

SUMMARY
For decades, two fusion modes were thought to control hormone and transmitter release essential to life; one facilitates release via fusion pore dilation and flattening (full collapse), and the other limits release by closing a narrow fusion pore (kiss-and-run). Using super-resolution stimulated emission depletion (STED) microscopy to visualize fusion modes of dense-core vesicles in neuroendocrine cells, we find that facilitation of release is mediated not by full collapse but by shrink fusion, in which the Ω-profile generated by vesicle fusion shrinks but maintains a large non-dilating pore. We discover that the physiological osmotic pressure of a cell squeezes, but does not dilate, the Ω-profile, which explains why shrink fusion prevails over full collapse. Instead of kiss-and-run, enlarge fusion, in which Ω-profiles grow while maintaining a narrow pore, slows down release. Shrink and enlarge fusion may thus account for diverse hormone and transmitter release kinetics observed in secretory cells, previously interpreted within the full-collapse/kiss-and-run framework.
In Brief
Shin et al. discover two fusion modes; one involves fused vesicle shrinking that employs a large pore to facilitate content release, and the other involves vesicle enlargement with a small pore that slows down release. Shrinking is energetically preferred over the generally assumed full-collapse fusion, because osmotic pressure squeezes fused vesicles.
Graphical Abstract
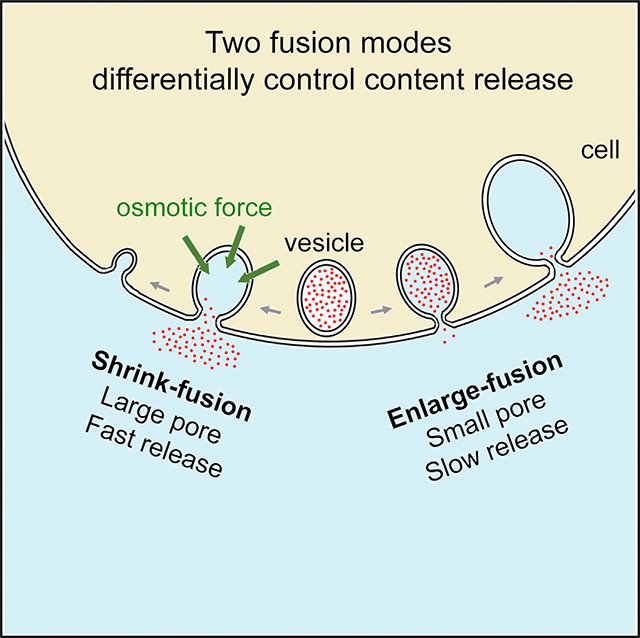
INTRODUCTION
Hormone and transmitter release by endocrine cells and neurons mediates many important functions, such as stress responses, immune response, control of blood glucose with relevance to diabetes, and synaptic transmission, which is essential for cognition and coordinated motor activity (Jahn and Fasshauer, 2012; Saheki and De Camilli, 2012; Alabi and Tsien, 2013; Wu et al., 2014; Chang et al., 2017; Sharma and Lindau, 2018). Regulation of the amount and rate of release is physiologically crucial, and much research has addressed the mechanisms involved. From decades of research, a widely held view emerged that content release by neurons and endocrine cells is controlled by two modes of fusion: (1) full collapse, in which the fusion pore dilates while the vesicle flattens into the membrane, resulting in rapid and complete release; and (2) kiss-and-run, when a narrow fusion pore opens and then closes, giving a slow and/or partial release of contents (Jahn and Fasshauer, 2012; Saheki and De Camilli, 2012; Alabi and Tsien, 2013; Wu et al., 2014; Chang et al., 2017; Sharma and Lindau, 2018).
This view has been questioned in two respects. First, inconsistent with the assumed narrow pore, fusion pore conductance measurements revealed large conductance during some kiss-and-run events (Alés et al., 1999; He et al., 2006), and a recent imaging study directly visualized pores of ~60–490 nm during some kiss-and-run events (Shin et al., 2018). Second, while full collapse was proposed on the basis of electron microscopy (EM) data (Heuser and Reese, 1981), it has not been observed in live cells. Imaging in endocrine cells shows shrinking fusion spots rather than growing fusion spots that subsequently disappear, as would be expected from full collapse (Chiang et al., 2014; Wen et al., 2016). It was proposed that the shrinking spots indicated a fusion mode termed shrink fusion, in which the vesicle shrinks while retaining its shape without pore dilation, and shrink fusion was suggested to be mediated by F-actin-dependent plasma membrane (PM) tension (Wen et al., 2016).
While these observations imply the possibility of replacing full collapse with shrink fusion, direct evidence of shrink fusion is lacking. To demonstrate shrink fusion would require visualization of vesicle membrane profile shape changes and evidence that the pore does not dilate. Such visualization is also required to eliminate other possibilities, including rapid budding of clathrin-coated vesicles at the fusion-generated Ω-profile as recently suggested (Bittner et al., 2013; Abbineni et al., 2018). Direct visualization is indeed feasible, as shown in recent studies that visualized Ω--shaped membrane profiles and their pores (Zhao et al., 2016; Shin et al., 2018). However, these studies did not investigate Ω-profile shrinking or pore dynamics during shrinking.
The proposal of shrink fusion in replacement of full collapse raises the questions of why shrinking is preferred to the intuitively appealing full-collapse pathway, whether shrink fusion promotes release, and by what mechanism. The observation that kiss-and-run may not limit content release (Alés et al., 1999; Shin et al., 2018) raises the question of whether there is a fusion mode specifically for the limiting of content release. Here, we addressed these fundamental questions by super-resolution stimulated emission depletion (STED) imaging of vesicular membrane profiles and fusion pore dynamics, confocal imaging of content release, electron microscopy (EM) and realistic mathematical modeling in neuroendocrine chromaffin cells. We found that facilitation of content release is mediated not by full collapse but by shrink fusion with a large fusion pore and that inhibition of release is mediated not by kiss-and-run but by enlarge-fusion mode, in which Ω-profiles increase in size but maintain a narrow pore. We found that cells’ physiological osmotic pressure squeezes, but does not dilate, the Ω-profile, explaining why shrink fusion rather than full collapse is the default fusion mode for merging vesicles with the PM and promoting content release. We conclude that shrink and enlarge fusion control the yin and yang of exocytotic content release.
RESULTS
Direct Observation of Fused Vesicle Shrinking and Enlargement in Live Cells
To study how vesicles merge with the PM, we transfected bovine adrenal chromaffin cells in primary cultures with EGFP or mNeonGreen attached to the phospholipase C delta PH domain (PHG; either PH-EGFP or PH-mNeonGreen). PHG binds to PtdIns(4,5)P2 (PIP2) at the PM cytosolic-facing leaflet and thus labels PM (Figure 1A) (Zhao et al., 2016; Shin et al., 2018). We added Atto 532 (A532) in the bath (Figure 1A), which enters the fusing Ω-profile to confirm the structural changes revealed by PHG imaging (Chiang et al., 2014; Shin et al., 2018). We induced vesicle fusion using 1-s depolarization from −80 to +10 mV (depol1 s) through a pipette in the whole-cell configuration, which induced calcium currents and capacitance changes reflecting robust exo- and endocytosis (Figure 1A). STED imaging of A532 and PHG was performed in the microscope xz plane at a fixed y location near the cell bottom every 26–300 ms (xz/yfix imaging) before and after depol1 s for ~20–30 s. Each cell was subjected to only one depol1 s, which induced fusion mostly during and ~3 s after depol1 s (Chiang et al., 2014; Shin et al., 2018).
Figure 1. Visualization of Ω-Shaped Membrane Profile Shrinking and Enlargement.

(A) Top: setup drawing. The cell membrane is labeled with PHG (green) and bath labeled with A532 (red). Calcium current (ICa) and capacitance (Cm) are recorded at the whole-cell voltage-clamp configuration. Bottom: Sampled ICa and Cm change induced by depol1 s.
(B–H) PH-Ω fluorescence (FPH; normalized to baseline), A532 spot fluorescence (F532; normalized to baseline), PH-Ω height (H; circles), PH-Ω width (W; triangles), and sampled images at times indicated with lines showing different modes of fusion: (B–D) shrink fusion (B, slow; C, fast; and D, tubular intermediates observed); (E and F) partial shrink fusion (E, reduced-size Ω-profile; and F, reduced-size tubular structure); (G) same-size fusion; and (H) enlarge fusion. The inset in (G) and (H) is shown at the same timescale (right). When PH-Ω was too dim or smaller than our resolution, PH-Ω height and width were not measured. Images were acquired with STED xz/yfix scanning.
(I) The percentage of shrink fusion, partial shrink fusion, same-size fusion, and enlarge fusion (total PH-Ω number: 236; from 202 cells).
See also Figures S1 and S2.
Depol1 s induced PHG-labeled Ω-shape profiles (PH-Ωs) filled with A532 (Figures 1B–1H), reflecting diffusion of PHG and A532 from PM and bath into the fusion-generated Ω-profile. PH-Ω without A532, which reflected hemi-fusion (Zhao et al., 2016), was not analyzed here. In ~20% of cells, we observed a depol1 s-induced, fusion-generated A532-filled PH-Ω with a clear membrane outline. Following PH-Ω appearance (236 events, 202 cells; Figure S1A), the Ω-profile (1) shrank while maintaining an Ω-shape until it became undetectable (shrink fusion, 45 events; Figures 1B–1D); (2) shrank partially and then maintained a reduced-size Ω-profile (partial shrink fusion, 24 events; Figures 1E and 1F); (3) maintained its Ω-profile and A532 spot size (same-size fusion, 136 events; Figure 1G); or (4) increased in size while maintaining its Ω-shape (enlarge fusion, 31 events; Figure 1H; summarized in Figure 1I). Figure S1B illustrates these four fusion modes schematically. The PH-Ω width (see Figure S1C for measurements) at the fusion onset was similar for these four fusion modes (Figure S1D).
