

Abstract
Nicotinamide adenine dinucleotide (NAD+) is an essential biomolecule involved in many critical processes. Its role as both a driver of energy production and a signaling molecule underscores its importance in health and disease. NAD+ signaling impacts multiple processes that are dysregulated in cancer, including DNA repair, cell proliferation, differentiation, redoxregulation, and oxidative stress. Distribution of NAD+ is highly compartmentalized, with each subcellular NAD+ pool differentially regulated and preferentially involved in distinct NAD+-dependent signaling or metabolic events. Emerging evidence suggests that targeting NAD+ metabolism is likely to repress many specific mechanisms underlying tumor development and progression, including proliferation, survival, metabolic adaptations, invasive capabilities, heterotypic interactions with the tumor microenvironment, and stress response including notably DNA maintenance and repair. Here we provide a comprehensive overview of how compartmentalized NAD+ metabolism in mitochondria, nucleus, cytosol, and extracellular space impacts cancer formation and progression, along with a discussion of the therapeutic potential of NAD+-targeting drugs in cancer.
Keywords: NAD+, PARP, SIRT, CD38, NAMPT, NMNAT
1. Introduction
A critical hallmark in the multistep development and progression of cancer is the reprogramming of energy metabolism (Hanahan & Weinberg, 2011). NAD+ is a fundamental signaling cofactor that regulates cancer metabolism through its coenzymatic function in redox reactions underlying essential bioenergetic pathways including glycolysis, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation. Continuous supplementation of NAD+ is essential for the functionality of glyceraldehyde-3-phosphate dehydrogenase in glycolysis (Busso et al., 2008). Serine biosynthesis from glucose requires NAD+-dependent 3-phosphoglycerate dehydrogenase that can be genomically amplified in cancer (Murphy et al., 2018; Possemato et al., 2011). NAD+ regulates multiple signaling pathways, including poly (ADP-ribose) polymerase (PARP)-mediated ADP-ribosylation and sirtuin (SIRT)-mediated deacetylation, both of which contribute to metabolic regulation, DNA damage repair, cell cycle progression, and epithelial-to-mesenchymal transition (EMT) (Imai, Armstrong, Kaeberlein, & Guarente, 2000; Schreiber, Dantzer, Ame, & De Murcia, 2006). NAD+ can also be converted by glycohydrolase CD38 (cluster of differentiation 38) to cyclic ADP-ribose (cADPR), a Ca2+-mobilizing second messenger that regulates multiple aspects of cancer biology, including cell survival, apoptosis, and inflammation (Camacho-Pereira et al., 2016; Chini, Chini, Netto, de Oliveira, & van Schooten, 2018). It is important to note that when NAD+ functions as a coenzyme, in redox reactions for example, NAD+ is converted to NADH and can be regenerated. In contrast, when NAD+ functions as a signaling molecule in NAD+-dependent signaling processes, NAD+ is continuously and irreversibly catabolized by cleavage of the glycosidic bond between nicotinamide (NAM) and the ADP-ribose moiety. Therefore, replenishment of NAD+ pool is particularly challenging for the rapidly dividing cancer cells. The emergence of NAD+ as a key molecule in tumorigenesis has prompted the development of therapeutic interventions targeting the NAD+ biosynthetic and consuming enzymes (Chiarugi, Dölle, Felici, & Ziegler, 2012; Chini, 2009; Ju et al., 2016; Sharif et al., 2019).
In mammalian cells, NAD+ has a relatively short half-life, about 1 h (Rechsteiner, Hillyard, & Olivera, 1976), and a limited diffusion radius due to its charge and cellular membrane barriers (Van Roermund, Elgersma, Singh, Wanders, & Tabak, 1995). It is conceivable that NAD+ concentration is not uniform within the cell and regulation of NAD+ concentration is “local”. This notion is supported by the finding that many enzymes involved in the biosynthesis or consumption of NAD+ are highly compartmentalized at subcellular level (Fig. 1). For example, nicotinamide mononucleotide adenylyltransferase (NMNAT), the last enzyme in the NAD+ synthetic pathway, has three isoforms in mammals that are distinctly localized: NMNAT1, enriched in the nucleus; NMNAT2, in the cytosol and membrane structures; and NMNAT3, reported to be in the mitochondria (Berger, Lau, Dahlmann, & Ziegler, 2005; Brazill, Li, Zhu, & Zhai, 2017). Since most of the bioenergetic and NAD+-dependent signaling pathways occur within distinct subcellular compartments, locally accessible NAD+ is required for these metabolic processes (Alano et al., 2007; Nikiforov, Kulikova, & Ziegler, 2015). Several approaches have been attempted to visualize subcellular distribution of NAD+ pools. For example, targeted expression of PARP-1 can be used as a molecular NAD+ detector since the presence of NAD+ can be visualized by the formation of immunodetectable poly-ADP-ribose (Dölle, Niere, Lohndal, & Ziegler, 2010; VanLinden, Niere, Nikiforov, Ziegler, & Dölle, 2017). Moreover, several genetically encoded NAD+ biosensors have been developed, which contain a bipartite NAD+-binding domain derived from bacterial NAD+-dependent DNA ligase fused to a circularly permuted Venus fluorescent protein (cpVenus) (Cambronne et al., 2016). These NAD+ biosensors can be expressed either in nucleus or in cytoplasm and exhibit reduction in their fluorescent signals upon binding to NAD+. With these powerful molecular tools, NAD+ has been detected in the nucleus and almost all cytoplasmic organelles, including mitochondria, peroxisomes, endoplasmic reticulum (ER), and Golgi complex (Cambronne et al., 2016; Dölle et al., 2010; VanLinden et al., 2017).
Fig. 1.
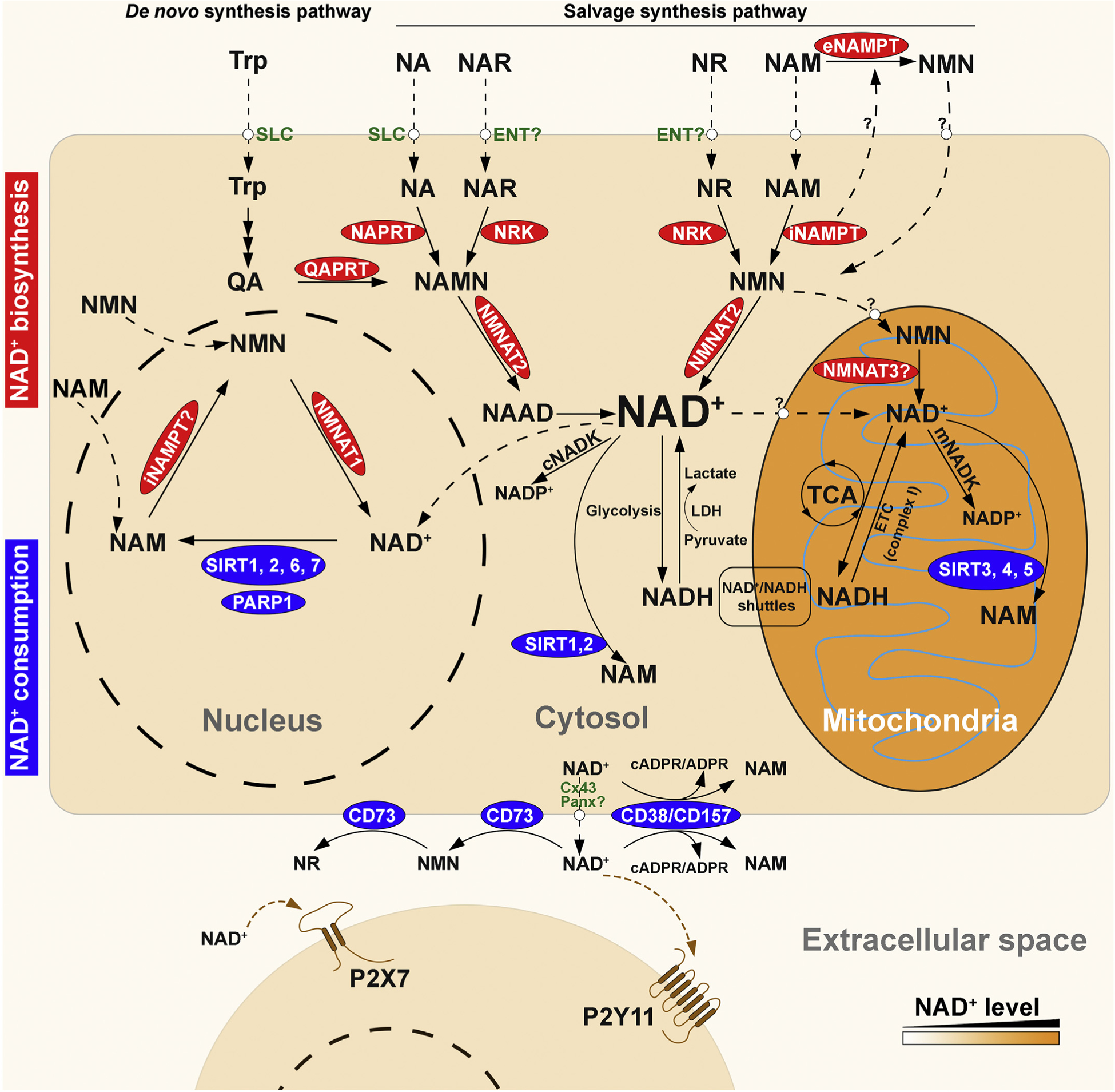
Overview of the compartmentalized NAD+ metabolome in humans. NAD+ is synthesized via two major pathways: the de novo biosynthesis pathway from tryptophan (Trp), and the salvage pathway from various extracellular and intracellular precursors including nicotinic acid (NA), NA riboside (NAR), nicotinamide (NAM), and NAM riboside (NR). The precursors can cross cell membrane via different transporters, including solute carrier (SLC) transporters for Trp and NA, and equilibrative nucleoside transporters (ENTs) for NAR and NR. In the de novo synthesis pathway, Trp is degraded to quinolinic acid (QA), which can be converted to NA mononucleotide (NAMN) by quinolinic acid phosphoribosyltransferase (QAPRT). In the salvage synthesis pathway, NA and NR can be converted to their corresponding mononucleotides NAMN and nicotinamide mononucleotide (NMN) by nicotinic acid phosphoribosyltransferase (NAPRT) and nicotinamide phosphoribosyltransferase (NAMPT), respectively. NAMN and NMN can also be generated from phosphorylation of the nucleosides, NAR and NR, by NR kinases (NRKs). NMN may be generated from NAM by extracellular NAMPT (eNAMPT), although the secretion and function eNAMPT has not been elucidated, and whether or not NMN can enter cells is a subject of some debate in the field. The formation of dinucleotide NAAD or NAD+ from NAMN or NMN is catalyzed by nicotinamide mononucleotide adenylyltransferases (NMNATs). Human NMNAT comprises three isoforms that are localized in the nucleus (NMNAT1), cytosol (NMNAT2), and mitochondria (NMNAT3), although the presence and function of NMNAT3 is still under question. In cytosol, NAD+ can be reduced to NADH in glycolysis, and electrons from NADH can enter the mitochondrial electron transport chain (ETC) via glycerol-3-phosphate shuttle or malate-aspartate shuttle. NADH is oxidized to NAD+ when pyruvate is converted to lactate by lactate dehydrogenase (LDH). In mitochondria, NAD+ is reduced to NADH by tricarboxylic acid (TCA) cycle, and can be regenerated from NADH by the ETC complex I. Whether NAD+ or its precursors can enter mitochondria is still unclear. Cytosolic and mitochondrial NAD+ can be converted to NADP+ by cytosol NAD+ kinase (cNADK) and mitochondrial NADK (mNADK), respectively. NAD+ is consumed by poly (ADP-ribose) polymerases (PARPs) and sirtuins (SIRTs) at different subcellular compartments. Intracellular NAD+ is released from cells through connexin 43 (Cx43) and possibly pannexin (Panx) hemi channels. Extracellular NAD+ can be degraded by membrane-bound glycohydrolases CD38/CD157 to cyclic ADP-ribose (cADPR) and ADPR, or by CD73 to NMN and further to NR. Extracellular NAD+ can modulate inflammatory response by activation of P2X7 and P2Y11 purinergic receptors.
In this review, we will focus on the compartmentalized regulation of NAD+ biosynthesis and metabolism in cancer development and progression, as well as on currently available small molecules targeting NAD+ and its associated biomolecules for cancer treatment. For a comprehensive understanding of the NAD+ metabolome in tumorigenesis, we refer readers to several excellent reviews recently published that cover the topic extensively (Demarest et al., 2019; Sharif et al., 2019; Yaku, Okabe, Hikosaka, & Nakagawa, 2018).
2. Compartmentalized NAD+ pools in cancer
2.1. Mitochondrial NAD+ pool
Mitochondria house the largest NAD+ pool with concentrations of ~250 μM, which account for 50–70% of total cellular NAD+ in cardiac myocytes and neurons (Alano et al., 2007; Cambronne et al., 2016). Maintaining mitochondrial NAD+ levels is crucial to cell survival, especially when nuclear and cytosolic pools of NAD+ become depleted under stress conditions (Yang et al., 2007). Although the cytosolic NAD+ and mitochondrial NAD+ pools are connected by glycolysis and oxidative phosphorylation (Ying, 2008), the exact mechanism of mitochondrial NAD+ regeneration remains unclear. There are two possible sources of mitochondria NAD+, imported from cytosol and locally synthesized in mitochondrial matrix. Numerous studies have gathered evidence supporting each source, although detailed mechanisms are still lacking. The most prominent outstanding question regarding the imported NAD+ is how NAD+ and its precursors, including NAM, nicotinamide riboside (NR), nicotinic acid (NA), nicotinic acid riboside (NAR), and nicotinamide mononucleotide (NMN), cross the mitochondrial membrane. One study shows that cytosolic NMN can be taken up by mitochondria and converted to mitochondrial NAD+ by NMNAT3 (Nikiforov, Dölle, Niere, & Ziegler, 2011), and partly supporting this notion is the fact that, so far, no mitochondrial NAD+ transporter has been identified in mammalian cells (Barile, Passarella, Danese, & Quagliariello, 1996; VanLinden et al., 2015). Additionally, supplementation of NR increases mitochondrial NAD+ level both in cultured cells and the mouse liver (Cantó et al., 2012). However, another group shows that providing precursors like NAM, NR, NA or NMN does not affect mitochondrial NAD+ content (Felici, Lapucci, Ramazzotti, & Chiarugi, 2013). Despite the lack of a confirmed transporter, supplying exogenous NAD+ has been shown to significantly increase mitochondrial NAD+ content (Felici et al., 2013; Pittelli et al., 2011), and a recent study using isotopic labeling of NR and NAR indicates that cytosolic NAD+ can be directly transported into mitochondria (Davila et al., 2018). Similar questions remain outstanding regarding the source of NAD+ from mitochondrial local synthesis. For example, the presence of mitochondrially localized enzymes essential for NAD+ synthesis remain unconfirmed. Nicotinamide phosphoribosyltransferase (NAMPT) catalyzes the rate-limiting step in the NAD+ salvage pathway. One study reported the localization of NAMPT in mitochondria (Yang et al., 2007), while another group could not confirm these results (Pittelli et al., 2010). Given a lack of consensus supporting the presence of mitochondrial NAD+ transporters or NAMPT in mammals, it is proposed that mitochondrial NAD+ is produced through NMN shuttling from the cytoplasm followed by NMNAT3 catalysis (Sharif et al., 2019; VanLinden et al., 2015). NMNAT3 is reported to have a mitochondrial localized splice variant FKSG76 and the ectopically expressed NMNAT3 localizes to mitochondria (Felici et al., 2013). However, although the mRNA transcripts can be detected, the presence of the endogenous protein is yet to be confirmed (Felici et al., 2013). Although one study suggests that NMNAT3 overexpression increases mitochondrial NAD+ (Son, Kwon, Son, & Cho, 2016), other studies have reported that NMNAT3 is dispensable for the maintenance of mitochondrial NAD+ level (Hikosaka et al., 2014; Yamamoto et al., 2016). It is likely that mitochondrial NAD+ comes from both local synthesis as well as transport from the cytosol. Each source serves as the backup for the other, and both sources together contribute to the tightly maintained NAD+ concentrations within mitochondria. Since local synthesis also requires the transport of NAD+ precursors, the concentration of mitochondrial NAD+ would be closely connected to the availability of cytosolic NAD+ and/or its precursors.
