

Abstract
The intracellular levels of the cytoprotective enzyme heme oxygenase-1 (HO-1) are tightly controlled. Here, we reveal a novel mechanism preventing the exaggerated expression of HO-1. The analysis of mice with a knock-out in the ubiquitin E3 ligase seven in absentia homolog 2 (SIAH2) showed elevated HO-1 protein levels in specific organs such as heart, kidney and skeletal muscle. Increased HO-1 protein amounts were also seen in human cells deleted for the SIAH2 gene. The higher HO-1 levels are not only due to an increased protein stability but also to elevated expression of the HO-1 encoding HMOX1 gene, which depends on the transcription factor nuclear factor E2-related factor 2 (NRF2), a known SIAH2 target. Dependent on its RING (really interesting new gene) domain, expression of SIAH2 mediates proteasome-dependent degradation of its interaction partner HO-1. Additionally SIAH2-deficient cells are also characterized by reduced expression levels of glutathione peroxidase 4 (GPX4), rendering the knock-out cells more sensitive to ferroptosis.
Subject terms: Biocatalysis, Chemical modification, Proteolysis
Introduction
Ubiquitin E3 ligases regulate the activity and turnover of many target proteins, thus controlling key features such as metabolism, stress signaling and cell cycle progression1. The RING family of ubiquitin E3 ligases comprises the SIAH family. The human genome encodes SIAH1 and the homologous SIAH2 protein as well as SIAH3, which lacks a functional RING domain and is only expressed in a limited subset of cancer cell lines2. SIAH1 and SIAH2 proteins have a largely divergent N-terminal part, but are highly conserved in the RING domain. Both E3 ligases have overlapping and distinct substrate binding abilities3. The enzymatic activity of SIAH proteins typically leads to poly-ubiquitination and subsequent proteasomal degradation of its client proteins, but also SIAH-mediated mono- and di-ubiquitination has been described4–6. SIAH function can be regulated at several levels, as these E3 ligases can form homo- and heterodimers, and also associate with regulatory proteins4,7. The substrate binding ability of SIAH proteins is additionally regulated by phosphorylation, which can be mediated by several kinases8,9. The intracellular levels of SIAH proteins are typically very low, due to ongoing auto-ubiquitination and degradation, as well as by association with ubiquitin carboxyl-terminal hydrolase 13 (USP13)10. The SIAH2 protein has been implicated in the regulation of many different biological processes including the control of cell metabolism by degradation of α-ketoglutarate dehydrogenase, thus shifting glutamine metabolism from oxidation to reductive carboxylation under hypoxic conditions11. SIAH2 contributes to control several hypoxia-regulated pathways exerted by the p53 family member p73, Hippo signaling and homeodomain-interacting protein kinase 2 (HIPK2)8,12,13. Several studies have shown that SIAH2 indirectly controls the abundance of the hypoxia-induced transcription factor HIF-1α by SIAH2-mediated degradation of hypoxia regulators, such as prolyl hydroxylase (PHD)1 and PHD314. SIAH2 is also a relevant regulator of reactive oxygen species (ROS) metabolism, as this E3 ligase does not only regulate the cell response to low oxygen and the stability of several metabolic enzymes, but also of transcription factors such as NRF1 and NRF2, which contribute to the expression of antioxidant proteins15,16.
One NRF2-regulated anti-oxidative protein is heme oxygenase 1 (HO-1), an inducible and rate-limiting enzyme in the catabolism of heme. Pro-oxidative conditions, inflammation and further adverse conditions lead to increased HO-1 expression to allow HO-1-mediated removal of the pro-oxidant heme17. HO-1 catalyses the oxidative cleavage of heme to biliverdin, ferrous iron (Fe2+), and carbon monoxide18. This enzymatic activity is important to mediate the cytoprotective effect of HO-1 against oxidative injury. While HO-1 mostly acts as a pro-survival factor, it can also promote ferroptosis, an iron- and lipid peroxidation-dependent form of cell death19,20. HO-1 is found as a membrane protein in the smooth endoplasmic reticulum (sER) with most of the protein oriented towards the cytosol. Adverse conditions lead to the C-terminal cleavage of HO-1, allowing its release from the sER and the diffusion to various intracellular localizations, including the nucleus and mitochondria21,22.
Here, we reveal a novel role of SIAH2 as controller of oxygen homeostasis by the identification of HO-1 as SIAH2 target. The stabilization of HO-1 and the downregulation of GPX4 make SIAH2-deficient cells more vulnerable to ferroptosis.
Results
SIAH2 regulates HO-1 protein levels
The contribution of SIAH2 and also HO-1 in oxygen-dependent signalling raised the question of a mutual cross-regulation between both proteins. As a starting point, we tested HO-1 protein levels in immortalized mouse cardiac fibroblasts (i-MCF) derived from control or SIAH2-knock-out animals. Western blot analysis showed increased HO-1 protein levels in i-MCF cells derived from SIAH2 knock-out mice (Fig. 1A). As the mouse and human SIAH-encoding genes show differences in sequence and gene organization23,24, we investigated whether this type of cross-regulation between SIAH2 and HO-1 also occurs in human cells. We used the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system to interfere with SIAH2 expression in human embryonic kidney (HEK)293T cells, thus generating two independent cell clones harbouring the same Indel mutant resulting in expression of only the first 7 amino acids of human SIAH2 (Fig. 1B). Comparison of these cells with wild-type controls showed an inverse correlation of HO-1 and SIAH2 protein abundance (Figs. 1C and 2A). This increase of HO-1 protein was also seen in cells expressing inducible SIAH2-specific shRNA (Fig. 2B). Collectively, these data show that reduction of SIAH2 expression results in increased HO-1 protein levels, irrespective of the method of SIAH2 downregulation or species.