Shrink fusion was rapid, with a 20%–80% shrinking time mostly <2 s (Figures 1B–1D). During shrinking episodes (both shrink fusion and partial shrink fusion), in most cases, the Ω shape was preserved as the width and height decreased proportionally (Figures 1B, 1C, and 1E). However, in some instances of shrink fusion, PH-Ω had an extended tubular shape with a height considerably greater than the width (Figures 1D and 1F). We did not observe vesicle budding off the PH-Ω during shrink fusion (n = 45), excluding this as the mechanism that shrinks the PH-Ω.
Enlarge fusion was slower than shrink fusion, with a 20%–80% time of ~2–20 s (Figure 1H). Enlarge fusion was detected if the PH-Ω’s width at 20–30 s after fusion onset (516 ± 18 nm) increased by >15% of that at the fusion onset (352 ± 13 nm, n = 31; t test, p < 0.001). The width of PH-Ω at 20–30 s after the onset of enlarge fusion (516 ± 18 nm, n = 31) was significantly larger than that of same-size fusion (374 ± 7 nm, n = 136; t test, p < 0.001). The width and height of the PH-Ω increased approximately proportionally during enlarge fusion (Figure 1H). Additional vesicle fusion at the PH-Ω was not observed during enlarge fusion (n = 31), excluding compound fusion as the origin of enlarge fusion.
Three sets of evidence showed that shrinking or enlargement was not due to Ω-profile movement in the xy plane. First, random Ω-profile movement generally involves displacements in both the x and y directions, whereas during shrinking or enlargement captured by STED xz/yfix imaging, the Ω-profile did not shift in the x direction (n = 69; Figures 1B–1H). Second, STED xy-plane imaging at a fixed z-focal plane (xy/zfix imaging) showed that depol1 s induced the appearance of PH-labeled rings containing A532 spots (Figures S2A–S2C), which reflected fusion-generated Ω-profiles induced by depol1 s (Chiang et al., 2014; Shin et al., 2018). These PH-labeled rings shrank, remain unchanged or enlarged but did not move in the xy plane (Figures S2A–S2C; 102 events, 7 cells). Third, in cells treated with high-potassium solution (70 mM, 45 s) and fixed with 4% paraformaldehyde, all PH-Ω (59 PH-Ω in 39 cells) did not change in width during 20–30 s STED xz/yfix imaging (Figure S2D). Thus, PH-Ω size changes observed in live cells are not due to microscopic y axis drift.
To verify that the PH-Ω size changes reflect post-fusion structural changes, we loaded vesicles with fluorescent false neurotransmitter FFN511 via bath application (Figure 2A) (Gubernator et al., 2009). We observed the appearance of PH-Ω that released FFN511, as detected with STED xz/yfix imaging of PHG and FFN511 (Figures 2A–2E; n = 92 events, 71 cells) (Shin et al., 2018). Similar to STED PHG/A532 imaging, we observed shrink fusion (n = 8), partial shrink fusion (n = 13), same-size fusion (n = 53), and enlarge fusion (n = 18; Figures 2B–2E). The 20%–80% FFN511 release time depended on the fusion mode (Figure 2F), which will be further described later. We concluded that the PH-Ω size changes reflect post-fusion vesicle size changes.
Figure 2. Observation of Ω-Shaped Membrane Profile Shrinking and Enlargement while Releasing FFN511.

(A) Setup diagram. Vesicles were loaded with FFN511 (red, pseudo-color), the PM was labeled with PHG (green), and ICa and Cm were recorded via a pipette.
(B–E) FPH, FFN511 fluorescence (FFFN; normalized to baseline), PH-Ω height (H; circles) and width (W; triangles), and sampled images at times indicated with lines showing release of FFN511 for shrink fusion (B), partial shrink fusion (C), same-size fusion (D), and enlarge fusion (E).
(F) 20%–80% decay time of FFN511 (TFFN_decay, mean + s.e.m.) for shrink fusion (8 events), partial shrink fusion (13 events), same-size fusion (53 events), and enlarge-fusion (18 events). Data are from 71 cells under STED xz/yfix imaging of PHG/FFN511. ***p < 0.001; ANOVA test.
Consistent with Ω-profile shrinking and enlargement (Figure 1), EM revealed Ω-profiles of various sizes, some of which approached a tubular shape (Figures 3A and 3B); widths ranged from 56 to 784 nm, and larger width was correlated with larger height (Figure 3C, top). The height/width ratio of most Ω-profiles was >1 (85% of cases; Figure 3C, bottom), indicating that Ω-profiles were mostly somewhat elongated with some approaching a tubular shape, consistent with STED observation of mostly elongated Ω-profiles (e.g., Figures 1B–1H and 3D–3H). The pore size range measured with EM was similar to the STED-measured visible pore (Porev) range, as shown in a previous study (Shin et al., 2018).
Figure 3. Fusion Pore Dynamics during Shrink, Partial Shrink, Same-Size, and Enlarge Fusion.

(A and B) Electron microscopic images of Ω-profiles (A) or elongated Ω-profiles resembling cylinders (B) with different sizes. Cells were in 70 mM KCl for 90 s.
(C) The width (W) and the ratio between the height (H) and width (H/W) of Ω-profiles observed with EM (each circle: 1 Ω-profile; 84 Ω-profiles from 800 cell cross sections). The line (upper panel) is a linear regression fit (correlation coefficient: 0.81). Dotted line (lower panel) indicates H/W = 1.
(D–H) FPH (PH-Ω fluorescence), F532 (A532 fluorescence), H (PH-Ω height, circles), W (PH-Ω width, triangles), pore (PH-Ω pore size, squares), and sampled images at times indicated with lines showing pore dynamics during shrink fusion (D and E), partial shrink fusion (F), same-size fusion (G), and enlarge fusion (H). Gray squares (G and H), pore remained open, but below our STED resolution of ~60 nm.
(I) The percentage of observing a Porev (Porev%, mean + s.e.m.) during shrinkrelated fusion (including shrink fusion and partial shrink fusion), same-size fusion, and enlarge fusion (total fusion event number: 236; from 202 cells). *p < 0.05, ANOVA test.
See also Figure S3.
As the PH-Ω shrank, remained unchanged in size or enlarged, in some cases the pore closed. This was reflected as decay of the spot A532 fluorescence (F532; strong excitation) to the baseline while PH-Ω size remained unchanged due to pore closure that prevented exchange of the bleached A532 (caused by strong excitation) with fluorescent A532 in the bath (Figure S3A; STAR Methods) (for details, see Chiang et al., 2014; Zhao et al., 2016).
Resolving Fusion Pore during Vesicle Shrinking or Enlargement
At fusion onset, sampled every 26–300 ms, ~21% of PH-Ω (51 of 236 PH-Ω) showed a Porev larger than ~60 nm, our STED resolution (Figures 3D–3H; see Figure S3B for Porev measurement). The pore in the remaining PH-Ω could not be resolved, mostly because it was below STED resolution (Shin et al., 2018). The percentage of Porev (Porev%) for shrink-related fusion (shrinkrelated fusion), which denotes both shrink fusion (e.g., Figures 3D and 3E) and partial shrink fusion (e.g., Figure 3F), was significantly larger than that for enlarge fusion (e.g., Figure 3H) but not significantly larger than that for same-size fusion (e.g., Figure 3G) (summarized in Figure 3I). Results similar to those in Figure 3I were obtained when pore closure events were excluded (Figure S3C). Thus, fusion pores are larger for shrinkrelated fusion than enlarge fusion. This finding suggests that shrinkrelated fusion releases contents faster than enlarge fusion, a conjecture that was experimentally verified, as described in the next section.
We noticed that in 3 of 14 shrink-fusion events with a Porev, following substantial shrinking of the Ω-profile so that the width had become smaller than the pore size, the Ω-shape changed into a Λ-shape during the final stages (e.g., Figure 3E). Such Λ-profiles were not observed during shrink fusion with no Porev (31 out of 31 events; e.g., Figures 1B–1D). Thus, only ~7% of shrink-fusion events showed a late-stage Λ-profile. These results suggest that as the shrinking Ω-profile approaches the size of its own pore, a conversion to a Λ-profile occurs before finally merging with the flat PM.
Shrink Fusion Releases Contents Much Faster than Enlarge Fusion
To determine whether shrink fusion and enlarge fusion are associated with different release kinetics, we overexpressed a vesicular lumen protein neuropeptide Y-EGFP (NPY-EGFP) and performed confocal imaging of NPY-EGFP with Alexa Fluor 647 (A647; in the bath) in the xy/zfix configuration ~200 nm above the cell bottom (Figure 4A). Depol1 s induced NPY-EGFP spot release (218 spots, 28 cells; Figures 4B–4G), accompanied by sudden A647 spot appearance and fluorescence (F647) increase due to A647 diffusion from the bath to the fusion-generated Ω-profile (Chiang et al., 2014).
Figure 4. Imaging NPY-EGFP Release and Structural Changes of A647-Labeled Fusing Vesicles.

(A) Schematic drawing of a cell containing NPY-EGFP-labeled vesicles (green) bathed with a solution containing A647 (red).
(B–G) F647 (A647 spot fluorescence), FNPY (NPY-EGFP spot fluorescence), A647 spot width (W; full-width-half-maximum), and A647/NPY-EGFP confocal xy-images at times indicated with lines for various rates of NPY-EGFP release at various fusion modes: (B) shrink fusion (inset shows traces in larger timescale); (C) partial shrink fusion; (D) partial shrink fusion followed by pore closure; (E) same-size fusion; (F) same-size fusion followed by pore closure; and (G) enlarge fusion (inset shows traces in larger timescale).
