

SUMMARY
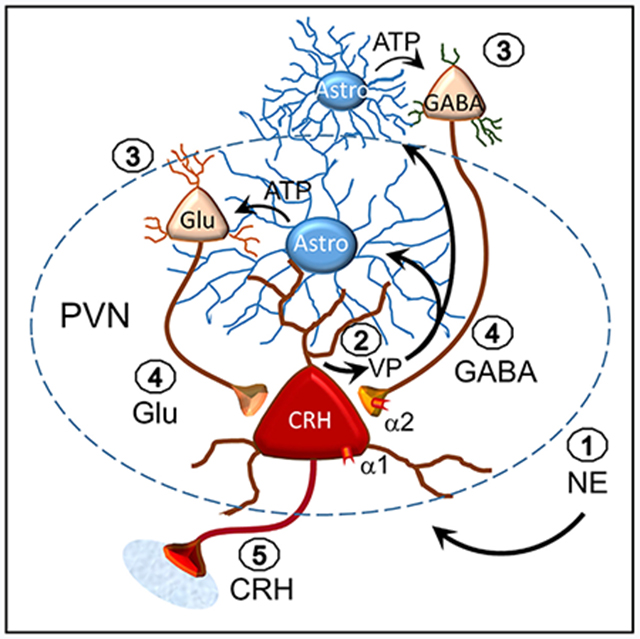
In addition to their support role in neurotransmitter and ion buffering, astrocytes directly regulate neurotransmission at synapses via local bidirectional signaling with neurons. Here, we reveal a form of neuronal-astrocytic signaling that transmits retrograde dendritic signals to distal upstream neurons in order to activate recurrent synaptic circuits. Norepinephrine activates α1 adrenoreceptors in hypothalamic corticotropin-releasing hormone (CRH) neurons to stimulate dendritic release, which triggers an astrocytic calcium response and release of ATP; ATP stimulates action potentials in upstream glutamate and GABA neurons to activate recurrent excitatory and inhibitory synaptic circuits to the CRH neurons. Thus, norepinephrine activates a retrograde signaling mechanism in CRH neurons that engages astrocytes in order to extend dendritic volume transmission to reach distal presynaptic glutamate and GABA neurons, thereby amplifying volume transmission mediated by dendritic release.
In Brief
Norepinephrine is a primary driver of the stress response. Chen et al. show that norepinephrine activates hypothalamic CRH neurons by engaging a dendritic signaling mechanism that recruits astrocytes to activate upstream neurons. This retrograde neuronal-glial signaling allows neurons to control distal presynaptic partners via astrocyte amplification of dendritic volume transmission.
Graphical Abstract
INTRODUCTION
The functional role of astrocytes is not limited to buffering extracellular ions and neurotransmitters but also includes an active involvement in neurotransmission. Astrocytes interact with neurons by responding to neurotransmitters and by releasing gliotransmitters (Verkhratsky and Nedergaard, 2018). However, the neuron-astrocyte interaction has been largely limited to the tripartite synapse or neuronal pre- and postsynaptic elements and surrounding astrocytes (Araque et al., 1999; Halassa et al., 2007; Perea et al., 2009). Astrocytic calcium signals can be transmitted through branched astrocyte arbors (Charles et al., 1991; Cornell-Bell et al., 1990; Porter and McCarthy, 1996) or can be localized to subdomains within branches (Haustein et al., 2014; Shigetomi et al., 2013), giving astrocytes the potential to signal remotely from the response generation site, and electrical coupling by gap junctions extends this capacity beyond the limits of the astrocytic arbor (Finkbeiner, 1992; Giaume et al., 2010). Whether astrocytes transmit neuron-derived signals distally is currently not known.
Volume neurotransmission is mediated by diffusion of the neurotransmitter away from its release site and can be orthograde, between the presynaptic terminal and postsynaptic soma/dendrite, or retrograde, from the postsynaptic dendrite to the presynaptic neuron. It is spatially constrained by astrocytic buffering and enzymatic degradation (Di et al., 2013; Son et al., 2013; Wu and Tasker, 2017). Pioneering research on hypothalamic neuroendocrine cells demonstrated retrograde volume transmission of the neuropeptides oxytocin and vasopressin, which exerts localized paracrine actions on presynaptic neurotransmitter release (de Kock et al., 2003; Kombian et al., 1997; Ludwig et al., 2002; Oliet et al., 2007). We reported that vasopressin released from vasopressin neuron dendrites activates a calcium signal in astrocytes that leads to stimulation of presynaptic GABA neurons (Haam et al., 2014).
The hypothalamic-pituitary-adrenal (HPA) axis comprises the main neuroendocrine stress response, producing systemic glucocorticoid secretion from the adrenals following corticotropin-releasing hormone (CRH) secretion from the hypothalamic paraventricular nucleus (PVN) and adrenocorticotrophic hormone secretion from the pituitary. Brainstem noradrenergic systems provide a major excitatory drive to the HPA axis. Anatomical and physiological studies indicate that CRH neurons receive direct noradrenergic innervation (Cole and Sawchenko, 2002; Cunningham and Sawchenko, 1988; Flak et al., 2009, 2014; Helmreich et al., 2001; Herman et al., 2004; Itoi et al., 1994, 1999; Plotsky, 1987; Sawchenko and Swanson, 1982; Woulfe et al., 1990; Ziegler et al., 2012) and medial parvocellular neurons express α1 adrenoreceptors (Cummings and Seybold, 1988; Day et al., 1997). In contrast, electrophysiological studies indicate that norepinephrine excites parvocellular neurons indirectly via activation of local synaptic circuits (Daftary et al., 2000; Han et al., 2002). Thus, despite multiple efforts, the mechanism of noradrenergic activation of the HPA axis remains elusive.
Here, we investigated the cellular mechanisms of the norepinephrine excitation of CRH neurons by using brain-slice electrophysiology, calcium imaging, and optogenetics in transgenic mice. We found that norepinephrine elicits the dendritic release of vasopressin from the CRH neurons to activate an astrocytic relay to presynaptic glutamate and GABA neurons, revealing a previously unrecognized astrocytic amplification of retrograde volume transmission.
RESULTS
Norepinephrine Activation of CRH Neurons
Extracellular loose-seal, cell-attached patch clamp recordings were performed in CRH neurons (Figure S1) in hypothalamic slices to record spontaneous spiking activity. Bath application of norepinephrine (100 μM, 5 min) caused an increase in spiking frequency that was diminished by blocking glutamate receptors with 6,7-Dinitroquinoxaline-2,3-dione disodium salt (DNQX)/DL-2-Amino-5-phosphonopentanoic acid (APV) and enhanced by blocking GABAA receptors with picrotoxin (PTX;two-way ANOVA, F (2, 33) = 3.522, p = 0.04) (Figures 1A–1C). Post hoc Bonferroni’s comparisons showed a significant increase in spiking in response to norepinephrine in control artificial cerebrospinal fluid (aCSF) (p < 0.01) and in PTX (p < 0.01). The norepinephrine response in glutamate receptor blockers did not reach statistical significance (p = 0.37). Thus, norepinephrine caused an increase in spiking in CRH neurons that was largely dependent on glutamate receptor activation and restrained by GABAA receptor activation.
Figure 1. Norepinephrine Regulation of CRH Neurons via Local Synaptic Circuits.
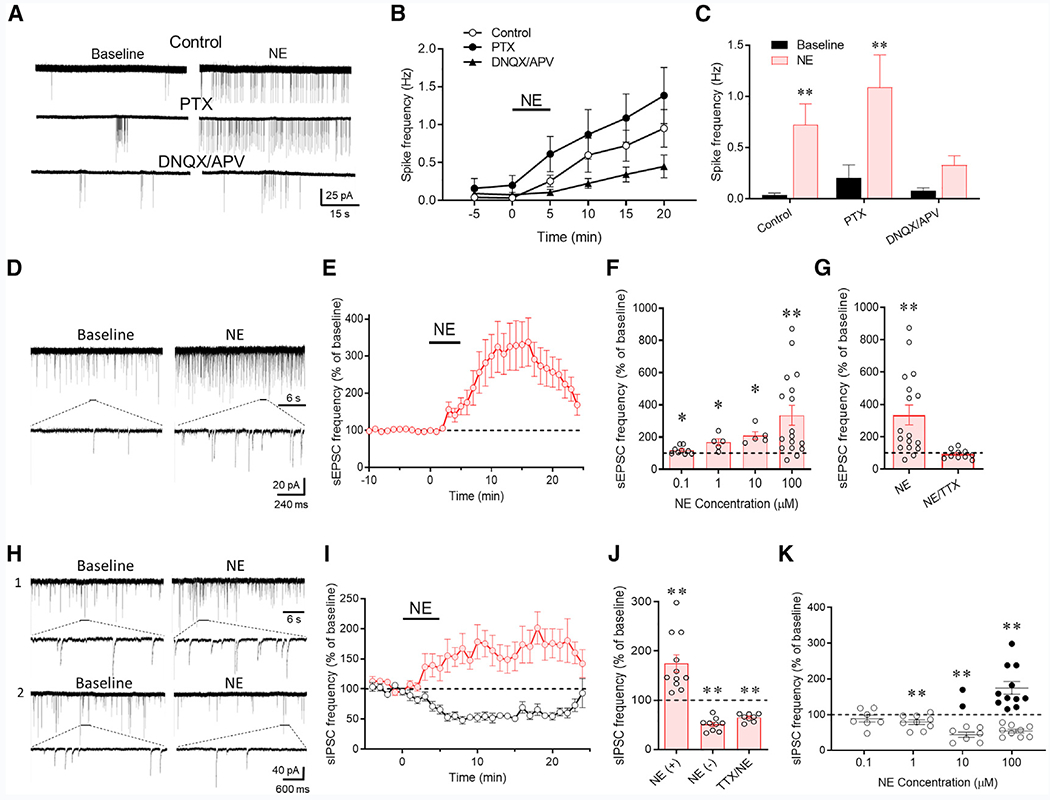
(A) Loose-seal patch clamp recordings of the spiking responses of CRH neurons to bath application of norepinephrine (100 μM) in control aCSF and in GABAA (PTX) and ionotropic glutamate receptor blockers (DNQX/APV).
(B) Time series of changes in spike frequency prior to (−5 to 0 min), during (0–5 min), and after norepinephrine application (100 μM) in control solution, PTX, and DNQX/APV (n = 9–14 cells/group).
(C) Mean spike frequency responses to norepinephrine without (control) and with the GABAA (PTX) and glutamate receptor antagonists (DNQX/APV) (n = 9–14 cells/group).
(D) Whole-cell recording of the effect of norepinephrine (100 μM) on sEPSCs in a CRH neuron recorded in picrotoxin to block sIPSCs.
(E) Time plot of the norepinephrine-induced increase in the frequency of sEPSCs (n = 17 cells).
(F) Concentration dependence of the norepinephrine-induced increase in sEPSC frequency (n = 5–17 cells/group).
(G) The norepinephrine-induced increase in sEPSC frequency was blocked by TTX (n = 10 cells), suggesting action potential dependence.
(H) Whole-cell recording of the norepinephrine (100 μM) effect on sIPSCs in a CRH neuron recorded in DNQX/APV to block sEPSCs. Norepinephrine caused an increase in sIPSCs in some cells (1) and a decrease in sIPSCs in others (2).
(I) Time plots of the normalized increase and decrease in mean sIPSC frequency in response to norepinephrine in different cohorts of CRH neurons (n = 11 and 9 cells, respectively).
(J) Mean norepinephrine-induced increase (NE (+)) and decrease (NE (−) in sIPSC frequency relative to baseline (n = 9 and 8 cells, respectively). The norepinephrine-induced increase, but not decrease, in sIPSC frequency was blocked by TTX (n = 8 cells), suggesting the facilitation (NE (+)), but not the suppression (NE (−)), of sIPSCs by norepinephrine was spike dependent.
(K) Norepinephrine concentration dependence showing that the two sIPSC responses to norepinephrine had different concentration dependencies. The norepinephrine-induced suppression of sIPSCs (open circles) was dominant at lower concentrations, whereas the norepinephrine-induced facilitation of sIPSCs (closed circles) was seen in some but not all recorded cells at higher concentrations (n = 7–9 cells [suppression] and 2–11 cells [facilitation]). *p < 0.05; **p < 0.01.
