

Abstract
Diabetic Retinopathy (DR), is a significant public health issue and the leading cause of blindness in working-aged adults worldwide. The vision loss associated with DR affects patients’ quality of life and has negative social and psychological effects. In the past, diabetic retinopathy was considered a vascular disease; however, it is now recognized to be a neuro-vascular disease of the retina. Current therapies for DR, such as laser photocoagulation and anti-VEGF therapy, treat advanced stages of the disease, particularly the vasculopathy and have adverse side effects. Unavailability of effective treatments to prevent the incidence or progression of DR is a major clinical problem. There is a great need for therapeutic interventions capable of preventing retinal damage in DR patients. A growing body of evidence shows that neurodegeneration is an early event in DR pathogenesis. Therefore, studies of the underlying mechanisms that lead to neurodegeneration are essential for identifying new therapeutic targets in the early stages of DR. Deregulation of the polyamine metabolism is implicated in various neurodegenerative diseases, cancer, renal failure, and diabetes. Spermine Oxidase (SMOX) is a highly inducible enzyme, and its dysregulation can alter polyamine homeostasis. The oxidative products of polyamine metabolism are capable of inducing cell damage and death. The current review provides insight into the SMOX-regulated molecular mechanisms of cellular damage and dysfunction, and its potential as a therapeutic target for diabetic retinopathy. Structural and functional changes in the diabetic retina and the mechanisms leading to neuronal damage (excitotoxicity, loss of neurotrophic factors, oxidative stress, etc.) are summarized in this review. Furthermore, existing therapies and new approaches to neuroprotection are discussed.
Introduction
Diabetes Mellitus (DM) has become an epidemic health problem in the modern world, affecting predominantly working age group population. Globally, it is accounted that 451 million people are affected by diabetes mellitus in the year 2017 and has been predicted as further increase to 693 million diabetic cases by 2045 [1]. DM is characterized by a group of complex metabolic syndromes like insulin resistance, defective insulin signaling, altered beta cell function, abnormally high blood glucose level, modified lipid metabolism, inflammation and induced oxidative stress [2]. The condition of diabetes causes a deleterious effect on the various organs of the body, including the retina.
Diabetic Retinopathy (DR) is one of the major complications of diabetes causing progressive damage to the retina that leads to vision loss. The vision loss associated with DR affects patients’ quality of life and has negative social and psychological effects. Unavailability of effective treatments to prevent the incidence or progression of DR is a major problem in vision research. Current therapies for DR treat advanced stages of the disease particularly vasculopathy and have adverse side effects.
In the past, diabetic retinopathy was considered as a vascular disease; however, it is now recognized that neurodegeneration plays a significant part in the progression of DR [3–6]. Though DR is clinically considered as a symptomatically recognizable microvasculature defect, several studies implicated that neurodegeneration occurs before the microcapillaries are affected [6–14]. Early retinal function tests in DR patients and cellular, molecular and functional studies in animal models suggest that retinal neurons are vulnerable to damage shortly after diabetes onset [15]. Elucidating molecular mechanisms that lead to neuronal damage in the diabetic retina has the potential to uncover new therapeutic targets aimed at attenuating the neurodegeneration in DR.
Several studies have demonstrated that oxidative stress has a critical role in the pathogenesis of DR [16–19] .The oxidative stress produced by the accumulation of ROS is considered as one of the significant mechanisms responsible for the damages in the retina which eventually progress to DR [20]. Several biochemical pathways have been studied to be associated with the elevated oxidative stress in the diabetic retina.
Deregulation of the polyamine metabolic pathway is implicated in various neurodegenerative diseases, cancer, renal failure and diabetes [21–25]. The oxidative products of polyamine metabolism can induce cellular damage and cell death [26]. Polyamine metabolism is regulated by the combined actions of various enzymes. Spermine Oxidase (SMOX) is a highly inducible enzyme in the polyamine metabolic pathway, and its dysregulation can alter polyamine homeostasis. Studies from our laboratory are the first to demonstrate that polyamine oxidation is fundamentally involved in mediating neuronal and vascular damage in the retina [27, 28]. Increased expression of SMOX was observed in response to hyperoxia-induced neuronal damage in the retina [27] as well as excitotoxicity-induced retinal neurodegeneration[29]. Treatment with SMOX inhibitor significantly reduced neuronal death and retinal degeneration in both models.
The current review emphasizes the impact of retinal neurodegeneration in the DR progression, the major pathways of oxidative stress involved, and the potential of SMOX as a therapeutic target for DR. Factors affecting DR progression, the existing treatment strategies and the need for new therapies are also discussed.
Prevalence and risk factors of DR
The prevalence of DR is increasing proportionately worldwide at the rate of one-third case of diabetic mellitus [30, 31], and one-tenth of DR cases are diagnosed with vision loss. It is estimated that individual lifetime risk of DR is up to 90% in patients with type 1 diabetes mellitus and 50–60% in those with type 2 diabetes mellitus [32–35]. DR largely affect the working age group (20-65 years old), and accounts for 4.8% of vision loss cases [36]. A pooled meta-analysis study of 35 population-based (mostly from developed countries) reported that more than 90 million individuals are estimated to have DR[31]. Incidence and progression of DR is affected by various factors, including age, duration of diabetes, pregnancy, hyperglycemia, systolic blood pressure, hyperlipidemia, obesity, smoking etc. Several studies have reported the contribution of these factors on the onset of diabetic retinopathy. The major association between these factors and DR are summarized in Table 1.
Table1.
Factors affecting diabetic retinopathy
| Factors | Correlation with DR | Modifiable | Comments | Reference | |
|---|---|---|---|---|---|
| Yes | No | ||||
| Age | Positive | No | Patient age (≥50 years) is associated with increased frequency of DR | [38] | |
| Duration of diabetes | Positive | No | Longer duration of diabetes (≥20 years) increases the risk of DR | [39];[40] | |
| Pregnancy | Positive | No | Pregnancy increase the risk of DR | [41];[42] | |
| Hyperglycemia | Positive | Yes | HbA1c > 7% would increase the risk of DR development | [43, 44] | |
| Systolic blood pressure | Positive | Yes | Every increase of 10mmHg in systolic blood pressure leads to 10% risk of early DR and 15% risk of PDR or DME. | [32];[45] | |
| Hyperlipidemia | Indeterminate | Yes | Inconsistent results on the effect of lipid associated with the development and progression of DR and DME | [45];[46] | |
| Obesity | Positive | Yes | Increased waist-hip ratio and waist circumference are positively associated with DR | [47];[40] | |
| Smoking | Positive | Yes | Non-significant positive trend | [48];[49];[50] | |
| Physical activity | Moderate | Yes | Slightly protective effect | [51];[52] | |
| Male Sex | Positive | No | Increased incidence in male | [48];[53] | |
| Myopia | Negative | No | Protective effect towards DR | [54];[55] | |
Neurodegeneration in the diabetic retina
The concept of diabetic retinopathy is now considered as a complex diabetic complication in which neurodegeneration plays a significant role [30, 37]. A growing body of literature on experimental models as well as clinical evidence from patients suggest that neurodegeneration is an early event in the pathogenesis of DR. Therefore, identifying the molecular mechanisms leading to degeneration of neurons in the diabetic retina is essential for the development of new and effective therapeutic strategies. The hallmarks of diabetes-induced neurodegeneration include reduced retinal neuronal function, apoptosis of retinal neurons and thinning of inner retinal layers and nerve fiber layer.
Damage to the neuroretina produces functional abnormalities and these alterations can be detected by Electroretinography (ERG) analysis. ERG can detect functional deficits in the diabetic retina at the earlier stages of the disease. Impairment in ERG is now considered as a characteristic feature of DR. Studies have shown that retinal function is severely affected in DR patients and experimental models [56, 57]. Alterations in ERG have been demonstrated in diabetic patients even before microvascular lesions can be detected [58, 59]. An earlier study has reported ERG abnormalities including a reduction in the amplitude and a delay in the latency of the oscillatory potentials in patients with type 1 diabetes without any evidence of microvascular abnormality [60]. Multifocal ERG (mfERG) has emerged to provide a successful prediction of any new development in eyes with and without retinopathy at baseline [61]. It has been shown that a delayed mfERG implicit time predicts the development of early microvascular abnormalities [62–64]. Localized retinal dysfunction described by mfERG was observed in DM1 patients even in the absence of DR clinical signs [65]. Significant inner retinal dysfunction is documented in DM2 patients without retinopathy [66]. Deficits in the ERG have been observed as early as two weeks after the onset of diabetes in streptozotocin (STZ) treated diabetic rats [67], four weeks in STZ treated mice [68] and three months in the spontaneously diabetic Ins2Akita model [69]. A recent study has shown reduced b-wave amplitude in diabetic mice four months after STZ induction [70], STZ induced diabetic rats after seven weeks of diabetes [71].
Several studies have demonstrated loss of RGCs in diabetic patients [72–74], and animal models studied [75, 76]. It has been identified that retinal ganglion cells are the earliest cells to undergo apoptosis in the diabetic retina [77, 78]. Immunostaining experiments using retinal sections from diabetic patients show an increased level of caspase-3, caspase-9, and Bax in the RGCs [74]. Degenerative changes such as synaptic loss, axonal beading, and morphological changes precede neuronal death in diabetic retina. Severe impairment of synaptic connectivity and loss of amacrine and ganglion cells are reported in 9-month-old Ins2Akita mice [69]. An earlier study conducted on the same model showed RGC loss and dendritic abnormalities within 3 months of diabetes [79]. Similar results are reported in STZ induced diabetic (3 months) rats [80], followed by the outer nuclear layer (photoreceptors) and retinal pigment epithelium (RPE) [81]. Furthermore, studies have been reported that the retinal neurons including bipolar, horizontal, amacrine and photoreceptor cells undergo degeneration during the progression of DR [82]. Presence of an increased number of glial cells and thinning of the optic nerve were also demonstrated through immunohistochemistry studies in animal and diabetic donor eyes [83].
Progressive retinal thinning is another key feature of DR in patients [84, 85]. With the invent of Optical Coherence Tomography (OCT) it is now quite possible to assess the neuroretinal layer thickness in both type 1 and type 2 diabetes with or without DR. Numerous studies have documented structural changes in animal models of DR, diabetic patients, and post-mortem human retinas [72, 86, 87]. The SD-OCT has become an indispensable tool for identifying early signs of neurodegeneration in diabetic patients. Structural studies using OCT demonstrated reduced retinal thickness in T1DM patients [88]. Thinning of the inner retina or NFL has been reported in diabetic patients, without DR or even before visible vascular signs of DR, supporting the presence of neurodegeneration and warrants neuroprotective intervention to prevent chronic neurodegeneration [89, 90]. Recent studies on experimental models using optical coherence tomography reflect these clinical data. A significant reduction in inner retinal thickness was observed in Ins2Akita model between 3-6 weeks of diabetes [69] and STZ diabetic mice five weeks post onset of diabetes[91].
Mechanisms of neurodegeneration in the diabetic retina
Several reviews have detailed the various molecular mechanisms underlying neurodegeneration in the diabetic retina [30, 92–96]. Some of the major mechanisms including the glutamate excitotoxicity, oxidative stress, and imbalance in neurotrophins are described here.
Glutamate excitotoxicity:
Glutamate neurotoxicity is known as a major mechanism of neurodegeneration in the CNS. The excessive synaptic release of glutamate causes the activation of post-synaptic glutamate receptors and mediates excitotoxic cell death. Glutamate is a major neurotransmitter in the retina, engaged in sending an excitatory signal from photoreceptors to bipolar cells and from bipolar cells to ganglion cells [95, 97] in the retina. Increased level of glutamate is found in the retina of experimental animal models [86, 98, 99], and vitreous fluid of diabetic patients [100, 101] and correlated with the inability of macroglia to metabolize the glutamate [98]. Muller cell dysfunction, a characteristic feature of the diabetic retina, is associated with the altered glutamate metabolism [102], and the excess glutamate stimulation generates an uncontrolled intracellular calcium response in postsynaptic neurons resulting in cell death [101].