(H and I) Cumulative frequency (H; normalized) or the mean (I; mean + s.e.m.) of TNPY_decay (20%–80% FNPY decay time, top) and T647_rise (20%–80% F647 rise time, bottom) for shrinkrelated fusion (69 events, 28 cells), same-size fusion (111 events, 28 cells), and enlarge fusion (38 events, 28 cells). ***p < 0.001, ANOVA test.
(J) 20%–80% decay time of NPY-EGFP (TNPY_decay) and A647 fluorescence during shrink fusion (mean + SEM, 41 events). A647 fluorescence decay was measured after the peak was reached. **p < 0.01, t test.
See also Figure S4.
Under strong excitation, we observed four distinct patterns of evolution of A647 spots (218 spots, 28 cells) (see Chiang et al., 2014 for systematic characterizations). First, shrink fusion was reflected as A647 spot width shrinking until it was undetectable while F647 decayed to the baseline (Figure 4B; 41 events). Second, the A647 spot width could shrink partially with partial F647 decrease and then maintain a spot of reduced size, which reflected partial shrink fusion (Figures 4C and 4D; 28 events). The reduced-size spot was sometimes followed by spot F647 decay to baseline while the spot size did not change further, reflecting pore closure that prevented exchange of bleached A647 in the spot with fluorescent A647 in the bath (partial shrink fusion with pore closure; Figure 4D) (Chiang et al., 2014). Third, A647 spot size and F647 could remain unchanged (same-size fusion; Figure 4E; 111 events), sometimes followed by F647 decay to baseline without spot size change (same-size fusion with pore closure; Figure 4F). Fourth, spot width and F647 could increase (enlarge fusion; Figure 4G; 38 events), occasionally followed by spot F647 decay to baseline without further change in spot size (enlarge fusion with pore closure; not shown). Consistent with STED xy/zfix imaging (Figure S3A), we used these A647 spot behaviors to identify shrink fusion, partial shrink fusion, same-size fusion, and enlarge fusion (for detail, see Chiang et al., 2014).
The percentages of shrink fusion (19% ± 4%, n = 28 cells) and enlarge fusion (17% ± 5%, n = 28 cells) were not significantly different compared to those without NPY-EGFP overexpression (shrink fusion: 18% ± 3%; enlarge fusion: 10% ± 2%, n = 60 cells). Thus, NPY-EGFP overexpression is not crucial in determining whether a vesicle undergoes shrink or enlarge fusion.
NPY-EGFP release was detected as decay of NPY-EGFP fluorescence (FNPY; Figures 4B–4G) (Taraska et al., 2003; Perrais et al., 2004). The FNPY 20%–80% decay time (TNPY_decay) ranged from 12 ms to 12 s, in most cases less than ~2 s. Of 218 spots, 6 did not release or else partially released NPY-EGFP due either to rapid pore closure (5 out of 95 pore closure events) or to a narrow pore (1 out of 82 same-size fusion or enlarge fusion without pore closure) (Shin et al., 2018). These 6 events were excluded from further analysis.
TNPY_decay for shrinkrelated fusion was much shorter than that for same-size fusion, whereas enlarge fusion showed a much longer TNPY_decay than that of either same-size fusion or shrinkrelated fusion (Figures 4H and 4I, top; 212 events, 28 cells). Similar results were obtained when pore closure events were excluded (Figures S4A and S4B; 122 events, 28 cells), or partial shrink fusion was excluded from shrinkrelated fusion (Figures S4C and S4D). Thus, shrinkrelated fusion and enlarge fusion are associated with faster and slower content release, respectively.
STED xz/yfix imaging of FFN511 release further confirmed that shrinkrelated fusion and enlarge fusion are associated with faster and slower content release, respectively. The 20%–80% decay time of FFN511 (TFFN_decay) during shrinkrelated fusion, as observed with STED xz/yfix imaging, was much shorter than that for same-size fusion, whereas enlarge fusion observed with STED xz/yfix imaging showed the longest TFFN_decay (e.g., Figures 2B–2E, summarized in Figure 2F; 92 events, 71 cells). These results were analogous to different TNPY_decay observed during shrinkrelated, same-size, and enlarge fusion (Figure 4I).
Three sets of evidence suggest that to facilitate content release, shrinkrelated fusion employs a larger fusion pore than enlarge fusion. First, as mentioned above, Porev% of shrinkrelated fusion was significantly larger than that of enlarge fusion (Figure 3I). Second, release of either NPY-EGPF or FFN511 was fastest for shrinkrelated fusion, slower for same-size fusion, and the slowest for enlarge fusion (Figures 2F and 4I, top), supporting that these different release time courses are controlled by the fusion pore size differences. Third, the F647 20%–80% rise time (T647_rise) was shortest for shrinkrelated fusion, significantly longer for same-size fusion, and longest for enlarge fusion (Figures 4H and 4I, bottom), mirroring the order of the respective TNPY_decay values for the three classes of events (Figures 4H and 4I, top). The same order for the time course of content release and A647 entry into the fusing vesicle further suggests that this order reflects the fusion pore size differences.
Both TNPY_decay and T647_rise were shorter for shrinkrelated fusion than for same-size fusion (Figures 4H and 4I), suggesting that shrinkrelated fusion employs a larger fusion pore than same-size fusion. We could not detect a difference in Porev% values for shrinkrelated fusion and same-size fusion (Figure 3I), likely due to our limited STED resolution (~60 nm). Although shrink fusion was associated with fast NPY release, shrinking was apparently not the cause of this fast release, because NPY release time, TNPY_decay (0.14 ± 0.03 s, n = 41), was much faster than the time for the vesicle to shrink, the 20%–80% decay time of F647 during shrink fusion (0.80 ± 0.25 s, n = 41, t test, p < 0.01; e.g., Figure 4B, summarized in Figure 4J).
Mechanisms Underlying Shrink Fusion
What mechanism leads to shrink fusion, and why is this pathway preferred to the full-collapse pathway envisaged in the classical picture? To address this, we mathematically modeled the merging of a fused vesicle with the chromaffin cell PM (Figure 5A; STAR Methods, Mathematical modeling) and verified the model’s predictions with experimental data.
Figure 5. Mathematical Model of Vesicle Evolution during Exocytosis.

(A) Schematic of the model (not to scale). Cells maintain an outward (swelling) osmotic pressure ΔP=Pcell – Pext (green arrows), with cell pressure Pcell exceeding extracellular pressure Pext. Intact vesicles maintain swelling pressure (red arrow), with vesicle pressure Pves >Pcell. Following fusion with PM, rapid equilibration between the vesicle lumen and extracellular medium is assumed, so Pves =Pext. The vesicle osmotic pressure then equals ΔP but is now an inward squeezing pressure. The model calculates the vesicle tension, γves, while the PM tension, γPM, and the adhesion energy εadhesion to the actin cortex (maroon layer adjacent to PM) are taken from experiment (see Table 1).
(B) Predicted shrink fusion sequence. Computed vesicle shapes and free energies for squeezing pressure ΔP = 100 Pa and the indicated effective diameters D (such that vesicle area equals πD2). A transition occurs at D = 56 nm from Ω to Λ shape (defined as a profile lacking overhang).
(C and D) Free energy (C) and vesicle membrane tension (D) plotted versus diameter for the vesicles in (B).
(E) Predicted tubular shapes for two squeezingpressures (100 or 200 Pa). Tubular vesicles are predicted for large squeezing pressure or large vesicle area.
(F) Three classes of vesicle shape are predicted (Ω, tubular, and Λ), each corresponding to one sector in the vesicle width-height plane. Predicted isobars are shown for the indicated osmotic squeezing pressures. The EM data of Figure 3C are also plotted. Each experimental point belongs to one isobar, representing the predicted squeezing pressure for the observed vesicle.
(G) Shrink fusion mechanism predicted by themodel. Osmotic squeezing deflates the vesicle and abolishes its membrane tension, so the membrane is reeled into the PM by the PM tension and PM adhesion to the actin cortex.
See also Figure S5
The vesicle membrane is treated as a continuous surface, with bending and osmotic pressure energy contributions following the well-established Helfrich theory (Helfrich, 1973). The membrane connects to the PM in a small area assumed free of actin cortex (Table 1; Figure 5A), and elsewhere, the PM is assumed adhered to the actin cortex, likely via actin-binding transmembrane and membrane-associated proteins (Dai and Sheetz, 1999; Borghi and Brochard-Wyart, 2007; Porat-Shliom et al., 2013; Wen et al., 2016; Shi et al., 2018). Using a method similar to a previous study (Jenkins, 1977), we obtained differential equations whose solution yielded the minimum energy shape and volume (see STAR Methods, Mathematical modeling). All model parameters were fixed by experiment, either from the present study or published data (Table 1).
Table 1.
Model Parameters
| Symbol | Meaning | Value | Reference |
|---|---|---|---|
| κ | membrane bending modulus | 20 kBT | Rawicz et al., 2000 |
| w | diameter of actin cortex-free zone | 80 nm | Figure 3Aa |
| γPM | PM tension | 0.04 pN nm−1 | Tinevez et al., 2009 |
| εadhesion | membrane-cortex adhesion energy density | 0.02 pN nm−1 | Dai and Sheetz, 1999 |
Model solutions with w = 80 nm had ∼30-nm-diameter fusion pores, consistent with the experimental Ω-shapes of Figure 3A. The solutions exhibited a Ω-to-Λ transition at vesicle diameter 56 nm (slightly less than the STED resolution limit), consistent with the finding that 93% of shrink fusion events lacked a visible Λ-sequence in STED measurements.
