

Abstract
The EFSA Panel on Contaminants in the Food Chain (CONTAM) established a tolerable daily intake (TDI) for T2 and HT2 of 0.02 μg/kg body weight (bw) per day based on a new in vivo subchronic toxicity study in rats that confirmed that immune‐ and haematotoxicity are the critical effects of T2 and using a reduction in total leucocyte count as the critical endpoint. An acute reference dose (ARfD) of 0.3 μg for T2 and HT2/kg bw was established based on acute emetic events in mink. Modified forms of T2 and HT2 identified are phase I metabolites mainly formed through hydrolytic cleavage of one or more of the three ester groups of T2. Less prominent hydroxylation reactions occur predominantly at the side chain. Phase II metabolism involves conjugation with glucose, modified glucose, sulfate, feruloyl and acetyl groups. The few data on occurrence of modified forms indicate that grain products are their main source. The CONTAM Panel found it appropriate to establish a group TDI and a group ARfD for T2 and HT2 and its modified forms. Potency factors relative to T2 for the modified forms were used to account for differences in acute and chronic toxic potencies. It was assumed that conjugates (phase II metabolites of T2, HT2 and their phase I metabolites), which are not toxic per se, would be cleaved releasing their aglycones. These metabolites were assigned the relative potency factors (RPFs) of their respective aglycones. The RPFs assigned to the modified forms were all either 1 or less than 1. The uncertainties associated with the present assessment are considered as high. Using the established group, ARfD and TDI would overestimate any risk of modified T2 and HT2.
Keywords: T2, HT2, modified forms, group health based guidance values
Summary
Following a request from the European Commission, the EFSA Panel on Contaminants in the Food Chain (CONTAM) assessed whether it is appropriate and feasible to set a group health based guidance value (group HBGV) for T‐2 toxin (T2) and HT‐2 toxin (HT2) and their modified forms related to their presence in food and feed, and to consider, whether it would be appropriate to use the parent compound as a marker for toxicity. Modified forms of mycotoxins comprise all metabolites of the parent molecule, which are formed in the fungus, infested plant and animals used for food and feed. It is increasingly realised that not only mycotoxins itself, but also their modified forms may contribute to the overall toxicity. Modified forms include phase I metabolites formed through oxidation, reduction or hydrolysis of the parent toxin, as well as phase II metabolites arising from conjugation with endogenous molecules. In the plant, these are particularly conjugates with glucose or sulfate and in animals' glucuronic acid.
Previous risk assessments from EFSA on T2/HT2 and on modified mycotoxins have been used as a starting point for the present assessment. In addition, a systematic literature search has been carried out to obtain up‐to‐date and comprehensive information on T2/HT2 and its modified forms. In this opinion, the general principles for risk assessment were followed.
Before assessing whether modified forms of T2/HT2 could be included in a group HBGV for T2/HT2, the CONTAM Panel decided to review new relevant data on T2/HT2 published after the European Food Safety Authority (EFSA) assessment in 2011 to evaluate whether the established tolerable daily intake (TDI) for T2/HT2 needed to be revised and if in addition there was a need also to set an acute reference dose (ARfD) for T2/HT2.
T2 and HT2 are members of the type A group trichothecenes, which are tetracyclic sesquiterpenoids produced by many species of Fusarium infesting crop plants. A large number of modified forms (phase I and phase II metabolites) of T2/HT2 generated by fungi, plants and mammals have been identified and characterised. These may occur together with T2 and HT2 as residues in food of plant and animal origin. The predominant metabolic pathways are the hydrolytic cleavage of one or more of the three ester groups of T2 yielding to neosolaniol (NEO), T2‐triol and eventually T2‐tetraol. Hydroxylation reactions at various positions of T2 and of hydrolytic metabolites (predominantly at C‐19, but also at C‐20 and C‐8) appear to be less pronounced. Another phase I reaction, mostly performed by intestinal and ruminal microflora, is de‐epoxidation resulting in loss of toxicity. Phase II metabolism involves conjugation of the parent compounds and their phase I metabolites with glucose, modified glucose, and sulfate, feruloyl and acetyl groups.
There are appropriate analytical methods for T2, HT2 and their phase II metabolites and these are mainly based on liquid chromatography–tandem mass spectrometry (LC–MS/MS) and appear to be adequate in terms of analytical performance. Methods such as gas chromatography–mass spectrometry (GC–MS) or liquid chromatography‐fluorescence detection (LC‐FLD) are still in use in control laboratories. Both approaches require excessive sample purification and a derivatisation step prior to the analysis, thus affecting the overall recovery and sensitivity. The analytical methods available for phase I metabolites of T2 and HT2 need improvement regarding their sensitivity. There is a lack of commercially available standards and reference materials.
There are few data on occurrence of modified forms of T2 and HT2 in plant‐derived food products, but grain products appear to be the main source of modified forms of T2 and HT2. The proportion of their occurrence relative to the respective parent compounds, in the few samples where this has been determined, varies widely.
There is a lack of occurrence data on modified forms of T2 and HT2 in food of animal origin.
Upon ingestion, T2 appears to be rapidly absorbed. It is rapidly hydrolysed to HT2 and distributed to the liver, kidney and other organs without accumulation. No studies on absorption or distribution of modified forms of T2 or HT2 have been identified. There are no data on absorption of phase I metabolites from food. They are assumed to be easily absorbed, distributed and entering metabolism as described for T2. Neither are there data available on the absorption of phase II metabolites of T2 and HT2. It is assumed that such phase II metabolites are hydrolysed in the gastrointestinal tract after ingestion, releasing their aglycones, which can then be absorbed. This assumption is based on the fact that conjugates of other trichothecenes may undergo intestinal hydrolysis followed by absorption. It is expected that phase I metabolites will subsequently be subject to phase II metabolism with conjugation to glucuronic acid or sulfate. Mammals excrete the phase I and phase II metabolites of T2/HT2, and presumably also ingested modified forms within 2–3 days in urine and faeces.
Since the previous opinion in 2011, new in vivo acute toxicity studies (supported by subacute studies) show that T2 and HT2 have anorectic effects upon short‐term exposure. Emetic events observed in mink upon single oral and intraperitoneal exposure to both T2 and HT2 were identified as critical effects for setting an acute HBGV in the present opinion. A benchmark dose (BMD) analysis was performed using a combined data set on the incidence of emetic events in mink exposed to either T2 or HT2 in two independent experiments. This approach was justified considering that the two toxins show similar acute toxicity, are equipotent with regard to emetic response and appear to work via the same mode of action. In addition, T2 is rapidly metabolised to HT2 in the organism. Using a benchmark response (BMR) of 10% resulted in a benchmark dose 10% (BMD10) total confidence interval (BMDL10–BMDU10) of 2.97–49.8 μg T2 or HT2/kg body weight (bw) per day across the applied models. Using an uncertainty factor (UF) of 10, the CONTAM Panel established a group ARfD of 0.3 (rounded from 0.297) μg T2 and HT2/kg bw. The phase I metabolite NEO showed equal emetic potency and was therefore included in a group ARfD with T2 and HT2 with the same, molarity‐based relative potency factor (RPF) of 1. In the absence of toxicity data on phase II metabolites of T2, HT2 and NEO, the CONTAM Panel assumed that these conjugates can be hydrolysed in the intestine and therefore included them in the group ARfD and used the same RPFs of 1 as for their parent/modified phase I metabolites.
In its previous opinion of 2011, the Panel concluded that T2 inhibits protein, DNA and ribonucleic acid (RNA) synthesis, causes apoptosis, necrosis and lipid peroxidation and induces haemato‐ and myelotoxicity. Reduction in antibody response to a specific antigen in a subacute 21‐day pig study was identified as the critical effect for human risk assessment. This effect was accompanied by other haematotoxic effects including a reduced number of leucocytes. Since then, a new in vivo subchronic toxicity study with rats (duration 90 days) has become available overall confirming that immune‐ and haematotoxicity are the critical effects of T2 upon repeated exposure. In this study, a dose‐dependent reduction in total leucocyte count, mainly caused by a reduced number of lymphocytes, was observed. The CONTAM Panel identified this as the critical effect for setting a chronic health based guidance value in the present opinion. Using a BMR of 10% on the basis of biological considerations, the Panel calculated a BMDL10‐BMDU10 confidence interval of 3.33–27.6 μg T2/kg bw per day across the applied models. The CONTAM Panel used the BMDL10 and an UF of 200, taking into account 10 for interspecies and 10 for intraspecies variation and a factor of 2 because it was a subchronic study and the toxic effect progressed during the whole study period reaching no plateau at the end of the study. A TDI of 0.02 (rounded from 0.017) μg T2 and HT2/kg bw per day was established.
Haematotoxicity, in particular with reduced production of leucocytes but also erythrocytes and platelets, is the critical effect of T2. The underlying mode of action is protein synthesis inhibition, induction of ribotoxic stress and apoptosis. Since T2 is rapidly metabolised to HT2 the toxicity of T2 might partly be attributed to HT2. No in vivo studies on haematotoxicity of modified forms of T2 and HT2 have been identified. However, it can be assumed that phase I metabolites of T2 and HT2 work via a similar mode of action, because some have been shown to cause protein synthesis inhibition. The CONTAM Panel therefore considered it appropriate to include the modified forms NEO, T2‐triol and T2‐tetraol in a group TDI with T2 and HT2 assuming dose addition as a model of joint action. Because phase I metabolites show different potencies in inhibition of protein synthesis and other toxic effects, it was decided to assign molarity‐based RPFs for their inclusion in any risk assessment. These RPFs were rounded up to half orders of magnitude to avoid spurious accuracy and are 1 for T2 and HT2 and 19‐HO‐T2, and, based on significantly lower toxic potency, 0.3 for NEO and 19‐HO‐HT2 and 0.1 for T2‐triol and T2‐tetraol. Since phase II metabolites of T2, HT2, NEO, T2‐triol and T2‐tetraol can be hydrolysed to their aglycones after ingestion, they were included in the Group TDI. Thus, T2‐3‐Glc, T2‐3‐diGlc, T2‐3‐Sulf, T2‐3‐GlcA, 3‐Ac‐T2, 3‐Fer‐T2, HT2‐3‐Glc, HT2‐diGlc, HT2‐GlcA and HT2‐MalGlc were included in the group TDI by applying an RPF of 1. NEO‐Glc was included using a factor 0.3 and T2‐triol‐Glc and T2‐tetraol‐Glc by applying a factor of 0.1. The CONTAM Panel noted that none of the modified form of T2 and HT2 had a higher toxic potency than T2 or HT2.
The overall uncertainty associated with including modified forms of T2 and HT2 into a group ARfD and a group TDI in the present assessment is considered as high and its application would rather overestimate than underestimate the risk. Uncertainty could be reduced, provided more data are made available, particularly on the in vivo haematotoxicity of modified forms of T2 and HT2. For the group, ARfD data on the ability of several modified forms to induce an emetic response are needed. Also more data on the absorption and bioavailability of the major phase II metabolites present in plant products used for food and feed are needed. Further chemical characterisation of not fully characterised modified forms should be performed. Certified reference materials and standards for the modified forms of T2 and HT2 should be made available and more data on the occurrence of modified forms of T2 and HT2 in food are needed in order to characterise risks using the group ARfD and group TDI and the RPFs established in this opinion.
1. Introduction
1.1. Background and Terms of Reference as provided by the requestor
Following a request from the European Commission, the risks to human and animal health related to modified forms of the Fusarium toxins zearalenone, nivalenol, T‐2 and HT‐2 toxins, and fumonisins were evaluated in the scientific opinion on the risks for human health related to the presence of modified forms of certain mycotoxins in food and feed,1 adopted by the EFSA Panel on Contaminants in the Food Chain (CONTAM) on 25 November 2014.
The CONTAM Panel considered it appropriate to assess human exposure to modified forms of the various toxins in addition to the parent compounds, because many modified forms are hydrolysed into the parent compounds or released from the matrix during digestion. In the absence of specific toxicity data, toxicity equal to the parent compounds was assumed for modified mycotoxins. Risk characterisation was done by comparing exposure scenarios with reference doses of the parent compounds.
The regulatory follow‐up to this scientific opinion was discussed at the Expert Committee ‘Agricultural contaminants’ on 15 January 2015. The Standing Committee on Plants, Animals, Food and Feed has been informed thereof at its meeting on 11 February 2015.2
Before taking regulatory measures as regards the modified mycotoxins, it was agreed that it is appropriate to request the European Food Safety Authority (EFSA) to assess whether it is appropriate and feasible to set a group health based guidance value (HBGV) for the parent compound and its modified forms and to consider, if relevant, the appropriateness to use the parent compound as a marker for the presence and toxicity of the parent compound and its modified forms.
1.2. Terms of Reference as provided by the requestor
In accordance with Art. 29 (1) (a) of Regulation (EC) No 178/2002, the Commission asks EFSA for scientific opinions to assess whether it is appropriate and feasible to set a group HBGV for the parent compound and its modified forms for zearalenone, fumonisins, nivalenol, and T‐2 and HT‐2 toxin and to consider, if relevant, the appropriateness to use the parent compound as a marker for the presence and toxicity of the parent compound and its modified forms for these mycotoxins.
The four requested scientific opinions are:
assessment whether it is appropriate and feasible to set a group HBGV for zearalenone and its modified forms identified in the CONTAM opinion on the risks for human health related to the presence of modified forms of certain mycotoxins in food and feed, and to consider, if relevant, the appropriateness to use the parent compound as a marker for presence and toxicity of zearalenone and its modified forms.
assessment whether it is appropriate and feasible to set a group HBGV for fumonisin B1 and B2 and their modified forms identified in the CONTAM opinion on the risks for human health related to the presence of modified forms of certain mycotoxins in food and feed and to consider, if relevant, the appropriateness to use the parent compounds as a marker for the presence and toxicity of fumonisin B1 and B2 and their modified forms.
assessment whether it is appropriate and feasible to set a group HBGV for nivalenol and its modified forms identified in the CONTAM opinion on the risks for human health related to the presence of modified forms of certain mycotoxins in food and feed and to consider, if relevant, the appropriateness to use the parent compound as a marker for the presence and toxicity of nivalenol and its modified forms.
assessment whether it is appropriate and feasible to set a group HBGV for T‐2 and HT‐2 toxin and their modified forms identified in the CONTAM opinion on the risks for human health related to the presence of modified forms of certain mycotoxins in food and feed and to consider, if relevant, the appropriateness to use the parent compound as a marker for the presence and toxicity of T‐2 and HT‐2 toxin and their modified forms.
1.3. Introduction to modified mycotoxins
Mycotoxins are secondary metabolites of filamentous fungi. They are usually low molecular weight compounds and serve no function in the intermediary metabolism of the fungus, but provide advantages with respect to its competition for nutrients and habitat. Consequently, many mycotoxins are toxic for bacteria and other microorganisms. As mycotoxins are also toxic for humans and animals, their presence in food and feed may pose a health risk.
Numerous mycotoxins have been characterised. Even though some of these may be metabolites of the other, they are recognised as separate mycotoxins, e.g. HT‐2 toxin (HT2) is a metabolite of T‐2 toxin (T2). However, it is increasingly realised that also modified forms of these mycotoxins occur in food and feed and that these modified forms should be taken into account for risk assessment, because they may contribute to the toxicity of the parent toxin.
According to a recent definition, modified forms of mycotoxins comprise all biologically, chemically and physically modified derivatives of the parent molecule, which are formed in the fungus, infested plant and mammalian organism (Rychlik et al., 2014). This includes inter alia phase I metabolites formed through oxidation, reduction or hydrolysis of the parent toxin, as well as phase II metabolites arising from conjugation with endogenous molecules. Phase II metabolites formed in the plant through conjugation with polar low molecular weight molecules, such as glucose or sulfate, have also been called ‘masked’ mycotoxins because they were hard to capture by routine analysis (Rychlik et al., 2014). However, after intake with the food or feed such conjugates may be hydrolysed in the digestive tract, thereby releasing the parent toxin which may add to the total uptake of toxin. Therefore, phase II metabolism in the plant or fungi is of paramount importance for the risk assessment of mycotoxins.
In the context of risk assessment of mycotoxins in food and feed, modified mycotoxins comprise all metabolites of a given mycotoxin that occur in food or feed. These include phase I and II metabolites formed in the fungus, infested plant used for food and feed or food (and feed) products of animal origin. It does not include metabolites formed in humans, even if these may be similar.
1.4. Legislation
Article 2 of Council Regulation (EEC) No 315/933 stipulates that food containing a contaminant in amount unacceptable for public health shall not be placed on the market, that contaminant levels should be kept as low as can reasonably be achieved and that, if necessary, the European Commission may establish maximum levels for specific contaminants. These maximum levels are laid down in the Annex of Commission Regulation (EC) No 1881/20064 and may include maximum levels (MLs) for the same contaminants in different foods, analytical detection limits and reference to the sampling and analysis methods to be used. Neither for T2 and HT2 nor for their metabolites have MLs been set in this regulation.
Commission Recommendation 2013/165/EU5 states that referring to the respective EFSA opinion (EFSA CONTAM Panel, 2011) since the exposure estimates for T2 and HT2 are below the tolerable daily intake (TDI) there is no immediate public health concern. However, in order to assess changes and trends in exposure, it stipulates that the European Union (EU) Member States, with active involvement of feed and food business operators, monitor the presence of T2 and HT2 in cereals and cereal products and that any results should be provided on a regular basis to EFSA. It furthermore provides indicative levels for the sum of T2 and HT2 in cereals and cereal products, from which onwards/above investigations should be performed, certainly in the case of repetitive findings. These indicative levels range from 15 μg/kg in cereal‐based foods for infants to 2,000 μg/kg in oat milling products (husks).
1.5. Interpretation of Terms of Reference
The CONTAM Panel took the assumption that the previous risk assessment of T2 and HT2 in food and feed (EFSA CONTAM Panel, 2011) is comprehensively covering all relevant aspects of the compound and therefore used it together with the recent opinion on modified fusarium toxins (EFSA CONTAM Panel, 2014) as a starting point for the present assessment.
The CONTAM Panel decided to review the new relevant data on T2 and HT2 (i.e. published after 2010) to evaluate whether the TDI established for T2 and HT2 in 2011 needs to be revised and if in addition there is a need also to set an acute reference dose for T2 and HT2.
The Panel then decided to present the modified forms of T2 and HT2 identified to date and reviewed the appropriateness of the methods currently available for their analysis.
In line with the previous EFSA opinion on modified mycotoxins (EFSA CONTAM Panel, 2014), modified T2 and HT2 occurring in plants (arising from both plant and fungal metabolism), formed as a consequence of food processing and transfer from feed to livestock were considered for possible inclusion in group HBGVs.
In order to assess whether it was appropriate to include the modified forms of T2 and HT2 in group HBGVs with T2 and HT2, all data available and relevant for that task were evaluated.
2. Data and methodologies
2.1. Methodology for data collection and study appraisal
The CONTAM Panel considered the previous assessments on T2 and HT2 (EFSA CONTAM Panel, 2011) and on modified fusarium toxins including modified T2 and HT2 (EFSA CONTAM Panel, 2014) as comprehensive, covering all relevant publications on T2 and HT2 and its modified forms, respectively, until those dates. All publications referenced therein have been considered, wherever appropriate, also for the present evaluation.
In order to cover also new publications not considered in these previous assessments, a systematic and comprehensive search for literature was conducted for peer‐reviewed original research pertaining to T2 and HT2 and its modified forms published after 2010 including scientific literature dealing with analytical determination, chemistry, occurrence, toxicokinetics and toxicity of T2 and HT2 and/or its modified forms. Studies on analytical methods, chemistry and occurrence of the parent compounds T2 and HT2 only, however, were excluded since not considered of relevance for the present assessment.
Date, search strings, data bases used and numbers of publications retrieved and used for assessment are presented in detail in Appendix A. In total, 1,087 citations/abstracts were obtained. Those considered relevant by expert judgement were included in the present assessment. Only papers in English language were considered for inclusion in the assessment.
Since the 2011 opinion on T2 and HT2 did not include information on metabolites, in order to obtain information on the toxicity of T2 metabolites additional publications on T2‐tetraol and neosolaniol were collected using the same databases as above, search terms T2‐tetraol AND tox* and neosolaniol AND tox*, but applying no time limit. This search yielded a total of 335 publications of which 56 were considered as potentially relevant for the present assessment.
During the development of the opinion, additional relevant publications not retrieved in the above‐mentioned literature evaluation have been identified and considered for the assessment when relevant.
2.2. Methodology applied for hazard assessment
The CONTAM Panel applied the general principles of the risk assessment process for chemicals in food as described by the World Health Organization (WHO, 2009), which include hazard identification and characterisation, exposure assessment and risk characterisation. In addition to the principles described by WHO (2009), EFSA guidance pertaining to risk assessment (EFSA Scientific Committee, 2009) and on default values (EFSA Scientific Committee, 2012) have been applied for the present assessment.
3. Previous assessments
In the Scientific Opinion on the risks for animal and public health related to the presence of T2 and HT2 in food and feed (EFSA CONTAM Panel, 2011), it was noted that the pig is among the most sensitive species towards T2 and that immunological and haematological effects are the most sensitive/critical endpoints. Using the data obtained on reduction in antibody response to a specific antigen (for which a lowest‐observed‐adverse‐effect level (LOAEL) of 29 μg T2/kg body weight (bw) per day was observed while a no no‐observed‐adverse‐effect level (NOAEL) could be identified) in a benchmark dose (BMD) analysis a BMDL05 of 10 μg T2/kg bw per day was derived for T2. Since T2 is rapidly metabolised to HT2 and the toxicity of T2 might at least partly be attributed to HT2, a group TDI of 0.1 μg/kg bw per day was established for the sum of T2 and HT2 by applying an uncertainty factor (UF) of 100 to the BMDL05.
In the Scientific Opinion on the risks for human and animal health related to the presence of modified forms of certain mycotoxins in food and feed (EFSA CONTAM Panel, 2014), no specific information on the toxic effects of the modified forms of T2 and HT2 could be identified. However, the chemistry and toxicokinetics of T2 and HT2 as well as general considerations of biotransformation suggested that T2 and HT2 conjugates may be cleaved in the gastrointestinal tract releasing T2 and HT2. Taking a pragmatic approach until more information became available, the CONTAM Panel assumed that modified forms of T2 and HT2 have the same toxicological profile and potency as their parent compounds. Based on occurrence data available at that time (2014), it was then assumed that modified forms of T2 and HT2 add another 10% to the exposure to T2 and HT2.
