

Abstract
Cellular senescence represents the state of irreversible cell cycle arrest during cell division. Cellular senescence not only plays a role in diverse biological events such as embryogenesis, tissue regeneration and repair, ageing and tumour occurrence prevention, but it is also involved in many cardiovascular, renal and liver diseases through the senescence‐associated secretory phenotype (SASP). This review summarizes the molecular mechanisms underlying cellular senescence and its possible effects on a variety of renal diseases. We will also discuss the therapeutic approaches based on the regulation of senescent and SASP blockade, which is considered as a promising strategy for the management of renal diseases.
Keywords: acute renal injury, cellular senescence, diabetic nephropathy, glomerulonephritis, kidney transplanation, renal diseases, renal fibrosis, senescence‐associated secretory phynotype
1. INTRODUCTION
Cellular senescence refers to an irreversible cell cycle arrest. Senescent cells exhibit a series of changes in cell morphology and epigenetics, including the changes in cell cyclins and increased expression of β‐galactosidase1; although there is replicative exhaustion in senescent cells, these cells still have metabolic activity.1 The inhibition of DNA replication and rupture of its double‐strand are typical features of cellular senescence.2 There are many causes of cellular senescence including genomic damage, activation of oncogenes and inflammation.3
Cellular senescence plays a critical role in normal embryonic development in humans and animals and is involved in wound healing.4 However, enhanced cellular senescence is also found in multiple organs in the process of ageing or after injury, suggesting that there should be different effects of cellular senescence depending on the pathophysiological context.5 More importantly, cellular senescence has been verified as a fundamental cause for the development of many diseases, such as cardiovascular, liver and kidney diseases (Figure 2).4 Additional studies confirm that the number of senescent cells increases in multiple anatomical sites in the kidney during ageing and kidney diseases.5, 6 Therefore, preventing cellular senescence may be a potentially important approach to inhibit the development of chronic kidney disease (CKD).7 In this review, we will focus on the mechanisms of cellular senescence and the relationship between senescence and renal diseases.
Figure 2.

A schematic view of the effects of cellular senescence in renal diseases. In renal diseases, various types of cells may experience senescence such as endothelial cells and renal tubular epithelial cells (RTECs). Although senescence may have protective effects in the development of autosomal dominant polycystic kidney disease (ADPKD), acute phase of acute kidney disease (AKI) and renal fibrosis, senescence can promote the progression of several renal diseases, including AKI, diabetic nephropathy, glomerulonephritis, renal fibrosis and dysfunction of transplant kidney
2. GENERAL CONCEPT OF CELLULAR SENESCENCE AND SENESCENCE‐ASSOCIATED SECRETORY PHENOTYPE
In 1961, cellular senescence was first described as a consequence of replicative exhaustion in cultured human fibroblasts.8 Senescent cells are alive but display typical features of an enlarged, flattened morphology, senescence‐associated heterochromatin marks, accumulation of lipofuscin granules, expression of β‐galactosidase and lack of mitogenic responses.1 Cellular senescence can be classified into two different types according to the presence or the absence of telomere shortening: replicative senescence caused by shortening of telomeres and premature senescence caused by other stress signals such as aberrant oncogene activation and genomic damage.9
An important characteristic of senescent cells is their production of a series of proteins named as the senescence‐associated secretory phenotype (SASP).10 Through the SASP, senescent cells can affect surrounding cells by secreting various inflammatory cytokines, chemokines, growth factors and extracellular matrix remodelling factors such as interleukin 1α (IL‐1α), interleukin 6 (IL‐6), plasminogen activator inhibitor‐1 (PAI‐1), TGF‐β, connective tissue growth factor (CTGF) and monocyte chemoattractant protein‐1 (MCP‐1).10, 11 The expression of SASP genes is up‐regulated during senescence, mainly via the actions of NF‐κB and C/EBPβ.12 mTOR signalling is also essential for NF‐κB activation and the secretion of pro‐inflammatory SASP genes.13 Although the SASP was initially thought to be similar in all senescent cells, the function and specific composition of SASP varies greatly with the different types of stressors, different cell types and environments.
Senescent cells can exert beneficial or harmful effects through SASP and significantly influence their local microenvironment. These SASP factors induce senescence of adjacent cells through a paracrine fashion and contribute to inflammation, which in turn helps to remove senescent cells,14 and facilitate tissue repair and remodelling. Some factors promote the tumour‐suppressive function of cellular senescence due to their crucial role for the onset of stable cell cycle arrest.15 The benefit of SASP usually exists in a transient situation such as in acute wound‐healing events. However, when senescent cells exist permanently, they may induce serious problems with long‐term harmful consequences due to the chronic secretion of SASP factors such as IL‐1α and MCP‐1. Meanwhile, the increased expression of cytokines and chemokines by senescent cells, such as IL‐6 and MCP‐1, can attract immune cells to improve tissue recovery by secreting more SASP factors and removing harmful factors.16 If tissue repair fails, the increased SASP factors may accelerate the process of senescence, ultimately leading to ageing‐associated damage.
3. THE MECHANISMS AND RELATED PATHWAYS OF CELLULAR SENESCENCE
3.1. Oxidative stress and inflammation
Increasing evidence suggests that persistent DNA damage response triggers cellular senescence.17 Oxidative stress can induce DNA damage, which is an important mechanism related to stress‐induced cellular senescence.17 Intracellular ROS comes from mitochondria, cell membranes and endoplasmic reticulum. For example, reduction in the nicotinamide‐adenine dinucleotide‐ubiquinone oxidases and dysfunction of the mitochondrial electron transport chain are the main sources of ROS.18 Highly reactive oxygen molecules may damage lipids, DNA and proteins. Furthermore, it has been shown that several signalling pathways can be activated by ROS, such as p53/p21 signalling and mitogen‐activated protein kinase pathways, resulting in enhanced apoptosis, inflammation and stress‐induced senescence.19 For example, a study has shown that ROS caused telomere‐associated genomic instability and senescence in mesenchymal stem cells.20
At the same time, persistent and chronic inflammation may induce senescence, although activation of inflammatory responses is necessary to remove pathogens and mediate tissue repair. For example, several pro‐inflammatory cytokines, especially IL‐6 and IL‐8, are important mediators in the induction of premature senescence. Activation of these two cytokines and their receptors initiates senescence, whereas inhibition of these cytokines blocks senescence.21 Also, an intense inflammatory process, including innate and adaptive immune responses, has been shown to regulate kidney senescence.22
3.2. Increased expression of cyclin‐dependent kinase inhibitors
It is known that cellular senescence is tightly correlated with up‐regulation of cyclin‐dependent kinase (CDKs) inhibitors. The INK4α/ARF locus (the cyclin‐dependent kinase inhibitor 2A (CDKN2A) gene) encodes two different proteins, p16INK4a (hereafter referred as p16) and ARF, through an alternative splicing mechanism. The expression of p16 is primarily regulated by environmental stress‐induced DNA damage,23 which is predominant in stress‐induced premature senescence (SIPS). SIPS can be induced by many stress signals, including radiation, oxidative stress, chemical toxicants, DNA damage, oncogenic mutation and nutrient deficiency.9 The stimuli‐induced DNA damage response (DDR) is involved in the activation of p16/phosphorylated retinoblastoma (pRb) pathway, especially in epithelial cells.