Oxidative stress:
For decades, oxidative stress has been extensively studied as a major contributor to DR pathophysiology. The role of oxidative stress in diabetic retinopathy has been well established. However, the specific mechanisms of oxidative stress-induced neuronal abnormalities in the diabetic retina are not understood. Numerous reports are available for the unusual generation of oxidative stress by the deregulated metabolic pathways in the diabetic retinas [18, 103, 104]. It has been confirmed that metabolic pathways including AGEs, polyol, hexosamine, PKC are activated under the diabetic condition, and accumulation of its byproducts can induce oxidative stress through the formation of ROS and nitrogen oxygen species [15, 97, 105]. In the diabetic state, the excitatory amino acids stimulate the ROS/RNO formation. The produced ROS/RNO can potentially attack the retinal neurons by upregulating several apoptotic pathways [106]. Similarly, the influx of calcium ions observed in ischemic condition could cause accumulation of calcium ion-dependent glutamate [107] to produce glutamate toxicity and at the same time reducing glutathione level favors the condition to generate oxidative stress [107]. In another study, a reduced level of glutathione was observed in the retina than in the brain of diabetic rat induced with STZ [108]. Also, many lipids undergo photooxidation upon exposure with light to produce oxidized lipids which cause the toxic effect to retinal cells [109]. The defective L-glutamate/ L–aspartate transporter (GLAST) was observed with overproduction of ROS to promote excitotoxicity [102]. All these findings provide an understanding of how various mechanisms of oxidative stress are activated to execute neurodegeneration in the retina at the early stage of diabetic retinopathy.
NADPH-oxidase (NOX) enzymes are another known source for ROS production in the diabetic retina. [110];[111] Indeed, it has been shown that NOX enzymes including NOX2, NOX4 and NOX1 are involved in DR pathology. [110, 112–114] In a genome-wide association study of type 2 diabetic patients with severe DR, a strong association of NOX4 gene with the severe DR was observed. [115] In a recent study using the rat model of ischemic retinopathy, the dual blockade of NOX1 and NOX4 using GKT137831 significantly reduced neuroglial injury, leukocyte adherence and hypoxia-induced inflammation. [116] Several mechanisms have been reported to be involved in the NOX-induced injury in DR, however the majority of them are focused on vascular damage. Diabetes-induced premature endothelial cell senescence was shown that it could be induced through NOX 2 activation in retinal endothelial cells. [114] The blockade of NOX2 or its downstream signaling, arginase, was protective against EC premature senescence. [115] NOX2 was also reported to mediate the permeability effect of the bioactive lipid metabolite, 12-Hydroxyeicosatetraenoic acid (12-HETE) in diabetic mice. [117] These studies did not only introduce NOX enzymes as major sources for ROS production, but also as key players in inducing inflammation and vascular injury in DR.
Mitochondrial dysfunction:
Mitochondria are the one of the major targets of oxidative insults, and oxidative damage-mediated mitochondrial dysfunction is a major mechanism for neuronal damage in neurodegenerative diseases.[118, 119] Oxidative stress-mediated mitochondrial dysfunction is well studied in DR associated vascular dysfunction.[104, 120] Mitochondrial respiration is a major non-enzymatic source for ROS production, shown to be implicated in diabetic retinopathy. Indeed, diabetes was shown to increase the superoxide formation in retinal mitochondria and it is believed that complex III is one of the sources for increased superoxide formation which led to increased mitochondrial permeability. [104, 120] Another mechanism of ROS-induced mitochondrial dysfunction is through affecting the mitochondrial DNA (mtDNA), which is highly sensitive to oxidative damage. In a rat model of diabetes, it was reported that poor control of diabetes-induced the damage of retinal mtDNA, compromised the electron transport chain proteins encoded by that DNA and elevated DNA repair enzymes. [121] Mitochondrial dysfunction is linked to neurodegeneration in several retinal diseases. [122] Recently, a study has reported no marked dysfunction of mitochondria activity, mitochondrial protein expression or alterations in the mtDNA or mtDNA damage in diabetic retinas in relation to retinal neuronal changes. [123], suggesting that the early retinal neurodegeneration in DR could be mediated by non-mitochondrial mechanisms. More studies are needed to confirm these findings and elucidate the mechanisms as several therapeutic agents have shown neuroprotective effects in DR through the regulation of the mitochondrial function. [124–127] Fenofibrate was shown to reduce retinal neuronal cell death by improving mitochondrial efficiency and decreasing oxidative stress. [125] Ultrastructural changes in mitochondria, and neuronal death were evident in diabetic rat retina [128]. Imbalance in mitochondrial dynamics, where the balance between mitochondrial fission and fusion is shifted towards fission, resulting in mitochondrial fragmentation and activation of cell death is demonstrated in other neurodegenerative diseases [129].
Compromised antioxidant defense mechanism:
Elevated oxidative stress in the diabetic retina is not only because of the increased production of ROS and other free radicles, but it also due to the compromised antioxidants defense mechanisms with diabetes. More than two decades ago, it was shown that several antioxidants enzymes are affected by diabetes. [130] The enzymatic activity of glutathione reductase, glutathione peroxidase, superoxide dismutase and catalase were significantly decreased in the diabetic retina. [130, 131] In patients with proliferative diabetic retinopathy, the total antioxidant status in the serum was significantly lower compared to diabetic patients with no retinopathy. [132] The decrease of the different antioxidant enzymes has been linked to retinal apoptosis in diabetes. [128, 133] When different antioxidants compounds were used for DR treatment in experimental models, it was shown that they could prevent retinal apoptosis by restoring some of the antioxidant enzymes. [134–136] Several antioxidants are being supplemented to treat DR, however further studies are needed to confirm the efficiency of antioxidant therapy.
Neurotoxic and neuroprotective factors:
Loss of neuroprotective factors or the reduction in their effectiveness in the retina is another cause for neurodegeneration. Among the neuroprotective or neurotrophic factors pigment epithelial-derived factor (PEDF), somatostatin (SST) and erythropoietin seem to play an essential role [94]. PEDF is a potent neuroprotective and anti-angiogenic factor that is downregulated in DR [94]. Using an experimental model of DR, it is shown that PEDF can protect neurons from glutamate-mediated neurodegeneration, by upregulating glutamine synthetase levels in the early phase of DR [137]. PEDF has shown to increase neuronal survival in models of ischemia/reperfusion injury [138], excessive light exposure [139] and a glutamate-induced model of neurodegeneration [140].
Higher expression of homocysteine is related to neurodegenerative diseases like DR [141, 142]. Elevated homocysteine levels have shown to induce RGC death in vivo and in vitro models [143, 144]. It is metabolized by enzyme methionesynthetase in the presence of cofactors vitamin B12 and folate. A recent study demonstrated a correlation between increased homocysteine levels and a decrease in RNFL thickness and increased severity of diabetic retinopathy [145]. Increased homocysteine levels are found in the vitreous of DR patients [146], and intravitreal treatment of homocysteine resulted in retinal neurodegeneration in a mouse model [147].
Previous studies from our laboratory have demonstrated that diabetes induces arginase expression and activity in the retina and reduce the availability of arginine for nitric oxide synthase to reduce the nitric oxide concentration but generating superoxide at the same time [148]. The superoxide produces more amount of peroxynitrite in the retina to cause neuronal and vascular damages in the diabetic retina. [105, 148]. Recent investigations have shown the involvement of spermine oxidase (a polyamine oxidase) in elevating oxidative damages in neurodegenerative diseases including diabetic retinopathy. In this review, we summarize the literature on polyamine oxidases and their downstream mediators in causing diabetes-induced retinal neuronal damage and dysfunction.
Polyamine metabolism and neurodegeneration
Polyamine metabolism is a major intracellular regulatory system which plays a crucial role in the regulation and maintenance of polyamine homeostasis [149, 150]. Polyamines (spermine, spermidine, and putrescine) are involved in multiple cellular functions and can modulate potassium and calcium trafficking by altering ion channel and receptor function and are indispensable in mitochondrial stabilization [30, 151–154]. Metabolism of polyamines is regulated by the combined actions of multiple enzymes involved (Fig. 1). Spermine Oxidase (SMOX), Spermine Spermidine Acetyl Transferase (SSAT) and Acetyl Polyamine Oxidase (APAO) are major enzymes regulating the backward conversion of polyamines. While SMOX and SSAT are highly inducible, APAO is constitutively expressed. Dysregulated polyamine metabolism has been associated with various neurodegenerative diseases. Altered levels of polyamines have been implicated in neurological disease conditions such as Alzheimer’s disease, [30, 31, 155] Parkinson’s disease, [37, 156, 157] traumatic brain injury [24] and in the pathogenesis of ischemic brain damage [158–160]. Alterations in the expression and activity of enzymes in polyamine metabolic pathway, as well as changes in individual polyamine levels, have been observed in mental disorders, such as schizophrenia, mood disorders, anxiety and suicidal behavior [150]. Even though these evidence suggest an important role for polyamines in neurodegeneration, the mechanisms whereby they participate in neuronal death have yet to be elucidated. Spermine oxidase (SMOX), a FAD-dependent enzyme, is an integral part of the polyamine interconversion cycle, and is localized in the cytoplasm and nucleus of mammalian cells [161, 162]. It is a key enzyme in polyamine catabolism that plays an essential role in maintaining polyamine homeostasis [37, 150]. Increasing evidence shows the involvement of SMOX in neurodegenerative diseases [163],[164]. SMOX selectively oxidizes spermine to produce spermidine and the reactive oxygen species hydrogen peroxide (H2O2) and the aldehyde 3-aminopropanal (3-AP), each with the potential to induce cellular damage and pathologies [21, 165, 166]. The reactive aldehyde, 3-AP is capable of causing damage to RNA, DNA, proteins and membranes while the highly diffusible H2O2 can cause similar damage, once converted to the reactive hydroxyl radical through Fenton or Fenton-like catalysis [22]. Furthermore, the 3-AP generated by SMOX spontaneously converts into the highly reactive unsaturated aldehyde known as acrolein. Acrolein is highly toxic and a potent mediator of oxidative damage. Mechanisms of acrolein-induced toxicity include DNA adduction, inflammation, membrane disruption, protein adduction, endoplasmic reticulum stress etc.[118]. Acrolein causes protein modifications by reacting with their nucleophilic sites [118]. Acrolein conjugates with lysine residues to form acrolein-lysine adducts, [N«-(3-formyl-3, 4-dehydropiperidino)] lysine called FDP-Lysine [167].
Figure 1.
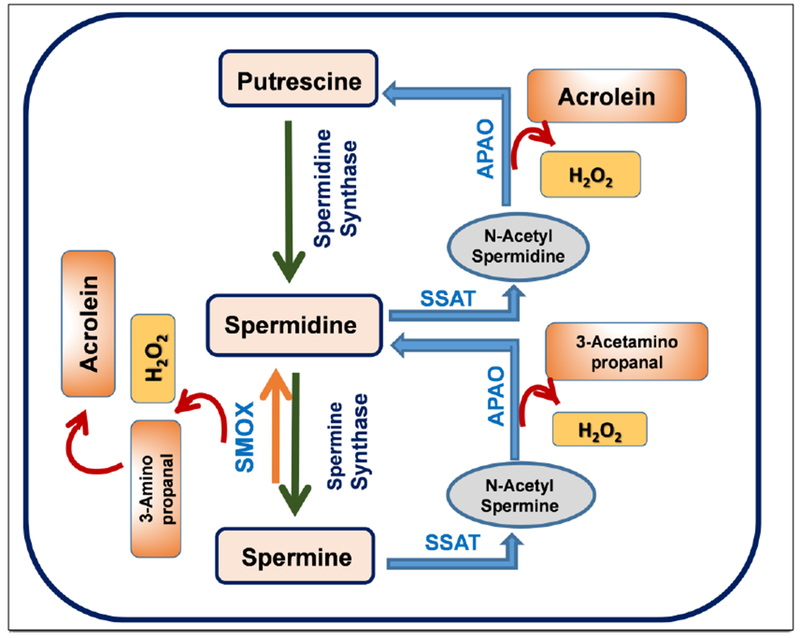
The polyamine metabolic pathway. A diagrammatic representation of the synthesis and oxidation of polyamines. Abbreviations used: SMOX, spermine oxidase; APAO, N (1)-acetyl polyamine oxidase; SSAT, spermine spermidine acetyl transferase.
