

Abstract
Dilated cardiomyopathy (DCM) is one of the leading causes of heart failure. A large proportion of genetic cause remains unexplained, especially in idiopathic DCM. We performed target next-generation sequencing of 102 genes which were known causes or candidate genes for cardiomyopathies and channelpathies in 118 prospectively recruited Han Chinese patients with idiopathic DCM. 41 of the 118 patients carried 40 pathogenic or likely pathogenic variants, providing a molecular diagnosis in 34.7% of patients. 32 of these variants were novel. TTN truncating variants were predominant, with a frequency of 31.0%, followed by variants of LMNA (14.3%), RBM20 (4.8%), and NEXN (4.8%). These 4 genes accounted for over half variants identified. No significant difference in clinical characteristics or rates of reaching the composite end point (cardiac transplantation and death from cardiac causes) between pathogenic or likely pathogenic variant carriers and noncarriers (hazard ratio 1.11, 95% CI: 0.41 to 3.00), or between patients with TTN truncating variants or without (hazard ratio 0.49, 95% CI: 0.36 to 6.10). In our prospective study, we first determined the overall genetic profiles and genotype-phenotype correlations in Han Chinese idiopathic DCM patients, which could provide insight for genetic diagnosis of DCM in this population.
Subject terms: Cardiology, Diseases
Introduction
Dilated cardiomyopathy (DCM) is the one of the leading causes of heart failure and sudden death, and the most common cause of heart transplantation, affecting approximately 1 in 250 individuals1. DCM is a progressive disease, with 50% of patients reported to die within 5 years of diagnosis without transplantation2. DCM frequently has a genetic etiology, and multiple causative genes have been discovered. The genetic basis of DCM is highly diverse; over 30 genes have been identified as the potentially disease-causing genes1,3. About 25–30% of individuals with DCM have a familial form of the disease1,4. Truncating variants in TTN, which encodes titin, account for up to 25% of familial DCM5. A large proportion of genetic cause of DCM remains unexplained, especially in idiopathic DCM6.
Next-generation sequencing (NGS) approaches have enabled rapid genetic testing, particularly for large genes such as TTN which are hard to sequence with traditional methods. Using NGS, researchers have characterized the genetic atlas of DCM in Caucasian population7,8. Zhao and colleagues performed NGS of 25 genes in 21 Chinese patients9, but the number of genes and patients were limited, and the most commonly pathogenic gene in DCM—TTN was not included in their sequencing panel. Also, understanding the potential genotype-phenotype correlations may identify high-risk patients in this condition. In this study, we developed a custom “cardiomyopathy panel” containing 102 genes which were known causes or candidate genes for cardiomyopathies and channelpathies. We prospectively recruited 118 unrelated patients with idiopathic DCM and performed target NGS in this cohort to determine the molecular characterization of this cohort and to examine the genotype-phenotype correlations.
Results
Clinical characteristics
Our study consisted of 118 unrelated DCM patients of Han Chinese origin. Baseline characteristics of these patients are summarized in Table 1. Of the 118 DCM patients, 75% were male, and the mean age at diagnosis was 55.9 ± 14.7 years. The mean left ventricular ejection fraction (LVEF) was 30.2 ± 6.8%. Beta receptor blocker was used in 81% of patients, angiotensin converting enzyme inhibitor or angiotensin receptor blocker in 82% of patients, and aldosterone antagonists in 81% of patients, which indicated that most of these patients received standard therapy for heart failure. Thirty-one percent of patients received implantable cardioverter defibrillator (ICD) or cardiac resynchronization therapy with defibrillator (CRTD). Female patients had similar clinical characteristics as compared with male patients, except that the rate of smoking was lower than male patients.
Table 1.
Patient characteristics stratified by variation status.
