toxicology Program), 2014. Report on carcinogens, 13th Edition. Research Triangle Park, NC: U.S. Department of Health and Human Services, Public Health Service. Available online: http://ntp.niehs.nih.gov/ntp/roc/content/profiles/furan.pdf
- Nunes J, Martins IL, Charneira C, Pogribny IP, de Conti A, Beland FA, Marques MM, Jacob CC and Antunes AM, 2016. New insights into the molecular mechanisms of chemical carcinogenesis: in vivo adduction of histone H2B by a reactive metabolite of the chemical carcinogen furan. Toxicology Letters, 264, 106–113. 10.1016/j.toxlet.2016.10.018 [DOI] [PubMed] [Google Scholar]
- Okada Y and Okajima H, 1998. Scavenging activity of furan derivatives against hydroxyl radical generated by fenton system. Yakugaku Zasshi‐Journal of the Pharmaceutical Society of Japan, 118, 226–230. [DOI] [PubMed] [Google Scholar]
- Owczarek‐Fendor A, De Meulenaer B, Scholl G, Adams A, Van Lancker F, Yogendrarajah P, Eppe G, De Pauw E, Scippo ML and De Kimpe N, 2010a. Furan formation from vitamin C in a starch‐based model system: influence of the reaction conditions. Food Chemistry, 121, 1163–1170. 10.1016/j.foodchem.2010.01.065 [DOI] [Google Scholar]
- Owczarek‐Fendor A, De Meulenaer B, Scholl G, Adams A, Van Lancker F, Yogendrarajah P, Uytterhoeven V, Eppe G, De Pauw E, Scippo ML and De Kimpe N, 2010b. Importance of fat oxidation in starch‐based emulsions in the generation of the process contaminant furan. Journal of Agricultural and Food Chemistry, 58, 9579–9586. 10.1021/jf101671u [DOI] [PubMed] [Google Scholar]
- Owczarek‐Fendor A, De Meulenaer B, Scholl G, Adams A, Van Lancker F, Eppe G, De Pauw E, Scippo ML and De Kimpe N, 2012. Furan formation in starch‐based model systems containing carbohydrates in combination with proteins, ascorbic acid and lipids. Food Chemistry, 133, 816–821. 10.1016/j.foodchem.2012.01.098 [DOI] [Google Scholar]
- Palmen NGM and Evelo CTA, 1996. Glutathione depletion in human erythrocytes and rat liver: a study on the interplay between bioactivation and inactivation functions of liver and blood. Toxicology in Vitro, 10, 273–281. 10.1016/0887-2333(96)00002-1 [DOI] [PubMed] [Google Scholar]
- Palmers S, Grauwet T, Kebede BT, Hendrickx ME and Van Loey A, 2014. Reduction of furan formation by high‐pressure high‐temperature treatment of individual vegetable purees. Food and Bioprocess Technology, 7, 2679–2693. 10.1007/s11947-014-1300-3 [DOI] [Google Scholar]
- Palmers S, Grauwet T, Buve C, Van de Vondel L, Kebede BT, Hendrickx ME and Van Loey A, 2015a. Furan formation during storage and reheating of sterilised vegetable purees. Food Additives and Contaminants: Part A, 32, 161–169. 10.1080/19440049.2014.999720 [DOI] [PubMed] [Google Scholar]
- Palmers S, Grauwet T, Celus M, Kebede BT, Hendrickx ME and Van Loey A, 2015b. Furan formation as a function of pressure, temperature and time conditions in spinach puree. LWT‐Food Science and Technology, 64, 565–570. 10.1016/j.lwt.2015.06.028 [DOI] [Google Scholar]
- Palmers S, Grauwet T, Celus M, Wibowo S, Kebede BT, Hendrickx ME and Van Loey A, 2015c. A kinetic study of furan formation during storage of shelf‐stable fruit juices. Journal of Food Engineering, 165, 74–81. 10.1016/j.jfoodeng.2015.05.006 [DOI] [Google Scholar]
- Palmers S, Grauwet T, Vanden Avenne L, Verhaeghe T, Kebede BT, Hendrickx ME and Van Loey A, 2016. Effect of oxygen availability and pH on the furan concentration formed during thermal preservation of plant‐based foods. Food Additives and Contaminants: Part A, 33, 612–622. 10.1080/19440049.2016.1154613 [DOI] [PubMed] [Google Scholar]