Figure 1.

Increased levels of HO-1 protein in SIAH2-deficient cells. (A) i-MCFs from wild-type (WT) and SIAH2 knock-out mice were lysed and equal amounts of protein contained in cell lysates was tested by Western blotting for the expression of the indicated proteins using specific antibodies. The positions of molecular weight (MW) markers are indicated. (B) The genomic DNA of two independent 293T cell clones engineered by CRISPR-Cas9 to contain an Indel mutation in the first SIAH2-encoding exon and defect on SIAH2 protein expression were isolated. PCR amplification of the relevant genomic region and sequencing showed the same mutation in both cell clones designated SIAH2-KO-1 and SIAH2-KO-2. The consequences of this mutation on protein translation are shown, the region undergoing a frame shift is indicated by red. (C) Equal numbers of 293T wild-type cells and two independently obtained derivatives thereof with a mutation in the SIAH2 gene were lysed and tested for the expression of the indicated proteins by immunoblotting (left). The positions of non-specific bands are indicated by asterisks. The right part shows a quantification of HO-1 expression from four independent experiments. Protein amounts of HO-1 and β-Actin were quantified using the ChemiDoc Imaging System. Relative protein amounts were normalized to β-Actin and HO-1 expression in wild-type cells was set as one, the median and 25–75% quantiles are indicated. SE: short exposure, LE: long exposure.
Figure 2.
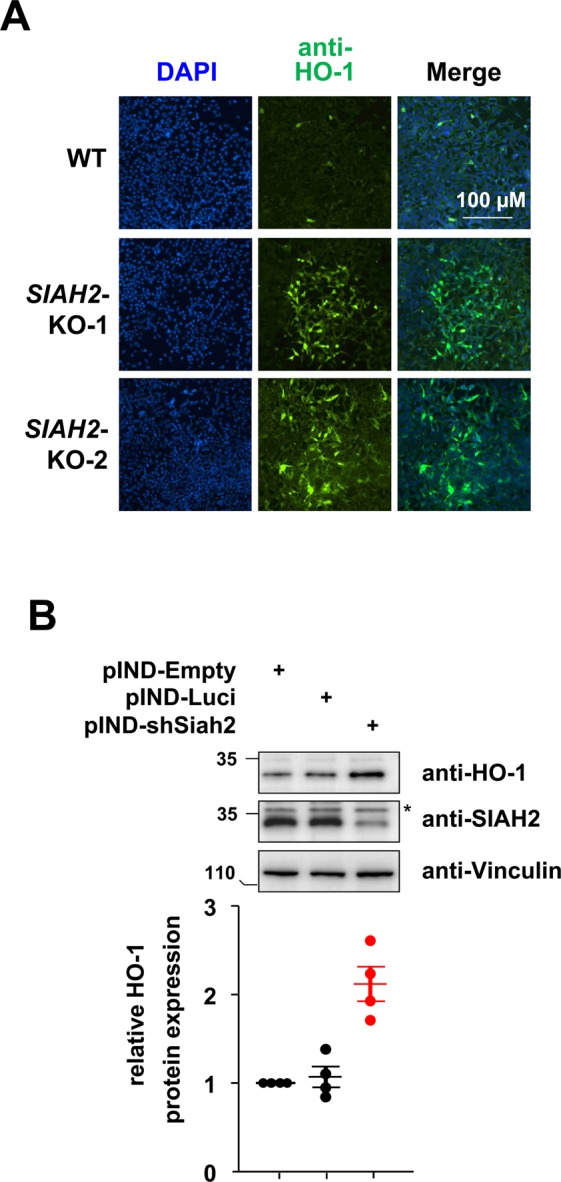
Inverse correlation of HO-1 and SIAH2 abundance. (A) The indicated 293T wild-type cells and two SIAH2-deleted cell clones were stained for HO-1 expression by indirect immunofluorescence, the nuclear DNA was stained with DAPI. (B) Cell clones with a stably integrated pINDUCER plasmid allowing doxycycline-inducible downregulation of luciferase (control) or SIAH2 were treated for 4 days with Dox (1 μg/ml) and analysed for expression of HO-1 and SIAH2 by Western blotting, the asterisk indicates a non-specific band. The lower part shows a quantification of four independent experiments.
SIAH2-mediated restriction of HO-1 abundance is organ-specific
Next, we determined whether SIAH2-mediated control of HO-1 protein abundance occurs in all organs. Heart, kidney and liver samples from SIAH2 knock-out mice presented with elevated HO-1 abundance compared to wild-type organs (Fig. 3A), consistent with the results we obtained from murine i-MCFs. A quantitative analysis of Western blot signals from multiple experiments showed only slightly increased HO-1 levels in skeletal muscle, but unchanged HO-1 abundance in the lung and brain of SIAH2-deficient animals (Fig. 3B). Collectively, these data suggest that the suppressive effects of SIAH2 on HO-1 protein abundance are confined to specific organs and probably also cell types.
Figure 3.