| Characteristics | All | Variants present | Variants absent | P value | TTN variants present | TTN variants absent | P value | LMNA variants present | LMNA variants absent | P value |
|---|---|---|---|---|---|---|---|---|---|---|
| Number | 118 | 41 | 77 | 13 | 105 | 6 | 112 | |||
| Male (%) | 89 (75%) | 29 (70.7%) | 60 (77.9%) | 0.50 | 11 (85%) | 78 (74%) | 0.52 | 6 (100%) | 83 (74%) | 0.33 |
| Age of diagnosis (years) | 55.9 ± 14.7 | 53.9 ± 14.0 | 56.9 ± 15.1 | 0.30 | 54.4 ± 15.6 | 56.1 ± 14.7 | 0.69 | 45.3 ± 15.5 | 56.5 ± 14.6 | 0.07 |
| Smoking (%) | 41 (35%) | 14 (34.1%) | 27 (35.1%) | 1.0 | 6 (46%) | 35 (33%) | 0.37 | 4 (667%) | 37 (33%) | 0.18 |
| Diabetes (%) | 20 (17%) | 9 (21.9%) | 11 (14.3%) | 0.32 | 3 (23%) | 17 (16%) | 0.46 | 2 (33%) | 18 (16%) | 0.31 |
| Systolic BP (mmHg) | 123.2 ± 19.9 | 120.4 ± 18.7 | 123.2 ± 20.5 | 0.96 | 119.5 ± 19.8 | 123.7 ± 19.9 | 0.49 | 111.2 ± 9.2 | 123.9 ± 20.1 | 0.13 |
| Diastolic BP (mmHg) | 77.3 ± 15.6 | 80.3 ± 14.9 | 75.8 ± 15.8 | 0.15 | 74.3 ± 12.8 | 77.6 ± 15.9 | 0.49 | 72.7 ± 9.2 | 77.5 ± 15.9 | 0.46 |
| β receptor blocker (%) | 96 (81%) | 32 (78.0%) | 64 (83.1%) | 0.60 | 11 (85%) | 85 (81%) | 1.0 | 5 (83%) | 91 (81%) | 0.52 |
| ACEI/ARB (%) | 97 (82%) | 31 (75.7%) | 66 (85.7%) | 0.21 | 10 (77%) | 87 (85%) | 0.70 | 6 (100%) | 91 (81%) | 1.0 |
| Diuretic (%) | 110 (92%) | 32 (78.0%) | 68 (88.3%) | 0.18 | 13 (100%) | 97 (92%) | 0.60 | 6 (100%) | 104 (93%) | 1.0 |
| Aldosterone antagonists (%) | 95 (81%) | 34 (82.9%) | 61 (79.3%) | 0.81 | 10 (77%) | 85 (81%) | 0.72 | 6 (100%) | 89 (79%) | 0.61 |
| Digoxin (%) | 38 (32%) | 14 (34.1%) | 24 (31.1%) | 0.84 | 4 (31%) | 34 (32%) | 1.0 | 2 (33%) | 36 (32%) | 1.0 |
| ICD/CRTD (%) | 36 (31%) | 10 (24.3%) | 26 (33.8%) | 0.40 | 5 (38%) | 31 (30%) | 0.53 | 2 (33%) | 34 (30%) | 1.0 |
| LVEF (%) | 30.2 ± 6.8 | 29.2 ± 6.3 | 30.7 ± 6.9 | 0.27 | 29.2 ± 7.2 | 30.3 ± 6.7 | 0.58 | 30.8 ± 6.4 | 30.2 ± 6.8 | 0.81 |
| LVEDD (cm) | 7.1 ± 0.9 | 7.08 ± 0.76 | 7.04 ± 0.91 | 0.82 | 6.9 ± 1.0 | 7.1 ± 0.9 | 0.44 | 7.2 ± 0.5 | 7.0 ± 0.9 | 0.58 |
| IVSTD (cm) | 0.9 ± 0.6 | 0.97 ± 0.72 | 0.91 ± 0.52 | 0.63 | 0.9 ± 0.1 | 0.9 ± 0.6 | 0.64 | 0.82 ± 0.1 | 0.94 ± 0.6 | 0.64 |
| LAD (cm) | 5.2 ± 0.9 | 5.02 ± 0.92 | 5.21 ± 0.83 | 0.26 | 5.01 ± 0.8 | 5.21 ± 0.8 | 0.33 | 5.86 ± 1.5 | 5.15 ± 0.7 | 0.02 |
Data are mean ± standard deviation, number, or percent.
ACEI, angiotensin converting enzyme inhibitor; ARB, angiotensin receptor blocker; BP, blood pressure; CRTD, cardiac resynchronization therapy (with defibrillator); DCM, dilated cardiomyopathy; DM, diabetes mellitus; ICD, Implantable cardioverter defibrillator; IVSTD, interventricular septal end-diastolic thickness; LAD, left atrial diameter; LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction.
Cardiomyopathy panel coverage
We performed deep sequencing with the Cardiomyopathy panel covering the coding exons and splice junctions of 102 genes (Table S1). A total of 300 Mb of sequence was yielded per sample. The NGS captured 99.5% of the target region, and 60% of all reads are mapped to our designed target regions. A mean coverage of 281 × was reached and an average of 93.7% of target regions were covered to a depth of at least 20×. We provided the bed file for capture targets, reads covered <20 × in ~20% samples, and the percentage of patients with a <20 × depth in target regions in supplementary datasets (datasets 1 to 3).
