

Abstract
Background:
Theoretical models have emphasized systems-level abnormalities in Major Depressive Disorder (MDD). For unbiased yet rigorous evaluations of pathophysiological mechanisms underlying MDD, it is critically important to develop data-driven approaches that harness whole-brain data to classify MDD and evaluate possible normalizing effects of targeted interventions. Here, using an experimental therapeutics approach coupled with machine-learning we investigated the effect of a pharmacological challenge aiming to enhance dopaminergic signaling on whole-brain’s response to reward-related stimuli in MDD.
Methods:
Using a double-blind placebo-controlled design, functional magnetic resonance imaging (fMRI) data from 31 unmedicated MDD participants receiving a single dose of 50 mg amisulpride (MDDAmisulpride), 26 MDD participants receiving placebo (MDDPlacebo), and 28 healthy controls receiving placebo (HCPlacebo) recruited through two independent studies were analyzed. An importance-guided machine learning technique for model selection was used on whole-brain fMRI data probing reward anticipation and consumption to identify features linked to MDD (MDDPlacebo vs. HCPlacebo) and dopaminergic enhancement (MDDAmisulpride vs. MDDPlacebo).
Results:
Highly predictive classification models emerged that distinguished MDDPlacebo from HCPlacebo (AUC=0.87) and MDDPlacebo from MDDAmisulpride (AUC=0.89). Although reward-related striatal activation and connectivity were among the most predictive features, the best truncated models based on whole-brain features were significantly better relative to models trained using striatal features only.
Conclusions:
Results indicate that, in MDD, enhanced dopaminergic signaling restores abnormal activation and connectivity in a widespread network of regions. These findings provide new insights into the pathophysiology of MDD and pharmacological mechanism of antidepressants at the system level in addressing reward processing deficits among depressed individuals.
Keywords: Depression, Dopamine, Biomarker, Machine Learning, Biotypes, fMRI
Introduction
Major depressive disorder (MDD) is a debilitating disorder, often characterized by anhedonia (1), which is poorly addressed by current treatments (1, 2). Converging evidence across species suggests that mesocorticolimbic dopaminergic pathways involving the striatum are essential for reward processing (3–5). Dysfunction in this circuit has been associated with deficits in reward processing across psychiatric diseases (6). In MDD, neuroimaging studies have documented decreased striatal activation and reduced functional connectivity between the striatum and other nodes of the brain reward system in response to reward-related stimuli (7–9). Notably, some of these abnormalities were found to be acutely restored by pharmacologically-induced dopaminergic enhancement (10).
Despite advancements in our understanding of the pathophysiology of MDD, an unresolved issue is how enhanced dopaminergic signaling might modulate large-scale whole-brain activation and functional coordination in MDD. Besides the striatum, other brain regions, including the orbitofrontal cortex, amygdala, and anterior cingulate cortex, have been implicated in reward processing (11–14). Given that antidepressant treatments aiming to increase dopaminergic signaling might have faster therapeutic onsets (15, 16), it is important to investigate the effects of dopaminergic enhancement to better understand the potential neural mechanism through which these interventions may address reward processing deficits in MDD. Thus, we identified several needs to address in this study, including developing and evaluating: 1) a robust, data-driven, multivariate approach to analyze whole-brain data in order to probe the purported distributed nature of the reward system, 2) an approach to assess MDD-related abnormalities and putative normalization of those abnormalities, and 3) comparisons between a multivariate approach and a hypothesis-driven approach to evaluate whether a broad set of regions beyond the striatum does indeed better highlight reward-related abnormalities.
Towards these goals, we used a machine learning based approach to analyze whole-brain functional magnetic resonance imaging (fMRI) data collected from a double-blind placebo-controlled study, in which unmedicated individuals with MDD and healthy controls (HCs) performed a monetary incentive delay (MID) task after being randomized to either a single low dose of amisulpride (50 mg) or placebo. Amisulpride, a selective dopamine D2/D3 receptor antagonist, was selected because of its high affinity to block presynaptic autoreceptors at lower doses, thereby increasing dopamine release (17). In a first step, to identify the effects of enhanced dopaminergic transmission on reward-related brain activity, whole-brain fMRI data were entered into an importance-guided model selection procedure (based on the logistic regression with elastic net regularization; Fig. 1) to identify brain regions in which reward-related metrics were most predictive of differences between the MDD individuals receiving amisulpride vs. placebo. Next, to investigate the potential normalizing effect of enhanced dopaminergic transmission on MDD-related abnormalities, brain regions from the above step were compared with those most predictive of differences between MDD and HC group receiving placebo. The regions with MDD-related abnormalities that also demonstrated an MDD amisulpride effect constitute a potential multivariate signature that we used to assess amisulpride-induced BOLD normalization in MDD patients. Based on prior findings (7,10,18–22), we hypothesized that (1) under placebo, MDD would be associated with widespread reward-related abnormalities along the brain’s reward pathway and (2) transient DA enhancement would rescue such abnormalities. We further compared whole-brain and hypothesis-driven approaches.
Figure 1:
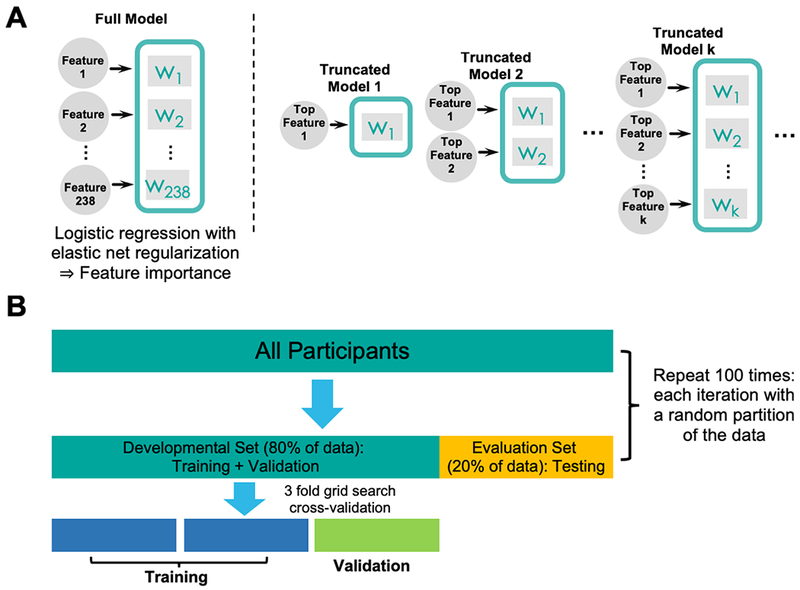
a) An illustration of the importance-guided sequential model selection procedure used to find the optimal set of features. First, a full model including all features is trained using logistic regression with elastic net regularization to determine relative importance of individual features. Next, a series of truncated models were trained based on a progressively increasing set of top features rank ordered by the full model. The set of features in the best truncated model on the evaluation set were deemed as the optimal feature set. b) An illustration of the nested cross-validation procedure used to train, validate, and test the models. A grid search procedure with 3-fold cross-validation was implemented on the developmental set to determine the best model parameters. The resulting model was further tested on the evaluation set, which contained an independent set of participants not used in training and validation. The entire procedure was repeated on 100 different random partitioning of the data to allow for stable model performance.