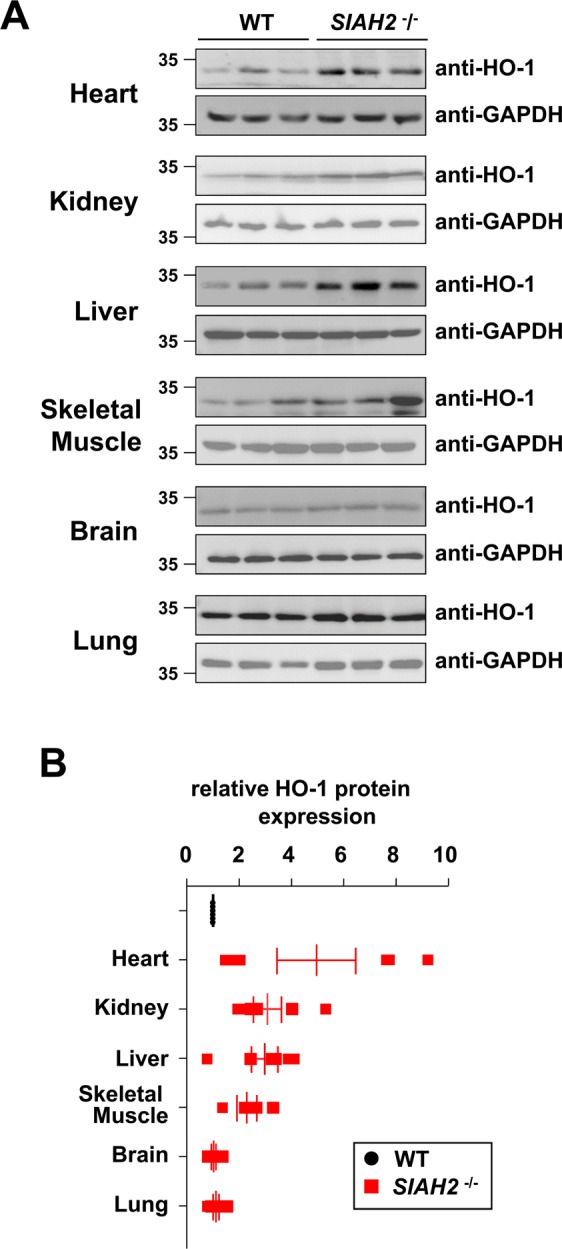
Organ-specific consequences of SIAH2 knock-out on HO-1 abundance. Control wild-type mice or SIAH2−/− mice were sacrificed and tissue extracts were prepared from the indicated different organs. Western blot analysis was performed to reveal relative expression of HO-1. The results in (A) show immunoblots from samples tested in triplicates on the same gel. (B) This quantification shows relative HO-1 expression from 6 different samples per genotype, relative protein amounts were normalized to GAPDH. HO-1 expression in the respective wild-type tissues was set as one, the median and 25–75% quantiles are indicated.
SIAH2 loss results in increased HO-1 mRNA and protein abundance
To test whether the increased HO-1 protein levels correlate with increased mRNA levels, we performed RT-qPCR experiments. We found elevated HO-1 encoding mRNAs in SIAH2-deficient i-MCFs and 293T cells (Fig. 4A,B), suggesting an impact of SIAH2 on the expression of the HMOX1 gene. In order to test a potential influence of SIAH2 on HO-1 protein stability, de novo protein synthesis was blocked by Anisomycin and HO-1 decay monitored over 9 h. The quantitative analysis of these experiments detected increased protein stability of HO-1 in SIAH2-deficient cells (Fig. 4C). As SIAH2 can act as an ubiquitin E3 ligase, we tested whether HO-1 levels are controlled by the ubiquitin/proteasome system. Inhibition of the proteasome by lactacystin dose-dependently resulted in increased HO-1 protein levels (Fig. 4D), showing the importance of constitutive HO-1 degradation by the ubiquitin/proteasome system.
Figure 4.

Increased levels of HO-1 mRNA and elevated HO-1 protein stability in SIAH2-deficient cells. (A) i-MCFs from wild-type and SIAH2 knock-out mice were analysed for expression levels of HMOX1 mRNAs by RT-qPCR, results from 4 experiments are shown. (B) The experiment was done as in (A) with the difference that HMOX1 mRNAs from wild-type and SIAH2 knock-out 293T cells were used. (C) The indicated cell lines were treated with Anisomycin (5 µM) for different periods as shown and protein expression of HO-1 was analysed by immunoblotting, a long exposure (LE) and short exposure (SE) is displayed. The lower part shows HO-1 decay curves from three experiments, standard deviations are shown. (D) 293T cells were treated for 8 h with lactacystin and HO-1 expression was determined by immunoblotting.
SIAH2 targets HO-1 for degradation
To test whether SIAH2 expression leads to HO-1 degradation, 293T cells were transfected to express Flag-tagged HO-1 alongside increasing amounts of HA-SIAH2. We found a dose-dependent reduction of HO-1 protein levels inversely correlated with increasing amounts of SIAH2 (Fig. 5A). SIAH1 and SIAH2 have overlapping but also distinct substrates. To test if SIAH1 is also capable of controlling HO-1 abundance, we expressed increasing SIAH1 alongside HO-1. Similar to SIAH2, SIAH1 is able in a dose-dependent fashion to reduce HO-1 protein levels (Fig. 5B), although HO-1 degradation was less efficient and complete. SIAH2-mediated target degradation commonly depends on an intact RING domain, which binds E2 proteins and is important for auto- and trans-ubiquitination25. Expression of the wild-type, but not of a RING-mutated version of SIAH2 (SIAH2 RM), resulted in the reduced abundance of HO-1 (Fig. 5C), albeit the SIAH2 RM being expressed at much higher levels due to defective auto-ubiquitination. These data suggest that SIAH2 causes, dependent on the functionality of its RING domain, reduced HO-1 levels, raising the possibility that it serves as a direct ubiquitin E3 ligase for HO-1. To test for a possible interaction between SIAH2 and HO-1, co-immunoprecipitation experiments were performed. Immunoprecipitation of the endogenous SIAH2 protein allowed the specific detection of co-immunoprecipitated HO-1, while vice versa SIAH2 was found in association with the immunoprecipitated endogenous HO-1 protein (Fig. 5D). Together, this data shows that SIAH2 and HO-1 interact, and this interaction results in the proteasomal degradation of the HO-1 protein.