Landscape of genetic alterations in DCM
In total, 956 unique genetic variants were identified in 118 DCM patients. An average of 298 variants was detected in each patient in the target region with over 20 × coverage. Variants that were rare (defined here by MAF <0.01% in the ExAC and gnomAD databases) and altered protein sequences (truncating, missense, in-frame insertions/deletions [indels]) in a set of 102 cardiomyopathy-associated genes were evaluated. After filtering, a total of 65 different rare variants in 28 genes were found. Among these 65 rare variants, 43 were missense variants, 6 were nonsense variants, 14 were frameshift indels, and 2 were in-frame indels (Table S2). Of the 118 patients’ samples, 59 (50%) harbored at least one rare variant. Eighteen of the 43 (41.8%) rare missense variants were predicted to be “damaging” according to various prediction programs. Specially, the Combined Annotation Dependent Depletion (CADD) scores of all these 18 variants were higher than 20, indicating rarity and deleteriousness of these variants. Altogether, 40 variants were considered as pathogenic or likely pathogenic in our study (Table 2 and Table 3), 8 of which were recorded in HGMD or the ClinVar database and/or supported by published data and the remaining 32 were novel. All pathogenic or likely pathogenic variants were heterozygous, and most of these variations were found in our study are private, except 2 variants (LMNA: c.568 C > T p.R190W; RBM20: c.2017C > T p.R673W). Each of these LMNA and RBM20 variants were shared in 2 unrelated DCM patients and published in other populations10,11.
Table 2.
Pathogenic and likely pathogenic truncating variants or in-frame insertions/deletions in Chinese DCM cohort (22 variants).
| Gene | Transcript | Exon | Nucleotide Change | Amino Acid Change | Effect | Publication | MAF gnomAD | MAF ExAC | TTN band |
|---|---|---|---|---|---|---|---|---|---|
| ADRB1 | NM_000684 | 1 | c.763_764delGT | p.255_255delV | Frameshift | — | 0 | 0 | — |
| ANK2 | NM_001148 | 38 | c.5772_5773insAAAAC | p.K1924fs | Frameshift | — | 0 | 0 | — |
| CBL | NM_005188 | 9 | c.1363_1364insATG | p.Y455delinsYD | Nonframeshift | — | 0 | 0 | — |
| EMD | NM_000117 | 6 | c.596 C > G | p.S199X | Nonsense | — | 0 | 0 | — |
| LMNA | NM_170707 | 8 | c.1477 C > T | p.Q493X | Nonsense | — | 0 | 0 | — |
| LMNA | NM_170707 | 9 | c.1590delC. | p.L530fs | Frameshift | — | 0 | 0 | — |
| MYBPC3 | NM_000256 | 24 | c.2541 C > G | p.Y847X | Nonsense | Yes | 0 | 0 | — |
| NEXN | NM_144573 | 12 | c.1587_1589delAAG | p.529_530del | Nonframeshift | — | 0 | 0 | — |
| TTN | NM_001267550 | 352 | c.98650_98651insT | p.S32884fs | Frameshift | Yes | 0 | 0 | A band |
| TTN | NM_001267550 | 258 | c.48325_48326insT | p.L16112fs | Frameshift | — | 0 | 0 | A band |
| TTN | NM_001267550 | 326 | c.78749 T > A | p.L26250X | Nonsense | — | 0 | 0 | A band |
| TTN | NM_001267550 | 335 | c.89855delT | p.L29952fs | Frameshift | — | 0 | 0 | A band |
| TTN | NM_001267550 | 358 | c.101000_101001delAT | p.Y33667fs | Frameshift | — | 0 | 0 | A band |
| TTN | NM_001267550 | 342 | c.94931delA | p.E31644fs | Frameshift | — | 0 | 0 | A band |
| TTN | NM_001267550 | 274 | c.52154 C > A | p.S17385X | Nonsense | — | 0 | 0 | A band |
| TTN | NM_001267550 | 248 | c.46051delA | p.R15350fs | Frameshift | — | 0 | 0 | I band |
| TTN | NM_001267550 | 49 | c.14251delT | p.S4751fs | Frameshift | — | 0 | 0 | I band |
| TTN | NM_001267550 | 255 | c.47843_47844insT | p.I15948fs | Frameshift | — | 0 | 0 | A band |
| TTN | NM_001267550 | 246 | c.45550 C > T | p.Q15184X | Nonsense | — | 0 | 0 | I band |
| TTN | NM_001267550 | 226 | c.41377delG | p.V13793fs | Frameshift | — | 0 | 0 | I band |
| TTN | NM_001267550 | 326 | c.71024_71027del | p.23675_23676del | Frameshift | — | 0 | 0 | A band |
| VCL | NM_003373 | 6 | c.632delT | p.I211fs | Frameshift | — | 0 | 0 | — |
Table 3.
Pathogenic and likely pathogenic missense variants in Chinese DCM cohort (18 variants).