To test whether the excitation of CRH neurons is mediated by norepinephrine modulation of excitatory and inhibitory synaptic inputs, we recorded spontaneous excitatory postsynaptic currents (sEPSCs) and spontaneous inhibitory postsynaptic currents (sIPSCs) in the CRH neurons. The sEPSCs in CRH neurons had a mean basal frequency of 1.56 ± 0.10 Hz, amplitude of 19.13 ± 0.50 pA, and decay time of 2.18 ± 0.06 ms (n = 47); bath application of norepinephrine (100 μM, 5 min) evoked a robust increase in the frequency of sEPSCs (335.2% ± 61.5% of baseline at 100 μM, n = 17, p < 0.01) (Figures 1D and 1E), without causing a change in sEPSC amplitude (p = 0.14) or decay time (p = 0.58), which suggested a possible presynaptic site of action. The norepinephrine stimulation of excitatory synaptic inputs to CRH neurons was concentration dependent, with a threshold concentration of ~ 100 nM (118.7% ± 8.0% of baseline, n = 9, p = 0.047) (Figure 1F). It was also action-potential-dependent because the blockade of voltage-gated Na+ channels with tetrodotoxin (TTX; 1 μM) abolished the norepinephrine-induced increase in sEPSC frequency (92.4% ± 9.0% of baseline, n = 9, p = 0.42) (Figure 1G).
The sIPSCs in CRH-EGFP neurons had a mean basal frequency of 0.86 ± 0.10 Hz, amplitude of 51.23 ± 3.82 pA, and decay time of 12.72 ± 0.54 ms (n = 20). Norepinephrine (100 μM, 5 min) induced two distinct effects on sIPSCs in separate cohorts of CRH neurons: 11 of 20 CRH neurons (55%) showed a ~75% increase in sIPSC frequency (p < 0.01) and 9 of 20 neurons (45%) showed a ~50% decrease in sIPSC frequency (p < 0.01) (Figure 1H and 1I), with no effect on sIPSC amplitude (p = 0.53) or decay time (p = 0.12). The norepinephrine-induced increase in sIPSC frequency was not seen in any CRH neurons recorded in the presence of TTX, with all of the cells showing a decrease in the sIPSC frequency (65.1% ± 2.8% of baseline, n = 8, p < 0.01) (Figure 1J). Thus, the norepinephrine facilitation of inhibitory synaptic inputs to CRH neurons is action-potential dependent, whereas the norepinephrine-induced suppression of inhibitory synaptic inputs is action-potential independent, and the facilitatory effect of norepinephrine on GABA release in some cells (55%) masked the norepinephrine-induced suppression of GABA release, which was present, therefore, in all the CRH neurons tested. The absence of the spike-dependent sIPSC facilitatory response to norepinephrine in some CRH neurons suggested that the norepinephrine activation of local inhibitory inputs to those cells was compromised by the slicing process.
The two effects of norepinephrine on synaptic inhibition had different concentration sensitivities. Nearly all the CRH neurons (14/16 cells) responded to norepinephrine with either no response or a suppression of sIPSCs at the lowest concentrations tested (100 nM and 1 μM), and the proportion of cells that responded with a facilitation of sIPSCs increased at higher norepinephrine concentrations (2/9 cells [22%] at 10 μM and 11/20 cells [55%] at 100 μM) (Figure 1K). Thus, the spike-dependent norepinephrine facilitation of GABA release had a higher threshold (~10 μM) than the spike-independent norepinephrine suppression of GABA release (~100 nM). Note that the threshold for facilitation of excitatory synaptic inputs (~100 nM) was over an order of magnitude lower than the threshold for norepinephrine facilitation of inhibitory synaptic inputs to the CRH neurons. Additionally, the norepinephrine activation of inhibitory synaptic inputs was less robust than the activation of excitatory synaptic inputs. Together, these findings account for the net excitatory effect of norepinephrine on CRH neuron spiking and the relatively weak restraint of the norepinephrine-induced spiking by GABAA receptor activation shown in Figures 1A–1C.
Adrenoreceptor Dependence of the Norepinephrine Modulation of Synaptic Inputs to CRH Neurons
We next investigated the adrenoreceptor dependence of the norepinephrine facilitation of excitatory synaptic inputs to CRH neurons in recordings of sEPSCs in the presence of bicuculline. Bath application of the α1 adrenoreceptor antagonist prazosin (10 μM) alone caused a small decrease in sEPSC frequency (85.2% ± 5.5% of baseline, n = 9, p = 0.027), suggesting a tonic activation of α1 adrenoreceptors by ambient endogenous norepinephrine (or epinephrine). Prazosin abolished the norepinephrine-induced increase in sEPSC frequency (p = 0.35, n = 9) (Figures 2A and 2C). Bath application of the α1 adrenoreceptor agonist phenylephrine (100 μM) for 5 min caused an increase in sEPSC frequency (424.2% ± 106.2% of baseline, n = 8, p = 0.019) similar to that induced by norepinephrine (norepinephrine versus phenylephrine, p = 0.45), although it did not reverse with a 20-min washout (Figures 2B and 2C). Taken together, these results indicate that the norepinephrine-induced facilitation of sEPSCs in CRH neurons is mediated by α1 adrenoreceptor activation.
Figure 2. Adrenoreceptor Dependence of Norepinephrine Effects on Excitatory and Inhibitory Synaptic Inputs to CRH Neurons.
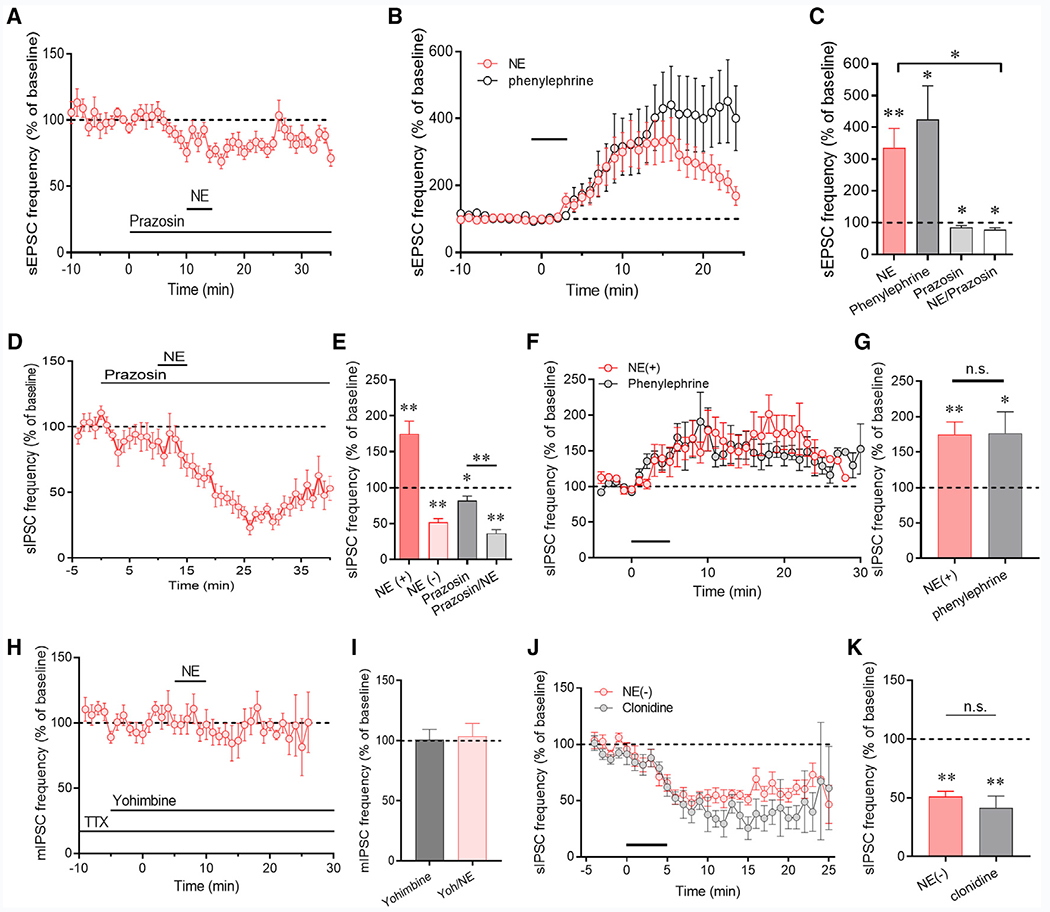
(A) The norepinephrine-induced facilitation of sEPSCs was blocked by the α1 receptor antagonist prazosin (n = 8 cells).
(B) The α1 receptor agonist phenylephrine increased the sEPSC frequency (n = 8 cells), similar to the response to norepinephrine (data from Figure 1E redisplayed for comparison).
(C) Summary of α1 receptor agonist and antagonist effects on sEPSC frequency (n = 8–9 cells/group).
(D) The norepinephrine-induced increase in sIPSC frequency was blocked by the α1 receptor antagonist prazosin, revealing the norepinephrine-induced decrease in sIPSC frequency (n = 9 cells).
(E) Summary of α1 receptor antagonist effect on the norepinephrine-induced facilitation (NE(+)) and suppression (NE(–)) of sIPSCs (n = 8–11 cells/group).
(F) The α1 receptor agonist phenylephrine increased sIPSC frequency (n = 8 cells), similar the sIPSC facilitatory response to norepinephrine (data from Figure 1) redisplayed for comparison).
(G) Summary of α1 receptor facilitatory effect on sIPSC frequency (n = 11 and 8 cells).
(H) The α2 receptor antagonist yohimbine blocked the norepinephrine-induced decrease in sIPSC frequency isolated in TTX (n = 9 cells).
(I) Summary of α2 receptor antagonist blockade of the norepinephrine-induced suppression of sIPSCs (n = 9 and 8 cells).
(J) The α2 receptor agonist clonidine decreased the sIPSC frequency (n = 5 cells), similar to the norepinephrine sIPSC suppression (redisplayed for comparison).
(K) Summary of the norepinephrine- and clonidine-induced decrease in sIPSC frequency (n = 9 and 5 cells). *p < 0.05; **p < 0.01.
In recordings of sIPSCs in the presence of DNQX and AP5, bath application of the α1 adrenoreceptor antagonist prazosin (10 μM) alone caused a small decrease in the sIPSC frequency (81.8% ± 6.3% of baseline, n = 8, p = 0.02), again suggesting a tonic activation of α1 adrenoceptors and facilitation of GABA release by endogenous norepinephrine. Norepinephrine (100 μM) failed to elicit an increase in sIPSC frequency following preapplication of prazosin, but all 8 CRH neurons recorded in prazosin responded with a significant decrease in sIPSC frequency (36.3% ± 5.0% of baseline, n = 8, p < 0.01) (Figures 2D and 2E). Bath application of the α1 adrenoreceptor agonist phenylephrine (20 μM) for 5 min caused a significant increase in sIPSC frequency (176.6% ± 30.2% of baseline, n = 8, p = 0.03) similar to that induced by norepinephrine (NE (+) versus phenylephrine, p = 0.95) (Figures 2F and 2G). This indicated that the norepinephrine facilitation of sIPSCs, like the facilitation of sEPSCs, is mediated by α1 adrenoreceptor activation, whereas the norepinephrine-induced suppression of sIPSCs is α1-receptor independent. The norepinephrine-induced suppression of sIPSCs was greater in the presence of prazosin (to 51% of baseline without prazosin versus 36% of baseline with prazosin), and this difference showed a strong trend toward significance (p = 0.05) (Figure 2E), supporting a model of opposing norepinephrine regulation of GABA release onto the CRH neurons.
Because the norepinephrine facilitation, but not suppression, of GABA release was spike dependent, we blocked the facilitatory response with TTX (1 μM) to isolate the norepinephrine-induced suppression of GABA release. In the presence of TTX, preapplication of the α2 adrenoreceptor antagonist yohimbine (20 μM) alone had no effect on sIPSC frequency but completely blocked the norepinephrine-induced decrease in sIPSC frequency (103.8% ± 10.6% of baseline, n = 8, p = 0.86) (Figures 2H and 2I). Bath application of the α2 adrenoreceptor agonist clonidine (50 μM) for 5 min caused a significant decrease in sIPSC frequency (41.4% ± 10.2% of baseline, n = 5, p < 0.01) similar to that induced by norepinephrine (NE versus clonidine, p = 0.33) (Figures 2J and 2K). These two experiments indicated that the norepinephrine-induced decrease in IPSC frequency is mediated by α2 adrenoreceptor activation. Thus, at lower concentrations, norepinephrine activates presynaptic α2 adrenoreceptors to suppress GABA release, and at higher concentrations, it also activates α1 adrenoreceptors to stimulate spiking in presynaptic GABA neurons and an increase in spike-dependent GABA release onto PVN CRH neurons.