Studies from our laboratory are the first to demonstrate the specific role of SMOX in retinal neurodegeneration [27]. We have shown increased expression of SMO in a mouse model of ischemic retinopathy [27] and recently in a model of excitotoxicity [29]. Using the mouse model of oxygen-induced retinopathy (OIR), we showed that hyperoxia-induced neuronal degeneration is associated with significant increases in the expression and activity of SMOX along with an increase in spermidine, decrease in spermine and increased the formation of H2O2 suggesting a significant role for SMOX in the neuronal injury [27]. Our recent study showed that SMOX expression is upregulated in the model of excitotoxicity-induced neuronal damage [29]. In both models, treatment with SMOX inhibitor significantly reduced neuronal death and degeneration in the retina. These results suggest that SMOX function is critically involved in retinal neurodegeneration. In general SMOX expression is increased during cellular stress conditions, like inflammation and DNA damage [150]. Earlier studies have shown that SMOX activity is induced rapidly in various cell types, indicating that the increase in activity is resulting from mRNA and protein synthesis, rather than due to modification of the pre-existing protein [168–170]. Studies have shown that TNFα can mediate ROS formation through the upregulation of SMOX [168, 171]. A recent study from our lab investigating the impact of MDL 72527 in the model of excitotoxicity showed the involvement of ERK and Akt signaling pathways in PAO mediated neurodegeneration [29]. Our earlier study using hyperoxia-induced neural death demonstrated that inhibition of SMO (using MDL 72527) protected retinal neurons via the regulation of JNK/SAPK and FASL mediated signaling. [27] Studies performed on human retinal pigmented epithelial cells, ARPE-19, treated with HG showed increased VEGF and cytokine production mediated through TGFβ pathway [172]. Studies by Vidro-Kotchan and group [173] showed acrolein-induced release of cytokine signal through TGFβ signaling. Acrolein is primarily metabolized through GHS, and therefore it contributes directly to cellular oxidative stress through depletion of glutathione.
Spermine oxidase and diabetic retinopathy.
Even though several studies have addressed the critical involvement of polyamine oxidation in neurodegenerative diseases, not much information is available so far in the field of diabetic retinopathy. An earlier study has reported altered levels of serum SMOX in patients with insulin-dependent diabetes mellitus and microvascular complications [181]. Another study conducted in DR patients exhibited altered polyamine levels in the vitreous samples from PDR patients [182]. Our laboratory has observed increased expression of SMOX in the streptozotocin-induced diabetic retina as well as the spontaneously diabetic Akita retina (study under preparation). Other laboratories have reported the role of acrolein (generated during SMOX activity) in the DR progression [71, 172, 176, 183]. As described earlier, acrolein is a highly reactive aldehyde and reacts with the nucleophilic sites of proteins, causing protein modifications. Acrolein conjugates with lysine residues to form acrolein-lysine adducts, called FDP-Lysine. Zhang et al. [184] reported that serum and hemoglobin levels of FDP-lysine were significantly elevated in diabetic patients compared with control individuals. However, no significant correlation was observed between serum FDP-lysine levels and the severity of diabetic retinopathy. In contrast, increased levels of hemoglobin FDP-lysine were observed in patients with proliferative retinopathy compared to those without retinopathy or with non-proliferative retinopathy suggesting that hemoglobin FDP-lysine may be a useful risk marker for the development of proliferative diabetic retinopathy.
Not much information is available on the impact of SMOX in DR induced vascular damage. Murata et al [175] showed the presence of FDP-lysine in the fibrovascular tissue from PDR patients. An earlier study from our laboratory using the Oxygen Induced Retinopathy model of ischemic retinopathy showed a significant involvement of polyamine oxidation in vascular damage. Further, treatment with MDL 72527 significantly reduced OIR-induced vascular injury and retinal inflammation [28]. A recent study using fibrovascular tissue from patients showed FDP-Lys staining in the vascular components containing CD34-positive cells and alpha smooth muscle actin (α-SMA)-positive cells. Furthermore, immunofluorescent staining depicted the subcellular localization of FDP-Lys in the nucleus and cytoplasm of the cells [176]. In a study using an experimental rat model of diabetes, markedly increased FDP-lysine immunoreactivity was observed in the Müller glia, in which initial accumulation of FDP-lysine occurred within Müller glial endfeet and later spread distally throughout the inner radial processes [177]. A recent report from the same research group showed that in vivo administration of 2-HDP (2-hydrazino-4, 6-dimethylpyrimidine, a potent acrolein scavenger) reduced Müller cell accumulation of FDP-lysine in rats (3 months) rendered diabetic with STZ treatment [71]. Furthermore, diabetes-induced retinal expression of inflammatory markers, inflammatory signaling molecules, and activation of microglial cells was markedly reduced in 2-HDP-treated animals. Retinal functional defects in diabetic animals, as indicated by changes in the ERG (7 weeks post diabetes), were also reduced by 2-HDP treatment. Studies using ARPE cells, showed elevated acrolein levels in response to hyperglycemia [172]. Acrolein is also capable of depleting cellular anti-oxidant levels including glutathione, by conjugating with thiol groups [167], [185] and can also increase oxidative damage by protein carbonylation [186, 187]. Recent reports show that protein carbonyl levels are elevated in DR patients [188, 189]. However, the mechanisms by which the FDP-lysine or protein carbonyls are elevated in the diabetic retina are unknown. Acrolein is released as a byproduct during lipid peroxidation as well [118, 190]. A recent investigation using Neuro 2A cells show that acrolein toxicity is reduced with a decline in polyamine oxidases [191]. Acrolein-induced mitochondrial dysfunction has been demonstrated in rat brain [192, 193] and retinal pigment epithelial cells [172, 194, 195]. Carbonylation of mitochondrial proteins by acrolein has been demonstrated as a mechanism of neuronal death in neuron-like PC12 cells [196]. So far, no studies have been performed on acrolein-induced oxidative damage and mitochondrial dysfunction in causing neuronal injury in DR. A proposed mechanism for SMOX-induced neuronal damage and dysfunction is presented in Figure 2.
Figure 2.
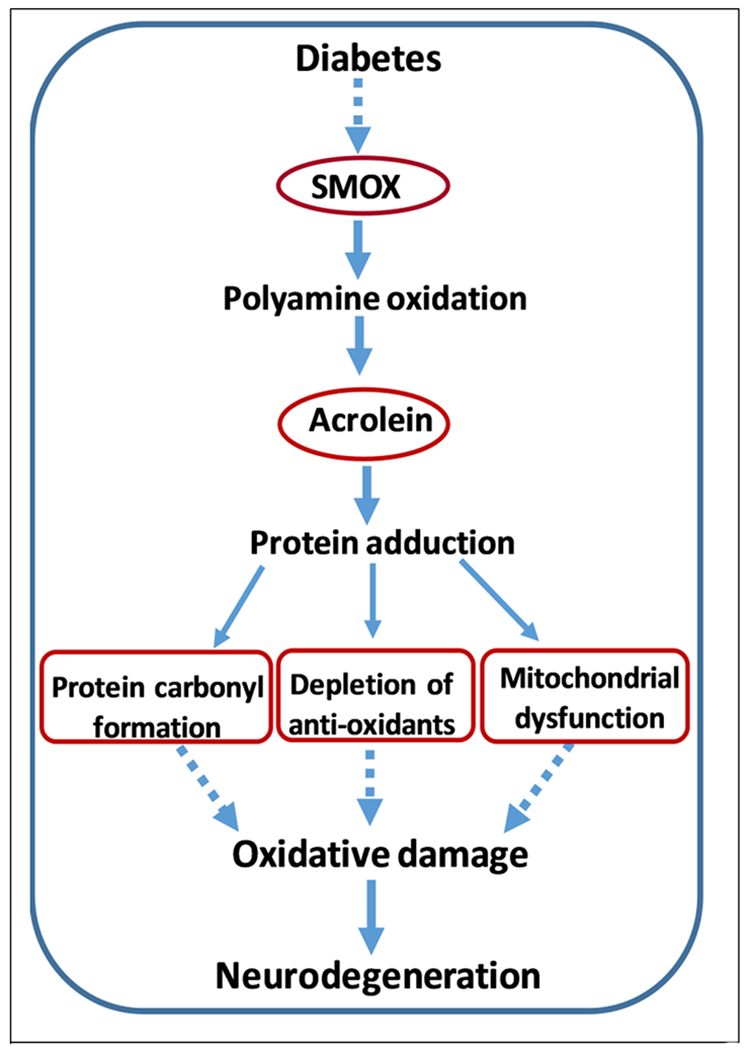
A proposed mechanism for SMOX-induced neurodegeneration in the diabetic retina.
Spermine oxidase inhibitors.
Considering the crucial role SMOX plays in neurodegenerative diseases, blocking its activity would be significant in developing therapies. MDL 72527 is a competitive irreversible inhibitor of SMO and APAO [197]. Studies in a rat model of cerebral ischemia have shown that MDL 72527 treatment significantly reduced brain edema, ischemic injury volume and polyamine levels [197, 198]. Furthermore, blockade of the polyamine oxidation using MDL 72527 was found to be protective against edema and necrotic cavitation after traumatic brain injury [197]. Our study in the mouse OIR model showed for the first time that treatment with MDL 72527 limits the oxidative stress and reduces retinal neurodegeneration, confirming the role of SMOX in hyperoxia-induced neuronal damage [27]. Utilizing the same model, we further showed that MDL 72527 treatment protected the hyperoxia-induced vascular injury, inflammatory signaling and glial activation [28]. Our recent study demonstrated the potential of MDL 72527 against NMDA-induced retinal excitotoxicity[29]. The marked decrease in the neuronal survival in the RGC layer observed in the excitotoxic was significantly improved in response to MDL 72527 treatment. Furthermore, excitotoxicity induced neurodegeneration was also demonstrated by reduced levels of synaptophysin, degeneration of inner retinal neurons and TUNEL labeling of apoptotic cells in the NMDA treated retinas. These changes were abolished in excitotoxic retinas with MDL 72527 treatment [29]. Considering excitotoxicity as a major mechanism of diabetes-induced retinal neurodegeneration and the critical role of SMOX in DR, MDL 72727 is a promising therapeutic target for diabetic retinopathy. Preclinical studies are in progress in our laboratory, investigating the impact of MDL 72527 in diabetes-induced neurodegeneration and impaired retinal function.
It should be noted that in addition to SMOX inhibition, MDL-72527 has been used as a chemotherapeutic strategy for treating cancer [199, 200]. The cytotoxic effect is associated with the rapid formation of numerous lysosomally derived vacuoles [201,202]. However, the cytotoxicity is demonstrated only at very high concentrations, much greater than those required for PAO inactivation[203]. As described earlier, MDL 72527 is a common inhibitor for SMOX and APAO. There is a great need for the development of a specific SMO inhibitor. Spermine analogs (which compete with spermine uptake), CPENspem (N1-ethyl-N11-(cyclopropyl)-methyl-4, 8-diazaundecane) and BENspem (bis(ethyl) norspermine) have been tested to inhibit SMOX activity in a breast cancer model [150, 204]. However, these inhibitors display a poor affinity for the enzyme. Among all the inhibitors studied so far, none of these possesses desired affinity and specificity to be used as a selective inhibitor [150]. Therefore, the search for specific SMOX inhibitors is still an important open issue.