- Parmar D and Burka LT, 1993. Studies on the interaction of furan with hepatic cytochrome P‐450. Journal of Biochemical Toxicology, 8, 1–9. 10.1002/jbt.2570080103 [DOI] [PubMed] [Google Scholar]
- Perez Locas C and Yaylayan VA, 2004. Origin and mechanistic pathways of formation of the parent furan ‐ a food toxicant. Journal of Agricultural and Food Chemistry, 52, 6830–6836. 10.1021/jf0490403 [DOI] [PubMed] [Google Scholar]
- Peterson LA, Naruko KC and Predecki DP, 2000. A reactive metabolite of furan, cis ‐2‐butene‐1,4‐dial, is mutagenic in the ames assay. Chemical Research in Toxicology, 13, 531–534. 10.1021/tx000065f [DOI] [PubMed] [Google Scholar]
- Peterson LA, Cummings ME, Vu CC and Matter BA, 2005. Glutathione trapping to measure microsomal oxidation of furan to cis‐2‐butene‐1,4‐dial. Drug Metabolism and Disposition, 33, 1453–1458. 10.1124/dmd.105.004432 [DOI] [PubMed] [Google Scholar]
- Peterson LA, Cummings ME, Chan JY, Vu CC and Matter BA, 2006. Identification of a cis‐2‐butene‐1,4‐dial‐derived glutathione conjugate in the urine of furan‐treated rats. Chemical Research in Toxicology, 19, 1138–1141. 10.1021/tx060111x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LA, Phillips MB, Lu D and Sullivan MM, 2011. Polyamines are traps for reactive intermediates in furan metabolism. Chemical Research in Toxicology, 24, 1924–1936. 10.1021/tx200273z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MB, Sullivan MM, Villalta PW and Peterson LA, 2014. Covalent modification of cytochrome c by reactive metabolites of furan. Chemical Research in Toxicology, 27, 129–135. 10.1021/tx400368r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuong J, Kim S, Thomas R and Zhang LP, 2012. Predicted toxicity of the biofuel candidate 2,5‐dimethylfuran in environmental and biological systems. Environmental and Molecular Mutagenesis, 53, 478–487. 10.1002/em.21702 [DOI] [PubMed] [Google Scholar]
- Pino JA and Marbot R, 2001. Volatile flavor constituents of acerola (Malpighia emarginata DC.) fruit. Journal of Agricultural and Food Chemistry, 49, 5880–5882. 10.1021/jf010270g [DOI] [PubMed] [Google Scholar]
- Pluskota‐Karwatka DM, Hoffmann M, Kuta M, Kronberg L, 2015. Studies on the reactions between the DNA bases and a model alpha, beta‐unsaturated oxoaldehyde. New Journal of Chemistry, 39, 9171–9180. 10.1039/c5nj01149c [DOI] [Google Scholar]
- Pouli AE, Hatzinikolaou DG, Piperi C, Stavridou A, Psallidopoulos MC and Stavrides JC, 2003. The cytotoxic effect of volatile organic compounds of the gas phase of cigarette smoke on lung epithelial cells. Free Radical Biology and Medicine, 34, 345–355. [DOI] [PubMed] [Google Scholar]
- Poulin P and Krishnan K, 1998. A quantitative structure‐toxicokinetic relationship model for highly metabolised chemicals. Atla‐Alternatives to Laboratory Animals, 26, 45–59. [PubMed] [Google Scholar]
- Ramm S, Limbeck E and Mally A, 2016. Functional and cellular consequences of covalent target protein modification by furan in rat liver. Toxicology, 361–362, 49–61. 10.1016/j.tox.2016.06.018 [DOI] [PubMed] [Google Scholar]
- Ramos B, Pinho O and Ferreira IMPLVO, 2009. Volatile profile and sensory characteristics of creams used as fillers of bakery products: home made egg creams vs industrial creams. Italian Journal of Food Science, 21, 255–268. [Google Scholar]
- Ravindranath V and Boyd MR, 1985. Metabolic‐activation of 2‐methylfuran by rat microsomal systems. Toxicology and Applied Pharmacology, 78, 370–376. 10.1016/0041-008x(85)90242-x [DOI] [PubMed] [Google Scholar]
- Ravindranath V and Boyd MR, 1991. Effect of modulators of glutathione synthesis on the hepatotoxicity of 2‐methylfuran. Biochemical Pharmacology, 41, 1311–1318. [DOI] [PubMed] [Google Scholar]