Localization of Adrenoreceptors to Pre- and Postsynaptic Loci
Our data suggest that norepinephrine causes a spike-dependent increase in excitatory and inhibitory synaptic inputs to PVN CRH neurons by acting at α1 adrenoreceptors on local presynaptic glutamate and GABA neurons, respectively. However, considerable immunohistochemical evidence exists for noradrenergic synapses directly on CRH neurons (Flak et al., 2009; Liposits et al., 1986) and for α1 adrenoceptor expression by CRH neurons (Day et al., 1999). Here, we tested for the dependence of the norepinephrine effect on the activation of postsynaptic receptors by including a broad-spectrum G-protein inhibitor, GDP-β-S (1 mM), in the patch electrode. The norepinephrine-induced increase in sEPSC frequency was blocked in 7 of 10 CRH neurons (70%) recorded with GDP-β-S-containing electrodes (123.4% ± 36.6% of baseline, n = 10, p = 0.54) (Figures 3A and 3B), which suggested that the α1-adrenoceptor-induced facilitation of excitatory synaptic inputs to the CRH neurons has a postsynaptic locus of action. With GDP-β-S in the patch solution and glutamate receptors blocked, only 15% of CRH neurons (2/13) responded to norepinephrine (100 μM) with an increase in sIPSC frequency, whereas 85% of CRH neurons (11/13) responded with a decrease in sIPSC frequency (71.5% ± 10.9% of baseline, n = 13, p = 0.023) (Figures 3C and 3D). The shift in the distribution of the two sIPSC responses caused by postsynaptic G-protein blockade (facilitation: 55% without GDP-βs to 15% with GDP-βs; suppression: 45% without GDP-βs to 85% with GDP-βs) was significant (p = 0.023, chi-square). This suggested that the norepinephrine-induced facilitation of GABA release, like that of glutamate release, is dependent on postsynaptic G protein activation, whereas the norepinephrine-induced suppression of GABA release is not. Therefore, the norepinephrine facilitation of excitatory and inhibitory synaptic inputs to CRH neurons share a common mechanism: postsynaptic α1 receptor activation that results in spike-dependent release from presynaptic glutamate and GABA neurons, respectively. The norepinephrine-induced suppression of inhibitory synaptic inputs to the CRH neurons, on the other hand, is mediated by the activation of presynaptic α2 adrenoreceptors and is spike independent, indicating that it occurs at presynaptic GABA terminals. The reliance of the norepinephrine facilitation of glutamate and GABA inputs on a postsynaptic G-protein-dependent mechanism suggests the recruitment of a retrograde signaling mechanism.
Figure 3. Norepinephrine Regulation of Synaptic Inputs to CRH Neurons Involves the Dendritic Release of a Retrograde Signal.
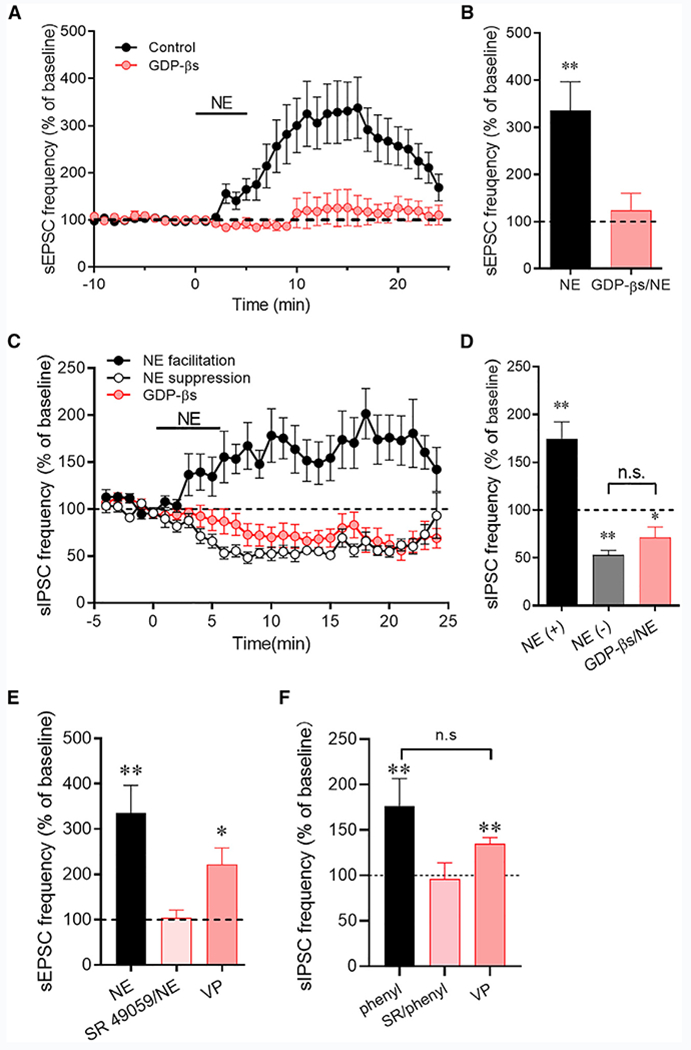
(A) The norepinephrine-induced increase in sEPSC frequency was blocked by intracellular application of a G-protein blocker, GDP-βs, in the CRH neurons (n = 11 cells).
(B) Average sEPSC frequency changes in response to norepinephrine in control CRH neurons (NE) and in CRH neurons in which G-protein activity was blocked with GDP-βs (GDP-βs/NE) (n = 10 cells).
(C) The norepinephrine-induced increase in sIPSC frequency was blocked by blocking postsynaptic G-protein activity, revealing the norepinephrine-induced decrease in sIPSC frequency (n = 13 cells), which was resistant to postsynaptic G-protein blockade.
(D) Average sIPSC frequency facilitation (NE(+)) and suppression (NE(−)) to norepinephrine without and with GDP-βs in the patch pipettes (n = 10–13 cells/group). The facilitation of sIPSCs by norepinephrine was blocked, but the suppression of sIPSCs was maintained during postsynaptic G-protein inhibition.
(E) The vasopressin (VP) V1a receptor antagonist SR 49059 blocked the norepinephrine-induced increase in sEPSC frequency (n = 6 cells) and focal application of VP elicited an increase in sEPSC frequency (n = 6 cells).
(F) The VP V1a receptor antagonist SR 49059 blocked the phenylephrine-induced increase in sIPSC frequency (SR/phenyl) (n = 6 cells) and focal application of VP elicited an increase in sIPSC frequency (n = 5 cells). *p < 0.05; **p < 0.01.
The Norepinephrine-Induced Facilitation of Excitatory and Inhibitory Synaptic Inputs to CRH Neurons Is Mediated by Dendritic Release
The activation of presynaptic glutamate and GABA neurons by a postsynaptic α1-adrenoreceptor-dependent mechanism implicates the dendritic release of an excitatory retrograde messenger. We first tested for the dependence of the α1-receptor-mediated effect on dendritic CRH release. Preincubation of the slices in the corticotropin-releasing hormone receptor type 1 (CRHR1) antagonist antalarmin (100 μM) had no effect on basal sEPSC frequency and failed to block the norepinephrine-induced increase in sEPSC frequency (Figure S2A). We next tested for the dependence of the norepinephrine effect on nitric oxide (NO) release. Preincubation of the slices in the extracellular NO scavenger hemoglobin (10 μM) had no effect on basal sEPSC frequency and failed to block the norepinephrine-induced increase in sEPSC frequency (Figure S2A). Similarly, bath application of the NO donors S-nitroso-N-acetylpenicillamine (SNAP, 100 μM) and N-[4-[1-(3-Aminopropyl)-2-hydroxy-2-nitro-sohydrazino]butyl-1,3-propanediamine (spermine NONOate) (100 μM) did not reproduce the norepinephrine-induced increase in sEPSC frequency (Figure S2B).
We showed previously that vasopressin is released as a retrograde messenger from the dendrites of vasopressinergic magnocellular neurons in the PVN and triggers a retrograde signaling mechanism that increases spike-dependent GABAergic inputs to the vasopressin neurons (Haam et al., 2014). Vasopressin is co-expressed in CRH neurons and released from CRH neuron axon terminals in the median eminence in response to different stressors and central α1 adrenoreceptor activation (Whitnall et al., 1993; de Goeij et al., 1991). Therefore, we tested for the vasopressin dependence of the α1-adrenoreceptor-mediated synaptic facilitation. Although without an effect on basal sEPSCs (p = 0.42) or sIPSCs (p = 0.98), preincubation of slices in the vasopressin V1a receptor antagonist SR 49059 (10 μM) completely blocked the norepinephrine-induced increase in sEPSC frequency (104.5% ± 17.1% of baseline, n = 6, p = 0.80) (Figure 3E) and the phenylephrine-induced increase in sIPSC frequency (96.4% ± 17.4% of baseline, n = 5, p = 0.66) (Figure 3F). We then tested for an agonist effect of vasopressin on sEPSC and sIPSC frequencies with pressure application of vasopressin (20 μM) on the surface of the slices close to the recorded CRH neurons (8–20 psi, 30 s). We used focal pressure application of vasopressin because of the rapid desensitization of the response to bath application of vasopressin. Vasopressin caused a robust increase in the sEPSC frequency (221.9% ± 36.6% of baseline, n = 6, p = 0.021) (Figure 3E) and the sIPSC frequency (134.9% ± 6.9% of baseline, n = 5, p < 0.01) (Figure 3F). The facilitatory effects of vasopressin on sEPSCs and sIPSCs were not significantly different from the norepinephrine-induced increases in sEPSC and sIPSC frequencies. Pressure application of vehicle (aCSF) on the surface of slices with the same parameters as a control for the mechanical effects of the drug application had no effect on synaptic currents. These experiments together suggested vasopressin as the retrograde messenger that is released dendritically from the CRH neurons in response to norepinephrine and that activates presynaptic glutamate and GABA neurons.
The release of vasopressin from CRH neuron dendrites, however, has not been described before and was unexpected, so we tested for the possible release of vasopressin from neighboring vasopressin neurons in the PVN being responsible for the norepinephrine effect in CRH neurons. In a previous study, we found that ghrelin elicits the dendritic release of vasopressin from vasopressin neurons in the PVN, resulting in an increase in TTX-sensitive synaptic inputs to the vasopressin neurons (Haam et al., 2014). We, therefore, tested for an effect of ghrelin on sEPSCs in CRH neurons to control for vasopressin neurons as the source of vasopressin and the vasopressin-receptor-dependent effect in the CRH neurons. Bath application of ghrelin (100 nM) had no effect on the sEPSCs recorded in CRH neurons (frequency: 104.5% ± 11.5% of baseline, n = 10, p = 0.71) (Figure S2B), which supports the idea that the V1a-receptor-dependent response to norepinephrine is mediated by vasopressin release from the CRH neuron dendrites.
The Norepinephrine Facilitation of Synaptic Inputs to CRH Neurons Is Dependent on Retrograde Gliotransmission
The spike dependence of the norepinephrine retrograde facilitation of excitatory and inhibitory synaptic inputs to the CRH neurons suggested that the dendritic messenger acts at a distal presynaptic somato-dendritic site to excite upstream glutamate and GABA neurons. We tested for a role of astrocytes as an intermediate in the relay of the retrograde signal to presynaptic excitatory and inhibitory neurons. Fluorocitric acid (FCA) is preferentially taken up by glia and reversibly blocks glial metabolic function by impairing the Krebs cycle (Paulsen et al., 1987; Swanson and Graham, 1994), which inhibits glial signaling. Preincubation of slices in FCA (100 μM) for 2–4 h prior to recordings significantly blunted the norepinephrine-induced increase in sEPSC frequency (144.3% ± 16.2% of baseline, n = 13, p = 0.018 compared to baseline, p < 0.01 compared to norepinephrine effect in untreated slices) (Figures 4A and 4B). We also tested whether the norepinephrine-induced increase in sEPSC frequency is dependent on the release of the gliotransmitter ATP by blocking purinergic receptors. Bath application of pyridoxalphosphate-6-azophenyl-2’,4’-disulfonic acid tetrasodium salt (PPADS, 100 μM), a non-selective P2 receptor antagonist, decreased both the sEPSC frequency (87.4% ± 5.3% of baseline, n = 7, p = 0.031) and amplitude (84.6% ± 2.3%, n = 7, p < 0.01), and blocked the NE-induced increase in sEPSC frequency (NE/PPADS: 120.8% ± 37.7% compared to PPADS baseline, n = 7, p = 0.42; NE versus NE/PPADS, p = 0.04) (Figure 4C). Bath application of the P2X-receptor-selective antagonist 2′,3′-O-(2,4,6-Trinitrophenyl)adenosine-5′-triphosphate tetra(triethylammonium) salt (TNP-ATP, 10 μM) also blocked the norepinephrine-induced increase in sEPSC frequency (108.9% ± 8.0%, n = 5, p = 0.33) (Figure 4C). Next, to confirm the ATP activation of local glutamate circuits and test whether the ATP actions were downstream of astrocyte activation, following preincubation of slices in the gliotoxin FCA, we puff-applied ATP-γ-S (100 μM, 30 s, 20 psi), a non-hydrolysable analog of ATP (i.e., preventing ATP conversion to adenosine), to the surface of slices near the recorded CRH neurons. ATP-γ-S application caused a robust increase in the frequency of sEPSCs (n = 7, p < 0.01) (Figure S3). The rescue of the excitatory synaptic response by ATP-γ-S following astrocyte inactivation indicated that the ATP stimulation of local glutamate circuits was downstream of astrocyte activation in the retrograde signaling pathway.