4. Chemistry
T‐2 toxin and HT‐2 toxin (Figure 1) are members of the trichothecene family, which represents the largest group of Fusarium mycotoxins and comprises more than 150 compounds. The common structure of all trichothecenes is a tetracyclic sesquiterpene with a spiro‐epoxide group at C‐12 and C‐13 and an olefinic double bond between C‐9 and C‐10. According to the substituents of the tetracyclic ring system, trichothecenes are grouped into different types (A–D). T‐2 and HT‐2 toxin belong to the type A trichothecenes, which are characterised by an esterified or free hydroxyl group at C‐8, or an unsubstituted C‐8. In contrast, all type B trichothecenes, e.g. nivalenol and deoxynivalenol (DON), carry a keto group at C‐8. Type A and B compounds constitute the majority of trichothecene contaminants in food and feed.
Figure 1.
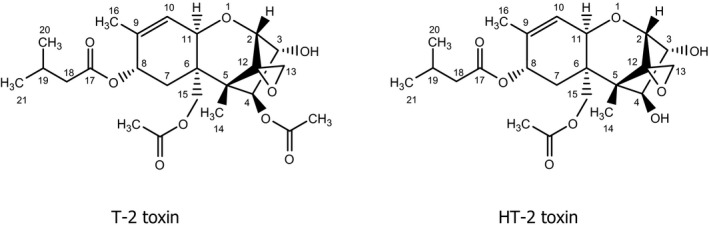
Chemical structures of T‐2 toxin (left) and HT‐2 toxin (right)
The biosynthesis of T‐2 and HT‐2 toxin involves the initial cyclisation of farnesyl phosphate to the bicyclic sesquiterpene trichodiene, which does not contain oxygen, followed by a complex sequence of hydroxylation, epoxidation, further cyclisation, and acylation reactions (for review, see McCormick et al., 2011). HT‐2 toxin arises through hydrolysis of the 4‐acetoxy group of T‐2 toxin and is a metabolite of T‐2 toxin formed in fungi, plants and animals.
In this opinion, the nomenclature according to the International Union of Pure and Applied Chemistry (IUPAC) rules was applied. It should be noted that the numbering of the isovaleroyl group of T‐2 and HT‐2 toxin is not consistent in the literature. Instead of 17–19 (see Figure 1), some publications use 1′ to 3′, respectively.
In general, plants, microorganisms and animals biotransform mycotoxins by phase I metabolism, which includes oxidation, reduction and hydrolysis of the parent (fungal) compounds, and phase II metabolism, which comprises all conjugation reactions. While phase II metabolites formed in the mammalian organism are usually excreted via urine and/or bile, plants store conjugated metabolites in vacuoles and/or attach them to structures of the cell wall. The latter processes are sometimes referred to as phase III metabolism or compartmentation.
4.1. Parent compounds and phase I metabolites
T‐2 toxin, in the older literature also referred to as fusariotoxin T‐2, insariotoxin, or mycotoxin T‐2, has the IUPAC name (2α,3α,4ß,8α)‐4,15‐bis(acetoxy)‐3‐hydroxy‐12,13‐epoxytrichothec‐9‐en‐8‐yl 3‐methylbutanoate (Chemical Abstracts Service, CAS No 21259‐20‐1, C24H34O9, molecular weight (MW) 466). It is a colourless crystalline solid with a melting point (m.p.) of 151–152°C (Bamburg et al., 1968).
HT‐2 toxin has the IUPAC name (2α,3α,4ß,8α)‐15‐acetoxy‐3,4‐dihydroxy‐12,13‐epoxytrichothec‐9‐en‐8‐yl 3‐methylbutanoate (CAS No 26934‐87‐2, C22H32O8, MW 424). It is a colourless crystalline solid with a m.p. of 151–152°C (Bamburg et al., 1968).
The solubility of T‐2 toxin and HT‐2 toxin is low in water and petroleum ether, but good in ethanol, ethyl acetate, chloroform, dimethylsulfoxide and other organic solvents.
Although the trivial names of T‐2 toxin and HT‐2 toxin are short, they are frequently abbreviated in the scientific literature as T2 and HT2, respectively. These abbreviations are also used in this Opinion, mainly because they are of advantage for abbreviating the more complex names of the phase II metabolites of T2 and HT2 (see Section 4.2).
As discussed in more detail in Sections 6 and 7.3.1, numerous phase I metabolites of T2 and HT2 have been identified in fungal cultures and plants, as well as in studies with experimental animals. They are listed in Table 1, and the chemical structures of the key metabolites arising from the major metabolic pathways are depicted in Figure 2. As T2 and its metabolites lack a strong chromophore, the analytical method of choice is often liquid chromatography–mass spectrometry (LC–MS) analysis, which has the advantage of an efficient separation from complex matrices and high sensitivity (see Section 5). Therefore, the element formulas and molecular weights of the metabolites are given in Table 1. It should be noted that some of the phase I metabolites are isomeric (e.g. the regioisomers of the monohydroxylated metabolites and their de‐epoxidation products), which has to be taken into account in LC–MS analysis.
Table 1.
T2 and HT2 and their phase I metabolites
| Compounda | Element formula | Molecular weight |
|---|---|---|
| T2 | C24H34O9 | 466 |
| 19‐HO‐T2b (TC‐1) | C24H34O10 | 482 |
| iso‐TC‐1c | C24H34O10 | 482 |
| 20‐HO‐T2d | C24H34O10 | 482 |
| DE‐T2 | C24H34O8 | 450 |
| 15‐Deacetyl‐T2 | C22H32O8 | 424 |
| DE‐19‐HO‐T2b | C24H34O9 | 466 |
| 15‐Deacetyl‐19‐HO‐T2 | C22H32O9 | 440 |
| HT2 | C22H32O8 | 424 |
| 7‐HO‐HT2 | C22H32O9 | 440 |
| 19‐HO‐HT2b (TC‐3) | C22H32O9 | 440 |
| 20‐HO‐HT2d | C22H32O9 | 440 |
| 7,19‐diHO‐HT2e | C22H32O10 | 456 |
| DE‐HT2 | C22H32O7 | 408 |
| DE‐19‐HO‐HT2b | C22H32O8 | 424 |
| Neosolaniol (NEO) | C19H26O8 | 382 |
| 4,8‐Diacetyl‐T2‐tetraol (NT‐1 toxin)f | C19H26O8 | 382 |
| 15‐Deacetyl‐NEO (NT‐2 toxin) | C17H24O7 | 340 |
| 4‐Deacetyl‐NEO (TMR‐1 toxin) | C17H24O7 | 340 |
| T2‐triol | C20H30O7 | 382 |
| 19‐HO‐T2‐triolb | C20H30O8 | 398 |
| DE‐T2‐triol | C20H30O6 | 366 |
| DE‐19‐HO‐T2‐triolb | C20H30O7 | 382 |
| T2‐tetraol | C15H22O6 | 298 |
| 15‐Acetyl‐T2‐tetraol | C17H24O7 | 340 |
| DE‐15‐Acetyl‐T2‐tetraol | C17H24O6 | 324 |
The trivial designations used for some of the metabolites are also given. The structural formulas are depicted in Appendix B.
HO: hydroxyl; DE: de‐epoxy; NEO: neosolaniol.
Also denoted as 3′ in the literature due to different numbering system.
Regioisomer of TC‐1 with acetoxy group at C‐3 and free hydroxy group at C‐4.
Also denoted as 4′.
Also denoted as 3′,7.
Regioisomer of NEO with acetoxy group at C‐8 and free hydroxy group at C‐15.
Figure 2.
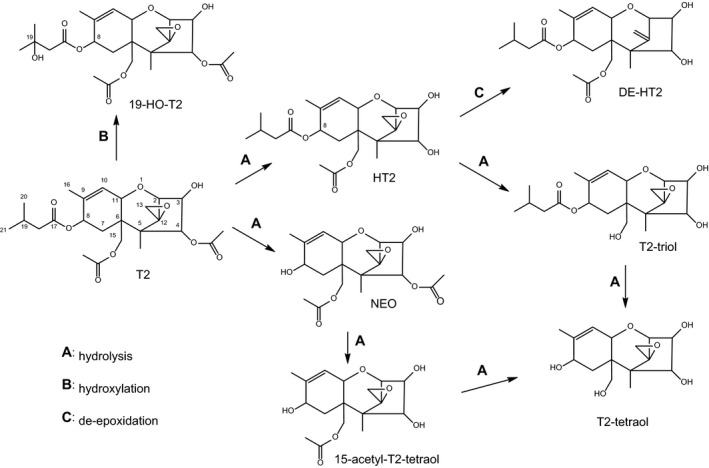
Major metabolic phase I pathways of T2 and HT2
The pathways of biotransformation of T2 and HT2 in fungi, plants and mammals are essentially the same and are depicted in Figure 2. Essentially, the predominant metabolic pathway A is the hydrolytic cleavage of one or more of the three ester groups of T2. Pathway B involves hydroxylation reactions at various positions of T2 and of hydrolytic metabolites. Hydroxylation occurs predominantly at C‐19, but also at C‐20, C‐8 and others. Pathway C constitutes the reduction of the epoxide group at C‐12 and C‐13 to an olefinic double bond.
4.2. Phase II metabolites
In most phase II metabolic pathways, water‐soluble conjugates are formed through covalent binding of the parent compound or a phase I metabolite with a highly polar molecule, e.g. a carbohydrate or sulfate from the metabolising organism. In most cases, activation of the conjugating moiety and a transferase enzyme are needed to form the covalent bond. Sulfate is used as a conjugate moiety by fungi, plants and animals. Different carbohydrates are also utilised by plants and fungi, the major carbohydrate employed for conjugation is glucose, whereas animals use glucuronic acid. The glucose can be further modified in plants, e.g. by esterification with another hexose or with malonic acid. Moreover, plants can use acetate and ferulate for conjugation. The major conjugating moieties in plants are depicted in Figure 3.
Figure 3.
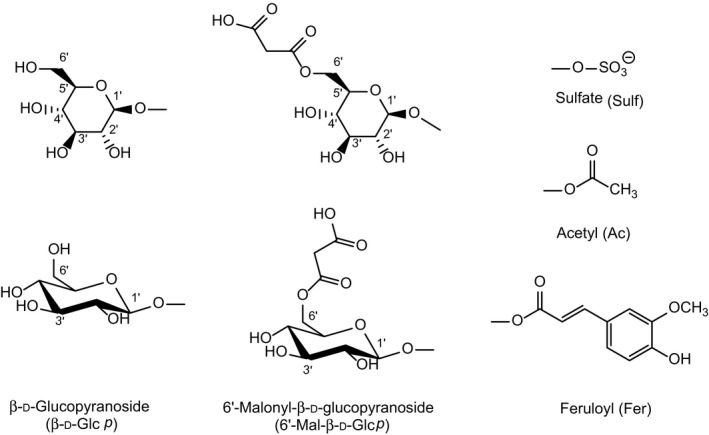
Glucose (depicted in two different stereochemical representations), modified glucose, and sulfate, feruloyl and acetyl groups used for the conjugation of T2 and HT2
For the abbreviation of the carbohydrates and their modified forms used in phase II metabolism, the same nomenclature as in the Scientific Opinion on the risks for human and animal health related to the presence of modified forms of certain mycotoxins in food and feed (EFSA CONTAM Panel, 2014) and in the Scientific Opinion on the appropriateness to set a group‐based guidance value for zearalenone and its modified forms (EFSA CONTAM Panel, 2016) will be used. These abbreviations, which are common in carbohydrate chemistry, clearly designate the specific carbohydrate (e.g. Glc for glucose, Man for mannose, Xyl for xylose, etc.) and its oxidation state (e.g. GlcA for glucuronic acid). For a complete designation, also the type of ring (p for pyranose or f for furanose), the configuration (d or l), and the type of glycosidic bond (α or β) could be given, if known. For further details, see Scientific Opinion on the risks for human and animal health related to the presence of modified forms of certain mycotoxins in food and feed (EFSA CONTAM Panel, 2014).
The phase II metabolites of T2, HT2 and their phase I metabolites identified in fungal and yeast cultures, plants and mammals are summarised in Table 2. The microbial and plant metabolites are discussed in more detail in Section 6. Conjugation of with glucuronic acid does not occur to an appreciable extent in plants and fungi but is a major pathway of phase II metabolism in animals and humans (Section 7.3.2).
Table 2.
Phase II metabolites of T2 and HT2 detected in fungi, yeast and plants
| Metabolitea | Element formula | Molecular weight | Comment |
|---|---|---|---|
| T2‐3‐α‐Glc | C30H44O14 | 628 | Fully characterised (1H and 13C NMR) |
| T2‐3‐β‐Glc | C30H44O14 | 628 | Fully characterised (1H and 13C NMR) |
| T2‐3‐diGlc | C36H54O19 | 790 | Exact chemical structure unknown |
| T2‐3‐GlcA | C30H42O15 | 642 | Fully characterised (1H and 13C NMR) |
| T2‐Sulf | C24H34O12S | 546 | Fully characterised (1H and 13C NMR) |
| 3‐Ac‐T2 | C26H36O10 | 508 | Chemically synthesised, no NMR spectra |
| 3‐Fer‐T2 | C34H42O12 | 642 | Exact chemical structure unknown |
| HT2‐3‐Glc | C28H42O13 | 586 | Exact chemical structure unknown |
| HT2‐diGlc | C34H52O18 | 748 | Exact chemical structure unknown |
| HT2‐3‐GlcA | C28H40O14 | 600 | Fully characterised (1H and 13C NMR) |
| HT2‐4‐GlcA | C28H40O14 | 600 | Fully characterised (1H and 13C NMR) |
| HT2‐MalGlc | C31H44O16 | 672 | Exact chemical structure unknown |
| HO‐HT2‐Glc | C28H42O14 | 602 | Exact chemical structure unknown |
| HO‐HT2‐MalGlc | C31H44O17 | 688 | Exact chemical structure unknown |
| 3‐Ac‐HT2 (iso‐T2) | C24H34O9 | 466 | Regioisomer of T2, tentatively identified |
| T2‐Triol‐Glc | C26H40O12 | 544 | Exact chemical structure unknown |
| T2‐Tetraol‐Glc | C21H32O11 | 460 | Exact chemical structure unknown |
| 15‐Ac‐T2‐tetraol‐Glc | C23H34O12 | 502 | Exact chemical structure unknown |
| NEO‐Glc | C25H36O13 | 544 | Exact chemical structure unknown |
The structural formulas of the fully characterised metabolites are presented in Appendix C.
Abbreviations of conjugate moieties: Glc: glucose; diGlc: diglucose; GlcA: glucuronic acid; Sulf: sulfate; Ac: acetyl; Fer: feruloyl; MalGlc: malonylglucose; NEO: neosolaniol; NMR: nuclear magnetic resonance.
The vast majority of the metabolites listed in Table 2 have only been tentatively identified, usually based on liquid chromatography–high resolution mass spectrometry (LC–HRMS), liquid chromatography–tandem mass spectrometry (LC–MS/MS) and enzymatic cleavage to release the aglycones. Chemical syntheses have been reported for T2‐3‐Glc, T2‐Sulf and 3‐Ac‐T2, while T2‐3‐GlcA, HT2‐3‐GlcA and HT2‐4‐GlcA have been prepared by enzymatic glucuronidation of the respective aglycones using pig liver microsomes.
T2‐3‐Glc is a major conjugate of T2 and formed in fungi, yeast and plants (see Section 6). Glycosides can exist in two stereoisomeric forms (α and β) which differ in the configuration of the glycosidic C‐atom. The α‐anomer of T2‐3‐Glc was obtained from incubations of T2 with three species of the yeast Blastobotrys (McCormick et al., 2012), and the β‐anomer of T2‐3‐Glc has been chemically synthesised (McCormick et al., 2015). Both anomers have been fully characterised by 1H and 13C NMR spectroscopy. When LC–MS/MS was used to compare both anomers to the T2‐3‐Glc isolated from wheat and oats naturally infected with Fusarium fungi, the latter was found to be the α‐anomer, based on high‐performance liquid chromatography (HPLC) retention time and tandem mass spectrometry (MS/MS) (McCormick et al., 2015).
Fruhmann et al. (2014) have recently synthesised the ammonium salt of T2‐3‐Sulf and characterised it by HRMS, 1H and 13C nuclear magnetic resonance (NMR), in order to obtain a reference compound for this metabolite. To date, however, no report has yet appeared on the detection of T2‐3‐Sulf in food or feed, which is to be expected in view of the reported formation of sulfates of closely related trichothecenes such as DON (Warth et al., 2015).
3‐Ac‐T2 is accessible by simple acetylation of T2 (Nathanail et al., 2015), but its full spectroscopic characterisation has not yet been reported.
Welsch and Humpf (2012) have isolated T2‐3‐GlcA, HT2‐3‐GlcA and HT2‐4‐GlcA from incubations of T2 with pig liver microsomes and activated glucuronic acid. In addition to direct glucuronidation of T2, which has only one hydroxyl group, the microsomes catalysed the deacetylation of T2 to HT2 and the subsequent formation of two regioisomeric HT2 glucuronides, which could be separated by HPLC. All three glucuronides were purified by preparative HPLC and characterised by NMR and MS.
5. Analytical methods
Methods for analysis of T2 and HT2 are well established and can be applied for the analysis of both food and feed (in particular cereals) and biological samples (such as body fluids).
The CONTAM Panel has extensively reviewed the analytical methods developed for T2 and HT2 in the Scientific Opinion on the risks for animal and public health related to the presence of T‐2 and HT‐2 toxin in food and feed (EFSA CONTAM Panel, 2011), thus this opinion will consider only those methods published since 2011.
T2 and HT2 are usually analysed together with some other Fusarium toxins such as DON or zearalenone. As for other Fusarium mycotoxins, sample extraction is based on mixtures of acetonitrile/water or methanol/water, acidified with acetic or formic acid (De Girolamo et al., 2013). Especially, when compound feed is considered, sample clean‐up is often performed to reduce matrix effects (Chen et al., 2013; De Girolamo et al., 2013). Recently, the use of liquid–liquid partition or ‘Quick, Easy, Cheap, Effective, Rugged and Safe’ (QuEChERS) clean‐up has been reported when matrices different from grains are considered (Arroyo‐Manzanares et al., 2013; Azaiez et al., 2014).
Methods, such as gas chromatography–mass spectrometry (GC–MS) or liquid chromatography‐fluorescence detection (LC‐FLD), are still in use in control laboratories. Both approaches require excessive sample purification and a derivatisation step prior to the analysis, thus affecting the overall recovery and sensitivity (Burek et al., 2011; Amelin et al., 2013a,b).
Similarly to other Fusarium toxins, the method used for the analysis of T2, HT2 and some of their modified forms (NEO, T2 and HT2 glucosides) has been LC–MS/MS, often as a multitoxin approach. However, no validated quantification methods for modified forms have been reported so far. Apart from NEO, modified forms are mainly detected with screening or semiquantitative methods, without properly optimised analytical parameters. Such methods might be inadequate with regard to recovery and sensitivity.
The two studies in which conjugated forms were determined were based on acetonitrile/water or methanol/water extraction followed by LC–MS/MS quantification, using a multitoxin approach (Busman et al., 2011; De Boevre et al., 2012). In both studies, conjugated forms were included as target analytes based on theoretical MS transitions, due to the lack of commercial standards. Similarly, the quantification of T2‐Glc and HT2‐Glc was based on extrapolation of chromatographic peak areas from T2 and HT2. This could strongly affect data reliability and accuracy. Only in one study so far, both anomeric forms of T2‐Glc were synthesised and used as reference compound for LC–MS/MS analysis (McCormick et al., 2015).
A more recent approach is based on ambient mass spectrometry, such as direct‐analysis‐in‐real‐time mass spectrometry (DART‐MS). This method, which requires the use of an isotope‐labelled internal standard for quantification, allows for an extremely rapid analysis with very good sensitivity (Busman and Maragos, 2015). However, the authors reported a sixfold lower sensitivity for HT2 than for T2, the limit of quantification being 300 μg/kg and 50 μg/kg, respectively.
Immunochemical methods have been proposed for the simultaneous determination of T2 and T2‐Glc. In a study by Maragos et al. (2013), a monoclonal antibody was specifically designed and validated towards T2 and T2‐Glc. Very low cross‐reactivity towards HT2 was seen. When used in an enzyme‐linked immunosorbent assay, the antibody gave a median inhibition concentration in the low ng/mL range.
When both anomeric forms of T2Glc were considered, the cross‐reactivity of the antibody towards the α‐ and β‐forms was 100% and 57% (McCormick et al., 2015). However, the system was not yet applied to real samples.
6. Occurrence
The occurrence of T2 and HT2 in food and feed has been previously considered by the CONTAM Panel (EFSA CONTAM Panel, 2011). Modified forms of T2 and HT2 have been described in food from plant origin, as a consequence of the biotransformation performed by plants and/or fungi. The chemistry of T2 and HT2 and their phase I and II metabolites is described in detail in Section 4, including the chemical structures of the major phase I metabolites including their interrelationships, which are depicted in Figure 2 of Section 4. The formation of phase I and phase II metabolites in mammals is described in Section 7.3. The chemical structures of all phase I and phase II metabolites of T2 and HT2 are presented in Appendices B and C, respectively.
In Table 3, the origin of the major modified forms of T2 and HT2 that might occur in food or feed and reported so far in the literature are listed.
Table 3.
Occurrence of modified forms of T2 and HT2 in food (plants, fungi and mammals)
| Modified form | Formed in plants | Formed in fungi | Formed in mammals |
|---|---|---|---|
| NEO | Torp and Langseth (1999) | Wu et al. (2011) | |
| T2‐Triola | Torp and Langseth (1999) | Wu et al. (2014) | |
| T2‐Tetraola | Torp and Langseth (1999) | Wu et al. (2014) | |
| T2‐Glcb | McCormick et al. (2012) | Busman et al. (2011) | |
| 3‐Ac‐T2a | Nathanail et al. (2015) |
McCormick et al. (2012) Nathanail et al. (2016) |
|
| 3‐Fer‐T2a | Nathanail et al. (2015) | ||
| HT2‐3‐Glc | Nathanail et al. (2015) | Busman et al. (2011) | |
| HT2‐diGlc | Veprikova et al. (2012) | ||
| HT2‐Sulf | Nathanail et al. (2016) | ||
| T2‐diGlc | Veprikova et al. (2012) | ||
| HT2‐MalGlca | Nathanail et al. (2015) | ||
| HO‐HT2‐Glca | Nathanail et al. (2015) | ||
| HO‐HT2‐MalGlca | Nathanail et al. (2015) | ||
| 3‐Ac‐HT2a | Nathanail et al. (2015) | ||
| T2‐Triol‐Glca | Nathanail et al. (2015) | ||
| T2‐Tetraol‐Glca | Nathanail et al. (2015) | ||
| 15‐Ac‐T2‐tetraol‐Glca | Nathanail et al. (2015) | ||
| NEO‐Glc | Nakagawa et al. (2013a) | Lattanzio et al. (2013) |
Glc: glucose; Ac: acetyl; Fer: feruloyl; diGlc: diglucose; Sulf: sulfate; MalGlc: malonylglucose; NEO: neosolaniol.