Methods and Materials
Participants
Participants were recruited by the Center for Depression, Anxiety and Stress Research at McLean Hospital using online advertisements, mailing and flyers within the Boston metropolitan areas for two independent studies using identical procedures that each enrolled individuals with MDD and healthy controls.
Across the first (ClinicalTrials.gov identifier: ) and second () study, 62 unmedicated individuals with MDD (34 randomized to amisulpride, 28 randomized to placebo) and 63 demographically-matched healthy controls (placebo: N=30, amisulpride: N=33) were run in the imaging session. For the current analyses, we focused on analyses aiming at classifying case vs. controls (MDDPlacebo vs. HCPlacebo model) and classifying the potential normalizing effects of dopaminergic enhancement (MDDPlacebo vs. MDDAmisulpride model); thus, 92 participants were considered. Among these 92, 85 had useable fMRI data. A subset of participants (46 MDD, 23 randomized to amisulpride, 23 to placebo; 20 HC controls randomized to placebo) were included in a recent study that used a region-of-interest (ROI) approach to probe the effects of MDD and amisulpride on striatal activation and functional connectivity (10). Groups were matched for age, gender, ethnicity, and years of education (Table 1). General inclusion criteria were: right-handedness, age between 18-45, no MRI contraindications, no lifetime substance dependence, no past-year substance abuse, and no serious medical conditions. For the MDD groups, a diagnosis of MDD according to the Structured Clinical Interview for DSM-IV-TR Axis I Disorders (SCID) (2) was required, and exclusion criteria included: psychotropic medication in the past 2 weeks (6 weeks for fluoxetine, 6 months for dopaminergic drugs or antipsychotics) and any other axis I disorders (however, social anxiety disorder, simple phobia, or generalized anxiety disorder were allowed if secondary to MDD). For HC, exclusion criteria were: any medication in the last 3 weeks, current or past psychiatric illnesses (SCID), and first-degree familial psychiatric illness. Participants received $15/hour in addition to earnings in the fMRI task. The two protocols were approved by Partners Human Research Committee, and all participants provided written informed consent.
Table 1:
Clinical and demographic characteristics of the participants
| MDDAmisulpride N = 31 |
MDDPlacebo N = 26 |
HCPlacebo N = 28 |
||||
|---|---|---|---|---|---|---|
| Characteristic | Mean | SD | Mean | SD | Mean | SD |
| Age (years) | 27.2 | 7.7 | 25.6 | 5.0 | 25.1 | 6.1 |
| Education (years) | 15.4 | 2.2 | 16.8 | 3.0 | 15.2 | 2.9 |
| Beck Depression Inventory 2nd Ed. | 26.3 | 7.9 | 26.7 | 7.9 | 1.8 | 2.7 |
| Hamilton Depression Rating Scale | 15.6 | 3.7 | 16.7 | 5.3 | 1.0 | 1.2 |
| Mood and Anxiety Symptom Questionnaire | ||||||
| Total Score | 168.5 | 22.9 | 174.1 | 21.7 | 91.5 | 13.3 |
| General Distress Anxiety Subscore | 23.6 | 5.1 | 25.4 | 6.6 | 12.3 | 1.2 |
| General Distress Depression Subscore | 37.9 | 9.4 | 39.0 | 9.3 | 13.9 | 2.0 |
| Anxious Arousal Subscore | 24.0 | 6.0 | 25.6 | 6.4 | 18.4 | 2.0 |
| Anhedonic Depression Subscore | 82.9 | 11.2 | 84.1 | 9.1 | 47.0 | 11.3 |
| Snaith-Hamilton Pleasure Scale | 31.7 | 4.7 | 31.4 | 7.0 | 22.8 | 6.7 |
| Duration of Current Major Depressive Episode (months) | 17.3 | 20.0 | 17.6 | 31.9 | N/A | N/A |
| Number of Past Depressive Episodes | 3.2 | 2.6 | 3.3 | 3.2 | N/A | N/A |
| N | % | N | % | N | % | |
| Female | 28 | 90.3 | 19 | 73.1 | 22 | 81.5 |
| Caucasian | 20 | 64.5 | 13 | 50.0 | 13 | 48.1 |
| Current Comorbid Anxiety Disorders | 10 | 32.3 | 11 | 42.3 | N/A | N/A |
| Past Comorbid Anxiety Disorders | 13 | 41.9 | 12 | 46.2 | N/A | N/A |
Note: Groups were matched for age, gender, race, and years of education (one-way ANOVA; χ2-test). All participants were right-handed. Between the MDDAmisulpride and MDDPlacebo group, participants were matched for current and past comorbid anxiety disorders, as well as clinical scale measures (χ2-test; two-sample t-test).
Procedure
The two studies followed identical procedures, pharmacological challenge, and MRI acquisition. In the first session, a PhD- or Masters-level clinician administered the SCID to determine eligibility, and participants filled out self-report scales (Table 1 and Supplement). In the second session, participants performed the MID task during fMRI scanning after receiving a single dose of amisulpride or placebo. The MID task was started one hour after pill administration due to pharmacokinetic data indicating that plasma concentration of amisulpride has a first peak approximately 1-1.5 hours after administration (17).
fMRI Task
The MID has been described in detail (10, 23). Briefly, the task includes anticipation and receipt of monetary rewards (and penalties), which robustly recruit mesocorticolimbic regions (12, 13) and has been used to uncover reward-related abnormalities in both magnitude of activation and functional connectivity in MDD (7, 9, 10, 22, 24).
Data Acquisition and Preprocessing
For both studies, MRI data were acquired at the McLean Imaging Center using a Siemens Tim Trio 3T MR scanner equipped with a 32-channel head coil. Data collection for the two studies overlapped in time. See Supplementary Methods for acquisition parameters and preprocessing.