Whether transported or locally synthesized, mitochondrial NAD+ serves an essential role in tumorigenesis. Mitochondrial NAD+ is not only essential for bioenergetic processes underlying cell growth, proliferation and survival such as TCA cycle, oxidative phosphorylation, and fatty acid oxidation, but also serves as an obligatory co-substrate for mitochondrial-localized SIRTs (SIRT3, 4 and 5) that have been implicated as master regulators of metabolic homeostasis (George & Ahmad, 2016; Stein & Imai, 2012). For example, SIRT3 has been shown to physically interact Complex I subunit 9 (NDUFA9, NADH:Ubiquinone Oxidoreductase Subunit A9) and selectively deacetylate and augment Complex I activity to maintain basal ATP production by oxidative phosphorylation (Ahn et al., 2008). SIRT4 or SIRT5 knockout animals exhibit significantly dysregulated glycolysis, gluconeogenesis, and fatty acid metabolism (Du et al., 2018; Wood et al., 2018). Interestingly, all mitochondrial SIRTs have potential dual functions in cancer in that they can serve as either tumor suppressors or tumor promoters (Demarest et al., 2019).
SIRT3 is predominantly localized in the mitochondria, but is also reported to be a nuclear NAD+-dependent histone deacetylase that is translocated into mitochondria upon etoposide and UV-induced DNA damage putatively leading to derepression of nuclear genes involved in mitochondrial function (Scher, Vaquero, & Reinberg, 2007). SIRT3 has been shown to possess oncogenic function, for instance it inhibits apoptosis and promotes cell growth and division by enhancing pyruvate dehydrogenase activity in glycolysis (Ozden et al., 2014) (Fig. 2), facilitating mtDNA repair (Cheng et al., 2013), preserving mitochondrial membrane integrity (Kim et al., 2010; Yang et al., 2016), and increasing cell resilience to environmental stress (Tao et al., 2010; Torrens-Mas, Pons, Sastre-Serra, Oliver, & Roca, 2017). SIRT3 deacetylates and stabilizes hydroxymethyltransferase 2 (SHMT2), a mitochondrial enzyme that catalyzes the reversible reaction of serine and tetrahydrofolate to generate glycine and 5,10-methylene tetrahydrofolate, promoting colorectal carcinogenesis (Wei et al., 2018). Silencing SIRT3 has been shown to increase ROS production and sensitize breast cancer cells to cytotoxic compounds (Torrens-Mas et al., 2017).
Fig. 2.
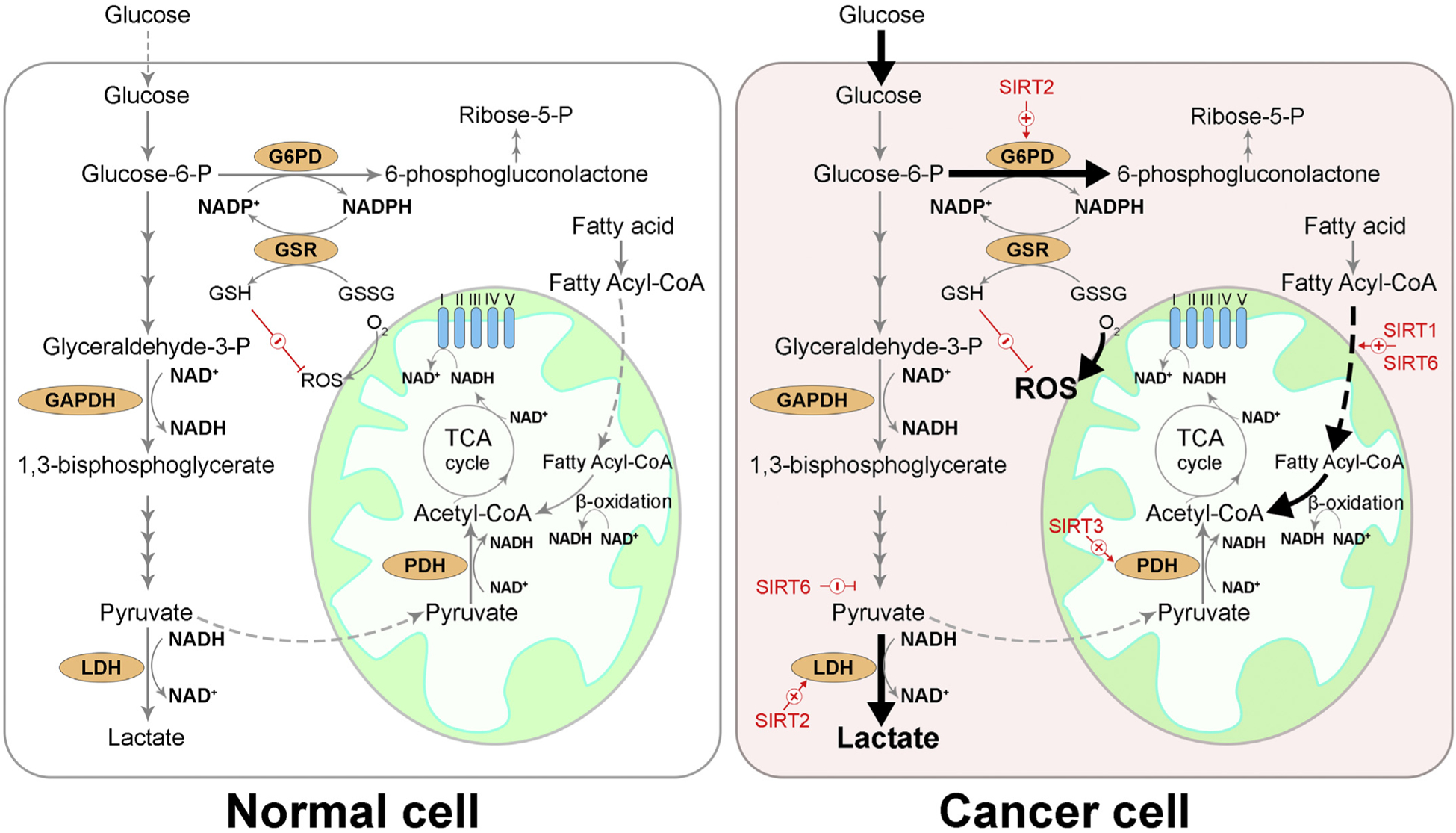
Overview of NAD+ in cancer metabolic reprogramming. Under normal conditions, glucose is metabolized by glycolysis and generates pyruvate, which is preferentially transported into mitochondria. Pyruvate dehydrogenase (PDH) converts pyruvate to acetyl-CoA, which is further metabolized by tricarboxylic acid (TCA) cycle, concomitantly converting NAD+ to NADH. NADH and FADH2 deliver high energy electrons to the electron transport chain (ETC) complexes (I~V) and power the production of ATP. Cancer cells adapt their metabolism to fulfill their increased demand for energy, biosynthetic intermediates, and to counter aerobic respiration-induced oxidative stress by: (1) increasing glucose uptake, (2) preferentially converting pyruvate to lactate by lactate dehydrogenase (LDH) even under aerobic conditions (Warburg effect) to provide metabolic building blocks, (3) diverting glycolysis to pentose phosphate pathway (PPP), where NADPH is produced to neutralize reactive oxygen species (ROS) and ribose-5-phosphate (ribose-5-P) is produced to support nucleic acid synthesis, (4) promoting fatty acid oxidation to provide acetyl-CoA. GAPDH: glyceraldehyde-3-phosphate dehydrogenase, G6PD: glucose-6-phosphate dehydrogenase, GSR: glutathione-disulfide reductase, GSSG: glutathione disulfide, GSH: glutathione.
On the other hand, SIRT3 can also function to suppress tumor growth by negatively regulating the Warburg effect, destabilizing hypoxia-inducible factor-1α (HIF1α), and alleviating ROS burden (Bell, Emerling, Ricoult, & Guarente, 2011; Finley et al., 2011). A recent study shows that SIRT3 promotes FOXO3A by attenuating Wnt/β-catenin pathway, thereby suppressing EMT in prostate cancer cells (Li, Quan, & Xia, 2018). Additionally, SIRT3 can downregulate 2-hydroxyglutarate (2HG) production through deacetylation of mitochondrial isocitrate dehydrogenase 2 (IDH2) to reverse the tumor-promoting mitochondrial metabolic profile and reduce colony formation and tumorigenesis of breast cancer cells (Dvorak, Zelenka, Smolkova, Vitek, & JeZek, 2017; Zou et al., 2017). Upregulation of mitochondrial NAD+ by overexpressing mitochondrial enzyme nicotinamide nucleotide transhydrogenase (NNT) is accompanied by increased SIRT3 activity and compromised clonogenicity of glioblastoma tumor-initiating cells (Son et al., 2017), while downregulation of mitochondrial NAD+ through dysregulation of mitochondrial Ca2+ signaling inhibits SIRT3, leading to deactivation of superoxide dismutase 2 (SOD2) and increased reactive oxygen species (ROS) production, facilitating metastasis of hepatocellular carcinoma cells (Ren et al., 2017).
SIRT4 mainly functions as an ADP-ribosyltransferase. Similar to SIRT3, SIRT4 has both oncogenic and tumor-suppressive activities (Huang & Zhu, 2018). It has been shown that SIRT4 promotes tumorigenic transformation when induced under oncogenic stresses such as DNA damage and ER stress (Jeong, Hwang, & Seong, 2016), and maintains metabolic homeostasis (Wood et al., 2018). In contrast, evidence supporting the tumor-suppressive activity of SIRT4 comes from studies that show downregulation of SIRT4 inhibits apoptosis and desensitizes colorectal cancer cells to chemotherapy (Zhu et al., 2018), and that SIRT4 suppresses tumor growth by inhibiting glutamine metabolism (Csibi et al., 2013; Jeong et al., 2013; Wang et al., 2018).
SIRT5 plays an integral role in metabolic reprogramming during tumorigenesis (Bringman-Rodenbarger, Guo, Lyssiotis, & Lombard, 2018). SIRT5 is an inefficient deacetylase; instead it regulates its target protein activity by NAD+-dependent removal of malonyl, glutaryl, or succinyl groups from lysine residues (Du et al., 2011; Tan et al., 2014). For example, SIRT5 confers chemotherapy resistance to colorectal carcinoma through demalonylation of succinate dehydrogenase complex subunit A (SDHA), leading to accumulation of the oncometabolite succinate (Du et al., 2018). Additionally, SIRT5 deglutarylates and activates glutamate dehydrogenase 1 (GLUD1), permitting the entry of glutamine into the TCA cycle to support colorectal tumorigenesis (Wang et al., 2018). Moreover, SIRT5 desuccinylates and activates mitochondrial serine hydroxymethyltransferase 2 (SHMT2) that catalyzes serine catabolism, driving cancer cell proliferation (Yang et al., 2018). Notably, SIRT5-mediated desuccinylation also inhibits peroxisomal acyl-CoA oxidase 1 (ACOX1) activity, reduces oxidative stress, and protects against the development of hepatocellular carcinoma (Chen et al., 2018).
Recent studies highlight the role of mitochondrial NAD+ in stem cell function. It has been shown that NAD+ repletion by NR supplementation improves mitochondrial function and enhances stem cell function in aged mice (Zhang et al., 2016). Overexpressing NNT and NMNAT3 to restore mitochondrial NAD+ levels increases the lifespan of human mesenchymal stem cells (MSCs) by inhibiting cell senescence and also increases the reprogramming efficiency of aged somatic cells (Son et al., 2016). MSCs comprise tumor stroma, a key part of the tumor microenvironment, and are known to promote tumor progression and metastasis (Ridge, Sullivan, & Glynn, 2017). MSCs also possess immunomodulatory properties, including suppression of T-cell proliferation, potentially contributing to immunosuppression (Krueger, Thorek, Meeker, Isaacs, & Brennen, 2019; Shi et al., 2018). Thus, targeting mitochondrial NAD+ synthesis could potentially eliminate the tumor-promoting stromal cells that contribute to tumor progression and metastasis.
2.2. Nuclear NAD+ pool
The nuclear NAD+ pool is estimated to be smaller (~70–100 μM) than the mitochondrial NAD+ pool (Cambronne et al., 2016; Fjeld, Birdsong, & Goodman, 2003; Nakagawa, Lomb, Haigis, & Guarente, 2009). Nuclear NAD+ is thought to be mainly synthesized by nuclear NMNAT1, the most ubiquitously expressed NMNAT isoform (Lau, Niere, & Ziegler, 2009). Two-photon excitation microscopy shows specific elevation of NAD+ in the nucleus with ectopic expression of NMNAT1 (Zhang et al., 2012). Mutations in NMNAT1 have been found to cause Leber congenital amaurosis (LCA)9, a childhood blindness (Falk et al., 2012; Koenekoop et al., 2012). It was proposed that reduced NAD+ production is associated with the photoreceptor pathology in LCA9 (Koenekoop et al., 2012).
The nuclear role of NAD+ is centered around PARP-mediated ADP-ribosylation and SIRT- mediated deacetylation. PARP1 is the most abundantly expressed member of the PARP protein family and has been extensively studied in cancer, including DNA damage detection and repair, telomerase protection, chromatin modification, transcriptional regulation, p53-mediated apoptosis, and programmed necrosis (Brown, O’Carrigan, Jackson, & Yap, 2017; Chaudhuri & Nussenzweig, 2017; Gomez et al., 2006; Kim, Zhang, & Kraus, 2005; Sosna et al., 2014). Upon DNA damage, PARP1 activation consumes up to 80% nuclear NAD+ for massive ADP-ribosylation, especially poly ADP-ribosylation (PARylation) (Yu et al., 2002). A recent study suggests PARP1 has dual functions in colorectal tumorigenesis, where it suppresses tumor initiation following DNA alkylation but paradoxically enhances inflammation-driven tumor progression (Dörsam et al., 2018). Mechanistic investigation reveals that PARP1 poly-ADP-ribosylates transcription factor E2F1 and restrains E2F1-induced apoptosis in cancer cells (Kumari et al., 2015). Activated PARP1 was also reported to possess a transcriptional-regulatory function (Schiewer et al., 2012; Schiewer & Knudsen, 2014) that promotes E2F1-mediated induction of DNA repair factors, thereby promoting progression to aggressive and treatment-resistant prostate cancer (Schiewer et al., 2018). For an in-depth discussion of the localization and function of other PARP enzymes, we refer readers to this review (Bai, 2015).
In addition to PARP, nuclear NAD+ also regulates many oncogenes and tumor suppressor genes through SIRTs. Through varies targets, nuclear SIRTs (SIRT 1, 6 and 7) carry out multifaceted functions in regulating chromatin structure, genomic stability, cellular metabolism, and immune response that go awry in cancer cells (Chalkiadaki & Guarente, 2015; Zhu et al., 2018). Specifically, SIRT1 binds and deacetylates p53 upon DNA damage, resulting in repression of p53 transcriptional activity and subsequent disinhibition of cell cycle arrest (Vaziri et al., 2001). SIRT1 also deacetylates and inactivates E2F1, promoting abnormal cell proliferation and malignant transformation (Wang et al., 2006). Moreover, SIRT1 activity is required for O-GlcNAcylation-mediated cancer invasion and metastasis (Ferrer et al., 2017). Paradoxically, SIRT1 mutant mice exhibit compromised DNA repair, genome instability, and enhanced tumorigenesis (Wang et al., 2008), whereas SIRT1-overexpressing mice show delayed appearance of K-RasG12V-driven lung adenocarcinomas and extended survival (Costa-Machado et al., 2018), indicating a tumor-suppressive role of SIRT1. Notably, SIRT1 can shuttle between nuclear and cytoplasm as an adaptation to environmental stress. For example, in human bronchial epithelial cells where SIRT1 is enriched in cytoplasm under normal conditions, cigarette smoke exposure induces nuclear translocation of SIRT1, contributing to cell survival under oxidative stress but also likely promoting malignant transformation (Yanagisawa et al., 2018). Conversely, in primary fibroblast cells, where SIRT1 is predominantly nuclear localized, malignant transformation induces aberrant translocation of SIRT1 to cytoplasm, likely through regulation by PI3K (phosphoinositide 3-kinase)/IGF (insulin-like growth factor)-1R signaling (Byles et al., 2010). Inhibition of cytoplasmic SIRT1 promotes caspase-2-dependent apoptosis in breast cancer cells by promoting dissociation of its negative regulator, 14-3-3ζ (Andersen et al., 2011).
