

Abstract
Due to the massive data sets available for drug candidates, modern drug discovery has advanced to the big data era. Central to this shift is the development of artificial intelligence approaches to implementing innovative modeling based on the dynamic, heterogeneous, and large nature of drug data sets. As a result, recently developed artificial intelligence approaches such as deep learning and relevant modeling studies provide new solutions to efficacy and safety evaluations of drug candidates based on big data modeling and analysis. The resulting models provided deep insights into the continuum from chemical structure to in vitro, in vivo, and clinical outcomes. The relevant novel data mining, curation, and management techniques provided critical support to recent modeling studies. In summary, the new advancement of artificial intelligence in the big data era has paved the road to future rational drug development and optimization, which will have a significant impact on drug discovery procedures and, eventually, public health.
Keywords: artificial intelligence, big data, deep learning, machine learning, rational drug design, computer-aided drug discovery
INTRODUCTION
Drug research and development is a complex, expensive, time-consuming procedure and has a high attrition rate (1). Drug attritions that happen in clinical studies induce great resource loss, and currently, nine out of ten drug candidates fail between phase I clinical trials and regulatory approval (2). Compared to traditional animal models, both in vitro and in silico approaches have great potential to lower the cost of drug discovery. The application of in vitro and in silico protocols in the early stages of the drug research and development procedure can reduce the number of drug attritions by identifying drug candidates with suitable therapeutic activities and excluding unsuitable compounds with undesirable side effects (3–6). However, the results of in vitro and in silico testing normally have low correlations to drug activities in vivo, especially for efficacy and complex side effects (7, 8).
Artificial intelligence (AI), which is sometimes presented as machine intelligence, refers to the ability of computers to learn from existing data. Computational modeling based on AI is a promising method to evaluate compounds for their potential biological activities and toxicities. Existing computational models, such as those based on quantitative structure-activity relationship (QSAR) approaches (9), can be used to quickly predict large numbers of new compounds for various biological end points. The existing models (e.g., those available in commercial drug discovery software) can make predictions of simple physicochemical properties (e.g., logP and solubility) and thus are relatively precise in predicting the pharmacokinetic properties of new compounds with simple mechanisms; however, the models for complex biological properties (e.g., drug efficacy and side effects) are far from optimal (8, 10) (Figure 1). Critical issues existed in previous QSAR modeling studies such as the use of small training sets (11), experimental data errors in training sets (12, 13), and a lack of experimental validations (14). The resulting QSAR model predictions of new compounds were questionable due to their coverage of a limited chemical space (15), existing activity cliffs (16), and overfitting (17, 18). The primary hypothesis of QSAR modeling (i.e., similar compounds will have similar activities) sometimes proved to be flawed (10, 19–21), indicating that training sets with only chemical structure information and target activity are not enough to answer the above challenges.
Figure 1.
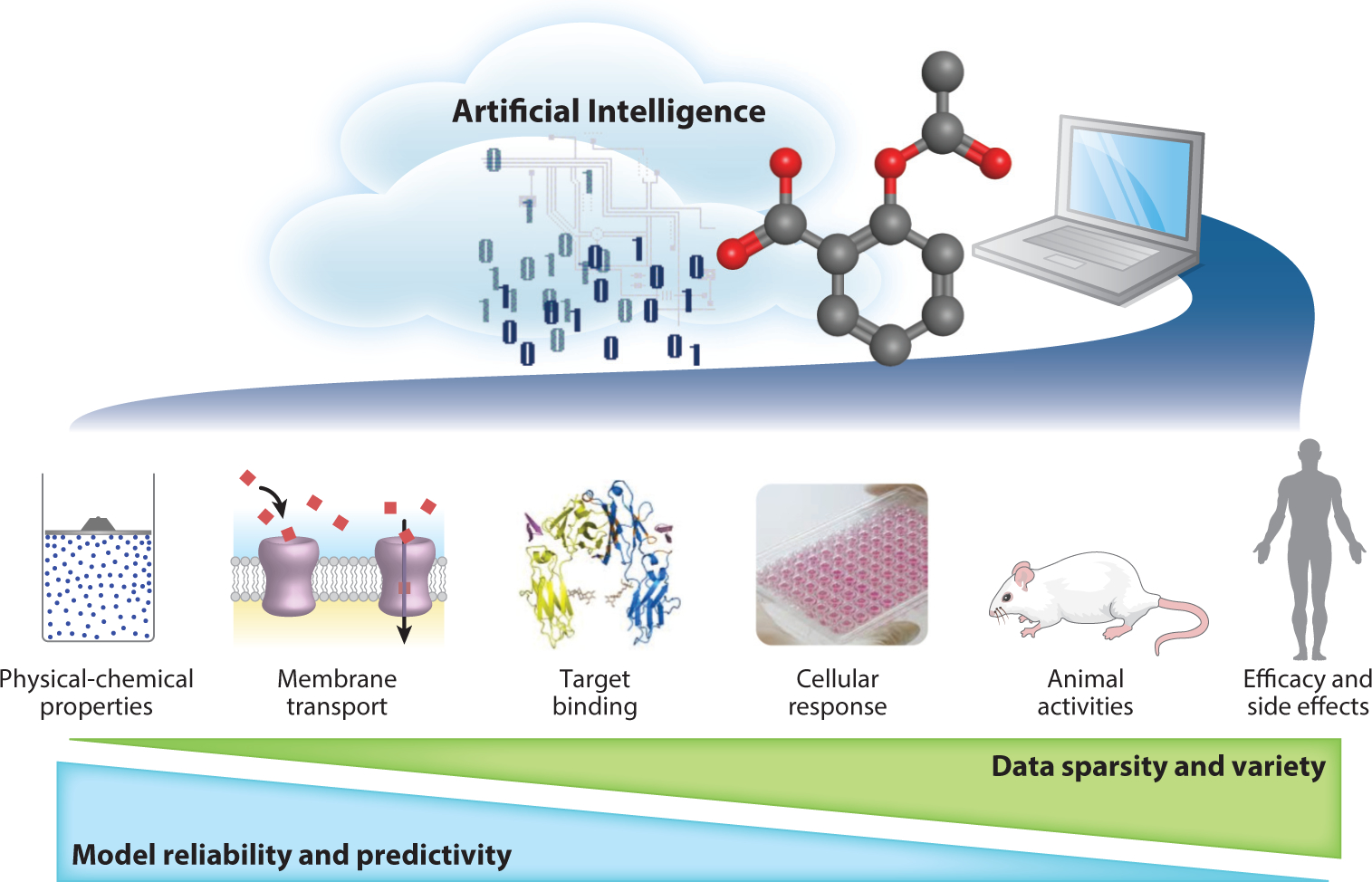
Challenges of data-driven artificial intelligence modeling in modern, computer-aided drug discovery.
With the great progress of combinatorial chemistry since the 1990s, large chemical libraries have become the major source of new chemical development procedures (22, 23). Over the past ten years, this effort has also stimulated the development of high-throughput screening (HTS) techniques (24–26). HTS is a process that screens thousands to millions of compounds using a rapid and standardized protocol. Current HTS techniques are usually combined with robotic methods and require few resources to test a chemical library. Parallel HTS data processing and assay miniaturization have become increasingly popular in pharmaceutical industries and regulatory agencies as they greatly reduce the cost of experimental testing (27, 28). The chemical-response data obtained from HTS keep growing daily and contribute to the current big data environment. Facilitated by the combined efforts of HTS and combinatorial chemical synthesis, modern screening programs produce enormous amounts of biological data, especially regarding drug responses on specific targets (29, 30).
The challenges raised by big data are known as the “four Vs”: volume (scale of data), velocity (growth of data), variety (diversity of sources), and veracity (uncertainty of data) (31, 32). The data sets available for drug development, especially in pharmaceutical industries, may involve many compounds (e.g., from 100,000 to several million) that were tested against many targets (33), and traditional QSAR modeling and machine learning approaches are not always suited to dealing with these types of data under these conditions. Furthermore, the uncertainty of available data (or data sparsity) is one of the major obstacles of using big data (32). Unfortunately, when coupled with more complex biological mechanisms such as drug responses, the sparsity and variety of the resulting data increased dramatically from in vitro to in vivo studies (Figure 1). This big data scenario necessitated the development of new computational approaches to deal with high-volume, multidimensional, and high-sparsity data sources to predict drug efficacy and side effects in animals and/or humans.