Feature Extraction
The features used in our classifiers consisted of coefficients from the single-subject level general linear models (GLM) averaged according to the AAL template (25). To obtain these features, for each participant, we first fitted a GLM to the fMRI data during the MID task (see 10 for more details). Next, for each regressor in the GLM, the estimated coefficients were averaged according to the AAL template, producing one averaged coefficient for each ROI. ROIs for the left and right nucleus accumbens (NAcc) were further extracted according to a manually segmented MNI-152 brain (26) and added to the existing AAL ROIs, resulting in 118 ROIs. The following BOLD contrasts were included as features in our classification models to represent reward anticipation and consumption, respectively: 1) reward cue minus neutral cue and 2) reward outcome minus no-change outcome following reward cue. In addition, two striatal connectivity features emerging from (10) were included in our classification models, representing the psychophysiological interaction (PPI) under the reward outcome condition between 1) caudate and dorsal anterior cingulate cortex and 2) NAcc and mid-cingulate cortex. In total, 238 features (118 ROIs x 2 contrasts + 2 PPIs) were included in the classification models. Modeling was also done without the PPI regressors to establish if they brought any additional predictive information (see Supplementary Materials). All features were standardized to zero mean and unit variance before entered into the models.
Classification and Importance-guided Sequential Model Selection
Two main classifiers were built to classify 1) MDDPlacebo vs. HCPlacebo and 2) MDDPlacebo vs. MDDAmisulpride. These were designed to capture features linked to 1) MDD, and 2) the effect of acute dopaminergic enhancement on whole-brain BOLD activation in individuals with MDD. To further test the hypothesis that dopaminergic enhancement transiently normalized reward-related abnormalities in MDD, a third classifier was built to classify MDDAmisulpride vs. HCPlacebo. Across analyses, we used logistic regression with elastic net regularization (27) for classification. The elastic net regularization is well-suited for problems where the number of features is much greater than the number of observations (27). The models were trained and tested via the following nested cross-validation procedure. First, we performed model training on a development set containing 80% of the participants via a 3-fold grid search cross-validation procedure (stratified using class labels; Fig. 1b). Then, the model with the best regularization parameters was further tested on the evaluation set containing an independent set of 20% participants which the model had not seen during the training and validation phases. The above procedure was repeated 100 times to ensure stable performance was obtained on a large number of development-evaluation splits. The area under the receiver operating characteristics curve (AUC) was selected as the metric to quantify model performance, and reported AUCs are only from testing on the independent evaluation set.
To identify the set of most predictive features for each classifier (i.e., MDDPlacebo vs. HCPlacebo and MDDPlacebo vs. MDDAmisulpride), we adopted the following importance-guided sequential model selection procedure (Fig. 1a). Specifically, we first rank-ordered the features using the mean model weights across 100 implementations as a measure of predictability. Then, we built a series of truncated models such that each model only took the top k most predictive features as inputs to perform the classification tasks, with k varying from the top 1 most predictive feature to the number of participants involved in a given classifier. Imposing the number of participants as the upper limit was to ensure that models’ performance was not mainly driven by the regularization term. All truncated models underwent the nested cross-validation procedure described above and the test performance from each truncated model on the independent evaluation set was obtained. The set of features used by the truncated model achieving the highest AUC on the evaluation set were deemed as the optimal feature set.
After identifying the best truncated models for the classifiers, we compared the feature sets – both the selected regions and the regression weight signs (positive/negative) as they indicated the direction of the BOLD difference (greater for one class over another). Based on how we set up the classifiers, those regions shared by the MDDPlacebo vs. HCPlacebo and MDDPlacebo vs. MDDAmisulpride classifiers with convergent regression signs constitute a potential multivariate signature that we can use to assess amisulpride-induced BOLD normalization in MDD patients. We calculated signed BOLD sum scores by summing up the BOLD values of the convergent features multiplied by the regression weight sign to assess normalization. The convergent features should largely be absent in the set of highly-differentiating features of the MDDAmisulpride vs. HCPlacebo classifier if they have been normalized with amisulpride.
Statistical Analysis
The significance of the models’ performances against chance level was tested using a random permutation test scheme in which the truncated model based on the optimal feature set were re-trained on label shuffled training data (28). The entire test procedure was iterated 1000 times to empirically construct the null distribution of test AUCs. The p-values were obtained by comparing the AUC from the best truncated model based on unshuffled data against the empirical null distribution. The performances between models were statistically compared via Mann-Whitney U tests. Effect sizes between two distributions were calculated using Cohen’s d.
Results
Classification Performances
The best truncated models selected by the importance-guided model selection procedure (Fig. 1) based on most predictive features from whole-brain BOLD activations and striatal connectivity achieved high predictive performances (Table 2; see Fig. S1 for model performance as a function of top features). For both MDDPlacebo vs. HCPlacebo and MDDPlacebo vs. MDDAmisulpride, the AUC of the best truncated models were significantly above chance level (MDDPlacebo vs. HCPlacebo: mean AUC = 0.87, permutation testing p = 0.004; MDDPlacebo vs. MDDAmisulpride: mean AUC = 0.89, p = 0.002; Fig. 2a, b; Fig. S2). Predictive features displayed some collinearity, but collinearity did not account for the diminishing AUC returns of the lower-ranked predictive features (Supplementary Results; see Fig. S3 and Fig. S4). Compared with models trained using striatal features only (Supplementary Methods), the performances of the best truncated models based on whole-brain features were significantly better for both contrasts (p’s < 0.001, Mann-Whitney U test). The histograms of sum scores created by summing up the top feature values while taking into account the sign of the corresponding model weights demonstrated high separability between MDDPlacebo and HCPlacebo as well as between MDDPlacebo and MDDAmisulpride (Fig. 2c, d). Overall, these results indicate that our models were able to extract highly predictive information embedded in the whole-brain BOLD signal.