The cell cycle inhibitor p16 binds to cyclin‐dependent kinase 4 and 6 (CDK4/6) complex and inhibits its activity, thereby resulting in dephosphorylation of pRb and suppression of G1 phase progression24 (Figure 1). The p16/pRb pathway may be essential for the initiation and maintenance of senescence. In addition, the expression of p16 is often used to identify senescent cells due to the expression of p16 in most (though not all) senescent cells.25 Therefore, control of p16 expression may be a promising treatment to inhibit cellular senescence.
Figure 1.
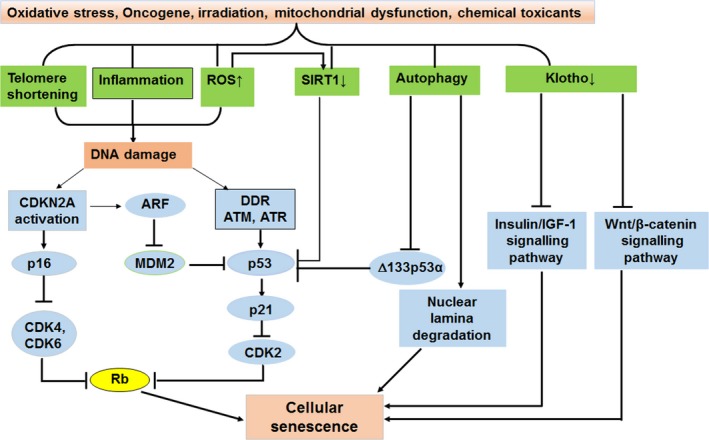
The mechanisms and related pathways of cellular senescence. A variety of stressors induce telomere shortening, increase ROS production, enhance inflammation and autophagy and decrease expression of SIRT1 and Klotho. Telomere shortening, ROS and inflammation usually lead to DNA damage that induces activation of p16/pRb and p53/p21 pathways. Activation of p16 and p21 leads to inhibition of cyclin‐dependent kinase (CDK) complexes and retinoblastoma protein (Rb); thus, senescence is present. Lower expression of SIRT1 causes activation of p53 and induces senescence. Decreased expression of Klotho leads to reduced inhibitory effects on insulin/insulin‐like growth factor‐1(IGF‐1) signalling and Wnt/β‐catenin signalling, resulting in cellular senescence. In addition, autophagy induces cellular senescence through targeting Δ133p53α or lamin B. ROS, reactive oxygen species; DDR, DNA damage response; MDM2, murine double minute 2
p53 and p21Waf1/Cip1 (hereafter referred as p21) are also CDK inhibitors. Phosphorylation of p53 and enhanced p21 expression are associated with the telomere shortening‐induced senescence and SIPS. p21 can contribute to inhibition of various CDKs, especially CDK2, leading to cell cycle arrest and replicative senescence26 (Figure 1). In addition, ARF binds and sequesters the murine double minute 2 oncogene protein into the nucleoli, which leads to inhibition of p53 ubiquitination and degradation, thus resulting in cell cycle arrest through p53/p21 signalling pathway (Figure 1). Evidence strongly suggests that p53/p21 signalling is involved in the initiation of cellular senescence, but p16pRb signalling is necessary to maintain cellular senescence.27
3.3. Telomere shortening
Telomeres are highly conserved non‐coding repetitive TTAGGG sequences of DNA. These specialized structures protect genetic material from damage by preventing fusion or damage to the chromosome ends. Telomerase is an enzyme that maintains telomeres and sustains chromosomal stability. As there is no telomerase expression in most human cells, except in the cells with highly proliferative capacity (bone marrow, skin and germ cells),28 telomeres shorten progressively as cells divide. Telomere shortening causes genetic instability and cell apoptosis, and is the main trigger of replicative senescence.29 Replicative senescence is found in many kinds of cells such as fibroblast, keratinocytes, endothelial cells, lymphocytes, adrenocortical cells and chondrocytes.30 In multiple cells, telomeres shorten progressively with successive cell division. In addition to replicative senescence, cellular senescence induced by various harmful stimuli can also result in shortening of the telomeres.29 A persistent DDR is activated when telomeres shorten to a critical length, which leads to the down‐regulation of genes involved in cell cycle progression and the up‐regulation of growth suppressors such as p53 and p16 (Figure 1).