The challenges to using big data discussed above and the involvement of new types of data (e.g., images) have demanded the recent development of novel AI approaches to advance predictive modeling in modern drug discovery (34–36). The popular AI approaches in the current big data era are based on deep learning (3, 4, 37). One of the early efforts of applying deep learning in the drug discovery process in pharmaceutical industries was the 2012 QSAR machine learning challenge supported by Merck (38). In this challenge, deep learning models showed significantly better predictivity than traditional machine learning approaches for 15 absorption, distribution, metabolism, and excretion (ADME) and toxicity data sets for drug candidates developed at Merck. Since then, and with the development of neural network approaches [e.g., convolutional neural networks (CNNs)], deep learning has been widely applied to drug discovery approaches. Although still viewed as a black box algorithm (39, 40), the current progress of AI supported by deep learning has shown great promise in rational drug discovery in this era of big data. The big data challenges; relevant AI developments; and modeling for drugs and drug candidates, especially those studies using deep learning and other new techniques, are the primary focus of this review.
BIG DATA IN DRUG DISCOVERY
The term big data describes a collection of data sets that are so large and complex that they are too difficult to process with traditional data analysis tools (41). Big data is gaining increasing recognition in clinical studies and other research areas driven by biological data (42, 43). As one of the fields generating a massive amount of data, modern drug discovery has moved into the big data era. The need for novel computational techniques, including data mining/generation, curation, storage, and management, brings new challenges and opportunities to the research community.
Several data-sharing projects, in parallel with the developments of HTS techniques in various screening centers, were also initiated in the past ten years. For example, PubChem is a public repository for chemical structures and their biological properties (44–46). In ten years, the number of PubChem compounds increased from 25 million in 2008 (46) to 96 million in 2018 (47). During the same period, the number of bioassays that were deposited into PubChem increased from 1,197 in 2008 (46) to over a million in 2018 (47). The current statistics of PubChem indicate that the repository contains 97.3 million compounds and 1.1 million bioassays (https://pubchem.ncbi.nlm.nih.gov). The tremendous amount of PubChem bioassay data that are updated daily constitutes a publicly accessible big data resource for compounds, including most drugs and drug candidates, with a variety of target response information. Similar to PubChem, ChEMBL is a database containing binding, functional, ADME, and toxicity data for numerous compounds (48). Compared to PubChem, ChEMBL contains a large amount of manually curated data from the literature. Currently, the ChEMBL database consists of over 2.2 million compounds tested against over 12,000 targets, resulting in activity data for 15 million compound-target pairs (https://www.ebi.ac.uk/chembl/).
Several other data sources are specifically designed for drugs and drug candidates. For example, DrugBank (https://www.drugbank.ca) is a publicly available database containing all approved drugs with their mechanisms, interactions, and relevant targets (49). The latest release of DrugBank (version 5.1.2, released December 20, 2018) contains 12,110 drug entries, including 2,553 approved small-molecule drugs, 1,280 approved biotech (protein/peptide) drugs, 130 nutraceuticals, and over 5,842 experimental drugs. DrugMatrix (https://ntp.niehs.nih.gov/results/drugmatrix/index.html), on the other hand, focuses on the toxicogenomic data of drugs to reduce the time to formulate a xenobiotic’s potential for toxicity. The current DrugMatrix database contains large-scale gene expression data from tissues of rats administered over 600 drugs, mostly targeting several major organs (e.g., liver). The Binding Database (BindingDB) is a public, web-accessible resource of drug-target binding data, shown as measured binding affinities (50). The targets included in BindingDB are proteins/enzymes that are considered drug targets. BindingDB currently contains 1,587,753 binding data for 7,235 protein targets and 710,301 small molecules (https://www.bindingdb.org/bind/index.jsp).
The public big data sources can also be characterized by the size of electronic files for these data sets. For example, the current PubChem bioassay database has around 240 million bioactivities, which are contained in 30 GB of XML files. Instead of using personal computers with central processing units, the use of new hardware techniques such as cloud computation (41, 51) and graphics processing units (GPUs) (52) is necessary to process and analyze these available big data.
BIG DATA MODELING CHALLENGES: MISSING DATA AND BIASED DATA
The response profiles of 2,118 approved drugs tested against 531 PubChem assays (each assay having at least 25 active responses among these drug molecules) are shown in Figure 2. The results were generated using an in-house automatic data profiling tool (http://ciipro.rutgers.edu/) (53). There are more than a million data points in this response profile. Nevertheless, many responses in this profile were shown as missing data (Figure 2). Furthermore, the ratio of active versus inactive responses is also biased (approximately 1:6 in this profile). For example, two well-known drugs were included in this profile: acetaminophen (CAS 103-90-2), which has 16 active and 213 inactive responses, and acetylsalicylic acid (aspirin, CAS 50-78-2), which has 14 active and 237 inactive responses. Due to the nature of the HTS techniques, the HTS data normally consist of much fewer active than inactive responses (21, 54), especially for the drugs. In an early review of pharmacological space based on 4.8 million unique compounds, only 275,000 of them showed one (or more) active response when tested against 1,036 targets (55), indicating that most of the testing results were negative. Notably, the drugs that showed the most active responses in public big data sets are for chemotherapy purposes, which normally have critical side effects and other off-target interactions. For example, bortezomib (CAS 179324-69-7) is a chemotherapy drug used to treat multiple myeloma and mantle cell lymphoma. It has the most active responses (258 actives and 49 inactives) in the response profile of Figure 2.
Figure 2.
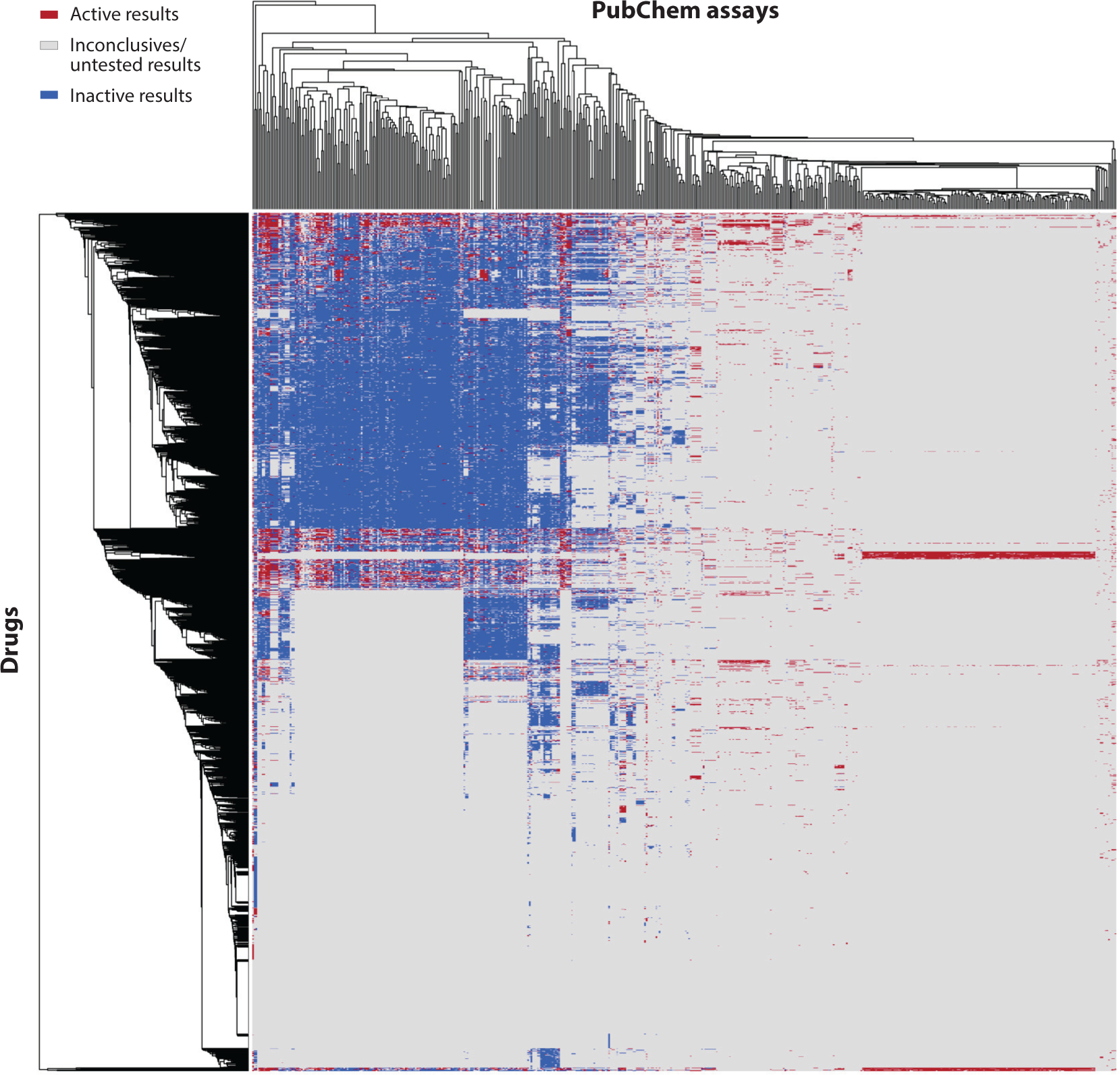
Bioprofile of 2,118 approved drugs from DrugBank (x axis) represented by the response data obtained from 531 PubChem assays(y axis). Each assay against all drug molecules (one column) has at least 25 active responses (red spots). Data from DrugBank (https://www.drugbank.ca) and PubChem (https://pubchem.ncbi.nlm.nih.gov).