Figure 4. Astrocyte Dependence of the Norepinephrine Regulation of CRH Neurons.
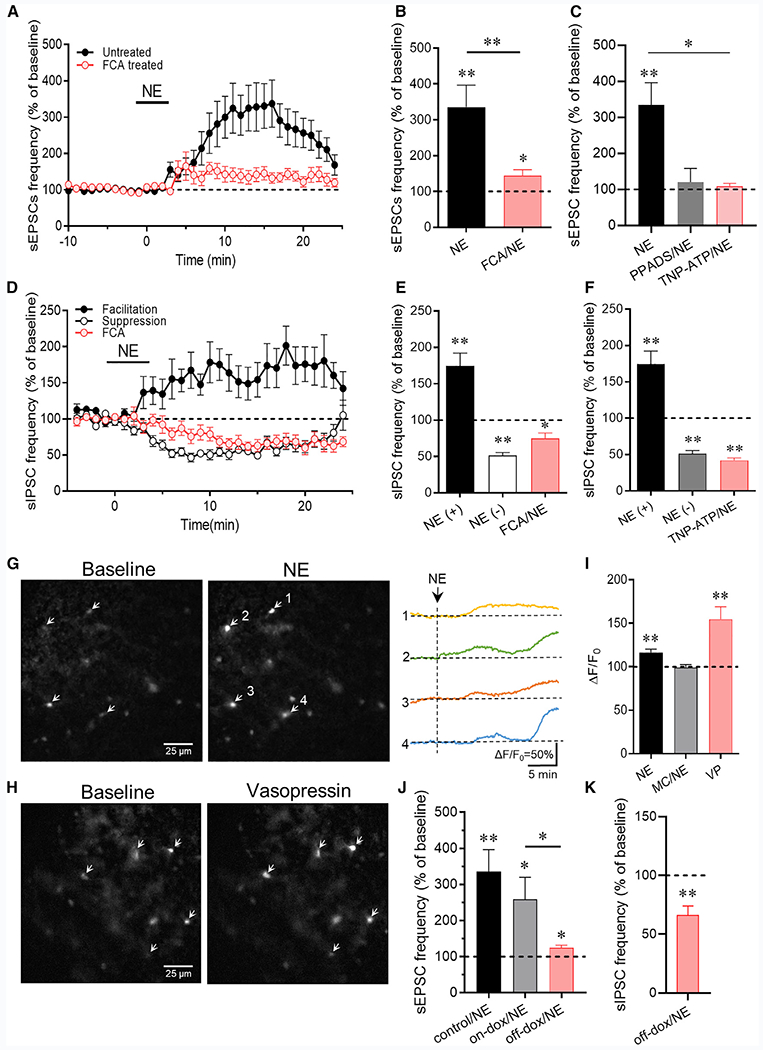
(A) The norepinephrine-induced increase in sEPSC frequency is inhibited by preincubation of slices in the gliotoxin fluorocitric acid (FCA) (n = 13 cells).
(B) Mean sEPSC frequency changes in norepinephrine relative to baseline in control slices and slices incubated in FCA (n = 17 and 13 cells).
(C) The norepinephrine-induced increase in sEPSC frequency was blocked by the purinergic receptor antagonist PPADS (n = 7 cells) and by the P2x-selective antagonist TNP-ATP (n = 5 cells).
(D) The norepinephrine-induced facilitation of sIPSC frequency was blocked by preincubation of slices in the gliotoxin FCA, but the norepinephrine-induced suppression of sIPSCs was retained following FCA preincubation (n = 10 cells).
(E) Mean norepinephrine-induced increase (NE(+)) and decrease (NE(−)) in sIPSC frequency relative to baseline following FCA treatment. The norepinephrine-induced increase in sIPSC frequency (NE(+)), but not the decrease in sIPSC frequency (NE(−)), was blocked by FCA preincubation (n = 9–11 cells/group).
(F) The norepinephrine-induced increase in sIPSC frequency was blocked by the P2X-selective purinergic receptor antagonist TNP-ATP but the norepinephrine-induced decrease in sIPSC frequency was not (n = 6 cells).
(G) Fluorescence image showing the glial Ca2+ response to norepinephrine in the PVN with the glia-specific Rhod-2/AM Ca2+ indicator. The changes in Ca2+ signal in 4 of the cells in the slice (arrows) are shown in the image and in the fluorescence recordings (ΔF/F0) on the right.
(H) The glial Ca2+ response to VP imaged with the glia-specific Rhod-2/AM Ca2+ indicator.
(I) Quantification of the glial Ca2+ responses to norepinephrine (NE) (n = 30 cells), which was blocked by the VP receptor blocker Manning compound (NE/MC) (n = 13 cells), and to VP (n = 9 cells).
(J) The norepinephrine-induced increase in sEPSC frequency (control/NE) was retained in CRH neurons from dnSNARE mice maintained on the dox diet (on-dox/NE) (n = 6 cells) but was lost in CRH neurons of mice taken off the dox diet (off-dox/NE) (n = 7 cells).
(K) The norepinephrine-induced increase in sIPSC frequency was blocked, but the sIPSC suppression was not affected, in CRH neurons from mice taken off the dox diet (off-dox/NE) (n = 10 cells). *p < 0.05; **p < 0.01.
Preincubation of slices in the gliotoxin also blocked the norepinephrine-induced increase in sIPSC frequency but not the decrease in sIPSC frequency (74.7% ± 7.8% of baseline, n = 10, p = 0.01) (Figures 4D and 4E). Inhibition of P2X purinergic receptors with TNP-ATP (10 μM) also blocked the norepinephrine-induced increase in sIPSC frequency but not the norepinephrine-induced decrease in sIPSC frequency (NE(+) versus TNP-ATP/NE: p < 0.01; NE(−) versus TNP-ATP/NE: p = 0.12) (Figure 4F), which was consistent with the dependence of the norepinephrine-induced facilitation, but not suppression, of GABA release on the retrograde neuronal-glial signaling mechanism. Together, these findings suggested that the norepinephrine-induced facilitation of both excitatory and inhibitory synaptic inputs to the CRH neurons is dependent on astrocyte activity.
To further test for the astrocyte participation in the norepinephrine-induced facilitation of excitatory and inhibitory synaptic inputs, we conducted calcium imaging experiments using the glia-specific calcium indicator Rhod-2/AM (Mulligan and MacVicar, 2004) to determine whether norepinephrine causes a calcium response in glial cells that is dependent on vasopressin receptor activation. Slices were pre-incubated in Rhod-2/AM (1–3 μM) for 30–60 min to bulk load the calcium fluorophore into astrocytes. Bath application of norepinephrine (100 μM, 5 min) resulted in a significant increase in the relative fluorescence intensity in imaging experiments with standard fluorescence microscopy (ΔF/F0 = 116.4% ± 3.9%, n = 30 cells in 5 slices, p < 0.01) (Figures 4G and 4I) and in experiments performed with two-photon microscopy (Figure S4; ΔF/F0 = 143.4% ± 8.1%, n = 5 cells in 4 slices, p < 0.01). The norepinephrine-induced increase in astrocytic calcium was blocked by preincubation of slices in the vasopressin receptor antagonist Manning compound (MC; kindly provided by Professor Maurice Manning, University of Toledo) (Figure 4I), revealing the vasopressin-receptor dependence of the norepinephrine effect. We did not use the selective V1a receptor antagonist here because it induced a fluorescence response in our brain slices, whereas the MC did not. We next tested whether vasopressin also activates a calcium response in astrocytes. Bath application of vasopressin (200 nM) caused a robust increase in the relative fluorescence signal (ΔF/F0 = 154.7% ± 14.4%, n = 9 cells in 2 slices, p < 0.01) (Figures 4H and 4I). These results together suggest that norepinephrine activates PVN astrocytes via a vasopressin-receptor-dependent mechanism.
The recruitment of glia into the retrograde signaling mechanism triggered by norepinephrine and the involvement of ATP as a gliotransmitter in the activation of upstream glutamate and GABA neurons suggested that ATP may be released as a gliotransmitter from astrocytes in the response to norepinephrine. We tested this hypothesis by using a transgenic mouse model in which exocytosis is suppressed in astrocytes by the conditional Tet-off expression of a dominant-negative synaptotagmin (dnSNARE) under the control of the glial fibrillary acidic protein promoter (kindly provided by Dr. Phillip Haydon, Tufts University) (Figure S5). We crossed the dnSNARE mouse with the CRH-EGFP mouse to allow us to target EGFP-expressing CRH neurons for patch-clamp recording. PVN CRH neurons from CRH-EGFP,dnSNARE mice taken off the doxycycline (dox) diet (5 weeks, to induce dnSNARE expression) were tested for their sEPSC and sIPSC responses to norepinephrine, and CRH neurons from mice maintained on the dox diet served as controls. There was no difference in the basal sEPSC frequency in CRH neurons from mice on dox compared to those from mice taken off the dox diet (on-dox: 1.94 ± 0.23 Hz, off-dox: 1.48 ± 0.20 Hz, p = 0.17); similarly, there was no difference in the basal sEPSC frequency in CRH neurons from dnSNARE mice compared to those from the CRH-EGFP mice (on-dox dnSNARE versus CRH-eGFP: p = 0.23; off-dox dnSNARE versus CRH-EGFP: p = 0.75). However, the norepinephrine-induced increase in sEPSC frequency was significantly suppressed in CRH neurons from CRH-EGFP,dnSNARE mice taken off dox (124.4% ± 7.3% of baseline, n = 7) compared to CRH neurons from mice maintained on dox (258.7% ± 60.9% of baseline, n = 6) (Figure 4J). Similarly, there was no difference in the basal sIPSC frequency in CRH neurons from mice maintained on the dox diet compared to mice taken off the dox diet (on-dox: 1.37 ± 0.75 Hz, off-dox: 1.01 ± 0.13 Hz, p = 0.43), or in the CRH neurons from dnSNARE mice compared to CRH neurons from control CRH-EGFP mice (on-dox versus CRH-EGFP: p = 0.53; off-dox versus CRH-EGFP: p = 0.77). None of the CRH neurons from dnSNARE mice taken off dox showed a norepinephrine-induced increase in sIPSC frequency, and all the cells showed a decrease in sIPSC frequency (66.3% ± 7.7% of baseline, n = 10) (Figure 4K). A norepinephrine-induced increase in sIPSCs (170.8% ± 5.3% of baseline, p = 0.048) was recorded in 2 of 3 CRH neurons and a decrease (to 35% of baseline) in the third CRH neuron from dnSNARE mice maintained on dox. These findings together support a mechanism for the norepinephrine-induced facilitation of excitatory and inhibitory synaptic inputs to CRH neurons that is dependent on ATP exocytosis from astrocytes.
Modulation of Excitatory and Inhibitory Synaptic Inputs to CRH Neurons by Endogenous Norepinephrine
To determine whether endogenous norepinephrine release exerts a similar modulatory effect on excitatory and inhibitory synaptic inputs to PVN CRH neurons, we applied an optogenetic strategy to activate noradrenergic afferent inputs to CRH neurons from the nucleus of the solitary tract (NTS). An AAV9-expressing Cre-dependent channelrhodopsin (ChR2) and mCherry was injected bilaterally into the NTS of CRH-EGFP,TH-Cre mice. Following confirmation of mCherry expression in cell bodies of the NTS and in axon terminal fields in the PVN with confocal microscopy (Figures 5A and 5B), we tested for an excitatory synaptic response in CRH-EGFP neurons to photostimulation of ChR2-expressing axons in the PVN with blue light (490 nm, 2 min continuous). CRH neurons responded to photostimulation with an increase in sEPSC frequency (645% ± 101.2% of baseline, n = 20, p < 0.01) (Figures 5C and 5D) but not amplitude or decay (p = 0.99 and 0.89, respectively). The increase in sEPSC frequency was suppressed by >50% by the α1 adrenoreceptor antagonist prazosin (10 μM) (p = 0.041), but a significant α1-adrenoreceptor-independent EPSC response to the photostimulation remained (243.4% ± 44.7% of baseline, n = 7, p = 0.018) (Figure 5D). The increase in sEPSC frequency to photostimulation was also similarly suppressed by blocking postsynaptic G protein activity in the recorded cells with GDP-β-S application (1 μM) via the patch pipette (p = 0.03), but a significant G-protein-independent residual sEPSC response remained (252.5% ± 35.2%, n = 6, p < 0.01) (Figure 5D). The α1-receptor- and G protein-independent response suggested that glutamate may be co-released from the activated noradrenergic afferents. We tested this by blocking action potentials with TTX (1 μM), which should inhibit the norepinephrine-induced retrograde activation of local glutamate circuits but not monosynaptic glutamate release stimulated by strong direct ChR2 depolarization of the noradrenergic axons. TTX also significantly suppressed the photostimulation-induced increase in sEPSC frequency (photostimulation versus photo/TTX: p = 0.048) but did not abolish the response (297.3% ± 74.68% of baseline, n = 8) (Figure 5D). These findings further suggest that the noradrenergic afferents co-release glutamate to activate the CRH neurons monosynaptically, in addition to the norepinephrine activation of local glutamate circuits.