3.4. Down‐regulation of Klotho or Sirtuin 1 expression
In 1997, Klotho was discovered as a transmembrane protein usually expressed in proximal and distal renal tubules, which was subsequently identified as an anti‐ageing factor and shown to exert inhibitory effects on insulin/insulin‐like growth factor‐1 signalling (Figure 1), oxygen free radicals and phosphate/calcium homoeostasis.31 Multiple ageing‐associated diseases develop in mice with genetic deficiency of Klotho, including osteoporosis, stroke and arteriosclerosis. Conversely, mice with overexpression of Klotho gene exhibit longer lifespan.32 If Klotho is exogenously administered, the senescence of endothelial cell can be significantly reduced.33 Interestingly, the levels of Klotho were significantly decreased in old patients (80‐89 years) with CKD compared to patients with CKD at the age of 60‐69; serum Klotho levels were inversely correlated with age, suggesting that Klotho may serve a biomarker of ageing progression.34
Sirtuin 1(SIRT1) is also recognized as a key modulator of ageing. SIRT1 can deacetylate histone as well as non‐histone proteins such as FOXOs, p53 and NF‐κB,35, 36 thereby influencing several important signalling pathways associated with cellular stress, metabolism and longevity. It has been shown that SIRT1 can inhibit cellular senescence of endothelial cells.37 Down‐regulation of SIRT1 in endothelial cells leads to premature senescence‐like phenotype by enhancing acetylation of p53. On the contrary, increased expression and activity of SIRT1 prevent cells from entering into the state of senescence by inhibiting p53 activity.37 Furthermore, it has been shown that inhibition of miR‐570‐3p in small airway epithelial cells from COPD patients restores SIRT1 expression, leading to SIRT1‐dependent inhibition of senescence markers and cellular rejuvenation.38
3.5. Other mechanisms
Another possible mechanism involved in cellular senescence is autophagy. Autophagy is a key catabolic cellular process, which degrades damaged proteins as well as organelles. Autophagy often exerts protective effects under various stress conditions.39 Recently, autophagy was considered as an effective mechanism involved in the induction of cellular senescence.40 During senescence, autophagy is induced and activated, consequently facilitating the process of senescence. In addition, the senescence phenotype could be delayed if autophagy is inhibited.41 Furthermore, autophagy can cause cellular senescence by targeting Δ133p53α, a p53 isoform that inhibit full‐length p53, or contributing to nuclear lamina degradation under various circumstances42, 43(Figure 1).
Senescent cells have been shown to be resistant to apoptosis.44 The BCL‐2 protein family, including the anti‐apoptotic proteins BCL‐2, BCL‐W and BCL‐XL, is essential for senescent cell resistance to apoptosis.45 Increased expression of BCL‐W and BCL‐XL is involved in senescent cell resistance to apoptosis, and inhibition of BCL‐W and BCL‐XL causes death of senescent cells. Furthermore, a small‐molecule inhibitor targeting the BCL‐2, BCL‐W and BCL‐XL proteins (ABT‐737) contributed to apoptosis of senescent cells both in vivo and in vitro.45
In addition, Wnt/β‐catenin signalling is involved in the pathogenesis of cellular senescence (Figure 1). Wnt/β‐catenin signalling is a conserved signalling pathway in organ development that remains silent in normal adult kidneys.46 According to a recent study,47 Wnt/β‐catenin signalling and renin‐angiotensin system (RAS) activity were up‐regulated in ageing kidneys. Moreover, inhibition of Wnt/β‐catenin signalling significantly protected the normal structure and function of mitochondria, which reduced age‐related renal fibrosis. At the same time, ectopic expression of Klotho, an antagonist of endogenous Wnt/β‐catenin activity, eliminated renal fibrosis in d‐lactose‐induced accelerated ageing mouse model by alleviating cellular senescence and mitochondrial dysfunction.47
4. MARKERS FOR DETECTION OF SENESCENCE
Currently, it is difficult to find unique markers to identify and quantify senescent cells, especially under in vivo conditions. Senescent cells are classically arrested at G1/S transition of the cell cycle triggered by eroded telomeres in ageing cells, thus typically display a DNA content characteristic of the G1 phase and express distinct cell cycle inhibitors such as p16, p21 and p53.48 Subsequently, p21 has been shown to mediate permanent cell arrest in G2/M transition induced by DNA damage through inhibiting mitotic CDK complexes and pRb phosphorylation.49 Assessment of these cell cycle inhibitor expression is very helpful for senescence evaluation.
In addition to aforementioned CDK inhibitors, other well‐known phenotypic markers are also considered to be indicators of cellular senescence. The secretion of SASP factors and the mitochondrial dysfunction50 are consequences of senescence, which stabilize the senescent state and promote paracrine senescence of neighbouring cells. Therefore, monitoring the alterations of mitochondrial dynamics and the secretome may be used to estimate senescent cell burden. Increased expression and activity of senescence‐associated β‐galactosidase (SA‐β‐gal, a lysosomal hydrolase) are commonly applied to identify senescent cells.51 Also, telomere shortening inducing replicative limits can also indirectly, but effectively, identify cellular senescence. Increased production of senescence‐associated DNA damage foci (SADF) and senescence‐associated heterochromatic foci (SAHF) in the nucleus are also regarded as senescence markers. SADFs contain important proteins for induction of senescence.52 Overexpression of proteins related to SAHF formation can also induce senescence and inhibit the expression of genes associated with proliferation.53 Another common senescence marker is γ‐H2AX, which is produced by the phosphorylation of the histone H2AX. Additionally, senescent cells often demonstrate distinctive changes in nuclear morphology associated with loss of lamin B1 (Figure 1), which is also a senescence‐associated biomarker.54 In fact, until now, there is no single marker can be used to entirely define cellular senescence. Therefore, aforementioned markers have been used together to determine senescent cells in different disease and ageing contexts.
In addition, DcR2, a decoy receptor for tumour necrosis factor‐related apoptosis‐inducing ligand, is a marker of senescence.55 Previous studies found that DcR2 is highly expressed in senescent tumour cells and senescent hepatic stellate cell.55 Recently, it has been shown that DcR2 is exclusively expressed in renal tubular epithelia and co‐expressed with senescent markers in patients with diabetic nephropathy, and the extracellular portion of DcR2 is detectable in urine.56
5. SENESCENCE IN RENAL DISEASES
5.1. Type of renal senescent cells
During ageing and renal diseases, senescent cells can be observed in the cortex and medulla. Generally, proximal tubular cells in the cortex are the majority of senescent cells. They are predominant G2‐arrested senescent cells.57 Although senescent cells are mainly present in tubular epithelial cells, other kinds of cells may become senescent. For example, increased p16 expression was described in the glomerular cells during glomerular diseases, such as glomerulonephritis, membranous nephropathy, diabetic nephropathy and focal segment glomerular sclerosis.58, 59 In addition, vascular and interstitial cells may undergo senescence during glomerular diseases and hypertension.58 Therefore, different stressors that occur on different site may decide the localization and type of senescent cells.