A key model assumption is that during shrink fusion, the vesicle and extracellular pressures are equal, because fusion pores are large enough to allow rapid pressure equilibration, unhindered by vesicular lumen contents (Figure 5A). Several observations support this assumption. First, ~26% of shrink-fusion events involved pores >60 nm (Figure 3I). Second, shrink-fusion events permitted rapid ~0.14 s release of the ~4 nm NPY-EGFP (Palm et al., 1997) (Figure 4J), which is much faster than the Ω-profile shrinking time (~0.80 s, Figure 4J, p < 0.01). Thus, content release precedes Ω-profile shrinking. Consistent with this, chromogranin A, a major vesicular matrix component, was released in ~0.1 s during high potassium application (Abbineni et al., 2019), which might induce mostly shrink fusion (Wen et al., 2016). Once the contents are gone, pressure equilibration is almost instantaneous; even for a small 4-nm-diameter pore, we estimate unhindered pressure equilibration requires only milliseconds (see STAR Methods, Mathematical modeling).
Pressure equalization has significant consequences; since cells maintain a swelling osmotic pressure (ΔP = intracellular osmotic pressure – extracellular osmotic pressure) (Stewart et al., 2011), it follows that the fused vesicle experiences a squeezing osmotic pressure, ΔP, equal to the cell swelling pressure but acting in reverse (Figure 5A). Computed shapes are shown in Figure 5B for a typical squeezing pressure of ΔP = 100 Pa and vesicle sizes up to effective diameter D = 400 nm, typical for shrink-fusion onset. The sequence follows a shrink-fusion trajectory of mildly prolate Ω-shapes, with height/width ratios of ~1.2–1.5, which is close to our EM measurements (Figure 3C).
We calculated the free energies of the vesicle states in Figure 5B and found the free energy monotonically decreased with decreasing vesicle size, demonstrating that the shrink-fusion trajectory is indeed selected (Figures 5B and 5C; STAR Methods, Mathematical modeling). The decrease originated in the osmotic squeezing force, which deflated the vesicle and abolished its tension to negative values (Figures 5D and S5A). Thus, a powerful membrane tension gradient reels in membrane, from the tensionless vesicle into the PM, driven by the PM tension and the PM-actin cortex adhesion forces (Figure 5G). For a membrane area ΔA reeled onto the PM, we find a free energy decrease ΔF = −(εadhesion + γPM − γves) ΔA, where εadhesion, γPM, γves are the membrane-cytoskeleton adhesion energy, the PM tension, and the vesicle membrane tension, respectively (STAR Methods, Mathematical modeling).
In the late stages of the shrink-fusion sequence (Figure 5B), the model generated Λ shapes similar to those in STED images (Figure 3E). The Ω→Λ shape transition depended on the diameter w of the actin cortex-free zone where the vesicle joins the PM (Figure 5A). For example, if w = 80 nm, Ω→Λ transition occurred as vesicle diameter D shrank to ~56 nm (Figure 5B); the transition was delayed to D = 14 nm if w = 20 nm (Figures S5B–S5F). The merging of a Λ-shaped membrane with the PM is a full-collapse subsequence; thus, full-collapse shapes are realized only in the latest stages of the trajectory.
From the computed shapes (Figure 5B), we measured pore diameters, defined as the minimum width near the PM for Ω shapes, or the vesicle width at a fixed height for Λ shapes (Figure S5F). Down to a height of ~200 nm in the shrink-fusion sequence of Figure 5B, the pore size increases gradually from 16 to 23 nm and then increases more sharply to 42 nm before decreasing to zero during the Λ-shape sequence as the vesicle flattens into the PM (Figure S5F). The qualitative behavior is similar for larger diameters of the actin cortex-free zone w, but with somewhat larger pores (Figures S5B–S5F). These pore size changes were not observed with STED imaging, likely in part due to limited STED resolution. When PH-Ω’s pore was detected with STED microscopy, Porev often constricted during shrinkrelated fusion (Figures 3D–3F). This was likely due to dynamin-mediated constriction of the fusion pore, as recently reported (Shin et al., 2018), and/or changes in w during shrink fusion, both of which are beyond the scope of the present model, which focused on modeling shrinking of the entire Ω-profile.
For even larger vesicles (e.g., D > 400 nm at ΔP = 100 Pa), the model reproduced the remarkable tubular shapes that we observed in cells, a vivid consequence of squeezing forces (Figure 5E). Tubular vesicles had large height-to-width ratios and widths that decreased with height, asymptoting a pressure-dependent width for large heights (Figure 5F).
Agreeing with experiment, the model identified three vesicle classes: Ω-shaped, tubular-shaped, and Λ-shaped. In Figure 5F, model-predicted isobars (constant ΔP) are plotted in the vesicle height-width plane, together with the EM data of Figure 3C. Each experimental point belongs to one of the three vesicle shape sectors and to a certain isobar, illustrating how measurement of vesicle shape enables the model to predict the squeezing pressure on that vesicle. The data in Figure 5F suggest vesicles in cells were subject to squeezing pressures from ~40 to ~400 Pa.
Four specific model predictions are confirmed by experimental observations, considerably strengthening our model’s conclusions. (1) During shrink fusion, predicted vesicle shapes have mild prolateness (height greater than width) and a late transition to Λ-shape before flattening into the PM (Figure 5B). This is experimentally confirmed (Figures 1 and 3). (2) Tubular shapes are predicted for large squeezing pressures or vesicle size, a vivid manifestation of osmotic squeezing closely related to the spherical-to-prolate first-order transition previously predicted for closed vesicles under high squeezing pressure (Jenkins, 1977). Tubular-shaped vesicles were predicted to be shrinking (Figure 5E), consistent with our observations of large shrinking tubular vesicles (Figures 1 and 3). (3) Lowering the osmotic squeezing driving force by increasing extracellular osmolarity or (4) lowering the reeling-in force by compromising adhesion between the PM and the actin cortex is predicted to reduce the shrink-fusion frequency (Figure 5A). Both predictions were confirmed in a recent study, where increased extracellular osmolarity or inhibition of F-actin by latrunculin A, cytochalasin D, or actin knockout significantly reduced the percentage of shrink fusion detected by confocal imaging of fusion spots (Wen et al., 2016).
DISCUSSION
Recent imaging of dense-core vesicle fusion in chromaffin cells shows that fusion spots may shrink or enlarge in size and that F-actin-dependent PM tension is needed for shrinking (Chiang et al., 2014; Wen et al., 2016). The present work advanced over these studies in three aspects. First, with STED imaging of PHG-labeled fused vesicular membrane dynamics and the dynamics of FFN511 release from vesicular lumen in chromaffin cells, we directly visualized, and thus verified, two fusion modes associated with size changes: shrink fusion (or shrinkrelated fusion) and enlarge fusion (Figures 1, 2, and 3). Second, we found that shrinkrelated fusion is associated with a large pore that promotes rapid content release, whereas enlarge fusion is associated with a narrow pore that slows down content release (Figures 2, 3, and 4). These two fusion modes may contribute to explain diverse release rates reported in decades of studies that had interpreted release under the framework of full-collapse and kiss-and-run fusion modes (Saheki and De Camilli, 2012; Alabi and Tsien, 2013; Wu et al., 2014). Third, we discovered the underlying mechanism of shrink fusion; the swelling osmotic pressure maintained by cells squeezes the Ω-profile and abolishes the Ω-profile membrane tension, producing a tension gradient (from PM to Ω-profile) that reels Ω-profile membrane into the PM (Figure 5). The requirement of the PM-to-Ω-profile tension gradient explains why F-actin-dependent PM tension is needed to mediate shrink fusion, as suggested recently (Wen et al., 2016). The squeezing pressure is equal in magnitude to the swelling osmotic pressure of the cell (Diz-Muñoz et al., 2010; Boulant et al., 2011; Stewart et al., 2011; Tsujita et al., 2015; Wen et al., 2016), once the fused vesicle equilibrates with the extracellular medium (Figure 5A). With squeezing force and membrane-reeling-in force, Ω-profile shrinking is free energetically favored over full collapse (Figures 5, S5G, and S5H), challenging the traditional view that full collapse is the default fusion mode (Alabi and Tsien, 2013; Wu et al., 2014).
Our findings as summarized above, together with a recent finding that most fusion pore closure events (kiss-and-run) on average do not slow down content release (Shin et al., 2018), lead us to suggest that shrink and enlarge fusion are the yin and yang of fusion modes that control the rate of content release. This suggestion challenges the classical view that full collapse and kiss-and-run serve as the yin and yang fusion modes accounting for rapid and slow release observed in endocrine cells and neurons (Saheki and De Camilli, 2012; Alabi and Tsien, 2013; Wu et al., 2014). Decades of studies on the release rates (Saheki and De Camilli, 2012; Alabi and Tsien, 2013; Wu et al., 2014) that interpret exocytosis with the classical view may need to be re-examined with the fusion modes reported here.
Our finding that cells use shrink fusion instead of full collapse to promote content release has broad implications for two reasons. First, swelling osmotic pressure and membrane-actin cortex adhesion, both required for shrink fusion (Figure 5), are general properties of cells (Dai and Sheetz, 1999; Diz-Muñoz et al., 2010; Boulant et al., 2011; Stewart et al., 2011; Tsujita et al., 2015; Wen et al., 2016). Consequently, shrink fusion may serve as the fusion mode that facilitates exocytotic content release in many cell types. Second, shrink fusion may describe exocytosis of various sizes of vesicles, because Ω-profile shrinking was observed down to our STED resolution limit of ~60 nm (Figure 1). A recent study had speculated that as the width of the Ω-profile shrinks to an extent that is smaller than its pore, the Ω-profile may appear as a collapse-like Λ-shaped profile before flattening at the PM (Wen et al., 2016). Modeling and imaging in the present work showed that during the final stage of shrinking, the Ω-profile undergoes a transition to a Λ-profile (Figures 3 and 5). Indeed, such Λ-profiles, observed with EM, are the basis for the full-collapse fusion hypothesis (Heuser and Reese, 1981; Alabi and Tsien, 2013; Wu et al., 2014). Thus, we propose a shrink-collapse fusion mode in which Ω-profile shrinking is followed by a transition to a Λ-profile and then flattening to unify the apparently contradictory observations of shrink fusion and full collapse.