Table 2:
Classification performance for the best truncated models
| MDDPlacebo vs. HCPlacebo | MDDPlacebo vs. MDDAmisulpride | MDDPlacebo vs. HCPlacebo: Striatum Only | MDDPlacebo vs. MDDAmisulpride: Striatum Only | |||||
|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| AUC | 0.87 | 0.12 | 0.89 | 0.09 | 0.59 | 0.14 | 0.61 | 0.17 |
| Accuracy | 0.77 | 0.12 | 0.80 | 0.10 | 0.59 | 0.13 | 0.59 | 0.13 |
| Sensitivity | 0.84 | 0.18 | 0.89 | 0.11 | 0.58 | 0.25 | 0.65 | 0.19 |
| Specificity | 0.72 | 0.22 | 0.67 | 0.24 | 0.59 | 0.22 | 0.50 | 0.28 |
| Number of Features | 48 | 44 | 6 | 11 | ||||
Figure 2:
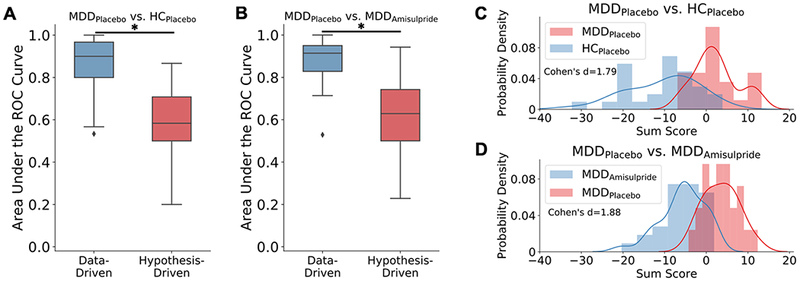
Comparing classification performance between the data-driven models based on features selected from the whole-brain and the hypothesis-driven models based only on striatal features for a) MDDPlacebo vs. HCPlacebo and b) MDDPlacebo vs. MDDAmisulpride classifications. Asterisks denote significantly different median area under the Receiver Operating Characteristic (ROC) curve measures between the data-driven and hypothesis-driven models as assessed by the Mann-Whitney U test. The black markers denote outliers falling outside the ±1.5 interquartile range. The histogram of the signed sum score from the model-identified most predictive brain regions show high separability between c) MDDPlacebo vs. HCPlacebo and d) MDDPlacebo vs. MDDAmisulpride.
Brain Regions Specific to Reward Anticipation
Positive model weights.
The best truncated model for MDDPlacebo vs. MDDAmisulpride identified the lateral orbitofrontal cortex (lOFC), visual cortex, anterior cingulate cortex (ACC), dorsomedial prefrontal cortex (dmPFC), mid-cingulate cortex (MCC), and precuneus as most predictive features with positive weights during reward anticipation (Fig. 3a; Table S1). This indicates that, within the MDD group, BOLD activation in these regions related to the contrast of reward cue minus neutral cue was reduced following administration of amisulpride compared to placebo. Critically, the lOFC, visual cortex, and MCC were also selected by the best MDDPlacebo vs. HCPlacebo model as top features having positive weights (Fig. 3b; Table S2), and at the same time these regions, except a right occipital region, were not among the most predictive features in the MDDAmisulpride vs. HCPlacebo model (Fig. S5). Collectively, these findings indicate that, within the MDD group, amisulpride largely normalized the heightened BOLD activation in these regions toward reward cues. Other regions with positive weights in the MDDPlacebo vs. HCPlacebo classification included the thalamus, supplementary motor area (SMA), and the ventromedial prefrontal cortex (vmPFC). Again, these regions were not among the top features in the MDDAmisulpride vs. HCPlacebo model (Fig. S5), suggesting that amisulpride mitigated the hyperactivation in these regions within the MDD group.
Figure 3:
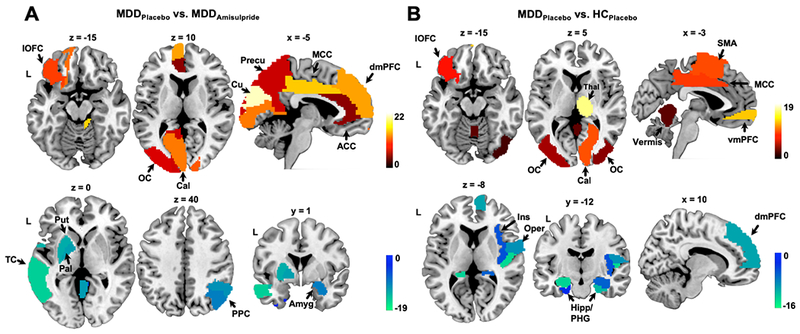
Weight maps showing the most predictive brain regions for the contrast of the reward minus neutral cue conditions. a) Weight map for the MDDPlacebo vs. MDDAmisulpride model. Positive weights indicate higher BOLD in the MDDPlacebo group relative to the MDDAmisulpride group and negative weights indicate the opposite direction. b) Weight map for the MDDPlacebo vs. HCPlacebo model, with positive weights indicating higher BOLD in the MDDPlacebo group relative to the HCPlacebo group and vice versa. ACC: anterior cingulate cortex; Amyg: amygdala; Cal: calcarine sulcus; Cu: cuneus; dmPFC: dorsomedial prefrontal cortex; Hipp: hippocampus; Ins: insula; lOFC: lateral orbitofrontal cortex; MCC: middle cingulate cortex; OC: occipital cortex; Oper: operculum; Pal: pallidum; PHG: parahippocampal gyrus; PPC: posterior parietal cortex; Precu: precuneus; Put: putamen; SMA: supplementary motor area; TC: temporal cortex; vmPFC: ventromedial prefrontal cortex.
Negative model weights.
Regions selected by the best MDDPlacebo vs. MDDAmisulpride model with negative model weights included the putamen, pallidum, amygdala, posterior parietal cortex (PPC), and temporal cortex (Fig. 3a; Table S1). The negative weights observed in the putamen and pallidum were consistent with the hypothesis that amisulpride might have increased dopaminergic signaling in the basal ganglia in MDD (10, 14). This effect is rather pronounced as the MDDAmisulpride vs. HCPlacebo model showed that the contrast of reward cue minus neutral cue evoked higher activation in the putamen in the MDDAmisulpride group even compared with the HCPlacebo group (Fig. S5). Within the MDDPlacebo group, reduced activation in the operculum, hippocampus, parahippocampal gyrus (PHG), and dmPFC was observed relative to HCs during reward anticipation (features in the MDDPlacebo vs. HCPlacebo model with negative weights; Fig. 3b; Table S2). The reduced activation in the hippocampus and operculum persisted in the MDDAmisulpride vs. HCPlacebo model (Fig. S5), indicating that amisulpride had limited effects in these regions.
Brain Regions Specific to Reward Consumption
Positive model weights.