SIRT6 functions as a master regulator of metabolic programming and maintains genome integrity during tumorigenesis (Desantis, Lamanuzzi, & Vacca, 2018). SIRT6 has high tumorigenic activity, and high expression of SIRT6 in colon cancer samples has been shown to correlate with poor prognosis (Geng et al., 2018). SIRT6 regulates glucose homeostasis and suppresses the Warburg effect through inhibition of pyruvate kinase M2 (PKM2), the final rate-limiting step of glycolysis (Bhardwaj & Das, 2016; Sebastián et al., 2012) (Fig. 2). In an acidic tumor microenvironment, SIRT1 and SIRT6-mediated histone deacetylation downregulate acetyl-CoA carboxylase 2, (ACC2), a mitochondria-anchored enzyme that normally inhibits degradation of newly synthesized fatty acids, thereby facilitating a shift of the source of acetyl-CoA from glycolysis-derived pyruvate to fatty acid oxidation (Corbet et al., 2016) (Fig. 2). Additionally, SIRT6 can be mobilized to DNA damage sites by lamin A (Ghosh, Liu, Wang, Hao, & Zhou, 2015) or c-Jun N-terminal kinase (Van Meter et al., 2016), and promotes DNA repair by activating and recruiting PARP1 (Mao et al., 2011; Van Meter et al., 2016) and by mediating the rate-limiting step of DNA base excision repair through the 5′-deoxyribose phosphate (dRP) lyase activity of DNA polymerase β (Mostoslavsky et al., 2006). Ubiquitin-protein ligase E3A (UBE3A)-mediated SIRT6 degradation epi-genetically activates annexin A2 and promotes cancer cell proliferation and invasion (Kohli, Bhardwaj, Kumari, & Das, 2018). Moreover, SIRT6-β-catenin signaling is involved in the EMT of ovarian cancer cells (Bae et al., 2018). Interestingly, NAMPT has been found to be a direct substrate of SIRT6 - SIRT6-mediated deacetylation activates NAMPT and boosts intracellular NAD+ level in cancer cells, conferring resistance to oxidative stress (Sociali et al., 2018).
The pathophysiological role of SIRT7 in cancer is relatively poorly understood, due to its low enzymatic activity and few identified substrates (Blank & Grummt, 2017). SIRT7 is known to regulate ribosome biogenesis and RNA metabolism. For example, SIRT7 activates RNA polymerase II transcription by deacetylating cyclin dependent kinase 9 (CDK9), a subunit of the elongation factor P-TEFb, exhibiting oncogenic property (Blank et al., 2017). SIRT7 serves as a prognostic factor in colorectal cancer as it activates Raf-MEK-ERK pathway and induces EMT, promoting cell proliferation and invasion (Yu et al., 2014). However, SIRT7 also shows tumor-suppressive activity, as it inhibits breast cancer metastasis by promoting SMAD4 degradation and antagonizing TGF-β signaling (Tang et al., 2017).
The extensive network of PARP and SIRT proteins and their regulatory targets indicate the profound impact of nuclear NAD+. As PARP and SIRT are NAD+-consuming enzymes, their activity requires the replenishment of nuclear NAD+, mainly through nuclear NAD+ synthesis. It is therefore conceivable to influence PARP- or SIRT-dependent activity by manipulating nuclear NAD+ levels. Indeed, PARP1 activity is tightly regulated by the availability and accessibility of nuclear NAD+. It has recently been shown that under normal conditions, PARP1 has limited NAD+ access that accounts for the basal unstimulated PARP1 activity; while upon DNA damage, PARP1 interaction with DNA strand breaks leads to full NAD+ access to its catalytic site and promotes allosteric activation of PARP1 (Langelier, Zandarashvili, Aguiar, Black, & Pascal, 2018). Additionally, NMNAT1 can be recruited to target gene promotors by PARP1, and generates NAD+ to support PARP1 catalytic activity (Zhang et al., 2012). A recent study shows that NAD+ can directly regulate PARP1 activity by regulating the binding of PARP1 to the protein DBC1 (Deleted in Breast Cancer 1), a potential tumor suppressor with an NAD+ binding pocket. Depletion of NAD+ promotes the binding of PARP1 to DBC1, which inhibits PARP1 activity and leads to DNA damage accumulation (Li et al., 2017). This process may serve as a negative feedback mechanism that prevents excessive depletion of NAD+ by PARP1 upon DNA damage (Li et al., 2017). Likewise, SIRT1 activity can be directly regulated by nuclear NAD+. Increasing cellular NAD+ level by overexpressing NAMPT directly promotes the recruitment of SIRT1 catalytic domain to target gene and enhances transcriptional regulation (Revollo, Grimm, & Imai, S.-i., 2004).
2.3. Cytosolic NAD+ pool
The cytosolic NAD+ pool serves as a central hub that connects and regulates other NAD+ pools (Fig. 1). Cytosolic NAD+ is mainly synthesized by NMNAT2, which has been reported to be localized in the Golgi apparatus (Berger et al., 2005; Mayer et al., 2010). Cytosolic and nuclear NAD+ pools are closely connected as the levels of nuclear NAD+ and cytosolic NAD+ are comparable, and the small molecule NAMPT inhibitor FK866 produces a similar reduction of nuclear NAD+ and cytosolic NAD+ (Cambronne et al., 2016). It is likely that the NAD+ precursors, NAM and NMN, are shared between nucleus and cytosol. The final step, converting NMN to NAD+, is catalyzed locally by either the nuclear or cytosolic NMNAT. Recent studies found that depleting NMNAT2 significantly reduces cytosolic NAD+ but minimally affects nuclear NAD+, whereas nuclear NAD+ cannot completely replenish cytosolic NAD+ when NMNAT2 is depleted (Cambronne et al., 2016). These observations suggest that nuclear and cytosolic NAD+ pools are locally and specifically regulated by NMNAT1 and NMNAT2, respectively, in a compartment-specific manner.
Cytosolic NAD+/NADH together with other metabolic intermediates, including NADP+/NADPH (nicotinamide adenine dinucleotide phosphate) and FAD+/FADH2 (flavin adenine dinucleotide), serve as key metabolic currency that is controlled by various oncogenes and tumor suppressors during metabolic reprogramming of tumors and host immune cells (Pearce, Poffenberger, Chang, & Jones, 2013). Cytosolic NAD+ is pivotal for glycolysis in glucose metabolism, where glyceraldehyde-3-phosphate dehydrogenase converts glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate and NAD+ is reduced to NADH (Fig. 2). Since NADH cannot cross mitochondrial membrane, electrons from NADH can enter the mitochondrial electron transport chain via glycerol-3-phosphate shuttle where NAD+ is regenerated from reducing dihydroxyacetone phosphate to glycerol-3-phosphate, or via malate-aspartate shuttle, where NAD+ is regenerated from reducing oxaloacetate to malate (MacDonald, 1981; MacDonald, 1982). Cytosolic NAD+ is essential for cancer cell growth due to the elevated level of glycolytic flux for increased metabolic demands. However, cancer cells shift their glucose metabolism to aerobic glycolysis (the aforementioned Warburg effect), where they enhance glucose uptake but reduce oxidative phosphorylation, and energy is generated from cytosolic lactate fermentation (Warburg, Wind, & Negelein, 1927). This metabolic alteration provides several benefits. First, NAD+ can be regenerated from NADH during the conversion of pyruvate to lactate by lactate dehydrogenase (LDH) (Fig. 1), thereby maintaining active aerobic glycolysis (Vander Heiden, Cantley, & Thompson, 2009). Second, the Warburg effect minimizes ROS production from oxidative phosphorylation in mitochondria, promoting cell survival (Wang et al., 2019). Third, increased glucose uptake activates pentose phosphate pathway (PPP) and generates glycolytic intermediates, such as nucleic acids, amino acids, and lipids, as metabolic building blocks to meet the anabolic needs of cancer cell proliferation (Liberti & Locasale, 2016) (Fig. 2).
Increased demand of cytosolic NAD+ in glucose metabolism during cancer development indicate cancer cells need to prioritize the replenishment of cytosolic NAD+ pools (Vander Heiden et al., 2009), which can be achieved by balancing the activities of different NMNATs. A recent study shows that NMNAT1 and NMNAT2 compete for NMN, their common substrate, to promote the compartmentalized regulation of nuclear and cytosolic NAD+ pools. Specifically, in undifferentiated adipocytes, NMN is mostly converted by NMNAT1 to maintain nuclear NAD+ levels, required to activate PARP1 and inhibit transcriptional events involved in cell differentiation. During adipogenesis, NMNAT2 supports glucose metabolism by producing cytosolic NAD+ and the reduction of nuclear NAD+ disinhibits transcriptional program, permitting adipogenic differentiation (Ryu et al., 2018). Interestingly, the NMNAT1 gene in cancerous tissue seems to be mostly mutated or deleted (source: www.cbioportal.org) (Cerami et al., 2012). Cancer cells with decreased NMNAT1 expression exhibit low concentrations of nuclear NAD+ and reduced PARylation of multifunctional nuclear protein CCCTC-binding factor, leading to epigenetic silencing of tumor suppressor genes (Henderson, Miranda, & Emerson, 2017). NMNAT1 has been reported to function as a DNA damage response gene and its inhibition delays DNA repair, thus potentially sensitizing cancer cells to DNA damage-inducing stressors; however it also increases rRNA transcription which contributes to cellular transformation, suggesting a context-dependent dual role in cancer (Song et al., 2013). Low levels of NMNAT1 are considered a poor prognostic marker for renal cancer (Human Protein Atlas, www.proteinatlas.org) (Uhlen et al., 2017; Uhlén et al., 2015). By contrast, the NMNAT2 gene appears to be amplified in most cancer types (source: www.cbioportal.org) (Cerami et al., 2012). Specifically, NMNAT2 upregulation in colorectal cancer correlates with the invasive depth and TNM stage (Cui et al., 2016; Qi et al., 2018). The upregulated NMNAT2 enzymatic activity in non-small cell lung cancer (NSCLC) can be triggered by SIRT3-mediated deacetylation (Li et al., 2013) or p53 signaling (Pan et al., 2014). Depletion of NMNAT2 via shRNA in neuroblastoma cells inhibits cell growth, and reducing glucose availability decreases NMNAT2 expression and activates PARP1 (Ryu et al., 2018). These observations support that putative mechanisms involving metabolic regulation of compartmentalized NAD+ synthesis could serve as adaptations during tumorigenesis to support glucose metabolism and fuel rapid cell growth and division.
SIRT2, a major cytosolic NAD+ consuming enzyme, was originally considered a tumor suppressor (Park et al., 2012). SIRT2 deacetylates and inhibits the peroxidase activityofperoxiredoxin-1(Prdx-1), thereby sensitizing breast cancer cells to oxidative stress (Fiskus et al., 2016). Additionally, SIRT2 deacetylates HIF1α in the cytosol, promoting hydroxylation and degradation of HIF1α and suppressing hypoxia-dependent tumorgrowth (Seoetal.,2015). Mutations in SIRT2 lead to genomic instability and promote tumorigenesis (Head et al., 2017; Kim et al., 2011). However, growing evidence demonstrates that SIRT2 promotes cancer growth by stabilizing the Myc family of oncoproteins (Liu et al., 2013) and potentiating Notch signaling (Zhao et al., 2014). Thiomyristoyl, a SIRT2-selective inhibitor, promotes c-Myc oncoprotein ubiquitination and degradation, exhibiting broad anticancer activity with minimal effect on non-cancerous cell lines and tumor-free mice (Jing et al., 2016).
Recent studies highlight the role of SIRT2 as a cytoplasmic nutrient sensor that directs metabolic activity to match the energy need for cancer growth, typically through regulating glycolysis and fatty acid synthesis. In glycolysis, many SIRT2 downstream deacetylation targets have been identified, including aldolase, enolase, LDH, and pyruvate kinase (Park et al., 2016). For example, SIRT2 deacetylates and activates LDH-A to accelerate glycolysis and lactate production, promoting pancreatic cancer growth (Zhao et al., 2013) (Fig. 2). Additionally, SIRT2 regulates PPP by activating glucose-6-phosphate dehydrogenase (G6PD) to boost NADPH production, thereby supporting leukemia cell proliferation (Xu, Wang, Li, & Wang, 2016) (Fig. 2). Moreover, SIRT2 mediates tetramerization of pyruvate kinase and regulates breast cancer progression (Park, Ozden, et al., 2016). In rapidly proliferating cells that undergo fatty acid synthesis, citrate produces from TCA cycle can be exported from mitochondria to cytosol and converted by ATP citrate lyase (ACLY) to cytosolic acetyl coenzyme A (acetyl-CoA). In lung cancer, upregulated acetylation of ACLY increases its protein stability, while SIRT2 deacetylates ACLY and promotes its ubiquitination, thereby inhibiting de novo lipid synthesis and cell proliferation (Lin et al., 2013). Since SIRT2 activity can be pharmacologically regulated by modulation of intracellular NAD+/NADH ratio (Seo et al., 2015), it becomes a promising target to control nutrient sensing and metabolism in cancer cells typically when its level is dysregulated in tumorigenesis.
Cytosolic de novo NAD+ synthesis from tryptophan metabolism (via the kynurenine pathway, KP) (Fig. 1) appears to be important for cells comprising the tumor microenvironment, including innate immunity-associated cells, such as macrophages, which can promote anti-tumor responses and enhance therapeutic outcomes (Moynihan & Irvine, 2017). The tryptophan-metabolizing enzyme indoleamine 2,3 dioxygenase 1 (IDO1) that catalyzes the initial step of KP is frequently overexpressed and activated in cancer and immunoregulatory cells (Prendergast, Malachowski, DuHadaway, & Muller, 2017). Macrophage effector responses have been shown to rely on the KP-produced NAD+ levels as their metabolic switch, and decreases in NAD+ are likely to at least partially underlie innate immune dysfunction (Minhas et al., 2019). Interestingly, KP activation induced by immune challenge paradoxically reduces NAD+ synthesis by inhibiting the conversion of quinolinic acid (QA) to NAD+ (Minhas et al., 2019). KP metabolites promote colorectal cancer (CRC) growth by activating PI3K-Akt signaling (Bishnupuri et al., 2019). Collectively these findings underscore the importance of cytosolic de novo NAD+ synthesis in cancer development and the clinical implications of IDO1 modulators in anti-cancer therapies.
NADPH is in high demand for cancer cell growth as it functions as a key cofactor in multiple metabolic pathways, and it neutralizes ROS burden during rapid cancer cell growth (Patra & Hay, 2014). Under oxidative or hypoxic stress, promoting NADPH production through TIGAR (TP53-induced glycolysis and apoptosis regulator)-mediated diversion of glycolysis into PPP increases cell survival (Bensaad et al., 2006; Ko et al., 2016; Wanka, Steinbach, & Rieger, 2012) (Fig. 2). Recent studies have highlighted the cancer-promoting role of NAD+ kinase (NADK) that converts cytosolic and mitochondrial NAD+ to NADP+, which can be further reduced to NADPH by G6PD in PPP (Tedeschi et al., 2016). NADK has been found to be localized in both the cytosol (cNADK) and mitochondria (mNADK) (Ohashi, Kawai, & Murata, 2012). Activating mutations in cNADK has been reported to drive malignant transformation of pancreatic cells (Tsang et al., 2016). An NADK inhibitor prodrug, thionicotinamide, has shown potent cytotoxic effects to cancer cells by decreasing the cytosolic NADPH pool, and can help to sensitize cancer cells to other chemotherapeutic agents (Tedeschi et al., 2015). NADK activity is also likely to be dampened by conditions that decrease NAD+ concentrations (either by increased consumption or by inhibiting its synthesis), and the reduced level of NADP+ can potentially lead to replication stress, including compromised nucleotide synthesis and accompanying tumoricidal DNA breaks (Chiarugi et al., 2012).