In the progression of kidney diseases, various types of cells, including renal tubular epithelial cells, podocytes, endothelial cells, interstitial cells, immune cells and mesangial cells (Figure 2), may become senescent and secrete a lot of factors, which are collectively named the CASP (CKD‐associated secretory phenotype).57 SASP and CASP share many similarities, which might be the mediator of the crosstalk between cellular senescence and CKD.57
5.2. Acute renal injury
Acute renal injury (AKI) is the primary driver of renal injury and CKD. AKI is characterized by a sudden deterioration of kidney function and high morbidity and mortality. During AKI, the kidneys fail to eliminate metabolic wastes, concentrate the urine and maintain water and electrolyte balance.60 Due to a rise in the elderly population, the incidence of AKI has significantly increased in recent years because age‐related renal functional and morphological changes as well as accompanying comorbidities may promote the vulnerability of the elderly population to AKI.61 Increased susceptibility to injury and lack of sufficient repair in ageing and ischaemic kidneys may be associated with telomere shortening and other senescence‐related mechanisms.62
Cell cycle arrest may serve as a protective mechanism following AKI by allowing the cell to avoid the replication of damaged DNA. A study described that p21 knockout mice were more susceptible to ischaemia‐induced acute renal failure.63 On the other hand, it has been shown that mitotic arrest at the G2/M phase plays a crucial role in response to AKI, where it drives maladaptive repair and progressive fibrosis; this suggests that renal recovery after injury would be hampered due to increased numbers of senescent cells and reduced regenerative capacity of aged kidneys. Senescent tubular cells will reduce cell proliferation for repairing injury and contribute to the progression of renal fibrosis.64 A study demonstrated that sustained epithelial Notch activation may trigger a pro‐senescent state and maladaptive repair after ischaemia/reperfusion damage, leading to more inferior outcome of old kidneys after injury.65 Therefore, the role of cell senescence in AKI is complex which needs to be elucidated in different time points of AKI or different models of AKI.
5.3. Diabetic nephropathy
Diabetic nephropathy is a major cause of end‐stage nephropathy. During the development of diabetic nephropathy, proteinuria is positively correlated with high‐glucose‐induced glomerular cell senescence, which is related to the increased production of ROS.66 A study demonstrated that proximal tubule epithelial cells (PTECs) are the major target for impaired glucose‐induced metabolic disorders.67 Also, Tsai et al confirmed that high glucose can induce the senescence of PTEC by increasing the expression of microRNA‐378i, a potential biomarker of kidney damage in diabetic nephropathy.68 Meanwhile, kidney interstitial injury is also critical to the development of diabetic nephropathy.66
Increased p16 expression and SA‑β‑gal activity were observed in tubule cells, mesangial cells, podocytes and endothelial cells from patients with type 2 diabetic nephropathy59 and mice with streptozotocin‐induced type 1 diabetes mellitus.69 Furthermore, several studies have demonstrated a direct link between hyperglycaemia and the induction of senescence in vitro in cultured proximal tubule cells69 and mesangial cells70 as well as in vivo in a mouse model of type 1 diabetes mellitus.69 Overall, these results indicate that hyperglycaemia is a crucial driver of cellular senescence, which may contribute to the progression of diabetic nephropathy.
5.4. Glomerulonephritis
Recent studies show that cellular senescence is closely related to glomerular lesions and the degree of glomerular ageing is also related to the progression of nephropathy.58 For example, during IgA nephropathy (IgAN), disease progression is related to telomere shortening71 as well as several other senescence‐related alterations such as elevated SA‑β‑gal activity and increased expression of p16 and p21.72 These findings suggest that cellular senescence may play a crucial role in the development of IgAN. In this case, however, it remains to be clarified whether senescence is only associated with tissue damage or whether it promotes disease progression. Through a series of experiments and tests, Chen et al found that mice with lupus nephropathy (LN) and severe proteinuria showed enhanced SA‐β‐gal expression and administration of dexamethasone (DEX) reduced SA‐β‐gal expression and renal dysfunction, which suggested that accelerated senescence of the glomerulus was related to the development of LN.73 Another study showed that bone marrow (BM)‐derived mesenchymal stem cells (BM‐MSCs) exhibited signs of senescence in systemic lupus erythematosus (SLE) patients and MRL/lpr mice. The senescent phenotype of BM‐MSCs was involved in the pathogenesis of LN in MRL/lpr mice, which can be reversed by rapamycin.74 In addition, Chuang et al found that the reduction in podocyte SIRT1 led to senescence of podocytes along with reduced activation of peroxisome proliferator‐activated receptor(PPAR)‐a coactivator‐1(PGC1a)/PPAR‐γ, forkhead box O (FOXO)3, FOXO4 and p65 NF‐κB, resulting in aggravated ageing‐induced glomerulosclerosis.75
5.5. Renal fibrosis
Renal fibrosis is common in end‐stage renal failure and may develop from a variety of kidney diseases. Eventually, renal fibrosis leads to loss of kidney function. Recent studies indicate that the kidney with renal fibrosis is more prone to develop CKD due to incomplete recovery after AKI.76 Health regeneration or recovery after AKI should result in recovery of normal structure with differentiated tubule epithelium. However, due to severe damage, recovery of normal structure is often incomplete which contributes to the development of focal tubulointerstitial fibrosis.76 Renal fibrosis may be caused by the accelerated senescence of tubular cells; for example, Wnt9a induced senescent tubular cells to produce TGF‐β1, driving proliferation and activation of rat kidney fibroblasts.77 In addition, TGF‐β1 may induce premature senescence of mesangial cells and myofibroblast‐like phenotype transformation, which may contribute to development of glomerulosclerosis.78 At the same time, it was reported that G2/M‐arrested proximal tubular cells activated c‐jun NH(2)‐terminal kinase (JNK) signalling, promoting pro‐fibrotic cytokine production.79 The inactivation of p16 alleviated nephron atrophy and interstitial fibrosis in kidney transplant experiments.2 Therefore, senescent cells play a crucial part in the development of renal fibrosis.
The role of cellular senescence in renal interstitial fibrosis appears to be complex. Senescent cells may contribute to pro‐fibrotic circumstances through their SASP in ageing and kidney diseases.16 But the specific role of cellular senescence in renal fibrosis has not yet been elucidated. Contrary to the general opinion about the detrimental effect of cellular senescence, there is evidence regarding a beneficial role of senescence in renal fibrosis. It has been reported that p16 inactivation promoted renal interstitial fibrosis in normal mice and a mouse model of unilateral ureteral occlusion.80 These seemingly contradictory findings demonstrated dual role of cellular senescence in different kidney injury processes. Further studies are needed to elucidate whether different effector molecules are involved in the action of senescent renal tubular cells.