Examining features selected from the contrast of reward minus no change outcomes in the MDDPlacebo vs. MDDAmisulpride model revealed that the lOFC, PPC, superior frontal gyrus, and the pre- and post-central gyrus were selected as most predictive features with positive weights (Fig. 4a, Table S3). This indicates reduced activation in these regions during reward consumption in MDDAmisulpride compared with MDDPlacebo. Of note, the lOFC and PPC emerged as among the most predictive features with positive weights in the MDDPlacebo vs. HCPlacebo model (Fig. 4c, Table S4). Additionally, while the lOFC hyperactivation was still observed in the MDDAmisulpride vs. HCPlacebo model, the PPC was not identified as a predictive feature (Fig. S5). Overall, these results suggest that, under placebo, the MDD group was characterized by increased BOLD activity in these regions during reward consumption relative to HC and that the hyperactivation was reduced by amisulpride. Other brain regions identified as most predictive features with positive weights in the MDDPlacebo vs. HCPlacebo model included the inferior frontal gyrus, PCC, precuneus, and MCC. The lack of predictability from these regions between MDDAmisulpride and HCPlacebo (Fig. S5) again suggests a mitigating effect of amisulpride on the hyperactivation in these regions.
Figure 4:
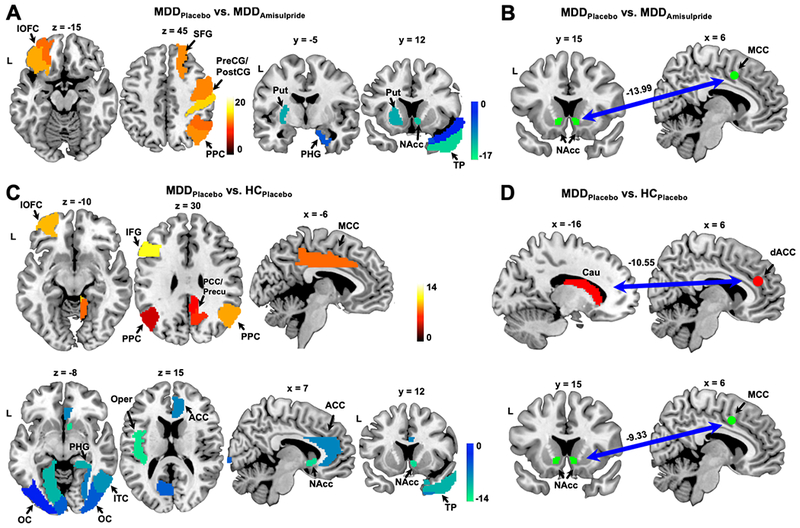
Weight maps showing the most predictive brain regions/connectivity for the contrast of reward minus no-change outcomes. a) Weight map for the MDDPlacebo vs. MDDAmisulpride model, with positive weights indicating higher BOLD in the MDDPlacebo group relative to the MDDAmisulpride group and vice versa. b) Negative weight assigned to the NAcc-MCC connectivity in the MDDPlacebo vs. MDDAmisulpride model. c) Weight map for the MDDPlacebo vs. HCPlacebo model. Positive weights indicate higher BOLD in the MDDPlacebo group relative to the MDDAmisulpride group and vice versa. d) Negative weights assigned to the Caudate-dACC and NAcc-MCC connectivity features by the MDDPlacebo vs. HCPlacebo model. Abbreviations followed those used in Fig. 2. Cau: caudate; dACC: dorsal anterior cingulate cortex; IFG: inferior frontal gyrus; ITC: inferior temporal cortex; PCC: posterior cingulate cortex; PreCG/PostCG: pre- and post-central gyrus; SFG: superior frontal gyrus; TP: temporal pole.
Negative model weights.
The most predictive regions from the contrast of reward minus no change outcomes with negative weights in the MDDPlacebo vs. MDDAmisulpride model included the putamen, NAcc, PHG, and temporal pole (Fig. 4a, Table S3), as well as the connectivity between the NAcc and MCC (Fig. 4b). This suggests that, within the MDD group, amisulpride increased BOLD activation and corticostriatal connectivity to reward feedback in these regions. Highlighting again convergence, the NAcc, PHG, temporal pole, and the NAcc-MCC connectivity were also selected as most predictive features having negative weights in the MDDPlacebo vs. HCPlacebo classification (Fig. 4c, d, Table S4), and none of these regions was selected as among the top predictive features in the MDDAmisulpride vs. HCPlacebo model (Fig. S5). Thus, in MDD, amisulpride normalized both hypoactivation and hypoconnectivity in response to rewards in these regions. Other most predictive features with negative weights in the MDDPlacebo vs. HCPlacebo model included the visual cortex, inferior temporal cortex, operculum, ACC, and the connectivity between the caudate and dACC. These features, except the caudate-dACC connectivity, were not identified as among the top features in the MDDAmisulpride vs. HCPlacebo model (Fig. S5), indicating increased activation to rewards in these regions following amisulpride administration in the MDD group. The fact that amisulpride did not normalize the hypoconnectivity between caudate and dACC in the MDD group is consistent with previously published ROI-based results obtained on a subset of the participants (10).
A Multivariate Signature of Normalization
The signed BOLD sum scores calculated from the convergent features across the MDDPlacebo vs. HCPlacebo and MDDPlacebo vs. MDDAmisulpride classifiers showed that the multivariate neural signature is significantly greater in the MDDPlacebo than in either MDDAmisulpride or HCPlacebo groups (Fig. 5; p’s < 0.001, Mann-Whitney U test) while the latter two groups were statistically equivalent based on equivalence testing (p = 0.01, see Supplementary Material for more information). Taken together, these results suggest that amisulpride normalized MDD-related abnormalities.
Figure 5: Multivariate signatures across groups demonstrated amisulpride-based brain normalization.