Although vesicles smaller than 60 nm, such as synaptic vesicles (~20–60 nm), are beyond our detection limit, it is likely that these vesicles display shrink collapse, with shrinking as the main component for larger vesicles (near 60 nm) and collapse as the main component for smaller vesicles (near 20 nm). It would be of great interest to verify this possibility in the future. The forces involved in mediating shrink fusion, as described here (Figure 5), could not account for same-size and enlarge fusion. Additional unknown mechanisms must be involved in mediating same-size and enlarge fusion.
The suggestion that shrink and enlarge fusion regulate content release by controlling the fusion pore is further supported by our finding that shrinkrelated fusion facilitates FFN511 and NPY-EGFP release, whereas enlarge-fusion slows down release of them (Figures 2 and 4). While we emphasized these two fusion modes in controlling fusion pore size and content release rate, other factors may also regulate fusion pore and release. For example, some fusion pore closure events (kiss-and-run) may release only a fraction of large protein molecules, such as NPY-EGFP and tissue plasminogen activator (Taraska et al., 2003; Perrais et al., 2004; Shin et al., 2018), although a large fraction of kiss-and-run events fully release contents (Alés et al., 1999; Shin et al., 2018). Fusion pore dynamics and lifetime are regulated by many factors, such as the vesicular lumen protein NPY, chromogranin A, tissue plasminogen activator, the vesicular membrane protein synaptotagmin, and the GTPase dynamin (Rao et al., 2014; Weiss et al., 2014; Rao et al., 2017; MacDougall et al., 2018; Shin et al., 2018; Abbineni et al., 2019). Whether and how these regulators affect shrink and enlarge fusion are unclear. Consequently, our mathematical model did not include these potential regulators but focused on elucidating the impacts of the biophysical properties of the vesicle and PM on shrink fusion. Although beyond the scope of the present work, it would be of interest to study how shrink and enlarge fusion are regulated by factors known to regulate fusion pore. The present work showed that overexpression of NPY-EGFP does not significantly affect the percentage of shrink and enlarge fusion, ruling out overexpressed NPY-EGFP as a regulator of shrink or enlarge fusion.
Various sizes of Ω-, tubular, and Λ-profiles have been reported in electron microscopy studies (Heuser and Reese, 1973, 1981). The present work revealed that these structures can originate from the dynamic processes of shrink, partial shrink, and enlarge fusion. For example, large Ω-profiles were previously presumed to result from bulk endocytosis, phagocytosis, or compound exocytosis of large vesicles formed by vesicle-vesicle fusion (Saheki and De Camilli, 2012; Wu et al., 2014; Kononenko and Haucke, 2015). We showed that they may alternatively originate from enlarge fusion. Enlarge fusion may not be limited to chromaffin cells; capacitance recordings in mast cells show that the capacitance up-step is sometimes followed by a larger down-step, suggesting that retrieved vesicles can be larger than fused vesicles (Monck et al., 1990). Fusion pore closure after enlarge fusion may thus be a mechanism to generate large endosome-like structures previously attributed to bulk endocytosis. What controls vesicle size is largely unclear; our work suggests that fusion pore closure after partial shrink fusion and enlarge fusion may produce different sizes of vesicles. The origin of tubular membrane structures is unclear and often attributed to bulk endocytosis (Saheki and De Camilli, 2012; Wu et al., 2014; Kononenko and Haucke, 2015). The present work reveals that partial shrink fusion, mediated by the osmotic squeezing forces, may generate tubular membrane shapes. Pore closure of these tubular structures may thus constitute a mechanism to generate tubular vesicles widely observed.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Ling-Gang Wu (wul@ninds.nih.gov). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary cell culture
Fresh adult (21 – 27 months old) male bovine adrenal glands purchased from a local abattoir (J. W. Treuth and Sons, Inc.) were used to prepare primary chromaffin cell culture, which are widely used for the study of exo- and endocytosis (Wu et al., 2014; Sharma and Lindau, 2018).
METHOD DETAILS
Chromaffin cell culture
We prepared primary bovine adrenal chromaffin cell culture as described previously (Chiang et al., 2014). Fresh adult (21 – 27 months old) bovine adrenal glands (from a local abattoir) were immersed in pre-chilled Locke’s buffer on ice containing: NaCl, 145 mM; KCl, 5.4 mM; Na2HPO4, 2.2 mM; NaH2PO4, 0.9 mM; glucose, 5.6 mM; HEPES, 10 mM (pH 7.3, adjusted with NaOH). Glands were perfused with Locke’s buffer, then infused with Locke’s buffer containing collagenase P (1.5 mg/ml, Roche), trypsin inhibitor (0.325 mg/ml, Sigma) and bovine serum albumin (5 mg/ml, Sigma), and incubated at 37°C for 20 min. The digested medulla was minced in Locke’s buffer, and filtered through a 100 μm nylon mesh. The filtrate was centrifuged (48 xg, 5 min), re-suspended in Locke’s buffer and re-centrifuged until the supernatant was clear. The final cell pellet was re-suspended in pre-warmed DMEM medium (GIBCO) supplemented with 10% fetal bovine serum (GIBCO).
Electroporation and plating
Cells were transfected by electroporation using Basic Primary Neurons Nucleofector Kit (Lonza), according to the manufacturer’s protocol and plated onto glass coverslips with mouse Laminin coating over PDL layer (Neuvitro). The cells were incubated at 37°C with 9% CO2 and used within 5 days.
Plasmids and fluorescent dyes
The PH-EGFP (phospholipase C delta PH domain attached with EGFP) was obtained from Dr. Tamas Balla. NPY-EGFP was purchased from Addgene. PH-mNeonGreen construct was created by replacing the EGFP tag of PH-EGFP with mNeonGreen (Allele Biotechnology) (Shaner et al., 2013). Both PH-EGFP and PH-mNeonGreen are abbreviated as PHG. For imaging, the dye concentration in the bath solution was: Atto 532 (A532, Sigma), 30 μM; Alexa 647 (A647, Sigma), 30 μM. For FFN511 (Abcam) imaging, cells were bathed with FFN511 (5–10 μM) for 10 min and images were performed after washing out FFN511 in the bath solution.
Overexpression of PHG or NPY-EGFP did not significantly affect the basic properties of exo- and endocytosis, because 1) whole-cell capacitance measurements and imaging show robust exo- and endocytosis, and similar percentages of close-fusion and non-close-fusion as control (Chiang et al., 2014; Zhao et al., 2016), and 2) Ω-profile’s pore could also be resolved by imaging extracellularly applied mCLING-A488 or by EM (Shin et al., 2018).
Electrophysiology
At room temperature (20 – 22°C), whole-cell voltage-clamp and capacitance recordings were performed with an EPC-10 amplifier together with the software lock-in amplifier (PULSE, HEKA, Lambrecht, Germany) (Lindau and Neher, 1988; Chiang et al., 2014). The holding potential was −80 mV. For capacitance measurements, the frequency of the sinusoidal stimulus was 1000 – 1500 Hz with a peak-to-peak voltage ≤ 50 mV. The bath solution contained 125 mM NaCl, 10 mM glucose, 10 mM HEPES, 5 mM CaCl2, 1 mM MgCl2, 4.5 mM KCl, 0.001 mM TTX and 20 mM TEA, pH 7.3 adjusted with NaOH. The pipette (2 – 4 MΩ) solution contained 130 mM Cs-glutamate, 0.5 mM Cs-EGTA, 12 mM NaCl, 30 mM HEPES, 1 mM MgCl2, 2 mM ATP, and 0.5 mM GTP, pH 7.2 adjusted with CsOH. These solutions pharmacologically isolated calcium currents.
For stimulation, we used a 1 s depolarization from the holding potential of −80 mV to +10 mV (depol1 s). We used this stimulus, because it induces robust exo-endocytosis as reflected in capacitance recordings (Figure 1A) (Engisch and Nowycky, 1998; Perrais et al., 2004; Chiang et al., 2014). In a fraction of experiments during FFN511 imaging, we used 10 pulses of 400-ms depolarization from −80 mV to +10 mV at 2 Hz.
Confocal image acquisition
Imaging with A647 and NPY-EGFP were performed with an inverted confocal microscope (TCS SP5II, Leica, Germany, 100 × oil objective, numerical aperture: 1.4), where A647 (30 μM in bath, Sigma) and NPY-EGFP were excited by a Diode laser at 640 nm (maximum power: 40 mW) and an Argon laser at 488 nm (maximum power: 50 mW), respectively. The 640 nm laser was set at 50% of the maximum power, which could bleach A647 inside the Ω-profile within a few seconds once the Ω-profile closed (Chiang et al., 2014). The 488 nm laser was set at 1.5–2% to avoid significant bleaching of NPY-EGFP. A647 fluorescence was collected with a photomultiplier at 650 – 720 nm, whereas NPY-EGFP, at 498 – 580 nm. Both excitation and fluorescence collection were done simultaneously at the XY/Zfix scanning mode at the cell bottom (XY-plane with Z focal plane fixed). Confocal imaging area was ~70 – 160 μm2 at the XY plane with a fixed Z axis focal plane ~100–200 nm above the cell-bottom membrane. Images were collected every 10–33 ms at 50 – 70 nm per pixel at the XY/Zfix scanning mode.
Fusion mode identification during confocal imaging
During confocal imaging of A647 (strong excitation, 50% maximal power) and NPY-EGFP (weak excitation: 1.5%–2% maximal power) at the cell-bottom, we identified fusion modes based on A647 imaging as characterized in detail in the main text (for detail, see Chiang et al., 2014). Upon fusion pore closure, continuous strong excitation of A647 gradually bleached A647 inside the closed Ω-profile, resulting in F647 decay to baseline with a time constant of ~2.9 s, during which the spot size remained unchanged (Chiang et al., 2014). Although shrink-fusion also reduced spot F647 to baseline, F647 decay was generally much faster (time constant: ~1.1 s), and the decay was accompanied by the spot width decrease (Figure 4B) (Chiang et al., 2014).