Figure 5.

SIAH2-mediated downregulation of its interaction partner HO-1. (A) 293T cells were transfected to express increasing amounts of HA-SIAH2 along with Flag-tagged HO-1 as shown. Two days later cells were harvested and analysed by Western blotting as shown. (B) The experiment was done as in (A) with the exception that increasing amounts of HA-SIAH1 were expressed. (C) 293T cells were transfected to express HA-tagged HO-1, either alone or together with wild-type SIAH2 or a SIAH2 RING domain mutant (RM). The relative expression of proteins is shown, please note that the SIAH2 RM shows an increased expression level due to the lack of auto-ubiquitination and degradation. (D) Lysates from 293T cells were used for immunoprecipitation (IP) using antibodies for SIAH2 (upper) or HO-1 (lower) in combination with control IgGs. The precipitated samples were analysed by Western Blotting using appropriate antibodies, input samples ensured correct protein expression.
Functional consequences of SIAH2-mediated HO-1 degradation
To elucidate the physiological and functional consequences of the SIAH2/HO-1 interaction, we first investigated the hypoxic response pathway as both proteins have been shown to be involved. 293T wild-type and SIAH2-deficient cells were treated with the iron chelator deferoxamine (DFO), a chemical hypoxia-mimetic. DFO-dependent stabilization of HIF-1α was only seen in wild-type cells, but not in SIAH2-deficient cells (Fig. 6a), in line with previous publications26. Additionally, consistent with the literature we also observed a reduced HO-1 expression in DFO-treated cells27, which also occurred in SIAH2-deficient cells (Fig. 6a). These results suggest that SIAH2 is unlikely to participate in DFO-dependent HO-1 degradation.
Figure 6.
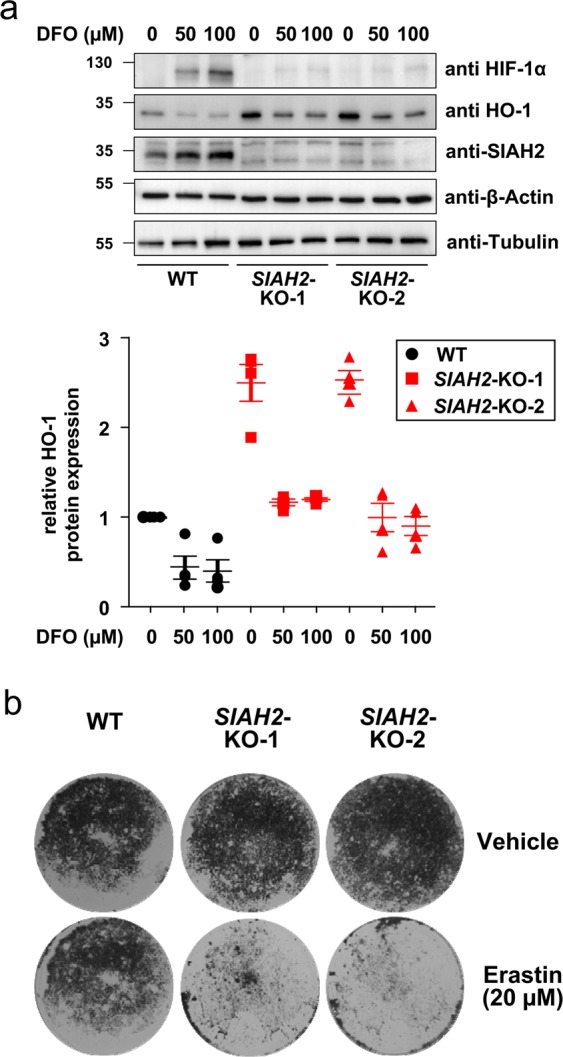
Functional analysis of SIAH2-mediated HO-1 degradation. (a) 293T wild-type and SIAH2 knock-out cells were treated with the indicated DFO concentrations for 8 h, followed by analysis of HIF-1α, SIAH2 and HO-1 expression by Western blotting. The lower part shows a quantification of HO-1 expression from three independent experiments. (b) The indicated cell lines were treated with DMSO as a control or with erastin to induce ferroptosis. After two days, the surviving cells were grown to colonies and stained with crystal violet, one out of three independent experiments with similar results is shown.
Another HO-1 regulated process is ferroptosis, a regulated but non-apoptotic form of cell death that can be induced experimentally by compounds such as erastin28. Ferroptosis is characterized by high ROS levels derived from iron metabolism and lipid peroxidation. Depending on the stimulus and cell type, HO-1 can mitigate or enhance ferroptosis19,29,30. To test the effects of SIAH2 deletion on erastin-induced ferroptosis, wild-type and SIAH2-deficient cells were used. Two days after induction of ferroptosis, dead cells were aspirated off and the surviving cells were grown to colonies. Trypan blue staining of the surviving cell clones showed that SIAH2-deficient cells were more susceptible to ferroptosis (Fig. 6B), raising the possibility that elevated HO-1 levels contribute to enhanced ferroptosis in the knock-out cells. Similar results were obtained after induction of ferroptosis with RSL3, an agent that induces ferroptosis via inhibition of GPX4 (Suppl. Fig. S1)31. The occurrence of ferroptosis in response to erastin and RSL3 treatment was confirmed by the cell protective function of Ferrostatin (Suppl. Fig. S2), a known suppressor of this form of cell death32.