Figure 5. Endogenous Norepinephrine Regulation of Excitatory and Inhibitory Synaptic Inputs to CRH Neurons.
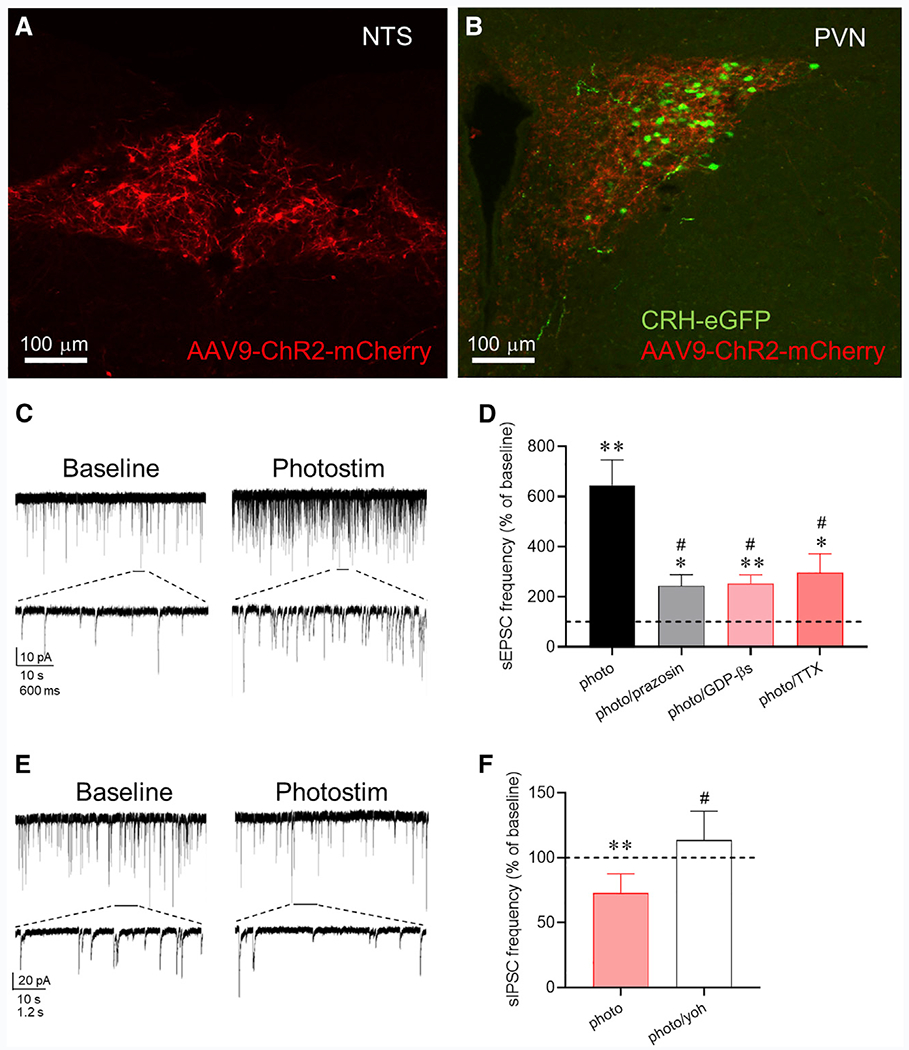
(A) ChR2-mCherry expression in tyrosine hydroxylase (TH)-expressing neurons in the NTS.
(B) EGFP expression in CRH neurons and ChR2-mCherry expression in TH axons in the PVN.
(C) Whole-cell recording of sEPSCs in a CRH-EGFP neuron in the PVN before (baseline) and after photostimulation of ChR2 (photostim) in the presence of GABAA receptor antagonist.
(D) Summary sEPSC frequency response to photostimulation relative to baseline before (n = 20 cells) and after blocking α1 receptors with prazosin (n = 7 cells), postsynaptic G-protein activity with intracellular GDP-βs (n = 6 cells), and action-potentialdependent release with TTX (n = 8 cells).
(E) Whole-cell recording of sIPSCs in a CRH-EGFP neuron before (baseline) and after photostimulation of ChR2 (photostim) in the presence of glutamate receptor antagonists.
(F) Summary sIPSC frequency response to photostimulation relative to baseline in control aCSF (photo, n = 7 cells) and in the α2 receptor antagonist yohimbine (photo/yoh, n = 5 cells). *p < 0.05; **p <0.01 versus baseline; # p < 0.05 versus photostimulation group.
We also tested for the modulation of GABAergic inputs to the CRH neurons by endogenous norepinephrine release in the presence of glutamate receptor antagonists. Photostimulation of ChR2-expressing noradrenergic axons failed to elicit an increase in sIPSC frequency but caused a decrease in sIPSC frequency in all the cells tested (73.0% ± 5.6% of baseline, n = 7, p < 0.01) (Figure 5E). The photostimulation-induced decrease in sIPSC frequency was blocked by the β2 adrenoceptor antagonist yohimbine (113.6% ± 10.0%, n = 5, p < 0.01 compared to control response) (Figure 5F).
DISCUSSION
We found that norepinephrine stimulates CRH neurons by a dendritic signaling mechanism that activates upstream presynaptic glutamate and GABA neurons via retrograde transmission through astrocytes. The astrocyte participation in the retrograde signaling significantly expands the spatial domain of the dendritic signal to more distal targets than would be reached by dendritic volume transmission alone. This retrograde neuronal-glial signaling mechanism is not spatially restricted to the three components of the tripartite synapse, i.e., the presynaptic terminal, the postsynaptic dendrite, and the peri-synaptic astrocyte branch (Perea and Araque, 2002; Perea et al., 2009), but spans a potentially significant distance to the presynaptic somata and/or dendrites of upstream glutamate and GABA neurons to drive action potential generation. Indeed, the spatial reach of the retrograde signal could attain even greater distances if the astrocytic calcium signal was transmitted to distal astrocytes by gap junction coupling. This needs to be further studied but could explain why we saw a less robust norepinephrine facilitation of inhibitory than excitatory synaptic inputs because the presynaptic GABA neurons are thought to be located outside the PVN (Boudaba et al., 1996; Herman et al., 2004; Ulrich-Lai and Herman, 2009) (but see also Jiang et al., 2018), whereas the presynaptic glutamate neurons may be intrinsic to the PVN and, thus, more proximal to the CRH neurons (Daftary et al., 1998, 2000; Wittmann et al., 2005; Hrabovszky et al., 2005, 2005). Consistent with a difference in the relative distances of presynaptic glutamate and GABA neurons from the CRH neurons, the facilitation of EPSCs was seen in over 90% of recorded CRH neurons, whereas the facilitation of IPSCs occurred in 55% of the neurons, suggesting that the presynaptic GABA neurons are more distal and fewer retained intact axonal projections to the CRH neurons in our slices. Also, the weaker facilitation of IPSCs than EPSCs by norepinephrine (x ≈ 75% versus 300%, respectively, at 100 μM) may be due to the lower fidelity of the retrograde signaling to presynaptic GABA neurons because of their remote extranuclear location. Signaling to neurons outside the PVN is likely to require transmission through more than one astrocyte because PVN astrocytes appear to be contained largely within the nucleus (unpublished data). We found that optogenetics also support a difference in the locations of the presynaptic glutamate and GABA neurons. Photostimulation of noradrenergic axons elicited an α1-receptor-dependent increase in glutamatergic synaptic inputs but failed to elicit the α1-receptor-induced facilitation of GABAergic inputs, which is likely due to a higher threshold of activation of the retrograde signaling pathway to the presynaptic GABA neurons because of their extranuclear location and extended neuronal-glial pathway. The rightward shift in the norepinephrine concentration required to activate GABAergic inputs compared to glutamatergic inputs is consistent with a higher threshold to activate the presynaptic GABA neurons.
We found converging lines of evidence for an astrocyte intermediate in the retrograde transmission to presynaptic glutamate and GABA neurons. First, both norepinephrine and vasopressin elicited calcium responses in astrocytes. Second, the norepinephrine facilitation of synaptic inputs to the CRH neurons was lost following pretreatment of brain slices with the gliotoxin FCA. Third, the norepinephrine responses were also lost in CRH neurons from transgenic mice in which exocytosis was conditionally suppressed in astrocytes. Finally, the norepinephrine responses were blocked by antagonists of the conventional gliotransmitter ATP. The sensitivity of the responses to the P2X receptor antagonists and to TTX, and their dependence on astrocytic exocytosis, suggest that astrocytic ATP release drives action potential generation in presynaptic glutamate and GABA neurons by activating ionotropic purinergic receptors. Thus, the evidence for the retrograde transmission to presynaptic glutamate and GABA neurons by way of one or more intercalated astrocytes is compelling. Previous studies reported a norepinephrine facilitation of postsynaptic glutamate responses in PVN magnocellular neurons via astrocyte α1-receptor-induced ATP release (Gordon et al., 2005, 2009), which suggests that the noradrenergic signaling in PVN magnocellular neurons is distinct from that of CRH neurons.
A surprising finding of this study was that vasopressin appears to be the retrograde transmitter, which was indicated by the blockade of both the norepinephrine-induced synaptic response in CRH neurons and the calcium response in astrocytes by vasopressin receptor antagonists, but not by a CRHR1 antagonist or NO scavenger, as well as by the vasopressin-induced synaptic response in the CRH neurons and calcium response in astrocytes. Astrocytes express vasopressin V1a receptors (Hatton et al., 1992; Yamazaki et al., 1997), and we showed previously, as well as here, that vasopressin generates a calcium response in PVN astrocytes (Haam et al., 2014). The source of the vasopressin could be neighboring PVN vasopressin neurons, but this was largely ruled out because ghrelin, which stimulates vasopressin release from vasopressin neuron dendrites (Haam et al., 2014), did not elicit an increase in sEPSCs in the CRH neurons.
CRH neurons express vasopressin mRNA and peptide in their somata and axons basally (Whitnall et al., 1987; Lightman and Young, 1988) and increase their vasopressin expression following acute stress, chronic stress (Bartanusz et al., 1993a, 1993b; Ma et al., 1997), and adrenalectomy (Kovács et al., 2000). Vasopressin is co-released with CRH in the median eminence and facilitates adrenocorticotrophic hormone secretion from the pituitary (Whitnall et al., 1985). Our findings support previous studies showing that α1 adrenoceptor activation stimulates a vasopressin-containing subpopulation of CRH neurosecretory cells (Cummings and Seybold, 1988; Whitnall et al., 1993) and suggest that vasopressin is also released from CRH neuron dendrites. Nevertheless, this unexpected finding awaits further confirmation by direct demonstration of CRH neuron dendritic release of vasopressin.
Norepinephrine induced an α2-receptor-dependent suppression of sIPSCs in 45% of recorded neurons, which increased to 100% of the CRH neurons when the α1-receptor-mediated facilitation was blocked with TTX. Thus, the α2 receptor suppression of GABA release was masked in a subset of CRH neurons (55%) at a high norepinephrine concentration by α1-receptor-dependent stimulation of presynaptic GABA neurons with intact afferent axons. The two GABA responses to norepinephrine had differing concentration dependencies, with the GABA suppression effective at a lower concentration (1 μM) and the facilitation emergent at higher concentrations (>10 μM). The threshold concentration for the norepinephrine facilitation of excitatory inputs was lower than both, near 100 nM. The results of optogenetic stimulation are consistent with the lower threshold of norepinephrine activation of local excitatory circuits compared to inhibitory circuits because photostimulation of the noradrenergic axons elicited an α1-receptor-dependent facilitation of excitatory, but not inhibitory, synaptic inputs and an α2-receptor-dependent suppression of inhibitory synaptic inputs to the CRH neurons. The combined α1 facilitation of excitatory inputs and α2 suppression of inhibitory inputs by endogenous norepinephrine would lead to a robust activation of the CRH neurons and the HPA axis. The higher threshold for the α1-eceptor-mediated facilitation of GABA inputs may result in the recruitment of inhibitory circuits with more robust activation of the ascending noradrenergic afferents, possibly to prevent excessive activation of the HPA axis.