The compound has been found in model plants (i.e. wheat, barley) after artificial fungal inoculation, but there is no data about occurrence in the field.
Elucidated as T2‐3‐α‐Glc in plants and T2‐3‐β‐Glc in microorganisms (McCormick et al., 2015).
Of particular importance for the health assessment of modified forms of mycotoxins in food and feed is the question where the phase I and II metabolites are formed, i.e. in the fungi, plants or animals and enter the food chain. In contrast to animals, which tend to eliminate water‐soluble metabolites through urine and faeces, phase I and phase II metabolites formed in the fungus and/or plant must be expected to remain in the infested plant and may therefore reach the consumer via food items containing the contaminated plant material.
Modified forms of T2 and HT2 obtained by phase I metabolism have been less frequently reported in the literature, with the only exception of NEO that has been described as fungal metabolite in many commodities. This is likely due to the fact that NEO is the only phase I metabolite of T2 and HT2 commercially available as standard.
The formation of phase I metabolites T2‐triol and T2‐tetraol have been reported in fungal culture (Torp and Langseth, 1999). Several yeast strains have been reported to be able to biotransform T2 and HT2 to their phase I metabolites. McCormick et al. (2012) investigated several Trichomonascus clade (n = 23) and Blastobotrys (n = 19) yeast strains. The formation of 3‐Ac‐T2, NEO and T2‐Glc were reported. Some species performed more than one type of biotransformation reaction. Very recently, Nathanail et al. (2016) identified 3‐Ac‐T2 and HT2‐Sulf as products by Saccharomyces pastorianus in brewer's wort.
Phase II conjugates of T2 and HT2 are mainly formed in plants. However, T2‐Glc and HT2‐Glc glucosides may also result from fungal metabolism. Busman et al. (2011) were the first to detect and tentatively identify the 3‐O‐Glc of T2 and HT2 in cultures of Fusarium sporotrichioides as well as in wheat and oats. The formation of HT2‐Glc glucoside and NEO‐Glc in Fusarium langsethiae isolates cultured on durum wheat was reported by Lattanzio et al. (2013).
McCormick et al. (2015) demonstrated that T2‐Glc produced by fungi and by plant can be anomerically different as fungal biotransformations have been shown to preferentially result in the α‐Glc, while plant enzymatic pathways yield the β‐Glc (McCormick et al., 2015).
Besides conjugation with glucose, T2 and HT2 can be further biotransformed in plants following other phase II pathways resulting in other conjugates. In particular, Nathanail et al. (2015) and Meng‐Reiterer et al. (2015) studied the metabolism of T2 and HT2 in wheat and barley, respectively, by applying uniformly 13C‐labelled toxins to the flowering ears of wheat plants and profiling the metabolites using untargeted screening and quantitative LC–HRMS analysis. A total of 12 T2‐derived and 11 HT2‐derived biotransformation products were detected. Previously reported mono‐ and diGlcs were confirmed, and several malonylglucosylated, acetylated and feruloylated metabolites were reported for the first time. In addition, the time courses of metabolite formation were determined. Phase II metabolism proceeded rapidly and was almost completed within 1 week after a single exposure in flowering wheat ears.
6.1. Occurrence of modified forms of T2 and HT2 in food of plant origin
6.1.1. Phase I metabolites
Although plants and fungi may produce phase I metabolites of T2 and HT2, the CONTAM Panel could only identify very few relevant data for their occurrence in food. Among phase I metabolites, only NEO and T2‐tetraol were reported in food commodities. Recently, García‐Moraleja et al. (2015) reported the occurrence of NEO in brewed coffee samples (n = 169). According to the authors, NEO was found in 13% of the samples in the concentration range of 29–314 μg/kg (mean: 86.7 μg/kg). The same authors reported the occurrence of T2 and HT2 in 29% and 9% of the samples (mean concentration: 2.4 and 12.2 μg/kg), respectively. Pereira et al. (2015) reported the occurrence of NEO (87 μg/kg) and T2‐tetrol (112 μg/kg) in one out of nine baby food samples containing cereals, using GC–MS as an analytical method. NEO was also found at trace level in barley field malt samples from northern Italy (Lattanzio et al., 2015).
6.1.2. Phase II metabolites
Many conjugated forms of T2 and HT2 have been reported in the literature so far as a result of plant metabolism. However, the few studies addressing the occurrence of such forms in grains and food products thereof analysed only occurrence of T2‐3‐Glc and HT2‐3‐Glc.
Lattanzio et al. (2012) estimated the occurrence of T2‐3‐Glc and HT2‐3‐Glc in artificially inoculated wheat and oats. Collected data reported amounts of HT2‐3‐Glc and T2‐3‐Glc of up to 27% and 24% of their parent compounds, respectively (mean: 10%). The same authors found up to 37% HT2‐3‐Glc compared to parent compound, depending on the harvest season (Lattanzio et al., 2013). In a more recent survey, the occurrence of T2 and HT2 glucosides was reported in field barley (Lattanzio et al., 2015). In this study, HT2‐3‐Glc was detected at levels of up to 163 μg/kg in 17 out of 18 barley samples, whereas T2‐3‐Glc was detected in only a few samples and at low μg/kg levels. The sum of T2 and HT2 conjugates ranged from 2% to 280% in comparison with the total of the parent forms.
Veprikova et al. (2012) detected monoglycosylated forms of T2 and HT2 in naturally contaminated barley, wheat and oats, and also documented for the first time the existence of one diglucoside of T2 and two diglucosides of HT2 in barley. Diglucosidic conjugates of T2 were reported in corn by Nakagawa et al. (2013b). The same authors reported the detection of a conjugate of the phase I metabolite NEO in corn (Nakagawa et al., 2013a).
In a survey, De Boevre et al. (2012) collected 30 samples from a variety of food and feed matrices including maize (n = 6), wheat (n = 6), oats (n = 6), cornflakes (n = 6) and bread (n = 6) from the Belgian market. In 67% of the samples, T2 and HT2 were detected at a trace level. One oat sample was contaminated with 118 μg/kg HT2 and 34 μg/kg T2, while the mean values in oats were 31 and 15 μg/kg, respectively. Although T2‐3‐Glc and HT2‐3‐Glc were included among the monitored compounds, they were not detected in any sample in this survey, most probably because of the low sensitivity of the methods applied.
A review on the occurrence of Fusarium mycotoxins in unprocessed cereals has been published by Broekaert et al. (2015). The concentrations of conjugated T2 and HT2 were substantially lower than those of T2 and HT2 in the samples where both T2 and HT2 and conjugated T2 and HT2 were detected. The total number of samples containing conjugated T2 and HT2 was higher than samples containing unconjugated T2 and HT2. For T2‐3‐Glc and HT2‐3‐Glc, the numbers of positive samples were 73% and 80%, respectively (n = 15), with a mean and maximum concentration of 2.4 μg/kg and 11 μg/kg T2‐3‐Glc, and of 5.1 μg/kg and 15 μg/kg HT2‐3‐Glc. For T2 and HT2, the fractions of positive samples were 45% and 54%, respectively (n = 321), with a mean and maximum concentration of 16.7 μg/kg and 377 μg/kg T2, and of 61 μg/kg and 834 μg/kg HT2. Overall, the mean concentrations of conjugated T2 and HT2 were substantially lower than those of T2 and HT2, with an average percentage of 8.3% for HT2‐Glc and 14.4% for T2‐Glc with respect to the parent compounds.
Nathanail et al. (2015) studied the occurrence of major Fusarium toxins and their modified forms in oats (n = 31), barley (n = 34), and wheat (n = 30) from Finland. The method was calibrated for T2, HT2 and HT2‐3‐Glc because of the available standard compounds. T2, HT2 and HT2‐3‐Glc were found in 21%, 35% and 53% of the barley samples, and the mean values were 10.7, 20 and 10.8 μg/kg, respectively. As far as oats are concerned, T2, HT2 and HT2‐3‐Glc were found in 61.3%, 74.2% and 58.1% of the samples, and the mean values were 60.1, 159 and 41.4 μg/kg, respectively. Finally, in wheat, T2, HT2 and HT2‐3‐Glc were found in 46.7%, 63.3%, and 43.3% of the samples, and the mean values were 4.2, 9 and 15 μg/kg, respectively. Maximum concentrations were particularly high in oats, reaching maximum values of 548, 1,830 and 300 μg/kg for T2, HT2 and HT2‐3‐Glc, respectively.
In contrast to the preliminary data on the occurrence of T2 and HT2 conjugates in grains indicating an incidence of up to 10% of the parent compounds as presented in the previous opinion (EFSA CONTAM Panel, 2014), the studies presented above reported higher occurrence rates of conjugates in comparison with the parent compounds. This can be ascribed to improvements of analytical methods, as well as to the variability in sampling time and harvest season conditions (i.e. grain varieties, climate).
Overall, the proportion of occurrence of modified forms of T2 and HT2 relative to the respective parent compounds, in the few samples where this has been determined, varies widely.
6.2. Occurrence of modified forms of T2, HT2 and their modified forms in food of animal origin
No relevant information about the occurrence of T2, HT2 or their modified forms in food of animal origin has been identified by the CONTAM Panel. It can generally be assumed that at least a part of them will be excreted (after conjugation in the case of phase I metabolites) by the animal prior to slaughter and processing to food items. This assumption is supported by studies indicating that animals rapidly excrete T2 and its metabolites in urine and faeces, mainly as glucuronides (EFSA CONTAM Panel, 2011; Welsch and Humpf, 2012).
7. Toxicokinetics of T2 and HT2 and their modified forms
No studies on the toxicokinetics of modified forms (phase I metabolites and phase II metabolites) of T2 or HT2 have been identified. The toxicokinetics of T2 and HT2 have been reviewed by the Joint FAO/WHO Expert Committee on Food Additives (FAO/WHO, 2001) and EFSA (EFSA CONTAM Panel, 2011). The following sections will briefly summarise the major findings and discuss in more detail pertinent studies published since 2011. There are also some recent review papers on trichothecenes including T2 and HT2 (Wu et al., 2014; Broekaert et al., 2015).
7.1. Absorption
Studies on the in vivo absorption of T2 and HT2 in animals after oral administration are very scarce. To date, the bioavailability of T2 and HT2 has not been quantified. However, when tritium‐labelled T2 was intraduodenually administered to bile duct‐cannulated male rats, 45% and 57% of the dosed radioactivity was excreted in the bile within 4 and 8 h, respectively (Coddington et al., 1989). Likewise, about 40% of the total radioactive dose appeared in the effluent blood within 50 min after injection of tritium‐labelled T2 into isolated autoperfused jejunal segments of rats (Conrady‐Lorck et al., 1988). Only minute amounts of T2 were observed in the bile and blood in these studies, suggesting an extensive hydrolysis to HT2 and other metabolites during the rapid intestinal absorption of T2. The presumed rapid absorption is consistent with the fact that the excretion of total radioactivity in the urine and faeces of rats was completed 48 h after a single oral dose of tritium‐labelled T2 administered by gavage (Pfeiffer et al., 1988).
No studies on the absorption of modified forms (phase I and phase II metabolites) of T2 or HT2 have been identified.
7.2. Distribution
In rats and mice orally dosed with tritium‐labelled T2, radioactivity was rapidly distributed to the liver, kidney and other organs without accumulation in any organ (Matsumoto et al., 1978; EFSA CONTAM Panel, 2011). Radioactivity was also detected in the fetuses of late pregnancy rats following oral administration of tritium‐labelled T2 (Lafarge‐Frayssinet et al., 1990). Wang et al. (2014) have recently studied the transfer mechanisms of T2 and HT2 in human placental choriocarcinoma (BeWo) cells, which are derived from human choriocarcinoma cells and considered as an in vitro model for maternal–fetal exchange. Using LC–MS/MS analysis, an active transport mechanism was suggested for the uptake of T2 into BeWo cells, whereas passive diffusion was observed for HT2.
No studies on the distribution of modified forms (phase I and phase II metabolites) of T2 or HT2 have been identified.
7.3. Metabolism
The complex metabolism of T2 and HT2 in animals and humans has been addressed in the previous risk assessment of T2 and HT2 in food and feed (EFSA CONTAM Panel, 2011) and are also discussed in a recent review (Wu et al., 2014). No information on the biotransformation of modified forms of T2/HT2 from food has been identified.
7.3.1. Phase I metabolism
Most of the numerous phase I metabolites of T2 and HT2 known to date have been reported prior to 2011, and only few minor metabolites have been identified since then. As depicted in Figure 2 (Section 4.1), the phase I metabolites arise from three different biotransformation reactions, i.e. (A) hydrolysis of one or more of the ester groups, (B) hydroxylation at various positions and (C) de‐epoxidation. These metabolic reactions may also occur in combination, and the resulting phase I metabolites are listed in Table 1 (see Section 4.1).
Various enzymes are involved in the phase I metabolism of T2 and HT2 in mammals (Wu et al., 2014). While the hydrolytic pathway A is mediated by carboxylesterases located in hepatic microsomes and white and red blood cells, a variety of cytochrome P450 (CYP) isoforms has been implicated in the hydroxylation reactions of pathway B. The enzymes accounting for the de‐epoxidation pathway C have yet to be clarified.
A few minor metabolites (not included in Table 1) were tentatively identified more recently in Wistar rats in vitro and in vivo (Yang et al., 2013). These are 15‐deacetyl‐T2, 15‐deacetyl‐19‐hydroxy‐T2, 7,19‐dihydroxy‐T2 and a regioisomer, 7‐hydroxy‐HT2 and a regioisomer, as well as another regioisomer of hydroxy‐T2 and dihydroxy‐T2 not further identified.
No in vivo studies on the metabolism of T2 and HT2 in humans are available, but data from in vitro experiments indicate that the same metabolic pathways as observed for T2 and HT2 in animal studies operate in humans. After incubation of T2 with primary renal proximal tubule epithelial cells, primary human lung fibroblasts or HT‐29 cells (a cell line derived from human colon carcinoma cells), several of the hydrolytic and monohydroxylated T2 metabolites (as shown in Figure 2) were identified using LC–MS (Weidner et al., 2012). HT2 and NEO were the major metabolites, whereas minor amounts of 19‐hydroxy‐T2, T2‐triol, and 4‐deacetyl‐NEO were detected.
7.3.2. Phase II metabolism
The glucuronides of T2 and HT2, which appear to be the most prevalent mammalian phase II metabolites of these toxins, are listed in Table 2 of Section 4. Several older in vivo and in vitro studies in animals provide indirect evidence for the formation of glucuronides of T2, HT2, 3‐hydroxy‐T2 and 3‐hydroxy‐HT2 by demonstrating the release of the respective aglycones from a polar metabolite fraction, assumed to represent glucuronides, upon treatment with the enzyme β‐glucuronidase (reviewed by Weidner et al., 2012). More recently, LC–MS together with reference compounds has been used to identify T2‐GlcA and HT2‐3‐GlcA in two human cell types after incubation with T2 (Weidner et al., 2012). When hepatic microsomes of rat, mouse, pig and human were incubated with T2 in the presence of uridine 5′‐diphospho‐glucuronic acid (UDPGA) and the pattern of glucuronides determined by LC–MS/MS, human microsomes exhibited a higher activity for glucuronidation than rodent or pig microsomes. Of the two regioisomers of HT2‐GlcA, the 3‐isomer was exclusively formed in rat, mouse and human microsomes, and only pig hepatic microsomes generated both isomers. A mixture of HT2‐3‐GlcA and HT2‐4‐GlcA was also demonstrated in the urine of female pigs dosed orally or by intravenous (i.v.) injection with T2 (Weidner et al., 2012).
7.4. Excretion
No study on the excretion of T2 or HT2 or their metabolites has been published since 2011. As stated in the previous EFSA opinion, animals rapidly excrete T2 and its metabolites in urine and faeces, mainly as glucuronides, with the urine to faeces ratio depending on the species (EFSA CONTAM Panel, 2011; Welsch and Humpf, 2012). For example, almost all the radioactivity after a single dose of tritium‐labelled T2 given by intubation was excreted by a lactating cow within 72 h (Yosizawa et al., 1981); urinary excretion was completed after 48 h and accounted for 30% of the dose. Similar results were obtained after a single oral dose of tritium‐labelled T2 administered to mice or rats (Matsumoto et al., 1978; Pfeiffer et al., 1988). In guinea pigs, 75% of the radioactivity was excreted in the urine and faeces at a ratio of 4 to 1 after 5 days, albeit after a single intramuscular injection of tritiated T2 (Pace et al., 1985).
8. Toxicity
8.1. In vivo toxicity data on T2 an HT2
8.1.1. Study used for establishing the TDI by EFSA 2011
In their previous opinion (EFSA CONTAM Panel, 2011), the Panel concluded that T2 induces haemato‐ and myelotoxicity, and noted that T2 was positive in some clastogenicity tests but mainly at concentrations known to inhibit protein and DNA synthesis and produced cytotoxicity. The Panel concluded at that time that T2 inhibited protein‐, DNA‐ and RNA synthesis and that there were studies indicating that T2 causes apoptosis, necrosis and lipid peroxidation.
A subacute study (21 days) with pigs (Rafai et al., 1995) was identified as the most appropriate one to derive a HBGV for T2 despite the shortness of the study and the fact that the purity of T2 was only 90%. The toxin was prepared by fermentation of Fusarium tricinctum. The calculated amount of T2 was used to prepare a corn meal‐based premix, which was then added to the daily ration. Nine to ten piglets per group (gender not reported, mean weight of animals at beginning of study about 9 kg) were fed diets containing 0, 0.5, 1, 2 or 3 mg T2/kg diet (average daily feed intakes were 0, 0.38, 0.81, 1.24 and 1.43 mg, respectively) over a period of 21 days, resulting in doses of 0, 29, 62, 105 or 129 μg T2/kg bw per day, respectively. The CONTAM Panel noted that in this study the feed intake was decreased significantly (by 13%) already at the lowest dose which was also reflected in the decreased bodyweight gain observed. No pair fed controls was investigated, which might have led to biased observations of other effects. Decreased stimulation of lymphocytes by concanavalin A (ConA), decreased leucocyte and T‐cell counts as well as reduced anti‐horse immunoglobulin titre and histological changes in the thymus, spleen and lymph nodes were observed. Reduction in antibody response to a specific antigen was identified as the critical effect for human risk assessment (a LOAEL of 29 μg T2/kg bw per day was identified while no NOAEL could be identified in this study) and used for a BMD analysis. A BMDL05 of 10 μg T2/kg bw per day was established. Since T2 is rapidly metabolised to HT2 and the toxicity of T2 might at least partly be attributed to HT2, a group TDI of 0.1 μg/kg bw per day was established for the sum of T2 and HT2 by applying an UF of 100 to the BMDL05.
8.1.2. In vivo toxicity studies with T2 and HT2 toxin published after 2011
The following subchapters provide an overview on oral in vivo studies with T2 and HT2 published after publication of the EFSA opinion of 2011. When the doses tested were reported only in mg/kg diet, they have been calculated to mg/kg bw per day according to respective EFSA guidances (EFSA FEEDAP Panel, 2012; EFSA Scientific Committee, 2012).
8.1.2.1. Acute toxicity studies
Mice
Wu et al. (2015) used a mouse model to compare the anorectic potencies of T2 with HT2. Groups of six female B6C3F1 mice were dosed with 0, 0.01, 0.1, 0.5 and 1 mg T2 or HT2/kg bw by single oral gavage and were then immediately given preweighed food pellets. Food intakes were measured 0.5, 1, 2, 3, 6, 16, 24 and 48 h after exposure. T2 and HT2 induced anorectic responses (marked reduction in feed consumption) that lasted up to 48 h. The authors identified a LOAEL of 0.1 mg/kg bw and a NOAEL of 0.01 mg/kg bw for both compounds. It is notable that, when the animals were administered T2 and HT2 at the same doses intraperitoneal (i.p.), anorectic responses were induced at the same dose (LOAEL of 0.1 mg/kg bw) lasting up to 96 h after injection.
Mink
Fasted female mink (Neovison vison, four animals/group) were administered T2 or HT2 (0, 0.001, 0.01, 0.05 and 0.25 mg/kg bw) (experiment 1) or emetine (0, 0.5, 1, 2.5 and 5 mg/kg bw) by i.p. injection, and T2 or HT2 (0, 0.005, 0.05, 0.25 and 0.5 mg/kg bw) or emetine (0, 0.5, 1, 2.5 and 5 mg/kg bw) (experiment 2) by oral gavage (Wu et al., 2016). Thirty minutes before gavage, 50 g of feed were given to the animals. Then they were monitored for emetic events for 3 h. During this period, the incidence of emesis, the latency to emesis, duration of emesis and the number of emetic events were screened. An emetic event was described as either vomiting or retching. Wu et al. (2016) defined vomiting as a rhythmic abdominal contraction with oral expulsion of solid or liquid material. Retching is defined as a reaction that mimicked vomiting but without voiding material. Latency to emesis is the time from application of the substance to the first emetic event. Emesis duration is the time from the first until the last emesis. The lowest dose that induced an emetic response by oral dosing was 0.05 mg/kg bw with an incidence of three out of four animals for both T2 and HT2. At all the effective doses, both retching and vomiting took place, with retching as the more frequent event occurring. Following i.p. administration the incidence was lower, one out of four animals at the same dose. Emetic events occurred in four animals at a dose of 0.25 mg/kg bw by both administration routes. The latency decreased while duration and frequency of emetic events increased with dose. Oral administration of T2 and HT2 elicited marked elevations in plasma concentrations of the anorectic peptide pancreatic peptide YY3‐36 (PYY3‐36) and 5‐hydroxytryptamine (5‐HT), hormones known to be implicated in emesis. According to the authors the effective dose (ED) resulting in emetic events in 50% (ED50) of the animals for oral exposure to T2 and HT2 was 30 μg/kg bw.
For the ipecacuanha alkaloid emetine, that was used as a positive control in this study. The ED50 was 1.03 mg/kg bw. Emetine is a major alkaloid in ipecacuanha syrup, which has been used to induce vomiting in humans. The dose of emetine effective in humans is in the same range as that given to the mink and the mink has been suggested to be used as a model for emesis in drug testing (Gordon, 1985; Zhang et al., 2006; Percie du Sert et al., 2012). The CONTAM Panel therefore concludes that the mink is an appropriate animal model to investigate vomiting in humans.