To monitor the differential sensitivity to ferroptosis by an alternative experimental approach, we determined the levels of glutathione peroxidase 4 (GPX4), which inhibits ferroptosis and is a widely used marker for this process31. Increased sensitivity of SIAH-2-deficient cells to ferroptosis was mirrored at the level of diminished GPX4 protein expression in untreated, erastin-treated (Fig. 7A) and RSL3-treated SIAH2-deficient cells (Suppl. Fig. S3). This reduced expression was not reflected at the mRNA level (Suppl. Fig. S4), suggesting SIAH2-dependent GPX4 regulation at the posttranscriptional level. Differential GPX4 expression also occurred in wild-type and SIAH2 knock-out i-MCFs (Fig. 7B), showing that SIAH2-dependent expression changes of GPX4 is reproducible across species and cell lines. Re-expression of GPX4 largely rescued SIAH2-deficient cells from increased ferroptosis (Fig. 7C), suggesting a contribution of GPX4 for the ferroptosis-antagonizing function of SIAH2.
Figure 7.

Effect of SIAH2-regulated GPX4 expression on ferroptosis. (A) The indicated cells were treated with DMSO or erastin and two days later cell extracts were prepared to measure expression of the indicated proteins by immunoblotting. The lower part shows a quantification of GPX4 expression. (B) The experiment was done as in (A) with the exception that GPX4 expression was determined in untreated wildtype and SIAH2 knock-out i-MCFs. (C) The indicated cells were transfected to express GFP-GPX4. The next day, a fraction of the cells was lysed and used for immunoblotting to determine GFP-GPX4 expression (lower). The other cells were treated with erastin for 40 h, followed by determination of cell death using the PrestoBluecell viability reagent (upper). Error bars show SEM from two experiments measured in triplicates.
Discussion
Here, we show that elimination or downregulation of SIAH2 expression by genetic knock-out, CRISPR-Cas9-mediated inactivation or shRNA-mediated knockdown leads to elevated HO-1 protein levels. This increase is also reflected at the mRNA level and readily explained by increased stability of transcription factor NRF2, a known SIAH2 target and master regulator of HO-1 expression16,33. It will be interesting to study whether SIAH2 only contributes to the control of basal HO-1 turnover in unstressed cells, or whether it also participates in signal-regulated HO-1 degradation pathways.
Although HO-1 per se is mainly cytoprotective, permanently elevated levels have negative physiological effects, as revealed by the analysis of transgenic mice overexpressing this enzyme. Lung-specific overexpression of HO-1 causes lower amounts of LPS-induced cytokine levels, thus impairing the immune response34. In addition, HO-1 overexpression causes neurological disorders and enhances carcinogenesis35–37. Also lack of HO-1 expression is detrimental, as mice with a global HO-1 knock-out show a reduced stress defense and iron reutilization38,39, showing the necessity to appropriately balance HO-1 expression and protein abundance. This is achieved by multiple layers of regulation, including control of de novo transcription, but also through regulation of HO-1 mRNA deadenylation and turnover40. HO-1 protein is regulated at various levels, including the occurrence of posttranslational modifications, such as phosphorylation and ubiquitination41,42. In addition, the protein stability of HO-1 is positively regulated by the 14-3-3-ζ protein43 and downregulated by valproic acid, an anti-epileptic drug that inhibits histone deacetylases44.
This study shows that also overexpression of SIAH1 causes a reduction in HO-1 protein levels, but it remains unclear whether this effect is mediated by its known ability to associate with SIAH23. The differential contribution of various distinct ubiquitin E3 ligase and the occurrence of further mechanisms controlling HO-1 protein homeostasis could potentially explain the surprising finding that SIAH2 knock-out results in organ-specific effects on HO-1 protein levels and potentially occurs with a high volatility. This finding could also be attributable to differences in the relative amount or intracellular localization of HO-1, as for example mitochondrial HO-1 would escape from degradation of the SIAH2 protein, which locates to the cytosol and nucleus. Alternatively, these organ-specific effects could also be due to differences in relative SIAH2 protein expression. The low expression of the SIAH2 protein did not allow its mass spectrometric detection in mouse organs (https://www.ebi.ac.uk/gxa/home), while SIAH2 and SIAH1a/b encoding mRNAs were present in all organs at different levels (Suppl. Fig. S5) (http://biogps.org),
The increased vulnerability of SIAH2 knock-out cells to ferroptosis is probably due to several factors, including the increased expression of pro-ferroptotic HO-1 and the decreased expression of GPX4, a key factor for this iron-catalysed necrotic pathway. Ferroptosis is specifically suited for the elimination of cancer cells, which are more vulnerable to ferroptosis, as they typically have increased iron demand compared with normal, non-cancer cells45. As inhibition of GPX4 activity exhibit a high selectivity and potency in killing of specific cancers, such as clear-cell carcinomas46, an increased understanding of the mechanisms controlling GPX4 levels will be important.
Materials and Methods
Antibodies, plasmids and treatments
All the information is given in the Supplementary Table 1.
Cell culture and transfections
293T (ATCC CRL-3216), Phoenix ECO (ATCC CRL-3214) and i-MCF cells (generated for this study by immortalisation of primary mouse cells as described below) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) medium supplemented with 10% fetal calf serum (FCS), Glutamine, Penicillin and Streptomycin (P/S) and occasionally in MycoZapPlus. Cells were grown at 37 °C and 5% CO2 and counted prior to plating using a LUNA II automated cell counter according to manufacturer’s instructions. All the transfections were performed using linear polyethylenimine (PEI) as published47.
RNA extraction and qRT-PCR analysis
RNA was extracted using RNeasy kit from Qiagen as described in the manufacturer’s instructions. Concentration of RNA was measured using Eppendorf-Biophotometer, 1 µg of RNA was used for cDNA synthesis using the PrimeScript RT Master Mix from Takara Bio Inc. Generated cDNA was diluted 1:10 using sterile RNase free water and equal volumes were used as a template for amplification in a qRT-PCR. Amplification was performed using intron-flanking primers in 2X Absolute qPCR SYBR Green ROX mix (Thermo) using a Applied Biosystems 7300 device. Every reaction was performed as duplicates and quantified with the ΔΔCT-method. Therefore, threshold cycles (CT) of target genes were normalized to a housekeeping gene (ACTB). The resulting ΔCT were compared to control samples and relative mRNA expression was calculated by R = 2−ΔΔCT.