- Ravindranath V, Burka L and Boyd M, 1984. Reactive metabolites from the bioactivation of toxic methylfurans. Science, 224, 884–886. 10.1126/science.6719117 [DOI] [PubMed] [Google Scholar]
- Ravindranath V, McMenamin MG, Dees JH and Boyd MR, 1986. 2‐Methylfuran toxicity in rats ‐ role of metabolic‐activation in vivo . Toxicology and Applied Pharmacology, 85, 78–91. 10.1016/0041-008x(86)90389-3 [DOI] [PubMed] [Google Scholar]
- Recio L, Phillips SL, Maynor T, Waters M, Jackson AF and Yauk CL, 2013. Differential expression of long noncoding RNAs in the Livers of female B6C3F1 mice exposed to the carcinogen furan. Toxicological Sciences, 135, 369–379. 10.1093/toxsci/kft153 [DOI] [PubMed] [Google Scholar]
- Reynolds S, Stowers S, Patterson R, Maronpot R, Aaronson S and Anderson M, 1987. Activated oncogenes in B6C3F1 mouse liver tumors: implications for risk assessment. Science, 237, 1309–1316. 10.1126/science.3629242 [DOI] [PubMed] [Google Scholar]
- Roberts D, Crews C, Grundy H, Mills C and Matthews W, 2008. Effect of consumer cooking on furan in convenience foods. Food Additives and Contaminants, 25, 25–31. 10.1080/02652030701551842 [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Burruezo A, Kollmannsberger H, González‐Mas MC, Nitz S and Fernando N, 2010. HS‐SPME comparative analysis of genotypic diversity in the volatile fraction and aroma‐contributing compounds of Capsicum Fruits from the annuum−chinense−frutescens complex. Journal of Agricultural and Food Chemistry, 58, 4388–4400. 10.1021/jf903931t [DOI] [PubMed] [Google Scholar]
- Román‐Leshkov Y, Barrett CJ, Liu ZY and Dumesic JA, 2007. Production of dimethylfuran for liquid fuels from biomass‐derived carbohydrates. Nature, 447, 982–985. 10.1038/nature05923 [DOI] [PubMed] [Google Scholar]
- Santonicola S and Mercogliano R, 2016. Occurrence and production of furan in commercial foods. Italian Journal of Food Science, 28, 155–177. [Google Scholar]
- Scholl G, Huybrechts I, Humblet MF, Scippo ML, De Pauw E, Eppe G and Saegerman C, 2012a. Risk assessment for furan contamination through the food chain in Belgian children. Food Additives and Contaminants. Part A, 29, 1219–1229. 10.1080/19440049.2012.686456 [DOI] [PubMed] [Google Scholar]
- Scholl G, Humblet MF, Scippo ML, De Pauw E, Eppe G and Saegerman C, 2012b. Risk assessment of Belgian adults for furan contamination through the food chain. Food Additives and Contaminants Part A‐Chemistry Analysis Control Exposure and Risk Assessment, 29, 345–353. 10.1080/19440049.2011.637240 [DOI] [PubMed] [Google Scholar]
- Selmanoglu G, Karacaoglu E, Kilic A, Kockaya EA and Akay MT, 2012. Toxicity of food contaminant furan on liver and kidney of growing male rats. Environmental Toxicology, 27, 613–622. 10.1002/tox.20673 [DOI] [PubMed] [Google Scholar]
- Senyuva HZ and Gokmen V, 2005. Analysis of furan in foods. Is headspace sampling a fit‐for‐purpose technique? Food Additives and Contaminants, 22, 1198–1202. 10.1080/02652030500337310 [DOI] [PubMed] [Google Scholar]
- Seok YJ, Her JY, Kim YG, Kim MY, Jeong SY, Kim MK, Lee JY, Kim CI, Yoon HJ and Lee KG, 2015. Furan in thermally processed foods ‐ a review. Toxicological Research, 31, 241–253. 10.5487/tr.2015.31.3.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MY, Liu Q, Jia HB, Jiang YJ, Nie SP, Xie JH, Li C and Xie MY, 2016. Simultaneous determination of furan and 2‐alkylfurans in heat‐processed foods by automated static headspace gas chromatography‐mass spectrometry. LWT‐Food Science and Technology, 72, 44–54. 10.1016/j.lwt.2016.04.030 [DOI] [Google Scholar]