The missing data issue is a common problem of big data modeling (56). In previous studies, a common solution was to develop QSAR models for individual assays and use the resulting models to predict target compounds that were not tested against these assays (19, 20, 57). This approach was applicable only when the predicted data used for model development had simple biological mechanisms (e.g., logPs or structural rigid target bindings). However, this process still introduced uncertainty into the modeling process due to the prediction errors from QSAR models (57). When dealing with heterogeneous and complex data (e.g., clinical data), advanced statistical methods such as multiple imputations are needed (58, 59). To reflect the biased nature of HTS data, emphasis should be given to active rather than inactive results during modeling procedures (53). Early-stage computational studies normally used pharmacophore modeling to identify chemical features that were responsible for relevant bioactivities (60–62). The later modeling projects using machine learning approaches needed the biased training sets to be preprocessed by using various methods such as downsampling to balance active and inactive results (63–65).
ADVANCING ARTIFICIAL INTELLIGENCE FROM MACHINE LEARNING TO DEEP LEARNING
The historical progress of AI coupled with the increase of the data size used for model development and hardware improvement in drug discovery is summarized in Figure 3. The concept of AI was born in the 1950s (66) and was used in drug discovery after the first study of QSAR was presented in the 1960s (67). In the early stage of drug discovery (e.g., before the 1990s), the common computational approaches used for model developments were linear regressions (68). In these early studies, the chemical descriptors used for modeling were also limited to chemical structural features, such as atomic type and fragmental descriptors (69, 70). The advancement of AI in drug discovery was first facilitated by the development of novel chemical descriptors such as topological descriptors (71) and molecular fingerprints (72, 73), which greatly increased the size/categories of descriptors calculated from training sets. Instead of using all available descriptors, descriptor selection was integrated into the modeling procedure, e.g., the genetic algorithm (74, 75) and simulated annealing (76). Instead of using linear regression, new machine learning approaches, which were developed based on nonlinear modeling algorithms such as k-nearest neighbors (77), support vector machines (78), and random forest (79, 80), were used frequently in modeling studies from the 1990s to the 2000s. In the same period, model validation was emphasized and treated as a must-have component of modeling (81). Instead of only showing self-correlations, the developed models using these new machine learning approaches were always validated using cross-validations, external validations, and/or experimental validations (14, 63, 82, 83). In addition, the applicability domain became standard practice for model development (17, 84–86). In the early 2000s, QSAR modeling, together with relevant studies (e.g., docking), became a well-developed workflow based on the progress of AI discussed above (Figure 3). These milestones of AI in drug discovery are emphasized in other reviews (9, 87–91).
Figure 3.
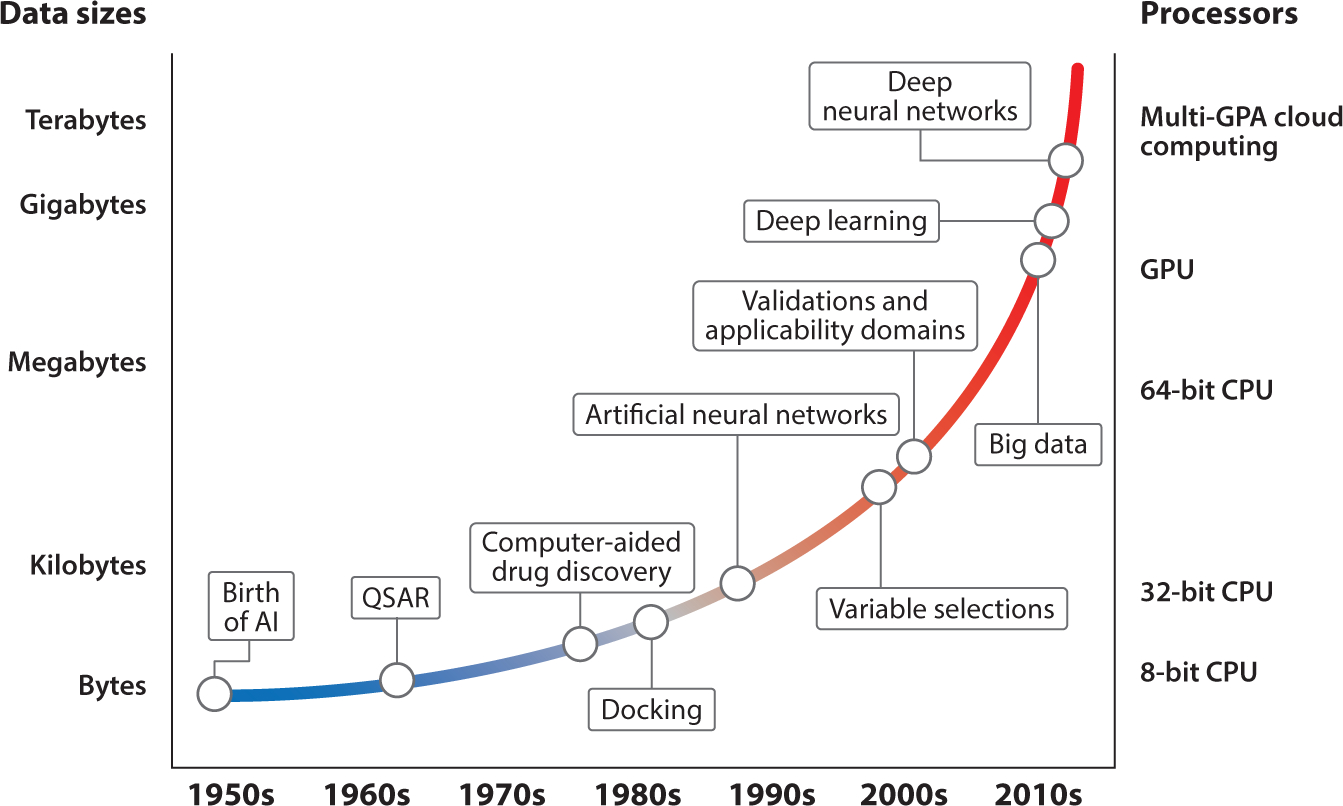
The historical progress of artificial intelligence in drug discovery coupled with increasing data size and computer power (shown as processor improvement).
In addition to the development of AI, the computational power of hardware and the available data for modeling were also significantly improved to facilitate this progress (Figure 3). The early-stage computational modeling of small training sets by simple algorithms (e.g., linear regressions) did not require significant computational power. The advancement of computational power and the availability of biological data for drugs enabled the application of novel modeling techniques such as large-scale networks to address challenges in drug discovery. The first application of the neural network, which was designed as a computational tool in the 1980s (92), in drug discovery was reported in 1989 (93). Since then, various neural network approaches have been applied to drug discovery (90, 94). The first popular approach was the artificial neural network (ANN) (95, 96), which focuses on the variable selection procedure (97). This approach is a machine learning algorithm inspired by biological neural networks such as those in the human brain. With several variables as the input (e.g., chemical descriptors), ANN approaches form hundreds of artificial neurons, which are connected with relationships (quantified as weights) in the form of a network. A single neuron might have some effectiveness in predicting output, but the actual predictions are made by the network consisting of hundreds or even thousands of neurons. Since they learn from the input data, ANNs represent an excellent machine learning approach for constructing nonlinear relationships among the variables and the target biological activities (98). The advanced computational models using various machine learning approaches, such as ANNs, required powerful computers and benefited directly from the hardware developments in the 1990s (Figure 3).