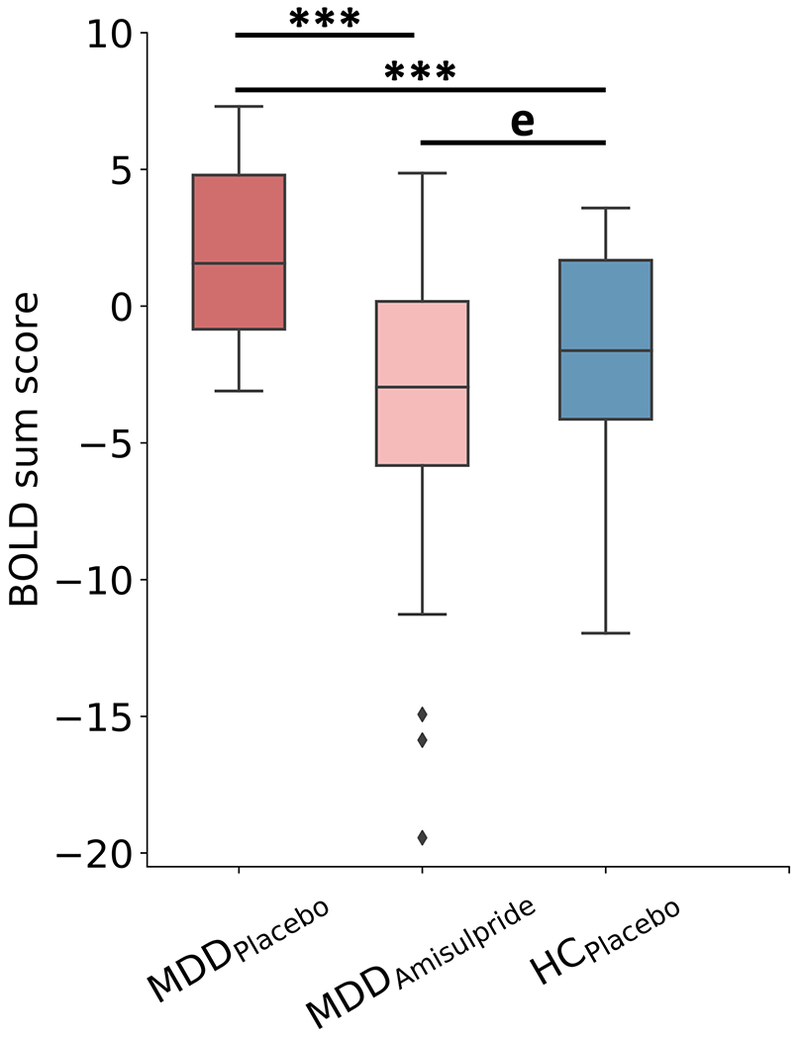
The signed BOLD sum scores calculated across the convergent features of MDDPlacebo vs. MDDAmisulpride and MDDPlacebo vs. HCPlacebo models suggest normalization of MDD-related abnormalities following amisulpride administration. MDDPlacebo patients had overall greater multivariate neural signatures compared to HCPlacebo or MDDAmisulpride (p<0.001 for both tests). Equivalence testing demonstrated that HCPlacebo and MDDAmisulpride had statistically equivalent (denoted using “e” in plot) scores (p=0.01).
Discussion
This study used a machine learning based approach to identify reliable brain-wide features that delineated MDD-related abnormalities as well as features linked to their normalization after an acute dopaminergic pharmacological challenge. In addition to increased striatal activation in the MDDAmisulpride relative to MDDPlacebo group (which is consistent with ROI-based conventional analyses of a smaller subset of the participants included here, 10), the classification model also identified an extensive set of reward-related brain regions differentiating these groups, which provided additional predictive power over striatal regions alone. Converging of features between the MDDPlacebo vs. MDDAmisulpride model and the MDDPlacebo vs. HCPlacebo model suggested that amisulpride had a bi-directionally normalizing effect on reward-related activation and functional connectivity of brain regions spanning the lOFC, NAcc, PHG, MCC, PPC, and areas of the visual cortex among depressed individuals. Taken together, these results highlight the unique contribution of machine learning-based approaches to examine brain-wide circuit engagement and potential normalization after a single dose. Such mechanistic evidence can help evaluate novel compounds before pursuing longer efficacy-oriented clinical trials with a compound. Overall, this study provided novel evidence for the mechanism through which (transient) dopaminergic enhancement might restore system-level activity during reward processing among individuals with MDD.
Amisulpride appeared to have bi-directional normalizing effects on brain activation and functional coordination among depressed individuals. Within the striatum, consistent with previous ROI-based analyses based on a subset of the participants used here (10), results from our classification models showed that decreased striatal/basal ganglia activation and corticostriatal connectivity among depressed individuals were enhanced following acute administration of amisulpride (see (29) for conceptually similar imaging findings using a single dose of the novel D2 antagonist lurasidone). This supports the validity of the importance-guided model selection procedure and fits the view that lower doses of amisulpride enhance dopaminergic signaling in the striatum (17).
Among regions outside the striatum, one notable finding was that increased lOFC activation during reward anticipation in MDD was reduced after administration of amisulpride. Neurophysiological evidence has shown that subpopulations of neurons in the lOFC respond to non-reward/unpleasant events and maintain elevated firing rate after such events (30). This led to the theory implicating overly reactive and prolonged activation of the lOFC non-reward circuit as a potential mechanism underlying depression (31). Previous studies have documented increased lOFC activation in MDD (32), and our result fits this theoretical view. In the MDDAmisulpride group, reduced lOFC activation suggests that amisulpride may normalize reward processing by decreasing lOFC hyperactivation, consistent with previous reports that improvements in depressive symptoms were accompanied by reduced lOFC activation (33) and electrical stimulation of the lOFC acutely improved depressive symptoms (34).
In addition to effects in frontostriatal circuitry, amisulpride restored hypoactivation in the parahippocampal gyrus and temporal pole in MDD. The hippocampus and parahippocampal complex connect with the medial OFC and are hypothesized to facilitate the formation of episodic memory regarding reward (35). Decreased hippocampal activation has emerged in MDD and prolonged/repeated depressive episodes have been linked to reduced hippocampal volume (36, 37). These abnormalities have been linked to dysfunctions in both memory encoding and retrieval characteristic of MDD, even after treatment (38, 39). The fact that amisulpride restored parahippocampal and temporal pole activation suggests that interventions aiming to increase dopaminergic signaling might improve encoding and retrieval of positive memories in MDD. However, it should be noted that hippocampal activation did not differentiate between the MDDAmisulpride and MDDPlacebo group, suggesting that the effects on memory might be limited following a single acute pharmacological challenge.
Hyperactivation in the mid-cingulate cortex towards the reward cue was also reduced among depressed individuals after amisulpride. Moreover, amisulpride also reduced reward cue-evoked activations in adjacent ACC and dmPFC. The supracallosal part of the cingulate cortex receives neuronal projections from the lOFC and is thought to also encode non-reward and punishing events such as physical and social pain (40, 41). A recent study has identified a nociceptive pathway between the mid-cingulate cortex and posterior insula responsible for generating a hypersensitive state for pain, providing a mechanism for the increased pain sensitivity by psychosocial factors (42). The reduced hyperactivation in these regions following amisulpride administration may indicate decreased sensitivity to negative affective states among individuals with MDD and therefore priming or biasing them toward reward.