Pore closure detected with spot F647 bleaching by strong excitation is not due to a narrow pore smaller than A532 molecule size, because after spot dimming, bath application of an acid solution cannot quench the pH-sensitive VAMP2-EGFP or VAMP2-pHluorin overexpressed at the same spot, indicating that the spot is impermeable to H+ or OH−, the smallest molecules, and thus is closed (Chiang et al., 2014; Zhao et al., 2016). Furthermore, the closure time course calculated from spot dimming matches approximately with whole-cell endocytosis time course (Chiang et al., 2014), and inhibition of dynamin by dynamin inhibitors, dynamin dominant-negative mutant dynamin 1-K44A or dynamin knockdown blocks not only whole-cell endocytosis but also pore closure detected with the spot dimming method (Chiang et al., 2014; Zhao et al., 2016). These results further confirm that spot dimming under strong excitation reflects pore closure.
STED imaging
STED images were acquired with Leica TCS SP8 STED 3 × microscope that is equipped with a 100 × 1.4 NA HC PL APO CS2 oil immersion objective and operated with the LAS-X imaging software. Excitation was with a tunable white light laser and emission was detected with hybrid detectors. In time-gated STED mode, PH-EGFP and A532 were sequentially excited at 470 and 532 nm, respectively, with the 592 nm STED depletion beam, and their fluorescence collected at 475–525 nm and 540–587 nm, respectively. PH-mNeonGreen and A532 were sequentially excited at 485 and 540 nm, respectively, with the 592 nm STED depletion beam, and their fluorescence collected at 490–530 nm and 545–587 nm, respectively. PH-mNeonGreen and FFN511 were sequentially excited at 505 and 442 nm, respectively, with the 592 nm STED depletion beam, and their fluorescence collected at 510–587 nm and 447–490 nm, respectively.
The excitation power for A532 was 10% of the maximum, at which fluorescent A532 can be bleached within a few seconds. This feature was used to distinguish whether the fusion pore is closed or not, because pore closure prevents bleached A532 (caused by strong excitation) from exchange with fluorescent A532 in the bath, resulting in A532 spot fluorescence decay (Chiang et al., 2014). In contrast, an open pore would not cause A532 spot fluorescence decay, because an open pore allows for continuous exchange of bleached A532 in the Ω-profile with fluorescent A532 in the bath (Chiang et al., 2014).
STED imaging generally causes more photobleaching and phototoxicity. Severe phototoxicity could cause loss of the whole-cell giga seal during patch-clamp recording (Chiang et al., 2014). We avoided severe phototoxicity by applying only one depol1 s and imaging for only ~30–40 s per cell. With this setting, we have not noticed significant differences in the exo- and endocytosis properties obtained under confocal and STED imaging conditions (Chiang et al., 2014; Zhao et al., 2016). For imaging of PHG and A532, continuous exchange of bleached PHG or A532 with fluorescent ones from non-imaging areas lessened the photobleaching problem.
STED scanning modes
STED images were acquired at the cell bottom with the XZ (perpendicular to the coverslip) scanning mode with the y axis focal plane fixed at about the cell center (Figure 1A, XZ/Yfix scanning mode), at which images were acquired every 26–300 ms at 15 nm per pixel in an XZ area of 19.4 μm x 0.7–1.5 μm (Shin et al., 2018). We used 300 ms per XZ frame at the beginning of this project, but used 26–100 ms as we realized the importance of catching fast events with fast imaging rates.
The STED resolution for imaging PHG (PH-EGFP or PH-mNeonGreen) in our conditions was ~60 nm on the microscopic X- and y axis (parallel to cell-bottom membrane or coverslip), and ~150–200 nm on the microscopic Z axis. We observed on average about 0.2 PH-Ω-profile with clear outline per XZ/Yfix scan after depol1 s. STED images were deconvolved using Huygens software (Scientific Volume Imaging).
Electron microscopy
Bovine chromaffin cells were stimulated for 90 s using a solution containing 70 mM KCl, 60 mM NaCl, 10 mM glucose, 10 mM HEPES, 2 mM CaCl2, and 1 mM MgCl2 (pH 7.3). The cells were then immediately fixed with 2% glutaraldehyde, 2% paraformaldehyde and 4% tannic acid in 0.1 M cacodylate buffer (pH 7.4) solution for 15 min, followed by the same fixative without tannic acid for additional 15 min. The cells were washed first with a solution of 100 mM glycine in 0.1 M sodium cacodylate buffer for 1 min at room temperature, and then with 0.1 M sodium cacodylate. After post-fixation in 1% OsO4 for 1 h on ice, the samples were dehydrated in ethanol series. During dehydration the specimens were stained with 2% uranyl acetate in 70% ethanol for 30 min following 70% ethanol step. All preparations were embedding in Embed-812 resin (EMS). 70–90 nm ultrathin sections were cut in an ultramicrotome with Diamond knife (Diatome). The sections were placed onto pioloform-coated 200 mesh grids. Images were collected at up to 73,000x magnification in a JEOL JEM-200CX transmission electron microscope (voltage used 120kV) equipped with an AMT XR-100 CCD camera.
Mathematical modeling
Estimating pressure equilibration time
We estimate here an upper bound of the time required, during shrink fusion, for the pressure inside the fused vesicle to decrease and become equal to the extracellular pressure. The equilibration is assumed unhindered by vesicular contents, which are assumed released before shrink fusion commences (see main text). During this equilibration, a small volume of fluid flows out of the vesicle and the membrane tension of the fused vesicle decreases simultaneously. This tension change is due to an increase in lipid density at a constant number of lipids, as lipid flow from the vesicle to the plasma membrane is much slower than the flow of the content fluid (Shi et al., 2018). We calculate the timescale of tension relaxation below, which is the same as the timescale for pressure relaxation as the two are related according to Laplace’s law.
We first estimate the volume change ΔVves needed to let the vesicle membrane tension relax to zero. We assume a linear stress-strain relation for the membrane tension (Hochmuth et al., 1973):
where Em is the area expansion modulus of the vesicle membrane and ρ0 is the lipid density under zero tension.
We assume the total number of lipids does not change as stated earlier, so the area of the membrane is inversely proportional to lipid density. Then the area change is given by ΔAves = − AiΔγ/Em, where Ai is the initial membrane area. For simplicity we assume the vesicle remains a sphere, so the corresponding change in volume ΔVves to completely abolish the tension is given by
where Di is the initial vesicle diameter and γi the pre-fusion vesicle tension. This volume change is relatively small compared to the initial vesicle volume Vi as ΔVves/Vi≈γi/Em ≪ 1.
The flux Q of the content fluid through a fusion pore with a diameter Dpore is given by , where is the mean vertical velocity across the cross-section of the fusion pore. This flux is caused by the high initial vesicle pressure ΔPi, set by initial tension γi following Laplace’s law ΔPi = 4γi=Di. The mean velocity is estimated by the Stokes equation (ignoring inertial terms) , where η is the viscosity of the content fluid. Thus, the flux is given by
The pressure relaxation time τrelax is given by
We use the following parameter values from experiment: Di = 350 nm, the mean diameter of large dense-core vesicles in chromaffin cells (Plattner et al., 1997); η = 1.1 cP, the measured cytoplasmic viscosity in fibroblast cells (Bicknese et al., 1993); and Em = 250 pN nm−1, measured expansion modulus of phosphatidylcholine bilayers (Rawicz et al., 2000). For a relaxation time of ~1 ms, Dpore ~4 nm. This is an upper bound for the relaxation time as larger pores have been observed (Figure 3D). These have much smaller relaxation times as .
Calculation of fused vesicle shapes
Our procedure yields the shape of a fused vesicle corresponding to an area Aves, an actin cortex-free zone of diameter w, and a squeezing pressure ΔP (Table 1). Such a shape has a local balance of forces and moments, which are produced by osmotic pressure ΔP, membrane tension γves, and force q that arises from membrane bending. The membrane bending energy is given by (the integral is over the entire surface of the vesicle) (Helfrich, 1973), where k is the bending modulus and H is the mean curvature. These force and moment balance equations stated below were derived by Jenkins (1977).
We assume the vesicle shape is axisymmetric with the vertical axis being the symmetry axis. We numerically solve a system of six first order equations for the height coordinate z, the cross-sectional radius r, the angle between the tangent plane and the z axis ϕ, mean curvature H, force that arises from bending q, and membrane tension γves. All the six variables (except ϕ) as well as the parameter ΔP are nondimensionalized as
where . These quantities are obtained as a function of an angle S as defined by Jenkins (Jenkins, 1977), which ranges from 0 to π, where S = 0 and π denote the top and the bottom of the fused vesicle respectively (the vesicle membrane joins the cytoskeleton-attached plasma membrane at the bottom). The six first order equations are
for 0≤S≤π. Of the following six boundary conditions, the first is vanishing force at and the others are geometric constraints.
Here, is the normalized diameter of the cortex-free zone.
These differential equations are solved using the solver ‘bvp4c’ in MATLAB® using an absolute error tolerance of 10−4 and a relative error tolerance of 10−2 (MathWorks, Natick, MA). We get the sequence of shapes and tensions along one isobar by solving these equations for different Rves values, and by repeating this procedure for different ΔP values, several isobars were achieved (Figure 5F). For one isobar, the vesicle adopts Ω shapes for small sizes and tubular ones for large sizes. The Ω-tubular boundary is defined as points where the vesicle width is a maximum for a given pressure value (Figure 5F, dashed line).