Current treatments for diabetic retinopathy and their limitations
Since vascular abnormalities are the most visible changes, DR is clinically classified as microvascular disease, and the existing treatments are based on fixing the vasculature. The disease may develop either as non-proliferative DR (NPDR) or proliferative DR (PDR). In NPDR, the retinal microvasculature is compromised and loses its control on permeability and becomes susceptible to microaneurysm and microhemorrhages [37, 205]. This hampers blood supply to the retina, resulting in ischemic areas, known as cotton-wool spots [206]. In PDR, new but weak and leaky blood vessels are generated through the abnormal higher expression of angiogenic factors such as VEGF, angiopoietin (Ang-1, Ang-2) [158]. These capillaries start to grow on the retinal surface and extend into the inner limiting membrane and further go down to vitreous. As the blood vessels are not strong enough, blood and other exudates ooze out to manifest vitreous hemorrhage [207]. Additionally, thickening of the retinal basement membrane also identified during the progression of DR with an affected blood-retinal barrier (BRB) that mostly correlated with Muller cells. The edematous feature in the macula, called the diabetic macular edema (DME) also observed in most case of advanced DR. The DME could result either as focal edema due to leakage at microaneurysm and hard exudate or diffuse edema by leakage due to dilated blood vessels [37]. Although the treatment options of diabetic retinopathy have been widely expanded, there is an emerging need for new therapies for DR. The existing treatments aim at reducing the microvascular complications and preventing vision loss. As depicted in Figure 3, this section summarizes the current treatments, their limitations and novel strategies for treating diabetic retinopathy.
Figure 3:
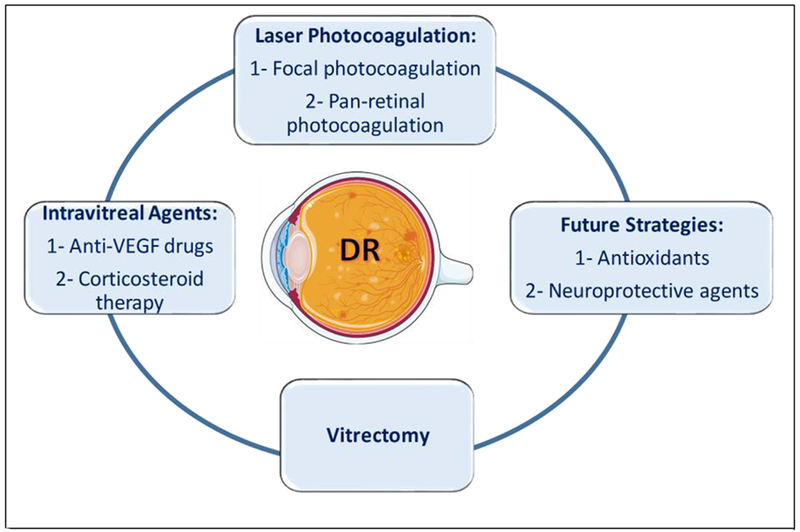
Current treatments and future strategies for DR. Treatment can be implemented in the non-proliferative or the proliferative stages of DR based on the disease course and progression. The three treatment modalities currently being used are: laser photocoagulation, vitrectomy and intravitreal treatment with anti-VEGF agent or corticosteroid therapy. Future strategies that are currently under investigation include antioxidants and neuroprotective agents. The figure was created using Servier Medical Art https://smart.servier.com/
Laser Photocoagulation:
Laser photocoagulation is performed either as the pan-retinal photocoagulation for high-risk proliferative DR or as the focal laser photocoagulation which is used mainly for DME when the central vision is affected. [208] Pan-retinal photocoagulation (PRP), also sometimes referred to as scatter laser, has been the gold standard for proliferative DR treatment in most of the cases before the approved use of anti-VEGF drugs. PRP refers to the use of high energy light (laser) which is absorbed by the retina tissue to destroy abnormal and leaky blood vessels which cause visual impairment.
Focal laser photocoagulation, as mentioned earlier here, has been used for the treatment of DME as demonstrated thirty years ago by the Early Treatment Diabetic Retinopathy Study (ETDRS)[209]. Another report published by the same study group, found that PRP is not effective in reducing the rate of moderate visual loss in DME [97]. In the focal laser approach, ophthalmologists locate the leaky blood vessels in the macula using fluorescein angiography and other examination tools. Then, the less intense laser is applied directly to those spots to treat the leaky vessels and resolve the fluids accumulated in the macula. In the present time, focal laser photocoagulation is used in conjunction with anti-VEGF agents for reducing visual loss in DME patients.
In the updated DR treatment guidelines published by the American Academy of Ophthalmology in 2017, PRP is usually administered in the high-risk proliferative diabetic retinopathy and in special cases of severe nonproliferative DR or non-high-risk-proliferative DR [15, 210]. However, a recent study has shown that administering photocoagulation at an earlier stage of retinopathy (severe non-proliferative diabetic retinopathy) could be more cost-effective than delaying it until the high risk-PDR stage [105]. Given its destructive nature, PRP may cause permanent damage to retinal tissue causing several adverse effects. These include transient central vision loss from macular edema, reduced night vision, complete loss of peripheral vision, vitreous hemorrhage if neovascularization is present and pupillary dilation (mydriasis). [106, 107, 211, 212]
Vitrectomy:
Vitrectomy is the surgical removal of the vitreous in certain cases of DR. It was established in the mid-1980s after the Diabetic Retinopathy Vitrectomy Study (DRVS) has shown that early vitrectomy is beneficial for severe vitreous hemorrhage in type 1 diabetic patients but not in type 2 diabetic patients. A higher percentage of early vitrectomy group had visual acuity of 20/40 or better compared to deferred vitrectomy group at two years after the surgery [17, 102, 105, 108, 213, 214]. The common indications for vitrectomy in PDR include vitreous hemorrhage and tractional retinal detachment. In non-proliferative DR, DME is a clear indication for vitrectomy. Vitrectomy does not only remove the opacities from the media, but it can also affect the retina. A recent clinical study, investigating the effect of vitrectomy surgery on outer retinal layers using SD-OCT found that while vitrectomy improved visual acuity, it also reduced hyperreflective foci which is an indicator for photoreceptor damage in outer retinal layers in DME patients [215, 216]. It has also been reported previously that vitrectomy can increase the risk of open-angle glaucoma. However, further studies with a larger sample number should be conducted to investigate the correlation between vitrectomy and increased intraocular pressure.
Intravitreal pharmacotherapy agents
A). Anti-VEGF drugs:
Laser photocoagulation remained the mainstay treatment for DR for decades before introducing the VEGF as a therapeutic target to reduce DR progression. In clinical trials of Anti-VEGF medications for DME, they showed better efficacy in improving visual acuity compared to focal laser photocoagulation. To date, four anti-VEGF drugs (Pegaptanib, Bevacizumab, Ranibizumab, and Aflibercept) have been using for treating DR and DME, and three of them are FDA approved. Pegaptanib (Macugen), the VEGF antagonist binds to VEGF and prevents its binding to the VEGF receptor, which leads to decreased angiogenesis and reduced vascular permeability and inflammation. It was approved in 2004 by the Food and drug administration (FDA) for the treatment of wet (age)-related macular degeneration. In 2006, the Macugen Diabetic Retinopathy Study Group published that subjects assigned to Pegaptanib showed regression of retinal neovascularization by week 36 in patients with diabetes mellitus [217, 218]. Pegaptanib was predominantly used for DME. [105] However, the use of Pegaptanib in DR has been deferred by the wide use of VEGF antibodies discussed here. Ranibizumab (Lucentis) is a recombinant human monoclonal antibody that binds to the receptor binding site of VEGF-A. This prevents the binding of VEGF-A to its receptors (VEGFR1 and VEGFR2) which reduces endothelial cell proliferation, vascular leakage, and angiogenesis. It was originally approved by the FDA in 2006 as an intravitreal injection for the treatment of patients with neovascular (wet) age-related macular degeneration.[219] Following this, it was also approved for the treatment of Macular Edema Following Retinal Vein Occlusion (RVO) and Diabetic Macular Edema (DME). In 2015, the FDA approved Ranibizumab for DR in patients with DME. Recently, in 2017, it was approved for all stages of DR [219]. Aflibercept (Eylea) is a recombinant fusion protein consisting of portions of human VEGF receptors 1 and two extracellular domains fused to the Fc portion of human IgG1. Aflibercept acts like a soluble decoy receptor that binds to VEGF-A and placental growth factor and reduces their binding to their receptors leading to decreased neovascularization and vascular permeability. It was initially approved by the FDA in 2011 for neovascular (wet) age-related macular degeneration. Later on, it was approved for the treatment of DR in patients with DME [220]. Recently, Regeneron Pharmaceuticals, Inc. has submitted supplemental Biologics License Application (sBLA) of EYLEA® (aflibercept) Injection for the treatment of DR without DME to the FDA for review. This was based on the results of Phase 3 PANORAMA trial investigating EYLEA as a treatment for patients with moderately severe to severe non-proliferative DR. Bevacizumab (Avastin) is a recombinant humanized IgG1 antibody that binds to VEGF and inhibits its biological activity. It was originally approved by the FDA in 2004 for the treatment of metastatic colorectal cancer. Till now, it is not approved by the FDA for the treatment of DR or DME. However, Avastin is currently used as an adjunct therapy with laser photocoagulation based on the positive outcomes shown in several clinical trials evaluating the efficacy and safety of Avastin in DR and DME patients [221–224].
Even though anti-VEGF agents have been recognized as a fundamental tool in treating proliferative DR and DME, these drugs have limitations. VEGF-A is not only identified as a proangiogenic factor, but it was also shown that VEGF A is an important neuroprotective factor. VEGF-A has both a direct and indirect neuroprotective role. VEGF-A indirectly protects the neurons by increasing the blood flow to the neuronal layers of the retina. VEGF-A also induces direct neuroprotection as shown by in ex-vivo retinal cultures [225]. It was shown that VEGF-A is involved as survival and neuroprotective factor in the adaptive response to ischemic injury in the retina [225]. In another study, the gene transfer of a transcriptional factor that induces VEGF-A expression protected against neuropathy in a rat model of diabetes [226]. Knowing that DR is not only a vascular disease and it does involve a neurodegeneration component, the anti-VEGF drugs do not only decrease vascular permeability, but it can also affect the neurons. Indeed, a recent clinical study has shown as the association between the thinning of the retinal nerve fiber layer and the repeated injections of anti-VEGF in age macular degeneration patients [227]. Another study has shown that the retinal ganglion cell layer is also affected in the long-term anti-VEGF treatment in AMD patients [228]. In murine models, repeated intravitreal administration of anti-VEGF induced neurodegeneration in the retinas of Akita mice [229]. These studies emphasis the neurodegeneration adverse effect of the sustained administration of anti-VEGF agents.
Several adverse effects (including both local and systemic) have been reported after the intravitreal injection of anti-VEGF agents. Common local side effects include the transient increase in the intraocular pressure (IOP) after the injection [230–233]. This increase in the IOP is usually observed 30 minutes after the intravitreal injection of Bevacizumab and Ranibizumab due to vitreal expansion. This effect is generally reversible, and the IOP starts to decrease significantly in the first-day post-injection in non-glaucomatous patients [230, 231]. Another common side effect is tractional retinal detachment after the anti-VEGF intravitreal injections which is more common with Bevacizumab [234, 235]. A possible mechanism of this is the misbalance between angiogenic factors and fibrotic factors. When VEGF and other angiogenic factors are inhibited with the anti-VEGF agents, the connective tissue growth factor (CTGF) levels are increased which promotes fibrosis [236, 237]. Acute endophthalmitis is another local side effect of anti-VEGF injection which is increased with frequent injections [238, 239]. Other local side effects include cataract, ocular hemorrhage, and ocular inflammation. Some systemic adverse effects have been reported after the anti-VEGF intravitreal injection including acute elevation of systemic blood pressure, myocardial infarctions, iliac artery aneurysms, and cerebrovascular accidents [240].
B). Corticosteroid therapy:
Inflammation has been identified as an important competent of the DR pathophysiology. Alleviating or preventing inflammation in the eye has been a novel strategy in treating proliferative DR. As previously reported, corticosteroids are strong anti-inflammatory agents that have been widely used for various disease conditions. Corticosteroids reduce inflammation by modulating interleukin-1β (IL-1β), tumor necrosis factor (TNF-α) and prostaglandins which are upregulated in DR [241]. It has also been reported that corticosteroids have angiostatic effects regardless of their glucocorticoid or mineralocorticoid effects[242]. Corticosteroids inhibit the gene expression of VEGF [243].