In MDD, amygdalar activation evoked by reward cues was enhanced following amisulpride. Reduced amygdalar response to positive and rewarding stimulus, coupled with heightened amygdalar activation toward negative stimulus, are well-documented findings in MDD, which highlights an imbalanced reactivity toward emotionally-salient cues (43). Antidepressant treatment has been shown to address this imbalance by partially normalizing the bi-directional abnormal amygdalar activation (43, 44). These findings were further bolstered by the recent report that enhanced amygdalar response toward positive memories through real-time fMRI neurofeedback was associated with reduction in depressive symptoms (45). The increased amygdalar activation evoked by reward cues is consistent with these studies and implicates improved sensitivity toward reward following acute dopaminergic enhancement.
While several regions showed predictive power following the administration of amisulpride, it is difficult to assess whether changes in these regions reflected a direct modulation resulting from the enhanced dopaminergic signaling or alternatively reflected secondary responses through network interactions. Future studies could utilize network analysis and/or neural perturbation methods to further dissociate direct vs. indirect effects (34). In addition, amisulpride also has 5-HT7 antagonism (46), which has been hypothesized to contribute to its antidepressant property. While we cannot rule out that the effects observed here may be partially caused by this off-target mechanism, additional research is needed to distinguish the effect of dopaminergic enhancement vs. 5-HT7 antagonism of amisulpride. Lastly, we only focused on investigating the effects of dopaminergic enhancement on reward processing among depressed individuals. Future studies could seek to examine the effect of enhanced dopamine on whole-brain fMRI activity in depression under additional conditions; additionally, based on hypotheses of shared mesocorticolimbic dopaminergic abnormalities, this molecule could be tested in other disorders such as addiction or schizophrenia (e.g., 47, 48).
Supplementary Material
Acknowledgements
This project was supported by R01 MH068376, R37 MH068376 and R01MH095809 from the National Institute of Mental Health (Dr. Pizzagalli). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
ClinicalTrials.gov identifier: and
Competing Interests
Y.L., M.M., and P.A. are current or previous full-time employees at Blackthorn Therapeutics Inc. Over the past 3 years, D.A.P. has received consulting fees from Akili Interactive Labs, BlackThorn Therapeutics, Boehringer Ingelheim, Posit Science, and Takeda Pharmaceuticals and an honorarium from Alkermes for activities unrelated to the current review. All other authors report no biomedical financial interests.
Data Availability
Data are available at the NIMH Data Archive (https://nda.nih.gov/)
Code Availability
Analysis scripts are available upon request.
References
- 1.Calabrese JR, Fava M, Garibaldi G, Grunze H, Krystal AD, Laughren T, et al. (2014): Methodological approaches and magnitude of the clinical unmet need associated with amotivation in mood disorders. J Affect Disorders 168: 439–451. [DOI] [PubMed] [Google Scholar]
- 2.American Psychiatric Association (2000): Diagnostic and Statistical Manual of Mental Disorders, 4th ed, Text Revision: DSM-IV-TR. Washington, DC, American Psychiatric Publishing. [Google Scholar]
- 3.Wise RA (2004): Dopamine, learning and motivation. Nat Rev Neurosci 5: 483–494. [DOI] [PubMed] [Google Scholar]
- 4.Berridge KC, Kringelbach ML (2015): Pleasure systems in the brain. Neuron 86: 646–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Der-Avakian A, Markou A (2012): The neurobiology of anhedonia and other reward-related deficits. Trends Neurosci 35: 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Husain M, Roiser JP (2018): Neuroscience of apathy and anhedonia: a transdiagnostic approach. Nat Rev Neurosci 19: 470–484. [DOI] [PubMed] [Google Scholar]
- 7.Pizzagalli DA, Holmes AJ, Dillon DG, Goetz EL, Birk JL, Bogdan R, et al. (2009): Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am J Psychiatry 166: 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pizzagalli DA (2014): Depression, stress, and anhedonia: toward a synthesis and integrated model. Clin Psychology 10: 393–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Admon R, Nickerson LD, Dillon DG, Holmes AJ, Bogdan R, Kumar P, et al. (2014): Dissociable cortico-striatal connectivity abnormalities in major depression in response to monetary gains and penalties. Psychol Med 45: 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Admon R, Kaiser RH, Dillon DG, Beltzer M, Goer F, Olson DP, et al. (2017): Dopaminergic enhancement of striatal response to reward in major depression. Am J Psychiatry 174: 378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McClure SM, York MK, Montague RP (2004): The neural substrates of reward processing in humans: the modern role of fMRI. Neurosci 10: 260–268. [DOI] [PubMed] [Google Scholar]
- 12.Oldham S, Murawski C, Fornito A, Youssef G, Yücel M, Lorenzetti V (2018): The anticipation and outcome phases of reward and loss processing: a neuroimaging meta‐analysis of the monetary incentive delay task. Hum Brain Mapp 39: 3398–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson RP, Colizzi M, Bossong MG, Allen P, Kempton M, MTAC, et al. (2018): The neural substrate of reward anticipation in health: a meta-analysis of fMRI findings in the monetary incentive delay task. Neuropsychol Rev 28: 496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schott BH, Minuzzi L, Krebs RM, Elmenborst D, Lang M, Winz OH, et al. (2008): Mesolimbic functional magnetic resonance imaging activations during reward anticipation correlate with reward-related ventral striatal dopamine release. J Neurosci 28: 14311–14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amore M, Jori M (2001): Faster response on amisulpride 50 mg versus sertraline 50–100 mg in patients with dysthymia or double depression: a randomized, double-blind, parallel group study. Int Clin Psychopharm 16: 317–324. [DOI] [PubMed] [Google Scholar]
- 16.Cassano G, Jori M (2002): Efficacy and safety of amisulpride 50 mg versus paroxetine 20 mg in major depression: a randomized, double-blind, parallel group study. Int Clin Psychopharm 17: 27–32. [DOI] [PubMed] [Google Scholar]
- 17.Rosenzweig P, Canal M, Patat A, Bergougnan L, Zieleniuk I, Bianchetti G. A review of the pharmacokinetics, tolerability and pharmacodynamics of amisulpride in healthy volunteers. Hum Psychopharmacol. 2002. January;17(1):1–13. [DOI] [PubMed] [Google Scholar]