2.4. Extracellular NAD+ pool
Under physiological conditions, the extracellular NAD+ concentration in mammalian serum is reported to be maintained between 0.1 and 0.5 μM (Davies et al., 1999; O’Reilly & Niven, 2003). Extracellular and intracellular NAD+ pools are intricately connected (Fig. 1). First, extracellular NAD+ can be augmented by liberation of intracellular NAD+ upon cell damage or lysis (Adriouch et al., 2007; Haag et al., 2007). Intracellular NAD+ has been observed to be released from neuronal terminals by exocytosis, and functions as a neurotransmitter (Hwang et al., 2011; Mutafova-Yambolieva et al., 2007). A recent study demonstrates that prostate cancer cells release intracellular NAD+ into the culture media, indicating that NAD+ may serve as an autocrine or paracrine signaling molecule for nearby cancerous or noncancerous cells (Mottahedeh et al., 2018). Second, extracellular NAD+ helps to restore intracellular NAD+, as supplementation of extracellular NAD+ significantly elevates intracellular NAD+ level and counteracts cell death induced by the NAMPT inhibitor FK866 (Billington et al., 2008). It has been suggested that the direct exchange of intracellular and extracellular NAD+ pools can be achieved by connexin 43 hemi channels (Bruzzone, Guida, Zocchi, Franco, & De Flora, 2001), although the presence of connexin 43 hemi channels on the plasma membrane under physiological conditions has not been confirmed. On the other hand, pannexin hemi channels are possible candidates that would allow the exchange of NAD+ because of their channel conductance and permeability under physiological conditions (Wang & Dahl, 2018). Alternatively, extracellular NAD+ can be degraded to NMN, which is further metabolized by NR kinases (NRKs) to NR that can be imported as NAD+ precursors, likely through equilibrative nucleoside transporters (ENTs) (Nikiforov et al., 2011; Ratajczak et al., 2016).
Consumption of extracellular NAD+ mainly occurs through CD38, a ubiquitously expressed transmembrane NAD+ glycohydrolase (Chini et al., 2018). CD38 has a highly conserved homolog CD157 that may likely arise from a gene duplication event (Quarona et al., 2013). Induction of CD38 expression lowers NAD+ levels in prostate cancer cells and delays cancer development by arresting cell cycle progression and decreasing cell metabolism (Chmielewski et al., 2018). Increased expression of CD38 during aging may account for the age-related NAD+ decline and mitochondrial dysfunction (Camacho-Pereira et al., 2016). CD38 downregulates NAD+ in multiple ways. First, CD38 converts NAD+ to cADPR, an intracellular Ca2+-mobilizing messenger. Interestingly, CD38 has two opposing orientations, with its carboxyl-terminal catalytic domain facing extracellularly (type III) or intracellularly (type II). Therefore, CD38 is able to mobilize either extracellular or intracellular NAD+ pools, depending on its membrane topological transitions (Zhao, Lam, & Lee, 2012). Notably, a recent study shows that in primary and metastatic prostate cancer cells, CD38 expressions pecifically depletes extracellular NAD+ without altering the level of intracellular NAD+ (Mottahedeh et al., 2018). Second, CD38 also degrades extracellular NMN and generates NAM, which can cross plasma membrane and be converted to NAD+ through NAMPT and NMNAT (Camacho-Pereira et al., 2016).
Another important membrane-bound enzyme that degrades extracellular NAD+ is CD73. CD73 successively degrades NAD+ to NMN, and further to NR (Garavaglia et al., 2012; Grozio et al., 2013) (Fig. 1). The conversion of extracellular NMN to NR is important to support intracellular NAD+ biosynthesis, and the downregulation or pharmacological inhibition of CD73 enhances FK866-induced cytotoxicity to cancer cells (Grozio et al., 2013; Sociali et al., 2016). Notably, CD73 also functions as an adhesion molecule that mediates cancer cell migration and invasion (Sadej & Skladanowski, 2012).
Extracellular NAD+ plays key roles in immune modulation. Inflammation can trigger the release of intracellular NAD+ into extracellular space, which in turn finetunes the immune response (Adriouch et al., 2007). Specifically, extracellular NAD+ acts as a pro-inflammatory cytokine that stimulates granulocytes though activation of P2Y11, augmenting chemotaxis (Moreschi et al., 2006). Additionally, evidences suggest the key regulatory role of extracellular NAD+ in anti-tumor T cell response. First, extracellular NAD+ activates the P2X7 purinergic receptor through ADP-ribosyltransferase 2 (ART2)-mediated ADP-ribosylation, leading to apoptosis of naïve T cells while facilitating the expansion of primed T cells (Adriouch et al., 2007; Haag et al., 2007). Second, Foxp3+ regulatory T cells (Treg) are specifically susceptible to NAD+-induced cell death (NICD), and systemic administration of NAD+ selectively depletes Tregs, promoting anti-tumor responses in mouse tumor xenograft models (Hubert et al., 2010). Third, T cells with reduced surface expression of the CD38 exhibited higher level of NAD+ and superior anti-tumor response, including enhanced oxidative phosphorylation, higher glutaminolysis, and altered mitochondrial dynamics (Chatterjee et al., 2018). Therefore, targeting CD38/NAD+ axis can potentially boost adoptive T cell therapy (ACT) to control tumor growth.
3. Targeting NAD+ metabolism in cancer therapy
Given the essential role of NAD+ in cell growth and other cancer-relevant processes, many enzymes in the NAD+ synthetic and metabolic pathways have been evaluated as targets for anti-cancer therapy. In this section, we discuss the potential therapeutic outcomes of targeting NAD+ metabolism with the consideration of the distinct subcellular pools of NAD+ (Fig. 3).
Fig. 3.

Overview of pharmacological inhibitors targeting NAD+ metabolism. Nicotinamide phosphoribosyltransferase (NAMPT) catalyzes the transfer of the ribose 5-phosphate moiety of phosphoribosyl pyrophosphate (PRPP) to nicotinamide (NAM), producing nicotinamide mononucleotide (NMN) and pyrophosphate (PPi). Inhibitors targeting NAMPT include FK866, GMX1777, and GMX1778. Nicotinamide mononucleotide adenylyltransferase (NMNAT) catalyzes the transfer of the adenylate moiety of adenosine triphosphate (ATP) onto the NMN, producing NAD+ and PPi. An NAD+ analog, Vacor adenine dinucleotide (VAD), has shown potent inhibitory activity against NMNAT. NAD+ can be consumed by sirtuins (SIRTs) that catalyze the transfer of protein-bound acetyl (Ac) group onto the ADP-ribose moiety, producing O-acetyl-ADP ribose (OAcADPR). NAD+ can also be consumed by poly (ADP-ribose) polymerases (PARPs) that catalyze the transfer of poly (ADP-ribose) moiety (pADPr) onto target protein. Moreover, NAD+ can be degraded by CD38 into cyclic ADP-ribose (cADPR) and ADPR. Several inhibitors targeting PARPs (olaparib, rucaparib, niraparib, talazoparib) and CD38 (daratumumab) are FDA-approved for treatment of specific cancers (drugs in green).
3.1. Targeting NAD+ biosynthetic enzymes
3.1.1. NAMPT
Nicotinamide phosphoribosyltransferase (NAMPT) catalyzes the conversion of NAM to NMN, and is a rate limiting enzyme in NAD biosynthesis. NAMPT presents both intracellularly (iNAMPT) and extracellularly (eNAMPT). Intracellular NAMPT is reported to be localized in the nucleus and cytoplasm, while its presence in the mitochondria is controversial (Pittelli et al., 2010; Yang et al., 2007). It has been well-characterized that iNAMPT regulates intracellular NAD+ synthesis, thereby modulating cellular energetics and NAD+-dependent signaling (Garten et al., 2015). In contrast, the functional role of eNAMPT is poorly understood. Extracellular NAMPT not only has enzymatic activity that produces NMN, but also functions as a cytokine and known as pre-B cell colony-enhancing factor (PBEF) or visfatin that modulate immune response and lipid homeostasis (Grolla, Travelli, Genazzani, & Sethi, 2016; Jieyu et al., 2012). A wide range of stress signals, including ER and stress oxygen-glucose deprivation (OGD), promote eNAMPT excretion (Zhao et al., 2014). NAMPT has been recognized as a prognostic marker in cancer, where its expression level correlates with tumor behavior, stage, therapeutic efficacy, and patient survival (Gujar et al., 2016; Lucena-Cacace, Otero-Albiol, Jiménez-García, Muñoz-Galvan, & Carnero, 2018; Ohanna et al., 2018; Sharif et al., 2016). NAMPT signaling is also important for the tumor-promoting, pro-angiogenic function of tumor-associated neutrophils (TAN) (Pylaeva et al., 2019). Inhibition of NAMPT inhibits SIRT1 signaling and prevents transcription of pro-angiogenic genes in TAN (Pylaeva et al., 2019), potentially pointing to a targeting node in the tumor microenvironment. Additionally, a recent study indicates that NAMPT expression can be upregulated by the high mobility group A (HMGA) proteins, and that the enhanced HMGA/NAMPT/NAD+ axis suppresses p53-mediated inhibition of p38 MAPK, activating NF-κB signaling and promoting the pro-tumorigenic inflammatory senescence-associated secretory phenotype (SASP) (Nacarelli et al., 2019). Thus, targeting NAMPT may produce not only cell-autonomous tumor-inhibitory responses by reducing NAD+ availability in cancer cells but also inhibit the tumor-promoting paracrine signaling associated with the SASP. For the specific pathological roles of iNAMPT and eNAMPT in cancer, we refer readers to recent reviews with detailed discussions (Carbone et al., 2017; Dalamaga, Christodoulatos, & Mantzoros, 2018).
As NAMPT catalyzes the rate-limiting step in the synthesis of NAD+, inhibitors of NAMPT have been examined as anti-cancer agents. Three potent NAMPT inhibitors, FK866 (APO866/WK175), GMX1777, and GMX1778 (CHS-8280), have completed Phase I trials. By depletion of primarily cytosolic and nuclear NAD+, FK866 significantly attenuates glycolysis at the glyceraldehyde-3-phosphate dehydrogenase step (Tan et al., 2013), increases ROS production, induces apoptosis (Gehrke et al., 2014; Thakur et al., 2013), and inhibits the tumorigenicity of cancer stem cells (Gujar et al., 2016). FK866 delays tumor growth and increases radiation sensitivity in a mouse model of breast cancer through decreased NAD+ levels, pH and bioenergetics status (Muruganandham et al., 2005). FK866 combined with the protease inhibitor bortezomib synergistically depletes intracellular NAD+ to undetectable level in multiple myeloma cells and overcomes bortezomib resistance in anti-myeloma therapy (Cagnetta et al., 2013). GMX1777 is a prodrug of GMX1778, and has shown synergistic therapeutic benefits with other anti-cancer therapies in animal models, such as Pemetrexed in NSCLC (Chan et al., 2014) and radiation therapy in head and neck carcinoma (Kato et al., 2010).
Given the key role of NAMPT in NAD+ synthesis, inhibiting NAMPT is a double-edged sword, damaging both cancer and non-cancer cells that require NAD+. The impact on non-cancer cells results in unwanted on-target toxicity and indeed, all these NAMPT inhibitors have currently been discontinued for clinical evaluation due to systemic side effects, including thrombocytopenia, lymphopenia, cardiotoxicity, retinal toxicity, and gastrointestinal symptoms (Sampath, Zabka, Misner, O’Brien, & Dragovich, 2015). Despite these side effects, the potential of NAMPT as an anti-cancer target has yet to be fully realized, and several approaches have been considered to improve therapeutic outcome. First, several new NAMPT inhibitors have been developed, such as KPT-9274, LSN3154567, and OT-82, with broad spectrum anticancer activity as well as decreased toxicity to normal tissues (Aboud et al., 2016; Korotchkina et al., 2017; Zhao et al., 2017). Second, NAMPT inhibitors can be highly selective against certain cancer types. For example, FK866 exhibits selective toxicity towards gastric cancer cells marked by nicotinic acid phosphoribosyltransferase (NAPRT) deficiency and the EMT (Lee et al., 2018; Shames et al., 2013). Cancers carrying isocitrate dehydrogenase 1 (IDH1) mutations exhibit reduced level of NAPRT and exhibit enhanced sensitivity to NAD+ depletion by NAMPT inhibitors (Tateishi et al., 2015). Moreover, NSCLC with nucleotide excision repair (NER) deficiency shows exquisite sensitivity to NAMPT inhibition (Touat et al., 2018). Therefore, identification of predictive biomarkers may greatly help to improve therapeutic efficacy of these second-generation NAMPT inhibitors.
Combinatorial targeting may have particular benefit when targeting NAMPT, especially because cancer cells can develop resistance to NAMPT inhibitors through enhancing NAD+ de novo synthesis through upregulated quinolinic acid phosphoribosyltransferase (QAPRT), NAMPT mutations (Guo et al., 2017), or enhanced amino acid catabolism (such as tryptophan in de novo pathway and glutamine in salvage pathway) (Thongon et al., 2018). Moreover, therapeutic resistance has been attributed to the self-renewal and dedifferentiation abilities of tumor-initiating “cancer stem cells” (CSCs) residing within the bulk tumor (Gupta, Chaffer, & Weinberg, 2009). NAMPT overexpression in glioma cell lines has been shown to enrich the CD133-high/CD44-high population that marks glioma stem cells, and its expression correlates with high levels of stem cell markers such as Nanog in patient-derived glioblastoma specimen (Lucena-Cacace, Otero-Albiol, Jiménez-García, Peinado-Serrano, & Carnero, 2017), pointing to its role in maintaining the treatment-refractory component of tumors. The persistence of the CSC population under therapeutic challenge has been attributed to metabolic and oxidative stress resistance due to their metabolic plasticity; however, such CSCs have also been reported to be acutely sensitive to mitochondrial metabolism-targeting drugs (Peiris-Pages, Martinez-Outschoorn, Pestell, Sotgia, & Lisanti, 2016). Thus, mitochondrialspecific targeting of tumor-associated NAMPT activity could circumvent the observed systemic toxicity of the first-generation inhibitors and provide enhanced therapeutic index. Notably, the dedifferentiation ability and damage recovery of cancer cells that have undergone therapy-relevant stresses (oxidative stress, glucose deprivation, hypoxia) depends critically on the labile, mitochondrially generated signaling molecule, hydrogen sulfide (H2S) and its ability to upregulate NAMPT (Ostrakhovitch, Akakura, Sanokawa-Akakura, Goodwin, & Tabibzadeh, 2015; Sanokawa-Akakura, Ostrakhovitch, Akakura, Goodwin, & Tabibzadeh, 2014). The H2S-induced dedifferentiation of the stress-challenged cancer cells can be inhibited by NAMPT suppression or by inactivation of the H2S-producing enzymes, cystathionine beta synthase and cystathionase, which are also upregulated by NAMPT (Ostrakhovitch et al., 2015). The existence of such feedback regulation between NAMPT and other tumor-promoting signaling axes needs to be further investigated as they provide uniquely tumor-specific opportunities for combinatorial therapeutic targeting of NAMPT. Due to observed synergistic effects with FK866 and histone deacetylase complex (HDAC) inhibitors, recently dual NAMPT/HDAC inhibitors have been developed which possess nanomolar range IC50s (half maximal inhibitory concentration) for both molecules, and exhibit apoptosis and autophagy in cancer cell lines as well as tumoricidal activity in xenograft tumor models in vivo (Chen et al., 2018; Dong et al., 2017). This type of combinatorial targeting exploits cancer-specific requirements for epigenome remodeling to transcribe tumor-driving genes and the essential role of NAMPT and NAD+ metabolism in such processes. Combining FK866 with the clinically relevant PARP inhibitor, Olaparib, results in synthetic lethality through suppression of the PARP substrate β-NAD+ in triple negative breast cancer cell lines as well as in vivo models, with negligible systemic effects of the combinatorial treatment (Bajrami et al., 2012). Thus, the true utility of NAMPT as a cancer target may lie in judicious synergistic combinations that will enable more effective co-targeting of known cancer-driving pathways through leveraging NAD+-dependent biology underlying these pathways. The added advantage of such combinatorial approaches is that their tumor specificity may serve to abrogate issues of systemic toxicity associated with single agent NAMPT targeting, through reduced dose requirements or better therapeutic index in tumors vs. normal tissue.
3.1.2. NAPRT
NAPRT converts nicotinic acid (NA) to nicotinic acid mononucleotide (NAMN) and serves as an alternative source for NAD+ production. Similar to NAMPT, NAPRT is located in both nuclear and cytoplasm, but has not been detected in mitochondria (Nikiforov et al., 2011; Piacente et al., 2017). NAPRT gene amplification is present in various cancer types, including ovarian, pancreatic, and prostate cancer, and commonly confers resistance to NAMPT inhibitor treatment (Piacente et al., 2017). Notably, the efficacy of NAPRT inhibitors largely depends on the NAD+ metabolic signature of the cancer. NAPRT inhibitor 2-hydroxinicotinic acid (2-HNA) sensitizes ovarian cancer cells to FK866, producing a significant decrease of cell viability (Piacente et al., 2017). By contrast, triple-negative breast cancer cells are not sensitive to 2-HNA, as they have low NAPRT-mediated NAD+ biosynthesis activity and instead primarily rely on LDH in aerobic glycolysis for the resistance to FK866 (Thongon et al., 2018).