5.6. Kidney transplantation
CKD as a trend in a variety of kidney diseases can lead to end‐stage renal disease (ESRD) if the renal function gradually deteriorates. In this case, kidney function deteriorates and alternative therapies such as a kidney transplant are necessary for patients with ESRD.81 Elderly patients are more susceptible to the complications of kidney transplantation and the side effects of the immunosuppressive regimen, showing a lower survival rate after transplant.82 Cells from various tissue of the human body become senescent with age; therefore, ageing may also be one of the factors to affect the therapeutic effect of kidney transplantation. Other studies have demonstrated that ischaemia‐reperfusion injury during kidney transplantation causes oxidative stress, which in turn induces cellular senescence and accelerates dysfunction of the donor organ.83, 84 In experimental rat models, kidney transplant resulted in transient elevation of p21, sustained increase in p16, enhanced expression of SA‐β‐gal expression and accelerated telomere shortening.85 Furthermore, other studies showed that mice undergoing kidney transplant coupled with a loss of INK4a had a significantly better survival rate, less interstitial fibrosis and improved tubular cell proliferation, compared with mice that received a wild‐type transplant.2 Although kidney transplantation is the preferred treatment for ESRD,86 long‐term failure of kidney transplants remains an important clinical problem. Long‐term graft survival is affected by donor age. Ageing increases AKI and reduces renal regeneration capacity. Further, interstitial fibrosis and tubular atrophy were observed in ageing kidney graft. Therefore, kidney transplant senescence could promote graft loss. In addition, in human kidney transplant, CDKN2A expression in implantation biopsies was related to donor age and graft function.87 Telomere length assessed in biopsies collected in the peri‐transplant period can predict the long‐term kidney allograft function, and a significant shortening of telomere was observed in patients with delayed graft function, acute rejection and chronic allograft dysfunction.88 Therefore, all these findings demonstrated that senescence plays a crucial role in kidney transplantation, which may affect the outcome and prognosis of kidney transplant.
5.7. Polycystic kidney disease
The most common kind of polycystic kidney disease (PKD) is autosomal dominant polycystic kidney disease (ADPKD), which leads to ESRD in patients between 50 and 70 years.89 PKD belongs to the ciliopathies family and mainly influence the ciliated epithelial cells that line the renal tubules. PKD can also lead to tubule dilation by increasing cell proliferation.90 Reduced p21 levels in the kidney are found in a rat model of PKD and patients with ADPKD.91 Additionally, application of a CDK inhibitor, roscovitine, increases SA‑β‑gal activity, restores p21 expression, reduces proliferation of renal tubular cells and slows disease progression in an ADPKD mouse model.92 In contrast to aforementioned renal injury, these findings demonstrate that cellular senescence attenuates ADPKD progression.
6. THERAPEUTIC INTERVENTIONS FOR RENAL DISEASES BY REGULATING SENESCENCE
Due to the important role of senescence and SASP in kidney disease, they have been proposed to be therapeutic targets for the treatment of these diseases. It has been verified that some drugs (including glucocorticoids, rapamycin and resveratrol) have extensive pharmacological effects in the management of renal diseases. Notably, these drugs have been shown to reduce ROS levels and transcriptional activity of NF‐κB, thereby decreasing cellular senescence.
For instance, rapamycin reduces transcription and translation of SASP factors, eventually resulting in secretome depletion through inhibition of mTOR and NF‐κB transcriptional activity mediated by suppression of IL1A translation.13
Similar to the effects of rapamycin, pre‐treatment with dexmedetomidine also decreased the number of senescent tubular cells and weakened the protein expression of p53, p21 and p16,7 thus alleviating renal ischaemia/reperfusion‐induced AKI and chronic renal fibrosis in later stages.
The important role of senescence in multiple diseases contributes to the exploration of novel treatments that can regulate senescence. To overcome the detrimental effects of senescence on renal injury and ageing, the pharmacological effect of these novel treatments should focus on reducing the accumulation of senescent cells or increasing the clearance of those cells. Anti‐senescence compounds mainly include senolytics or senotherapeutics. Studies from animal experiments demonstrated that these compounds reversed ageing phenotypes and improved kidney function.93 ABT‐263(navitoclax), an inhibitor of Bcl‐2 family, has been confirmed as a senolytic, showing to remove senescent cells in aged mice and allow for tissue regeneration.94 However, the side effect of navitoclax lies in apoptosis and is not restricted to cellular senescence. Other BCL‐2 family inhibitors such as A1331852 and A1155463 were manifested to be toxic and did not target all senescent cells.95 Therefore, the benefit of removing senescent cells has not been completely clarified by these experiments. Feasibility of this treatment for each kidney disease should also be evaluated.
Kidney transplantation may be a suitable scenario for the application of senolysis. Anti‐senescent treatments should focus on the removal of sustained senescent cells but not on preventing the induction of a temporary cell cycle arrest at the early stage of kidney transplantation. The anti‐senescence compound FOXO4‐DRI has proven to be potent for therapy of the ageing kidney. In senescent cells, FOXO4 was observed to serve as a binding partner of p53, preventing these cells from p53‐induced pro‐apoptotic response. Therefore, FOXO4‐DRI can interrupt FOXO4‐p53 interaction and contribute to p53‐induced apoptosis of senescent cells.96
In addition, chronic treatment of the senolytic drug quercetin alleviated the expression of p16, p19 and p53, reducing renal fibrosis induced by obesity and dyslipidaemia.97 Interestingly, Leung et al found that the treatment of YAP inhibitor verteporfin inhibited activation of senescence‐associated genes, SASPs and activation of Stat3 as well as impeded the development of tubulointerstitial fibrosis.98 Furthermore, erythropoietin can preserve tubular epithelial cell regeneration and reduce renal fibrosis in mice with unilateral ureteral obstruction by inhibiting SIPS and epithelial‐to‐mesenchymal transition of renal epithelial cells.99
For diabetic nephropathy, it has been shown that rapamycin or resveratrol inhibits mesangial cells (MCs) senescence induced by high glucose through increasing the SIRT1 expression and activity, which was completely blocked by treatment with siRNA‐SIRT1.100
In addition, as mentioned, in an experimental LN study, intragastric administration of rapamycin alleviated symptoms of LN by reversing the senescence of BM‐MSCs in MRL/lpr mice through inhibition of the mTOR signalling pathway.74 Also, CDK inhibitors such as roscovitine may be a therapeutic approach for the treatment of PKD.92
7. SUMMARY/FUTURE PERSPECTIVES
Overall, cellular senescence and SASP are involved in kidney injury during both degenerative and hyperplastic diseases. In many renal diseases and in the ageing kidney, there is an accumulation of G1‐ and G2‐arrested senescent cells in multiple regions within the kidney, especially in the renal tubular cells of the cortex. Thus, renal function in failing or ageing kidneys may be improved by targeting senescent cells or SASP.