| Gene | Transcript | Exon | Nucleotide Change | Amino Acid Change | RsID | Effect | Publication | MAF gnomAD | MAF ExAC | SIFT score | PolyPhen2 HDIV score | MutationTaster score | CADD score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ANKRD1 | NM_014391 | 7 | c.682 A > G | p.R228G | — | Missense | — | 0 | 0 | 0.001 | 0.989 | 1 | 26.6 |
| CACNA1C | NM_000719 | 46 | c.6035 G > A | p.R2012Q | rs772606843 | Missense | Yes | — | 9.96e-06 | 0.048 | 1 | 1 | 24.3 |
| DES | NM_001927 | 4 | c.887 A > G | p.Y296C | — | Missense | — | 0 | 0 | 0 | 1 | 0.999 | 27.5 |
| DMD | NM_004010 | 59 | c.8480 T > A | p.L2827Q | — | Missense | — | 0 | 0 | 0.001 | 1 | 1 | 26 |
| DSG2 | NM_001943 | 15 | c.2959 G > T | p.V987F | rs141405267 | Missense | — | 6.46e-05 | 7.59e-05 | 0.041 | 0.986 | 1 | 23.8 |
| LMNA | NM_170707 | 3 | c.568 C > T | p.R190W | rs59026483 | Missense | Yes | 0 | 0 | 0 | 1 | 1 | 35 |
| LMNA | NM_170707 | 6 | c.1088 T > C | p.L363P | Missense | Yes | 0 | 0 | 0 | 1 | 1 | 28.7 | |
| LMNA | NM_170707 | 10 | c.1633C > A | p.R545S | Missense | — | 0 | 0 | 0.016 | 0.995 | 1 | 24.2 | |
| MYH6 | NM_002471 | 36 | c.5539 C > T | p.R1847W | rs752718246 | Missense | — | 8.165e-06 | 6.599e-05 | 0 | 1 | 1 | 34 |
| MYH7 | NM_000257 | 25 | c.3134 G > A | p.R1045H | rs397516178 | Missense | Yes | 0 | 3.295e-05 | 0 | 1 | 1 | 28.7 |
| MYPN | NM_032578 | 2 | c.468 C > G | p.D156E | — | Missense | — | 6.456e-05 | 0 | 0.012 | 1 | 1 | 24.5 |
| NEXN | NM_144573 | 8 | c.835 C > A | p.R279S | rs146245480 | Missense | — | 0 | 2.512e-05 | 0.007 | 0.987 | 1 | 29.6 |
| PKP2 | NM_001005242 | 1 | c.125 G > A | p.G42E | rs748880850 | Missense | — | 0 | 0 | 0.016 | 1 | 0.962 | 29.4 |
| PRDM16 | NM_022114 | 7 | c.1006 C > T | p.R336C | rs748880850 | Missense | — | 2.514e-05 | 6.466e-05 | 0.019 | 1 | 1 | 34 |
| RYR2 | NM_001035 | 18 | c.1748C > A | p.P583Q | — | Missense | — | 0 | 0 | 0.001 | 0.98 | 1 | 26.4 |
| RBM20 | NM_001134363 | 9 | c.2017C > T | p.R673W | rs397516599 | Missense | Yes | 9.699e-05 | 5.395e-05 | 0 | 1 | 0.998 | 28.9 |
| SCN5A | NM_001160161 | 25 | c.4357 C > A | p.Q1453K | — | Missense | — | — | — | 0.001 | 0.996 | 0.979 | 23.7 |
| TNNT2 | NM_000364 | 4 | c.472 C > T | p.R158W | rs730881123 | Missense | Yes | 0 | 0 | 0 | 1 | 1 | 35 |
In our cohort, 41 out of the 118 patients (34.7%) carried pathogenic or likely pathogenic variants. The distribution of these pathogenic or likely pathogenic variants was not equal among genes, as was presented in Table 2 and Table 3. TTN truncating variants were predominant, with a frequency of 31.0%, followed by variants of LMNA (14.3%), RBM20 (4.8%), and NEXN (4.8%). Other pathogenic or likely pathogenic variants present at low frequency (2.4%) in the study population were identified in ADRB1, ANK2, ANKRD1, CACNA1C, CBL, DES, DSG2, DMD, EMD, MYBPC3, MYH6, MYH7, MYPN, PKP2, PRDM16, RYR2, SCN5A, TNNT2 and VCL, each with 1 variant (Fig. 1). TTN truncating variants were observed in 13 of the 118 patients (11.0%). As expected, TTN truncating variants were nonrandomly distributed within titin5, with most variants located in the titin A-band region and others in I-band region (Table 2). All these TTN truncating variants are expressed in both the N2BA and N2B isoforms and constitutively expressed in the heart12. No patient carried multiple pathogenic or likely pathogenic variants.
Figure 1.

The distribution of pathogenic or likely pathogenic variants identified in the idiopathic dilated cardiomyopathy cohort.
Genotype–phenotype correlations
We compared the clinical characteristics of patients with and without pathogenic or likely pathogenic variants. As shown in Table 1, the age of diagnosis was similar between patients with or without variants. There were no significant differences in sex, treatment and dosage of common medical therapy for heart failure among these 2 groups. Also, patients present or absent with these variants had similar left ventricular ejection fractions, left ventricular end-diastolic diameter and other echocardiography parameters. In terms of the composite endpoint of cardiac death and heart transplantation, there was no significant difference between patients with and without pathogenic or likely pathogenic variants (hazard ratio 1.11, 95% confidence interval 0.41 to 3.00, P = 0.84, Fig. 2). We also made similar comparisons between patients who tested positive or negative with TTN truncating or LMNA variants in our study. No significant differences in clinical characteristics (Table 1) or follow-up endpoints were detected between DCM patients present with TTN truncating variants and those absent (hazard ratio 0.49, 95% confidence interval 0.36 to 6.10, P = 0.58, Fig. 3). LMNA genotype-positive subjects seem to have a younger age of diagnosis of DCM (45.3 ± 15.5 vs. 56.5 ± 14.6; P = 0.07) and larger left atrial (5.86 ± 1.5 vs. 5.15 ± 0.7; P = 0.02) than those negative patients. The outcome difference between these 2 groups was not significant and had wide confidence intervals (hazard ratio 2.04, 95% confidence interval 0.31 to 13.3, P = 0.45, Fig. 4). We did not compare clinical characteristics and outcomes between other individual gene variants because of limited number of patients with an individual variant in these genes.