The concept of deep learning was originally presented together with ANNs in the 1980s (4). However, neural networks did not show significant advantages over other machine learning approaches when data used for model development are limited (99, 100). From the 1990s to 2000s, computer hardware was still not adequate for training neural networks with many hidden layers and/or when the data sets for model development were large. In the 2010s, hardware development reached the milestone of using GPUs and cloud computing, which directly benefited neural network modeling studies (Figure 3). Advanced as one of the major interests of AI by various information technology companies, deep neural networks (DNNs), sometimes referred to as deep neural nets, with many hidden layers were developed to address challenging questions such as speech recognition (101). In the Google DeepMind project of 2015, an AI program based on a DNN with 13 hidden layers first mastered the game of Go, which has long been viewed as the most challenging of the classic games for AI (102). The milestone paper of deep learning was published at almost the same time (103), and the big data concept was proposed the next year (41, 104). Deep learning was immediately applied to the life sciences and demonstrated its capability to identify complex patterns in biological systems (4, 105). The first project in which deep learning approaches showed significantly better performance than other machine learning approaches for drug discovery was a QSAR machine learning challenge supported by Merck (38). Another similar effort organized by the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) was to model around 12,000 chemicals, including many drugs, for 12 different toxic effects (106). In this competition, DeepTox, a computational toxicity model based on DNNs outperformed other models based on machine learning approaches (107).
Besides the modeling challenges mentioned above, there have been various individual deep learning studies for drug discovery in the past three years. For example, Wen et al. (108) reported a deep learning model developed to predict interactions between drugs and their biological targets based on 15,524 drug-target pairs obtained from the DrugBank database. Another similar deep learning study was performed using transcriptome data obtained from the Library of Integrated Network-Based Cellular Signatures program (109). Furthermore, multitask learning based on DNNs is a modeling approach that allows multiple related tasks to be modeled simultaneously. Modeling several biologically related end points (i.e., bioactivities sharing similar mechanisms) for drug discovery purposes through multitask learning has shown superior performance to traditional QSAR models by reducing overfitting, solving issues of biased data, and identifying variables from related tasks (110–113). The high performance of these DNN models demonstrates the advantages of using deep learning approaches to model large data sets and select meaningful features. However, there were also recent reports that showed mixed results from the comparison between deep learning and machine learning modeling (114, 115). Since deep learning is a brand-new concept being applied to computer-aided drug discovery, there are no universal criteria for selecting relevant modeling parameters and/or constructing the modeling workflow (115).
OTHER AREAS OF COMPUTATIONAL MODELING UTILIZING ARTIFICIAL INTELLIGENCE FOR DRUG DISCOVERY
Rational Nanomaterials Design
Modern nanotechnology highly impacts drug discovery by offering biocompatible nanomaterials (e.g., nanomedicines with desirable therapeutic activities and low side effects) to the drug research and development process, especially as versatile yet reliable carriers for the delivery of drugs to treat systemic diseases such as cancers (116, 117). Early efforts of using AI in nanomodeling for drug discovery were based on molecular dynamic (MD) simulations. For example, several studies using MD simulations detected the insertion of nanoparticles in the plasma membranes of the recipient cells and an overall change in the cell membrane structure (118). Later, the same approach was used to estimate the affinity of carbon nanotubes to organic molecules (119). In another study, a set of nanoparticles was tested in vitro in four cell lines, and the potential membrane perturbation effects of these nanoparticles were studied (120). The reaction behaviors of individual nanoparticles were also investigated under certain conditions using MD simulations (e.g., interactions with or passing through membranes), along with the effects of the size, density, position, distribution, length, and type of surface ligands on the biological properties of the nanomaterials (121). The advantage of MD simulations is that they can precisely simulate molecular structures, but the clear disadvantages are that modeling procedures are computationally expensive and cannot provide rapid predictions for big databases due to the current limitations of computational resources. Another computational approach is to apply traditional QSAR modeling methods to nanomaterials. For example, the QSAR technique was used to create predictive models for nanoparticles with similar or different metal cores (122). Recently, membrane-nanoparticle interactions were modeled based on the atomization energy of the metal oxide, the period of the nanoparticle metal, and the primary size of the nanoparticle (123).
The current application of AI approaches in nanomodeling has been limited to designing new nanomaterials due to a lack of suitable chemical descriptors. Although descriptors calculated from only the surface ligands are useful in predicting specific bioactivities/properties of nanomaterials, as described above, the effects of the nanomaterial’s size/shape, density, position, distribution, length, and type of surface ligands were not considered in these studies. Some other nanomodeling studies have incorporated descriptors derived from experimental properties (e.g., nanoparticle size) (123, 124) or even biological data (e.g., proteomics data) (125, 126). Due to the diversity and complexity of nanomaterial structures, Puzyn et al. (127) argued that no universal nano-QSAR model can accurately predict the biological properties of variable nanomaterials. Figure 4 presents a recent methodology for nanostructure simulations in the modeling procedure (128). Briefly, the properties and bioactivities of nanomaterials were largely determined by their surface chemistry. To simulate the nano surface chemistry correctly, the surface ligand orientations and accessibility of functional groups needed to be considered in the calculations (Figure 4). For example, in an early modeling of nanohydrophobicity, the contributions of heavy atoms and functional groups to nanologP values were correlated with their accessibility by solvent molecules (128). In a recent study, an advanced method of integrating the solvent-accessible surface into calculations can be viewed as a universal nanologP calculator (129). A similar modeling strategy has been applied to model nanocellular uptake capacities (130) and several other nano bioactivities. The resulting models were utilized to design and synthesize several new nanoparticles with desired nano bioactivities (130).
Figure 4.
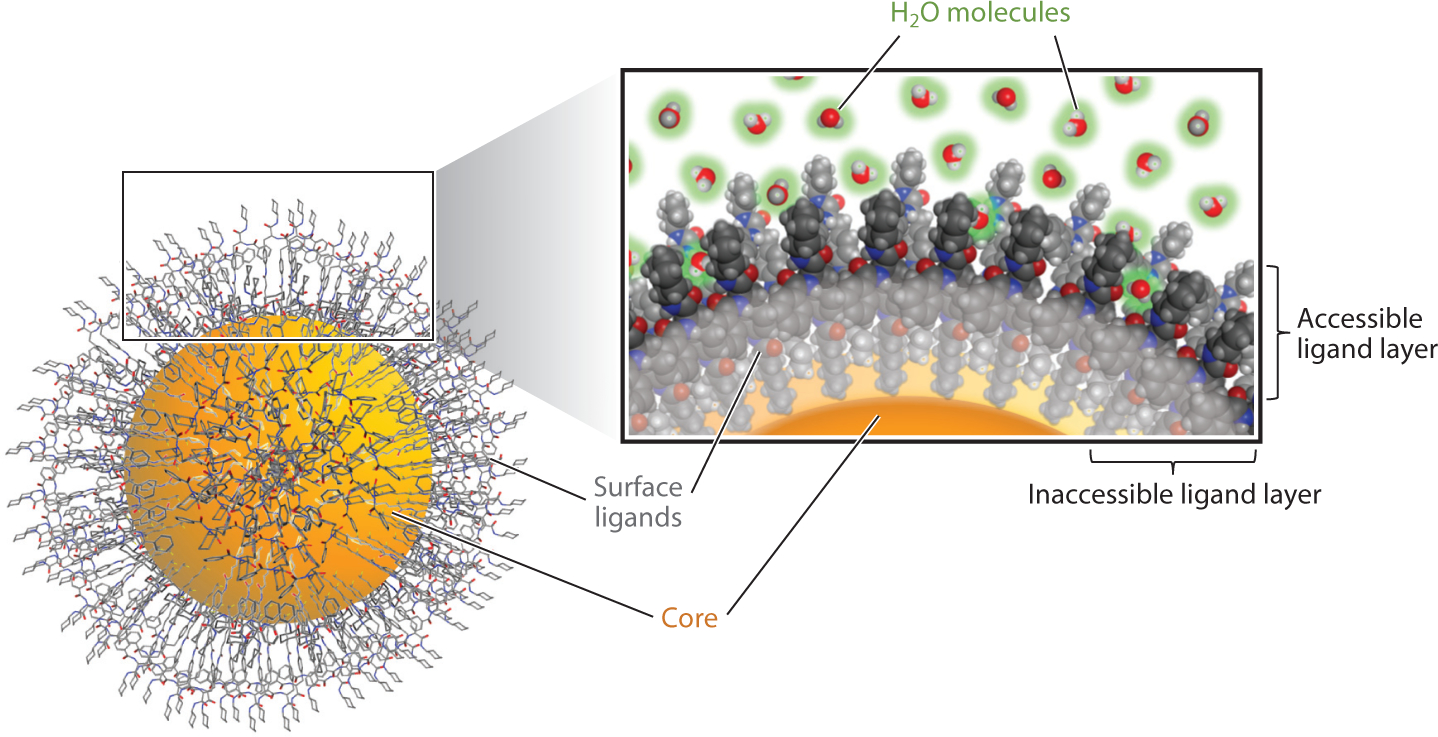
Nanomaterial surface simulations for computational modeling: surface ligand orientations and accessibility assessments.