During the development of renal disease, senescence plays a time‐dependent role in the progression of renal fibrosis, which is beneficial in the early stage but detrimental in the late stage with long‐term adverse consequences. Therefore, it is important to understand how cellular senescence plays a protective or a detrimental effect during different periods of renal injury and the mechanisms of senescence underlying the progression of CKD such as renal fibrosis. Further research is needed to clarify the characteristics of all senescent cells and the specific targets of senescent cells in renal diseases. Until now, most findings were acquired through studying animal models; therefore, the role of senescence in human renal diseases should be validated in future studies. Additionally, novel strategies to inhibit the progression of CKD by removing senescent cells in vivo, including targeted gene therapy to avoid side effects in other organs, need further exploration.
CONFLICT OF INTEREST
All authors declare that they have no conflicts of interests.
AUTHORS’ CONTRIBUTIONS
GA and PZ developed and organized this paper; YW and BZ mainly drafted the paper and created the figures. All other authors participated in revising the paper and finalizing the paper. All authors gave approval for publication.
ACKNOWLEDGEMENTS
This work was supported by a grant from Nature Science Foundation of China (No. 81873563 to Ying Wan); the Scientific Research Foundation from The Education Department Of Sichuan Province (12ZB059 to Ying Wan); and an Applied Basic Research Program of Sichuan Province (2015LZCYD‐S04(13/15) to Ping Zou). This work was also supported by the Hickam Endowed Chair, Gastroenterology, Medicine, Indiana University, the Senior Career Scientist Award from the United States Department of Veteran's Affairs. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs.
Zhou B, Wan Y, Chen R, et al. The emerging role of cellular senescence in renal diseases. J Cell Mol Med. 2020;24:2087–2097. 10.1111/jcmm.14952
Zhou and Wan are contributed equally to the manuscript.
Contributor Information
Gianfranco Alpini, Email: galpini@iu.edu.
Ping Zou, Email: lyzouping@163.com.
REFERENCES
- 1. Bernardes de Jesus B, Blasco MA. Assessing cell and organ senescence biomarkers. Circ Res. 2012;111:97‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Braun H, Schmidt BM, Raiss M, et al. Cellular senescence limits regenerative capacity and allograft survival. J Am Soc Nephrol. 2012;23:1467‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mo H, Wu Q, Miao J, et al. C‐X‐C chemokine receptor type 4 plays a crucial role in mediating oxidative stress‐induced podocyte injury. Antioxid Redox Signal. 2017;27:345‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Munoz‐Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482‐496. [DOI] [PubMed] [Google Scholar]
- 5. Valentijn FA, Falke LL, Nguyen TQ, Goldschmeding R. Cellular senescence in the aging and diseased kidney. J Cell Commun Signal. 2018;12:69‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol. 2018;80:309‐326. [DOI] [PubMed] [Google Scholar]
- 7. Li Q, Chen C, Chen X, et al. Dexmedetomidine attenuates renal fibrosis via alpha2‐adrenergic receptor‐dependent inhibition of cellular senescence after renal ischemia/reperfusion. Life Sci. 2018;207:1‐8. [DOI] [PubMed] [Google Scholar]
- 8. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585‐621. [DOI] [PubMed] [Google Scholar]
- 9. Kural KC, Tandon N, Skoblov M, et al. Pathways of aging: comparative analysis of gene signatures in replicative senescence and stress induced premature senescence. BMC Genom. 2016;17:1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coppe JP, Patil CK, Rodier F, et al. Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853‐2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matjusaitis M, Chin G, Sarnoski EA, Stolzing A. Biomarkers to identify and isolate senescent cells. Ageing Res Rev. 2016;29:1‐12. [DOI] [PubMed] [Google Scholar]
- 12. Lopes‐Paciencia S, Saint‐Germain E, Rowell MC, et al. The senescence‐associated secretory phenotype and its regulation. Cytokine. 2019;117:15‐22. [DOI] [PubMed] [Google Scholar]
- 13. Laberge RM, Sun Y, Orjalo AV, et al. MTOR regulates the pro‐tumorigenic senescence‐associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17:1049‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoare M, Narita M. Transmitting senescence to the cell neighbourhood. Nat Cell Biol. 2013;15:887‐889. [DOI] [PubMed] [Google Scholar]
- 15. Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell. 2008;133:1019‐1031. [DOI] [PubMed] [Google Scholar]
- 16. Tchkonia T, Zhu Y, van Deursen J, et al. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Venkatachalam G, Surana U, Clement MV. Replication stress‐induced endogenous DNA damage drives cellular senescence induced by a sub‐lethal oxidative stress. Nucleic Acids Res. 2017;45:10564‐10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sureshbabu A, Ryter SW, Choi ME. Oxidative stress and autophagy: crucial modulators of kidney injury. Redox Biol. 2015;4:208‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ashraf S, Cha BH, Kim JS, et al. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthr Cartilage. 2016;24:196‐205. [DOI] [PubMed] [Google Scholar]
- 20. Estrada JC, Torres Y, Benguria A, et al. Human mesenchymal stem cell‐replicative senescence and oxidative stress are closely linked to aneuploidy. Cell Death Dis. 2013;4:e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsirpanlis G. Cellular senescence and inflammation: a noteworthy link. Blood Purif. 2009;28:12‐14. [DOI] [PubMed] [Google Scholar]
- 22. Anders HJ, Schaefer L. Beyond tissue injury‐damage‐associated molecular patterns, toll‐like receptors, and inflammasomes also drive regeneration and fibrosis. J Am Soc Nephrol. 2014;25:1387‐1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729‐740. [DOI] [PubMed] [Google Scholar]