Protein extraction and Western blotting
Cells were washed in PBS and lysed in appropriate volume of RIPA buffer containing 1% (v/v) NP-40, 0.1% (w/v) SDS, 50 mM Tris/HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.5% (w/v) sodium deoxycholate and freshly added 1 mM phenylmethylsulfonylfluoride, 10 mM NaF, 0.5 mM leupeptin (10 µg/ml), aprotinin (10 µg/ml). After incubation for 20 min on ice, cells were sonicated twice for 20 seconds. Total protein quantification was performed using a PierceBCA protein assay kit, according to manufacturer’s instructions. The lysate was mixed with 5x SDS sample buffer and the extracts were heated for 5 min at 95 °C, followed by separation of proteins using SDS-PAGE and Western blotting as described previously47.
Tissues from wild-type mice on a C57BL/6J background or SIAH2 knock-out mice48 were homogenized in RIPA buffer containing protease and phosphatase inhibitor. Protein lysates were sonicated, cleared, and protein concentration measured. 20 μg of protein were loaded on a SDS-PAGE gel and further processed as described above. Organ harvesting was performed according to the regional authorities and ethics committees for animal research.
Isolation of mouse cardiac fibroblasts
Hearts were excised under deep anesthesia, transferred rapidly to ice-cold saline, and mounted on the cannula of a Langendorff perfusion system as described in greater detail previously49. Hearts were perfused first for 10 min in a non-re-circulating manner with a calcium-free perfusion buffer, then for 20–25 min in a re-circulating manner in a buffer supplemented with collagenase and 25 µmol/l calcium. Thereafter, ventricular tissue was minced and incubated for another 5 min in re-circulating buffer. The remaining cell solution was filtered through a 200 µm nylon mesh. The suspension was centrifuged at 25 × g for 10 min to pellet down the cardiomyocytes. The remaining cells (supernatant after centrifugation) were centrifuged at 250 × g for 10 min and the pellet was resuspended in 1 ml of endothelial cell basal medium (PromoCell) and incubated with magnetic beads (ThermoFisher) pre-coated with anti CD31 for 1 hour at 4 °C with end-to-end rotation. The microvascular endothelial cells coupled to magnetic beads were separated with a magnet, washed with endothelial cell basal medium, and seeded in 35 mm culture dishes. This procedure removed over 95% of endothelial cells from the mixture and the remaining cells were seeded as cardiac fibroblasts in M199 medium supplemented with 10% fetal calf serum (FCS). All animals were handled in accordance with the NIH Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, 1996).
Immortalization of primary mouse cardiac fibroblasts
Phoenix ECO cells were transfected with mTert-pBabe-puro, a retroviral plasmid encoding mouse telomerase reverse transcriptase (mTERT). Two days after transfection, the supernatant containing mTERT-encoding Retroviruses were passed through 0.45 µM sterile filters and polybrene was added to a final concentration of 5 µg/ml. The filtrate was pre-mixed with complete medium in different ratios (1:1, 2:1 and 4:1 respectively) and added to freshly isolated primary mouse cardiac fibroblasts. Two days after viral transduction cells were washed twice with PBS and further incubated in complete DMEM medium containing puromycin (2 µg/ml).
Immunofluorescence
Cells were washed with PBS and fixed in 4% (v/v) formaldehyde solution for 10 min, followed by 2x washing with PBS. Permeabilization of the fixed cells were performed using 0.5% (v/v) Triton X-100 for 10 min and subsequently washed twice with PBS and cells were blocked using 5% (w/v) BSA for 1 hour. After addition of the first antibody (1:500 dilution) in PBS containing 1% (w/v) BSA and incubation overnight at 4 °C, the cells were washed three times with PBS. After incubation with the anti-rabbit antibody conjugated with Alexa Fluor 488 dye (1:1000) in PBS containing 1% (w/v) BSA the cells were further incubated for 1 hr in the dark. After three more washing steps the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) contained in the mounting medium (VECTASHIELD Mounting Medium with DAPI). Microscopy was performed using a Nikon Eclipse TE2000-E Semi-Motorized inverted fluorescence microscope and data were processed using ImageJ software.
Co-immunoprecipitation
Cells were washed once with PBS and lysed with Pierce-IP lysis buffer. 10% of the lysate was taken as input, while the remaining material was used for co-immunoprecipitations using tosylactivated dynabeads coupled with 1 µg of the respective antibodies. After incubation in IP buffer containing 5 µM of the proteasome inhibitor lactacystin, the supernatant was removed on a magnetic stand and the pellet was washed three times with PBS containing 0.1% (W/V) Triton X-100. Bound proteins were eluted in 1x SDS sample buffer and further used for immunoblotting.
Generation of CRIPR-Cas9 knock-out cells
Oligonucleotides targeting the first exon of the human SIAH2 gene were cloned into pX459. These plasmids were transfected into 293T cells, followed by selection with Puromycin (1 µg/ml) for 3 days. Surviving cells were grown to colonies, picked and further expanded. Expression of SIAH2 and Cas9 was tested by Western blotting. Cell clones showing absent SIAH2 and Cas9 protein expression were further characterized by isolation of genomic DNA using the NucleoSpin Tissue kit according to the protocol of the manufacturer (Macherey-Nagel). Following PCR amplification of the genomic region encompassing the expected mutation, the PCR product was excised from an agarose gel and one of the PCR primers was directly used for sequencing of the PCR product.