Convolutional Neural Networks and Image Modeling
The CNN is a special network modeling approach inspired by neuroscience to imitate images within the visual cortex, where individual neurons respond to stimuli only in the receptive fields. Different neurons can partially overlap with each other to cover the entire receptive field. The CNN architecture is constructed in a way that hidden layers are particularly adept at screening multidimensional input such as the red, green, and blue saturation values obtained from thousands of pixels for an image. In the training process, the CNN approach uses kernels and grids of a predefined dimension to scan the image and learn to recognize certain critical features such as lines and contours for a human face. The concept of CNNs was proposed in the 1980s for image recognition purposes but did not draw great attention until the 2010s (4). This approach has become well known, as it has dominated all image recognition challenges since 2012, and it is now the base of image/speech recognition, video analysis, language understanding, and other relevant applications (131).
As one of the most popular deep learning approaches, CNNs have been used for image modeling in clinical diagnoses such as cancer (132), Alzheimer’s disease (133), and heart disease (134). In traditional drug discovery, CNNs were also applied to analyze image data obtained from experimental drug testing, such as HTS results (135). Due to its unique advantages in image recognition, CNNs were also used to recognize 3-D experimental and virtual images to predict ligand-protein interactions (136, 137). In some studies, CNNs were coupled with other computational approaches to realize specific goals. For example, CNNs were used as a new approach to recognize molecular features from drug molecular graphs (138). In this study, drug molecules were treated as 2-D graphs with atom features. The CNN was used to transform the input molecular graphs into new molecular features for training purposes. In another study, an advanced CNN approach called the survival convolutional neural network was used to predict the cancer outcomes of patients based on histological images and genomic biomarker data (139). Furthermore, CNNs were able to function as a text-mining technique to extract drug-drug interaction data from biomedical literature (140).
Personalized Medicine
A drug commonly interacts with multiple targets, including both on- and off-targets, and drug efficacy and side effects are greatly affected by this (141). The perturbation of an individual biological system (e.g., a patient) by a drug molecule is determined by various genetic, epigenetic, and environmental factors. To identify this hidden hierarchical information, personalized medicine was designed to respond to the individual characteristics of each patient (142). Personalized medicine strongly relies on a scientific understanding of how an individual patient’s unique characteristics, such as molecular and genetic profiles, make this patient vulnerable to a disease and sensitive to a therapeutic treatment. Driven by biomarker studies starting in the late 1990s, hundreds of genes have been identified for their contributions to human illness, and genetic variability in patients has been used to distinguish individual responses to dozens of treatments (142). Along with the huge amount of data generated by these studies, such as the Human Genome Project (143), computational modeling has become one of the most important tools for personalized medicine. Drug-target predictions (144), metabolic network modeling (145), and population genetics pattern identifications (146) are several recent advancements in this field that rely on computational modeling. Under the NIH Precision Medicine Initiative (147), many data generation and sharing initiatives and computational modeling efforts have arisen to support the expansion of precision medicine. For example, the Genomic Data Commons program of the National Cancer Institute aims to provide a data repository that enables data sharing across cancer genomic studies in support of precision medicine (148). So far, 33,549 case studies have been submitted and shared via this portal (https://gdc.cancer.gov/). Although it is not the focus of this review, genome sequencing analysis has been a widely applied approach involving AI techniques, and there are many reviews available on this popular bioinformatics topic (149–151).
CONCLUSIONS
AI is a promising method to greatly reduce the cost and time of drug discovery by providing evaluations of drug molecules in the early stages of development. In the current big data era, clinical and pharmaceutical data continue to grow at a rapid pace, and novel AI techniques to deal with big data sets are in high demand. The recent deep learning modeling studies have shown advantages compared to traditional machine learning approaches for this challenge. However, standard criteria and modeling workflows are still needed for deep learning models to be applicable. The applications of AI have been widely extended into all relevant areas beyond traditional drug discovery. Coupled with database curation, web portal development as data repository servers, and the improvement of computer hardware, AI and recent deep learning studies have paved the road to modern drug discovery.
ACKNOWLEDGMENTS
The author thanks Dr. Wenyi Wang, Daniel P. Russo, and Heather Ciallella for their contributions to figure and text editing.
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, et al. 2015. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov 14:475–86 [DOI] [PubMed] [Google Scholar]
- 2.Fleming N 2018. How artificial intelligence is changing drug discovery. Nature 557:S55–57 [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Tan J, Han D, Zhu H. 2017. From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discov. Today 22(11):1680–85 [DOI] [PubMed] [Google Scholar]
- 4.Gawehn E, Hiss JA, Schneider G. 2016. Deep learning in drug discovery. Mol. Inform 35:3–14 [DOI] [PubMed] [Google Scholar]
- 5.Beresford AP, Segall M, Tarbit MH. 2004. In silico prediction of ADME properties: Are we making progress? Curr. Opin. Drug Discov. Dev 7:36–42 [PubMed] [Google Scholar]
- 6.Hughes JP, Rees S, Kalindjian SB, Philpott KL. 2011. Principles of early drug discovery. Br. J. Pharmacol 162:1239–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins FS, Gray GM, Bucher JR. 2008. Transforming environmental health protection. Science 319:906–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, et al. 2014. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem 57:4977–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golbraikh A, Wang X, Zhu H, Tropsha A. 2016. Predictive QSAR modeling: methods and applications in drug discovery and chemical risk assessment In Handbook of Computational Chemistry, ed. Leszczynski J, Kaczmarek-Kedziera A, Puzyn T, Papadopoulos MG, Reis H, Shukla MK, pp. 2303–40. Dordrecht, Neth.: Springer [Google Scholar]
- 10.Zhu H, Bouhifd M, Donley E, Egnash L, Kleinstreuer N, et al. 2016. Supporting read-across using biological data. ALTEX 33:167–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roy PP, Leonard JT, Roy K. 2008. Exploring the impact of size of training sets for the development of predictive QSAR models. Chemometr. Intell. Lab 90:31–42 [Google Scholar]
- 12.Zhao L, Wang W, Sedykh A, Zhu H. 2017. Experimental errors in QSAR modeling sets: What we can do and what we cannot do. ACS Omega 2:2805–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fourches D, Muratov E, Tropsha A. 2010. Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model 50(7):1189–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tropsha A 2010. Best practices for QSAR model development, validation, and exploitation. Mol. Informat 29:476–88 [DOI] [PubMed] [Google Scholar]
- 15.Stouch TR, Kenyon JR, Johnson SR, Chen XQ, Doweyko A, Li Y. 2003. In silico ADME/Tox: why models fail. J. Comput.-Aided Mol. Des 17:83–92 [DOI] [PubMed] [Google Scholar]
- 16.Maggiora GM. 2006. On outliers and activity cliffs—why QSAR often disappoints. J. Chem. Inf. Model 46:1535. [DOI] [PubMed] [Google Scholar]
- 17.Tetko IV, Sushko I, Pandey AK, Zhu H, Tropsha A, et al. 2008. Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model 48:1733–46 [DOI] [PubMed] [Google Scholar]