Intravitreal triamcinolone acetenoid (IVTA) is a steroid derivative, which has been tested in several clinical trials to check if it has any beneficial effects alone or in combination with laser or anti-VEGF treatment. In protocol J published by Diabetic Retinopathy Clinical Research Network, they found that the addition of one IVTA in eyes receiving focal laser for DME significantly improved the visual acuity and decreased macular edema by 14 weeks [244]. However, larger clinical studies were needed to determine if longer treatment IVTA is beneficial or not [244]. In a five-year randomized clinical trial, eyes managed with Triamcinolone alone or with laser did not show a better long-term improvement of the vision compared to eyes receiving ranibizumab therapy for centerinvolving DME [245]. This derived the need to develop sustained release delivery forms of the steroids. In 2009, Ozurdex, intravitreal dexamethasone (DEX) implant was originally approved by the FDA for the treatment of macular edema after retinal vein occlusion. Later, the MEAD clinical trial has shown that Ozurdex was efficient in improving best-corrected visual acuity (BCVA) in DME patients with accepted safety profile leading to its FDA approval [246]. In the same year, the FDA also approved ILUVIEN® (fluocinolone acetonide intravitreal implant) based on the FAME clinical trial [247]. Cataract and increased intraocular pressure have been reported as common adverse effects for steroids intravitreal treatment [246, 247].
At the neuronal level, Triamcinolone intravitreal administration in early diabetic rate model inhibited the retinal apoptosis which could be mediated through the inhibition of p38MAPK pathway [248]. In another study utilizing a transgenic mouse model of Müller cell ablation, IVTA prevented photoreceptor degeneration and inhibited glial activation [249]. In a recent small-scale clinical trial, ranibizumab or triamcinolone decreased the RNFL thickness in DME patients [250]. Further studies are still needed to elucidate the neuroprotective role of corticosteroids in DR and DME.
Future treatment strategies
Antioxidants:
Oxidative stress has been extensively studied as a major component of DR pathology [16, 112, 148]. Several antioxidant agents have been used as a supplement to treat DR such as vitamin C, vitamin E, superoxide dismutase and magnesium [251]. However, no solid results were obtained to determine if the antioxidants intervention prevents or treats diabetic retinopathy. In a clinical trial, ubiquinone and combined antioxidant therapy were studied to determine if this would reduce oxidative stress markers or not in NPDR [252]. It was found that the treatment successfully reduced the oxidative stress markers, however, any clinical outcomes were not measured in patients especially that this study was done in NPDR patients. In another clinical trial published recently, it was observed that combined treatment of intravitreal ranibizumab with the addition of a dietary supplement rich in DHA (DOCOSAHEXAENOIC ACID) plus antioxidants vitamins, minerals and xanthophylls reduced the central subfield macular thickness after two years of follow-up compared to ranibizumab alone in DME patients [253]. Larger clinical trials are still needed to confirm the efficacy of antioxidants supplementation in proliferative DR and diabetes-induced neuronal damage and dysfunction.
In experimental diabetic models, several agents have been tested in relation to neurodegeneration. Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one) systemic administration, a free radical scavenger, significantly reduced retinal ganglion cell death in diabetic mice [68]. In vitro, L-carnitine attenuated high glucose-induced retinal ganglion cell death through reducing oxidative stress and mitochondrial dysfunction [254]. Dietary flavonoids have been widely known for their antioxidant and anti-inflammatory properties. They have also been introduced as a potential intervention to prevent neuronal damage and apoptosis in DR [255]. Mitochondria is a known source of oxidative stress in diabetic retina. Mitochondria- targeted peptide MTP- 131 is a novel antioxidant agent, which concentrates in the inner mitochondria membrane. It has been shown that MTP-131 significantly reduced hydrogen peroxide-induced cell death in the retinal ganglion cell line [256]. In a recent study, alpha-lipoic acid (ALA), a well-characterized antioxidant, treatment significantly protected against ganglion cell loss, preserved inner and outer retinal layers in diabetic mice [257]. These studies demonstrate the beneficial role of antioxidants in the treatment or prevention of diabetic retinopathy. However, further translational and clinical studies are still needed to confirm these findings.
Neuroprotective agents:
Although neurodegeneration signs are exhibited in the early stages of DR, most of the current treatment strategies of DR focus on resolving the microvascular abnormalities of DR. In a very recent report published by the EUROCONDOR (European Consortium for the Early Treatment of Diabetic Retinopathy), they found that neurodegeneration plays a role in DR pathology but not in all the type 2 diabetic patients [258]. A new approach in targeting DR is by elevating the levels of neurotrophic factors that are known to be downregulated during disease progression [259]. Among these factors are Pigment epithelium-derived factor (PEDF), somatostatin (SST), nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF). PEDF is an anti-angiogenic and neurotrophic factor which was shown to be reduced in diabetic human retina and vitreous [260]. In experimental diabetic retinopathy models, PEDF administration attenuated glial activation and vascular permeability through mediating NADPH-induced oxidative stress [261]. The application of PEDF derivative eye drops in Akita mice reduced retinal ganglion cell death and inner plexiform layer thinning [262]. The sustained delivery of PEDF to the eye using gene therapy in diabetic mice decreased inflammation and reduced glial activation [263]. SST is another neurotrophic factor which is downregulated in diabetes [264]. In a rat model of diabetes, the topical administration of SST significantly prevented neurodegeneration, glial activation, and ERG abnormalities [265]. The SST application also prevented glutamate accumulation and prevented diabetes-induced downregulation of its main transporter [265]. In a photoreceptor-like cell line, SST significantly protected against the high glucose-induced cell apoptosis [266]. In a small clinical trial, octreotide (SST analog) administration was shown to improve visual acuity in postsurgical cystoid macular edema patients with no difference in retinal thickness or vascular leakage [267]. In a clinical trial conducted by the EUROCONDOR (European Consortium for the Early Treatment of Diabetic Retinopathy), it was found that SST administration was only useful in certain subsets of diabetic patients with neuronal dysfunction [268]. Further studies with longer follow-up and neuroprotection assessment are needed to evaluate the clinical efficacy of SST in patients. NGF is a neurotrophic factor that is involved in the regulation of growth, proliferation, and survival of neurons. It has been reported that diabetes induces an imbalance between the pro form of NGF (proNGF) and NGF which could be related to neurovascular degeneration [269]. The exogenous administration of NGF significantly protected against RGC loss in diabetic rats [270]. In another study, authors looked at the vascular degeneration and found that the combination of Bevacizumab and NGF significantly prevented vascular leakage and limited inflammation and angiogenesis [271]. These studies introduced NGF as a preventive tool to limit DR progression through neuroprotection.
Although treatment strategies of DR have evolved a lot in the last two decades, there is still an emerging need for developing new therapies that prevent or limit the progression of neurovascular degeneration in DR with less invasive routes of administration and more compliant tools.
Conclusions
Degeneration of retinal neurons is an early event in DR pathogenesis and plays a crucial role in the disease progression. Unavailability of effective treatments to prevent the incidence or progression of DR is an unmet need in the field. Current therapies treat advanced stages of the disease, particularly the vasculopathy and have adverse side effects. Hence therapeutic approaches based on neuroprotection will be effective in DR progression. Deregulation of the polyamine metabolism is implicated in various neurodegenerative diseases. SMOX is a key enzyme in the polyamine metabolic pathway and its activation causes oxidative damages. Upregulation of SMOX and its downstream mediators are evident in the diabetic retina. Given the significance of neurodegeneration in DR, and the role of SMOX signaling in the diabetic retina it is anticipated that therapeutic strategies based on blocking SMOX function will be effective in preventing or delaying vision loss in diabetic patients.
Future directions:
Further investigations are required to better understand the role of SMOX in the diabetic retina, in order to develop effective strategies for targeting SMOX activity as a therapy for DR. Studies are needed to determine the specific cell types expressing SMOX in the retina and how they are altered during DR progression. Lack of specific inhibitor is a major drawback in the field and there is a great need for developing specific blockers for SMOX activity. Further studies are also required to elucidate the SMOX-mediated downstream signaling pathways in the diabetic retina.
Table 2:
Polyamine oxidase signaling in various retinal disorders.
| Disease model | Signaling/target | Reference |
|---|---|---|
| Ischemic retinopathy | SMOX | [27];[28] |
| Optic nerve injury | Spermidine | [174] |
| Diabetic retinopathy | Spermine | [175] |
| Retinal excitotoxicity | SMOX | [29] |
| Diabetic retinopathy | FDP-Lysine | [176];[177] |
| Glaucoma | Spermidine | [178] |
| RPE dysfunction | Spermidine | [179] |
| EAE | Spermidine | [180] |
| Diabetic retinopathy | Acrolein | [172];[71] |
Acknowledgments
Funding support
This work was supported by a grant from the National Institute of Health (R01EY028569 (SPN)).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors declare no conflict of interest.