Our findings reconcile discrepancies between previous neuroanatomical evidence of noradrenergic synapses directly on and α1 adrenoreceptor expression in CRH neurons (Liposits et al., 1986; Cummings and Seybold, 1988; Flak et al., 2009; Cunningham and Sawchenko, 1988; Sawchenko and Swanson, 1982; Day et al., 1999; Cunningham et al., 1990; Füzesi et al., 2007) and electrophysiological data showing an indirect norepinephrine regulation of PVN parvocellular neurons via activation of local glutamate and GABA circuits (Daftary et al., 2000; Han et al., 2002). What functional utility is served by a complex indirect regulation of CRH neurons via retrograde activation of local synaptic circuits? The local circuits may represent a common network by which both ascending physiological inputs and descending limbic inputs regulate the HPA axis. Although the ascending physiological inputs activate the circuits via retrograde signaling, the descending limbic inputs could activate the same local glutamate circuits directly. This remains to be tested. In any case, the complex heterotypic nature of the local neuronal-glial organization provides multiple levels for regulation of the circuit.
Retrograde dendritic signaling via transmission by astrocytes and astrocytic networks represents a powerful mechanism for the control by postsynaptic neurons of presynaptic neuronal ensembles and significantly extends the domain of influence of the postsynaptic neuron over its own upstream afferent circuits. Whether non-neuroendocrine neurons are also capable of dendritic volume transmission dependent on astrocyte activation is not known. Neuronal-glial signaling has been described in hippocampal neurons via excitatory endocannabinoid actions on astrocytes (Navarrete and Araque, 2008, 2010) and in dorsal root ganglion neurons via ATP activation of satellite cells “sandwiched” between DRG somata (Rozanski et al., 2013), but whether these forms of trans-cellular signaling can recruit distant presynaptic neurons remains to be determined.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffrey Tasker (tasker@tulane.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Three strains of adult male mice aged 6-10 weeks were used in these experiments:
CRH-eGFP mice: Transgenic mice expressing enhanced green fluorescent protein (eGFP) controlled by the CRH promoter were obtained from the Mutant Mouse Regional Resource Center (MMRRC) (stock: Tg(CRH-EGFP)HS57Gsat/Mm, RRID:MMRRC_017058-UCD) at the University of California at Davis and backcrossed with C57BL/6J mice (Nahar et al., 2015). The CRH-eGFP mice were genotyped following the supplier’s protocol with primers: 5′-CTGTCTTGTCGTGGGTGTCCGAT-3′ and 5′-TAGCGGCTGAAGCACTGCA-3′ to produce a 400 bp fragment.
CRH-eGFP::dnSNARE::GFAP.tTA mice: Two breeder lines, tetO-dnSNARE mice and GFAP-tTA mice, were generously provided by Dr. Philip Haydon, Tufts University, Boston MA. For the GFAP-tTA line, the tTA gene was under the control of the GFAP promotor. In the GFAP-tTA/tetO-dnSNARE line, only GFAP-positive glia express dnSNARE protein with tTA binding to tetO (Pascual et al., 2005). The two breeder lines were crossed with the CRH-eGFP line to obtain CRH-eGFP/tetO-dnSNARE and CRH-eGFP/GFAP-tTA mice, which were crossed to get the triple transgenic CRH-eGFP/tetO-dnSNARE/GFAP-tTA mice. The CRH-eGFP/dnSNARE/GFAP.tTA mice were genotyped using the following primer sets: for GFAP-tTA: 5′-ACT CAG CGC TGT GGG GCA TT-3′ (tTA forward), 5′-GGC TGT ACG CGG ACC CAC TT-3′(tTA reverse); for tetO-dnSNARE: 5′-TGG ATA AAG AAG CTC ATT AAT TGT CA-3′ (TSL Forward) 5′-GCG GAT CCA GAC ATG ATA AGA-3′ (TSL reverse). To prevent developmental issues, the crossed pups were maintained on a diet with doxycycline (dox), which binds with tTA and prevents it from binding with the tetO promotor, until 3-4 weeks of age to prevent dnSNARE expression. The experimental group was then taken off the dox diet for 5 weeks, while the control group was maintained on the dox diet for an equivalent duration until decapitation for slice preparation.
TH-cre/CRH-eGFP mice: TH-cre mice were purchased from Jackson lab (B6.Cg-7630403G23RikTg(Th-cre)1Tmd/J, IMSR Cat# JAX:008601, RRID:IMSR_JAX:008601) and were crossed with the CRH-eGFP mice to produce the TH-cre/CRH-eGFP mice. The TH-cre/CRH-eGFP mice were genotyped at 2–3 weeks of age using the primer sets listed with the Jackson Laboratory: 5′-AGT GGC CTC TTC CAG AAA TG-3′ (31704), 5′-TGC GAC TGT GTC TGA TTT CC-3′ (31705), 5′-GAG ACA GAA CTC GGG ACC AC-3′ (33122), 5′-AGG CAA ATT TTG GTG TAC GG-3′ (oIMR9074), to produce a ~300 bp fragment.
All mouse procedures were approved by the Tulane Institutional Animal Care and Use Committee. Mice were housed under controlled temperature and humidity in an AALAC-accredited animal facility on a 12:12 light/dark cycle and received food and water ad libitum.
Virus
A serotype 9 adeno-associated virus (AAV9) expressing cre-dependent channelrhodopsin (ChR2) and the mCherry fluorescent reporter under the control of the eukaryotic translation elongation factor 1 α1 (AAV9-EF1a-DIO-hChR2(H134R)-mCherry) was purchased from the University of Pennsylvania Vector Core (titer 5.6X1013 (vg/ml)).
METHOD DETAILS
Immunohistochemistry
To confirm that eGFP was expressed in CRH neurons in the CRH-eGFP transgenic mice, three 8-week-old CRH-eGFP mice were processed for CRH immunohistochemistry. Two days before sacrifice, each mouse received a stereotaxic injection of colchicine (8 mg/ml, 1.5 μg/g body weight, Sigma-Aldrich) into the lateral ventricle under ketamine/xylazine anesthesia.
To confirm dn-SNARE expression in astrocytes in the PVN, two 8-week-old GFAP-tTA/tetO-dnSNARE mice were taken off the doxycycline diet for five weeks prior to sacrifice for immunohistochemical double labeling for β-galactosidase, which is co-expressed in the transgene with the dn-SNARE and a marker for dn-SNARE expression (Pascual et al., 2005), and the astrocyte marker GFAP.
Mice were anesthetized with ketamine/xylazine and perfused transcardially with ice-cold phosphate buffered saline (PBS), followed by 4% paraformaldehyde in PBS. Their brains were dissected from the cranium and post-fixed with 4% paraformaldehyde at 4°C overnight, and then submerged successively in 15% and 30% sucrose in PBS. Thirty- or 40-μm coronal sections of the hypothalamus containing the PVN were cut on a cryostat. The sections were pre-incubated with a blocking solution containing 10% goat serum and 0.2% Triton X-100 in PBS for 1 h and then incubated with primary antibodies at 4°C overnight. The primary antibodies included rabbit anti-CRH (1:2000, T4037, Peninsula Laboratories, CA, USA), chicken anti-β-gal (1:500, Ab9361, Abcam, MA, USA), and rabbit anti GFAP (1:300, 620-GFAP, Phosphosolution, CO, USA). They were then incubated in the secondary antibodies dylight 550 goat anti-rabbit (1:1000, Thermo Scientific), Alexa Fluor 647 goat anti-chicken (1:1000, Thermo Scientific), and Alexa Fluor 594 goat anti-rabbit (1:1000, Thermo Scientific) at room temperature for 1 h. Sections were then mounted and imaged on a laser scanning confocal microscope (Nikon A1).
Brain slice preparation and electrophysiological recording
Acute brain slice preparation –
Whole-cell patch-clamp electrophysiological experiments were conducted in acutely-prepared hypothalamic slices. Mice were gently removed from their home cages and transferred to an adjacent room, immobilized in a plastic cone with a nose hole (DecapiCone, Braintree Scientific), and decapitated without anesthesia using a rodent guillotine, all within less than 2 minutes from removal from the home cages. Anesthetics activate the HPA axis and increase circulating ACTH and corticosterone levels (Vahl et al., 2005), which otherwise remain at basal levels in our hands for up to 3 minutes from the start of handling (unpublished observation). Following decapitation, the brain was quickly removed and cooled in an oxygenated, ice-cold artificial cerebrospinal fluid (aCSF) containing (in mM): 140 NaCl, 3 KCl, 1.3 MgSO4, 11 Glucose, 5 HEPES, 1.4 NaH2PO4, 3.25 NaOH, and 2.4 CaCl2, at pH 7.2-7.4 and osmolality of 290-300 mOsm. The base of the brain was then blocked, and the caudal face of the block was glued to the chuck of a vibratome, and two to three 300-μm coronal slices of the hypothalamus containing the PVN were sectioned in cooled aCSF, bisected down the midline, and transferred to a holding chamber. The slices were kept in the holding chamber in oxygenated aCSF at room temperature for at least 1 h to allow for recovery prior to electrophysiological recordings or calcium imaging.
Whole cell recording and CRH neuron identification –
Single hemi-slices were transferred from the holding chamber to a recording chamber, where they were submerged and perfused with aCSF at a rate of 2 ml/min. Whole-cell patch clamp recordings were performed at 30-32°C. eGFP-expressing CRH neurons in the PVN were first located under epifluorescence illumination, and then visualized with infrared light under differential interference contrast filters on a fixed-stage, upright microscope equipped with a water immersion 40x objective (Olympus BX51WI). Patch pipettes with a resistance of 3-6 MΩ were pulled from borosilicate glass (ID 1.2 mm, OD 1.65 mm) on a horizontal puller (Sutter P-97). The patch solution for excitatory postsynaptic current (EPSC) recordings contained (in mM): 120 potassium gluconate, 10 KCl, 1 NaCl, 1 MgCl2, 0.1 CaCl2, 5.5 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP; pH was adjusted to 7.3 with KOH; The osmolarity was adjusted to 300 mOsm with D-sorbitol. GABAA receptor antagonist picrotoxin (PTX, 50 μM) was bath-applied to isolate glutamatergic synaptic currents. The patch solution for inhibitory postsynaptic current (IPSC) recordings contained (in mM): 120 CsCl, 2 MgCl2, 1 CaCl2, 11 EGTA, 2 Mg-ATP, 0.3 Na-GTP and 30 HEPES. The ATP and GTP were excluded when GDP-β-s (1 mM, Sigma-Aldrich) was included in the patch solution to block G-protein activity. Glutamate receptor antagonists 6,7-dinitroquinoxaline-2,3-dione (DNQX, 15 μM) and DL-2-amino-5-phosphonopentanoic acid (AP5, 50 μM) were applied in the bath perfusion to isolate the GABA synaptic currents.
The parvocellular neuroendocrine cell identity of the recorded eGFP-expressing cells was confirmed at the beginning of the whole-cell recordings by testing for an A current-mediated transient outward rectification and a low-threshold calcium spike, the absence of both of which is characteristic of parvocellular neuroendocrine cells (Luther et al., 2000; Tasker and Dudek, 1991). To test for the A current in recordings with a cesium-based patch solution (i.e., during recordings of IPSCs), the pipette tip was first filled with the potassium gluconate-based patch solution and then back-filled with the cesium-based patch solution. We then tested for the A current using a voltage step protocol (Wu and Tasker, 2017) immediately after achieving the whole-cell configuration.
For loose-seal, cell-attached patch clamp recordings, glass pipettes with a resistance of 1-2 MΩ were used and cells were recorded at resting potential with minimal current injection, as previously described (Haam et al., 2012). Recordings were performed using a high-K+ extracellular solution to depolarize neurons and stimulate spontaneous spiking activity, consisting of (in mM): 133 NaCl, 10 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 2.4 CaCl2, 11 glucose, and 5 HEPES; pH adjusted to 7.2–7.4 with NaOH.
Calcium Imaging
After a recovery period, slices were bulk loaded with the glia-specific calcium indicator Rhod-2 AM (Life Technologies). A stock solution of Rhod-2 AM was reconstituted in 40 μl of DMSO and stored at −20°C until the day of experiments. The Rhod-2 AM stock solution was used within a week to avoid loss of cell loading capacity. The dye loading dish was prepared using a 30-mm culture plate insert (Millipore, Billerica, MA) placed in a 35-mm dish. One ml of aCSF was added inside and outside of the plate insert prior to moving one or two half slices onto the insert. Two to five μl of Rhod-2 AM stock solution were then dropped directly onto the PVN area of each hemi-slice. Slices were incubated in the dark at room temperature for 1 h, during which 100% O2 was continuously piped into the dish. After the incubation, the brain hemi-slices were washed for 30-240 min in dye-free medium to remove any unincorporated dye and to allow de-esterification of the intracellular acetoxymethyl (AM).
(1) Epifluorescence
Fluorescence images were acquired using 555 nm excitation/581 nm emission filters and an intensified CCD camera (QuantumEM CCD) at 1 frame/5 s and were captured using IP Lab 4.0 software (Scanalytics Inc.). We used 555 nm for excitation of Rhod-2 AM dye and quantified the fluorescence intensity of each cell using ImageJ software (NIH, Bethesda, MD), which allowed for simultaneous analysis of regions of interest in multiple individual cells. To calculate drug effects, we compared the average fluorescence intensity of the last 2 min (24 frames) of a 10-min drug application with the last 2 min of a 5-min control period immediately preceding the introduction of the drug.