- Shimoda M and Shibamoto T, 1990. Isolation and identification of headspace volatiles from brewed coffee with an on‐column GC MS method. Journal of Agricultural and Food Chemistry, 38, 802–804. 10.1021/jf00093a045 [DOI] [Google Scholar]
- Shinohara K, Kim E‐H and Omura H, 1986. Furans as the mutagens formed by amino‐carbonyl reactions. Developments in Food Science, 13, 353. [Google Scholar]
- Sirica AE, 1996. Biliary proliferation and adaptation in furan‐induced rat liver injury and carcinogenesis. Toxicologic Pathology, 24, 90–99. [DOI] [PubMed] [Google Scholar]
- Sirica AE, Gainey TW and Mumaw VR, 1994a. Ductular hepatocytes. Evidence for a bile ductular cell origin in furan‐treated rats. American Journal of Pathology, 145, 375–383. [PMC free article] [PubMed] [Google Scholar]
- Sirica AE, Cole SL and Williams T, 1994b. A unique rat model of bile ductular hyperplasia in which liver is almost totally replaced with well‐differentiated bile ductules. American Journal of Pathology, 144, 1257–1268. [PMC free article] [PubMed] [Google Scholar]
- Sirica AE, Radaeva S and Caran N, 1997. NEU overexpression in the furan rat model of cholangiocarcinogenesis compared with biliary ductal cell hyperplasia. American Journal of Pathology, 151, 1685–1694. [PMC free article] [PubMed] [Google Scholar]
- Stadler RH, 2012. Heat‐generated toxicants in foods: acrylamide, MCPD esters and furan. In: Schrenk D (ed.). Chemical Contaminants and Residues in Food, Woodhead Publishing Limited, Cambridge, UK. pp. 201–232. [Google Scholar]
- Stevens K and Madder A, 2009. Furan‐modified oligonucleotides for fast, high‐yielding and site‐selective DNA inter‐strand cross‐linking with non‐modified complements. Nucleic Acids Research, 37, 1555–1565. 10.1093/nar/gkn1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stich HF, Rosin MP, Wu CH and Powrie WD, 1981. Clastogenicity of furans found in food. Cancer Letters, 13, 89–95. 10.1016/0304-3835(81)90133-6 [DOI] [PubMed] [Google Scholar]
- Sviatenko L, Gorb L, Hovorun D and Leszczynski J, 2012. Interaction of 2’‐deoxyadenosine with cis‐2‐butene‐1,4‐dial: computational approach to analysis of multistep chemical reactions. Journal of Physical Chemistry. A, 116, 2333–2342. 10.1021/jp211911u [DOI] [PubMed] [Google Scholar]
- Sviatenko LK, Gorb LG, Hovorun DM and Leszczynski J, 2014. Interaction of 2 ‘‐deoxyguanosine with cis‐2‐butene‐1,4‐dial: computational approach to analysis of multistep chemical reactions. Biopolymers and Cell, 30, 239–246. [DOI] [PubMed] [Google Scholar]
- Takeoka GR, Flath RA, Guentert M and Jennings W, 1988. Nectarine volatiles: vacuum steam distillation versus headspace sampling. Journal of Agricultural and Food Chemistry, 36, 553–560. 10.1021/jf00081a037 [DOI] [Google Scholar]
- Tapia A, Villanueva F, Salgado MS, Cabanas B, Martinez E and Martin P, 2011. Atmospheric degradation of 3‐methylfuran: kinetic and products study. Atmospheric Chemistry and Physics, 11, 3227–3241. 10.5194/acp-11-3227-2011 [DOI] [Google Scholar]
- Taxak N, Kalra S and Bharatam PV, 2013. Mechanism‐based inactivation of cytochromes by furan epoxide: unraveling the molecular mechanism. Inorganic Chemistry, 52, 13496–13508. 10.1021/ic401907k [DOI] [PubMed] [Google Scholar]
- Terent'ev AP and Kazitsyna LA, 1949. Sulfonation and the sulfonic acids of acidophobic compounds. II. Sulfonation of furan homologs. Zhurnal Obshchei Khimii, 19, 531–537. [Google Scholar]