3.1.3. NMNAT
All three human NMNAT isoforms (NMNAT1–3) have enzymatic activities and convert the last step of NAD+ synthesis from NMN (Berger et al., 2005). The aberrant regulation and critical role of NMNATs in tumor metabolism are indicative of their potential in cancer therapy. However, only a few compounds targeting NMNAT have been identified and characterized. Gallotannin, a polyphenolic plant metabolite, inhibits enzymatic activity of all three NMNAT isoforms and is highly potent against NMNAT1 (Berger et al., 2005). Several NAD+ synthetic analogs also shown selective inhibition against the different NMNAT isoforms (Petrelli, Felczak, & Cappellacci, 2011). A recent study identified Vacor adenine dinucleotide (VAD), an NAD+ analog converted from Vacor by NAMPT and NMNAT2, as a potent inhibitor of NMNAT enzymatic activity (Buonvicino et al., 2018). VAD rapidly depletes NAD+ pools, blocks glycolysis through inhibition of NAD+-dependent dehydrogenases, and significantly reduces the growth of NMNAT2-proficient neuroblastoma and melanoma xenografts (Buonvicino et al., 2018). A phenotypic screen using the Meso Scale Discovery (MSD)-based platform has identified several positive and negative regulators of NMNAT2 (Ali, Bradley, & Lu, 2017). Of note, NMNAT not only functions as an enzyme, but also acts as a chaperone (Ruan, Zhu, Li, Brazill, & Zhai, 2015; Zhai et al., 2008), which could contribute to meeting the increased demand in maintaining proteostasis in rapidly adapting cancer cells (Calderwood & Murshid, 2017). Chaperone inhibitors have shown great promise both preclinically and clinically in cancer therapy (Chatterjee & Burns, 2017). Therefore, depleting NMNAT2 may produce enhanced benefits by both inhibiting NAD+-dependent energy metabolism and signaling and disrupting protein homeostasis, thus simultaneously targeting two tumor-promoting mechanisms.
3.2. Targeting NAD+ consuming enzymes
3.2.1. SIRT
The involvement of different SIRT isoforms highlight the complex relationship between epigenetic regulation, transcription, DNA repair, cell cycle regulation, and energy metabolism in cancer cells. Increasing evidence supports the therapeutic potential of SIRT modulators in the treatment of cancer. Several recent reviews have provided comprehensive discussions on SIRT-targeting compounds, including their chemical structures, selectivity in different SIRT isoforms, and biological effects (Carafa et al., 2016; Hu, Jing, & Lin, 2014; O’Callaghan & Vassilopoulos, 2017). More recently, several novel SIRT inhibitors have been identified that exhibit potent anticancer activities. For example, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and its derivatives, J11-C1 and J19, markedly inhibit SIRT1 activity and induce cell death in ovarian cancer cells (Tae et al., 2018). A quinoxaline-based compound 4bb selectively reduces colon cancer cell viability without affecting normal fibroblasts (Ghosh et al., 2017). Moreover, MHY2256, a potent SIRT1 inhibitor, significantly inhibits growth of endometrial and breast cancer cells (De et al., 2018; Park et al., 2016). Recently, a new SIRT7 inhibitor (ID: 97491) has been identified that shows potent inhibitory effect on cancer growth by upregulating apoptosis (Kim et al., 2019). All these compounds work partially via promoting acetylation and activation of p53, thereby driving cell cycle arrest and inducing apoptotic or autophagic cell death (De et al., 2018; Ghosh et al., 2017; Kim et al., 2019; Tae et al., 2018).
3.2.2. PARP
PARPs play pivotal roles in NAD+-dependent DNA damage repair and transcriptional regulation, both of which are highly essential for cancer development. Therefore, PARP inhibitors have been widely studied in the context of their anticancer action (Dulaney, Marcrom, Stanley, & Yang, 2017; Sisay & Edessa, 2017). Four compounds, olaparib (Audeh et al., 2010; Fong et al., 2009; Ledermann et al., 2012; Robson et al., 2017; Tutt et al., 2010), rucaparib (Coleman et al., 2017; Swisher et al., 2017), niraparib (Mirza et al., 2016; Sandhu et al., 2013), and talazoparib (de Bono et al., 2017; Litton et al., 2018) have been approved by the Food and Drug Administration (FDA) for treatment of gynecologic cancers (Fig. 3). Several other PARP inhibitors are under clinical trials for evaluation of efficacy and toxicity as monotherapies or in combination with radiation and other chemotherapeutics. PARP inhibitors are particularly effective in inducing synthetic lethality in BRCA1-and/or BRCA2-mutated cancers that are defective in homologous recombination repair (HRR) (Lord & Ashworth, 2017). The consequences of PARP inhibition are two-fold. First, PARP inhibition promotes progression of single-strand breaks to double-strand breaks, subsequently leading to cell death (Dziadkowiec, Gasiorowska, Nowak-Markwitz, & Jankowska, 2016). Second, PARP inhibition leads to deregulation of error-prone non-homologous end joining (NHEJ) which serves to introduce genomic instability (Patel, Sarkaria, & Kaufmann, 2011). Further mechanistic studies have shown that PARP inhibition reduces the availability of HRR factors by suppressing E2F1-mediated HR gene expression (Schiewer et al., 2018). Notably, PARP inhibitors can induce reversion mutations in HRR genes that restore HRR function and promote drug resistance (Edwards et al., 2008; Lin et al., 2019).
3.2.3. CD38
CD38 is a major NAD+ consumer, and its NAD+-dependent activity is required for cell growth. Recent studies indicated the role of CD38 in tumorigenesis. For example, CD38 promotes cervical cancer cell growth through reducing ROS levels and inhibiting apoptosis (Liao et al., 2017), and loss of CD38 in human lung adenocarcinoma cells inhibited cell growth, invasion, and xenograft growth in nude mice (Bu et al., 2017). CD38 also augments immunosuppression in cancer by inhibiting CD8+ T function (Chen et al., 2018; Feng et al., 2017). CD38 has been identified as a cell-surface marker for multiple myeloma (Chini et al., 2018), and CD38-targeting antibodies are specifically effective in relapsed/refractory multiple myeloma (RRMM) patients (Frerichs et al., 2018; van de Donk, Richardson, & Malavasi, 2018). Daratumumab is the first FDA-approved CD38-targeting drug with single-agent efficacy for RRMM (Dimopoulos et al., 2016; Lokhorst et al., 2015; Lonial et al., 2016; Mateos et al., 2018; Palumbo et al., 2016; Usmani et al., 2016). Other anti-CD38 antibodies, including isatuximab and MOR202, are currently in clinical trials, and preliminary data exhibit excellent infusion tolerability (Martin et al., 2017; Raab et al., 2016). CD38-targeting antibodies inhibit cancer development through multiple effector mechanisms. First, these antibodies possess classic Fc-dependent immune effector mechanisms for cancer cell lysis and removal, typically antibody-dependent cellular cytotoxicity (ADCC) (Frerichs et al., 2018; van de Donk et al., 2018). The effectiveness of the isatuximab-mediated ADCC correlates with cell-surface CD38 levels (Kriegsmann et al., 2018). Notably, since CD38 is highly expressed on multiple myeloma cells, with relatively low expression on normal myeloid or lymphoid cells (Chini et al., 2018; Krejcik et al., 2016), multiple myeloma cells are specifically susceptible to CD38 depletion. Second, targeting CD38 promotes T-cell expansion, enhances effector T cell function, and mitigates CD38+ Treg-mediated immunosuppression (Feng et al., 2017; Krejcik et al., 2016). Third, inhibition of CD38 expression on T cells upregulates NAD+ and activates T cells via promoting glutaminolysis, enhancing oxidative phosphorylation, and altering mitochondrial dynamics, thereby boosting the efficacy of adoptive T cell therapy (Chatterjee et al., 2018). Interestingly, a recent study demonstrates that cancer cells acquire resistance to PD-1/PD-L1 immune checkpoint inhibitors via upregulation of CD38 that blocks CD8+ T cell function through adenosine receptor signaling (Chen, Diao, et al., 2018), indicating potential benefits to employing CD38 inhibitors and PD-1/PD-L1 inhibitors in combination as anti-cancer therapy.
4. Concluding remarks
Decades of research has uncovered the key regulatory role of NAD+ in integrating multiple metabolic pathways underlying cancer pathophysiology. In particular, NAD+ functions at the nexus of a crucial signaling hub that interlinks DNA repair, gene transcription, and bioenergetics, processes that govern critical steps in tumorigenesis, including cell cycle regulation, apoptosis, autophagy, redox homeostasis, and immune response. Notably, recent advances on our understanding of the subcellular distribution of NAD+ shed light on how different NAD+ pools are dynamically maintained, how they preferentially influence NAD+-dependent bioenergetic and signaling processes, and how they engage in crosstalk with critical tumor-driving pathways. The interplay of these factors in cancer cells as well as the tumor microenvironment drive cancer development and metastasis. Identification of druggable targets in the NAD+ metabolome shows promise for cancer treatment, as tumors have altered metabolic requirements that are heavily reliant on NAD+ for survival and growth. Further in-depth mechanistic studies with the consideration of subcellular compartment-specific NAD+ metabolic profile of different cancer types will bear promise to achieve maximal drug efficacy and minimize unwanted side effects.
Acknowledgments
This research was supported by Lois Pope LIFE Foundation Fellows Program (Y.Z.), National Institutes of Health (NIH) funding (R01CA175086 to P.R.) and Taishan Scholar Project of Shandong Province (R.G.Z.).
Abbreviations:
- ADP
Adenosine diphosphate
- ATP
Adenosine triphosphate
- cADPR
Cyclic ADP-ribose
- CD38
Cluster of differentiation 38
- CSC
Cancer stem cell
- EMT
Epithelial-tomesenchymal transition
- ER
Endoplasmic reticulum
- FDA
Food and Drug Administration
- LDH
Lactate dehydrogenase
- MSC
Mesenchymal stem cells
- NA
Nicotinic acid
- NAD+
Nicotinamide adenine dinucleotide (oxidized)
- NADH
Nicotinamide adenine dinucleotide (reduced)
- NADP+
Nicotinamide adenine dinucleotide phosphate (oxidized)
- NADPH
Nicotinamide adenine dinucleotide phosphate (reduced)
- NAM
Nicotinamide
- NAMPT
Nicotinamide phosphoribosyltransferase
- NAPRT
Nicotinic acid phosphoribosyltransferase
- NAR
Nicotinic acid riboside
- NMN
Nicotinamide mononucleotide
- NMNAT
Nicotinamide mononucleotide adenylyltransferase
- NR
Nicotinamide riboside
- NSCLC
Non-small cell lung cancer
- HRR
Homologous recombination repair
- PARP
Poly (ADP-ribose) polymerase
- PPP
Pentose phosphate pathway
- ROS
Reactive oxygen species
- SIRT
Sirtuin
- TCA
Tricarboxylic acid
- Treg
Regulatory T cells
- VAD
Vacor adenine dinucleotide
Footnotes
Conflicts of interest statement
The authors declare that there are no conflicts of interest.
References
- Aboud OA, Chen CH, Senapedis W, Baloglu E, Argueta C, & Weiss RH (2016. September 1). Dual and specific inhibition of NAMPT and PAK4 by KPT-9274 decreases kidney cancer growth. Molecular Cancer Therapeutics 15(9), 2119–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriouch S, Hubert S, Pechberty S, Koch-Nolte F, Haag F, & Seman M (2007). NAD+ released during inflammation participates in T cell homeostasis by inducing ART2-mediated death of naive T cells in vivo. The Journal of Immunology 179, 186–194. [DOI] [PubMed] [Google Scholar]
- Ahn B-H, Kim H-S, Song S, Lee IH, Liu J, Vassilopoulos A, … Finkel T (2008). A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proceedings of the National Academy of Sciences 105, 14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alano CC, Tran A, Tao R, Ying W, Karliner JS, & Swanson RA (2007). Differences among cell types in NAD+ compartmentalization: A comparison of neurons, astrocytes, and cardiac myocytes. Journal of Neuroscience Research 85, 3378–3385. [DOI] [PubMed] [Google Scholar]
- Ali YO, Bradley G, & Lu H-C (2017). Screening with an NMNAT2-MSD platform identifies small molecules that modulate NMNAT2 levels in cortical neurons. Scientific Reports 7, 43846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JL, Thompson JW, Lindblom KR, Johnson ES, Yang C-S, Lilley LR, … Kornbluth S (2011). A biotin switch-based proteomics approach identifies 14-3-3ζ as a target of Sirt1 in the metabolic regulation of caspase-2. Molecular Cell 43, 834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, … Loman N (2010). Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. The Lancet 376, 245–251. [DOI] [PubMed] [Google Scholar]
- Bae JS, Noh SJ, Kim KM, Park S-H, Hussein UK, Park HS, … Chung MJ (2018). SIRT6 is involved in the progression of ovarian carcinomas via β-catenin-mediated epithelial to mesenchymal transition. Frontiers in Oncology 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P (2015). Biology of poly (ADP-ribose) polymerases: The factotums of cell maintenance. Molecular Cell 58, 947–958. [DOI] [PubMed] [Google Scholar]
- Bajrami I, Kigozi A, Van Weverwijk A, Brough R, Frankum J, Lord CJ, & Ashworth A (2012). Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Molecular Medicine 4, 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barile M, Passarella S, Danese G, & Quagliariello E (1996). Rat liver mitochondria can synthesize nicotinamide adenine dinucleotide from nicotinamide mononucleotide and ATP via a putative matrix nicotinamide mononucleotide adenylyltransferase. Biochemistry and Molecular Biology International 38, 297–306. [PubMed] [Google Scholar]
- Bell E, Emerling B, Ricoult S, & Guarente L (2011). SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 30, 2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, … Vousden KH (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120. [DOI] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, & Ziegler M (2005). Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. Journal of Biological Chemistry 280, 36334–36341. [DOI] [PubMed] [Google Scholar]
- Bhardwaj A, & Das S (2016). SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proceedings of the National Academy of Sciences 113, E538–E547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billington RA, Travelli C, Ercolano E, Galli U, Roman CB, Grolla AA, … Genazzani AA (2008). Characterization of NAD uptake in mammalian cells. Journal of Biological Chemistry 283, 6367–6374. [DOI] [PubMed] [Google Scholar]
- Bishnupuri KS, Alvarado DM, Khouri AN, Shabsovich M, Chen B, Dieckgraefe BK, & Ciorba MA (2019. March 15). IDO1 and kynurenine pathway metabolites activate PI3K-Akt signaling in the neoplastic colon epithelium to promote cancer cell proliferation and inhibit apoptosis. Cancer Research 79(6), 1138–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank MF, Chen S, Poetz F, Schnölzer M, Voit R, & Grummt I (2017). SIRT7-dependent deacetylation of CDK9 activates RNA polymerase II transcription. Nucleic Acids Research 45, 2675–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank MF, & Grummt I (2017). The seven faces of SIRT7. Transcription 8, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, … Smith DC (2017). Phase I, dose-escalation, two-part trial of the PARP inhibitor Talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discovery 7, 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazill JM, Li C, Zhu Y, & Zhai RG (2017). NMNAT: It’s an NAD(+) synthase… It’s a chaperone… It’s a neuroprotector. Current Opinion in Genetics & Development 44, 156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringman-Rodenbarger LR, Guo AH, Lyssiotis CA, & Lombard DB (2018). Emerging roles for SIRT5 in metabolism and cancer. Antioxidants & Redox Signaling 28, 677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JS, O’Carrigan B, Jackson SP, & Yap TA (2017). Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discovery 7, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone S, Guida L, Zocchi E, Franco L, & De Flora A (2001). Connexin 43 hemi channels mediate Ca2+−regulated transmembrane NAD+ fluxes in intact cells. The FASEB Journal 15, 10–12. [DOI] [PubMed] [Google Scholar]
- Bu X, Kato J, Hong JA, Merino MJ, Schrump DS, Lund FE, & Moss J (2017). CD38 knockout suppresses tumorigenesis in mice and clonogenic growth of human lung cancer cells. Carcinogenesis 39, 242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonvicino D, Mazzola F, Zamporlini F, Resta F, Ranieri G, Camaioni E, … Dölle C (2018). Identification of the nicotinamide salvage pathway as a new Toxification route for antimetabolites. Cell Chemical Biology 25, 471–482 (e477). [DOI] [PubMed] [Google Scholar]