- 18.Viviani R, Graf H, Wiegers M, Abler B (2013): Effects of amisulpride on human resting cerebral perfusion. Psychopharmacology 229: 95–103. [DOI] [PubMed] [Google Scholar]
- 19.Metzger CD, Wiegers M, Walter M, Abler B, Graf H (2016): Local and global resting state activity in the noradrenergic and dopaminergic pathway modulated by reboxetine and amisulpride in healthy subjects. Int J Neuropsychoph 19: pyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forbes EE, Hariri AR, Martin SL, Silk JS, Moyles DL, Fisher PM, et al. (2009): Altered striatal activation predicting real-world positive affect in adolescent major depressive disorder. Am J Psychiatry 166: 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar P, Waiter G, Ahearn T, Milders M, Reid I, Steele JD (2008): Abnormal temporal difference reward-learning signals in major depression. Brain 131: 2084–2093. [DOI] [PubMed] [Google Scholar]
- 22.Stoy M, Schlagenhauf F, Sterzer P, Bermpohl F, Hägele C, Suchotzki K, et al. (2012): Hyporeactivity of ventral striatum towards incentive stimuli in unmedicated depressed patients normalizes after treatment with escitalopram. J Psychopharmacol 26: 677–688. [DOI] [PubMed] [Google Scholar]
- 23.Knutson B, Westdorp A, Kaiser E, Hommer D (2000): FMRI visualization of brain activity during a monetary incentive delay task. Neuroimage 12: 20–27. [DOI] [PubMed] [Google Scholar]
- 24.Knutson B, Bhanji JP, Cooney RE, Atlas LY, Gotlib IH (2008): Neural responses to monetary incentives in major depression. Biol Psychiatry 63: 686–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. (2002): Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage 15: 273–289. [DOI] [PubMed] [Google Scholar]
- 26.Admon R, Holsen LM, Aizley H, Remington A, Whitfield-Gabrieli S, Goldstein JM, et al. (2015): Striatal hypersensitivity during stress in remitted individuals with recurrent depression. Biol Psychiatry 78: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou H, Hastie T (2005): Regularization and variable selection via the elastic net. J Royal Statistical Soc: Ser B Statistical Methodol 67: 301–320. [Google Scholar]
- 28.Ojala M, Garriga GC (2009): Permutation tests for studying classifier performance. 2009 Ninth IEEE Int Conf Data Min: 908–913. [Google Scholar]
- 29.Wolke SA, Mehta MA, O’Daly O, Zelaya F, Zahreddine N, Keren H, et al. (2019): Modulation of anterior cingulate cortex reward and penalty signalling in medication-naive young-adult subjects with depressive symptoms following acute dose lurasidone. Psychol Med. 49: 1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thorpe S, Rolls ET, Maddison S (1983): The orbitofrontal cortex: neuronal activity in the behaving monkey. Exp Brain Res 49: 93–115. [DOI] [PubMed] [Google Scholar]
- 31.Rolls ET (2016): A non-reward attractor theory of depression. Neurosci Biobehav Rev 68: 47–58. [DOI] [PubMed] [Google Scholar]
- 32.Cheng W, Rolls ET, Qiu J, Liu W, Tang Y, Huang C, et al. (2016): Medial reward and lateral non-reward orbitofrontal cortex circuits change in opposite directions in depression. Brain 139: 3296–3309. [DOI] [PubMed] [Google Scholar]
- 33.Brody AL, Saxena S, Mandelkern MA, Fairbanks LA, Ho ML, Baxter LR (2001): Brain metabolic changes associated with symptom factor improvement in major depressive disorder. Biol Psychiatry 50: 171–178. [DOI] [PubMed] [Google Scholar]
- 34.Rao VR, Sellers KK, Wallace DL, Lee MB, Bijanzadeh M, Sani OG et al. (2018): Direct electrical stimulation of lateral orbitofrontal cortex acutely improves mood in individuals with symptoms of depression. Curr Biol 28: 3893–3902. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki WA, Naya Y (2014): The perirhinal cortex. Annu Rev Neurosci 37: 39–53. [DOI] [PubMed] [Google Scholar]
- 36.Milne A, MacQueen GM, Hall GBC (2012): Abnormal hippocampal activation in patients with extensive history of major depression: an fMRI study. J Psychiatr Neurosci 37: 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKinnon MC, Yucel K, Nazarov A, MacQueen GM (2009): A meta-analysis examining clinical predictors of hippocampal volume in patients with major depressive disorder. J Psychiatry Neurosci Jpn 34: 41–54. [PMC free article] [PubMed] [Google Scholar]
- 38.Dillon DG (2015): The neuroscience of positive memory deficits in depression. Front Psychol 6: 1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dillon DG, Pizzagalli DA (2018): Mechanisms of memory disruption in depression. Trends Neurosci 41: 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grabenhorst F, Rolls ET (2011): Value, pleasure and choice in the ventral prefrontal cortex. Trends Cogn Sci 15: 56–67. [DOI] [PubMed] [Google Scholar]
- 41.Rotge JY, Lemogne C, Hinfray S, Huguet P, Grynszpan O, Tartour E, et al. (2015): A meta-analysis of the anterior cingulate contribution to social pain. Soc Cogn Affect Neur 10: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan L, Pelzer P, Heinl C, Tang W, Gangadharan V, Flor H, et al. (2017): A pathway from midcingulate cortex to posterior insula gates nociceptive hypersensitivity. Nat Neurosci 20: 1591–1601. [DOI] [PubMed] [Google Scholar]
- 43.Victor TA, Furey ML, Fromm SJ, Öhman A, Drevets WC (2010): Relationship between amygdala responses to masked faces and mood state and treatment in major depressive disorder. Arch Gen Psychiat 67: 1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu CH, Williams SCR, Cleare AJ, Brammer MF, Walsh ND, Kim J, et al. (2004): Attenuation of the neural response to sad faces in major depression by antidepressant treatment: a prospective, event-related functional magnetic resonance imaging study. Arch Gen Psychiatry 61: 877–889. [DOI] [PubMed] [Google Scholar]
- 45.Young KD, Siegle GJ, Zotev V, Phillips R, Misaki M, Yuan H, et al. (2017): Randomized clinical trial of real-time fMRI amygdala neurofeedback for major depressive disorder: effects on symptoms and autobiographical memory recall. Am J Psychiatry 174: 748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abbas AI, Hedlund PB, Huang XP, Tran TB, Meltzer HY, Roth BL (2009): Amisulpride is a potent 5-HT7 antagonist: relevance for antidepressant actions in vivo. Psychopharmacology 205: 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robison AJ, Nestler EJ (2011): Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 12: 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laviolette SR (2007): Dopamine Modulation of Emotional Processing in Cortical and Subcortical Neural Circuits: Evidence for a Final Common Pathway in Schizophrenia? Schizophrenia Bull. 33: 971–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.