- Terrell AN, Huynh M, Grill AE, Kovi RC, O'Sullivan MG, Guttenplan JB, Ho YY and Peterson LA, 2014. Mutagenicity of furan in female Big Blue B6C3F1 mice. Mutation Research. Genetic Toxicology and Environmental Mutagenesis, 770, 46–54. 10.1016/j.mrgentox.2014.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoolen B, Maronpot RR, Harada T, Nyska A, Rousseaux C, Nolte T, Malarkey DE, Kaufmann W, Kuttler K, Deschl U, Nakae D, Gregson R, Vinlove MP, Brix AE, Singh B, Belpoggi F and Ward JM, 2010. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicologic Pathology, 38, 5s–81s. 10.1177/0192623310386499 [DOI] [PubMed] [Google Scholar]
- Tryndyak V, de Conti A, Doerge DR, Olson GR, Beland FA and Pogribny IP, 2017. Furan‐induced transcriptomic and gene‐specific DNA methylation changes in the livers of Fischer 344 rats in a 2‐year carcinogenicity study. Archives of Toxicology, 91, 1233–1243. 10.1007/s00204-016-1786-8 [DOI] [PubMed] [Google Scholar]
- Van Lancker F, Adams A, Owczarek A, De Meulenaer B and De Kimpe N, 2009. Impact of various food ingredients on the retention of furan in foods. Molecular Nutrition and Food Research, 53, 1505–1511. 10.1002/mnfr.200800565 [DOI] [PubMed] [Google Scholar]
- Van Lancker F, Adams A, Owczarek‐Fendor A, De Meulenaer B and De Kimpe N, 2011. Mechanistic insights into furan formation in maillard model systems. Journal of Agricultural and Food Chemistry, 59, 229–235. 10.1021/jf102929u [DOI] [PubMed] [Google Scholar]
- Vercellotti JR, Kuan JCW, Liu RH, Legendre MG, St. Angelo AJ and Dupuy HP, 1987. Analysis of volatile heteroatomic meat flavor principles by purge‐and‐trap/gas chromatography‐mass spectrometry. Journal of Agricultural and Food Chemistry, 35, 1030–1035. 10.1021/jf00078a041 [DOI] [Google Scholar]
- Villanueva F, Cabanas B, Monedero E, Salgado S, Bejan I and Martin P, 2009. Atmospheric degradation of alkylfurans with chlorine atoms: product and mechanistic study. Atmospheric Environment, 43, 2804–2813. 10.1016/j.atmosenv.2009.02.030 [DOI] [Google Scholar]
- VKM (Norwegian Scientific Committee for Food Safety), 2012. Risk assessment of furan exposure in the Norwegian population; Opinion of the Panel on Food Additives, Flavourings, Processing Aids, Materials in Contact with Food and Cosmetics and the Panel on Contaminants of the Norwegian Scientific Committee for Food Safety. 107 pp.
- Von Tungeln LS, Walker NJ, Olson GR, Mendoza MC, Felton RP, Thorn BT, Marques MM, Pogribny IP, Doerge DR and Beland FA, 2017. Low dose assessment of the carcinogenicity of furan in male F344/N Nctr rats in a 2‐year gavage study. Food and Chemical Toxicology, 99, 170–181. 10.1016/j.fct.2016.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waizenegger J, Winkler G, Kuballa T, Ruge W, Kersting M, Alexy U and Lachenmeier DW, 2012. Analysis and risk assessment of furan in coffee products targeted to adolescents. Food Additives and Contaminants: Part A, 29, 19–28. 10.1080/19440049.2011.617012 [DOI] [PubMed] [Google Scholar]
- Wang E, Chen F, Hu X and Yuan Y, 2014a. Protective effects of apigenin against furan‐induced toxicity in mice. Food and Function, 5, 1804–1812. 10.1039/c4fo00038b [DOI] [PubMed] [Google Scholar]
- Wang K, Zheng LW, Peng Y, Song JE and Zheng J, 2014b. Selective and sensitive platform for function‐based screening of potentially harmful furans. Analytical Chemistry, 86, 10755–10762. 10.1021/ac502796x [DOI] [PubMed] [Google Scholar]
- Wang K, Li WW, Chen JM, Peng Y and Zheng J, 2015. Detection of cysteine‐ and lysine‐based protein adductions by reactive metabolites of 2,5‐dimethylfuran. Analytica Chimica Acta, 896, 93–101. 10.1016/j.aca.2015.09.017 [DOI] [PubMed] [Google Scholar]
- Weber K, 2017. Message to Grasl‐Kraupp B. 18 July 2017. Email.