Evaluation of free energy changes
We estimate the free energy change during shrink-fusion using the shapes and tensions of a fused vesicle at a constant squeezing pressure calculated as described above. For each shape which has area Aves and volume Vves, the squeezing pressure ΔP = Pcell − Pves and the vesicle membrane tension γves are related to partial derivatives of the membrane bending energy Fbend (Seifert et al., 1991),
The evaluation of the free energy change during shrink-fusion involves not only the membrane bending energy Fbend, but also the contribution from the vesicle interior (whose free energy is denoted as ), the cell interior , and the cell membrane . The total free energy F is the sum of the above four free energies, given by
where Vcell is the cell volume and Acell is the cell membrane area. We assume the chemical potential of the content molecules is the same between the vesicle and the extracellular solution, hence their free energy does not appear in F. The differential of F is given by
where γ is the sum of plasma membrane tension γPM and membrane-cortex adhesion energy density εadhesion (Table 1).
As total fluid volume and membrane area are conserved, dVves + dVcell = 0 and dAves + dAcell = 0. Thus, the above equation can be simplified as
Integrating, we get the free energy of a fused vesicle with area A
The reference area A0 is πD02 where D0 = 42 nm. We numerically calculated the free energy change as shown in Figure 5C using the above equation with the equilibrated vesicle shapes and tensions γves calculated as described before at a constant squeezing pressure ΔP = 100 Pa.
Free energy landscape of fusion pore dilation
We developed a Monte Carlo (MC) method to calculate the minimum energy shapes of vesicles with a fixed pore diameter, which allows determination of free energy landscape of fusion pore expansion by measuring the forces to expand or constrain the fusion pore. The contour of the axially symmetric vesicle is described by a function r(z) (r and z are defined same as in the previous section) and discretized into N points ((zi,ri), i = 1, 2, …N) which are uniformly distributed along the z axis. The MC method seeks the shape that minimizes the vesicle free energy
where two quadratic terms serve to constrain the vesicle membrane area Aves and fusion pore diameter dpore to their set values and (Fbend, Vves and ΔP are defined same as in the previous section). The vesicle membrane tension is given by and the force to constrain the fusion pore , which are obtained from partial derivatives of Fbend. As one expands the fusion pore, the variation of the free energy of the vesicle with respect to fusion pore diameter d is given by
where d0 is the fusion pore diameter of the reference state.
The simulation investigates the free energy landscape of fusion pore expansion of a fused vesicle with effective diameter D = 200 nm (Aves = πD2) with subject to a squeezing pressure ΔP = 100 Pa, and the reference state is dpore = 6 nm. The vesicle contour is discretised into N = 401 points. To ensure the calculated vesicle area and fusion pore diameter fall within ± 1% of the set value, kmembrane = 4 ×10−6 pN nm−3 and kpore = 103 pN nm−1 were chosen. The simulation runs at zero temperature and a move that generates a new shape will be accepted only if it leads to a lower free energy Fves. Each move is either a local radial change (neighboring ri) with a maximum displacement of 10−4 nm or a change in vesicle height of 10−4 nm (all zi scale with the same factor). All the simulations are equilibrated after 2×108 MC steps as in the final 2×107 steps Fves only decreases by ~0.05 kBT.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data selection
For every cell recorded with a pipette under the whole-cell configuration, the data within the first 2 min at the whole-cell configuration were used, which avoided rundown of endocytosis (gradual disappearance of endocytosis) as previously reported under the whole-cell configuration for a long time (Smith and Neher, 1997; Chiang et al., 2014). Cells expressed with PHG (or PH-mCherry) were used for visualization of PH-Ω. The criteria for selecting PH-Ωs for analysis during XZ/Yfix scanning are described in Figure S2 of Shin et al., 2018.
Visible pores of PH-Ω (Porev) containing A532 at the XZ frame are identified based on two criteria: 1) the Ω-profile pore is visible as judged by eyes and A532 spot is inside the Ω-profile, and 2) the FPH line profile across the pore region shows a V-shape valley with an amplitude at least three times larger than the standard deviation of the same FPH line profile before fusion (for detail, see Shin et al., 2018).
Analysis of PH-Ω structural changes
STED images of PH-Ω were analyzed with ImageJ and LAS X (Leica). While Ω-profiles labeled with PHG, but filled with no A532 may appear due to hemi-fusion (Zhao et al., 2016), we only analyzed Ω-profiles labeled with PH and filled with A532 throughout the work.
During XZ/Yfix scanning, some depol1 s-induced PH-Ω-profiles were out of the Yfix focal plane, as the outline of the Ω-profile was vague or unclear (for detail, see Shin et al., 2018). These out-of-focus Ω-profiles were not included for analysis. The criteria for the selection of the Ω-profile are described in detail in Figure S2 of Shin et al., 2018. PH-Ω’s width and height were measured as described in Figure S1C of the present work.
Pores labeled with PHG were identified based on the image and the fluorescence intensity line profile. We first identified the fluorescently labeled Ω-profiles with an open pore, the edge of which was continuous with the plasma membrane. The intensity line profile in the pore region should show a valley with a peak at least three times larger than the baseline fluctuation (standard deviation) in the non-pore region (for detail, see Shin et al., 2018). The full-width-half-maximum of the valley of the intensity line profile across the pore (Figure S3B) was proportional to the pore diameter, as shown with simulation (Shin et al., 2018). Thus, this method was used for pore size measurements.
The percentage of Porev was calculated as the number of PH-Ω showing a Porev divided by the total number of PH-Ω induced by a depol1 s during a XZ/Yfix scanning (~30–40 s) in each cell. Normally we observed 0 or 1 PH-Ω per cell during a XZ/Yfix scanning. Occasionally, we observed 2 PH-Ω. Thus, the percentage of Porev in a recorded cell was either 0%, 50% or 100%. These individual values were averaged to obtain the mean ± s.e.m. for the percentage of Porev.
During XZ/Yfix imaging, A532 was excited at a high laser power so that fluorescent A532 can be bleached with a time constant of 1.5–3.5 s. Pore closure was identified as the gradual dimming of the A532 spot fluorescence to baseline during XZ/Yfix PHG/A532 imaging while PHG image remained unchanged or dimmed gradually without changing the PH-Ω size (Figure S3A) (Shin et al., 2018).
Our STED XZ/Yfix sampling was 26–300 ms per frame. The width of the PH-mNG-labeled Ω-profile could be measured within 50–300 ms after fusion. Thus, we measured the PH-mNG-labeled Ω-profile’s width from 50–300 ms after the fusion onset to 20–30 s afterward. If the width was increased by more than 15% within the recording time period (~20–30 s), it was defined as the enlarge-fusion mode.
Confocal image analysis
Confocal images were analyzed with ImageJ and LAS X (Leica). The fluorescence intensity from an area covering the fluorescence spot was measured at every image frame. The spot width was measured as the full-width-half-maximum from intensity profiles of 1 – 4 lines across the spot center. The time course of NPY-EGFP release was quantified as the time the NPY-EGFP spot fluorescence decayed from 20% to 80% of the maximal decrease (TNPY_decay). Similarly, the 20%–80% risetime of A647 spot fluorescence (T647_rise) was quantified; the 20%–80% decay time of A647 spot fluorescence during shrink-fusion was quantified after A647 spot fluorescence reached the peak.
Statistical tests
Data were expressed as mean ± s.e.m. Replicates are indicated in results and figure legends. N represents the number of cells, pores, or Ω-profiles as indicated in results and figure legends. The statistical test used is t test or ANOVA. Although the statistics were performed based on the number of cells or pores, and Ω-profiles, each group of data was collected from at least four primary chromaffin cell cultures. Each culture was from at least two glands from one bovine.
DATA AND CODE AVAILABILITY
All experimental data produced for this manuscript are available from the Lead Contact (wul@ninds.nih.gov) upon reasonable request. All data and code for the computational analysis are available from the corresponding author (bo8@columbia.edu) upon reasonable request.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM medium | GIBCO | Cat#11885092 |
| Fetal bovine serum | GIBCO | Cat#10082147 |
| German glass coverslips with mouse Laminin coating over PDL layer | Neuvitro | GG-25-Laminin |
| Atto 532 | Sigma | 88793 |
| Alexa 647 | Sigma | A20502 |
| Trypsin inhibitor | Sigma | T9253; CAS: 9035-81-8 |
| Bovine serum albumin | Sigma | A2153; CAS: 9048-46-8 |
| Embed-812 | EMS | Cat#13940 |
| Collagenase P | Sigma | COLLP-RO Roche 11249002001 |
| Critical Commercial Assays | ||
| Basic Primary Neurons Nucleofector Kit | Lonza | Cat#VVPI-1003 |
| Experimental Models: Primary Cell Culture | ||
| Bovine adrenal chromaffin cell | J. W. Treuth and Sons, Inc. | Adrenal Glands |
| Recombinant DNA | ||
| PH-EGFP | Varnai and Balla, 1998 | Addgene Plasmid #51407 |
| mNeonGreen | Allele Biotechnology | ABP-FP-MNEONSB |
| PH-mNeonGreen | Shin et al., 2018 | Allele Biotechnology |
| NPY-EGFP | Taraska et al., 2003 | Addgene Plasmid #74629 |
| Software and Algorithms | ||
| Huygens Professional | Scientific Volume Imaging | https://svi.nl/Huygens-Professional |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| LAS X | Leica | https://www.leicabiosystems.com/ |
| Igor Pro | WaveMetrics | https://www.wavemetrics.com/products/igorpro |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Python v3.5.2 | Python Core Team | https://www.python.org |
Highlights.
Visualizing fused vesicular membrane shrinking and enlargement with STED microscopy
Shrinking is mediated by physiological osmotic pressure that squeezes fused vesicles
Vesicle shrinking facilitates content release by employing a large fusion pore
Vesicle enlargement slows down content release by employing a small fusion pore
ACKNOWLEDGMENTS
This work was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program (ZIA NS003009-15 and ZIA NS003105-10 to L.G.W.); the National Institute of General Medical Sciences of the National Institutes of Health (NIH) award number R01GM117046 to B.O.; the Swedish Research Council (grant 1501), Hjärnfonden, and Russian Science Foundation grant 16-15-10273 to O.S.; and the National Institute of Biomedical Imaging and Bioengineering Intramural Research Program (ZIA EB000015-12 to A.J.). G.A. received stipends and support from the Karolinska Institutet—NIH Doctoral Partnership Program in Neuroscience, Fernström’s Stiftelse, and Karolinska Institutet funds. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We acknowledge computing resources from Columbia University’s Shared Research Computing Facility project. We thank Carolyn Smith for stimulated emission depletion (STED) microscopy support and Susan Cheng (National Institute of Neurological Disorders and Stroke) and Christopher Bleck (National Heart, Lung, and Blood Institute) for electron microscopy (EM) support.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.044.