References
- 1.Cho NH, et al. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract, 2018. 138: p. 271–281. [DOI] [PubMed] [Google Scholar]
- 2.Wu T, et al. Metabolomics window into diabetic complications. J Diabetes Investig, 2018. 9(2): p. 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Moraes G and Layton CJ, Therapeutic targeting of diabetic retinal neuropathy as a strategy in preventing diabetic retinopathy. Clin Exp Ophthalmol, 2016. 44(9): p. 838–852. [DOI] [PubMed] [Google Scholar]
- 4.Imai H, et al. Neuroprotection for diabetic retinopathy. Dev Ophthalmol, 2009. 44: p. 56–68. [DOI] [PubMed] [Google Scholar]
- 5.Jindal V, Neurodegeneration as a primary change and role of neuroprotection in diabetic retinopathy. Mol Neurobiol, 2015. 51(3): p. 878–84. [DOI] [PubMed] [Google Scholar]
- 6.Kern TS, Interrelationships between the Retinal Neuroglia and Vasculature in Diabetes. Diabetes Metab J, 2014. 38(3): p. 163–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zlokovic BV, Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci, 2011. 12(12): p. 723–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zlokovic BV, Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci, 2005. 28(4): p. 202–8. [DOI] [PubMed] [Google Scholar]
- 9.Zlokovic BV, The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron, 2008. 57(2): p. 178–201. [DOI] [PubMed] [Google Scholar]
- 10.Zipser BD, et al. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol Aging, 2007. 28(7): p. 977–86. [DOI] [PubMed] [Google Scholar]
- 11.Zhong Z, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci, 2008. 11(4): p. 420–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul J, Strickland S, and Melchor JP, Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J Exp Med, 2007. 204(8): p. 1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalaria RN, Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr Rev, 2010. 68 Suppl 2: p. S74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown WR and Thore CR, Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol, 2011. 37(1): p. 56–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ola MS, et al. Neurodegeneration and neuroprotection in diabetic retinopathy. Int J Mol Sci, 2013. 14(2): p. 2559–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caldwell RB, et al. Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Targets, 2005. 6(4): p. 511–24. [DOI] [PubMed] [Google Scholar]
- 17.Kowluru RA, et al. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res, 2015. 48: p. 40–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowluru RA and Kanwar M, Oxidative stress and the development of diabetic retinopathy: contributory role of matrix metalloproteinase-2. Free Radic Biol Med, 2009. 46(12): p. 1677–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y, Tang L, and Chen B, Oxidative stress: implications for the development of diabetic retinopathy and antioxidant therapeutic perspectives. Oxid Med Cell Longev, 2014. 2014: p. 752387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin MT and Beal MF, Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 2006. 443(7113): p. 787–95. [DOI] [PubMed] [Google Scholar]
- 21.Seiler N, Oxidation of polyamines and brain injury. Neurochem Res, 2000. 25(4): p. 471–90. [DOI] [PubMed] [Google Scholar]
- 22.Casero RA and Pegg AE, Polyamine catabolism and disease. Biochem J, 2009. 421(3): p. 323–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaturvedi R, et al. Increased Helicobacter pylori-associated gastric cancer risk in the Andean region of Colombia is mediated by spermine oxidase. Oncogene, 2015. 34(26): p. 3429–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zahedi K, et al. Polyamine catabolism is enhanced after traumatic brain injury. J Neurotrauma, 2010. 27(3): p. 515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bjelakovic G, et al. Does polyamine oxidase activity influence the oxidative metabolism of children who suffer of diabetes mellitus? Mol Cell Biochem, 2010. 341(1-2): p. 79–85. [DOI] [PubMed] [Google Scholar]
- 26.Murray Stewart T, et al. Polyamine catabolism and oxidative damage. J Biol Chem, 2018. 293(48): p. 18736–18745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narayanan SP, et al. Arginase 2 deficiency reduces hyperoxia-mediated retinal neurodegeneration through the regulation of polyamine metabolism. Cell Death Dis, 2014. 5: p. e1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel C, et al. Treatment with polyamine oxidase inhibitor reduces microglial activation and limits vascular injury in ischemic retinopathy. Biochim Biophys Acta, 2016. 1862(9): p. 1628–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pichavaram P, et al. Targeting Polyamine Oxidase to Prevent Excitotoxicity-Induced Retinal Neurodegeneration. Front Neurosci, 2018. 12: p. 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simo R, Stitt AW, and Gardner TW, Neurodegeneration in diabetic retinopathy: does it really matter? Diabetologia, 2018. 61(9): p. 1902–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yau JW, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care, 2012. 35(3): p. 556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein R, et al. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XXII the twenty-five-year progression of retinopathy in persons with type 1 diabetes. Ophthalmology, 2008. 115(11): p. 1859–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein R, et al. The Wisconsin epidemiologic study of diabetic retinopathy. II. Prevalence and risk of diabetic retinopathy when age at diagnosis is less than 30 years. Arch Ophthalmol, 1984. 102(4): p. 520–6. [DOI] [PubMed] [Google Scholar]
- 34.Klein R, et al. The Wisconsin epidemiologic study of diabetic retinopathy. III. Prevalence and risk of diabetic retinopathy when age at diagnosis is 30 or more years. Arch Ophthalmol, 1984. 102(4): p. 527–32. [DOI] [PubMed] [Google Scholar]
- 35.Wong TY, et al. Diabetic retinopathy. Nat Rev Dis Primers, 2016. 2: p. 16012. [DOI] [PubMed] [Google Scholar]
- 36.Ting DS, Cheung GC, and Wong TY, Diabetic retinopathy: global prevalence, major risk factors, screening practices and public health challenges: a review. Clin Exp Ophthalmol, 2016. 44(4): p. 260–77. [DOI] [PubMed] [Google Scholar]
- 37.Lechner J, O’Leary OE, and Stitt AW, The pathology associated with diabetic retinopathy. Vision Res, 2017. 139: p. 7–14. [DOI] [PubMed] [Google Scholar]
- 38.Anwar SB, et al. Evaluation of multiple risk factors involved in the development of Diabetic Retinopathy. Pak J Med Sci, 2019. 35(1): p. 156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khan R, et al. Age of Onset of Diabetes and Its Comparison with Prevalence and Risk Factors for Diabetic Retinopathy in a Rural Population of India. Ophthalmic Res, 2019. 61(4): p. 236–242. [DOI] [PubMed] [Google Scholar]
- 40.Rajalakshmi R, et al. Prevalence and risk factors for diabetic retinopathy in Asian Indians with young onset type 1 and type 2 diabetes. J Diabetes Complications, 2014. 28(3): p. 291–7. [DOI] [PubMed] [Google Scholar]
- 41.Vestgaard M, et al. Pregnancy-induced sight-threatening diabetic retinopathy in women with Type 1 diabetes. Diabet Med, 2010. 27(4): p. 431–5. [DOI] [PubMed] [Google Scholar]
- 42.Rasmussen KL, et al. Progression of diabetic retinopathy during pregnancy in women with type 2 diabetes. Diabetologia, 2010. 53(6): p. 1076–83. [DOI] [PubMed] [Google Scholar]
- 43.Mohamed Q, Gillies MC, and Wong TY, Management of diabetic retinopathy: a systematic review. JAMA, 2007. 298(8): p. 902–16. [DOI] [PubMed] [Google Scholar]
- 44.Sasso FC, et al. High HDL cholesterol: A risk factor for diabetic retinopathy? Findings from NO BLIND study. Diabetes Res Clin Pract, 2019. 150: p. 236–244. [DOI] [PubMed] [Google Scholar]
- 45.Hainsworth DP, et al. Risk Factors for Retinopathy in Type 1 Diabetes: The DCCT/EDIC Study. Diabetes Care, 2019. 42(5): p. 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raman R, et al. Prevalence of Metabolic Syndrome and its influence on microvascular complications in the Indian population with Type 2 Diabetes Mellitus. Sankara Nethralaya Diabetic Retinopathy Epidemiology And Molecular Genetic Study (SN-DREAMS, report 14). Diabetol Metab Syndr, 2010. 2: p. 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawasaki R, et al. Factors associated with non-proliferative diabetic retinopathy in patients with type 1 and type 2 diabetes: the Japan Diabetes Complication and its Prevention prospective study (JDCP study 4). Diabetol Int, 2019. 10(1): p. 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Leiden HA, et al. Risk factors for incident retinopathy in a diabetic and nondiabetic population: the Hoorn study. Arch Ophthalmol, 2003. 121(2): p. 245–51. [DOI] [PubMed] [Google Scholar]
- 49.Klein R, et al. Relation of smoking, drinking, and physical activity to changes in vision over a 20-year period: the Beaver Dam Eye Study. Ophthalmology, 2014. 121(6): p. 1220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai X, et al. The association of smoking and risk of diabetic retinopathy in patients with type 1 and type 2 diabetes: a meta-analysis. Endocrine, 2018. 62(2): p. 299–306. [DOI] [PubMed] [Google Scholar]
- 51.Ren C, et al. Physical activity and risk of diabetic retinopathy: a systematic review and meta-analysis. Acta Diabetol, 2019. [DOI] [PubMed] [Google Scholar]
- 52.Bukht MS, et al. Association between physical activity and diabetic complications among Bangladeshi type 2 diabetic patients. Diabetes Metab Syndr, 2019. 13(1): p. 806–809. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, et al. Prevalence of diabetic retinopathy in the United States, 2005-2008. JAMA, 2010. 304(6): p. 649–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu Y, et al. Myopia and/or longer axial length are protective against diabetic retinopathy: a meta-analysis. Acta Ophthalmol, 2016. 94(4): p. 346–52. [DOI] [PubMed] [Google Scholar]
- 55.Wang X, et al. Myopia and diabetic retinopathy: A systematic review and meta-analysis. Diabetes Res Clin Pract, 2016. 111: p. 1–9. [DOI] [PubMed] [Google Scholar]
- 56.Jackson GR, et al. Inner retinal visual dysfunction is a sensitive marker of non-proliferative diabetic retinopathy. Br J Ophthalmol, 2012. 96(5): p. 699–703. [DOI] [PubMed] [Google Scholar]
- 57.Samuels IS, et al. Exclusion of aldose reductase as a mediator of ERG deficits in a mouse model of diabetic eye disease. Vis Neurosci, 2012. 29(6): p. 267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Di Leo MA, et al. Presence and further development of retinal dysfunction after 3-year follow up in IDDM patients without angiographically documented vasculopathy. Diabetologia, 1994. 37(9): p. 911–6. [DOI] [PubMed] [Google Scholar]
- 59.Ewing FM, et al. Seeing beyond retinopathy in diabetes: electrophysiological and psychophysical abnormalities and alterations in vision. Endocr Rev, 1998. 19(4): p. 462–76. [DOI] [PubMed] [Google Scholar]
- 60.Shirao Y and Kawasaki K, Electrical responses from diabetic retina. Prog Retin Eye Res, 1998. 17(1): p. 59–76. [DOI] [PubMed] [Google Scholar]
- 61.Bearse MA Jr., et al. A multifocal electroretinogram model predicting the development of diabetic retinopathy. Prog Retin Eye Res, 2006. 25(5): p. 425–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han Y, et al. Multifocal electroretinogram and short-wavelength automated perimetry measures in diabetic eyes with little or no retinopathy. Arch Ophthalmol, 2004. 122(12): p. 1809–15. [DOI] [PubMed] [Google Scholar]
- 63.Harrison WW, et al. Multifocal electroretinograms predict onset of diabetic retinopathy in adult patients with diabetes. Invest Ophthalmol Vis Sci, 2011. 52(2): p. 772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bearse MA Jr., et al. Local multifocal oscillatory potential abnormalities in diabetes and early diabetic retinopathy. Invest Ophthalmol Vis Sci, 2004. 45(9): p. 3259–65. [DOI] [PubMed] [Google Scholar]
- 65.Ziccardi L, et al. Early and localized retinal dysfunction in patients with type 1 diabetes mellitus studied by multifocal electroretinogram. Acta Diabetol, 2018. 55(11): p. 1191–1200. [DOI] [PubMed] [Google Scholar]
- 66.Adhikari P, et al. Multifocal electroretinogram responses in Nepalese diabetic patients without retinopathy. Doc Ophthalmol, 2014. 129(1): p. 39–46. [DOI] [PubMed] [Google Scholar]