- 24. Obexer P, Hagenbuchner J, Rupp M, et al. p16INK4A sensitizes human leukemia cells to FAS‐ and glucocorticoid‐induced apoptosis via induction of BBC3/Puma and repression of MCL1 and BCL2. J Biol Chem. 2009;284:30933‐30940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krishnamurthy J, Torrice C, Ramsey MR, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Okuma A, Hanyu A, Watanabe S, Hara E. p16(Ink4a) and p21(Cip1/Waf1) promote tumour growth by enhancing myeloid‐derived suppressor cells chemotaxis. Nat Commun. 2017;8:2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beausejour CM, Krtolica A, Galimi F, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212‐4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gadalla SM, Savage SA. Telomere biology in hematopoiesis and stem cell transplantation. Blood Rev. 2011;25:261‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bernadotte A, Mikhelson VM, Spivak IM. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging. 2016;8:3‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Magalhaes JP, Toussaint O. Telomeres and telomerase: a modern fountain of youth? Rejuvenation Res. 2004;7:126‐133. [DOI] [PubMed] [Google Scholar]
- 31. Alexander RT, Woudenberg‐Vrenken TE, Buurman J, et al. Klotho prevents renal calcium loss. J Am Soc Nephrol. 2009;20:2371‐2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kurosu H, Yamamoto M, Clark JD, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829‐1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buendia P, Carracedo J, Soriano S, et al. Klotho prevents NFkappaB translocation and protects endothelial cell from senescence induced by uremia. J Gerontol A Biol Sci Med Sci. 2015;70:1198‐1209. [DOI] [PubMed] [Google Scholar]
- 34. Koyama D, Sato Y, Aizawa M, et al. Soluble alphaKlotho as a candidate for the biomarker of aging. Biochem Biophys Res Commun. 2015;467:1019‐1025. [DOI] [PubMed] [Google Scholar]
- 35. Brunet A, Sweeney LB, Sturgill JF, et al. Stress‐dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011‐2015. [DOI] [PubMed] [Google Scholar]
- 36. Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF‐kappaB‐dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369‐2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ota H, Akishita M, Eto M, et al. Sirt1 modulates premature senescence‐like phenotype in human endothelial cells. J Mol Cell Cardiol. 2007;43:571‐579. [DOI] [PubMed] [Google Scholar]
- 38. Baker JR, Vuppusetty C, Colley T, et al. MicroRNA‐570 is a novel regulator of cellular senescence and inflammaging. FASEB J. 2019;33:1605‐1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stranks AJ, Hansen AL, Panse I, et al. Autophagy Controls Acquisition of Aging Features in Macrophages. J Innate Immun. 2015;7:375‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Young AR, Narita M, Ferreira M, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kalimuthu S, Se‐Kwon K. Cell survival and apoptosis signaling as therapeutic target for cancer: marine bioactive compounds. Int J Mol Sci. 2013;14:2334‐2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dou Z, Xu C, Donahue G, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Horikawa I, Fujita K, Jenkins LM, et al. Autophagic degradation of the inhibitory p53 isoform Delta133p53alpha as a regulatory mechanism for p53‐mediated senescence. Nat Commun. 2014;5:4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Childs BG, Baker DJ, Kirkland JL, et al. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014;15:1139‐1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yosef R, Pilpel N, Tokarsky‐Amiel R, et al. Directed elimination of senescent cells by inhibition of BCL‐W and BCL‐XL. Nat Commun. 2016;7:11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Y, Zhou CJ, Liu Y. Wnt signaling in kidney development and disease. Prog Mol Biol Transl Sci. 2018;153:181‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miao J, Liu J, Niu J, et al. Wnt/beta‐catenin/RAS signaling mediates age‐related renal fibrosis and is associated with mitochondrial dysfunction. Aging Cell. 2019;18:e13004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Campisi J. Replicative senescence: an old lives' tale? Cell. 1996;84:497‐500. [DOI] [PubMed] [Google Scholar]
- 49. Baus F, Gire V, Fisher D, et al. Permanent cell cycle exit in G2 phase after DNA damage in normal human fibroblasts. EMBO J. 2003;22:3992‐4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Correia‐Melo C, Marques FD, Anderson R, et al. Mitochondria are required for pro‐ageing features of the senescent phenotype. EMBO J. 2016;35:724‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363‐9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. d'Adda di Fagagna F, Reaper PM, Clay‐Farrace L, et al. A DNA damage checkpoint response in telomere‐initiated senescence. Nature. 2003;426:194‐198. [DOI] [PubMed] [Google Scholar]
- 53. Narita M, Narita M, Krizhanovsky V, et al. A novel role for high‐mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006;126:503‐514. [DOI] [PubMed] [Google Scholar]
- 54. Wang AS, Ong PF, Chojnowski A, et al. Loss of lamin B1 is a biomarker to quantify cellular senescence in photoaged skin. Sci Rep. 2017;7:15678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen J, Zhang WW, Chen KH, et al. Urinary DcR2 is a novel biomarker for tubulointerstitial injury in patients with diabetic nephropathy. Am J Physiol Renal Physiol. 2017;313:F273‐F281. [DOI] [PubMed] [Google Scholar]
- 57. Bonventre JV. Maladaptive proximal tubule repair: cell cycle arrest. Nephron Clin Pract. 2014;127:61‐64. [DOI] [PubMed] [Google Scholar]
- 58. Sis B, Tasanarong A, Khoshjou F, et al. Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int. 2007;71:218‐226. [DOI] [PubMed] [Google Scholar]
- 59. Verzola D, Gandolfo MT, Gaetani G, et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol Renal Physiol. 2008;295:F1563‐F1573. [DOI] [PubMed] [Google Scholar]
- 60. Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7:189‐200. [DOI] [PubMed] [Google Scholar]
- 61. Anderson S, Eldadah B, Halter JB, et al. Acute kidney injury in older adults. J Am Soc Nephrol. 2011;22:28‐38. [DOI] [PubMed] [Google Scholar]
- 62. Cheng H, Fan X, Lawson WE, et al. Telomerase deficiency delays renal recovery in mice after ischemia‐reperfusion injury by impairing autophagy. Kidney Int. 2015;88:85‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Megyesi J, Andrade L, Vieira JM Jr, et al. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 2001;60:2164‐2172. [DOI] [PubMed] [Google Scholar]
- 64. Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11:264‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sorensen‐Zender I, Rong S, Susnik N, et al. Renal tubular Notch signaling triggers a prosenescent state after acute kidney injury. Am J Physiol Renal Physiol. 2014;306:F907‐F915. [DOI] [PubMed] [Google Scholar]
- 66. Cheng NC, Hsieh TY, Lai HS, Young TH. High glucose‐induced reactive oxygen species generation promotes stemness in human adipose‐derived stem cells. Cytotherapy. 2016;18:371‐383. [DOI] [PubMed] [Google Scholar]