REFERENCES
- Abbineni PS, Axelrod D, and Holz RW (2018). Visualization of expanding fusion pores in secretory cells. J. Gen. Physiol 150, 1640–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbineni PS, Bittner MA, Axelrod D, and Holz RW (2019). Chromogranin A, the major lumenal protein in chromaffin granules, controls fusion pore expansion. J. Gen. Physiol 151, 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabi AA, and Tsien RW (2013). Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Annu. Rev. Physiol 75, 393–422. [DOI] [PubMed] [Google Scholar]
- Alés E, Tabares L, Poyato JM, Valero V, Lindau M, and Alvarez de Toledo G (1999). High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nat. Cell Biol 1, 40–44. [DOI] [PubMed] [Google Scholar]
- Bicknese S, Periasamy N, Shohet SB, and Verkman AS (1993). Cytoplasmic viscosity near the cell plasma membrane: measurement by evanescent field frequency-domain microfluorimetry. Biophys. J 65, 1272–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner MA, Aikman RL, and Holz RW (2013). A nibbling mechanism for clathrin-mediated retrieval of secretory granule membrane after exocytosis. J. Biol. Chem 288, 9177–9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghi N, and Brochard-Wyart F (2007). Tether extrusion from red blood cells: integral proteins unbinding from cytoskeleton. Biophys. J. 93, 1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S, Kural C, Zeeh JC, Ubelmann F, and Kirchhausen T (2011). Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat. Cell Biol 13, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CW, Chiang CW, and Jackson MB (2017). Fusion pores and their control of neurotransmitter and hormone release. J. Gen. Physiol 149, 301–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HC, Shin W, Zhao WD, Hamid E, Sheng J, Baydyuk M, Wen PJ, Jin A, Momboisse F, and Wu LG (2014). Post-fusion structural changes and their roles in exocytosis and endocytosis of dense-core vesicles. Nat. Commun 5, 3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, and Sheetz MP (1999). Membrane tether formation from blebbing cells. Biophys. J 77, 3363–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diz-Muñoz A, Krieg M, Bergert M, Ibarlucea-Benitez I, Muller DJ, Paluch E, and Heisenberg CP (2010). Control of directed cell migration in vivo by membrane-to-cortex attachment. PLoS Biol. 8, e1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engisch KL, and Nowycky MC (1998). Compensatory and excess retrieval: two types of endocytosis following single step depolarizations in bovine adrenal chromaffin cells. J. Physiol 506, 591–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubernator NG, Zhang H, Staal RG, Mosharov EV, Pereira DB, Yue M, Balsanek V, Vadola PA, Mukherjee B, Edwards RH, et al. (2009). Fluorescent false neurotransmitters visualize dopamine release from individual presynaptic terminals. Science 324, 1441–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Wu XS, Mohan R, and Wu LG (2006). Two modes of fusion pore opening revealed by cell-attached recordings at a synapse. Nature 444, 102–105. [DOI] [PubMed] [Google Scholar]
- Helfrich W (1973). Elastic properties of lipid bilayers: theory and possible experiments. Z. Naturforsch C 28, 693–703. [DOI] [PubMed] [Google Scholar]
- Heuser JE, and Reese TS (1973). Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J. Cell Biol 57, 315–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, and Reese TS (1981). Structural changes after transmitter release at the frog neuromuscular junction. J. Cell Biol 88, 564–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochmuth RM, Mohandas N, and Blackshear PL Jr. (1973). Measurement of the elastic modulus for red cell membrane using a fluid mechanical technique. Biophys. J 13, 747–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, and Fasshauer D (2012). Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins JT (1977). Static equilibrium configurations of a model red blood cell. J. Math. Biol 4, 149–169. [DOI] [PubMed] [Google Scholar]
- Kononenko NL, and Haucke V (2015). Molecular mechanisms of presynaptic membrane retrieval and synaptic vesicle reformation. Neuron 85, 484–496. [DOI] [PubMed] [Google Scholar]
- Lindau M, and Neher E (1988). Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflugers Arch. 411, 137–146. [DOI] [PubMed] [Google Scholar]
- MacDougall DD, Lin Z, Chon NL, Jackman SL, Lin H, Knight JD, and Anantharam A (2018). The high-affinity calcium sensor synaptotagmin-7 serves multiple roles in regulated exocytosis. J. Gen. Physiol 150, 783–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monck JR, Alvarez de Toledo G, and Fernandez JM (1990). Tension in secretory granule membranes causes extensive membrane transfer through the exocytotic fusion pore. Proc. Natl. Acad. Sci. USA 87, 7804–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm GJ, Zdanov A, Gaitanaris GA, Stauber R, Pavlakis GN, and Wlodawer A (1997). The structural basis for spectral variations in green fluorescent protein. Nat. Struct. Biol 4, 361–365. [DOI] [PubMed] [Google Scholar]
- Perrais D, Kleppe IC, Taraska JW, and Almers W (2004). Recapture after exocytosis causes differential retention of protein in granules of bovine chromaffin cells. J. Physiol 560, 413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner H, Artalejo AR, and Neher E (1997). Ultrastructural organization of bovine chromaffin cell cortex-analysis by cryofixation and morphometry of aspects pertinent to exocytosis. J. Cell Biol 139, 1709–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat-Shliom N, Milberg O, Masedunskas A, and Weigert R (2013). Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell. Mol. Life Sci. 70, 2099–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TC, Passmore DR, Peleman AR, Das M, Chapman ER, and Anantharam A (2014). Distinct fusion properties of synaptotagmin-1 and synaptotagmin-7 bearing dense core granules. Mol. Biol. Cell 25, 2416–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TC, Santana Rodriguez Z, Bradberry MM, Ranski AH, Dahl PJ, Schmidtke MW, Jenkins PM, Axelrod D, Chapman ER, Giovannucci DR, and Anantharam A (2017). Synaptotagmin isoforms confer distinct activation kinetics and dynamics to chromaffin cell granules. J. Gen. Physiol 149, 763–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawicz W, Olbrich KC, McIntosh T, Needham D, and Evans E (2000). Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J 79, 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheki Y, and De Camilli P (2012). Synaptic vesicle endocytosis. Cold Spring Harb. Perspect. Biol 4, a005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert U, Berndl K, and Lipowsky R (1991). Shape transformations of vesicles: Phase diagram for spontaneous- curvature and bilayer-coupling models. Phys. Rev. A 44, 1182–1202. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, et al. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 10, 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, and Lindau M (2018). The fusion pore, 60 years after the first cartoon. FEBS Lett. 592, 3542–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Graber ZT, Baumgart T, Stone HA, and Cohen AE (2018). Cell membranes resist flow. Cell 175, 1769–1779.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin W, Ge L, Arpino G, Villarreal SA, Hamid E, Liu H, Zhao WD, Wen PJ, Chiang HC, and Wu LG (2018). Visualization of membrane pore in live cells reveals a dynamic-pore theory governing fusion and endocytosis. Cell 173, 934–945.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, and Neher E (1997). Multiple forms of endocytosis in bovine adrenal chromaffin cells. J. Cell Biol 139, 885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MP, Helenius J, Toyoda Y, Ramanathan SP, Muller DJ, and Hyman AA (2011). Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 469, 226–230. [DOI] [PubMed] [Google Scholar]
- Taraska JW, Perrais D, Ohara-Imaizumi M, Nagamatsu S, and Almers W (2003). Secretory granules are recaptured largely intact after stimulated exocytosis in cultured endocrine cells. Proc. Natl. Acad. Sci. USA 100, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Schulze U, Salbreux G, Roensch J, Joanny JF, and Paluch E (2009). Role of cortical tension in bleb growth. Proc. Natl. Acad. Sci. USA 106, 18581–18586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujita K, Takenawa T, and Itoh T (2015). Feedback regulation between plasma membrane tension and membrane-bending proteins organizes cell polarity during leading edge formation. Nat. Cell Biol 17, 749–758. [DOI] [PubMed] [Google Scholar]
- Varnai P, and Balla T (1998). Visualization of Phosphoinositides That Bind Pleckstrin Homology Domains: Calcium- and Agonist-induced Dynamic Changes and Relationship to Myo-[3H]inositol-labeled Phosphoinositide Pools. J Cell Biol. 143, 501–510. 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss AN, Anantharam A, Bittner MA, Axelrod D, and Holz RW (2014). Lumenal protein within secretory granules affects fusion pore expansion. Biophys. J 107, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PJ, Grenklo S, Arpino G, Tan X, Liao HS, Heureaux J, Peng SY, Chiang HC, Hamid E, Zhao WD, et al. (2016). Actin dynamics provides membrane tension to merge fusing vesicles into the plasma membrane. Nat. Commun 7, 12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Hamid E, Shin W, and Chiang HC (2014). Exocytosis and endocytosis: modes, functions, and coupling mechanisms. Annu. Rev. Physiol 76, 301–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WD, Hamid E, Shin W, Wen PJ, Krystofiak ES, Villarreal SA, Chiang HC, Kachar B, and Wu LG (2016). Hemi-fused structure mediates and controls fusion and fission in live cells. Nature 534, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental data produced for this manuscript are available from the Lead Contact (wul@ninds.nih.gov) upon reasonable request. All data and code for the computational analysis are available from the corresponding author (bo8@columbia.edu) upon reasonable request.