- Busso N, Karababa M, Nobile M, Rolaz A, Van Gool F, Galli M, … De Smedt T (2008). Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PLoS One 3, e2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byles V, Chmilewski LK, Wang J, Zhu L, Forman LW, Faller DV, & Dai Y (2010). Aberrant cytoplasm localization and protein stability of SIRT1 is regulated by PI3K/IGF-1R signaling in human cancer cells. International Journal of Biological Sciences 6, 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnetta A, Cea M, Calimeri T, Acharya C, Fulciniti M, Tai YT, & Nencioni A (2013. August 15). Intracellular NAD+ depletion enhances bortezomib-induced anti-myeloma activity. Blood 122(7), 1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood SK, & Murshid A (2017). Molecular chaperone accumulation in cancer and decrease in Alzheimer’s disease: The potential roles of HSF1. Frontiers in Neuroscience 11, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho-Pereira J, Tarragó MG, Chini CC, Nin V, Escande C, Warner GM, … Galina A (2016). CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metabolism 23, 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, … Goodman RH (2016). Biosensor reveals multiple sources for mitochondrial NAD+. Science 352, 1474–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, … Cettour-Rose P (2012). The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metabolism 15, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, … Altucci L (2016). Sirtuin functions and modulation: From chemistry to the clinic. Clinical Epigenetics 8, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone F, Liberale L, Bonaventura A, Vecchie A, Casula M, Cea M, … Montecucco F (2017). Regulation and function of extracellular nicotinamide phosphoribosyltransferase/Visfatin. Comprehensive Physiology 7, 603–621. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, … Larsson E (2012). The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data (AACR). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, & Guarente L (2015). The multifaceted functions of sirtuins in cancer. Nature Reviews Cancer 15, 608. [DOI] [PubMed] [Google Scholar]
- Chan M, Gravel M, Bramoullé A, Bridon G, Avizonis D, Shore GC, & Roulston A (2014. November 1). Synergy between the NAMPT inhibitor GMX1777 (8) and pemetrexed in non–small cell lung cancer cells is mediated by PARP activation and enhanced NAD consumption. Cancer Research 74(21), 5948–5954. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, & Burns T (2017). Targeting heat shock proteins in cancer: A promising therapeutic approach. International Journal of Molecular Sciences 18, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, … Kang I (2018). CD38-NAD+ axis regulates immunotherapeutic anti-tumor T cell response. Cell Metabolism 27, 85–100 (e108). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri AR, & Nussenzweig A (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nature Reviews Molecular Cell Biology 18, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, … Tan L (2018). CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discovery 8, 1156–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Dong G, Wu Y, Zhang W, Miao C, & Sheng C (2018). Dual NAMPT/HDAC inhibitors as a new strategy for multitargeting antitumor drug discovery. ACS Medicinal Chemistry Letters 9, 34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XF, Tian MX, Sun RQ, Zhang ML, Zhou LS, Jin L, … Gao C (2018. May 1). SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Reports 19(5), e45124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Ren X, Gowda AS, Shan Y, Zhang L, Yuan Y, … Yang J (2013). Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death & Disease 4, e731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A, Dölle C, Felici R, & Ziegler M (2012). The NAD metabolome—A key determinant of cancer cell biology. Nature Reviews Cancer 12, 741. [DOI] [PubMed] [Google Scholar]
- Chini EN (2009). CD38 as a regulator of cellular NAD: A novel potential pharmacological target for metabolic conditions. Current Pharmaceutical Design 15, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini EN, Chini CC, Netto JME, de Oliveira GC, & van Schooten W (2018. April 1). The pharmacology of CD38/NADase: An emerging target in cancer and diseases of aging. Trends in Pharmacological Sciences 39(4), 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielewski JP, Bowlby SC, Wheeler FB, Shi L, Sui G, Davis AL, … Sirintrapun SJ (2018). CD38 inhibits prostate cancer metabolism and proliferation by reducing cellular NAD+ pools. Molecular Cancer Research 16, 1687–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, … Scambia G (2017). Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial Lancet (London, England: ) 390, 1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbet C, Pinto A, Martherus R, de Jesus JPS, Polet F, & Feron O (2016). Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metabolism 24, 311–323. [DOI] [PubMed] [Google Scholar]
- Costa-Machado LF, Martín-Hernández R, Sanchez-Luengo MÁ, Hess K, Vales-Villamarin C, Barradas M, … de Cabo R (2018). Sirt1 protects from K-Ras-driven lung carcinogenesis. EMBO Reports 19, e43879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csibi A, Fendt S-M, Li C, Poulogiannis G, Choo AY, Chapski DJ, … Morrison T (2013). The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153, 840–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Qi J, Deng Q, Chen R, Zhai D, & Yu J. J. B. r. i. (2016). Nicotinamide mononucleotide adenylyl transferase 2: a promising diagnostic and therapeutic target for colorectal cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalamaga M, Christodoulatos GS, & Mantzoros CS (2018. May 1). The role of extracellular and intracellular Nicotinamide phosphoribosyl-transferase in cancer: Diagnostic and therapeutic perspectives and challenges. Metabolism 82, 72–87. [DOI] [PubMed] [Google Scholar]
- Davies CA, Perrett D, Zhang Z, Nielsen BR, Blake DR, & Winyard PG (1999). Simultaneous analysis of nitrite, nitrate and the nicotinamide nucleotides by capillary electrophoresis: Application to biochemical studies and human extracellular fluids. Electrophoresis: An International Journal 20, 2111–2117. [DOI] [PubMed] [Google Scholar]
- Davila A, Liu L, Chellappa K, Redpath P, Nakamaru-Ogiso E, Paolella LM, … Baur JA (2018). Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. Elife 7, e33246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Donk N, Richardson PG, & Malavasi F (2018). CD38 antibodies in multiple myeloma: Back to the future. Blood 131, 13–29. [DOI] [PubMed] [Google Scholar]
- De U, Son J, Sachan R, Park Y, Kang D, Yoon K, … Kim H (2018). A new synthetic histone deacetylase inhibitor, MHY2256, induces apoptosis and autophagy cell death in endometrial cancer cells via p53 acetylation. International Journal of Molecular Sciences 19, 2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarest TG, Babbar M, Okur MN, Dan X, Croteau DL, Fakouri NB, … Bohr VA (2019). NAD+ metabolism in aging and cancer. Annual Review of Cancer Biology 3 (null). [Google Scholar]
- Desantis V, Lamanuzzi A, & Vacca A (2018). The role of SIRT6 in tumors. Haematologica 103, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, … Suzuki K (2016). Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. The New England Journal of Medicine 375, 1319–1331. [DOI] [PubMed] [Google Scholar]
- Dölle C, Niere M, Lohndal E, & Ziegler M (2010). Visualization of subcellular NAD pools and intra-organellar protein localization by poly-ADP-ribose formation. Cellular and Molecular Life Sciences 67, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G, Chen W, Wang X, Yang X, Xu T, Wang P, … Sheng C (2017). Small molecule inhibitors simultaneously targeting cancer metabolism and epigenetics: Discovery of novel nicotinamide phosphoribosyltransferase (NAMPT) and histone deacetylase (HDAC) dual inhibitors. Journal of Medicinal Chemistry 60, 7965–7983. [DOI] [PubMed] [Google Scholar]
- Dörsam B, Seiwert N, Foersch S, Stroh S, Nagel G, Begaliew D, … Minneker V (2018). PARP-1 protects against colorectal tumor induction, but promotes inflammation-driven colorectal tumor progression. Proceedings of the National Academy of Sciences 115, E4061–E4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, … Choi BH (2011). Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Hu H, Qu S, Wang J, Hua C, Zhang J, … Liu P (2018). SIRT5 deacylates metabolism-related proteins and attenuates hepatic steatosis in ob/ob mice. EBioMedicine 36, 347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Liu X, Chen T, Gao W, Wu Z, Hu Z, … Li Q (2018). Targeting a Sirt5-positive subpopulation overcomes multidrug resistance in wild-type kras colorectal carcinomas. Cell Reports 22, 2677–2689. [DOI] [PubMed] [Google Scholar]
- Dulaney C, Marcrom S, Stanley J, & Yang ES (2017). Poly (ADP-ribose) polymerase activity and inhibition in cancer Paper presented at: Seminars in cell & developmental biology. Elsevier. [DOI] [PubMed] [Google Scholar]
- Dvorak A, Zelenka J, Smolkova K, Vitek L, & JeZek P (2017). Background levels of neomorphic 2-hydroxyglutarate facilitate proliferation of primary fibroblasts. Physiological Research 66, 293–304. [DOI] [PubMed] [Google Scholar]
- Dziadkowiec KN, Gasiorowska E, Nowak-Markwitz E, & Jankowska A (2016). PARP inhibitors: Review of mechanisms of action and BRCA1/2 mutation targeting. Przeglad Menopauzalny = Menopause Review 15, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, … Ashworth A (2008). Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451, 1111. [DOI] [PubMed] [Google Scholar]
- Falk MJ, Zhang Q, Nakamaru-Ogiso E, Kannabiran C, Fonseca-Kelly Z, Chakarova C, … Borman AD (2012). NMNAT1 mutations cause Leber congenital amaurosis. Nature Genetics 44, 1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felici R, Lapucci A, Ramazzotti M, & Chiarugi A (2013). Insight into molecular and functional properties of NMNAT3 reveals new hints of NAD homeostasis within human mitochondria. PLoS One 8, e76938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, … Anderson KC (2017. August 1). Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clinical Cancer Research 23(15), 4290–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer CM, Lu TY, Bacigalupa ZA, Katsetos CD, Sinclair DA, & Reginato MJ (2017). O-GlcNAcylation regulates breast cancer metastasis via SIRT1 modulation of FOXM1 pathway. Oncogene 36, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, … Clish CB (2011). SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 19, 416–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskus W, Coothankandaswamy V, Chen J, Ma H, Ha K, Saenz DT, … Huang P (2016). SIRT2 deacetylates and inhibits the peroxidase activity of peroxiredoxin-1 to sensitize breast cancer cells to oxidant stress inducing agents. Cancer Research, Canres 0126(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjeld CC, Birdsong WT, & Goodman RH (2003). Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proceedings of the National Academy of Sciences 100, 9202–9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, … O’Connor MJ (2009). Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England Journal of Medicine 361, 123–134. [DOI] [PubMed] [Google Scholar]
- Frerichs KA, Nagy NA, Lindenbergh PL, Bosman P, Marin Soto J, Broekmans M, … Stege C (2018). CD38-targeting antibodies in multiple myeloma: Mechanisms of action and clinical experience. Expert Review of Clinical Immunology 14, 197–206. [DOI] [PubMed] [Google Scholar]
- Garavaglia S, Bruzzone S, Cassani C, Canella L, Allegrone G, Sturla L, … Rizzi M (2012). The high-resolution crystal structure of periplasmic Haemophilus influenzae NAD nucleotidase reveals a novel enzymatic function of human CD73 related to NAD metabolism. Biochemical Journal 441, 131–141. [DOI] [PubMed] [Google Scholar]
- Garten A, Schuster S, Penke M, Gorski T, De Giorgis T, & Kiess W (2015). Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nature Reviews Endocrinology 11, 535. [DOI] [PubMed] [Google Scholar]
- Gehrke I, Bouchard ED, Beiggi S, Poeppl AG, Johnston JB, Gibson SB, & Banerji V (2014. September 15). On-target effect of FK866, a nicotinamide phosphoribosyl transferase inhibitor, by apoptosis-mediated death in chronic lymphocytic leukemia cells. Clinical Cancer Research 20(18), 4861–4872. [DOI] [PubMed] [Google Scholar]
- Geng CH, Zhang CL, Zhang JY, Gao P, He M, & Li YL (2018). Overexpression of Sirt6 is a novel biomarker of malignant human colon carcinoma. Journal of Cellular Biochemistry 119, 3957–3967. [DOI] [PubMed] [Google Scholar]
- George J, & Ahmad N (2016). Mitochondrial sirtuins in cancer: Emerging roles and therapeutic potential. Cancer Research 76, 2500–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Sengupta A, Seerapu GPK, Nakhi A, Ramarao EVS, Bung N, … Haldar D (2017). A novel SIRT1 inhibitor, 4bb induces apoptosis in HCT116 human colon carcinoma cells partially by activating p53. Biochemical and Biophysical Research Communications 488, 562–569. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Liu B, Wang Y, Hao Q, & Zhou Z (2015). Lamin a is an endogenous SIRT6 activator and promotes SIRT6-mediated DNA repair. Cell Reports 13, 1396–1406. [DOI] [PubMed] [Google Scholar]
- Gomez M, Wu J, Schreiber V, Dunlap J, Dantzer F, Wang Y, & Liu Y (2006). PARP1 is a TRF2-associated poly (ADP-ribose) polymerase and protects eroded telomeres. Molecular Biology of the Cell 17, 1686–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grolla AA, Travelli C, Genazzani AA, & Sethi JK (2016). Extracellular nicotinamide phosphoribosyltransferase, a new cancer metabokine. British Journal of Pharmacology 173, 2182–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozio A, Sociali G, Sturla L, Caffa I, Soncini D, Salis A, Raffaelli N, … Bruzzone S (2013. September 6). CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. Journal of Biological Chemistry 288(36), 25938–25949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujar AD, Le S, Mao DD, Dadey DY, Turski A, Sasaki Y, … Yuan L (2016). An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proceedings of the National Academy of Sciences 113, E8247–E8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Lam LT, Longenecker KL, Bui MH, Idler KB, Glaser KB, … Huang T-H (2017). Identification of novel resistance mechanisms to NAMPT inhibition via the de novo NAD+ biosynthesis pathway and NAMPT mutation. Biochemical and Biophysical Research Communications 491, 681–686. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Chaffer CL, & Weinberg RA (2009). Cancer stem cells: Mirage or reality? Nature Medicine 15, 1010–1012. [DOI] [PubMed] [Google Scholar]
- Haag F, Adriouch S, Braß A, Jung C, Möller S, Scheuplein F, … Koch-Nolte F (2007). Extracellular NAD and ATP: Partners in immune cell modulation. Purinergic Signalling 3, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, & Weinberg RA (2011). Hallmarks of cancer: The next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Head PE, Zhang H, Bastien AJ, Koyen AE, Withers AE, Daddacha WB, … David SY (2017. June 16). Sirtuin 2 mutations in human cancers impair its function in genome maintenance. Journal of Biological Chemistry 292(24), 9919–9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson DJ, Miranda JL, & Emerson BM (2017). The β-NAD+ salvage pathway and PKC-mediated signaling influence localized PARP-1 activity and CTCF poly (ADP) ribosylation. Oncotarget 8, 64698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka K, Ikutani M, Shito M, Kazuma K, Gulshan M, Nagai Y, … Nakagawa T (2014. May 23). Deficiency of nicotinamide mononucleotide adenylyltransferase 3 (nmnat3) causes hemolytic anemia by altering the glycolytic flow in mature erythrocytes. Journal of Biological Chemistry 289(21), 14796–14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Jing H, & Lin H (2014). Sirtuin inhibitors as anticancer agents. Future Medicinal Chemistry 6, 945–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, & Zhu G (2018). Sirtuin-4 (SIRT4), a therapeutic target with oncogenic and tumor-suppressive activity in cancer. OncoTargets and Therapy 11, 3395–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert S, Rissiek B, Klages K, Huehn J, Sparwasser T, Haag F, … Adriouch S (2010). Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2–P2X7 pathway. Journal of Experimental Medicine 207, 2561–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Durnin L, Dwyer L, Rhee P-L, Ward SM, Koh SD, … Mutafova-Yambolieva VN (2011). β-Nicotinamide adenine dinucleotide is an enteric inhibitory neurotransmitter in human and nonhuman primate colons. Gastroenterology 140, 608–617 (e606). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S-I, Armstrong CM, Kaeberlein M, & Guarente L (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795. [DOI] [PubMed] [Google Scholar]
- Jeong SM, Hwang S, & Seong RH (2016). SIRT4 regulates cancer cell survival and growth after stress. Biochemical and Biophysical Research Communications 470, 251–256. [DOI] [PubMed] [Google Scholar]
- Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, … German NJ (2013). SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell 23, 450–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jieyu H, Chao T, Mengjun L, Shalong W, Xiaomei G, Jianfeng L, & Zhihong L (2012). Nampt/Visfatin/PBEF: A functionally multi-faceted protein with a pivotal role in malignant tumors. Current Pharmaceutical Design 18, 6123–6132. [DOI] [PubMed] [Google Scholar]
- Jing H, Hu J, He B, Abril YLN, Stupinski J, Weiser K, … Giannakakou P (2016). A SIRT2-selective inhibitor promotes c-Myc oncoprotein degradation and exhibits broad anticancer activity. Cancer Cell 29, 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju H-Q, Zhuang Z-N, Li H, Tian T, Lu Y-X, Fan X-Q, … Chiao PJJCI (2016). Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer 379, 1–11. [DOI] [PubMed] [Google Scholar]