(2). Two-photon imaging
Fluorescence images were acquired using a Galvo multiphoton microscopy system microscopy system (Scientifica) equipped with a Ti:sapphire pulsed laser (Chameleon Ultra II; Coherent) laser. The laser was tuned to 810 nm for imaging of Rhod-2. Epifluorescence signals were captured through 60X, 1.0 NA objectives and a 1.4 NA oil immersion condenser (Olympus) at 0.2-0.5 frame/s using SciScan software. We quantified the fluorescence intensity of each cell using ImageJ software (NIH, Bethesda, MD), which allowed for simultaneous analysis of regions of interest in multiple individual cells. To calculate drug effects, we compared the average fluorescence intensity of the last 2 min of a 5-min drug application with the last 2 min of a 5-min control period preceding the introduction of the drug.
Stereotaxic Virus injection
Four to 6-week-old mice were used that were the product of crossing the homozygous CRH-eGFP mice with mice homozygous for expression of Cre recombinase under the control of the TH promoter (TH-cre mice: B6.Cg-7630403G23RikTg(Th-cre)1Tmd/J. Stock No: 008601) (TH-cre/CRH-eGFP mice). The mice were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (8 mg/kg) and placed in a stereotaxic frame. Each mouse received bilateral stereotaxic injections of virus in the NTS (400 nL at 100 nl/min) (coordinates: AP: 7.6 mm caudal to bregma, ML: ± 0.5 mm from the midline, DV: −4.6 mm from the brain surface) using a Hamilton syringe with 10 μL in the beveled tip connected to a microsyringe pump (UMP2; WPI) and controller (Micro4; WPI). The injection needle was maintained in place for 10 min following injections to minimize virus spread up the needle track. The virus used was a serotype 9 adeno-associated virus (AAV9) expressing cre-dependent channelrhodopsin (ChR2) and the mCherry fluorescent reporter under the control of the eukaryotic translation elongation factor 1 α1 (AAV9-EF1a-DIO-hChR2(H134R)-mCherry) (UPENN Vector Core, titer 5.6X1013 (vg/ml)). Channelrhodopsin expression was thus targeted to TH-expressing noradrenergic neurons in the NTS.
Mice were allowed to recover for 2-3 weeks. PVN slices were then produced to perform whole-cell patch clamp recordings. The fluorescent light to excite channelrhodopsin was delivered through the objective lens (light source: Osram HBO 103W/2 Mercury short arc, wavelength: filtered by Olympus U-MNB2 at 470-490 nm); each trial had a sustained 2-min photostimulation.
Drug application
The following drugs were kept at −20°C as stock solutions and dissolved in aCSF to their final concentrations on the day of experiments: L-(−)-norepinephrine (+)-bitartrate salt monohydrate (NE, 10-100 μM, Sigma-Aldrich, shielded from light exposure during preparation, storage and application), picrotoxin (PTX, 50 μM, Tocris), prazosin hydrochloride (10 μM, Sigma-Aldrich), (R)-(−)-phenylephrine hydrochloride (100 μM, Sigma-Aldrich), yohimbine hydrochloride (20 μM, Tocris), tetrodotoxin (TTX, 1 μM, Tocris), TNP-ATP triethylammonium salt (10 μM, Tocris), pyridoxalphosphate-6-azophenyl-2′,4’-disulfonic acid tetrasodium salt (PPADS, 100 μM, Tocris).
Fluorocitric acid (FCA, Sigma-Aldrich) was prepared on the day of experiments with DL-fluorocitric acid barium salt (Paulsen et al., 1987) dissolved in 1 mL of 0.1 M HCl. Two-to-three drops of 0.1 M Na2SO4 was added to precipitate the Ba2+, after which 2 mL of 0.1 M Na2HPO4 was added and the suspension was centrifuged at 1000 g for 5 min. The supernatant was added to the aCSF to achieve a concentration of 100 μM.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as the means ± standard errors of the mean. Postsynaptic currents were selected and analyzed with Minianalysis 6.0 (Synaptosoft Inc.). The frequency, amplitude and decay time were compared statistically using a two-tailed Student’s unpaired t test for two-group comparisons with SigmaPlot 11.0 (Systat Software, Inc.). A two-tailed Student’s paired t test was used to compare the baseline with the drug effect within cells, and a one-way analysis of variance and post hoc tests were used as listed to compare among multiple treatment groups. All statistical analyses are presented in the body of the Results section. A capital n refers to the number of animals and the small n refers to the number of cells in the dataset. No data were excluded, but data points from sIPSC analyses were divided into two groups based on the frequency response to norepinephrine, increased frequencies and decreased frequencies, as described in the Results.
DATA AND CODE AVAILABILITY
All software used in this study is available and listed in the Key Resources Table. This study did not generate any unique datasets or code.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-CRH | Peninsula | Cat# T4037; RRID:AB_518252 |
| chicken anti-β-gal | Abcam | Cat# Ab9361; RRID:AB_307210 |
| rabbit anti GFAP | Phosphosolution | Cat# 620-GFAP; RRID:AB_2492124 |
| goat anti-rabbit IgG | Thermo Fisher Scientific | Cat# A-11012; RRID:AB_2534079 |
| goat anti-rabbit IgG | Thermo Fisher Scientific | Cat# SA5-10033; RRID:AB_2556613 |
| goat anti-chicken IgY | Thermo Fisher Scientific | Cat# A32933; RRID:AB_2762845 |
| Bacterial and Virus Strains | ||
| AAV9-EF1a-DIO-hChR2(H134R)-mCherry | Vector Biolabs | Cat. No: VB4651 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rhod-2 AM | Life Technologies | Cat# R1244 |
| GDP-β-s | Sigma-Aldrich | Cat# G7637 |
| 6,7-dinitroquinoxaline-2,3-dione | Tocris | Cat# 0189 |
| DL-2-amino-5-phosphonopentanoic acid | Tocris | Cat# 0105 |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Experimental Models: Organisms/Strains | ||
| Tg(CRH-EGFP)HS57Gsat/Mm | MMRRC | RRID:MMRRC_017058-UCD |
| tetO-dnSNARE mouse | Haydon Lab | N/A |
| GFAP-tTA mouse | Haydon Lab | N/A |
| B6.Cg-7630403G23RikTg(Th-cre)1Tmd/J | Jackson Lab | RRID:IMSR_JAX:008601 |
| TH-cre/CRH-eGFP mouse | In house | N/A |
| Software and Algorithms | ||
| Prism v. 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Mini Analysis | Synaptosoft, Inc. | http://www.synaptosoft.com/MiniAnalysis/ |
Supplementary Material
Highlights.
Norepinephrine activates a retrograde neuronal-glial circuit to stimulate CRH neurons
Norepinephrine-induced CRH neuron dendritic release stimulates a calcium response in astrocytes
Activated astrocytes release ATP to signal distally to upstream neurons
Upstream glutamate and GABA neurons signal back to the CRH neurons
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health grants R01 MH069879 and R01 MH119283 and the Catherine and Hunter Pierson Chair in Neuroscience. We thank Dr. Laura Harrison for her help with genotyping and animal husbandry. We thank Prof. Maurice Manning for his generous donation of the vasopressin/oxytocin receptor antagonist, the Manning Compound, and for the many oxytocin/vasopressin compounds he generously makes available to us and to the whole field. We also thank Dr. Phillip Haydon for his generous donation of the dnSNARE mouse and guidance in the effective use of the mouse.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.092.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Araque A, Parpura V, Sanzgiri RP, and Haydon PG (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215. [DOI] [PubMed] [Google Scholar]
- Bartanusz V, Aubry JM, Jezova D, Baffi J, and Kiss JZ (1993a). Up-regulation of vasopressin mRNA in paraventricular hypophysiotrophic neurons after acute immobilization stress. Neuroendocrinology 58, 625–629. [DOI] [PubMed] [Google Scholar]
- Bartanusz V, Jezova D, Bertini LT, Tilders FJH, Aubry JM, and Kiss JZ (1993b). Stress-induced increase in vasopressin and corticotropin-releasing factor expression in hypophysiotrophic paraventricular neurons. Endocrinology 132, 895–902. [DOI] [PubMed] [Google Scholar]
- Boudaba C, Szabó K, and Tasker JG (1996). Physiological mapping of local inhibitory inputs to the hypothalamic paraventricular nucleus. J. Neurosci 16, 7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles AC, Merrill JE, Dirksen ER, and Sanderson MJ (1991). Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 6, 983–992. [DOI] [PubMed] [Google Scholar]
- Cole RL, and Sawchenko PE (2002). Neurotransmitter regulation of cellular activation and neuropeptide gene expression in the paraventricular nucleus of the hypothalamus. J. Neurosci 22, 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, Cooper MS, and Smith SJ (1990). Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science 247, 470–473. [DOI] [PubMed] [Google Scholar]
- Cummings S, and Seybold V (1988). Relationship of alpha-1- and alpha-2-adrenergic-binding sites to regions of the paraventricular nucleus of the hypothalamus containing corticotropin-releasing factor and vasopressin neurons. Neuroendocrinology 47, 523–532. [DOI] [PubMed] [Google Scholar]
- Cunningham ET Jr., and Sawchenko PE (1988). Anatomical specificity of noradrenergic inputs to the paraventricular and supraoptic nuclei of the rat hypothalamus. J. Comp. Neurol 274, 60–76. [DOI] [PubMed] [Google Scholar]
- Cunningham ET Jr., Bohn MC, and Sawchenko PE (1990). Organization of adrenergic inputs to the paraventricular and supraoptic nuclei of the hypothalamus in the rat. J. Comp. Neurol 292, 651–667. [DOI] [PubMed] [Google Scholar]
- Daftary SS, Boudaba C, Szabó K, and Tasker JG (1998). Noradrenergic excitation of magnocellular neurons in the rat hypothalamic paraventricular nucleus via intranuclear glutamatergic circuits. J. Neurosci 18, 10619–10628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daftary SS, Boudaba C, and Tasker JG (2000). Noradrenergic regulation of parvocellular neurons in the rat hypothalamic paraventricular nucleus. Neuroscience 96, 743–751. [DOI] [PubMed] [Google Scholar]
- Day HE, Campeau S, Watson SJ Jr., and Akil H (1997). Distribution of alpha 1a-, alpha 1b- and alpha 1d-adrenergic receptor mRNA in the rat brain and spinal cord. J. Chem. Neuroanat 13, 115–139. [DOI] [PubMed] [Google Scholar]
- Day HE, Campeau S, Watson SJ, and Akil H (1999). Expression of alpha(1b) adrenoceptor mRNA in corticotropin-releasing hormone-containing cells of the rat hypothalamus and its regulation by corticosterone. J. Neurosci 19, 10098–10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Goeij DCE, Kvetnansky R, Whitnall MH, Jezova D, Berkenbosch F, and Tilders FJH (1991). Repeated stress-induced activation of corticotropin-releasing factor neurons enhances vasopressin stores and colocalization with corticotropin-releasing factor in the median eminence of rats. Neuroendocrinology 53, 150–159. [DOI] [PubMed] [Google Scholar]
- de Kock CPJ, Wierda KDB, Bosman LWJ, Min R, Koksma J-J, Mansvelder HD, Verhage M, and Brussaard AB (2003). Somatodendritic secretion in oxytocin neurons is upregulated during the female reproductive cycle. J. Neurosci 23, 2726–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Popescu IR, and Tasker JG (2013). Glial control of endocannabinoid heterosynaptic modulation in hypothalamic magnocellular neuroendocrine cells. J. Neurosci 33, 18331–18342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S (1992). Calcium waves in astrocytes-filling in the gaps. Neuron 8, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Flak JN, Ostrander MM, Tasker JG, and Herman JP (2009). Chronic stress-induced neurotransmitter plasticity in the PVN. J. Comp. Neurol 517, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flak JN, Myers B, Solomon MB, McKlveen JM, Krause EG, and Herman JP (2014). Role of paraventricular nucleus-projecting norepinephrine/epinephrine neurons in acute and chronic stress. Eur. J. Neurosci 39, 1903–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füzesi T, Wittmann G, Liposits Z, Lechan RM, and Fekete C (2007). Contribution of noradrenergic and adrenergic cell groups of the brainstem and agouti-related protein-synthesizing neurons of the arcuate nucleus to neuropeptide-y innervation of corticotropin-releasing hormone neurons in hypothalamic paraventricular nucleus of the rat. Endocrinology 148, 5442–5450. [DOI] [PubMed] [Google Scholar]
- Giaume C, Koulakoff A, Roux L, Holcman D, and Rouach N (2010). Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci 11, 87–99. [DOI] [PubMed] [Google Scholar]
- Gordon GRJ, Baimoukhametova DV, Hewitt SA, Rajapaksha WRA, Fisher TE, and Bains JS (2005). Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat. Neurosci 8, 1078–1086. [DOI] [PubMed] [Google Scholar]