8.1.2.2. Subacute toxicity studies
Rats
T2 (20 mg/kg feed, equivalent to 2.4 mg/kg bw per day) was fed to groups of six male Wistar rats for 14 days (Chandratre et al., 2014). Animals showed significant haematological alterations (total thrombocyte count was 2.6 × 103/μL compared to 17.07 × 103/μL in controls; total leucocyte count was 1.29 × 103/μL compared to 14.17 × 103/μL in controls) and increased levels of biological markers of oxidative stress with concomitant decrease in levels of serum and tissue catalase and superoxide dismutase were observed which were associated with histopathological changes. No residual T2 was detected in any of the organs tested, suggesting that T2 does not accumulate in tissues even at such a high exposure level.
Kashin–Beck disease (KBD) is an endemic degenerative osteoarthropathy that has been observed in selenium‐deficient areas and is supposedly connected to T2 and HT2 exposure. The mechanisms underlying its pathogenesis remain unclear. Since publication of the EFSA opinion on T2 and HT2 (EFSA CONTAM Panel, 2011) several subacute studies have been carried out to elucidate the pathogenesis of this disease (Chen et al., 2012; Guan et al., 2013; Kang et al., 2013). The CONTAM Panel noted that these new studies are not suitable for hazard characterisation of T2/HT2 and are therefore not presented.
Pigs
Obremski et al. (2013) administered doses of 10 μg T2/kg bw per day to piglets for 42 days. Ileum sections were collected on days 14, 28 and 42. After 42 days, a significant drop in the messenger RNA (mRNA) level of interleukin (IL)‐10 in ileal Peyer's patches was observed. A gradual, non‐significant decrease in the amount of IL‐4 and interferon (IFN)‐gamma cytokine transcripts was found throughout the experiment. On days 14 and 42, a significant increase in the percentage of cluster of differentiation (CD)8+ T lymphocytes was observed, while it was decreased on day 28. Percentage of CD21+ B cells in the treated animals group decreased steadily and was significant on days 28 and 42. On days 14 and 28, the percentages of CD4+ and CD8+ T lymphocytes were lowered in the experimental animals as compared with the control group, reaching statistical significance on day 28. The CONTAM Panel noted that only one dose level was tested in this study and that there is no clear evidence that the effects observed were related to adversity.
Three groups of 10 growing pigs were fed for 21 days diets containing 0, 0.3 and 0.5 mg T2/kg feed equal to 0, 11.5 and 18.6 μg T2/kg bw per day (Rafai et al., 2013). T2 was prepared by the fermentation of F. tricinctum (NRLL 3299) in a liquid medium. T2 significantly impaired feed intake and growth rate of pigs. Mean daily weight gains were 497, 377 and 317 g for control, low and high dose, respectively. Neither treatment level of T2 caused consistent nor significant changes in the metabolic, immunological and other blood parameters tested. The authors suggest that the considerably lower T2 intake due to feed refusal seen in this study might explain that immunotoxicity was not observed unlike in their previous study (Rafai et al., 1995).
8.1.2.3. Subchronic toxicity studies
Rats
Raut et al. (2013) fed rats with feed containing 0, 0.25, 0.50 and 0.75 mg/kg T2, respectively (equivalent to 0, 23, 45 and 68 μg T2/kg bw per day, respectively) for 90 days. A significant, dose‐ and duration‐dependent thrombocytopenia was observed. Mean thrombocyte counts were reduced with increasing dose. Total erythrocyte counts and total leucocyte counts were reduced, albeit not statistically significantly. The non‐statistically significant reduction in total leucocyte counts was attributed by the authors to lymphocytopaenia as (not statistically significant) decreases in the proportion of lymphocytes of the total leucocyte counts were observed. Hypoproteinaemia was diagnosed (reduced level of albumin and globulin paralleled by increased alanine amino transferase (ALT), aspartate amino transferase (AST) activities and creatinine levels). Alkaline phosphatase (ALP) activity was reduced at higher doses. The liver and kidneys were pale and slightly enlarged in animals at the highest dose. The relative liver, kidney and brain weight increased while the testes, thymus and spleen weights were reduced. Lipid peroxidase (LPO) activity was increased whereas superoxide dismutase (SOD) and catalase activities decreased. The CONTAM Panel did not identify a NOAEL in this study and considered the lowest dose tested (23 μg/kg bw per day) as a LOAEL. The Panel noted that while the decrease in total thrombocyte counts is in line with findings in other studies, the absence of significant effects on total leucocyte count and total erythrocyte count is not. In addition, the CONTAM Panel noted that the reporting of units for blood cell count in this study is unclear. Therefore, it was decided not to consider this study for hazard characterisation.
Rahman et al. (2014) gave feed containing 0, 0.5, 0.75 and 1 mg T2/kg (equivalent to 0, 45, 68 and 90 μg T2/kg bw per day, respectively) to male Wistar rats (48 per group) via the diet, daily for a period of up to 12 weeks. Subgroups of eight animals were sacrificed after 2, 4, 6, 8, 10 and 12 weeks. Cultures of F. sporotrichioides var. sporotrichioides MTCC 1894, were utilised to produce T2 on partially ground maize and intact wheat grains. In rats treated for 90 days, a statistically significant dose‐dependent decrease in body weights was seen. Mean body weights were 264, 219, 183 and 159 g in groups applied 0, 45, 68 and 90 μg T2/kg bw per day, respectively. Significant decreases in haemoglobin, packed cell volume, total erythrocyte, total thrombocyte and total leucocyte counts, mean corpuscular volume (MCV), mean corpuscular haemoglobin, and percentages of lymphocytes were observed while the percentage of neutrophils increased. Overall, all these observations became more pronounced with study length. At the end of the study, the mean total erythrocyte count levels were 8.97, 5.85, 5.77, 4.65 × 106/μL in animals treated with 0, 45, 68 and 90 μg T2/kg bw per day, respectively, mean total leucocyte count levels were 14.83, 8.95, 6.92 and 5.20 × 103/μL for groups receiving 0, 45, 68 and 90 μg T2/kg bw per day, respectively, and mean total thrombocyte count levels were 123, 78, 57 and 38 × 103/μL in groups treated with 0, 45, 68 and 90 μg T2/kg bw per day, respectively. Overall, the authors concluded that in rats, T2 induces microcytic hypochromic anaemia and leukocytopaenia (due to lymphocytopaenia) and thrombocytopenia increasing with dose and duration of exposure. The Panel did not identify a NOAEL in this study and considered the lowest dose tested (45 μg/kg bw per day) as a LOAEL. The Panel noted that the adverse effects observed in this study (microcytic hypochromic anaemia, leukocytopaenia, and thrombocytopenia) are not only consistent with those of previously published in vitro and in vivo studies (see EFSA CONTAM Panel, 2011) but also that the total thrombocyte count values reported in the study are unusually low. The Panel noted that the purity of the substance was not specified and nominal concentrations reported might not correspond to those actually tested. Furthermore, it cannot be excluded that other mycotoxins were present in the tested material limitations that also apply to the studies of Raut et al. (2013) and Rafai et al. (1995), the latter used for establishment of a TDI in 2011.
8.1.2.4. Developmental toxicity studies
Mice
Tanaka et al. (2016) investigated the effects of T2 on post‐natal hippocampal neurogenesis in mice. Dams were given T2 at 0, 1, 3 or 9 mg/kg in the diet from gestation day (GD) 6 to day 21 corresponding to exposures of 0, 0.14, 0.40 and 1.18 mg T2/kg bw per day during gestation and to 0, 0.49, 1.39 and to 3.79 mg T2/kg bw per day during lactation. Offspring were maintained through postnatal day (PND) 77 without T2 exposure. At PND 21, changes in the hippocampus paralleled with increased apoptosis were seen in male offspring of dams of the two highest dose groups and reduced relative brain weight was seen in male offspring of dams treated with the highest dose. Neurogenesis‐related changes disappeared on PND 77, suggesting that T2 reversibly affects neurogenesis by inducing apoptosis. The authors identified a NOAEL of 140–490 μg/kg body weight per day for effects of T2 on offspring neurogenesis.
8.1.2.5. Concluding remarks
New acute toxicity and subacute studies on anorectic effects (e.g. feed refusal, retching, vomiting, reduced body weight gain) at low doses in several species (mouse, mink and pig) have become available, indicating that there is a need for establishing an ARfD. In this context, it is noted that nausea and emesis has been reported in humans consuming mouldy grain contaminated with T2 producing strains of Fusarium poae and F. sporotrichoides (EFSA CONTAM Panel, 2011).
The Panel concluded that the new subchronic toxicity studies investigating similar endpoints but in longer term studies than those performed in pigs and used as a basis in the previous assessment, confirmed immuno‐ and haematotoxicity of T2 and HT2 (see also EFSA CONTAM Panel, 2011).
8.2. Toxicity data on modified forms of T2 and HT2
For this section, all available relevant literature has been considered (i.e. also publications prior to 2011).
8.2.1. In vivo toxicity of phase I metabolites
Only few in vivo studies evaluating the toxicity of phase I metabolites relative to T2 and HT2 were identified. All studies were acute toxicity studies and are presented in Table 4. In order to compare the toxic potency of the different metabolites to that of T2, the effective dose of each metabolite has been expressed on a molar basis, and the effective molar dose of T2 has been set to 1 for all studies.
Table 4.
In vivo acute toxicity studies comparing the toxicity of T2 and their phase I metabolites
| Species/administration | Endpoint/dose range | Effective molar doses relative toxicity)a | Reference |
|---|---|---|---|
| Ducklings/s.c. | Minimum emetic dose/dose range not reported |
T2: 0.21 μmol/kg bw (1) HT2: 0.24 μmol/kg bw (0.88) NEO: 0.26 μmol/kg bw (0.81) |
Ueno et al. (1974) |
| Broiler chicks/p.o. |
LD50/7 doses per dose range 6.2–18.7 μmol/kg bw 7.1–32.5 μmol/kg bw 26.2–127 μmol/kg bw 26.2–110 μmol/kg bwb 33.6–235 μmol/kg bw |
T2: 10.7 μmol/kg bw (1) HT2: 17.0 μmol/kg bw (0.63) NEO: 65.1 μmol/kg bw (0.16) T2‐triol: 79.0 μmol/kg bw (0.14) T2‐tetraol: 113 μmol/kg bw (0.094) |
Chi et al. (1978) |
| Male mice/i.p. | LD50/dose range not reported |
T2: 11.2 μmol/kg bw (1) HT2: 21.2 μmol/kg bw (0.53) NEO: 38.0 μmol/kg bw (0.29) T2‐triol: 283 μmol/kg bw (0.04) |
Ueno et al. (1986) |
| Female mice/i.v. | Induction of apoptosis in thymus cells (%DNA fragmentation above negative control (2%)/dose of 1.56 mg toxin/kg bw |
T2 (3.35 μmol/kg bw): 22% (1) HT2 (3.68 μmol/kg bw): 3% (0.14) 19‐HO‐T2c (3.24 μmol/kg bw): 24% (1.1) 19‐HO‐HT2c (3.55 μmol/kg bw) 4% (0.2) NEO (4.05 μmol/kg bw): 0.5% (0.023) T2‐tetraol (5.23 μmol/kg bw): 0% |
Islam et al. (1998) |
bw: body weight; i.p.: intraperitoneal; i.v.: intravenous; p.o.: per os; s.c.: subcutaneous; LD50: median lethal dose; NEO: neosolaniol.
Relative toxicity (doses have been converted from weight to moles and the effective molar dose has been set as 1 for T2).
Only three doses tested.
In the original paper (Islam et al., 1998), these compounds were designated as 3′‐HO‐T2 and 3′‐a)HO‐HT2 due to a different numbering system.
In 1974, Ueno and co‐workers showed that the minimal subcutaneous (s.c.) dose of trichothecenes to induce vomiting in ducklings is 0.1 mg/kg bw for T2, HT2 and NEO.
Acute toxic effects of T2, HT2, NEO, T2‐triol, and T2‐tetraol were investigated in 1‐day‐old broiler chicks by applying single oral doses (Chi et al., 1978). According to the authors, the results of the present study and previous reports indicate that T2 is metabolised into less toxic compounds when consumed by chickens.
The median lethal dose (LD50) values of T2, HT2, NEO and T2‐triol were estimated by a single i.p. injection of toxins to male mice. Macroscopic observation revealed severe haemorrhage in the intestine and lung (Ueno et al., 1986).
Islam et al. (1998) investigated the effects of T2‐derived metabolites on induction of apoptosis of thymus cells in vivo in mice. The ranking order of apoptosis‐inducing activity was found to be T2 = 19‐HO‐T2 > HT2 = 19‐HO‐HT2 > NEO = T2‐tetraol = control. The authors concluded that both the acetyl group at C‐4 position and the isovaleroyl or 19‐hydroyisovaleroyl group at the C‐8 position of T2 appear to be involved in thymic apoptosis.
In addition, Chattopadhyay et al. (2013) evaluated the haematotoxicity of T2 and NEO in Sprague–Dawley rats and observed inter alia significant decreases in white blood cell and red blood cell count upon application of T2 and NEO (data not shown in Table 4). Based on unclear reporting of the experiments and the fact that baseline levels of some blood cell counts in control groups deviated strongly from those usually reported in rats, the CONTAM Panel decided not to consider the results of the study.
8.2.1.1. Concluding remarks
The CONTAM Panel identified only few in vivo studies in which the acute toxicity of modified T2 and HT2 (only phase I metabolites) was compared with T2 and HT2. Endpoints investigated were acute lethality, induction of apoptosis in thymus cells and vomiting. In most instances, all metabolites tested exerted these effects and were equally or less potent than T2 or HT2.
8.2.2. In vitro toxicity of phase I metabolites
T2 has been described to be cytotoxic to a range of blood cells (white blood cell progenitors, platelet progenitors, red blood cell progenitors, red blood cells) from different species (human, rat, guinea pig) at concentrations of 5.4 nM or higher (see EFSA CONTAM Panel, 2011).
Furthermore, T2 has been reported to inhibit the transpeptidation of peptide formation at the 60S ribosomal subunit by inhibiting the peptidyl transferase (EFSA CONTAM Panel, 2011). Concentrations of T2 exceeding 6 nM resulted in induction of apoptosis in a range of different cell lines (EFSA CONTAM Panel, 2011).
Table 5 summarises the identified in vitro studies on the toxicity of T2/HT2 and its phase I metabolites. Both studies already described in EFSA CONTAM Panel (2011) as well as more recent studies are included. For each of the different phase I metabolites, its relative molar toxicity with respect to T2 is given in each study (potency of T2 set as 1).
Table 5.
In vitro toxicity studies with T2 and HT2 and its phase I metabolites in various cell systems
| Cell system | Endpoint | Effective concentrations (relative molar toxicity)g | Reference |
|---|---|---|---|
| Primary human lymphocytes | Inhibition of mitogen‐induced blastogenesis; 72‐h exposure; EC50 |
T2: 3.2 nM (1) HT2: 8.3 nM (0.39) T2‐triol: 390 nM (0.008) T2‐tetraol: 500 nM (0.006) 19‐Hydroxy‐T2: 8.3 nM (0.39) 19‐Hydroxy‐HT2: 115 nM (0.028) |
Forsell et al. (1985) |
| Kidney epithelial cell line (Vero cells, African green monkey) | Inhibition of protein synthesis (3H‐leucin incorporation); 1‐h exposure; EC50 |
T2: 24 nM (1) hAcetyl‐T2: 19,500 nM (0.0012) T2‐tetraol: 22,800 nM (0.0011) |
Ehrlich and Daigle (1985) |
| Mouse erythroleukaemia cell line | Inhibition of protein synthesis (3H‐leucin incorporation), 1‐h exposure; EC50 |
T2: 6 nM (1) hAcetyl‐T2: 3,500 nM (0.0017) T2‐tetraol: 6,400 nM (0.00094) |
Ehrlich and Daigle (1985) |
| Kidney epithelial cell line (Vero cells, African green monkey) | Inhibition of protein synthesis (3H‐leucin incorporation): 1.5 h exposure; EC50 |
T2: 14 nM (1) HT2: 65 nM (0.22) NEO: 273 nM (0.051) T2‐triol: 1,120 nM (0.013) T2‐tetraol: 5,856 nM (0.0024) hAcetyl‐T2: 5,792 nM (0.0024) |
Thompson and Wannemacher (1986) |
| Primary rat spleenocytes | Inhibition of protein synthesis (3H‐leucin incorporation); 1.5‐h exposure; EC50 |
T2: 6 nM (1) HT2: 10 nM (0.60) NEO: 127 nM (0.047) T2‐triol: 151 nM (0.040) T2‐tetraol: 7,832 nM (0.0008) hAcetyl‐T2: 1,036 nM (0.006) |
Thompson and Wannemacher (1986) |
| Rabbit reticulocytes | Inhibition of protein synthesis (3H‐leucin incorporation); 1.5‐h exposure; EC50 |
T2: 21.5 nM (1) HT2: 70.8 nM (0.30) NEO: 654 nM (0.033) T2‐triol: 5,240 nM (0.004) T2‐tetraol: 8,390 nM (0.002) |
Ueno et al. (1986) |
| Tetrahymen pyroformis | Inhibition of protein synthesis (3H‐leucin incorporation); 1.5‐h exposure; EC50 |
T2: 53.6 nM (1) HT2: 118 nM (0.45) NEO: 1,310 nM (0.041) T2‐triol: 5,240 nM (0.010) T2‐tetraol: 8,390 nM (0.006) |
Ueno et al. (1986) |
| Human acute promyelocytic leukaemia cell line (HL 60) | Cell viability (trypan blue exclusion);concentrations required to kill 100% of the cells in 5 days |
T2: 17 nM (1) HT2: 19 nM (0.89) NEO: 340 nM (0.050) T2‐triol: 262 nM (0.065) T2‐tetraol: 470 nM (0.036) hAcetyl‐T2: 39 nM (0.44) h9,10‐Epoxy‐T2: 207 nM (0.082) h9,10‐Dihydro‐T2: 280 nM (0.061) |
Samara et al. (1987) |
| Brine shrimp (Artemia salina) bioassay | Cytotoxicity; 24‐h exposure of eggs; lethality of hatched shrimps; LC50 |
T2: 240 nM (1) HT2: 610 nM (0.39) T2‐triol: 3,600 nM (0.067) T2‐tetraol: 3,200 nM; (0.075) De‐epoxy‐HT2: > 14,700 nM (< 0.016) De‐epoxy‐T2‐triol: > 13,700 (< 0.018) De‐epoxy‐T2‐tetraol: > 21,300 (< 0.011) |
Swanson et al. (1987) |
| Adult human liver cell line (Chang liver) | Cell viability; 6‐day exposure; amido black 10B; EC50 |
T2: 5 nM (1) HT2: 10 nM (0.50) NEO: 200 nM (0.025) T2‐triol: 105 nM (0.048) T2‐tetraol: 800 nM (0.0062) hAcetyl‐T2: 26 nM (0.19) hTetraacetyl‐T2‐tetraol: 150 nM (0.033) |
von Milczewski (1987) |
| Human epidermoid carcinoma cell line (HEp‐2) | Cell viability; 6‐day exposure; amido black 10B; EC50 |
T2: 5 nM (1) HT2: 10 nM (0.5) NEO: 180 nM (0.028) T2‐triol: 86 nM (0.058) T2‐tetraol: 670 nM (0.0075) hAcetyl‐T2: 16 nM (0.31) hTetraacetyl‐T2‐tetraol: 150 nM (0.033) |
von Milczewski (1987) |
| Girardi human heart cell line (clone GHc7) | Cell viability; 6‐day exposure; amido black 10B; EC50 |
T2: 9 nM (1) HT2: 12 nM (0.75) NEO: 210 nM (0.043) T2‐triol: 76 nM (0.12) T2‐tetraol: 470 nM (0.019) hAcetyl‐T2: 70 nM (0.13) hTetraacetyl‐T2‐tetraol: 100 nM (0.090) |
von Milczewski (1987) |
| Pig kidney cell line (clone Amc6sc8) | Cell viability; 6‐day exposure; amido black 10B; EC50 |
T2: 10 nM (1) HT2: 11.3 nM (0.88) NEO: 3–6 nM (3.33–1.67)c T2‐triol: 39 nM (0.26) T2‐tetraol: 540 nM (0.019) hAcetyl‐T2: 50 nM (0.20) hTetraacetyl‐T2‐tetraol: 9.4 nM (1.06) |
von Milczewski (1987) |
| Mouse lymphoma cell line (L5178Y) | Cell viability; 3‐day exposure; EC50 |
T2: 3.9 nM (1) HT2: 7.1 nM (0.55) NEO: 105 nM (0.037) T2‐triol: 260 nM (0.015) T2‐tetraol: 340 nM (0.012) h3‐Acetyl‐T2: 40 nM (0.097) h8‐Acetyl‐NEO: 3.2 nM (1.22) h3‐Acetyl‐NEO: 47 nM (0.083) hTetraacetyl‐T2‐tetraol: 43 nM (0.091) h3,4,8‐Triacetyl‐T2‐tetraol: 190 nM (0.021) |
Anderson et al. (1989) |
| Human fibroblast cell line (GM5757) | Cell viability; 48‐h exposure; MTTe assay; EC50 |
T2: 122 nM (1) HT2: 383 nM (0.32) NEO: 342 nM (0.36) T2‐tetraol: 6,540 nM (0.019) hAcetyl‐T2: 1,970 nM (0.062) |
Kim et al. (1991) |
| Human liver cell line (HepG2) | Cell viability, 48‐h exposure; neutral red uptake; EC50 |
T2: 21 nM (1) HT2: 37 nM (0.57) T2‐triol: 1,040 nM (0.020) T2‐tetraol: 1,490 nM (0.014) |
Babich and Borenfreund (1991) |
| Baby hamster kidney cell line (BHK‐21 cells) | Cytotoxicity; 24‐h exposure; approximate LC100 |
T2: 10.7 nM (1) HT2: 240 nM (0.045) NEO: 26.2 nM (0.41) T2‐tetraol: 33,500 nM (0.0003) 19‐Hydroxy‐T2: 10.3 nM (1.03) h8‐Acetyl‐T2‐tetraol: 294,000 nM (0.00004) h4,8‐Diacetyl‐T2‐tetraolb: 2,620 nM (0.0041) h8‐Acetyl‐NEO: 1,180 nM (0.009) |
Senter et al. (1991) |
| Primary human renal proximal tubule epithelial cells | Cell viability; 48‐h exposure; CCK‐8f assay; EC50 |
T2: 200 nM (1) HT2: 800 nM (0.25) NEO: 3,000 nM (0.067) T2‐triol: 14,600 nM (0.014) T2‐tetraol: 25,100 nM (0.008) |
Königs et al. (2009) |
| Primary human lung fibroblasts | Cell viability; 48‐h exposure; CCK‐8f assay; EC50 |
T2: 500 nM (1) HT2: 700 nM (0.71) NEO: 2,000 nM (0.25) T2‐triol: 10,600 nM (0.047) T2‐tetraol: 8,300 nM (0.060) |
Königs et al. (2009) |
| Lung fibroblast cell line from male Chinese hamster (V79) | Cell viability; 48‐h exposure; neutral red uptake; EC50 |
T2: 3 nM (1) HT2: 14 nM (0.21) |
Behm et al. (2012) |
RPF: relative potency factor with respect to T2; EC50: Half‐maximal effective concentration; LC: left‐censored; NEO: neosolaniol.