- Kato H, Ito E, Shi W, Alajez NM, Yue S, Lee C, … Green D (2010. February 1). Efficacy of combining GMX1777 with radiation therapy for human head and neck carcinoma. Clinical Cancer Research 16(3), 898–911. [DOI] [PubMed] [Google Scholar]
- Kim H-S, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, … Owens KM (2010). SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-S, Vassilopoulos A, Wang R-H, Lahusen T, Xiao Z, Xu X, … Yu H (2011). SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 20, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kim D, Cho SJ, Jung KY, Kim JH, Lee JM, … Kim KR (2019). Identification of a novel SIRT7 inhibitor as anticancer drug candidate. Biochemical and Biophysical Research Communications 508, 451–457. [DOI] [PubMed] [Google Scholar]
- Kim MY, Zhang T, & Kraus WL (2005). Poly (ADP-ribosyl) ation by PARP-1:PAR-laying’NAD+ into a nuclear signal. Genes & Development 19, 1951–1967. [DOI] [PubMed] [Google Scholar]
- Ko Y-H, Domingo-Vidal M, Roche M, Lin Z, Whitaker-Menezes D, Seifert E, … Tassone P (2016). TP53-inducible glycolysis and apoptosis regulator (TIGAR) metabolically reprograms carcinoma and stromal cells in breast cancer. Journal of Biological Chemistry 291, 26291–26303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenekoop RK, Wang H, Majewski J, Wang X, Lopez I, Ren H, … Genead M (2012). Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nature Genetics 44, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli S, Bhardwaj A, Kumari R, & Das S (2018). SIRT6 is a target of regulation by UBE3A that contributes to liver tumorigenesis in an ANXA2-dependent manner. Cancer Research 78, 645–658. [DOI] [PubMed] [Google Scholar]
- Korotchkina L, Joshi S, Vujcic S, Toshkov I, Chernov M, Kazyulkin D, … Gudkov AV (2017). Unbiased efficacy-based screening of small molecules highly selective against hematological malignancies revealed inhibitor of NAD synthesis (AACR). [Google Scholar]
- Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, … Ahmadi T (2016. July 21). Daratumumab depletes CD38+ immune-regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 128(3), 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegsmann K, Dittrich T, Neuber B, Awwad MH, Hegenbart U, Goldschmidt H, … Müller-Tidow C (2018). Quantification of number of CD38 sites on bone marrow plasma cells in patients with light chain amyloidosis and smoldering multiple myeloma. Cytometry part B: Clinical cytometry. [DOI] [PubMed] [Google Scholar]
- Krueger TE, Thorek DLJ, Meeker AK, Isaacs JT, & Brennen WN (2019). Tumor-infiltrating mesenchymal stem cells: Drivers of the immunosuppressive tumor microenvironment in prostate cancer? The Prostate 79, 320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari A, Iwasaki T, Pyndiah S, Cassimere EK, Palani CD, & Sakamuro D (2015). Regulation of E2F1-induced apoptosis by poly(ADP-ribosyl)ation. Cell Death and Differentiation 22, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier M-F, Zandarashvili L, Aguiar PM, Black BE, & Pascal JM (2018). NAD+ analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nature Communications 9, 844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, Niere M, & Ziegler M (2009). The NMN/NaMN adenylyltransferase (NMNAT) protein family. Frontiers in Bioscience 14, 410–431. [DOI] [PubMed] [Google Scholar]
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, … Safra T (2012). Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. The New England Journal of Medicine 366, 1382–1392. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim H, Lee JE, Shin S-J, Oh S, Kwon G, … Paik S (2018). Selective cytotoxicity of the NAMPT inhibitor FK866 toward gastric cancer cells with markers of the epithelial-mesenchymal transition, due to loss of NAPRT. Gastroenterology 155, 799–814 (e713). [DOI] [PubMed] [Google Scholar]
- Li H, Feng Z, Wu W, Li J, Zhang J, & Xia T (2013). SIRT3 regulates cell proliferation and apoptosis related to energy metabolism in non-small cell lung cancer cells through deacetylation of NMNAT2. International Journal of Oncology 43, 1420–1430. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li J, Bonkowski MS, Moniot S, Zhang D, Hubbard BP, Ling AJ, … Gorbunova V (2017). A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science 355, 1312–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Quan Y, & Xia W (2018). SIRT3 inhibits prostate cancer metastasis through regulation of FOXO3A by suppressing Wnt/β-catenin pathway. Experimental Cell Research 364, 143–151. [DOI] [PubMed] [Google Scholar]
- Liao S, Xiao S, Chen H, Zhang M, Chen Z, Long Y, … Peng S (2017). CD38 enhances the proliferation and inhibits the apoptosis of cervical cancer cells by affecting the mitochondria functions. Molecular Carcinogenesis 56, 2245–2257. [DOI] [PubMed] [Google Scholar]
- Liberti MV, & Locasale JW (2016). The Warburg effect: How does it benefit cancer cells? Trends in Biochemical Sciences 41, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, … Mann E (2019. February 1). BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discovery 9(2), 210–219. [DOI] [PubMed] [Google Scholar]
- Lin R,Tao R,Gao X,Li T,Zhou X,Guan K-L,…Lei Q-Y(2013).AcetylationstabilizesATP-citratelyasetopromotelipidbiosynthesisandtumorgrowth.MolecularCell 51,506–518. [Google Scholar]
- Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, … Martin M (2018). Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. The New England Journal of Medicine 379, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Xu N, Malyukova A, Scarlett C, Sun Y, Zhang X, … Chang D (2013). The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death and Differentiation 20, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, … Palumbo A (2015). Targeting CD38 with daratumumab monotherapy in multiple myeloma. The New England Journal of Medicine 373, 1207–1219. [DOI] [PubMed] [Google Scholar]
- Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, … Mateos MV (2016). Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial Lancet (London, England: ) 387, 1551–1560. [DOI] [PubMed] [Google Scholar]
- Lord CJ, & Ashworth A (2017). PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucena-Cacace A, Otero-Albiol D, Jiménez-García MP, Muñoz-Galvan S, & Carnero A (2018). NAMPT is a potent oncogene in colon cancer progression that modulates cancer stem cell properties and resistance to therapy through Sirt1 and PARP. Clinical Cancer Research 24, 1202–1215. [DOI] [PubMed] [Google Scholar]
- Lucena-Cacace A, Otero-Albiol D, Jiménez-García MP, Peinado-Serrano J, & Carnero A (2017). NAMPT overexpression induces cancer stemness and defines a novel tumor signature for glioma prognosis. Oncotarget 8, 99514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald M (1981). High content of mitochondrial glycerol-3-phosphate dehydrogenase in pancreatic islets and its inhibition by diazoxide. Journal of Biological Chemistry 256, 8287–8290. [PubMed] [Google Scholar]
- MacDonald MJ (1982). Evidence for the malate aspartate shuttle in pancreatic islets. Archives of Biochemistry and Biophysics 213, 643–649. [DOI] [PubMed] [Google Scholar]
- Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, … Gorbunova V (2011). SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin T, Baz R, Benson DM, Lendvai N, Wolf J, Munster P, … Campana F (2017). A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood 129, 3294–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos MV, Dimopoulos MA, Cavo M, Suzuki K, Jakubowiak A, Knop S, … Kaplan P (2018). Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. The New England Journal of Medicine 378, 518–528. [DOI] [PubMed] [Google Scholar]
- Mayer PR, Huang N, Dewey CM, Dries DR, Zhang H, & Yu G (2010. December 17). Expression, localization, and biochemical characterization of nicotinamide mononucleotide adenylyltransferase 2. Journal of Biological Chemistry 285(51), 40387–40396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, … Coronado M (2019). Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nature Immunology 20, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, … Vergote I (2016). Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. The New England Journal of Medicine 375, 2154–2164. [DOI] [PubMed] [Google Scholar]
- Moreschi I, Bruzzone S, Nicholas RA, Fruscione F, Sturla L, Benvenuto F, … Zocchi E (2006). Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. Journal of Biological Chemistry 281, 31419–31429. [DOI] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, … Murphy MM (2006). Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329. [DOI] [PubMed] [Google Scholar]
- Mottahedeh J, Haffner MC, Grogan TR, Hashimoto T, Crowell PD, Beltran H, … Isaacs WB (2018). CD38 is methylated in prostate cancer and regulates extracellular NAD+. Cancer & Metabolism 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynihan KD, & Irvine DJ (2017). Roles for innate immunity in combination immunotherapies. Cancer Research 77, 5215–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JP, Giacomantonio MA, Paulo JA, Everley RA, Kennedy BE, Pathak GP, … Sharif T (2018). The NAD+ salvage pathway supports PHGDH-driven serine biosynthesis. Cell Reports 24, 2381–2391 (e2385). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muruganandham M, Alfieri AA, Matei C, Chen Y, Sukenick G, Schemainda I, … Koutcher JA (2005). Metabolic signatures associated with a NAD synthesis inhibitor-induced tumor apoptosis identified by 1H-decoupled-31P magnetic resonance spectroscopy. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 11, 3503–3513. [DOI] [PubMed] [Google Scholar]
- Mutafova-Yambolieva VN, Hwang SJ, Hao X, Chen H, Zhu MX, Wood JD, … Sanders KM (2007). β-Nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proceedings of the National Academy of Sciences 104, 16359–16364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S, … Schultz D (2019). NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nature Cell Biology 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Lomb DJ, Haigis MC, & Guarente L (2009). SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137, 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov A, Dölle C, Niere M, & Ziegler M (2011. June 17). Pathways and subcellular compartmentation of NAD biosynthesis in human cells: From entry of extracellular precursors to mitochondrial NAD generation. Journal of Biological Chemistry 286 (24), 21767–21778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov A, Kulikova V, & Ziegler M (2015). The human NAD metabolome: Functions, metabolism and compartmentalization. Critical Reviews in Biochemistry and Molecular Biology 50, 284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan C, & Vassilopoulos A (2017). Sirtuins at the crossroads of stemness, aging, and cancer. Aging Cell 16, 1208–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohanna M, Cerezo M, Nottet N, Bille K, Didier R, Beranger G, … Bertolotto C (2018. March 1). Pivotal role of NAMPT in the switch of melanoma cells toward an invasive and drug-resistant phenotype. Genes & Development 32(5–6), 448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Kawai S, & Murata K (2012). Identification and characterization of a human mitochondrial NAD kinase. Nature Communications 3, 1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly T, & Niven DF (2003). Levels of nicotinamide adenine dinucleotide in extracellular body fluids of pigs may be growth-limiting for Actinobacillus pleuropneumoniae and Haemophilus parasuis. Canadian Journal of Veterinary Research 67, 229. [PMC free article] [PubMed] [Google Scholar]
- Ostrakhovitch EA, Akakura S, Sanokawa-Akakura R, Goodwin S, & Tabibzadeh S (2015). Dedifferentiation of cancer cells following recovery from a potentially lethal damage is mediated by H2S–Nampt. Experimental Cell Research 330, 135–150. [DOI] [PubMed] [Google Scholar]
- Ozden O, Park S-H, Wagner BA, Song HY, Zhu Y, Vassilopoulos A, … Gius D (2014). SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radical Biology and Medicine 76, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, … Mateos MV (2016). Daratumumab, bortezomib, and dexamethasone for multiple myeloma. The New England Journal of Medicine 375, 754–766. [DOI] [PubMed] [Google Scholar]
- Pan L-Z, Ahn D-G, Sharif T, Clements D, Gujar S, & Lee PW (2014). The NAD+ synthesizing enzyme nicotinamide mononucleotide adenylyltransferase 2 (NMNAT-2) is a p53 downstream target. Cell Cycle 13, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EY, Woo Y, Kim SJ, Kim DH, Lee EK, De U, … Ha K-T (2016). Anticancer effects of a new SIRT inhibitor, MHY2256, against human breast cancer MCF-7 cells via regulation of MDM2-p53 binding. International Journal of Biological Sciences 12, 1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-H, Ozden O, Liu G, Song HY, Zhu Y, Yan Y, … Principe DR (2016). SIRT2-mediated deacetylation and tetramerization of pyruvate kinase directs glycolysis and tumor growth. Cancer Research 76, 3802–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-H, Zhu Y, Ozden O, Kim H-S, Jiang H, Deng C-X, … Vassilopoulos A (2012). SIRT2 is a tumor suppressor that connects aging, acetylome, cell cycle signaling, and carcinogenesis. Translational Cancer Research 1, 15. [PMC free article] [PubMed] [Google Scholar]
- Patel AG, Sarkaria JN, & Kaufmann SH (2011). Nonhomologous end joining drives poly (ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proceedings of the National Academy of Sciences 108, 3406–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra KC, & Hay N (2014). The pentose phosphate pathway and cancer. Trends in Biochemical Sciences 39, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, & Jones RG (2013). Fueling immunity: Insights into metabolism and lymphocyte function. Science 342, 1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris-Pages M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, & Lisanti MP (2016). Cancer stem cell metabolism. Breast Cancer Research: BCR 18, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli R, Felczak K, & Cappellacci L (2011). NMN/NaMN adenylyltransferase (NMNAT) and NAD kinase (NADK) inhibitors: Chemistry and potential therapeutic applications. Current Medicinal Chemistry 18, 1973–1992. [DOI] [PubMed] [Google Scholar]
- Piacente F, Caffa I, Ravera S, Sociali G, Passalacqua M, Vellone VG, … Ballestrero A (2017). Nicotinic acid phosphoribosyltransferase regulates cancer cell metabolism, susceptibility to NAMPT inhibitors and DNA repair. Cancer Research, Canres 3079 (2016). [DOI] [PubMed] [Google Scholar]
- Pittelli M, Formentin L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, … Chiarugi A (2011). Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair and apoptosis. Molecular Pharmacology, Mol 111(073916). [DOI] [PubMed] [Google Scholar]
- Pittelli M, Formentini L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, … Chiarugi A (2010. October 29). Inhibition of nicotinamide phosphoribosyltransferase: Cellular bioenergetics reveals a mitochondrial insensitive NAD pool. Journal of Biological Chemistry 285(44), 34106–34114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, … Jha AK (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast GC, Malachowski WP, DuHadaway JB, & Muller AJ (2017). Discovery of IDO1 inhibitors: From bench to bedside. Cancer Research 77, 6795–6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylaeva E, Harati MD, Spyra I, Bordbari S, Strachan S, Thakur BK, … Welte K (2019). NAMPT signaling is critical for the proangiogenic activity of tumor-associated neutrophils. International Journal of Cancer 144, 136–149. [DOI] [PubMed] [Google Scholar]
- Qi J, Cui C, Deng Q, Wang L, Chen R, Zhai D, … Yu J (2018). Downregulated SIRT6 and upregulated NMNAT2 are associated with the presence, depth and stage of colorectal cancer. Oncology Letters 16, 5829–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, … Malavasi F (2013). CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytometry Part B, Clinical Cytometry 84, 207–217. [DOI] [PubMed] [Google Scholar]
- Raab MS, Chatterjee M, Goldschmidt H, Agis H, Blau I, Einsele H, … Röllig C (2016). A phase I/IIa study of the CD38 antibody MOR202 alone and in combination with pomalidomide or lenalidomide in patients with relapsed or refractory multiple myeloma (Am Soc Hematology). [Google Scholar]
- Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, … Migaud ME (2016). NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nature Communications 7, 13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M, Hillyard D, & Olivera BM (1976). Turnover of nicotinamide adenine dinucleotide in cultures of human cells. Journal of Cellular Physiology 88, 207–217. [DOI] [PubMed] [Google Scholar]
- Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, … Yang H (2017). MCU-dependent mitochondrial Ca 2+ inhibits NAD+/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 36, 5897. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, & Imai S. i. (2004). The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. Journal of Biological Chemistry 279, 50754–50763. [DOI] [PubMed] [Google Scholar]