- Gordon GRJ, Iremonger KJ, Kantevari S, Ellis-Davies GCR, MacVicar BA, and Bains JS (2009). Astrocyte-mediated distributed plasticity at hypothalamic glutamate synapses. Neuron 64, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haam J, Popescu IRR, Morton LAA, Halmos KCC, Teruyama R, Ueta Y, and Tasker JGG (2012). GABA is excitatory in adult vasopressinergic neuroendocrine cells. J. Neurosci 32, 572–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haam J, Halmos KC, Di S, and Tasker JG (2014). Nutritional state-dependent ghrelin activation of vasopressin neurons via retrograde trans-neuronal-glial stimulation of excitatory GABA circuits. J. Neurosci 34, 6201–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, and Haydon PG (2007). The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol. Med 13, 54–63. [DOI] [PubMed] [Google Scholar]
- Han SK, Chong W, Li LH, Lee IS, Murase K, and Ryu PD (2002). Noradrenaline excites and inhibits GABAergic transmission in parvocellular neurons of rat hypothalamic paraventricular nucleus. J. Neurophysiol 87, 2287–2296. [DOI] [PubMed] [Google Scholar]
- Hatton GI, Bicknell RJ, Hoyland J, Bunting R, and Mason WT (1992). Arginine vasopressin mobilises intracellular calcium via V1-receptor activation in astrocytes (pituicytes) cultured from adult rat neural lobes. Brain Res. 588, 75–83. [DOI] [PubMed] [Google Scholar]
- Haustein MD, Kracun S, Lu X-H, Shih T, Jackson-Weaver O, Tong X, Xu J, Yang XW, O’Dell TJ, Marvin JS, et al. (2014). Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron 82, 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmreich DL, Itoi K, Lopez-Figueroa MO, Akil H, and Watson SJ (2001). Norepinephrine-induced CRH and AVP gene transcription within the hypothalamus: differential regulation by corticosterone. Brain Res. Mol. Brain Res 88, 62–73. [DOI] [PubMed] [Google Scholar]
- Herman JP, Mueller NK, and Figueiredo H (2004). Role of GABA and glutamate circuitry in hypothalamo-pituitary-adrenocortical stress integration. Ann. N Y Acad. Sci 1018, 35–45. [DOI] [PubMed] [Google Scholar]
- Hrabovszky E, Wittmann G, Turi GF, Liposits Z, and Fekete C (2005). Hypophysiotropic thyrotropin-releasing hormone and corticotropin-releasing hormone neurons of the rat contain vesicular glutamate transporter-2. Endocrinology 146, 341–347. [DOI] [PubMed] [Google Scholar]
- Itoi K, Suda T, Tozawa F, Dobashi I, Ohmori N, Sakai Y, Abe K, and Demura H (1994). Microinjection of norepinephrine into the paraventricular nucleus of the hypothalamus stimulates corticotropin-releasing factor gene expression in conscious rats. Endocrinology 135, 2177–2182. [DOI] [PubMed] [Google Scholar]
- Itoi K, Helmreich DL, Lopez-Figueroa MO, and Watson SJ (1999). Differential regulation of corticotropin-releasing hormone and vasopressin gene transcription in the hypothalamus by norepinephrine. J. Neurosci 19, 5464–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Rajamanickam S, and Justice NJ (2018). Local Corticotropin-Releasing Factor Signaling in the Hypothalamic Paraventricular Nucleus. J. Neurosci 38, 1874–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kombian SB, Mouginot D, and Pittman QJ (1997). Dendritically released peptides act as retrograde modulators of afferent excitation in the supraoptic nucleus in vitro. Neuron 19, 903–912. [DOI] [PubMed] [Google Scholar]
- Kovács KJ, Földes A, and Sawchenko PE (2000). Glucocorticoid negative feedback selectively targets vasopressin transcription in parvocellular neurosecretory neurons. J. Neurosci 20, 3843–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightman SL, and Young WS III. (1988). Corticotrophin-releasing factor, vasopressin and pro-opiomelanocortin mRNA responses to stress and opiates in the rat. J. Physiol 403, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liposits Z, Phelix C, and Paull WK (1986). Adrenergic innervation of corticotropin releasing factor (CRF)-synthesizing neurons in the hypothalamic paraventricular nucleus of the rat. A combined light and electron microscopic immunocytochemical study. Histochemistry 84, 201–205. [DOI] [PubMed] [Google Scholar]
- Ludwig M, Sabatier N, Bull PM, Landgraf R, Dayanithi G, and Leng G (2002). Intracellular calcium stores regulate activity-dependent neuropeptide release from dendrites. Nature 418, 85–89. [DOI] [PubMed] [Google Scholar]
- Luther JA, Halmos KC, and Tasker JG (2000).A slow transient potassium current expressed in a subset of neurosecretory neurons of the hypothalamic paraventricular nucleus. J. Neurophysiol 84, 1814–1825. [DOI] [PubMed] [Google Scholar]
- Ma XM, Levy A, and Lightman SL (1997). Rapid changes in heteronuclear RNA for corticotrophin-releasing hormone and arginine vasopressin in response to acute stress. J. Endocrinol 152, 81–89. [DOI] [PubMed] [Google Scholar]
- Mulligan SJ, and MacVicar BA (2004). Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431, 195–199. [DOI] [PubMed] [Google Scholar]
- Nahar J, Haam J, Chen C, Jiang Z, Glatzer NR, Muglia LJ, Dohanich GP, Herman JP, and Tasker JG (2015). Rapid Nongenomic Glucocorticoid Actions in Male Mouse Hypothalamic Neuroendocrine Cells Are Dependent on the Nuclear Glucocorticoid Receptor. Endocrinology 156,2831–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete M, and Araque A (2008). Endocannabinoids mediate neuron-astrocyte communication. Neuron 57, 883–893. [DOI] [PubMed] [Google Scholar]
- Navarrete M, and Araque A (2010). Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 68, 113–126. [DOI] [PubMed] [Google Scholar]
- Oliet SHR, Baimoukhametova DV, Piet R, and Bains JS (2007). Retrograde regulation of GABA transmission by the tonic release of oxytocin and endocannabinoids governs postsynaptic firing. J. Neurosci 27, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, and Haydon PG (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116. [DOI] [PubMed] [Google Scholar]
- Paulsen RE, Contestabile A, Villani L, and Fonnum F (1987). An in vivo model for studying function of brain tissue temporarily devoid of glial cell metabolism: the use of fluorocitrate. J. Neurochem 48, 1377–1385. [DOI] [PubMed] [Google Scholar]
- Perea G, and Araque A (2002). Communication between astrocytes and neurons: a complex language. J. Physiol. Paris 96, 199–207. [DOI] [PubMed] [Google Scholar]
- Perea G, Navarrete M, and Araque A (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431. [DOI] [PubMed] [Google Scholar]
- Plotsky PM (1987). Facilitation of immunoreactive corticotropin-releasing factor secretion into the hypophysial-portal circulation after activation of cate-cholaminergic pathways or central norepinephrine injection. Endocrinology 121 , 924–930. [DOI] [PubMed] [Google Scholar]
- Porter JT, and McCarthy KD (1996). Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J. Neurosci 16, 5073–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozanski GM, Li Q, and Stanley EF (2013). Transglial transmission at the dorsal root ganglion sandwich synapse: glial cell to postsynaptic neuron communication. Eur. J. Neurosci 37, 1221–1228. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, and Swanson LW (1982). The organization of noradrenergic pathways from the brainstem to the paraventricular and supraoptic nuclei in the rat. Brain Res. 257, 275–325. [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, Kracun S, Xu J, Sofroniew MV, Ellisman MH, and Khakh BS (2013). Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J. Gen. Physiol 141 , 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son SJ, Filosa JA, Potapenko ES, Biancardi VC, Zheng H, Patel KP, Tobin VA, Ludwig M, and Stern JE (2013). Dendritic peptide release mediates interpopulation crosstalk between neurosecretory and preautonomic networks. Neuron 78, 1036–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, and Graham SH (1994). Fluorocitrate and fluoroacetate effects on astrocyte metabolism in vitro. Brain Res. 664, 94–100. [DOI] [PubMed] [Google Scholar]
- Tasker JG, and Dudek FE (1991). Electrophysiological properties of neurones in the region of the paraventricular nucleus in slices of rat hypothalamus. J. Physiol 434, 271–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich-Lai YM, and Herman JP (2009). Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci 10, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahl TP, Ulrich-Lai YM, Ostrander MM, Dolgas CM, Elfers EE, Seeley RJ, D’Alessio DA, and Herman JP (2005). Comparative analysis of ACTH and corticosterone sampling methods in rats. Am. J. Physiol. Endocrinol. Metab 289, E823–E828. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, and Nedergaard M (2018). Physiology of Astroglia. Physiol. Rev 98, 239–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitnall MH, Mezey E, and Gainer H (1985). Co-localization of corticotropin-releasing factor and vasopressin in median eminence neurosecretory vesicles. Nature 317, 248–250. [DOI] [PubMed] [Google Scholar]
- Whitnall MH, Smyth D, and Gainer H (1987). Vasopressin coexists in half of the corticotropin-releasing factor axons present in the external zone of the median eminence in normal rats. Neuroendocrinology 45, 420–424. [DOI] [PubMed] [Google Scholar]
- Whitnall MH, Kiss A, and Aguilera G (1993). Contrasting effects of central alpha-1-adrenoreceptor activation on stress-responsive and stress-nonresponsive subpopulations of corticotropin-releasing hormone neurosecretory cells in the rat. Neuroendocrinology 58, 42–48. [DOI] [PubMed] [Google Scholar]
- Wittmann G, Lechan RM, Liposits Z, and Fekete C (2005). Glutamatergic innervation of corticotropin-releasing hormone- and thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus of the rat. Brain Res. 1039, 53–62. [DOI] [PubMed] [Google Scholar]
- Woulfe JM, Flumerfelt BA, and Hrycyshyn AW (1990). Efferent connections of the A1 noradrenergic cell group: a DBH immunohistochemical and PHA-L anterograde tracing study. Exp. Neurol 109, 308–322. [DOI] [PubMed] [Google Scholar]
- Wu N, and Tasker JG (2017). Nongenomic Glucocorticoid Suppression of a Postsynaptic Potassium Current via Emergent Autocrine Endocannabinoid Signaling in Hypothalamic Neuroendocrine Cells following Chronic Dehydration. Eneuro 4, ENEURO.0216-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki RS, Chen Q, Schreiber SS, and Brinton RD (1997). Localization of V1a vasopressin receptor mRNA expression in cultured neurons, astroglia, and oligodendroglia of rat cerebral cortex. Brain Res. Mol. Brain Res 45, 138–140. [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Edwards MR, Ulrich-Lai YM, Herman JP, and Cullinan WE (2012). Brainstem origins of glutamatergic innervation of the rat hypothalamic paraventricular nucleus. J. Comp. Neurol 520, 2369–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All software used in this study is available and listed in the Key Resources Table. This study did not generate any unique datasets or code.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-CRH | Peninsula | Cat# T4037; RRID:AB_518252 |
| chicken anti-β-gal | Abcam | Cat# Ab9361; RRID:AB_307210 |
| rabbit anti GFAP | Phosphosolution | Cat# 620-GFAP; RRID:AB_2492124 |
| goat anti-rabbit IgG | Thermo Fisher Scientific | Cat# A-11012; RRID:AB_2534079 |
| goat anti-rabbit IgG | Thermo Fisher Scientific | Cat# SA5-10033; RRID:AB_2556613 |
| goat anti-chicken IgY | Thermo Fisher Scientific | Cat# A32933; RRID:AB_2762845 |
| Bacterial and Virus Strains | ||
| AAV9-EF1a-DIO-hChR2(H134R)-mCherry | Vector Biolabs | Cat. No: VB4651 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rhod-2 AM | Life Technologies | Cat# R1244 |
| GDP-β-s | Sigma-Aldrich | Cat# G7637 |
| 6,7-dinitroquinoxaline-2,3-dione | Tocris | Cat# 0189 |
| DL-2-amino-5-phosphonopentanoic acid | Tocris | Cat# 0105 |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Experimental Models: Organisms/Strains | ||
| Tg(CRH-EGFP)HS57Gsat/Mm | MMRRC | RRID:MMRRC_017058-UCD |
| tetO-dnSNARE mouse | Haydon Lab | N/A |
| GFAP-tTA mouse | Haydon Lab | N/A |
| B6.Cg-7630403G23RikTg(Th-cre)1Tmd/J | Jackson Lab | RRID:IMSR_JAX:008601 |
| TH-cre/CRH-eGFP mouse | In house | N/A |
| Software and Algorithms | ||
| Prism v. 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Mini Analysis | Synaptosoft, Inc. | http://www.synaptosoft.com/MiniAnalysis/ |