- 18.Tetko IV. 1995. Neural network studies. 1. Comparison of overfitting and overtraining. J. Chem. Inf. Comput. Sci 35:826–33 [Google Scholar]
- 19.Wang WY, Kim MT, Sedykh A, Zhu H. 2015. Developing enhanced blood-brain barrier permeability models: integrating external bio-assay data in QSAR modeling. Pharm. Res 32:3055–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MT, Sedykh A, Chakravarti SK, Saiakhov RD, Zhu H. 2014. Critical evaluation of human oral bioavailability for pharmaceutical drugs by using various cheminformatics approaches. Pharm. Res 31:1002–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Hsieh JH, Zhu H. 2014. Profiling animal toxicants by automatically mining public bioassay data: a big data approach for computational toxicology. PLOS ONE 9:e99863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu R, Li X, Lam KS. 2017. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol 38:117–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. 2008. Application of combinatorial chemistry science on modern drug discovery. J. Comb. Chem 10:345–54 [DOI] [PubMed] [Google Scholar]
- 24.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, et al. 2006. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. PNAS 103:11473–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malo N, Hanley JA, Cerquozzi S, Pelletier J, Nadon R. 2006. Statistical practice in high-throughput screening data analysis. Nat. Biotechnol 24:167–75 [DOI] [PubMed] [Google Scholar]
- 26.Zhu H, Xia M. 2016. High-Throughput Screening Assays in Toxicology. New York: Springer [Google Scholar]
- 27.Zhu H, Zhang J, Kim MT, Boison A, Sedykh A, Moran K. 2014. Big data in chemical toxicity research: the use of high-throughput screening assays to identify potential toxicants. Chem. Res. Toxicol 27:1643–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broach JR, Thorner J. 1996. High-throughput screening for drug discovery. Nature 384:14–16 [PubMed] [Google Scholar]
- 29.Klekota J, Brauner E, Roth FP, Schreiber SL. 2006. Using high-throughput screening data to discriminate compounds with single-target effects from those with side effects. J. Chem. Inform. Model 46:1549–62 [DOI] [PubMed] [Google Scholar]
- 30.Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, et al. 2011. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov 10:188–95 [DOI] [PubMed] [Google Scholar]
- 31.Ciallella HL, Zhu H. 2019. Advancing computational toxicology in the big data era by artificial intelligence: data-driven and mechanism-driven modeling for chemical toxicity. Chem. Res. Toxicol 32(4):536–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee CH, Yoon HJ. 2017. Medical big data: promise and challenges. Kidney Res. Clin. Pract 36:3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, et al. 2017. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov 16:19–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheeder C, Heigwer F, Boutros M. 2018. Machine learning and image-based profiling in drug discovery. Curr. Opin. Syst. Biol 10:43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu Y, Bajorath J. 2013. Compound promiscuity: What can we learn from current data? Drug Discov. Today 18:644–50 [DOI] [PubMed] [Google Scholar]
- 36.Chatzidakis M, Botton GA. 2019. Towards calibration-invariant spectroscopy using deep learning. Sci. Rep 9:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jing Y, Bian Y, Hu Z, Wang L, Xie XQ. 2018. Deep learning for drug design: an artificial intelligence paradigm for drug discovery in the big data era. AAPS J. 20:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V. 2015. Deep neural nets as a method for quantitative structure-activity relationships. J. Chem. Inf. Model 55:263–74 [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, Beck MW, Winkler DA, Huang B, Sibanda W, et al. 2018. Opening the black box of neural networks: methods for interpreting neural network models in clinical applications. Ann. Transl. Med 6:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dayhoff JE, DeLeo JM. 2001. Artificial neural networks: opening the black box. Cancer 91:1615–35 [DOI] [PubMed] [Google Scholar]
- 41.Schadt EE, Linderman MD, Sorenson J, Lee L, Nolan GP. 2011. Cloud and heterogeneous computing solutions exist today for the emerging big data problems in biology. Nat. Rev. Genet 12:224. [DOI] [PubMed] [Google Scholar]
- 42.Marx V 2013. Biology: the big challenges of big data. Nature 498:255–60 [DOI] [PubMed] [Google Scholar]
- 43.Swarup V, Geschwind DH. 2013. Alzheimer’s disease: from big data to mechanism. Nature 500:34–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, et al. 2010. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 38:D5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. 2009. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 37:W623–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, et al. 2009. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 37:D5–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sayers EW, Agarwala R, Bolton EE, Brister JR, Canese K, et al. 2019. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 47:D23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, et al. 2012. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40:D1100–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, et al. 2018. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46:D1074–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gilson MK, Liu T, Baitaluk M, Nicola G, Hwang L, Chong J. 2016. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 44:D1045–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Armbrust M, Fox A, Griffith R, Joseph AD, Katz R, et al. 2010. A view of cloud computing. Commun. ACM 53:50–58 [Google Scholar]
- 52.Nickolls J, Dally WJ. 2010. The GPU computing era. IEEE Micro. 30:56–69 [Google Scholar]
- 53.Russo DP, Kim MT, Wang W, Pinolini D, Shende S, et al. 2017. CIIPro: a new read-across portal to fill data gaps using public large-scale chemical and biological data. Bioinformatics 33:464–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Russo DP, Strickland J, Karmaus AL, Wang W, Shende S, et al. 2019. Nonanimal models for acute toxicity evaluations: applying data-driven profiling and read-across. Environ. Health Perspect 127(4):47001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paolini GV, Shapland RHB, van Hoorn WP, Mason JS, Hopkins AL. 2006. Global mapping of pharmacological space. Nat. Biotechnol 24:805–15 [DOI] [PubMed] [Google Scholar]
- 56.Mirza B, Wang W, Wang J, Choi H, Chung NC, Ping PP. 2019. Machine learning and integrative analysis of biomedical big data. Genes 10(2):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim MT, Huang R, Sedykh A, Wang W, Xia M, Zhu H. 2016. Mechanism profiling of hepatotoxicity caused by oxidative stress using antioxidant response element reporter gene assay models and big data. Environ. Health Perspect 124:634–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pedersen AB, Mikkelsen EM, Cronin-Fenton D, Kristensen NR, Pham TM, et al. 2017. Missing data and multiple imputation in clinical epidemiological research. Clin. Epidemiol 9:157–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sterne JAC, White IR, Carlin JB, Spratt M, Royston P, et al. 2009. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ 338:b2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vadivelan S, Sinha BN, Rambabu G, Boppana K, Jagarlapudi SA. 2008. Pharmacophore modeling and virtual screening studies to design some potential histone deacetylase inhibitors as new leads. J. Mol. Graph. Model 26:935–46 [DOI] [PubMed] [Google Scholar]
- 61.Marriott DP, Dougall IG, Meghani P, Liu YJ, Flower DR. 1999. Lead generation using pharmacophore mapping and three-dimensional database searching: application to muscarinic M-3 receptor antagonists.J. Med. Chem 42:3210–16 [DOI] [PubMed] [Google Scholar]
- 62.Gussio R, Pattabiraman N, Kellogg GE, Zaharevitz DW. 1998. Use of 3D QSAR methodology for data mining the National Cancer Institute Repository of Small Molecules: application to HIV-1 reverse transcriptase inhibition. Methods 14:255–63 [DOI] [PubMed] [Google Scholar]