- Ridge SM, Sullivan FJ, & Glynn SA (2017). Mesenchymal stem cells: Key players in cancer progression. Molecular Cancer 16, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, … Armstrong A (2017). Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. The New England Journal of Medicine 377, 523–533. [DOI] [PubMed] [Google Scholar]
- Ruan K, Zhu Y, Li C, Brazill JM, & Zhai RG (2015). Alternative splicing of Drosophila Nmnat functions as a switch to enhance neuroprotection under stress. Nature Communications 6, 10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KW, Nandu T, Kim J, Challa S, DeBerardinis RJ, & Kraus WL (2018). Metabolic regulation of transcription through compartmentalized NAD+ biosynthesis. Science 360 (eaan5780). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadej R, & Skladanowski AC (2012). Dual, enzymatic and non-enzymatic, function of ecto-5′-nucleotidase (eN, CD73) in migration and invasion of A375 melanoma cells. Acta Biochimica Polonica 59. [PubMed] [Google Scholar]
- Sampath D, Zabka TS, Misner DL, O’Brien T, & Dragovich PS (2015). Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacology & Therapeutics 151, 16–31. [DOI] [PubMed] [Google Scholar]
- Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, … Omlin A (2013). The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. The Lancet Oncology 14, 882–892. [DOI] [PubMed] [Google Scholar]
- Sanokawa-Akakura R, Ostrakhovitch EA, Akakura S, Goodwin S, & Tabibzadeh S (2014). A H2S-Nampt dependent energetic circuit is critical to survival and cytoprotection from damage in cancer cells. PLoS One 9, e108537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher MB, Vaquero A, & Reinberg D (2007). SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes & Development 21, 920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, … Chinnaiyan AM (2012). Dual roles of PARP-1 promote cancer growth and progression. Cancer Discovery 2, 1134–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiewer MJ, & Knudsen KE (2014). Transcriptional roles of PARP1 in cancer. Molecular Cancer Research: MCR 12, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiewer MJ, Mandigo AC, Gordon N, Huang F, Gaur S, de Leeuw R, … Parsons T (2018). PARP-1 regulates DNA repair factor availability. EMBO Molecular Medicine 10, e8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame J-C, & De Murcia G (2006). Poly (ADP-ribose): Novel functions for an old molecule. Nature Reviews Molecular Cell Biology 7, 517. [DOI] [PubMed] [Google Scholar]
- Sebastián C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, … Toiber D (2012). The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151, 1185–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo K, Park J, Heo J, Jing K, Han J, Min K, … Kang G (2015). SIRT2 regulates tumour hypoxia response by promoting HIF-1α hydroxylation. Oncogene 34, 1354. [DOI] [PubMed] [Google Scholar]
- Shames DS, Elkins K, Walter K, Holcomb T, Du P, Mohl D, … Liederer B (2013). Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 19, 6912–6923. [DOI] [PubMed] [Google Scholar]
- Sharif T, Ahn D, Liu R, Pringle E, Martell E, Dai C, … Murphy J (2016). The NAD+ salvage pathway modulates cancer cell viability via p73. Cell Death and Differentiation 23, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif T, Martell E, Dai C, Ghassemi-Rad MS, Kennedy BE, Lee PW, & Gujar S (2019. February 20). Regulation of cancer and cancer-related genes via NAD+. Antioxidants & Redox Signaling 30(6), 906–923. [DOI] [PubMed] [Google Scholar]
- Shi Y, Wang Y, Li Q, Liu K, Hou J, Shao C, & Wang Y (2018). Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nature Reviews Nephrology 14, 493–507. [DOI] [PubMed] [Google Scholar]
- Sisay M, & Edessa D (2017). PARP inhibitors as potential therapeutic agents for various cancers: Focus on niraparib and its first global approval for maintenance therapy of gynecologic cancers. Gynecologic Oncology Research and Practice 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sociali G, Grozio A, Caffa I, Schuster S, Becherini P, Damonte P, … Raffaell N (2018. December 4). SIRT6 deacetylase activity regulates NAMPT activity and NAD(P)(H) pools in cancer cells. The FASEB Journal 33(3), 3704–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sociali G, Raffaghello L, Magnone M, Zamporlini F, Emionite L, Sturla L, … Nencioni A (2016). Antitumor effect of combined NAMPT and CD73 inhibition in an ovarian cancer model. Oncotarget 7, 2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son MJ, Kwon Y, Son T, & Cho YS (2016). Restoration of mitochondrial NAD+ levels delays stem cell senescence and facilitates reprogramming of aged somatic cells. Stem Cells 34, 2840–2851. [DOI] [PubMed] [Google Scholar]
- Son MJ, Ryu J-S, Kim JY, Kwon Y, Chung K-S, Mun SJ, & Cho YS (2017). Upregulation of mitochondrial NAD+ levels impairs the clonogenicity of SSEA1+ glioblastoma tumor-initiating cells. Experimental & Molecular Medicine 49, e344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song T, Yang L, Kabra N, Chen L, Koomen J, Haura EB, & Chen J (2013). The NAD+ synthesis enzyme nicotinamide mononucleotide adenylyltransferase (NMNAT1) regulates ribosomal RNA transcription. Journal of Biological Chemistry 288, 20908–20917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosna J, Voigt S, Mathieu S, Lange A, Thon L, Davarnia P, … Chan FK (2014). TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cellular and Molecular Life Sciences: CMLS 71, 331–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, & Imai S. -i. (2012). The dynamic regulation of NAD metabolism in mitochondria. Trends in Endocrinology & Metabolism 23, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, … O’Malley DM (2017). Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 part 1): An international, multicentre, open-label, phase 2 trial. The Lancet Oncology 18, 75–87. [DOI] [PubMed] [Google Scholar]
- Tae IH, Park EY, Dey P, Son JY, Lee S-Y, Jung JH, … Kim HS (2018). Novel SIRT1 inhibitor 15-deoxy-Δ12, 14-prostaglandin J2 and its derivatives exhibit anticancer activity through apoptotic or autophagic cell death pathways in SKOV3 cells. International Journal of Oncology 53, 2518–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B, Young DA, Lu Z-H, Wang T, Meier TI, Shepard RL, … Kuo M-S (2013). Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells metabolic basis and potential clinical implications. Journal of Biological Chemistry 288, 3500–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, … Zhang Y (2014). Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metabolism 19, 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Shi L, Xie N, Liu Z, Qian M, Meng F, … Zhu W-G (2017). SIRT7 antagonizes TGF-β signaling and inhibits breast cancer metastasis. Nature Communications 8, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Coleman MC, Pennington JD, Ozden O, Park S-H, Jiang H, … McDonald WH (2010). Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Molecular Cell 40, 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, … Zhang B (2015). Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell 28, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi PM, Bansal N, Kerrigan JE, Abali EE, Scotto KW, & Bertino JR (2016). NAD+ kinase as a therapeutic target in cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 22, 5189–5195. [DOI] [PubMed] [Google Scholar]
- Tedeschi PM, Lin H, Gounder M, Kerrigan JE, Abali EE, Scotto K, & Bertino JR (2015). Suppression of cytosolic NADPH pool by thionicotinamide increases oxidative stress and synergizes with chemotherapy. Molecular Pharmacology 88, 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur BK, Dittrich T, Chandra P, Becker A, Kuehnau W, Klusmann JH, … Welte K (2013). Involvement of p53 in the cytotoxic activity of the NAMPT inhibitor FK866 in myeloid leukemic cells. International Journal of Cancer 132, 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thongon N, Zucal C, D’Agostino VG, Tebaldi T, Ravera S, Zamporlini F, … Quattrone A (2018). Cancer cell metabolic plasticity allows resistance to NAMPT inhibition but invariably induces dependence on LDHA. Cancer & Metabolism 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrens-Mas M, Pons DG, Sastre-Serra J, Oliver J, & Roca P (2017). SIRT3 silencing sensitizes breast cancer cells to cytotoxic treatments through an increment in ROS production. Journal of Cellular Biochemistry 118, 397–406. [DOI] [PubMed] [Google Scholar]
- Touat M, Sourisseau T, Dorvault N, Chabanon RM, Garrido M, Morel D, … Frankum JR (2018). DNA repair deficiency sensitizes lung cancer cells to NAD+ biosynthesis blockade. The Journal of Clinical Investigation 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang YH, Dogruluk T, Tedeschi PM, Wardwell-Ozgo J, Lu H, Espitia M, … Chen F (2016). Functional annotation of rare gene aberration drivers of pancreatic cancer. Nature Communications 7, 10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, … Schmutzler RK (2010). Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. The Lancet 376, 235–244. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, … Asplund A (2015). Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, … Edfors F (2017). A pathology atlas of the human cancer transcriptome. Science 357 (eaan2507). [DOI] [PubMed] [Google Scholar]
- Usmani SZ, Weiss BM, Plesner T, Bahlis NJ, Belch A, Lonial S, … Chari A (2016). Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood 128, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, … Bohr VA (2016). JNK phosphorylates SIRT6 to stimulate DNA double-strand break repair in response to oxidative stress by recruiting PARP1 to DNA breaks. Cell Reports 16, 2641–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Roermund C, Elgersma Y, Singh N, Wanders R, & Tabak HF (1995). The membrane of peroxisomes in Saccharomyces cerevisiae is impermeable to NAD (H) and acetyl-CoA under in vivo conditions. The EMBO Journal 14, 3480–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, & Thompson CB (2009). Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanLinden MR, Dölle C, Pettersen IK, Kulikova VA, Niere M, Agrimi G, … Ziegler M (2015. November 13). Subcellular distribution of NAD+ between cytosol and mitochondria determines the metabolic profile of human cells. Journal of Biological Chemistry 290(46), 27644–27659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanLinden MR, Niere M, Nikiforov AA, Ziegler M, & Dölle C (2017). Compartment-specific poly-ADP-ribose formation as a biosensor for subcellular NAD pools Poly (ADP-ribose) polymerase (pp. 45–56). Springer. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Eaton EN, Imai S-I , Frye RA, Pandita TK, … Weinberg RA (2001). hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159. [DOI] [PubMed] [Google Scholar]
- Wang C, Chen L, Hou X, Li Z, Kabra N, Ma Y, … Cress WD (2006). Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nature Cell Biology 8, 1025. [DOI] [PubMed] [Google Scholar]
- Wang J, & Dahl G (2018). Pannexin1: A multifunction and multiconductance and/or permeability membrane channel. American Journal of Physiology Cell Physiology 315, C290–c299. [DOI] [PubMed] [Google Scholar]
- Wang K, Jiang J, Lei Y, Zhou S, Wei Y, & Huang C (2019). Targeting metabolic-redox circuits for cancer therapy. Trends in Biochemical Sciences 44(5), 401–414. 10.1016/j.tibs.2019.01.001. ISSN 0968-0004 [DOI] [PubMed] [Google Scholar]
- Wang R-H, Sengupta K, Li C, Kim H-S, Cao L, Xiao C, … Chilton B (2008). Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14, 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-Q, Wang H-L, Xu J, Tan J, Fu L-N, Wang J-L, … Chen Y-X (2018). Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nature Communications 9, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Du L, Liang X, Meng P, Bi L, Wang Y, … Tang B (2018). Sirtuin 4 Depletion Promotes Hepatocellular Carcinoma Tumorigenesis Through Regulating Adenosine-Monophosphate–Activated Protein Kinase Alpha/Mammalian Target of Rapamycin Axis in Mice. Hepatology 69, 1614–1631. [DOI] [PubMed] [Google Scholar]
- Wanka C, Steinbach JP, & Rieger J (2012). Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by upregulating respiration and improving cellular redox homeostasis. Journal of Biological Chemistry 287, 33436–33446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Wind F, & Negelein E (1927). The metabolism of tumors in the body. The Journal of General Physiology 8, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Song J, Wang G, Cui X, Zheng J, Tang Y, … Liu C-Y (2018). Deacetylation of serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal carcinogenesis. Nature Communications 9, 4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Schwer B, Wickremesinghe PC, Hartnett DA, Burhenn L, Garcia M, … Helfand SL (2018. February 13). Sirt4 is a mitochondrial regulator of metabolism and lifespan in Drosophila melanogaster. Proceedings of the National Academy of Sciences 115(7), 1564–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S-N, Wang T-S, Li X, & Wang Y-P (2016). SIRT2 activates G6PD to enhance NADPH production and promote leukaemia cell proliferation. Scientific Reports 6, 32734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaku K, Okabe K, Hikosaka K, & Nakagawa T (2018). NAD metabolism in cancer therapeutics. Frontiers in Oncology 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Hikosaka K, Mahmood A, Tobe K, Shojaku H, Inohara H, & Nakagawa T (2016). Nmnat3 is dispensable in mitochondrial NAD level maintenance in vivo. PLoS One 11, e0147037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa S, Baker JR, Vuppusetty C, Koga T, Colley T, Fenwick P, … Ito K (2018). The dynamic shuttling of SIRT1 between cytoplasm and nuclei in bronchial epithelial cells by single and repeated cigarette smoke exposure. PLoS One 13, e0193921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, … Rosenzweig A (2007). Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Nagasawa K, Münch C, Xu Y, Satterstrom K, Jeong S, … Zaganjor E (2016). Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell 167, 985–1000 (e1021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wang Z, Li X, Liu B, Liu M, Liu L, … Yu M (2018). SHMT2 desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Research 78, 372–386. [DOI] [PubMed] [Google Scholar]
- Ying W (2008). NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxidants & Redox Signaling 10, 179–206. [DOI] [PubMed] [Google Scholar]
- Yu H, Ye W, Wu J, Meng X, Liu RY, Ying X, … Huang W (2014. July 1). Overexpression of sirt7 exhibits oncogenic property and serves as a prognostic factor in colorectal cancer. Clinical Cancer Research 20(13), 3434–3445. [DOI] [PubMed] [Google Scholar]
- Yu S-W, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, … Dawson VL (2002). Mediation of poly (ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297, 259–263. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, & Bellen HJ (2008). NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 452, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, … Aebersold R (2016). NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. [DOI] [PubMed] [Google Scholar]
- Zhang T, Berrocal JG, Yao J, DuMond ME, Krishnakumar R, Ruhl DD, … Kraus WL (2012). Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD+ synthase. The Journal of Biological Chemistry 287, 12405–12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Mo Y, Li M-T, Zou S-W, Cheng Z-L, Sun Y-P, … Lei Q-Y (2014). NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. The Journal of Clinical Investigation 124, 5453–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Zou S-W, Liu Y, Zhou X, Mo Y, Wang P, … Lei Q-Y(2013). Lysine-5 acetylation negatively regulates lactate dehydrogenase a and is decreased in pancreatic cancer. Cancer Cell 23, 464–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Green CF, Hui YH, Prieto L, Shepard R, Dong S, … Johnson RL (2017. December 1). Discovery of a highly selective NAMPT inhibitor that demonstrates robust efficacy and improved retinal toxicity with nicotinic acid coadministration. Molecular Cancer Therapeutics 16(12), 2677–2688. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Liu XZ, Tian WW, Guan YF, Wang P, & Miao CY (2014). Extracellular visfatin has nicotinamide phosphoribosyltransferase enzymatic activity and is neuro-protective against ischemic injury. CNS Neuroscience & Therapeutics 20, 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YJ, Lam CMC, & Lee HC (2012). The membrane-bound enzyme CD38 exists in two opposing orientations. Sci Signal 5 (ra67–ra67). [DOI] [PubMed] [Google Scholar]
- Zhu S, Dong Z, Ke X, Hou J, Zhao E, Zhang K, … Cui H (2018). The role of Sirtuins family in cell metabolism during tumor development Paper presented at: Seminars in cancer biology. Elsevier. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Wang G, Li X, Wang T, Weng M, & Zhang Y (2018). Knockout of SIRT4 decreases chemosensitivity to 5-FU in colorectal cancer cells. Oncology Letters 16, 1675–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Zhu Y, Park S-H, Liu G, O’Brien J, Jiang H, & Gius D (2017. August 1). SIRT3-mediated dimerization of IDH2 directs cancer cell metabolism and tumor growth. Cancer Research 77(15), 3990–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]