Figure 2.
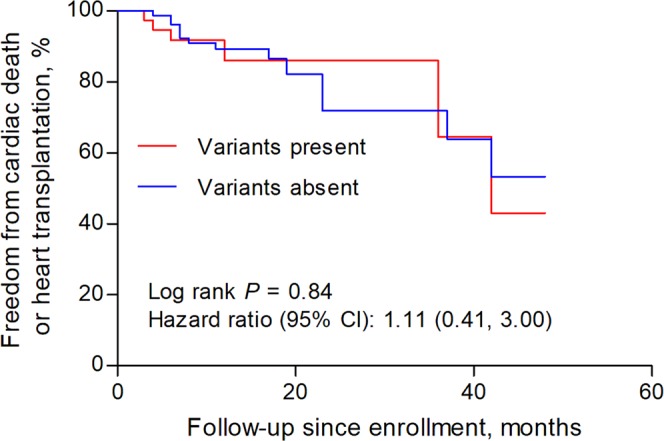
Survival curves comparing freedom from the composite endpoint of cardiac death and heart transplantation in patients with and without pathogenic or likely pathogenic variants.
Figure 3.
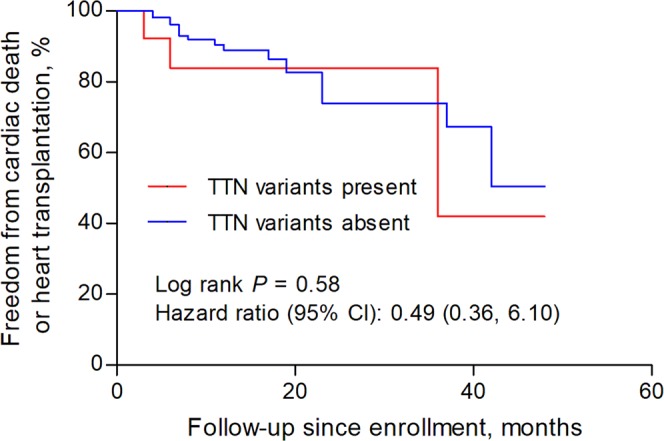
Survival curves comparing freedom from the composite endpoint of cardiac death and heart transplantation in patients with and without rare TTN truncating variants.
Figure 4.
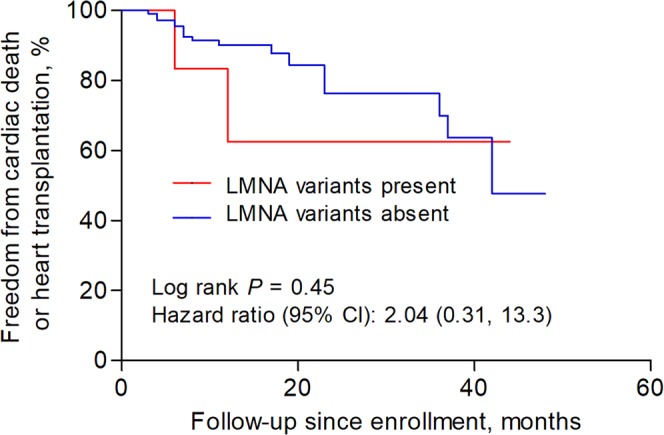
Survival curves comparing freedom from the composite endpoint of cardiac death and heart transplantation in patients with and without pathogenic or likely pathogenic LMNA variants.
Discussion
We developed and utilized a high-quality 102-gene targeted sequencing panel and sequenced 118 idiopathic DCM patients. To the best of our knowledge, this is the one of the very few genetic studies on idiopathic DCM with a prospective design. For the first time we revealed the distribution of disease-causing genes and the pathogenic or likely pathogenic variants of DCM patients in Chinese population. In our prospective cohort, 41of the 118 patients carried 40 pathogenic or likely pathogenic rare variants, with TTN, LMNA, RBM20 and NEXN being the 4 most frequently affected genes, accounting for over half these variants. We also revealed that no significant difference in baseline clinical characteristics or rates of reaching the composite end point (cardiac transplantation and death from cardiac causes) between pathogenic or likely pathogenic variant carriers and noncarriers, or between patients with TTN truncating variants and without. LMNA genotype-positive subjects seem to have a younger age of diagnosis of DCM and larger left atrial than those negative patients, but the outcome difference was not significant.