- 63.Zhang L, Fourches D, Sedykh A, Zhu H, Golbraikh A, et al. 2013. Discovery of novel antimalarial compounds enabled by QSAR-based virtual screening. J. Chem. Inf. Model 53:475–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ribay K, Kim MT, Wang W, Pinolini D, Zhu H. 2016. Hybrid modeling of estrogen receptor binding agents using advanced cheminformatics tools and massive public data. Front. Environ. Sci 4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bharti DR, Hemrom AJ, Lynn AM. 2019. GCAC: galaxy workflow system for predictive model building for virtual screening. BMC Bioinform. 19(Suppl. 13):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Russell SJ, Norvig P. 2003. Artificial Intelligence: A Modern Approach. Upper Saddle River, NJ: Prentice Hall/Pearson Ed. [Google Scholar]
- 67.Hansch C, Fujita T. 1964. ρ-σ-π Analysis. A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc 86:1616–26 [Google Scholar]
- 68.Martin YC. 2010. Quantitative Drug Design: A Critical Introduction. Boca Raton, FL: CRC Press. 2nd ed. [Google Scholar]
- 69.Zefirov NS, Palyulin VA. 2002. Fragmental approach in QSPR. J. Chem. Inform. Comput. Sci 42:1112–22 [DOI] [PubMed] [Google Scholar]
- 70.Labute P 2000. A widely applicable set of descriptors. J. Mol. Graph. Model 18:464–77 [DOI] [PubMed] [Google Scholar]
- 71.Gozalbes R, Doucet JP, Derouin F. 2002. Application of topological descriptors in QSAR and drug design: history and new trends. Curr. Drug Targets Infect. Disord 2:93–102 [DOI] [PubMed] [Google Scholar]
- 72.Willett P 2006. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 11:1046–53 [DOI] [PubMed] [Google Scholar]
- 73.McGregor MJ, Muskal SM. 1999. Pharmacophore fingerprinting. 1. Application to QSAR and focused library design. J. Chem. Inf. Comput. Sci 39:569–74 [DOI] [PubMed] [Google Scholar]
- 74.Leardi R, Boggia R, Terrile M. 1992. Genetic algorithms as a strategy for feature-selection. J. Chemomet 6:267–81 [Google Scholar]
- 75.Sheridan RP, SanFeliciano SG, Kearsley SK. 2000. Designing targeted libraries with genetic algorithms.J. Mol. Graph. Model 18:320–34 [DOI] [PubMed] [Google Scholar]
- 76.Sun L, Xie Y, Song X, Wang J, Yu R. 1994. Cluster analysis by simulated annealing. Comp. Chem 18:103–8 [Google Scholar]
- 77.Zheng W, Tropsha A. 2000. Novel variable selection quantitative structure–property relationship approach based on the k-nearest-neighbor principle. J. Chem. Inf. Comput. Sci 40:185–94 [DOI] [PubMed] [Google Scholar]
- 78.Burbidge R, Trotter M, Buxton B, Holden S. 2001. Drug design by machine learning: support vector machines for pharmaceutical data analysis. Comput. Chem 26:5–14 [DOI] [PubMed] [Google Scholar]
- 79.Sprague B, Shi Q, Kim MT, Zhang L, Sedykh A, et al. 2014. Design, synthesis and experimental validation of novel potential chemopreventive agents using random forest and support vector machine binary classifiers. J. Comput.-Aided Mol. Des 28:631–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Breiman L 2001. Random forests. Mach. Learn 45:5–32 [Google Scholar]
- 81.Golbraikh A, Tropsha A. 2002. Beware of q2! J. Mol. Graph. Model 20:269–76 [DOI] [PubMed] [Google Scholar]
- 82.Solimeo R, Zhang J, Kim M, Sedykh A, Zhu H. 2012. Predicting chemical ocular toxicity using a combinatorial QSAR approach. Chem. Res. Toxicol 25:2763–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gramatica P 2007. Principles of QSAR models validation: internal and external. QSAR Comb. Sci 26:694–701 [Google Scholar]
- 84.Zhu H, Martin TM, Ye L, Sedykh A, Young DM, Tropsha A. 2009. Quantitative structure-activity relationship modeling of rat acute toxicity by oral exposure. Chem. Res. Toxicol 22:1913–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu H, Tropsha A, Fourches D, Varnek A, Papa E, et al. 2008. Combinatorial QSAR modeling of chemical toxicants tested against Tetrahymena pyriformis. J. Chem. Inf. Model 48:766–84 [DOI] [PubMed] [Google Scholar]
- 86.Tropsha A, Golbraikh A. 2007. Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr. Pharm. Des 13:3494–504 [DOI] [PubMed] [Google Scholar]
- 87.Ekins S, Boulanger B, Swaan PW, Hupcey MA. 2002. Towards a new age of virtual ADME/TOX and multidimensional drug discovery. Mol. Divers 5:255–75 [DOI] [PubMed] [Google Scholar]
- 88.Muster W, Breidenbach A, Fischer H, Kirchner S, Muller L, Pahler A. 2008. Computational toxicology in drug development. Drug Discov. Today 13:303–10 [DOI] [PubMed] [Google Scholar]
- 89.Khedkar SA, Malde AK, Coutinho EC, Srivastava S. 2007. Pharmacophore modeling in drug discovery and development: an overview. Med. Chem 3:187–97 [DOI] [PubMed] [Google Scholar]
- 90.Duch W, Swaminathan K, Meller J. 2007. Artificial intelligence approaches for rational drug design and discovery. Curr. Pharm. Des 13:1497–508 [DOI] [PubMed] [Google Scholar]
- 91.Hecht D 2011. Applications of machine learning and computational intelligence to drug discovery and development. Drug Dev. Res 72:53–65 [Google Scholar]
- 92.Hopfield JJ. 1982. Neural networks and physical systems with emergent collective computational abilities. PNAS 79:2554–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aoyama T, Suzuki Y, Ichikawa H. 1989. Neural networks applied to pharmaceutical problems. 1. Method and application to decision-making. Chem. Pharm. Bull 37:2558–60 [PubMed] [Google Scholar]
- 94.Baskin II, Winkler D, Tetko IV. 2016. A renaissance of neural networks in drug discovery. Expert Opin. Drug Dis 11:785–95 [DOI] [PubMed] [Google Scholar]
- 95.Tetko IV, Villa AE, Aksenova TI, Zielinski WL, Brower J, et al. 1998. Application of a pruning algorithm to optimize artificial neural networks for pharmaceutical fingerprinting. J. Chem. Inf. Comput. Sci 38:660–68 [DOI] [PubMed] [Google Scholar]
- 96.Tetko IV, Tanchuk VY, Chentsova NP, Antonenko SV, Poda GI, et al. 1994. HIV-1 reverse transcriptase inhibitor design using artificial neural networks. J. Med. Chem 37:2520–26 [DOI] [PubMed] [Google Scholar]
- 97.Tetko IV, Villa AE, Livingstone DJ. 1996. Neural network studies. 2. Variable selection. J. Chem. Inf. Comput. Sci 36:794–803 [DOI] [PubMed] [Google Scholar]
- 98.Agatonovic-Kustrin S, Beresford R. 2000. Basic concepts of artificial neural network (ANN) modeling and its application in pharmaceutical research. J. Pharm. Biomed. Anal 22:717–27 [DOI] [PubMed] [Google Scholar]
- 99.Roy K, Roy PP. 2009. Comparative chemometric modeling of cytochrome 3A4 inhibitory activity of structurally diverse compounds using stepwise MLR, FA-MLR, PLS, GFA, G/PLS and ANN techniques. Eur. J. Med. Chem 44:2913–22 [DOI] [PubMed] [Google Scholar]
- 100.Simmons K, Kinney J, Owens A, Kleier D, Bloch K, et al. 2008. Comparative study of machine-learning and chemometric tools for analysis of in-vivo high-throughput screening data. J. Chem. Inform. Model 48:1663–68 [DOI] [PubMed] [Google Scholar]
- 101.Hinton G, Deng L, Yu D, Dahl GE, Mohamed AR, et al. 2012. Deep neural networks for acoustic modeling in speech recognition. IEEE Signal. Proc. Mag 29:82–97 [Google Scholar]
- 102.Silver D, Huang A, Maddison CJ, Guez A, Sifre L, et al. 2016. Mastering the game of Go with deep neural networks and tree search. Nature 529:484–89 [DOI] [PubMed] [Google Scholar]
- 103.LeCun Y, Bengio Y, Hinton G. 2015. Deep learning. Nature 521:436–44 [DOI] [PubMed] [Google Scholar]
- 104.Schadt EE, Linderman MD, Sorenson J, Lee L, Nolan GP. 2010. Computational solutions to large-scale data management and analysis. Nat. Rev. Genet 11:647–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xie L, Draizen EJ, Bourne PE. 2017. Harnessing big data for systems pharmacology. Annu. Rev. Pharmacol 57:245–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huang RL, Xia MH, Nguyen DT, Zhao TG, Sakamuru S, et al. 2016. Tox21 challenge to build predictive models of nuclear receptor and stress response pathways as mediated by exposure to environmental chemicals and drugs. Front. Env. Sci 3:85 [Google Scholar]
- 107.Mayr A, Klambauer G, Unterthiner T, Hochreiter S. 2016. DeepTox: toxicity prediction using deep learning. Front. Environ. Sci 3:80 [Google Scholar]
- 108.Wen M, Zhang Z, Niu S, Sha H, Yang R, et al. 2017. Deep-learning-based drug-target interaction prediction. J. Proteome Res 16:1401–9 [DOI] [PubMed] [Google Scholar]
- 109.Xie L, He S, Song X, Bo X, Zhang Z. 2018. Deep learning-based transcriptome data classification for drug-target interaction prediction. BMC Genom. 19:667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu Y, Pei J, Lai L. 2017. Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J. Chem. Inf. Model 57:2672–85 [DOI] [PubMed] [Google Scholar]