Also designated as 3‐acetyl‐HT2.
Also designated as NT‐1 toxin.
Considered an outlier (only range given, very different from results of numerous other studies on NEO).
Also designated as NT‐2 toxin.
MTT: 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide.
Cell Counting Kit‐8, using reduction of 2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium monosodium salt.
The doses used have been recalculated from weight to molar units. Relative toxicity (effective molar dose) has been set as 1 for T2.
Hypothetical metabolite (i.e. not identified occurring naturally).
Some of the compounds are hypothetical metabolites, i.e. they could be formed according to metabolic considerations but have not yet been detected as naturally occurring metabolites. Such compounds have been chemically synthesised and are included in Table 5 to obtain a broader basis for structure–activity relationships.
8.2.2.1. Concluding remarks
The studies listed in Table 5 support the data presented in EFSA CONTAM Panel (2011). The various phase I metabolites exhibit considerable differences in their toxic activity at two endpoints, viz. inhibition of protein synthesis and cell viability. Most data were available for the metabolites formed through consecutive hydrolysis of the ester groups of T2, i.e. HT2, NEO, T2‐triol and T2‐tetraol, while data on other phase I metabolites were very scarce or lacking. In vitro activity was always highest for T2. For the hydrolytic metabolites, HT2 was almost as active as T2, while NEO and T2‐triol were less active, and T2‐tetraol invariably exhibited the lowest toxicity. Hydroxylation of T2 at C‐19 had no significant effect on T2 toxicity. Thus, the ranking of toxicity derived from the in vitro studies (Table 5) is consistent with the ranking from in vivo studies (Table 4). For the other metabolites in Table 5, no clear ranking emerges from the few available studies.
8.2.3. Toxicity of phase II metabolites
The CONTAM Panel did not identify toxicity studies with phase II metabolites (e.g. glucosides, sulfates, glucuronides) of parent T2 and HT2 or from their phase I metabolites.
9. Mode of action for toxicity of T2 and HT2 toxins and their metabolites
Most of the data on the biochemical mode of action are concerning T2 with fewer studies available for HT2. The mode of action of T2 and HT2 has been reviewed review in the previous EFSA opinion (EFSA CONTAM Panel, 2011). The following chapter will briefly summarise the major finding and discuss in more detail pertinent studies since 2011.
9.1. Induction of ribotoxic stress and effect on nucleic acids
As other trichothecenes, T2 is a protein synthesis inhibitor known to target ribosome and binds to the 60S subunit of the ribosome. Recent crystallographic analysis show that the trichothecene DON more specifically targets the site A of the peptidyl transferase centre (Garreau de Loubresse et al., 2014). Binding to ribosomes, T2 interferes with the elongation step of protein synthesis, more specifically with the peptide bond formation (Garreau de Loubresse et al., 2014).
Transcriptomic analysis revealed that of the total genes affected by T2, 20% were related to transcription and 30% to translation pathways. Specifically, T2 reduces the activities of aminoacyl‐tRNA synthetases and adversely affects the translation initiation. The protein processing and folding are also influenced by T2 through the declined levels of sulfhydryl oxidases, disulfide isomerases and heat shock protein 90 (Hsp90) (Wan et al., 2015a).
Ribosome binding by T2 quickly activates mitogen‐activated protein kinase (MAPK) via a mechanism known as the ‘ribotoxic stress response’ (Yang et al., 2000; Wan et al., 2015b). MAPKs are important transducers of downstream signalling events related to immune response and apoptosis. T2 activates several MAPKs including p38, c‐Jun N‐terminal kinase (JNK), and extracellular signal‐regulated kinase 1 and 2 (ERK1/2).
DNA and RNA synthesis are also inhibited by T2; this could be secondary to the inhibition of protein synthesis (Rakkestad et al., 2010). Inhibition of DNA, synthesis by T2 has been reported at concentrations generally exceeding those that cause an inhibition of protein synthesis (EFSA CONTAM Panel, 2011).
9.2. Apoptosis
T2 also causes apoptosis in vitro in various cell types and in vivo in lymphoid organs, intestinal crypt, brain, skin and fetal tissues (EFSA CONTAM Panel, 2011). Apoptosis has been confirmed by histology, in situ detection of fragmented DNA, DNA agarose gel electrophoresis and flow cytometry.
As far as circulating blood cells are concerned, apoptosis of haematopoietic progenitors by T2, blocks the renewal of blood cells in the bone marrow. As the life time of leucocytes and platelets (approximately 10 days in humans) is shorter than the life time of red blood cells (120 days in humans), the most common symptom observed are decrease in leucocyte and thrombocytes count in circulating blood cells inducing leucopoenia and thrombocytopenia and increasing the risk of septicaemia and haemorrhage.
In vitro, T2 induced apoptosis has been associated with activation of several caspases, increased expression of genes involved in apoptosis such as c‐Jun and c‐fos, p53 and Bax and in some studies the decrease in the anti‐apoptotic factor Bcl‐xL (EFSA CONTAM Panel, 2011).
The mechanisms of T2 induced apoptosis have been controversially discussed and it has been hypothesised that DNA damage may be a secondary effect of the protein synthesis inhibition or oxidative stress that can in turn activate mitochondrial apoptotic pathways upon exposure to T2 and that apoptosis could be related to the induction of stress‐activated protein kinase (SAPK/JNK) and mitogen‐activated protein kinase (p38/MAPK) (Jaradat, 2005; EFSA CONTAM Panel, 2011; Wu et al., 2014).
9.3. Effects on membranes and oxidative stress
T2 is an amphophilic molecule, taken up into the cells bilayer membrane. It may then induce lipid peroxidation by generating free radicals, and thereby damaging cellular membranes. There is still some inconsistency among the results shown for the T2 mediated effects on oxidative stress markers. While several studies showed increased malondialdehyde (MDA) levels, others reported no changes. The extent to which reactive oxygen species (ROS) contribute to the clastogenicity and DNA strand breaks observed in some tests but not in others as reported in the previous opinion (EFSA CONTAM Panel, 2011) remains unclear. In 2011, the CONTAM Panel noted that these effects were observed primarily at concentrations also known to inhibit protein and DNA synthesis and producing cytotoxicity (EFSA CONTAM Panel, 2011).
9.4. Anorexia and vomiting
The mechanism by which T2 and HT2 induce anorexia/emesis is not yet fully understood. Two major mediators of anorexia/emesis, i.e. proinflammatory cytokines and satiety hormones, have been proposed. Of note, contrary to humans or mink, emesis cannot occur in rodents, but the abnormal food intake behaviour observed in mice is considered indicative of nausea‐induced anorexia (Yamamoto et al., 2002).
T2 upregulates mRNA levels of IL‐1 beta, IL‐6 and tumour necrosis factor (TNF)‐alpha in rodents (Agrawal et al., 2012; Gaigé et al., 2014). Upon injection, the these proinflammatory cytokines induce a set of symptoms characteristic of sickness behaviour, including anorexia (Kelley et al., 2003). It is therefore possible that anorexia induction by T2 is mediated in part by sickness responses to these cytokines (Gaigé et al., 2014).
In a study performed in mink, elicitation of emesis by T2 and HT2 corresponded to elevated plasma concentrations of PYY3‐36 and 5‐HT (Wu et al., 2016), two hormones known to be involved in emesis (Wu et al., 2013a; Lebrun et al., 2015). Similar effects were seen with DON, another trichothecene. In experiments performed with DON, pharmacological antagonism of neuropeptide Y2 receptor, which is activated by PYY3‐36, suppressed DON‐induced emesis in mink (Wu et al., 2013a). Similarly, blocking the 5‐HT3 receptor with granisetron completely inhibited induction of vomiting by DON. PYY3‐36 alone induces emesis that could be partially blocked with granisetron,6 thus indicating a possible upstream role for PYY3‐36 in 5‐HT release. Therefore, both PYY3‐36 and 5‐HT appear to contribute to emesis induction by DON (Wu et al., 2013a, 2016).
10. Structure–activity relationships for T2 and HT2 and their phase I and phase II metabolites
In contrast to the chemical structures of the numerous phase I metabolites of T2 and HT2, which have been well elucidated (see Section 7.3), only little information is available on the toxicological activity of these metabolites. Most of the studies on structure–activity relationships (SARs) of T2 metabolites have utilised various cell systems in vitro, and only very few in vivo studies have been conducted. Most of these SAR studies on T2 and its metabolites together with other trichothecenes have recently been reviewed by Wu et al. (2013b).
From the toxicological data of the phase I metabolites, none of them appears to exceed the potency of the parent toxin T2, unlike in the case of zearalenone, a mycotoxin that can be biotransformed to a metabolite of much higher oestrogenic potency (zearalenol) than its parent compound (EFSA CONTAM Panel, 2016).
As shown in Figure 2, the phase I metabolism of T2 essentially comprises three pathways, viz. stepwise hydrolysis of the three ester groups (pathway A), hydroxylation (pathway B) and de‐epoxidation (pathway C). In most metabolic in vivo and in vitro studies, the hydrolytic metabolites have been found to predominate.
10.1. Hydrolytic metabolites
The first hydrolysis of T2 leads preferentially to HT2. The toxic potencies of T2 and HT2 have been compared in several in vivo and numerous in vitro studies. As discussed in Section 8.1.2, HT2 is equipotent to T2 with respect to anorectic activity in mice and emetic activity in minks. The LD50 of HT2 in mice and broiler chicks is only slightly higher than the LD50 of T2 (Table 4). HT2 exhibited the same lethality as T2 when administered to the midbrain of rats (Bergmann et al., 1988). A marginally lower toxic activity of HT2 in comparison with T2 is suggested in many in vitro toxicity studies (Table 5), with relative molar toxicity mostly between 0.2 and 0.9.
The alternative product of the first hydrolysis of T2 is NEO (Figure 3). The LD50 of NEO is about three and five times higher than that of T2 in mice and broiler chicks, respectively (Table 4), and the relative molar toxicity in cellular assays ranges from 0.03 to 0.4 (Table 5).
The further hydrolysis of HT2 gives rise to T2‐triol (Figure 3). In mice and broiler chicks, the LD50 of T2‐triol is about 20 and six times higher than that of T2, respectively (Table 4), whereas its cerebral toxicity in rats was about the same as that of T2 (Bergmann et al., 1988). The relative moral toxicity of T2‐triol in cellular assays ranges from 0.01 to 0.1 (Table 5).
The final hydrolysis of the third ester group eventually leads to T2‐tetraol (Figure 3). In broiler chicks (Table 4) and in rat brain (Bergmann et al., 1988), T2‐tetraol was the least toxic of the hydrolysis products of T2. Its relative molar toxicity in most cellular assays was invariably below 0.07 and in most cases even below 0.01.
In conclusion, each consecutive cleavage of an ester group of T2 appears to reduce toxicity. This decrease is marginal for HT2 but pronounced for T2‐tetraol.
10.2. Hydroxylated metabolites
Hydroxylation of T2 and HT2 can occur at various positions, but C‐19 (located in the isovaleroyl group) appears to be preferred (see Section 7.3.1). The few studies available, e.g. induction of apoptosis in mice (Table 4) and studies in primary human lymphocytes and baby hamster kidney cells (Table 5) imply that 19‐hydroxylation of T2 does not reduce toxicity, whereas 19‐hydroxylation of HT2 does.
10.3. De‐epoxidised metabolites
De‐epoxidation of T2 and HT2 is believed to be primarily mediated by bacteria of the intestine and rumen (see Section 7.3.1). From the only study available, i.e. in the brine shrimp assay (Table 5), the de‐epoxidation of HT2, T2‐triol and T2‐tetraol leads to a marked reduction in cytotoxicity. This finding is in agreement with observations made with other 12,13‐epoxytrichothecenes, e.g. deoxynivalenol and nivalenol, for which de‐epoxidation has been clearly established as a metabolic detoxification pathway (Wu et al., 2013b).
10.4. Hypothetical metabolites
Various compounds, which may be generated in the metabolism of T2 according to expected pathways but have not been detected as naturally occurring metabolites to date, have been chemically synthesised and tested for their toxicity in vitro. Most of these hypothetical T2 metabolites are acetyl derivatives of T2 and its hydrolytic metabolites. Although the relative molar toxicities of most of these compounds are rather low (Table 5), there are not sufficient data or too much variabilities (e.g. from 0.4 to 0.01 for acetyl‐T2) to derive conclusions on SARs.
10.5. Conjugated metabolites
No studies have yet been conducted on the toxicity of the conjugates of T2 and HT2 with glucose, sulfate or glucuronic acid, formed as phase II metabolites in fungi, plants and mammals (see Sections 4.2 and 6). Studies with other bioactive compounds, e.g. phytochemicals, have shown that this type of phase II metabolites is, in general, devoid of biological activity per se. However, glucosides, sulfates and glucuronides may release the aglycone after ingestion due to enzymatic cleavage of the conjugated metabolite in the gastrointestinal tract.
10.6. Concluding remarks
Based on the limited data available, it is concluded that stepwise hydrolysis of the three ester groups of T2 leads to a concomitant decrease in toxic activity, while hydroxylation has little effect on toxicity. In addition to ester hydrolysis, de‐epoxidation constitutes a detoxifying metabolic pathway. Ester hydrolysis and de‐epoxidation are irreversible metabolic reactions. In contrast, formation of phase II metabolites through conjugation with glucose, sulfate or glucuronic acid, although presumably abolishing toxicity, is reversible and might release the aglycone upon ingestion.
11. Dose–response analysis
11.1. Acute effects
There are new studies reporting anorectic effects (e.g. feed refusal, retching, vomiting, reduced body weight gain) at low doses in several species (mouse, mink and pig). Therefore, the study of Wu et al. (2016) in mink, in which acute effects were observed at the lowest dose, was used by the CONTAM Panel as a basis for BMD analysis of acute effects of T2 and HT2. Following oral gavage in two independent tests, one with T2 and one with HT2, each with four animals/dose groups, identical results at identical doses were seen. Notably, the emetic events were associated with elevated plasma concentrations of PYY3‐36 and 5‐HT (Wu et al., 2016), two hormones known to be involved in emetic effects (Wu et al., 2013b; Lebrun et al., 2015).
Using a benchmark response (BMR) of 10% resulted in a benchmark dose 10% (BMD10) total confidence interval (BMDL10–BMDU10) of 2.97–49.8 μg T2 or HT2/kg bw per day across the applied models. The two independent tests, were combined to increase the statistical power of the BMD modelling (details on the calculations are provided in Appendix D). Due to the large dose spacing at the lower doses and the small number of animals used (low statistical power), the CONTAM Panel considers the BMDL10 as a conservative estimate (BMDL10 is below the lowest dose tested, 5 μg T2 or HT2/kg bw, and showing no effect). The CONTAM Panel used this value of 2.97 μg T2 or HT2/kg bw as a reference point for characterising the acute toxicity of T2 and HT2.
11.2. Chronic effects
Since the EFSA evaluation (EFSA CONTAM Panel, 2011), there has been no new evidence that other toxic effects including dermal toxicity, developmental and reproductive toxicity and neurotoxicity occur at doses lower than those causing immunotoxicity and haematotoxicity in pigs.
In its 2011 evaluation, EFSA noted that there was substantial evidence for the immunotoxicity and haematotoxicity of T2 in several species (EFSA CONTAM Panel, 2011). In 2011, EFSA identified a LOAEL of 0.029 mg/kg bw per day in a short‐term study with pigs (Rafai et al., 1995). Based on a BMD analysis of the decrease in anti‐horse globulin titre values, a 95% lower confidence limit for the benchmark dose response of 5% (BMDL05) of 10 μg T2/kg bw per day was calculated.
Since 2011, several subacute and subchronic toxicity studies have been published. In the 90‐day study in rats by Rahman et al. (2014), dose‐dependent decreases in total erythrocyte, leucocyte and thrombocyte counts as well as a decrease in the percentage of lymphocytes were observed. This study has been performed with T2 obtained from cultures of F. sporotrichioides var. sporotrichioides MTCC 1894. Data obtained by Rahman et al. (2014) are in accordance with haematotoxicity described for several other species, i.e. mice, pigs, humans and monkeys, in the previous EFSA assessments (EFSA CONTAM Panel, 2011). In the Rahman et al. (2014) study, the duration of exposure of animals (rats) to T2 is longer (90 days) in absolute terms, but also relative to species life time than in the Rafai et al. (1995) study, in pigs (21 days). Furthermore, it is noted that the toxic effect in the study of Rahman et al. (2014) progressed during the whole study period with no signs of reaching a plateau at the end.
In a similar 90‐day study (Raut et al., 2013), a strong decrease in total thrombocytes count and less decreases in erythrocyte and leucocyte counts were observed. It is noted that in the Rafai et al. (1995) study in addition to decreased leucocyte counts functional endpoints such as reduced anti‐horse globulin titre formation were observed at the lowest dose tested, 29 μg T2/kg bw per day. However, the Scientific Committee on Food (2001) based its assessment on a similar LOAEL for several immune parameters including a decrease in total leucocytes, EFSA (EFSA CONTAM Panel, 2011) used reduced anti‐horse globulin titre formation as basis for establishing a HBGV.
For the present assessment, the CONTAM Panel performed BMD analyses of several haematotoxic, immunotoxic as well as other endpoints in the studies by Rafai et al. (1995) and Rahman et al. (2014). These endpoints were: (i) total leucocyte count in the Rafai et al. (1995) study and (ii) total leucocyte count, total erythrocyte count, total thrombocyte count, haemoglobin (Hb) concentration and body weight in the Rahman et al. (2014) study. The Panel noted that, in essence, the effects observed (i.e. anorectic effects and effects on immune system and blood parameters) in the new (longer term) rat study were essentially similar to those seen in the pig study confirming the immune system and the blood cell production as target organs of T2 through species.
Therefore, the CONTAM Panel decided, considering the longer exposure duration of the study from Rahman et al. (2014) and its biological relevance, to use the total leucocytes count reported from this study for calculating a new BMD for T2 (for details on BMD calculations, see Appendix E). The CONTAM Panel used a BMR of 10% was used considering such a response in leucocyte counts to be within the individual physiological variation and negligible, and further noted that the selected BMR is slightly below the control standard deviation of the controls in the Rahman et al. study (14%). A 95% lower confidence limit for the benchmark dose response (BMDL10) of 3.3 μg T2/kg bw was derived. The CONTAM Panel used this value as a reference point for establishing a chronic HBGV for T2 and HT2. Additional BMD calculations have been carried out with several other potentially relevant endpoints (next to total leucocyte count in the Rahman et al. (2014) study) measured in the studies described above to gauge their appropriateness to derive a TDI. These alternative calculations are provided as supporting information to the present opinion.
12. Establishment of health based guidance values
12.1. Establishment of a group acute reference dose (ARfD) for T2 and HT2 and their modified forms
The CONTAM Panel decided to use the BMDL10 of 2.97 μg T2 or HT2/kg bw derived for emetic response in mink as a reference point for establishing a group ARfD for T2 and HT2. An UF of 10 for intraspecies variability was applied. No interspecies variability factor was applied because humans were not considered more sensitive than mink to the acute emetic effect of T2 or HT2, an assumption supported by studies on emesis with emetine showing that similar doses of the compound are effective both in humans and mink (Gordon, 1985; Zhang et al., 2006; Percie du Sert et al., 2012). An ARfD of 0.3 μg (rounded from 0.297 μg) T2 and HT2/kg bw was established.
With regard to phase I metabolites, NEO was equipotent with T2 and HT2 when tested for vomiting in ducklings (Ueno et al., 1974; see Table 4). Therefore, the CONTAM Panel decided that NEO can be included with a relative potency factor (RPF) of 1 (i.e. a molarity based potency factor relative to T2) together with T2 and HT2 in a group ARfD.
None of the other phase I metabolites have been tested with regard to emetic response. The data on comparative toxicity of T2 and HT2 with its metabolites presented in Section 8 cannot be related to emesis and therefore not be used to assess relative emetic potency of metabolites. Consequently, due to the lack of appropriate data, other phase I metabolites cannot be included in a group ARfD.
Since phase II metabolites of T2, HT2 and NEO are assumed to be hydrolysed to their parent compounds (aglycones) after ingestion, they are included in a group ARfD with the same potency as their parent compounds.
12.2. Establishment of group tolerable daily intake (TDI) for T2 and HT2 and their modified forms
The CONTAM Panel decided to use the BMDL10 of 3.3 μg T2/kg bw per day derived for reduction in the number of peripheral leucocytes in a subchronic study in rats as a basis for establishing a TDI for T2. An UF of 200 was used taking into account 10 for interspecies, 10 for intraspecies variability and two for extrapolation from subchronic to chronic exposure duration (see EFSA Scientific Committee, 2012) and for the fact that the toxic effect progressed during the whole study period with no signs of reaching a plateau at the end of the study. A TDI of 0.02 (rounded from 0.0165) μg T2/kg bw per day was established.
Haematotoxicity with reduced production of erythrocytes, leucocytes and platelets, is the critical chronic effect of T2. The underlying mode of action is inhibition of protein synthesis, induction of ribotoxic stress and apoptosis. Based on similar toxic profile and potency, structural similarity and the fact that HT2 is an immediate metabolite of T2 in agreement with the EFSA assessment of 2011, it was concluded that T2 and HT2 can be included in a group TDI with the same potency.
No in vivo studies on haematotoxicity of modified forms of T2 and HT2 have been identified. It is, however, assumed that phase I metabolites work via a similar mode of action as some have been shown to cause protein synthesis inhibition, and may as such induce haematotoxicity. The CONTAM Panel therefore considered it appropriate to include modified forms in a group TDI assuming dose addition as a model of joint action. Because the potencies of phase I metabolites differ with respect to the inhibition of protein synthesis and other toxic effects, it was decided to assign RPFs on a molar basis.