Our study showed that about one third DCM patients carried pathogenic or likely pathogenic variants, a frequency similar to those reported in in Japanese and Finish populations8,13. TTN truncating variants remain the most common variants in our Chinese cohort. The observed frequency of TTN truncating variants among our idiopathic DCM population (11.0%) lie around the lower limits of previously published cohorts, which reported a frequency between 12% and 27% mainly in Caucasian population5,7,8,14. As TTN truncating variant frequency varied with the disease severity of DCM, with a higher frequency in patients with severe, end-stage or clearly familial cases of DCM5, it is plausible that the relatively low frequency in our cohort was due to the exclusion of familial DCM and the inclusion of all DCM patients in hospital and the outpatient clinic in our analysis. Ethnic difference cannot be simply attributed to and needs further investigation, as no correlation between ethics and frequency of TTN truncating variants has been noted: a Caucasian population reported a frequency of 12.0%14, and a Japanese study reported 16.7%13.
Truncating TTN variants are not infrequent in the general population. It is estimated that the prevalence of Truncating TTN variants in the general population is ~0.4%15. Therefore, how to interpret truncating TTN variants is an open question. The etiological fraction for TTN truncating variants in DCM patients was ~97% when limiting to variants in exons that are constitutively expressed in the heart15,16, higher than the causative cut-off value of 0.9 or 0.95 recommended by consensus guidelines for variant interpretation in genetic testing17. All truncating variants in our DCM cohort lied in the constitutive exons, and therefore it is reasonable to classify them as pathogenic or likely pathogenic. Meanwhile, although most individuals with TTN truncating variants may not develop DCM over time, these variants are not necessarily phenotypically silent. In fact, there are studies showing morphological and functional abnormity in individuals in the general population with TTN truncating variants16.
Our study did not reveal a significant association between TTN truncating variants and the prognosis. Patients with TTN truncating variants had similar cardiac phenotypes and a similar risk for the clinical endpoint of cardiac transplantation and death from cardiac causes. This observation was in line with that reported by Tayal and colleagues, which showed a similar prognosis for DCM patients with TTN truncating variants, reaching the primary composite end point comprising cardiovascular mortality, major arrhythmic events, and major heart failure events14. Although the same study group showed an increased propensity to arrhythmia early in the disease course, the long-term arrhythmic events on follow-up proved similar14,18. Akinrinade and colleagues concluded in the Finnish population that adverse outcomes in patients with TTN truncating variants were indistinct from those other gene variant groups except LMNA variants8. In several other studies, however, patients with TTN truncating variants were reported to be less severe at presentation and to be associated with a favorable response to treatment than patients with LMNA variants or patients negative for TTN and LMNA genes13,19. Altogether, the published data on genotype-phenotype associations in DCM cannot provide a clear correlation between TTN truncating variants and clinical phenotype. Whether TTN truncating variants could predict clinical outcomes in the longer-term remain to be established in larger studies15. By contrast, accumulating evidence proved that DCM associated with LMNA variants had a higher risk for sudden cardiac death (SCD), cardiac transplantation, cardiac conduction disturbance, and atrial or ventricular arrhythmias20–22, indicating their potential role in risk stratification23. With limited power, our analysis did not show a genotype–phenotype correlation between LMNA variants and risk for the primary outcome.
Our study has several limitations. This study population is from a single, highly advanced center, patients may have been subjected to a selection bias. Second, we did not include familial DCM in our analysis and thus could not represent the general DCM cohort. Third, the genotype–phenotype findings should be interpreted with caution due to the limited statistical power and thus be considered as hypotheses generating; a larger number of cohorts are needed to establish these genotype–phenotype associations. Fourth, several newly discovered cardiomyopathy-related genes, including FLNC24 and FBXO3225, were not included in the study panel due to description of pathogenicity after the design of the study.
Conclusions
In our prospective idiopathic DCM cohort, about one third patients carried pathogenic or likely pathogenic variants, with TTN, LMNA, RBM20 and NEXN accounting for over half these variants. There is no significant difference in baseline clinical characteristics or rates of reaching the composite end point between pathogenic variant carriers and noncarriers, or between patients with TTN truanting variants or without.
Materials and Methods
Subjects
This study was conducted in accordance with the ethical guidelines of the Declaration of Helsinki. The Institutional Review boards at Nanjing University approved the study, and written informed consent was obtained from all participants or their legal representatives. The study population comprised 118 unrelated patients with idiopathic DCM. All patients were of Han Chinese origin, and were prospectively recruited to the affiliated Drum Tower Hospital, Nanjing University School of Medicine between 2011 and 2015.