- 111.Cai C, Guo P, Zhou Y, Zhou J, Wang Q, et al. 2019. Deep learning-based prediction of drug-induced cardiotoxicity. J. Chem. Inf. Model 59(3):1073–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wenzel J, Matter H, Schmidt F. 2019. Predictive multitask deep neural network models for ADME-Tox properties: learning from large data sets. J. Chem. Inf. Model 59:1253–68 [DOI] [PubMed] [Google Scholar]
- 113.Li X, Xu YJ, Lai LH, Pei JF. 2018. Prediction of human cytochrome P450 inhibition using a multitask deep autoencoder neural network. Mol. Pharm 15:4336–45 [DOI] [PubMed] [Google Scholar]
- 114.Russo DP, Zorn KM, Clark AM, Zhu H, Ekins S. 2018. Comparing multiple machine learning algorithms and metrics for estrogen receptor binding prediction. Mol. Pharm 15:4361–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou Y, Cahya S, Combs SA, Nicolaou CA, Wang J, et al. 2019. Exploring tunable hyperparameters for deep neural networks with industrial ADME data sets. J. Chem. Inf. Model 59:1005–16 [DOI] [PubMed] [Google Scholar]
- 116.Minko T, Rodriguez-Rodriguez L, Pozharov V. 2013. Nanotechnology approaches for personalized treatment of multidrug resistant cancers. Adv. Drug Deliv. Rev 65:1880–95 [DOI] [PubMed] [Google Scholar]
- 117.Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji WH, et al. 2017. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol 12:813–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu JZ, Hopfinger AJ. 2008. Identification of possible sources of nanotoxicity from carbon nanotubes inserted into membrane bilayers using membrane interaction quantitative structure-activity relationship analysis. Chem. Res. Toxicol 21:459–66 [DOI] [PubMed] [Google Scholar]
- 119.Liu J, Yang L, Hopfinger AJ. 2009. Affinity of drugs and small biologically active molecules to carbon nanotubes: a pharmacodynamics and nanotoxicity factor? Mol. Pharm 6:873–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shaw SY, Westly EC, Pittet MJ, Subramanian A, Schreiber SL, Weissleder R. 2008. Perturbational profiling of nanomaterial biologic activity. PNAS 105:7387–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu W, Wu Y, Wang C, Li HC, Wang T, et al. 2010. Impact of silver nanoparticles on human cells: effect of particle size. Nanotoxicology 4:319–30 [DOI] [PubMed] [Google Scholar]
- 122.Fourches D, Pu D, Tassa C, Weissleder R, Shaw SY, et al. 2010. Quantitative nanostructure-activity relationship modeling. ACS Nano 4:5703–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu R, Rallo R, George S, Ji ZX, Nair S, et al. 2011. Classification NanoSAR development for cytotoxicity of metal oxide nanoparticles. Small 7:1118–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Epa VC, Burden FR, Tassa C, Weissleder R, Shaw S, Winkler DA. 2012. Modeling biological activities of nanoparticles. Nano Lett. 12:5808–12 [DOI] [PubMed] [Google Scholar]
- 125.Chen R, Zhang Y, Monteiro-Riviere NA, Riviere JE. 2016. Quantification of nanoparticle pesticide adsorption: computational approaches based on experimental data. Nanotoxicology 10:1118–28 [DOI] [PubMed] [Google Scholar]
- 126.Pathakoti K, Huang MJ, Watts JD, He X, Hwang HM. 2014. Using experimental data of Escherichia coli to develop a QSAR model for predicting the photo-induced cytotoxicity of metal oxide nanoparticles. J. Photochem. Photobiol. B 130:234–40 [DOI] [PubMed] [Google Scholar]
- 127.Puzyn T, Leszczynska D, Leszczynski J. 2009. Toward the development of “nano-QSARs”: advances and challenges. Small 5:2494–509 [DOI] [PubMed] [Google Scholar]
- 128.Li S, Zhai S, Liu Y, Zhou H, Wu J, et al. 2015. Experimental modulation and computational model of nano-hydrophobicity. Biomaterials 52:312–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang WY, Yan XL, Zhao LL, Russo DP, Wang SQ, et al. 2019. Universal nanohydrophobicity predictions using virtual nanoparticle library. J. Cheminform 11:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang W, Sedykh A, Sun H, Zhao L, Russo DP, et al. 2017. Predicting nano–bio interactions by integrating nanoparticle libraries and quantitative nanostructure activity relationship modeling. ACS Nano 11:12641–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mnih V, Kavukcuoglu K, Silver D, Rusu AA, Veness J, et al. 2015. Human-level control through deep reinforcement learning. Nature 518:529–33 [DOI] [PubMed] [Google Scholar]
- 132.Chougrad H, Zouaki H, Alheyane O. 2018. Deep convolutional neural networks for breast cancer screening. Comput. Meth. Prog. Biomed 157:19–30 [DOI] [PubMed] [Google Scholar]
- 133.Lin WM, Tong T, Gao QQ, Guo D, Du XF, et al. 2018. Convolutional neural networks-based MRI image analysis for the Alzheimer’s disease prediction from mild cognitive impairment. Front. Neurosci 12:777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nirschl JJ, Janowczyk A, Peyster EG, Frank R, Margulies KB, et al. 2018. A deep-learning classifier identifies patients with clinical heart failure using whole-slide images of H&E tissue. PLOS ONE 13:e0192726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hofmarcher M, Rumetshofer E, Clevert DA, Hochreiter S, Klambauer G. 2019. Accurate prediction of biological assays with high-throughput microscopy images and convolutional networks. J. Chem. Inf. Model 59:1163–71 [DOI] [PubMed] [Google Scholar]
- 136.Stepniewska-Dziubinska MM, Zielenkiewicz P, Siedlecki P. 2018. Development and evaluation of a deep learning model for protein-ligand binding affinity prediction. Bioinformatics 34:3666–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ragoza M, Hochuli J, Idrobo E, Sunseri J, Koes DR. 2017. Protein-ligand scoring with convolutional neural networks. J. Chem. Inf. Model 57:942–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Altae-Tran H, Ramsundar B, Pappu AS, Pande V. 2017. Low data drug discovery with one-shot learning. ACS Cent. Sci 3:283–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mobadersany P, Yousefi S, Amgad M, Gutman DA, Barnholtz-Sloan JS, et al. 2018. Predicting cancer outcomes from histology and genomics using convolutional networks. PNAS 115:E2970–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhao ZH, Yang ZH, Luo L, Lin HF, Wang J. 2016. Drug drug interaction extraction from biomedical literature using syntax convolutional neural network. Bioinformatics 32:3444–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xie L, Ge XX, Tan HP, Xie L, Zhang YL, et al. 2014. Towards structural systems pharmacology to study complex diseases and personalized medicine. PLOS Comput. Biol 10:e1003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hamburg MA, Collins FS. 2010. The path to personalized medicine. N. Engl. J. Med 363:301–4 [DOI] [PubMed] [Google Scholar]
- 143.Collins FS, Morgan M, Patrinos A. 2003. The Human Genome Project: lessons from large-scale biology. Science 300:286–90 [DOI] [PubMed] [Google Scholar]
- 144.Sydow D, Burggraaff L, Szengel A, van Vlijmen HWT, IJzerman AP, et al. 2019. Advances and challenges in computational target prediction. J. Chem. Inf. Model 59:1728–42 [DOI] [PubMed] [Google Scholar]
- 145.Chang RL, Xie L, Xie L, Bourne PE, Palsson BO. 2010. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLOS Comput. Biol 6:e1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schrider DR, Kern AD. 2018. Supervised machine learning for population genetics: a new paradigm. Trends Genet. 34:301–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Collins FS, Varmus H. 2015. A new initiative on precision medicine. N. Engl. J. Med 372:793–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, et al. 2016. Toward a shared vision for cancer genomic data. N. Engl. J. Med 375:1109–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. 2000. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct 29:291–325 [DOI] [PubMed] [Google Scholar]
- 150.Vinga S, Almeida J. 2003. Alignment-free sequence comparison—a review. Bioinformatics 19:513–23 [DOI] [PubMed] [Google Scholar]
- 151.Lippmann C, Kringel D, Ultsch A, Lotsch J. 2018. Computational functional genomics-based approaches in analgesic drug discovery and repurposing. Pharmacogenomics 19:783–97 [DOI] [PubMed] [Google Scholar]