Colony formation assays
Equal numbers of 293T wild-type and SIAH2 knock-out cells were seeded in 6-well plates until they reached 60–70% confluency. The next day, cells were treated either with erastin (20 µM) or vehicle (DMSO) and further incubated for two days at 37 °C. After removal of dead cells and washing with PBS, surviving cells were further grown several days to colonies that were stained with crystal violet solution. Images of the dried cell culture plates were captured by placing them on Kaiser slimlite photo light device using rainbow-TV lens S6x11. Images were processed using deVision Software.
Determination of cell viability
Cell viability was evaluated using the PrestoBluecell viability reagent (Invitrogen) according to manufacturer’s instructions. Equal cell numbers were plated in flatbottom microtiter plates. After induction of cell death, 10 µl of 10-fold ready to use PrestoBluereagent was added to each well. The plate was incubated for 1 h at 37 °C in the dark and absorbance was measured using a Tecan Infinite Pro plate reader.
Statistical analysis
All data are expressed as median, ±SD of at least three or more independent biological replicates in different experimental set-ups. Relative amounts of proteins were determined using a ChemiDoc Imaging System using Image Lab Software. All normalizations were performed with the respective house keeping gene β-Actin, Tubulin, Vinculin or GAPDH and compared with the untreated or wild-type cells, accordingly.
Supplementary information
Acknowledgements
We are grateful to Dres. L.Y. Chau (Taipei, Taiwan) and P. Lehner (Cambridge, UK) for generously providing HO-1 expression plasmids. Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) SFB1213 - project number B06, and the Excellence Cluster Cardio-Pulmonary System (ECCPS, EXC 147/2) (to M.L.S., S.R. and M.K.). The work of M.L.S. is also supported by the IMPRS-HLR program of the Max-Planck Society. The work of M.K. is supported by the Deutsche Forschungsgemeinschaft KR1141/9-1 (KLIFO309).
Author contributions
S.C., R.B. and N.M. performed experiments, A.M. provided mouse models, A.M., M.K. and S.R. finalized the manuscript, S.C. and M.L.S. conceived the study, M.L.S. wrote the first version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Shashipavan Chillappagari, Email: shashipavan.chillappagari@innere.med.uni-giessen.de.
M. Lienhard Schmitz, Email: lienhard.schmitz@biochemie.med.uni-giessen.de.
Supplementary information
is available for this paper at 10.1038/s41598-020-59005-3.
References
- 1.Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nature reviews. Cancer. 2018;18:69–88. doi: 10.1038/nrc.2017.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pepper IJ, Van Sciver RE, Tang AH. Phylogenetic analysis of the SINA/SIAH ubiquitin E3 ligase family in Metazoa. BMC evolutionary biology. 2017;17:182. doi: 10.1186/s12862-017-1024-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gopalsamy A, Hagen T, Swaminathan K. Investigating the molecular basis of Siah1 and Siah2 E3 ubiquitin ligase substrate specificity. PloS one. 2014;9:e106547. doi: 10.1371/journal.pone.0106547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qi J, Kim H, Scortegagna M, Ronai ZA. Regulators and effectors of Siah ubiquitin ligases. Cell. Biochem Biophys. 2013;67:15–24. doi: 10.1007/s12013-013-9636-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee JT, Wheeler TC, Li L, Chin LS. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Human molecular genetics. 2008;17:906–917. doi: 10.1093/hmg/ddm363. [DOI] [PubMed] [Google Scholar]
- 6.Szargel R, et al. Synphilin-1A inhibits seven in absentia homolog (SIAH) and modulates alpha-synuclein monoubiquitylation and inclusion formation. The Journal of biological chemistry. 2009;284:11706–11716. doi: 10.1074/jbc.M805990200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santelli E, et al. Structural analysis of Siah1-Siah-interacting protein interactions and insights into the assembly of an E3 ligase multiprotein complex. The Journal of biological chemistry. 2005;280:34278–34287. doi: 10.1074/jbc.M506707200. [DOI] [PubMed] [Google Scholar]
- 8.Calzado MA, de la Vega L, Moller A, Bowtell DD, Schmitz ML. An inducible autoregulatory loop between HIPK2 and Siah2 at the apex of the hypoxic response. Nature cell biology. 2009;11:85–91. doi: 10.1038/ncb1816. [DOI] [PubMed] [Google Scholar]
- 9.Winter M, et al. Control of HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases ATM and ATR. Nature cell biology. 2008;10:812–824. doi: 10.1038/ncb1743. [DOI] [PubMed] [Google Scholar]
- 10.Scortegagna M, et al. USP13 enzyme regulates Siah2 ligase stability and activity via noncatalytic ubiquitin-binding domains. The Journal of biological chemistry. 2011;286:27333–27341. doi: 10.1074/jbc.M111.218214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun RC, Denko NC. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell metabolism. 2014;19:285–292. doi: 10.1016/j.cmet.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma B, et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nature cell biology. 2015;17:95–103. doi: 10.1038/ncb3073. [DOI] [PubMed] [Google Scholar]
- 13.Dulloo I, et al. Hypoxia-inducible TAp73 supports tumorigenesis by regulating the angiogenic transcriptome. Nature cell biology. 2015;17:511–523. doi: 10.1038/ncb3130. [DOI] [PubMed] [Google Scholar]
- 14.Möller A, et al. Inhibition of Siah ubiquitin ligase function. Oncogene. 2009;28:289–296. doi: 10.1038/onc.2008.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma B, et al. The SIAH2-NRF1 axis spatially regulates tumor microenvironment remodeling for tumor progression. Nature communications. 2019;10:1034. doi: 10.1038/s41467-019-08618-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baba K, Morimoto H, Imaoka S. Seven in absentia homolog 2 (Siah2) protein is a regulator of NF-E2-related factor 2 (Nrf2) The Journal of biological chemistry. 2013;288:18393–18405. doi: 10.1074/jbc.M112.438762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiological reviews. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 18.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annual review of pharmacology and toxicology. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 19.Chang LC, et al. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer letters. 2018;416:124–137. doi: 10.1016/j.canlet.2017.12.025. [DOI] [PubMed] [Google Scholar]