- 67. Phillips AO, Steadman R. Diabetic nephropathy: the central role of renal proximal tubular cells in tubulointerstitial injury. Histol Histopathol. 2002;17:247‐252. [DOI] [PubMed] [Google Scholar]
- 68. Tsai YC, Kuo PL, Kuo MC, et al. The interaction of miR‐378i‐Skp2 regulates cell senescence in diabetic nephropathy. J Clin Med. 2018;7(12):468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kitada K, Nakano D, Ohsaki H, et al. Hyperglycemia causes cellular senescence via a SGLT2‐ and p21‐dependent pathway in proximal tubules in the early stage of diabetic nephropathy. J Diabetes Complications. 2014;28:604‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang X, Chen X, Wu D, et al. Downregulation of connexin 43 expression by high glucose induces senescence in glomerular mesangial cells. J Am Soc Nephrol. 2006;17:1532‐1542. [DOI] [PubMed] [Google Scholar]
- 71. Lu YY, Yang X, Chen WQ, et al. Proteins induced by telomere dysfunction are associated with human IgA nephropathy. J Zhejiang Univ Sci B. 2014;15:566‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu J, Yang JR, He YN, et al. Accelerated senescence of renal tubular epithelial cells is associated with disease progression of patients with immunoglobulin A (IgA) nephropathy. Transl Res. 2012;159:454‐463. [DOI] [PubMed] [Google Scholar]
- 73. Yang C, Xue J, An N, et al. Accelerated glomerular cell senescence in experimental lupus nephritis. Med Sci Monit. 2018;24:6882‐6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gu Z, Tan W, Ji J, et al. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging. 2016;8:1102‐1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chuang PY, Cai W, Li X, et al. Reduction in podocyte SIRT1 accelerates kidney injury in aging mice. Am J Physiol Renal Physiol. 2017;313:F621‐F628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Venkatachalam MA, Griffin KA, Lan R, et al. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol. 2010;298:F1078‐F1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Luo C, Zhou S, Zhou Z, et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J Am Soc Nephrol. 2018;29:1238‐1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fu RG, Wu JJ, Xue RL, et al. Premature senescence and cellular phenotype transformation of mesangial cells induced by TGF‐B1. Ren Fail. 2013;35:1142‐1145. [DOI] [PubMed] [Google Scholar]
- 79. Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wolstein JM, Lee DH, Michaud J, et al. INK4a knockout mice exhibit increased fibrosis under normal conditions and in response to unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2010;299:F1486‐F1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Agarwal R. Defining end‐stage renal disease in clinical trials: a framework for adjudication. Nephrol Dial Transplant. 2016;31:864‐867. [DOI] [PubMed] [Google Scholar]
- 82. Kurschat C. Kidney transplantation in old age. Z Gerontol Geriatr. 2016;49:488‐493. [DOI] [PubMed] [Google Scholar]
- 83. van Willigenburg H, de Keizer PLJ, de Bruin RWF. Cellular senescence as a therapeutic target to improve renal transplantation outcome. Pharmacol Res. 2018;130:322‐330. [DOI] [PubMed] [Google Scholar]
- 84. Zhang Y, Unnikrishnan A, Deepa SS, et al. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1(‐/)(‐) mice is correlated to increased cellular senescence. Redox Biol. 2017;11:30‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Joosten SA, van Ham V, Nolan CE, et al. Telomere shortening and cellular senescence in a model of chronic renal allograft rejection. Am J Pathol. 2003;162:1305‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Panah F, Ghorbanihaghjo A, Argani H, et al. Ischemic acute kidney injury and klotho in renal transplantation. Clin Biochem. 2018;55:3‐8. [DOI] [PubMed] [Google Scholar]
- 87. Koppelstaetter C, Schratzberger G, Perco P, et al. Markers of cellular senescence in zero hour biopsies predict outcome in renal transplantation. Aging Cell. 2008;7:491‐497. [DOI] [PubMed] [Google Scholar]
- 88. Domanski L, Kloda K, Kwiatkowska E, et al. Effect of delayed graft function, acute rejection and chronic allograft dysfunction on kidney allograft telomere length in patients after transplantation: a prospective cohort study. BMC Nephrol. 2015;16:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287‐1301. [DOI] [PubMed] [Google Scholar]
- 90. Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384‐2398. [DOI] [PubMed] [Google Scholar]
- 91. Park JY, Schutzer WE, Lindsley JN, et al. p21 is decreased in polycystic kidney disease and leads to increased epithelial cell cycle progression: roscovitine augments p21 levels. BMC Nephrol. 2007;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bukanov NO, Smith LA, Klinger KW, et al. Long‐lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949‐952. [DOI] [PubMed] [Google Scholar]
- 93. de Keizer PL. The Fountain of Youth by Targeting Senescent Cells? Trends Mol Med. 2017;23:6‐17. [DOI] [PubMed] [Google Scholar]
- 94. Zhu Y, Tchkonia T, Fuhrmann‐Stroissnigg H, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl‐2 family of anti‐apoptotic factors. Aging Cell. 2016;15:428‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhu Y, Doornebal EJ, Pirtskhalava T, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL‐XL inhibitors, A1331852 and A1155463. Aging. 2017;9:955‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Baar MP, Brandt RMC, Putavet DA, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017;169(1):132–147.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kim SR, Jiang K, Ogrodnik M, et al. Increased renal cellular senescence in murine high‐fat diet: effect of the senolytic drug quercetin. Transl Res. 2019;213:112‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Leung JY, Wilson HL, Voltzke KJ, et al. Sav1 loss induces senescence and stat3 activation coinciding with tubulointerstitial fibrosis. Mol Cell Biol. 2017;37(12):1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tasanarong A, Kongkham S, Khositseth S. Dual inhibiting senescence and epithelial‐to‐mesenchymal transition by erythropoietin preserve tubular epithelial cell regeneration and ameliorate renal fibrosis in unilateral ureteral obstruction. Biomed Res Int. 2013;2013:308130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhang S, Cai G, Fu B, et al. SIRT1 is required for the effects of rapamycin on high glucose‐inducing mesangial cells senescence. Mech Ageing Dev. 2012;133:387‐400. [DOI] [PubMed] [Google Scholar]