- 67.Li Q, et al. Early retinal damage in experimental diabetes: electroretinographical and morphological observations. Exp Eye Res, 2002. 74(5): p. 615–25. [DOI] [PubMed] [Google Scholar]
- 68.Yuan D, et al. Edaravone protect against retinal damage in streptozotocin-induced diabetic mice. PLoS One, 2014. 9(6): p. e99219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hombrebueno JR, et al. Loss of synaptic connectivity, particularly in second order neurons is a key feature of diabetic retinal neuropathy in the Ins2Akita mouse. PLoS One, 2014. 9(5): p. e97970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li CP, et al. Long Noncoding RNA-Sox2OT Knockdown Alleviates Diabetes Mellitus-Induced Retinal Ganglion Cell (RGC) injury. Cell Mol Neurobiol, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McDowell RE, et al. Muller glial dysfunction during diabetic retinopathy in rats is reduced by the acrolein-scavenging drug, 2-hydrazino-4,6-dimethylpyrimidine. Diabetologia, 2018. 61(12): p. 2654–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barber AJ, et al. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest, 1998. 102(4): p. 783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Abu El-Asrar AM, et al. Expression of antiapoptotic and proapoptotic molecules in diabetic retinas. Eye (Lond), 2007. 21(2): p. 238–45. [DOI] [PubMed] [Google Scholar]
- 74.Oshitari T, et al. Mitochondria- and caspase-dependent cell death pathway involved in neuronal degeneration in diabetic retinopathy. Br J Ophthalmol, 2008. 92(4): p. 552–6. [DOI] [PubMed] [Google Scholar]
- 75.Martin PM, et al. Death of retinal neurons in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci, 2004. 45(9): p. 3330–6. [DOI] [PubMed] [Google Scholar]
- 76.Barber AJ, et al. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci, 2005. 46(6): p. 2210–8. [DOI] [PubMed] [Google Scholar]
- 77.Kern TS and Barber AJ, Retinal ganglion cells in diabetes. J Physiol, 2008. 586(18): p. 4401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park SH, et al. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia, 2003. 46(9): p. 1260–8. [DOI] [PubMed] [Google Scholar]
- 79.Gastinger MJ, et al. Dendrite remodeling and other abnormalities in the retinal ganglion cells of Ins2 Akita diabetic mice. Invest Ophthalmol Vis Sci, 2008. 49(6): p. 2635–42. [DOI] [PubMed] [Google Scholar]
- 80.Qin Y, Xu G, and Wang W, Dendritic abnormalities in retinal ganglion cells of three-month diabetic rats. Curr Eye Res, 2006. 31(11): p. 967–74. [DOI] [PubMed] [Google Scholar]
- 81.Aizu Y, et al. Degeneration of retinal neuronal processes and pigment epithelium in the early stage of the streptozotocin-diabetic rats. Neuropathology, 2002. 22(3): p. 161–70. [DOI] [PubMed] [Google Scholar]
- 82.Ahsan H, Diabetic retinopathy--biomolecules and multiple pathophysiology. Diabetes Metab Syndr, 2015. 9(1): p. 51–4. [DOI] [PubMed] [Google Scholar]
- 83.Scott TM, et al. Vascular and neural changes in the rat optic nerve following induction of diabetes with streptozotocin. J Anat, 1986. 144: p. 145–52. [PMC free article] [PubMed] [Google Scholar]
- 84.van Dijk HW, et al. Association of visual function and ganglion cell layer thickness in patients with diabetes mellitus type 1 and no or minimal diabetic retinopathy. Vision Res, 2011. 51(2): p. 224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Araszkiewicz A, et al. Neurodegeneration of the retina in type 1 diabetic patients. Pol Arch Med Wewn, 2012. 122(10): p. 464–70. [PubMed] [Google Scholar]
- 86.Lieth E, et al. Retinal neurodegeneration: early pathology in diabetes. Clin Exp Ophthalmol, 2000. 28(1): p. 3–8. [DOI] [PubMed] [Google Scholar]
- 87.Abu-El-Asrar AM, et al. Expression of apoptosis markers in the retinas of human subjects with diabetes. Invest Ophthalmol Vis Sci, 2004. 45(8): p. 2760–6. [DOI] [PubMed] [Google Scholar]
- 88.Ruiz-Ocana P, et al. Decreased Retinal Thickness in Type 1 Diabetic Children with Signs of Nonproliferative Diabetic Retinopathy. Int J Endocrinol, 2018. 2018: p. 1078531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chhablani J, et al. Neurodegeneration in Type 2 Diabetes: Evidence From Spectral-Domain Optical Coherence Tomography. Invest Ophthalmol Vis Sci, 2015. 56(11): p. 6333–8. [DOI] [PubMed] [Google Scholar]
- 90.Jonsson KB, Frydkjaer-Olsen U, and Grauslund J, Vascular Changes and Neurodegeneration in the Early Stages of Diabetic Retinopathy: Which Comes First? Ophthalmic Res, 2016. 56(1): p. 1–9. [DOI] [PubMed] [Google Scholar]
- 91.Barcelona PF, et al. p75NTR and Its Ligand ProNGF Activate Paracrine Mechanisms Etiological to the Vascular, Inflammatory, and Neurodegenerative Pathologies of Diabetic Retinopathy. J Neurosci, 2016. 36(34): p. 8826–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chakravarthy H and Devanathan V, Molecular Mechanisms Mediating Diabetic Retinal Neurodegeneration: Potential Research Avenues and Therapeutic Targets. J Mol Neurosci, 2018. 66(3): p. 445–461. [DOI] [PubMed] [Google Scholar]
- 93.Iwona BS, Growth Factors in the Pathogenesis of Retinal Neurodegeneration in Diabetes Mellitus. Curr Neuropharmacol, 2016. 14(8): p. 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Simo R, Hernandez C, and R. European Consortium for the Early Treatment of Diabetic, Neurodegeneration is an early event in diabetic retinopathy: therapeutic implications. Br J Ophthalmol, 2012. 96(10): p. 1285–90. [DOI] [PubMed] [Google Scholar]
- 95.Villarroel M, et al. Neurodegeneration: An early event of diabetic retinopathy. World J Diabetes, 2010. 1(2): p. 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hernandez C and Simo R, Neuroprotection in diabetic retinopathy. Curr Diab Rep, 2012. 12(4): p. 329–37. [DOI] [PubMed] [Google Scholar]
- 97.Ola MS and Alhomida AS, Neurodegeneration in diabetic retina and its potential drug targets. Curr Neuropharmacol, 2014. 12(4): p. 380–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lieth E, et al. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes, 1998. 47(5): p. 815–20. [DOI] [PubMed] [Google Scholar]
- 99.Kowluru RA, et al. Retinal glutamate in diabetes and effect of antioxidants. Neurochem Int, 2001. 38(5): p. 385–90. [DOI] [PubMed] [Google Scholar]
- 100.Ambati J, et al. Elevated gamma-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol, 1997. 115(9): p. 1161–6. [DOI] [PubMed] [Google Scholar]
- 101.Pulido JE, et al. A role for excitatory amino acids in diabetic eye disease. Exp Diabetes Res, 2007. 2007: p. 36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li Q and Puro DG, Diabetes-induced dysfunction of the glutamate transporter in retinal Muller cells. Invest Ophthalmol Vis Sci, 2002. 43(9): p. 3109–16. [PubMed] [Google Scholar]
- 103.Kowluru RA, Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxid Redox Signal, 2005. 7(11-12): p. 1581–87. [DOI] [PubMed] [Google Scholar]
- 104.Kowluru RA and Mishra M, Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim Biophys Acta, 2015. 1852(11): p. 2474–83. [DOI] [PubMed] [Google Scholar]
- 105.Kowluru RA and Chan PS, Oxidative stress and diabetic retinopathy. Exp Diabetes Res, 2007. 2007: p. 43603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zheng L and Kern TS, Role of nitric oxide, superoxide, peroxynitrite and PARP in diabetic retinopathy. Front Biosci (Landmark Ed), 2009. 14: p. 3974–87. [DOI] [PubMed] [Google Scholar]
- 107.Osborne NN, et al. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res, 2004. 23(1): p. 91–147. [DOI] [PubMed] [Google Scholar]
- 108.Kowluru R, Kern TS, and Engerman RL, Abnormalities of retinal metabolism in diabetes or galactosemia. II. Comparison of gamma-glutamyl transpeptidase in retina and cerebral cortex, and effects of antioxidant therapy. Curr Eye Res, 1994. 13(12): p. 891–6. [DOI] [PubMed] [Google Scholar]
- 109.Girotti AW and Kriska T, Role of lipid hydroperoxides in photo-oxidative stress signaling. Antioxid Redox Signal, 2004. 6(2): p. 301–10. [DOI] [PubMed] [Google Scholar]
- 110.Appukuttan B, et al. Effect of NADPH oxidase 1 and 4 blockade in activated human retinal endothelial cells. Clin Exp Ophthalmol, 2018. 46(6): p. 652–660. [DOI] [PubMed] [Google Scholar]
- 111.Sedeek M, et al. Oxidative stress, Nox isoforms and complications of diabetes--potential targets for novel therapies. J Cardiovasc Transl Res, 2012. 5(4): p. 509–18. [DOI] [PubMed] [Google Scholar]
- 112.Al-Shabrawey M, et al. Role of NADPH oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci, 2008. 49(7): p. 3239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiao W, et al. Activation of the NotchNox4reactive oxygen species signaling pathway induces cell death in high glucosetreated human retinal endothelial cells. Mol Med Rep, 2019. 19(1): p. 667–677. [DOI] [PubMed] [Google Scholar]
- 114.Rojas M, et al. NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence. Antioxidants (Basel), 2017. 6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meng W, et al. A genome-wide association study suggests new evidence for an association of the NADPH Oxidase 4 (NOX4) gene with severe diabetic retinopathy in type 2 diabetes. Acta Ophthalmol, 2018. 96(7): p. e811–e819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Deliyanti D and Wilkinson-Berka JL, Inhibition of NOX1/4 with GKT137831: a potential novel treatment to attenuate neuroglial cell inflammation in the retina. J Neuroinflammation, 2015. 12: p. 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ibrahim AS, et al. A lipidomic screen of hyperglycemia-treated HRECs links 12/15-Lipoxygenase to microvascular dysfunction during diabetic retinopathy via NADPH oxidase. J Lipid Res, 2015. 56(3): p. 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moghe A, et al. Molecular mechanisms of acrolein toxicity: relevance to human disease. Toxicol Sci, 2015. 143(2): p. 242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tully M and Shi R, New insights in the pathogenesis of multiple sclerosis--role of acrolein in neuronal and myelin damage. Int J Mol Sci, 2013. 14(10): p. 20037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kanwar M, et al. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci, 2007. 48(8): p. 3805–11. [DOI] [PubMed] [Google Scholar]
- 121.Madsen-Bouterse SA, et al. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal, 2010. 13(6): p. 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Barot M, Gokulgandhi MR, and Mitra AK, Mitochondrial dysfunction in retinal diseases. Curr Eye Res, 2011. 36(12): p. 1069–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Masser DR, et al. Functional changes in the neural retina occur in the absence of mitochondrial dysfunction in a rodent model of diabetic retinopathy. J Neurochem, 2017. 143(5): p. 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Silva KC, et al. Diabetic retinal neurodegeneration is associated with mitochondrial oxidative stress and is improved by an angiotensin receptor blocker in a model combining hypertension and diabetes. Diabetes, 2009. 58(6): p. 1382–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pearsall EA, et al. Neuroprotective effects of PPARalpha in retinopathy of type 1 diabetes. PLoS One, 2019. 14(2): p. e0208399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gaspar JM, et al. Tauroursodeoxycholic acid protects retinal neural cells from cell death induced by prolonged exposure to elevated glucose. Neuroscience, 2013. 253: p. 380–8. [DOI] [PubMed] [Google Scholar]
- 127.Chen K, et al. Taurine protects transformed rat retinal ganglion cells from hypoxia-induced apoptosis by preventing mitochondrial dysfunction. Brain Res, 2009. 1279: p. 131–8. [DOI] [PubMed] [Google Scholar]
- 128.Li X, Zhang M, and Zhou H, The morphological features and mitochondrial oxidative stress mechanism of the retinal neurons apoptosis in early diabetic rats. J Diabetes Res, 2014. 2014: p. 678123. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 129.Balog J, Mehta SL, and Vemuganti R, Mitochondrial fission and fusion in secondary brain damage after CNS insults. J Cereb Blood Flow Metab, 2016. 36(12): p. 2022–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kowluru RA, Kern TS, and Engerman RL, Abnormalities of retinal metabolism in diabetes or experimental galactosemia. IV. Antioxidant defense system. Free Radic Biol Med, 1997. 22(4): p. 587–92. [DOI] [PubMed] [Google Scholar]