- 20.Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget. 2015;6:24393–24403. doi: 10.18632/oncotarget.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin Q, et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. The Journal of biological chemistry. 2007;282:20621–20633. doi: 10.1074/jbc.M607954200. [DOI] [PubMed] [Google Scholar]
- 22.Slebos DJ, et al. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. American journal of respiratory cell and molecular biology. 2007;36:409–417. doi: 10.1165/rcmb.2006-0214OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Dickins RA, et al. The ubiquitin ligase component Siah1a is required for completion of meiosis I in male mice. Molecular and cellular biology. 2002;22:2294–2303. doi: 10.1128/mcb.22.7.2294-2303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Della NG, Senior PV, Bowtell DD. Isolation and characterisation of murine homologues of the Drosophila seven in absentia gene (sina) Development. 1993;117:1333–1343. doi: 10.1242/dev.117.4.1333. [DOI] [PubMed] [Google Scholar]
- 25.Hu G, Fearon ER. Siah-1 N-terminal RING domain is required for proteolysis function, and C-terminal sequences regulate oligomerization and binding to target proteins. Molecular and cellular biology. 1999;19:724–732. doi: 10.1128/mcb.19.1.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakayama K, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004;117:941–952. doi: 10.1016/j.cell.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Shibahara S, Han F, Li B, Takeda K. Hypoxia and heme oxygenases: oxygen sensing and regulation of expression. Antioxidants & redox signaling. 2007;9:2209–2225. doi: 10.1089/ars.2007.1784. [DOI] [PubMed] [Google Scholar]
- 28.Xie Y, et al. Ferroptosis: process and function. Cell. death and differentiation. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adedoyin O, et al. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. American journal of physiology. Renal physiology. 2018;314:F702–F714. doi: 10.1152/ajprenal.00044.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao JY, et al. A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell reports. 2019;26:1544–1556 e1548. doi: 10.1016/j.celrep.2019.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang WS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alam J, et al. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. The Journal of biological chemistry. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 34.Zampetaki A, Minamino T, Mitsialis SA, Kourembanas S. Effect of heme oxygenase-1 overexpression in two models of lung inflammation. Experimental biology and medicine. 2003;228:442–446. doi: 10.1177/15353702-0322805-02. [DOI] [PubMed] [Google Scholar]
- 35.Waza AA, Hamid Z, Ali S, Bhat SA, Bhat MA. A review on heme oxygenase-1 induction: is it a necessary evil. Inflammation research: official journal of the European Histamine Research Society… [et al.] 2018;67:579–588. doi: 10.1007/s00011-018-1151-x. [DOI] [PubMed] [Google Scholar]
- 36.Wang D, et al. Overexpression of heme oxygenase 1 causes cognitive decline and affects pathways for tauopathy in mice. Journal of Alzheimer’s disease: JAD. 2015;43:519–534. doi: 10.3233/JAD-140567. [DOI] [PubMed] [Google Scholar]
- 37.Chau LY. Heme oxygenase-1: emerging target of cancer therapy. Journal of biomedical science. 2015;22:22. doi: 10.1186/s12929-015-0128-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poss KD, Tonegawa S. Reduced stress defense in heme oxygenase 1-deficient cells. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:10925–10930. doi: 10.1073/pnas.94.20.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:10919–10924. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leautaud V, Demple B. Regulation of heme oxygenase-1 mRNA deadenylation and turnover in NIH3T3 cells by nitrosative or alkylation stress. BMC molecular biology. 2007;8:116. doi: 10.1186/1471-2199-8-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mertins P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akimov V, et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nature structural & molecular biology. 2018;25:631–640. doi: 10.1038/s41594-018-0084-y. [DOI] [PubMed] [Google Scholar]
- 43.Song J, et al. 14-3-3zeta inhibits heme oxygenase-1 (HO-1) degradation and promotes hepatocellular carcinoma proliferation: involvement of STAT3 signaling. Journal of experimental & clinical cancer research: CR. 2019;38:3. doi: 10.1186/s13046-018-1007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jez M, et al. Valproic acid downregulates heme oxygenase-1 independently of Nrf2 by increasing ubiquitination and proteasomal degradation. Biochemical and biophysical research communications. 2017;485:160–166. doi: 10.1016/j.bbrc.2017.02.041. [DOI] [PubMed] [Google Scholar]
- 45.Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer cell. 2019;35:830–849. doi: 10.1016/j.ccell.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 46.Zou Y, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nature communications. 2019;10:1617. doi: 10.1038/s41467-019-09277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seibert M, et al. CDK1-mediated phosphorylation at H2B serine 6 is required for mitotic chromosome segregation. The Journal of cell. biology. 2019;218:1164–1181. doi: 10.1083/jcb.201806057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frew IJ, et al. Generation and analysis of Siah2 mutant mice. Molecular and cellular biology. 2003;23:9150–9161. doi: 10.1128/mcb.23.24.9150-9161.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schlüter KD, Schreiber D. Adult ventricular cardiomyocytes: isolation and culture. Methods in molecular biology. 2005;290:305–314. doi: 10.1385/1-59259-838-2:305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.