12.3. Assignment of relative potency factors for chronic effects
When assigning different potency factors to the phase I metabolites of T2 and HT2, in vivo and in vitro studies on comparative toxicity in vivo and in vitro (see Tables 4 and 5) were used. The CONTAM Panel noted that none of the phase I metabolites examined were more potent than T2 of HT2. Since in vitro cell systems (presented in Table 5) may have a limited capacity for detoxification, results would in general overestimate toxicity of T2 because as compared to the in vivo situation. Therefore, in vivo data are preferentially used. When there are different values of relative potencies for one metabolite, the highest potency was selected to avoid underestimation of relative toxicity. Only metabolites that have been examined either in vivo or in vitro have been considered for establishing relative potencies as listed in Table 6. Since phase II metabolites are assumed to be hydrolysed to their parent compounds (aglycons) after ingestion, they should be included in a group TDI with the same potency as their parent compounds. The (RPFs have been calculated for each study on a molar basis (see Tables 4 and 5). They have been rounded up to half orders of magnitude, i.e. 1, 0.3 or 0.1 to avoid spurious accuracy but retaining a conservative approach.
Table 6.
Relative potency factors (RPFs) for chronic effects of modified forms of T2
| Compound | Relative potency factor (RPF)a | Reference |
|---|---|---|
| T2 | 1 | n.a. |
| T2‐3‐Glc | 1 | Hydrolysation to T2 assumed |
| T2‐3‐diGlc | 1 | Hydrolysation to T2 assumed |
| T2‐3‐Sulf | 1 | Hydrolysation to T2 assumed |
| T2‐3‐GlcA | 1 | Hydrolysation to T2 assumed |
| 3‐Ac‐T2 | 1 | Hydrolysation to T2 assumed |
| 3‐Fer‐T2 | 1 | Hydrolysation to T2 assumed |
| 19‐HO‐T2 | 1 | See Tables 4 and 5, Section 8 |
| HT2 | 1 | See Tables 4 and 5, Section 8 |
| HT2‐3‐Glc | 1 | Hydrolysation to HT2 assumed |
| HT2‐diGlc | 1 | Hydrolysation to HT2 assumed |
| HT2‐GlcA | 1 | Hydrolysation to HT2 assumed |
| HT2‐MalGlc | 1 | Hydrolysation to HT2 assumed |
| 19‐HO‐HT2 | 0.3 | See Tables 4 and 5, Section 8 |
| NEO | 0.3 | See Tables 4 and 5, Section 8 |
| NEO‐Glc | 0.3 | Hydrolysation to NEO assumed |
| T2‐triol | 0.1 | See Tables 4 and 5, section 8 |
| T2‐triol‐Glc | 0.1 | Hydrolysation to T2‐triol assumed |
| T2‐tetraol | 0.1 | See Tables 4 and 5, Section 8 |
| T2‐tetraol‐Glc | 0.1 | Hydrolysation to T2‐tetraol assumed |
n.a.: not applicable ; Glc: glucoside; diGlc: diglucose; Sulf: sulfate; GlcA: glucuronic acid; Ac: acetyl; Fer: feruloyl; MalGlc: malonylglucose; NEO: neosolaniol.
RPFs have been rounded up to half an order of magnitude, i.e. to either 1, 0.3 or 0.1.
According to information on SAR as described in Section 10.3, deperoxidation generally leads to a marked reduction in toxicity, which is in agreement with the effect of deperoxidation of other trichothecenes (Wu et al., 2013a). Deperoxidised metabolites where therefore not considered for inclusion in a group TDI.
In Appendix F, an example calculation on how the factors can be applied in practice for inclusion of modified forms of T2 and HT2 for a chronic risk assessment is provided.
13. Uncertainties
The CONTAM Panel identified a series of uncertainties in their evaluation of the appropriateness to set a group HBGVs for T2 and HT2 and its modified forms.
Upon reviewing new data, the TDI set for T2 in 2011 (EFSA CONTAM Panel, 2011) has been revised using a reduction in total leucocyte counts in a 90‐day rat study with T2 as a critical endpoint for establishing a chronic BMD10. The test compound was purified from fungal culture material and its final purity was not specified. It can therefore not be excluded that minor amounts of other mycotoxins including modified forms were present in the test material. Using a subchronic study for setting, a chronic HBGV is also associated with considerable uncertainty. Although no repeated dose studies are available on HT2, this toxin has been included in a group TDI with T2 based on similar toxic profile and potency, structural similarity and the fact that HT2 is an immediate metabolite of T2.
No subchronic toxicity data on total leucocyte counts with any of the T2 and HT2 phase I metabolites were available. The CONTAM Panel decided to include all phase I metabolites, for which either in vivo or in vitro studies comparing their acute toxicity to that of T2 or HT2 were available, in a group TDI together with T2 and HT2. The highest effective molar doses/concentrations of these metabolites relative to those of T2 were used to derive RPFs, which were then rounded up to half an order of magnitude of either 1, 0.3 or 0.1 to avoid spurious accuracy. Application of RPFs for chronic risk assessment on basis of results from acute studies was done assuming the same underlying mode of action, binding to ribosomes with inhibition of protein synthesis and ribotoxic stress, for both acute effects (other than emetic effects) and chronic effects. The Panel noted that the derivation of RPFs for the different phase I metabolites from acute toxicity studies for including phase I metabolites of T2 and HT2 is associated with a very significant uncertainty, probably overestimating the relative toxicity.
An ARfD for T2 and HT2 has been established based on a BMDL10 derived from observations of emesis in two similar acute studies with T2 and HT2. The large dose spacing at the lower doses in these studies and the small number of animals used lead to a BMDL10 calculation associated with considerable uncertainty.
NEO was included with the same RPF in a group ARfD with T2 and HT2 on the basis of a single in vivo study where emetic effects have been observed at the same dose for the three compounds.
In the absence of data on phase II metabolites (conjugates) of T2 and HT2 and their phase I metabolites, the CONTAM Panel assumed these release of their aglycones upon ingestion and that therefore phase II metabolites should be included in the group HBGVs and RPFs for their parent compounds be applied.
Dose additivity of T2 and HT2 and their modified forms is assumed, although antagonistic or less likely synergistic effects of their co‐occurrence cannot be excluded in principle.
There is a lack of analytical standards for detection of modified forms of T2 and HT2. Analytical detection of modified forms of T2 and HT2 are often based on semi‐ or relative quantification.
13.1. Summary of uncertainties
In Table 7, a summary of the uncertainty evaluation is presented, highlighting the main sources of uncertainty and indicating an estimate of whether the source of uncertainty leads to over/underestimation of the resulting risk.
Table 7.
Summary of the qualitative evaluation of the impact of uncertainties on the assessment
| Sources of uncertainty | Directiona |
|---|---|
| Purity of test compound in subchronic tests with T2 was not specified, thus it cannot be excluded that minor amounts of other mycotoxins or modified forms were also present | + |
| Inclusion of modified forms of T2 and HT2 in a group TDI based on subchronic toxic effects observed for T2 | + |
| Use of results from acute toxicity tests for derivation of relative potency factors and inclusion of metabolites in group TDI for chronic effects | +/− |
| Numerical values of RPFs are rounded to up to half an order magnitude | + |
| Assumption of complete cleavage of phase II metabolites to their parent compounds | + |
| Assumption of dose additivity for T2 and HT2 and its modified forms | +/− |
+: uncertainty with potential to cause overestimation of exposure/risk; −: uncertainty with potential to cause underestimation of exposure/risk; +/−: extent of potential over/underestimation might differ in direction.
The overall uncertainty associated with the inclusion of modified forms of T2 and HT2 into a group ARfD and a group TDI in the present assessment is considered as high and it would rather overestimate than underestimate any risk.
14. Conclusions
14.1. Introduction
T2 and HT2 are members of the type A group trichothecenes, which are tetracyclic sesquiterpenoids produced by many Fusarium species infesting crop plants. In addition to T2 and HT2, produced by Fusarium species, plants, fungi and mammals generate a large number of modified forms by phase I and phase II metabolism. Reliable analytical methods (based mainly on LC–MS/MS) for T2, HT2 and their phase II metabolites are available. Due to a lack of commercially available standards and reference materials, validated methods to quantify phase I metabolites are not available.
14.2. Occurrence of modified forms of T2 and HT2
There are few data on occurrence of modified forms of T2 and HT2, mainly conjugates, with grain products being their main source. The proportion of their occurrence relative to the respective parent compounds, in the few samples where this has been determined, varies widely. Recent reports show the frequent occurrence of conjugates, in particular glucosides, of T2 and HT2 even in samples were their parent compounds are not detected.
There are no data available on the occurrence of modified forms of T2 and HT2 in food from animal origin.
14.3. Toxicokinetics of T2 and HT2 and their modified forms
Upon ingestion, T2 appears to be rapidly absorbed. It is rapidly hydrolysed to HT2 and distributed to the liver, kidney and other organs without accumulation. No studies on absorption or distribution of modified forms of T2 or HT2 have been identified.
There are no data on absorption of phase I metabolites from food. They are assumed to be easily absorbed, distributed and entering metabolism as described for T2. No data are available on the absorption of phase II metabolites of T2 and HT2. It is assumed that such phase II metabolites are hydrolysed in the gastrointestinal tract after ingestion, releasing their aglycones, which can then be absorbed.
Phase I metabolism is predominated by hydrolysis of ester groups, leading to NEO, T2‐triol and eventually T2‐tetraol. Hydroxylation reactions appear to be less pronounced. Another phase I reaction, mostly in the ruminal microflora, is de‐epoxidation. Phase II metabolism comprises the formation of glucuronides in mammals.
Mammals excrete T2 and its phase I and phase II metabolites of T2 within 2–3 days in urine and faeces.
14.4. Toxicity of T2 and HT2 and its modified forms
In vivo acute toxicity studies (supported by subacute studies) show that T2 and HT2 have anorectic effects upon short‐term exposure. Emetic events observed upon single oral and i.p. exposures to both T2 and HT2 in mink were identified as the critical acute effects.
Only a few in vivo studies have been identified in which the acute toxicity of phase I metabolites of T2 and HT2 was compared with T2 and HT2. Effects investigated were lethality, induction of apoptosis and emesis. It was shown that the toxic potencies of the metabolites tested were either equal or less than that of T2 or HT2.
In vivo subacute and new subchronic tests show that T2 causes haematotoxicity (and myelotoxicity). Reduction in total leucocyte counts seen in a 90‐day rat study was identified as the critical chronic effect of T2.
No subacute, subchronic or chronic studies comparing the toxicity of T2 and HT2 to their modified forms have been identified. In the absence of repeated dose studies on HT2, it is assumed that HT2 exerts similar toxicity as T2. This is based on structural considerations and on the rapid metabolism of T2 to HT2.
A series of in vitro studies on toxicity of T2 and HT2 in comparison to its phase I metabolites have been identified, the endpoints being mainly inhibition of protein synthesis or cell viability. In vitro toxicity was always highest for T2. HT2 was almost as active as T2, whereas NEO and T2‐triol and 19‐OH‐HT2 were less active. T2‐tetraol invariably exhibited the lowest toxicity.
No relevant information has been identified on the toxicity of phase II metabolites of T2 and HT2 and their phase I metabolites.
14.5. Mode of action for toxicity
T2 interferes with the peptide bond formation and is a protein synthesis inhibitor and induces ribotoxic stress. It inhibits DNA and RNA synthesis but at concentrations higher than those inhibiting protein synthesis. T2 induces lipid peroxidation by generating free radicals. Rapidly proliferating tissues such as haematopoietic tissue are targets of T2. T2 induces apoptosis which can lead to leucopoenia and thrombocytopenia but the exact mechanism of induction is not fully understood.
Elicitation of emesis by T2 and HT2 in mink is mediated by systemic effects involving, i.e. the PYY3‐36) and 5‐HT. In rodents, emesis cannot occur, but the abnormal anorectic effect observed in mice following T2 and HT2 exposure is considered indicative of systemic induction of nausea by similar mechanisms.
No information on mode of action of toxicity of modified forms of T2 or HT2 were identified.
14.6. Structure–activity relationships
Stepwise hydrolysis of the three ester groups of T2 leads to a concomitant decrease in toxic activity, while hydroxylation has little effect on toxicity. De‐epoxidation constitutes a detoxifying metabolic pathway.
Formation of phase II metabolites through conjugation with glucose, sulfate or glucuronic acid, although presumably abolishing toxicity, is reversible and might release the aglycone upon ingestion.
14.7. Acute and chronic dose–response analysis
The CONTAM Panel selected the emetic effects of T2 and HT2 for the acute hazard characterisation. A BMD analysis was performed on a combined data set on the incidence of emetic events in mink exposed to either T2 or HT2.
The BMD modelling using a BMR of 10% resulted in a benchmark dose 95% lower and upper confidence interval (BMDL10–BMDU10) of 2.97–49.8 μg T2 or HT2/kg bw across the applied BMD models.
For chronic toxicity, the CONTAM Panel selected the decrease in total leucocyte count observed in a subchronic study on T2 in male Wistar rats as the critical effect. For this BMD analysis, the CONTAM Panel selected a BMR of 10%. A 95% BMDL10–BMDU10 confidence interval of 3.33–27.6 T2/kg bw per day across the applied models was calculated.
14.8. Group acute reference dose for T2 and HT2 and their modified forms
Based on new studies reporting anorectic effects (e.g. feed refusal, retching, vomiting, reduced body weight gain), the CONTAM Panel established a group ARfD for T2 and HT2 using a BMDL10 of 2.97 μg/kg bw for T2 and HT2 based on their emetic effects. Using an UF of 10, for intraspecies differences and an ARfD of 0.3 (rounded from 0.297) μg T2 and HT2/kg bw was established. An interspecies UF was not included because humans were not considered more sensitive to this endpoint than the mink.
NEO, a phase I metabolite showing emetic effects at a similar dose (at molar basis) as T2 and HT2, was included in the group ARfD with the same potency factor (i.e. 1).
In the absence of data on emetic effects of phase II metabolites, it is assumed that these will be hydrolysed in the intestine with the release of T2 or HT2 or their phase I metabolites. Therefore, the Panel found it appropriate to include these phase II metabolites with the same potency factor (i.e. 1, expressed on a molar basis) in the group ARfD.
14.9. Group tolerable daily intake for T2 an HT2 and their modified forms
The CONTAM Panel used a new 90‐day study in rats and decreases in leucocyte counts as the critical endpoint and derived a BMDL10 of 3.3 μg T2/kg bw. Based on rapid metabolism of T2 to HT2 and structural similarities, this value was used as a reference point for establishing a TDI for T2 and HT2. An UF of 200 was used taking into account 10 for interspecies and 10 for intraspecies variation and a factor of 2 since it was a subchronic study. A TDI of 0.02 (rounded from 0.017 μg T2 and HT2/kg bw) was established.
Haematotoxicity with reduced production of erythrocytes, leucocytes and platelets is the critical effect of T2. It is assumed that phase I metabolites exert toxicity via a similar mode of action, and therefore, the CONTAM Panel considered it appropriate on the assumption of dose addition, to include the modified forms NEO, T2‐triol and T2‐tetraol in a group TDI.
It was decided, to assign RPFs on a molar basis because of different potencies of phase I metabolites in inhibition of protein synthesis and other toxic effects. These are 1 for T2, HT2 and 19‐HO‐T2, 0.3 for NEO and 19‐HO‐HT2 and 0.1 for T2‐triol and T2‐tetraol.
Phase II metabolites of T2, HT2, NEO, T2‐triol and T2‐tetraol can be hydrolysed to their parent compounds after ingestion. They can be included in the Group TDI and be applied the same RPF. Thus, T2‐3‐Glc, T2‐3‐diGlc, T2‐3‐Sulf, T2‐3‐GlcA, HT2‐3‐Glc, HT2‐diGlc, HT2‐GlcA, HT2‐MalGlc can be included by applying an RPF of 1. NEO‐Glc and can be included using a factor 0.3 and T2 triol‐Glc and T2‐tetraol‐Glc by applying a factor of 0.1.
15. Recommendations
The chemical structures of the phase I and phase II metabolites of T2 and HT2, which have not been fully characterised, should be established.
Certified reference materials and standards for the modified forms of T2 and HT2 are needed.
More information on the occurrence of modified forms with appropriate standards of T2 and HT2 in feed and food is needed.
Studies on the toxicokinetics of modified forms of T2 and HT2, in particular major conjugates (glucosides) occurring in plant‐derived food items, should be conducted.
More data on toxicity are needed, in particular on emetic effects and on haematopoiesis of modified forms of T2/HT2 with appropriate study design and specified purity of the compounds applied.
Testing the dose additivity assumption is needed.
Comparative toxicity data on T2 and HT2 and their modified forms are needed.
Addendum
After adoption of this opinion, a study providing further information on the formation of modified forms of T2 and HT2 in cereals was published. Eight additional metabolites were described: One phase I metabolite, i.e. hydroxy‐HT2, and seven phase II metabolites, i.e. two isomers of hydroxy‐HT2‐diGlc, hydroxy‐HT2‐anhydro‐diGlc, HT2‐MalGlc, HT2‐anhydro‐diGlc, 15‐acetyl‐18,19‐dehydro‐T2‐tetraol‐Glc and 18,19‐dehydro‐HT2‐Glc.
Meng‐Reiterer J, Bueschl C, Rechthaler J, Berthiller F, Lemmens M and Schuhmacher R, 2016. Metabolism of HT‐2 toxin and T‐2 toxin in oats. Toxins, 8, 364–385, doi:10.3390/toxins8120364
Abbreviations
- 5‐HT
5‐hydroxytryptamine
- Ac
acetyl
- AIC
Akaike's information criterion
- ALP
alkaline phosphatase
- ALT
alanine amino transferase
- ARfD
acute reference dose
- AST
aspartate amino transferase
- BMD
benchmark dose
- BMD10
the benchmark response of 10% resulted in a benchmark dose 10%
- BMDL5
the 95th benchmark dose lower confidence limit
- BMDL10
the 90th benchmark dose lower confidence limit
- BMDU5
the 95th benchmark dose upper confidence limit
- BMDU10
the 95th benchmark dose upper confidence limit
- BMR
benchmark response
- BeWo cells
human placental choriocarcinoma cells
- bw
body weight
- CAS
Chemical Abstracts Service
- CD
cluster of differentiation
- ConA
concanavalin A
- CONTAM Panel
EFSA Panel on Contaminants in the Food Chain
- CYP
cytochrome P450
- DART‐MS
direct‐analysis‐in‐real‐time mass spectrometry
- DE
deoxy, de‐epoxy
- diGlc
diglucose
- DON
deoxynivalenol
- EC50
half maximal effective concentration
- ED50
median effective dose
- ERK
extracellular signal‐regulated kinase
- FAO
Food and Agriculture Organization of the United Nations
- Fer
feruloyl
- GC
gas chromatography
- GD
gestation day
- Glc
glucoside, glucose
- GlcA
glucuronic acid
- Hb
haemoglobin
- HBGV
health based guidance value
- HO
hydroxyl
- HPLC
high‐performance liquid chromatography
- HPLC‐FLD
high‐performance liquid chromatography–fluorescence detection
- HPLC–MS/MS
high‐performance liquid chromatography–tandem mass spectrometry
- Hsp90
heat shock protein 90
- HT2
HT2‐toxin
- IFN
interferon
- IL
interleukin
- i.p.
intraperitoneal
- i.v.
intravenous
- IUPAC
International Union of Pure and Applied Chemistry
- JNK
c‐Jun N‐terminal kinase
- KBD
Kashin–Beck disease
- LC
liquid chromatography/left‐censored
- LC‐FLD
liquid chromatography‐fluorescence detection
- LC–HRMS
liquid chromatography–high‐resolution mass spectrometry
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- LD50
median lethal dose
- LOAEL
lowest‐observed‐adverse‐effect‐level
- LPO
lipid peroxidase
- Mal
malonyl
- MalGlc
malonylglucose
- Man
mannose
- MAPK
mitogen‐activated protein kinase
- MCV
mean corpuscular volume
- MDA
malondialdehyde
- ML
maximum level
- m.p.
melting point
- mRNA
messenger RNA
- MS
mass spectrometry, mass spectrum
- MS/MS
tandem mass spectrometry
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- MW
molecular weight
- NEO
neosolaniol
- NMR
nuclear magnetic resonance
- NOAEL
no‐observed‐adverse‐effect‐level
- QuEChERS
Quick, easy, cheap, effective, rugged and safe
- PND
post‐natal day
- p.o.
Per os
- PYY‐36
pancreatic peptide YY3‐36
- RNA
ribonucleic acid
- ROS
reactive oxygen species
- RPF
relative potency factor
- SAPK
stress activated protein kinase
- SAR
structure–activity relationship
- s.c.
subcutaneous
- SCF
Scientific Committee on Food
- SE
standard error
- SOD
superoxide dismutase
- Sulf
sulfate
- T2
T2‐toxin
- TDI
tolerable daily intake
- TNF
tumour necrosis factor
- UDP
uridine 5′‐diphosphate
- UDPGA
uridine 5′‐diphosphate glucuronic acid
- UF
uncertainty factor
- UGT
UDP glucosyltransferase
- WHO
World Health Organization
- Xyl
xylose
Appendix A – Search for scientific literature on T2 and HT2 toxin and their modified forms
Table A.1.