Diagnosis of DCM
DCM was diagnosed according to the ESC (European Society of Cardiology) criteria26. Briefly, all patients had to have a reduced systolic function of the left ventricle (LVEF <45%) and a dilated left ventricle (left ventricular end-diastolic dimension >117% of the predicted value corrected for body surface area and age). Other identifiable causes such as hypertensive heart disease, primary valve disease, congenital heart disease, excess alcohol consumption and significant coronary artery disease were excluded. All recruited patients with DCM underwent family screening. Familial DCM was defined if at least 1 additional family member was diagnosed with DCM or encountered sudden cardiac death (up to third-degree relative). We did not include patients with familial DCM in our analyses.
Clinical data
Baseline demographic and clinical information was obtained from each subject during the index visit, through medical history interview, physical examination, electrocardiogram (ECG), and transthoracic echocardiography. Whole blood was collected for further genetic analysis. Follow-up data was collected from hospital care records and patient questionnaires by physicians blinded to the genetic data. The primary end point in this analysis was a composite of cardiac death and heart transplantation during the follow-up period.
Echocardiography
An experienced operator who was blinded of the genotype and clinical status of study subject performed the echocardiography test. We used a Philips Sonos 5500 ultrasound system to obtain the M-mode and 2-dimensional images, and acquire the Doppler recordings. We used the Simpson biplane method to calculate LVEF. We measured the left ventricular end-diastolic diameter and left ventricular septal and posterior wall thickness from 2-dimensional images.
Targeted next-generation sequencing
Genomic DNA was extracted from whole blood using the QIAamp DNA Blood Mini Kit (Qiagen). A panel consisting of 102 genes which were known causes or candidate genes for cardiomyopathies and channelpathies (Table S1) was designed. Targeted next-generation sequencing, which included library construction, capture, and sequencing, was carried out. Targeted gene enrichment was performed with the GenCap Custom Enrichment Kit according to the GenCap protocol, as described previously27–29. Captured DNA libraries were sequenced with the Illumina HiSeq. 2000 instrument (Illumina, San Diego, CA), producing 100-bp paired-end reads.
Variant classification
Mapping of the sequencing reads to the human genome reference sequence (hg19) was performed with the burrows-wheeler alignment tool (BWA, http://bio-bwa.sourceforge.net/)30. The Short Oligonucleotide Analysis Package (SOAPsnp) and the Genome Analysis Toolkit (GATK, https://www.broadinstitute.org/gatk/) were used to discover single-nucleotide polymorphism (SNP) and insertion-deletion (indel), respectively31,32. Gene related annotation was mainly done with ANNOVAR (http://wannovar.wglab.org/). Coding-sequence variants met quality metrics for mapping, read depth, and allelic balance were evaluated. The pathogenicity of a variant was determined based on frequency in the population and in silico prediction. We excluded synonymous variants, intronic variants outside of the flanking regions, and variants with a minor allele frequency greater than 0.01% in the National Heart, Lung, and Blood Institute ESP (Exome Sequencing Project), the 1000 Genomes database (http://browser.1000genomes.org), the dbSNP137 database (http://www.ncbi.nlm.nih.gov/snp), Exome Aggregation Consortium Browser (http://exac.broadinstitute.org/), the Genome Aggregation (gnomAD) databases (http://gnomad.broadinstitute.org/), and a cohort of 500 in-house whole-exome controls. All missense variants were subjected to in silico analysis with functional annotation algorithms including SIFT (http://sift.jcvi.org/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), GERP + + (http://mendel.stanford.edu/sidowlab/downloads/gerp/index.html), and MutationTaster (http://www.mutationtaster.org/). In addition, CADD scores were obtained to assess missense variant pathogenicity (https://cadd.gs.washington.edu/score). Variants were checked for known pathogenic relationships with cardiovascular diseases in the Human Gene Mutation Database (HGMD, http://www.hgmd.org). Rare coding-sequence variants resulting in premature truncation (frameshift insertions/deletions, stop gain, splice donor or acceptor site gain or loss) and rare missense variants declared to be disease-causing by analytical algorithms were considered as pathogenic or likely pathogenic in our study33. TTN missense variants were not considered likely pathogenic because they are common and present a challenge for bioinformatic classification, especially when informative families are not available34.
Statistical analysis
Variables were presented as number (percentage) or mean ± standard deviation, as appropriate. Categorical variables were compared using chi-square test or Fisher exact test, while continuous variables with independent sample’s t-test. The Kaplan-Meier curves were constructed, and differences between survival curves were compared with log-rank test. A two-sided p-value < 0.05 was considered significant. All statistical analyses were performed with SPSS version 17.0.
Supplementary information
Acknowledgements
This study was supported by the National Natural Science Foundation of China (NO. 81600312) and Fund for Distinguished Young Scholars of Nanjing (JQX15002). The funders had no role in the study design, data collection and analysis, writing of the report, and decision to submit the article for publication.
Author contributions
B.X. and W.X. conceived the study, X.Z., R.L., J.X. and L.K. conducted the study, X.Z. and L.W. analysed the results. All authors reviewed the manuscript.
Data availability
All materials were available in the manuscript and supplementary materials.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wei Xu, Email: 13390900868@163.com.