- Webster AF, Williams A, Recio L and Yauk CL, 2014a. Bromodeoxyuridine (BrdU) treatment to measure hepatocellular proliferation does not mask furan‐induced gene expression changes in mouse liver. Toxicology, 323, 26–31. 10.1016/j.tox.2014.06.002 [DOI] [PubMed] [Google Scholar]
- Webster AF, Williams A, Recio L and Yauk CL, 2014b. Gene expression analysis of livers from female B6C3F1 mice exposed to carcinogenic and non‐carcinogenic doses of furan, with or without bromodeoxyuridine (BrdU) treatment. Genomics Data, 2, 117–122. 10.1016/j.gdata.2014.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler MW and Bailer AJ, 2008. Model averaging software for dichotomous dose response risk estimation. Journal of Statistical Software, 26, 1–15.19777145 [Google Scholar]
- WHO/IPCS (World Health Organization/International Programme on Chemical Safety), 2008. Uncertainty and data quality in exposure assessment. Harmonisation project document No. 6. ISBN 978 92 4 156376 5.U.
- WHO/IPCS (World Health Organization/International Programme on Chemical Safety), 2009. Environmental Health Criteria 240. Principles and methods for the assessment of chemicals in food.
- Wiley RA, Traiger GJ, Baraban S and Gammal LM, 1984. Toxicity‐distribution relationships among 3‐alkylfurans in mouse liver and kidney. Toxicology and Applied Pharmacology, 74, 1–9. 10.1016/0041-008x(84)90263-1 [DOI] [PubMed] [Google Scholar]
- Williams JR, Rayburn JR, Cline GR, Sauterer R and Friedman M, 2014. Potential protective effect of L‐cysteine against the toxicity of acrylamide and furan in exposed Xenopus laevis embryos: an interaction study. Journal of Agricultural and Food Chemistry, 62, 7927–7938. 10.1021/jf5013743 [DOI] [PubMed] [Google Scholar]
- Wilson DM, Goldsworthy TL, Popp JA and Butterworth BE, 1992. Evaluation of genotoxicity, pathological lesions, and cell proliferation in livers of rats and mice treated with furan. Environmental and Molecular Mutagenesis, 19, 209–222. 10.1002/em.2850190305 [DOI] [PubMed] [Google Scholar]
- Witschi HP, Tryka AF, Mauderly JL, Haschek WM, Satterfield LC, Bowles ND and Boyd MR, 1985. Long‐term effects of repeated exposure to 3‐methylfuran in hamsters and mice. Journal of Toxicology and Environmental Health, 16, 581–592. [DOI] [PubMed] [Google Scholar]
- Yauk CL, Wade MG, Kuo B, Gill S, Williams A, Dong H and Curran IH, 2014. Toxicogenomic assessment of liver responses following subchronic exposure to furan in Fischer F344 rats cRNA. Archives of Toxicology, 90, 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauk CL, Buick JK, Williams A, Swartz CD, Recio L, Li HH and Fornace AJ Jr, Thomson EM and Aubrecht J, 2016. Application of the TGx‐28.65 transcriptomic biomarker to classify genotoxic and non‐genotoxic chemicals in human TK6 cells in the presence of rat liver S9. Environmental and Molecular Mutagenesis, 57, 243–260. 10.1002/em.22004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Chen D, Liu H, Wu S and Yan H, 2016. A comparison study of frying conditions on furan formation in 3 potato varieties. J Food Sci, 81, T2114–2121. 10.1111/1750-3841.13386 [DOI] [PubMed] [Google Scholar]
- Yuan Y, Wu SJ, Liu X and Zhang LL, 2013. Antioxidant effect of salidroside and its protective effect against furan‐induced hepatocyte damage in mice. Food and Function, 4, 763–769. 10.1039/c3fo00013c [DOI] [PubMed] [Google Scholar]
- Zeiger E, Anderson B, Haworth S, Lawlor T and Mortelmans K, 1992. Salmonella mutagenicity tests: V. results from the testing of 311 chemicals. Environmental and Molecular Mutagenesis, 19, 2–141. [DOI] [PubMed] [Google Scholar]
- Zoller O, Sager F and Reinhard H, 2007. Furan in food: headspace method and product survey. Food Additives and Contaminants, 24, 91–107. 10.1080/02652030701447389 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Occurrence data submitted to EFSA and dietary exposure assessment for humans