Search terms and information source for literature search on T2 and HT2 and their modified forms
| EFSA Systematic Review | |
|---|---|
| Chemistry and analysis | |
| Search terms | TOPIC: (T2‐toxin OR HT‐2 toxin) AND TOPIC: (chemistry OR analysis OR determination OR detection OR identification OR formation OR GC OR GC‐MS OR HPLC OR LC‐MS OR ICP‐MS) |
| Information source | Number of records retrieved |
| Web of Science | 705 |
| PubMed | 268 |
| Metabolism, Kinetics | |
| Search terms | TOPIC: (T2‐toxin OR HT‐2 toxin) AND TOPIC (toxicokinetic* OR metabolism OR distribution OR excretion OR absorption OR distribution OR biomarker OR mode of action OR biotransformation OR elimination OR reduction OR detoxification OR extraction) |
| Information source | Number of records retrieved |
| Web of Science | 471 |
| PubMed | 213 |
| Toxicity | |
| Search terms | TOPIC: (T2‐toxin OR HT‐2 toxin) AND TOPIC: (toxicity OR toxic* OR acute OR subacute OR subchronic OR chronic OR mutagen* OR carcino* OR genotox* OR reprotox* OR nephrotox* OR neurotox* OR hepatotox* OR immunotox* OR haemotox* OR hematotox* OR hemotox OR cytotox* OR develop* toxicity OR thyroid OR endocri* OR poisoning OR incidental poisoning OR rat OR mouse OR lab animal OR animal*) |
| Information source | Number of records retrieved |
| Web of Science | 966 |
| PubMed | 202 |
| Human data | |
| Search terms | TOPIC: (T2‐toxin OR HT‐2 toxin) AND TOPIC: (biomarker OR biological marker OR case study OR incidental poisoning OR poisoning OR human poisoning OR human OR epidemiol*) |
| Information source | Number of records retrieved |
| Web of Science | 586 |
| PubMed | 231 |
| Date accessed | January 2016 |
| Total number retrieved | 3,542 |
| Total number after duplicates removed | 1,087 |
Appendix B – Chemical structures of T2 and HT2 and their phase I metabolites
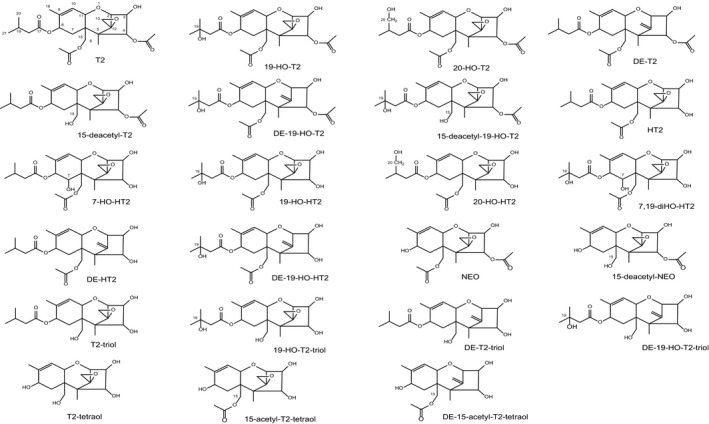
Appendix C – Chemical structures of phase II metabolites of T2 and HT2
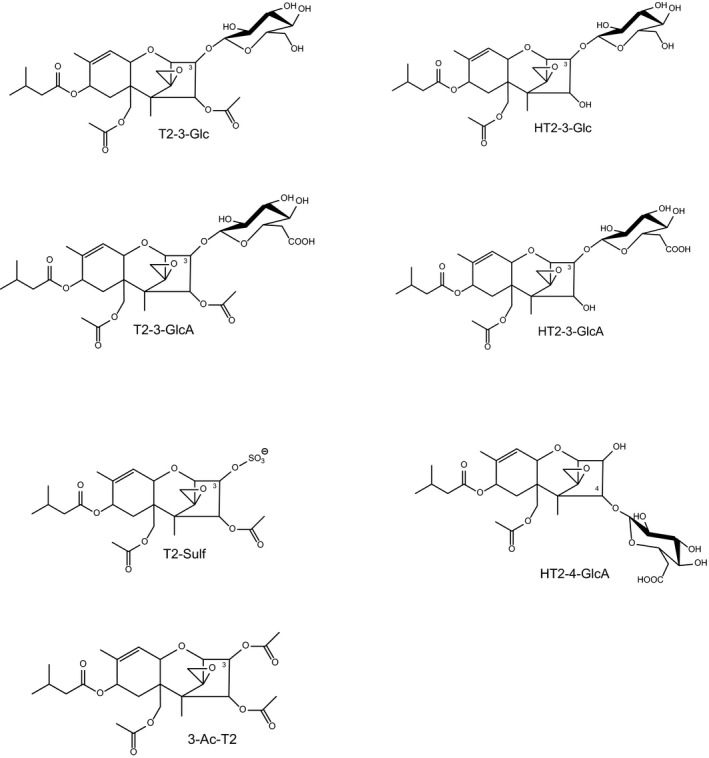
Appendix D – Derivation of a BMD for acute effects of T2 and HT2
The dose‐dependent incidences of emesis upon administration of T2 and HT2 in mink in the study from Wu et al. (2015) has been selected for derivation of a BMD for T2 and HT2 by applying expert knowledge and following the new EFSA guidance on the use of the BMD (EFSA Scientific Committee, 2017). The findings selected for deriving a BMD are described extensively in Sections 8, 11 and 13 of this opinion.
D.1. Data
| Substance | Dose (μg/kg bw) | Animals showing emesis | N | Sex |
|---|---|---|---|---|
| T2 | 0 | 0 | 4 | F |
| 5 | 0 | 4 | F | |
| 50 | 3 | 4 | F | |
| 250 | 4 | 4 | F | |
| 500 | 4 | 4 | F | |
| HT2 | 0 | 0 | 4 | F |
| 5 | 0 | 4 | F | |
| 50 | 3 | 4 | F | |
| 250 | 4 | 4 | F | |
| 500 | 4 | 4 | F |
bw: body weight.
N: number of animals.
D.2. BMR: 10% (extra risk)
D.3. Software used: Proast v38.9
D.4. Additional assumptions
The results from two independent experiments on T2 and HT2 were combined considering the experiments as covariate.
D.5. Table of results
| Models | No of parameters | Log likelihood | AICa | BMDL10 (μg/kg bw)b | BMD10 (μg/kg bw)b | BMDU10 (μg/kg bw)b |
|---|---|---|---|---|---|---|
| Full | 8 | −4.50e | 25.00 | – | – | – |
| Null | 2 | −27.73 | 59.05 | – | – | – |
| Gamma | 3 | −4.50 | 15.00 | 2.97 | 28.3 | 44.3 |
| Logistic | 2 | −4.50 | 13.00 | 12.30 | 42.7 | 49.8 |
| LogLogistic | 3 | −4.50 | 15.00 | 4.29 | 37.1 | 47.1 |
| LogProbit | 3 | −4.50 | 15.00 | 4.02 | 26.8 | 49.7 |
| Two‐stagec | 3 | −4.61 | 15.22b | NRd | NRd | NRd |
| Probite | 2 | −4.50 | 13.00 | 11.0 | 36.1 | NRd |
| Weibull | 3 | −4.50 | 15.00 | 3.02 | 29.9 | 47.9 |
AIC: Akaike's information criterion.
BMD: benchmark dose calculated at 10% extra risk. BMDL10: 95th lower confidence limit (one‐sided) of BMD; BMDU10: 95th upper confidence limit (one‐sided) of BMD.
Model not fulfilling the criterion (AIC ≤ AICmin +2).
NR: Not reported.
Calculated using BMDS v2.6.086, pooling data from the two experiments.
The overall BMDL–BMDU range is 2.97–49.8 μg/kg bw (when considering all models with AIC ≤ AICmin + 2).
D.6. Figures of fitted models
Figure D.1.
Dose–response curves with fitted models
D.7. Conclusion
Following the provisions of the EFSA guidance (EFSA Scientific Committee, 2017), a BMDL10 of 2.97 μg/kg bw was selected for further consideration as this was the lowest valid BMDL10.
Appendix E – Derivation of a BMDL for subchronic effects of T2
The dose‐dependent reduction in total lymphocyte counts upon administration of T2 in rats in the study from Rahman et al. (2014) has been selected for derivation of a BMDL for T2 for subchronic/chronic effects by applying expert knowledge and following the new EFSA guidance on the use of the BMD (EFSA Scientific Committee, 2017). Notably, a series of other potentially relevant effects seen in repeat dose experiments with T2 have been used for alternative calculations of a chronic BMD. The BMD derived as presented in this appendix is the most appropriate and has therefore been used for risk characterisation. The calculations with alternative endpoints are presented in the Annex A to this opinion.
E.1. Data
| Dose (μg/kg bw per day) | Mean total leucocyte count (× 103 per μL) | SEa | N | Sex |
|---|---|---|---|---|
| 0 | 14.83 | 0.73 | 8 | M |
| 45 | 8.95 | 0.36 | 8 | M |
| 68 | 6.92 | 0.83 | 8 | M |
| 90 | 5.2 | 0.73 | 8 | M |
SE: standard error.
E.2. BMR: 10%
A deviation from the default is appropriate both for statistical reasons (effects calculated at BMDL05–BMDU05 would fall under the standard deviation of total leucocyte count in the control group) and biological reasons since a 5% variation in total leucocyte counts would still fall under normal variations for the concerned endpoint.
E.3. Software used: PROAST 38.9
E.4. Additional assumptions
None.
E.5. Tables of results
| Model | No of parameters | Log Likelihood | AIC | BMDL10 | BMD10 | BMDU10 | |
|---|---|---|---|---|---|---|---|
| Null model | 1 | −21.99 | 45.98 | ||||
| Exponential | Model 3a | 3 | −1.14 | 8.28 | 3.30 | 11.52 | 23.75 |
| Model 5 | 4 | −1.14 | 10.28 | ||||
| Hill | Model 3a | 3 | −1.15 | 8.30 | 5.95 | 15.70 | 27.60 |
| Model 5 | 4 | −1.15 | 10.30 | ||||
| Full model | 4 | −1.14 | 10.28 |
AIC: Akaike's information criterion; BMD: benchmark dose calculated at 10% extra risk. BMDL10: 95th lower confidence limit (one‐sided) of BMD; BMDU10: 95th upper confidence limit (one‐sided) of BMD.
Model with lowest AIC.
The overall BMDL–BMDU range is 3.30–27.60 μg/kg bw (when considering all models with AIC ≤ AIC min + 2).
E.6. Figure of fitted models
Figure E.1.
Dose–response curves resulting from application of the Exponential and Hills models
E.7. Conclusion
Following the provisions of the EFSA guidance, a BMDL10 of 3.3 μg/kg bw per day was selected for further consideration as this was the lowest valid BMDL10.
Appendix F – Example calculation for inclusion of modified forms of T2 and HT2 in a chronic risk assessment
Example T2‐Triol:
Assumed is a total exposure of 0.03 μg T2‐triol/kg bw per day. The RPF for T2‐triol is 0.3.
MW of T2 is 466. MW of T2‐triol is 382. The resulting correction factor for molar weight is 466/382 = 1.22. The resulting total exposure to T2‐triol expressed as T2 equivalents is 0.03 × 1.2 × 0.3 = 0.0108 μg/kg bw per day (i.e. lower than the group TDI of 0.02 μg/kg bw per day).
Correction factors for molar weight for the different modified forms of T2 and HT2 are provided in Table F.1.
Table F.1.
Molecular weights and correction factors for molar weights to be applied
| Compound | Molar weight | Correction factor to be applied for differing molar weighta |
|---|---|---|
| T2 | 466 | 1.00 |
| T2‐3‐Glc | 628 | 0.74 |
| T2‐3‐diGlc | 790 | 0.59 |
| T2‐3‐Sulf | 546 | 0.85 |
| T2‐3‐GlcA | 642 | 0.73 |
| 3‐Ac‐T2 | 508 | 0.92 |
| 3‐Fer‐T2 | 642 | 0.73 |
| 19‐HO‐T2 | 482 | 0.97 |
| HT2 | 424 | 1.10 |
| HT2‐3‐Glc | 586 | 0.80 |
| HT2‐diGlc | 748 | 0.62 |
| HT2‐GlcA | 600 | 0.78 |
| HT2‐MalGlc | 672 | 0.69 |
| 19‐HO‐HT2 | 440 | 1.06 |
| NEO | 382 | 1.22 |
| NEO‐Glc | 544 | 0.86 |
| T2‐triol | 382 | 1.22 |
| T2‐triol‐Glc | 544 | 0.86 |
| T2‐tetraol | 298 | 1.56 |
| T2‐tetraol‐Glc | 460 | 1.01 |
Factors have been rounded to one significant figure.
Annex A – Additional calculations subchronic BMD
Annex A can be found in the online version of this output (‘Supporting information’ section): http://onlinelibrary.wiley.com/doi/10.2903/j.efsa.2017.4655/abstract
Description: Additional calculations subchronic BMD – PDF file supplemented by Subchronic calculation‐PROAST excel file.
Supporting information
Additional calculations subchronic BMD
Suggested citation: EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) , Knutsen H‐K, Barregård L, Bignami M, Brüschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl‐Kraupp B, Hogstrand C, Hoogenboom LR, Nebbia CS, Oswald I, Petersen A, Rose M, Roudot A‐C, Schwerdtle T, Vleminckx C, Vollmer G, Wallace H, Dall'Asta C, Gutleb A, Metzler M, Oswald I, Parent‐Massin D, Binaglia M, Steinkellner H and Alexander J, 2017. Scientific opinion on the appropriateness to set a group health based guidance value for T2 and HT2 toxin and its modified forms. EFSA Journal 2017;15(1):4655, 53 pp. doi: 10.2903/j.efsa.2017.4655
Requestor: European Commission
Question number: EFSA‐Q‐2015‐00229
Panel members: Jan Alexander, Lars Barregård, Margherita Bignami, Beat Bruschweiler (from 23 June 2016), Sandra Ceccatelli, Bruce Cottrill, Michael Dinovi, Lutz Edler, Bettina Grasl‐Kraupp, Christer Hogstrand, Laurentius (Ron) Hoogenboom, Helle Katrine Knutsen, Carlo Stefano Nebbia, Isabelle Oswald, Annette Petersen, Vera Maria Rogiers (until 9 May 2016), Martin Rose, Alain‐Claude Roudot, Tanja Schwerdtle, Christiane Vleminckx, Günter Vollmer and Heather Wallace.
Acknowledgements: The Panel wishes to thank the members of the Working Group on HBGV for mycotoxins and their modified forms: Jan Alexander, Chiara Dall'Asta, Arno Gutleb, Manfred Metzler, Isabelle Oswald and Dominique Parent‐Massin for the preparatory work on this scientific opinion, and the EFSA staff members: Marco Binaglia and Hans Steinkellner for the support provided to this scientific opinion.
Amendment: An editorial correction was carried out in the abstract, summary, Sections 12.1, 14.8 and 14.9, that does not materially affect the contents or outcome of this scientific output. To avoid confusion, the older version has been removed from the EFSA Journal, but is available on request, as is a version showing all the changes made.
Adopted: 23 November 2016
Amended: 16 May 2017
Notes
EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2014. Scientific Opinion on the risks for human and animal health related to the presence of modified forms of certain mycotoxins in food and feed. EFSA Journal 2014;12(12):3916, 107 pp. doi:10.2903/j.efsa.2014.3916 Available online: http://www.efsa.europa.eu/efsajournal
Summary report of the Standing Committee on Plants, Animals, Food and Feed, held in Brussels on 11 February 2015 (Section Toxicological Safety of the Food Chain), agenda item A.06. Report available at: http://ec.europa.eu/food/committees/regulatory/scfcah/toxic/docs/sum_20150211_en.pdf
Council Regulation (EEC) No 315/93 of February 1993 laying down Community procedures for contaminants in food. OJ L 37, 13.2.1993, p. 1–5.
Regulation (EC) No 1881/2006 of the European Parliament and the Council of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. OJ L 364, 20.12.2006, p. 5–24.
2013/165/EU: Commission Recommendation of 27 March 2013 on the presence of T‐2 and HT‐2 toxin in cereals and cereal products Text with EEA relevance OJ L 91, 3.4.2013, p. 12–15.
A 5‐HT3 receptor antagonist used as antiemetic.
References
- Agrawal M, Yadav P, Lomash V, Bhaskar A and Lakshmana P, 2012. Rao, T‐2 toxin induced skin inflammation and cutaneous injury in mice. Toxicology, 302, 255–265. [DOI] [PubMed] [Google Scholar]
- Amelin VG, Karaseva NM and Tretyakov AV, 2013a. Simultaneous determination of trichothecene micotoxins, ochratoxin A, and zearalenone in grain and products of its processing, feed premixes, and meat by gas chromatography ISSN 1061_9348. Journal of Analytical Chemistry, 68, 61–67. © Pleiades Publishing, Ltd., 2013a. Original Russian Text © V.G. Amelin, N.M. Karaseva, A.V. Tretyakov, 2013, published in Zhurnal Analiticheskoi Khimii, 2013, Vol. 68, No. 1, pp. 64–70. [Google Scholar]
- Amelin VG, Karaseva BNM and Tretyakov AV, 2013b. Combination of the QuEChERS method with dispersive liquid– liquid microextraction and derivatization in the determination of mycotoxins in grain and mixed feed by gas–liquid chromatography with an electron capture detector ISSN 1061_9348. Journal of Analytical Chemistry, 68, 552–557. © Pleiades Publishing, Ltd., 2013. Original Russian Text © V.G. Amelin, N.M. Karaseva, A.V. Tret'yakov, 2013, published in Zhurnal Analiticheskoi Khimii, 2013, Vol. 68, No. 6, pp. 612–618. [Google Scholar]
- Anderson DW, Black RM, Lee CG, Pottage C, Rickard RL, Sandford MS, Webber TD and Williams NE, 1989. Structure‐activity studies of trichothecenes: cytotoxicity of analogues and reaction products derived from T‐2 toxin and neosolaniol. Journal of Medicinal Chemistry, 32, 555–562. [DOI] [PubMed] [Google Scholar]
- Arroyo‐Manzanares N, García‐Campaña AM and Gámiz‐Gracia L, 2013. Multiclass mycotoxin analysis in Silybum marianum by ultra high performance liquid chromatography–tandem mass spectrometry using a procedure based on QuEChERS and dispersive liquid–liquid microextraction. Journal of Chromatography A, 1282, 11–19. [DOI] [PubMed] [Google Scholar]
- Azaiez I, Giusti F, Sagratini G, Mañes J and Fernández‐Franzón M, 2014. Multi‐mycotoxins analysis in dried fruit by LC/MS/MS and a modified QuEChERS procedure. Food Analytical Methods, 7:935–945. doi: 10.1007/s12161-013-9785-3 [DOI] [Google Scholar]
- Babich H and Borenfreund E, 1991. Cytotoxicity of T‐2 toxin and its metabolites determined with the neutral red cell viability assay. Applied and Environmental Microbiology, 57, 2101–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg JR, Riggs NV and Strong FM, 1968. The structures of toxins from two strains of Fusarium tricinctum . Tetrahedron, 24, 3329–3336. [DOI] [PubMed] [Google Scholar]
- Behm C, Föllmann W and Degen GH, 2012. Cytotoxic potency of mycotoxins in cultures of V79 lung fibroblast cells. Journal of Toxicology and Environmental Health, Part A, 75:19–20, 1226–1231, doi: 10.1080/15287394.2012.709170 [DOI] [PubMed] [Google Scholar]
- Bergmann F, Soffer D and Yagen B, 1988. Cerebral toxicity of the trichothecene toxin T‐2, of the products of its hydrolysis and of some related toxins. Toxicon, 26, 923–930. [DOI] [PubMed] [Google Scholar]
- Broekaert N, Devreese M, De Baere S, De Backer P and Croubels S, 2015. Modified Fusarium mycotoxins unmasked: from occurrence in cereals to animal and human excretion. Food and Chemical Toxicology, 80, 17–31. [DOI] [PubMed] [Google Scholar]
- Burek O, Wiśniewska–Dmytrow H and Żmudzki J, 2011. Determination of T‐2 and HT‐2 toxins in feedstuffs by high–performance liquid chromatography with fluorescence detector. Bulletin of the Veterinary Institute in Pulawy, 55, 737–740. [Google Scholar]
- Busman M and Maragos CM, 2015. Determination of T‐2 and HT‐2 toxins from maize by direct analysis in real time mass spectrometry. World Mycotoxin Journal, 8, 489–497. [Google Scholar]
- Busman M, Poling SM and Maragos CM, 2011. Observation of T‐2 toxin and HT‐2 tooxin glucosides from Fusarium sporotrichioides in liquid chromatography coupled to tandem mass spectrometry (LC‐MS/MS). Toxins, 3, 1554–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandratre GA, Telang AG, Badgujar PC, Raut SS and Sharma AK, 2014. Toxicopathological alterations induced by high dose dietary T‐2 mycotoxin and its residue detection in Wistar rats. Archives of Environmental Contamination and Toxicology, 67, 124–138. doi: 10.1007/s00244-014-0006-x [DOI] [PubMed] [Google Scholar]
- Chattopadhyay P, Upadhyay A, Agnihotri A, Karmakar S, Ghoyary D and Veer V, 2013. Comparative hematoxicity of Fusarium mycotoxin in experimental Sprague‐Dawley rats. Toxicology International, 20, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J‐H, Xue S, Li S, Wang Z‐L, Yang H, Wang W, Song D, Zhou X and Chen C, 2012. Oxidant damage in Kashin‐Beck disease and a rat Kashin‐Beck disease model by employing T‐2 toxin treatment under selenium deficient conditions. Journal of Orthopaedic Research, 30, 1229–1237. doi: 10.1002/jor.22073 [DOI] [PubMed] [Google Scholar]
- Chen D, Cao X, Tao Y, Wu Q, Pan Y, Peng D, Liu Z, Huang L, Wang Y, Wang X and Yuan Z, 2013. Development of a liquid chromatography–tandem mass spectrometry with ultrasound‐assisted extraction and auto solid‐phase clean‐up method for the determination of Fusarium toxins in animal derived foods. Journal of Chromatography A, 1311, 21–29. [DOI] [PubMed] [Google Scholar]
- Chi MS, Robison TS, Mirocha CJ and Reddy KR, 1978. Acute toxicity of 12,13‐epoxytrichothecenes in one‐day‐old broiler chicks. Applied and Environmental Microbiology, 35, 636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coddington KA, Swanson SP, Hassan AS and Buck WB, 1989. Enterohepatic circulation of T‐2 toxin metabolites in the rat. Drug Metabolism and Disposition, 17, 600–605. [PubMed] [Google Scholar]
- Conrady‐Lorck S, Gareis M, Feng XC, Amselgruber W and Forth Wand Fichtl B, 1988. Metabolism of T‐2 toxin in vascularly autoperfused jejunal loops of rats. Toxicology and Applied Pharmacology, 94, 23–33. [DOI] [PubMed] [Google Scholar]
- De Boevre M, Di Mavungu JD, Landschoot S, Audenaert K, Eeckhout M, Maene P, Haesaert G and De Saeger S, 2012. Natural occurrence of mycotoxins and their masked forms in food and feed products. World Mycotoxin Journal, 5, 207–219. [Google Scholar]
- De Girolamo AM, Solfrizzo VMT, Lattanzio J, Stroka A, Alldrick HP and van Egmond4 A, 2013. Visconti critical evaluation of LC‐MS‐based methods for simultaneous determination of deoxynivalenol, ochratoxin A, zearalenone, aflatoxins, fumonisins and T‐2/HT‐2 toxins in maize. World Mycotoxin Journal, 6, 317–334. [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2011. Scientific Opinion on the risks for animal and public health related to the presence of T‐2 and HT‐2 toxin in food and feed. EFSA Journal 2011;9(12):2481, 187 pp. doi: 10.2903/j.efsa.2011.2481. Available online: http://www.efsa.europa.eu/efsajournal [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2014. Scientific Opinion on the risks for human and animal health related to the presence of modified forms of certain mycotoxins in food and feed. EFSA Journal 2014;12(12):3916, 107 pp. doi: 10.2903/j.efsa.2014.3916 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2016. Scientific opinion on the appropriateness to set a group health‐based guidance value for zearalenone and its modified forms. EFSA Journal 2016;14(4):4425, 46 pp. doi: 10.2903/j.efsa.2016.4425 [DOI] [Google Scholar]