Biao Xu, Email: xubiao62@nju.edu.cn.
Supplementary information
is available for this paper at 10.1038/s41598-020-58984-7.
References
- 1.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 2.Towbin JA, Bowles NE. The failing heart. Nature. 2002;415:227–233. doi: 10.1038/415227a. [DOI] [PubMed] [Google Scholar]
- 3.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Invest. 2013;123:19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roura S, Bayes-Genis A. Vascular dysfunction in idiopathic dilated cardiomyopathy. Nat. Rev. Cardiol. 2009;6:590–598. doi: 10.1038/nrcardio.2009.130. [DOI] [PubMed] [Google Scholar]
- 5.Herman DS, et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenbaum, A. N., Agre, K. E. & Pereira, N. L. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol. (2019). [DOI] [PubMed]
- 7.Haas J, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015;36:1123–1135. doi: 10.1093/eurheartj/ehu301. [DOI] [PubMed] [Google Scholar]
- 8.Akinrinade O, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur. Heart J. 2015;36:2327–2337. doi: 10.1093/eurheartj/ehv253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Y, et al. Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int. J. Mol. Med. 2015;36:1479–1486. doi: 10.3892/ijmm.2015.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arbustini E, et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J. Am. Coll. Cardiol. 2002;39:981–990. doi: 10.1016/S0735-1097(02)01724-2. [DOI] [PubMed] [Google Scholar]
- 11.Pugh TJ, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014;16:601–608. doi: 10.1038/gim.2013.204. [DOI] [PubMed] [Google Scholar]
- 12.Roberts AM, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015;7:270r–276r. doi: 10.1126/scitranslmed.3010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tobita T, et al. Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. Sci. Rep. 2018;8:1998. doi: 10.1038/s41598-018-20114-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tayal U, et al. Phenotype and clinical outcomes of titin cardiomyopathy. J. Am. Coll. Cardiol. 2017;70:2264–2274. doi: 10.1016/j.jacc.2017.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ware JS, Cook SA. Role of titin in cardiomyopathy: from DNA variants to patient stratification. Nat. Rev. Cardiol. 2018;15:241–252. doi: 10.1038/nrcardio.2017.190. [DOI] [PubMed] [Google Scholar]
- 16.Schafer S, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat. Genetics. 2017;49:46–53. doi: 10.1038/ng.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tayal U, et al. Truncating variants in titin independently predict early arrhythmias in patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 2017;69:2466–2468. doi: 10.1016/j.jacc.2017.03.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jansweijer JA, et al. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur. J. Heart Fail. 2017;19:512–521. doi: 10.1002/ejhf.673. [DOI] [PubMed] [Google Scholar]
- 20.Hasselberg NE, et al. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2018;39:853–860. doi: 10.1093/eurheartj/ehx596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishiuchi, S. et al. Gene-based risk stratification for cardiac disorders in LMNA mutation carriers. Circ Cardiovasc Genet. 10 (2017). [DOI] [PubMed]
- 22.Kayvanpour E, et al. Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2017;106:127–139. doi: 10.1007/s00392-016-1033-6. [DOI] [PubMed] [Google Scholar]
- 23.Halliday BP, Cleland J, Goldberger JJ, Prasad SK. Personalizing risk stratification for sudden death in dilated cardiomyopathy: the past, present, and future. Circulation. 2017;136:215–231. doi: 10.1161/CIRCULATIONAHA.116.027134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Begay RL, et al. FLNC gene splice mutations cause dilated cardiomyopathy. JACC Basic. Transl. Sci. 2016;1:344–359. doi: 10.1016/j.jacbts.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Yacoub N, et al. FBXO32, encoding a member of the SCF complex, is mutated in dilated cardiomyopathy. Genome Biol. 2016;17:2. doi: 10.1186/s13059-015-0861-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mestroni L, et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur. Heart J. 1999;20:93–102. doi: 10.1053/euhj.1998.1145. [DOI] [PubMed] [Google Scholar]
- 27.Wu WF, Sun LY, Pan XD, Yang SW, Wang LY. Use of targeted exome sequencing in genetic diagnosis of Chinese familial hypercholesterolemia. PLoS One. 2014;9:e94697. doi: 10.1371/journal.pone.0094697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu J, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 2011;3:66r–92r. doi: 10.1126/scitranslmed.3002669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang XF, et al. Targeted exome sequencing identified novel USH2A mutations in Usher syndrome families. PLoS One. 2013;8:e63832. doi: 10.1371/journal.pone.0063832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li R, et al. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19:1124–1132. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blue GM, et al. Targeted next-generation sequencing identifies pathogenic variants in familial congenital heart disease. J. Am. Coll. Cardiol. 2014;64:2498–2506. doi: 10.1016/j.jacc.2014.09.048. [DOI] [PubMed] [Google Scholar]
- 34.Begay, R. L. et al. Role of titin missense variants in dilated cardiomyopathy. J Am Heart Assoc. 4 (2015). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All materials were available in the manuscript and supplementary materials.