- EFSA FEEDAP Panel (EFSA Panel on Additives and Products or Substances used in Animal Feed), 2012. Guidance for the preparation of dossiers for sensory additives. EFSA Journal 2012;10(1):2534, 26 pp. doi: 10.2903/j.efsa.2012.2534 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2009. Guidance of the Scientific Committee on transparency in the scientific aspects of risk assessments carried out by EFSA. Part 2: general principles. EFSA Journal 2009;7(5):1051, 22 pp. doi: 10.2903/j.efsa.2009.1051 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2012. Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data. EFSA Journal 2012;10(3):2579, 32 pp. doi: 10.2903/j.efsa.2012.2579 [DOI] [Google Scholar]
- EFSA Scientific Committee , Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen KH, More S, Mortensen A, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Silano V, Solecki R, Turck D, Aerts M, Bodin L, Davis A, Edler L, Gundert‐Remy U, Sand S, Slob W, Bottex B, Abrahantes JC, Marques DC, Kass G and Schlatter JR, 2017. Update: Guidance on the use of the benchmark dose approach in risk assessment. EFSA Journal 2017;15(1):4658, 41 pp. doi: 10.2903/j.efsa.2017.4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich KC and Daigle KW, 1985. Protein synthesis by mammalian cells treated with C‐3‐modified analogs of the 12,13‐epoxytrichothecenes T‐2 and T‐2 tetraol. Applied and Environmental Microbiology, 50, 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAO/WHO (Food and Agriculture Organization of the United Nations/World Health Organization), 2001. WHO FOOD ADDITIVES SERIES: 47, Safety evaluation of certain mycotoxins in food. Deoxynivalenol. Prepared by the Fifty‐sixth meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). pp. 419–528. Available online: http://www.inchem.org/documents/jecfa/jecmono/v47je01.htm
- Forsell JH, Kateley JR, Yoshizawa T and Pestka JJ, 1985. Inhibition of mitogen‐induced blastogenesis in human lymphocytes by T‐2 toxin and its metabolites. Applied and Environmental Microbiology, 49, 1523–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruhmann P, Skrinjar P, Weber J, Mikula H, Warth B, Sulyok M, Kska R, Adam G, Rosenberg E, Hametner C and Fröhlich J, 2014. Sulfation of deoxynivalenol, its acetylated derivatives, and T2‐toxin. Tetrahedron, 70, 5260–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaigé S, Djelloul M, Tardivel C, Airault C, Félix B, Jean A, Lebrun B, Troadec JD and Dallaporta M, 2014. Modification of energy balance induced by the food contaminant T‐2 toxin: a multimodal gut‐to‐brain connection. Brain, Behavior, and Immunity, 37, 54–72. [DOI] [PubMed] [Google Scholar]
- García‐Moraleja A, Font G, Mañes J and Ferrer E, 2015. Analysis of mycotoxins in coffee and risk assessment in Spanish adolescents and adults. Food and Chemical Toxicology, 86, 225–233. doi: 10.1016/j.fct.2015.10.014 [DOI] [PubMed] [Google Scholar]
- Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G and Yusupov M, 2014. Structural basis for the inhibition of the eukaryotic ribosome. Nature, 513, 517–522. [DOI] [PubMed] [Google Scholar]
- Gordon G, 1985. Ipecacuanha induced emesis in the treatment of self‐poisoned adults. Archives of Emergency Medicine, 2, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan F, Li S, Wang ZL, Yang H, Xue S, Wang W, Song D, Zhou X, Zhou W, Chen JH, Caterson B and Hughes C, 2013. Histopathology of chondronecrosis development in knee articular cartilage in a rat model of Kashin–Beck disease using T‐2 toxin and selenium deficiency conditions. Rheumatology International, 33, 157–166. doi: 10.1007/s00296-011-2335-7 [DOI] [PubMed] [Google Scholar]
- Islam Z, Nagase M, Ota A, Ueda S, Yoshizawa T and Sakato N, 1998. Structure‐function relationship of T‐2 toxin and its metabolites in inducing thymic apoptosis in vivo in mice. Bioscience, Biotechnology, and Biochemistry, 62, 1492–1497. [DOI] [PubMed] [Google Scholar]
- Jaradat ZW, 2005. T‐2 mycotoxin in the diet and its effects on tissues In: Watson RR, Preedy VR, (eds.). Reviews in Food and Nutrition Toxicity. Volume 4. CRC Press, Boca Raton, FL; London; New York; Singapore: pp. 173–212. [Google Scholar]
- Kang P, Yao Y, Yang J, Shen B, Zhou Z and Pei F, 2013. An animal model of KashineBeck disease induced by a low‐nutrition diet and exposure to T‐2 toxin. Osteoarthritis Cartilage, 21, 1108–1115. [DOI] [PubMed] [Google Scholar]
- Kelley KW, Bluthé RM, Dantzer R, Zhou JH, Shen WH, Johnson RW and Broussard SR, 2003. Cytokine‐induced sickness behavior. Brain, behavior, and immunity, 17, 112–118. [DOI] [PubMed] [Google Scholar]
- Kim V‐W, Sharma RP and Elsner V, 1991. Effects of T‐2 toxin and its congeners on membrane functions of cultured human fibroblasts. Mycotoxin Research, 7, 19–28. [DOI] [PubMed] [Google Scholar]
- Königs M, Mulac D, Schwerdt G, Gekle M and Humpf HU, 2009. Metabolism and cytotoxic effects of T‐2 toxin and its metabolites on human cells in primary culture. Toxicology, 258, 106–113. [DOI] [PubMed] [Google Scholar]
- Lafarge‐Frayssinet C, Chakor K, Lafont P and Frayssinet C, 1990. Transplacental transfer of T2‐toxin: pathological effects. Journal of Environmental Toxicology and Oncology, 10, 64–68. [PubMed] [Google Scholar]
- Lattanzio VM, Visconti A, Haidukowski M and Pascale M, 2012. Identification and characterization of new Fusarium masked mycotoxins, T2 and HT2 glycosyl derivatives, in naturally contaminated wheat and oats by liquid chromatography–high‐resolution mass spectrometry. Journal of Mass Spectrometry, 47, 466–475. [DOI] [PubMed] [Google Scholar]
- Lattanzio VMT, Ciasca B, Haidukowski M, Infantino A, Visconti A and Pascale M, 2013. Mycotoxin profile of Fusarium langsethiae isolated from wheat in Italy: production of type‐A trichothecenes and relevant glucosyl derivatives. Journal of Mass Specrometry, 48, 1291–1298. [DOI] [PubMed] [Google Scholar]
- Lattanzio VMT, Ciasca B, Terzi V, Ghizzoni R, McCormick SP and Pascale M, 2015. Study of the natural occurrence of T‐2 and HT‐2 toxins and their glucosyl derivatives from field barley to malt by high‐resolution Orbitrap mass spectrometry. Food Additives and Contaminants, Part A, 32, 1647–1655. [DOI] [PubMed] [Google Scholar]
- Lebrun B, Tardivel C, Félix B, Abysique A, Troadec JD, Gaigé S and Dallaporta M, 2015. Dysregulation of energy balance by trichothecene mycotoxins: mechanisms and prospects. Neurotoxicology, 49, 15–27. [DOI] [PubMed] [Google Scholar]
- Maragos CM, Kurtzman C, Busman M, Price N and McCormick S. 2013. Development and evaluation of monoclonal antibodies for the glucoside of T‐2 Toxin (T2‐Glc). Toxins, 5, 1299–1313. doi: 10.3390/toxins5071299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto H, Ito T and Ueno Y, 1978. Toxicological approaches to the metabolites of Fusaria. 12. Fate and distribution of T‐2 toxin in mice. Japanese Journal of Experimental Medicine, 48, 393–399. [PubMed] [Google Scholar]
- McCormick SP, Stanley AM, Stover NA and Alexander NJ, 2011. Trichothecenes: from simple to complex mycotoxins. Toxins, 3, 802–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick SP, Price NPJ and Kurtzman CP, 2012. Glucosylation and other biotransformations of T‐2 toxin by yeasts of the Trichomonascus clade. Applied Environmental Microbiology, 78, 8694–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick SP, Kato T, Maragos CM, Busman M and Lattanzio VMT. 2015. Anomericity of T‑2 toxin‐glucoside: masked mycotoxin in cereal crops. Journal of Agriculture and Food Chemistry, 63, 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng‐Reiterer J, Varga E, Nathanail AV, Bueschl C, Rechthaler J, McCormick SP, Michlmayr H, Malachová A, Fruhmann P, Adam G, Berthiller F, Lemmens M and Schuhmacher R, 2015. Tracing the metabolism of HT‐2 toxin and T‐2 toxin in barley by isotope‐assisted untargeted screening and quantitative LC‐HRMS analysis. Analytical and Bioanalytical Chemistry, 407, 8019–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Milczewski KE, 1987. Toxicity of epoxy trichothecenes in cultured mammalian cells. Mycotoxin Research, 3, 69–76. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Sakamoto S, Sago Y and Nagashima H, 2013a. Detection of masked mycotoxins derived from type A trichothecenes in corn by high‐resolution LC‐Orbitrap mass spectrometer. Food Additives and Contaminants: Part A, 30, 1407–1414. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Sakamoto S, Sago Y and Nagashima H, 2013b. Detection of type A trichothecene di‐glucosides produced in corn by high‐resolution liquid chromatography‐Orbitrap mass spectrometry. Toxins, 5, 590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanail AV, Varga E, Meng‐Reiterer J, Bueschl C, Michelmayr H, Malachova A, Fruhmann P, Jestoi M, Peltonen K, Adam G, Lemmens M, Schuhmacher R and Berthiller F, 2015. Metabolism of the Fusarium mycotoxins T‐2 toxin and HT‐2 toxin in wheat. Journal of Agricultural and Food Chemistry, 63, 7862–7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanail AV, Gibson B, Han L, Peltonen K, Ollilainen V, Jestoi M and Laitila A. 2016. The lager yeast Saccharomyces pastorianus removes and transforms Fusarium trichothecene mycotoxins during fermentation of brewer's wort. Food Chemistry, 203, 448–455. [DOI] [PubMed] [Google Scholar]
- Obremski K, Podlasz P, Zmigrodzka M, Winnicka A, Woźny M, Brzuzan P, Jakimiuk E, Wojtacha P, Gajecka M, Zielonka Ł and Gajecki M, 2013. The effect of T‐2 toxin on percentages of CD4+, CD8+, CD4+ CD8+ and CD21+ lymphocytes, and mRNA expression levels of selected cytokines in porcine ileal Peyer's patches. Polish Journal of Veterinary Sciences, 16, 341–349. [DOI] [PubMed] [Google Scholar]
- Pace JG, Watts MR, Burrows EP, Dinterman RE, Matson C, Hauer EC and Wannemacher RW Jr, 1985. Fate and distribution of 3H‐labeled T‐2 mycotoxin in guinea pigs. Toxicology and Applied Pharmacology, 80, 377–385. [DOI] [PubMed] [Google Scholar]
- Percie du Sert N, Holmes AM, Wallis R and Andrews PLR, 2012. Predicting the emetic liability of novel chemical entities: a comparative study. British Journal of Pharmacology, 165, 1848–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira VL, Fernandes JO and Cunha SC, 2015. Comparative assessment of three cleanup procedures after QuEChERS extraction for determination of trichothecenes (type A and type B) in processed cereal‐based baby foods by GC‐MS. Food Chemistry, 182, 143–149. doi: 10.1016/j.foodchem.2015.01.047 [DOI] [PubMed] [Google Scholar]
- Pfeiffer RL, Swanson SP and Buck WB, 1988. Metabolism of T‐2 toxin in rats – effects of dose, route, and time. Journal of Agricultural and Food Chemistry, 36, 1227–1232. [Google Scholar]
- Rafai P, Tuboly S, Bata Á, Tilly P, Ványi A, Papp Z, Jakab L and Túry E, 1995. Effect of various levels of T‐2 toxin in the immune system of growing pigs. The Veterinary Record, 136, 511–514. [DOI] [PubMed] [Google Scholar]
- Rafai P, Papp Z and Jakab L, 2013. Biotransformation of trichothecenes alleviates the negative effects of T‐2 toxin in pigs. Acta Veterinaria Hungarica, 61, 333–343. doi: 10.1556/AVet.2013.025 [DOI] [PubMed] [Google Scholar]
- Rahman S, Sharma AK, Singh ND, Telang AG, Azmi S and Prawez S, 2014. Clinico‐haematological changes in T‐2 toxicosis in Wistar rats. Indian Journal of Veterinary Pathology, 38, 22–28. [Google Scholar]
- Rakkestad KE, Skaar I, Ansteinsson VE, Solhaug A, Holme JA, Pestka JJ, Samuelsen JT, Dahlman HJ, Hongslo JK and Becher R, 2010. DNA damage and DNA damage responses in THP‐1 monocytes after exposure to spores of either Stachybotrys chartarum or Aspergillus versicolor or to T‐2 toxin. Toxicological Sciences, 115, 140–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raut SS, Sharma AK, Chandratre G and Telang AG, 2013. Experimentally induced sub‐chronic toxicity of T‐2 toxin in male Wistar rats. Indian Journal of Veterinary Pathology, 38, 41–48. [Google Scholar]
- Rychlik M, Humpf HU, Marko D, Dänicke S, Mally A, Berthiller F, Klaffke H and Lorenz N, 2014. Proposal of a comprehensive definition of modified and other forms of mycotoxins including “masked” mycotoxins. Mycotoxin Research, 30, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samara A, Yagen B, Agranat I, Rachmilewitz EA and Fibach E, 1987. Induction of differentiation in human myeloid leukemic cells by T‐2 toxin and other trichothecenes. Toxicology and Applied Pharmacology, 89, 415–428. [DOI] [PubMed] [Google Scholar]
- SCF (Scientific Committee on Food), 2001. Opinion of the Scientific Committee on Food on Fusarium toxins Part 5: T‐2 toxin and HT‐2 toxin, adopted on 30 May 2001. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_contaminants_catalogue_out88_en.pdf
- Senter LH, Sanson DR, Corley DG, Tempesta MS, Rottinghaus AA and Rottinghaus GE, 1991. Cytotoxicity of trichothecene mycotoxins isolated from Fusarium sporotrichioides (MC‐72083) and Fusarium sambucinum in baby hamster kidney (BHK‐21) cells. Mycopathologica, 113, 127–131. [DOI] [PubMed] [Google Scholar]
- Swanson SP, Rood HD, Behrens JC and Sanders PE, 1987. Preparation and characterization of the deepoxy trichothecenes: deepoxy HT‐2, deepoxy T‐2 triol, Deepoxy T‐2 tetraol, deepoxy 15‐monoacetoxyscirpenol, and deepoxy scirpentriol. Applied and Environmental Microbiology, 53, 2821–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Abe H, Kimura M, Onda N, Mizukami S, Yoshida T, and Shibutani M, 2016. Developmental exposure to T‐2 toxin reversibly affects postnatal hippocampal neurogenesis and reduces neural stem cells and progenitor cells in mice. Archives of Toxicology, 90, 2009–2024. doi: 10.1007/s00204-015-1588-4 [DOI] [PubMed] [Google Scholar]
- Thompson WL and Wannemacher RW JR, 1986. Structure ‐ function relationships of 12,13‐epoxytrichothecene mycotoxins in cell culture: comparison to whole animal lethality. Toxicon, 24, 985–994. [DOI] [PubMed] [Google Scholar]
- Torp M and Langseth W, 1999. Production of T‐2 toxin by a Fusarium resembling Fusarium poae . Mycopathologia, 147, 89–96. [DOI] [PubMed] [Google Scholar]
- Ueno Y, Kenji I, Norio S and Ohtsubo K, 1974. Toxicological approaches to the metabolites of Fusaria. VI. Vomiting factor from moldy corn infected with Fusarium spp. Japanese Journal of Experimental Medicine, 44, 123–127. [PubMed] [Google Scholar]
- Ueno Y, Nakayama K, Sato K, Ishii K, Komagata K, Minoda Y, Omori T and Hitokoto H, 1986. Biodegradation of trichothecence, T‐2 toxin In: Llewellyn GC. and O'Rear CE. (eds.). CAB International Farnham House, Farnham Royal, Slough SL2 3BN, United Kingdom. The Biodeterioration Society, Biodeterioration VI: pp. 248–254. [Google Scholar]
- Veprikova Z, Vaclavikova M, Lacina O, Dzuman Z, Zachariasova M and Hajslova J, 2012. Occurrence of mono‐ and di‐glycosylated conjugates of T‐2 and HT‐2 toxins in naturally contaminated cereals. World Mycotoxin Journal, 5, 231–240. [Google Scholar]
- Wan Q, Wu G, He Q, Tang H and Wang Y, 2015a. The toxicity of acute exposure to T‐2 toxin evaluated by the metabonomics technique. Molecular BioSystems, 11, 882–891. doi: 10.1039/c4mb00622d [DOI] [PubMed] [Google Scholar]
- Wan D, Wang X, Wu Q, Lin P, Pan Y, Sattar A, Huang L, Ahmad I, Zhang Y and Yuan Z, 2015b. Integrated transcriptional and proteomic analysis of growth hormone suppression mediated by trichothecene T‐2 toxin in rat GH3 cells. Toxicological Sciences, 147, 326–338. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang W, Cheng G, Huang L, Chen D, Tao Y, Pan Y, Hao H, Wu Q, Wan D, Liu Z, Wang Y and Yuan Z, 2014. High risk of embryo‐fetal toxicity: placental transfer of T‐2 toxin and its major metabolite HT‐2 toxin in BeWo cells. Toxicological Sciences, 137, 168–178. [DOI] [PubMed] [Google Scholar]
- Warth B, Fruhmann P, Wiesenberger G, Kluger B, Sarkanj B, Lemmens M, Hametner C, Fröhlich J, Adam G, Krska R and Schuhmacher R, 2015. Deoxynivalenol‐sulfates: identification and quantification of novel conjugated (masked) mycotoxins in wheat. Analytical and Bioanalytical Chemistry, 407, 1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner M, Welsch T, Hübner F, Schwerdt G, Gekle M and Humpf HU, 2012. Identification and apoptotic potential of T‐2 toxin metabolites in human cells. Journal of Agricultural and Food Chemistry, 60, 5676–5684. [DOI] [PubMed] [Google Scholar]
- Welsch T and Humpf HU, 2012. HT‐2 toxin 4‐glucuronide as new T‐2 toxin metabolite: enzymatic synthesis, analysis, and species specific formation of T‐2 and HT‐2 toxin glucuronides by rat, mouse, pig, and human liver microsomes. Journal of Agricultural and Food Chemistry, 60, 10170–10178. [DOI] [PubMed] [Google Scholar]
- Wu Q, Huang L, Liu Z, Yao M, Wang Y, Dai M and Yuan Z, 2011. A comparison of hepatic in vitro metabolism of T‐2 toxin in rats, pigs, chickens, and carp. Xenobiotica, 41, 863–873. [DOI] [PubMed] [Google Scholar]
- Wu Q, Dohnal V, Kuca K and Yuan Z, 2013a. Trichothecences: structure‐toxic activity relationships. Current Drug Metabolism, 14, 641–660. [DOI] [PubMed] [Google Scholar]
- Wu W, Bates MA, Bursian SJ, Flannery B, Zhou H‐R, Link JE, Zhang H and Pestka JJ, 2013b. Peptide YY3–36 and 5‐hydroxytryptamine mediate emesis induction by trichothecene deoxynivalenol (vomitoxin). Toxicological Sciences, 133, 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Xu W, Yang W, Nüssler AK, Xiong L, Kuca K, Dohnal V, Zhang X and Yuan Z, 2014. Oxidative stress‐mediated cytotoxicity and metabolism of T‐2 toxin and deoxynivalenol in animals and humans: an update. Archives of Toxicology, 88, 1309–1326. [DOI] [PubMed] [Google Scholar]
- Wu W, Zhou H, Xiao P and Pestka JJ, 2015. Comparison of anorectic potencies of the trichothecenes T‐2 toxin, HT‐2 toxin and satratoxin G to the Ipecac alkaloid emetine. Toxicology Reports, 2, 238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Zhou H, Bursian SJ, Link JE and Pestka JJ, 2016. Emetic responses to T‐2 toxin, HT‐2 toxin and emetine correspond to plasma elevations of peptide YY3‐36 and 5‐hydroxytryptamine. Archives of Toxicology, 90, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Matsunaga S, Matsui M, Takeda N and Yamatodani A, 2002. Pica in mice as a new model for the study of emesis. Methods and Findings in Experimental and Clinical Pharmacology, 24, 135–138. [DOI] [PubMed] [Google Scholar]
- Yang GH, Jarvis BB, Chung YJ and Pestka JJ. 2000. Apoptosis induction by the satratoxins and other trichothecene mycotoxins: relationship to ERK, p38 MAPK, and SAPK/JNK activation. Toxicology and Applied Pharmacology, 164, 149–160. [DOI] [PubMed] [Google Scholar]
- Yang S, Li Y, Cao X, Hu D, Wang Z, Wang Y, Shen J and Zhang S, 2013. Metabolic pathways of T‐2 toxin in in vivo and in vitro systems of Wistar rats. Journal of Agricultural and Food Chemistry, 61, 9734–9743. [DOI] [PubMed] [Google Scholar]
- Yosizawa T, Mirocha JC, Behrens JC and Swanson SP.1981. Metabolic fate of T‐2 toxin in a lactating cow. Food and Chemical Toxicology, 19, 31–39. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wang L, Yang ZH, Liu ZT and Yue W, 2006. Wang Yue value of mink vomit model in study of anti‐emetic drugs. World Journal of Gastroenterology, 12, 1300–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional calculations subchronic BMD