- 131.Obrosova IG, et al. Early diabetes-induced biochemical changes in the retina: comparison of rat and mouse models. Diabetologia, 2006. 49(10): p. 2525–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Siemianowicz K, et al. Blood antioxidant parameters in patients with diabetic retinopathy. Int J Mol Med, 2004. 14(3): p. 433–7. [PubMed] [Google Scholar]
- 133.Li W, et al. Altered mRNA levels of antioxidant enzymes in pre-apoptotic pericytes from human diabetic retinas. Cell Mol Biol (Noisy-le-grand), 1999. 45(1): p. 59–66. [PubMed] [Google Scholar]
- 134.Kumar B, et al. Hesperetin rescues retinal oxidative stress, neuroinflammation and apoptosis in diabetic rats. Microvasc Res, 2013. 87: p. 65–74. [DOI] [PubMed] [Google Scholar]
- 135.Dehdashtian E, et al. Diabetic retinopathy pathogenesis and the ameliorating effects of melatonin; involvement of autophagy, inflammation and oxidative stress. Life Sci, 2018. 193: p. 20–33. [DOI] [PubMed] [Google Scholar]
- 136.Kumar B, et al. Retinal neuroprotective effects of quercetin in streptozotocin-induced diabetic rats. Exp Eye Res, 2014. 125: p. 193–202. [DOI] [PubMed] [Google Scholar]
- 137.Shen X, et al. Effect of pigment epithelium derived factor on the expression of glutamine synthetase in early phase of experimental diabetic retinopathy. Ocul Immunol Inflamm, 2011. 19(4): p. 246–54. [DOI] [PubMed] [Google Scholar]
- 138.Takita H, et al. Retinal neuroprotection against ischemic injury mediated by intraocular gene transfer of pigment epithelium-derived factor. Invest Ophthalmol Vis Sci, 2003. 44(10): p. 4497–504. [DOI] [PubMed] [Google Scholar]
- 139.Imai D, et al. Intraocular gene transfer of pigment epithelium-derived factor rescues photoreceptors from light-induced cell death. J Cell Physiol, 2005. 202(2): p. 570–8. [DOI] [PubMed] [Google Scholar]
- 140.Bilak MM, et al. Pigment epithelium-derived factor (PEDF) protects motor neurons from chronic glutamate-mediated neurodegeneration. J Neuropathol Exp Neurol, 1999. 58(7): p. 719–28. [DOI] [PubMed] [Google Scholar]
- 141.Brazionis L, et al. Homocysteine and diabetic retinopathy. Diabetes Care, 2008. 31(1): p. 50–6. [DOI] [PubMed] [Google Scholar]
- 142.Lim CP, et al. Plasma, aqueous and vitreous homocysteine levels in proliferative diabetic retinopathy. Br J Ophthalmol, 2012. 96(5): p. 704–7. [DOI] [PubMed] [Google Scholar]
- 143.Ganapathy PS, et al. The role of N-methyl-D-aspartate receptor activation in homocysteine-induced death of retinal ganglion cells. Invest Ophthalmol Vis Sci, 2011. 52(8): p. 5515–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Moore P, et al. Apoptotic cell death in the mouse retinal ganglion cell layer is induced in vivo by the excitatory amino acid homocysteine. Exp Eye Res, 2001. 73(1): p. 45–57. [DOI] [PubMed] [Google Scholar]
- 145.Srivastav K, et al. Increased serum level of homocysteine correlates with retinal nerve fiber layer thinning in diabetic retinopathy. Mol Vis, 2016. 22: p. 1352–1360. [PMC free article] [PubMed] [Google Scholar]
- 146.Coral K, et al. Homocysteine levels in the vitreous of proliferative diabetic retinopathy and rhegmatogenous retinal detachment: its modulating role on lysyl oxidase. Invest Ophthalmol Vis Sci, 2009. 50(8): p. 3607–12. [DOI] [PubMed] [Google Scholar]
- 147.Chang HH, et al. Intravitreal homocysteine-thiolactone injection leads to the degeneration of multiple retinal cells, including photoreceptors. Mol Vis, 2011. 17: p. 1946–56. [PMC free article] [PubMed] [Google Scholar]
- 148.Patel C, et al. Arginase as a mediator of diabetic retinopathy. Front Immunol, 2013. 4: p. 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Seiler N, Polyamine oxidase, properties and functions. Prog Brain Res, 1995. 106: p. 333–44. [DOI] [PubMed] [Google Scholar]
- 150.Cervelli M, et al. Spermine oxidase: ten years after. Amino Acids, 2012. 42(2-3): p. 441–50. [DOI] [PubMed] [Google Scholar]
- 151.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 2012. 74(4): p. 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Volgyi B, et al. Gap junctions are essential for generating the correlated spike activity of neighboring retinal ganglion cells. PLoS One, 2013. 8(7): p. e69426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Runkle EA, et al. Occludin localizes to centrosomes and modifies mitotic entry. J Biol Chem, 2011. 286(35): p. 30847–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Qian H and Ripps H, Neurovascular interaction and the pathophysiology of diabetic retinopathy. Exp Diabetes Res, 2011. 2011: p. 693426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Krady JK, et al. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes, 2005. 54(5): p. 1559–65. [DOI] [PubMed] [Google Scholar]
- 156.Leasher JL, et al. Global Estimates on the Number of People Blind or Visually Impaired by Diabetic Retinopathy: A Meta-analysis From 1990 to 2010. Diabetes Care, 2016. 39(9): p. 1643–9. [DOI] [PubMed] [Google Scholar]
- 157.Paik MJ, et al. Polyamine patterns in the cerebrospinal fluid of patients with Parkinson’s disease and multiple system atrophy. Clin Chim Acta, 2010. 411(19-20): p. 1532–5. [DOI] [PubMed] [Google Scholar]
- 158.Wang W and Lo ACY, Diabetic Retinopathy: Pathophysiology and Treatments. Int J Mol Sci, 2018. 19(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Takano K, et al. Neuronal and glial responses to polyamines in the ischemic brain. Curr Neurovasc Res, 2005. 2(3): p. 213–23. [DOI] [PubMed] [Google Scholar]
- 160.Ivanova S, et al. Neuroprotection in cerebral ischemia by neutralization of 3-aminopropanal. Proc Natl Acad Sci U S A, 2002. 99(8): p. 5579–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cervelli M, et al. Mouse spermine oxidase gene splice variants. Nuclear subcellular localization of a novel active isoform. Eur J Biochem, 2004. 271(4): p. 760–70. [DOI] [PubMed] [Google Scholar]
- 162.Murray-Stewart T, et al. Nuclear localization of human spermine oxidase isoforms - possible implications in drug response and disease etiology. FEBS J, 2008. 275(11): p. 2795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cervelli M, et al. A New Transgenic Mouse Model for Studying the Neurotoxicity of Spermine Oxidase Dosage in the Response to Excitotoxic Injury. PLoS One, 2013. 8(6): p. e64810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Capone C, et al. A role for spermine oxidase as a mediator of reactive oxygen species production in HIV-Tat-induced neuronal toxicity. Free Radic Biol Med, 2013. 63: p. 99–107. [DOI] [PubMed] [Google Scholar]
- 165.Amendola R, et al. Direct oxidative DNA damage, apoptosis and radio sensitivity by spermine oxidase activities in mouse neuroblastoma cells. Biochim Biophys Acta, 2005. 1755(1): p. 15–24. [DOI] [PubMed] [Google Scholar]
- 166.Lucas DR and Newhouse JP, The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol, 1957. 58(2): p. 193–201. [DOI] [PubMed] [Google Scholar]
- 167.Furuhata A, et al. Thiolation of protein-bound carcinogenic aldehyde. An electrophilic acrolein-lysine adduct that covalently binds to thiols. J Biol Chem, 2002. 277(31): p. 27919–26. [DOI] [PubMed] [Google Scholar]
- 168.Wang Y, et al. Induction of human spermine oxidase SMO(PAOh1) is regulated at the levels of new mRNA synthesis, mRNA stabilization and newly synthesized protein. Biochem J, 2005. 386(Pt 3): p. 543–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Vujcic S, et al. Identification and characterization of a novel flavin-containing spermine oxidase of mammalian cell origin. Biochem J, 2002. 367(Pt 3): p. 665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cervelli M, et al. Heterologous expression and characterization of mouse spermine oxidase. J Biol Chem, 2003. 278(7): p. 5271–6. [DOI] [PubMed] [Google Scholar]
- 171.Babbar N and Casero RA Jr., Tumor necrosis factor-alpha increases reactive oxygen species by inducing spermine oxidase in human lung epithelial cells: a potential mechanism for inflammation-induced carcinogenesis. Cancer Res, 2006. 66(23): p. 11125–30. [DOI] [PubMed] [Google Scholar]
- 172.Grigsby J, et al. A possible role of acrolein in diabetic retinopathy: involvement of a VEGF/TGFbeta signaling pathway of the retinal pigment epithelium in hyperglycemia. Curr Eye Res, 2012. 37(11): p. 1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vidro-Kotchan E, et al. NBHA reduces acrolein-induced changes in ARPE-19 cells: possible involvement of TGFbeta. Curr Eye Res, 2011. 36(4): p. 370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Noro T, et al. Spermidine promotes retinal ganglion cell survival and optic nerve regeneration in adult mice following optic nerve injury. Cell Death Dis, 2015. 6: p. e1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Murata M, et al. Soluble Vascular Adhesion Protein-1 Mediates Spermine Oxidation as Semicarbazide-Sensitive Amine Oxidase: Possible Role in Proliferative Diabetic Retinopathy. Curr Eye Res, 2017. 42(12): p. 1674–1683. [DOI] [PubMed] [Google Scholar]
- 176.Dong Y, et al. Localization of Acrolein-Lysine Adduct in Fibrovascular Tissues of Proliferative Diabetic Retinopathy. Curr Eye Res, 2016: p. 1–7. [DOI] [PubMed] [Google Scholar]
- 177.Yong PH, et al. Evidence supporting a role for N-(3-formyl-3,4-dehydropiperidino)lysine accumulation in Muller glia dysfunction and death in diabetic retinopathy. Mol Vis, 2010. 16: p. 2524–38. [PMC free article] [PubMed] [Google Scholar]
- 178.Noro T, et al. Spermidine Ameliorates Neurodegeneration in a Mouse Model of Normal Tension Glaucoma. Invest Ophthalmol Vis Sci, 2015. 56(8): p. 5012–9. [DOI] [PubMed] [Google Scholar]
- 179.Ohashi K, et al. Spermidine Oxidation-Mediated Degeneration of Retinal Pigment Epithelium in Rats. Oxid Med Cell Longev, 2017. 2017: p. 4128061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guo X, et al. Spermidine alleviates severity of murine experimental autoimmune encephalomyelitis. Invest Ophthalmol Vis Sci, 2011. 52(5): p. 2696–703. [DOI] [PubMed] [Google Scholar]
- 181.Seghieri G, et al. Serum spermidine oxidase activity in patients with insulin-dependent diabetes mellitus and microvascular complications. Acta Diabetol Lat, 1990. 27(4): p. 303–8. [DOI] [PubMed] [Google Scholar]
- 182.Nicoletti R, et al. Vitreous polyamines spermidine, putrescine, and spermine in human proliferative disorders of the retina. Br J Ophthalmol, 2003. 87(8): p. 1038–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.McDowell RE, et al. Diabetes Impairs the Aldehyde Detoxifying Capacity of the Retina. Invest Ophthalmol Vis Sci, 2016. 57(11): p. 4762–71. [DOI] [PubMed] [Google Scholar]
- 184.Zhang X, et al. Evaluation of N (epsilon)-(3-formyl-3,4-dehydropiperidino)lysine as a novel biomarker for the severity of diabetic retinopathy. Diabetologia, 2008. 51(9): p. 1723–30. [DOI] [PubMed] [Google Scholar]
- 185.Qin WS, Deng YH, and Cui FC, Sulforaphane protects against acrolein-induced oxidative stress and inflammatory responses: modulation of Nrf-2 and COX-2 expression. Arch Med Sci, 2016. 12(4): p. 871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zarkovic N, et al. Pathophysiological relevance of aldehydic protein modifications. J Proteomics, 2013. 92: p. 239–47. [DOI] [PubMed] [Google Scholar]
- 187.Galligan JJ, et al. Protein carbonylation in a murine model for early alcoholic liver disease. Chem Res Toxicol, 2012. 25(5): p. 1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gehl Z, et al. Diabetes-induced oxidative stress in the vitreous humor. Redox Biol, 2016. 9: p. 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Loukovaara S, et al. Elevated protein carbonyl and HIF-1alpha levels in eyes with proliferative diabetic retinopathy. Acta Ophthalmol, 2014. 92(4): p. 323–7. [DOI] [PubMed] [Google Scholar]
- 190.Takabe W, et al. Oxidative stress promotes the development of transformation: involvement of a potent mutagenic lipid peroxidation product, acrolein. Carcinogenesis, 2001. 22(6): p. 935–41. [DOI] [PubMed] [Google Scholar]
- 191.Uemura T, et al. Decrease in acrolein toxicity based on the decline of polyamine oxidases. Int J Biochem Cell Biol, 2016. 79: p. 151–157. [DOI] [PubMed] [Google Scholar]
- 192.Picklo MJ and Montine TJ, Acrolein inhibits respiration in isolated brain mitochondria. Biochim Biophys Acta, 2001. 1535(2): p. 145–52. [DOI] [PubMed] [Google Scholar]
- 193.Luo J and Shi R, Acrolein induces oxidative stress in brain mitochondria. Neurochem Int, 2005. 46(3): p. 243–52. [DOI] [PubMed] [Google Scholar]
- 194.Jia L, et al. Acrolein, a toxicant in cigarette smoke, causes oxidative damage and mitochondrial dysfunction in RPE cells: protection by (R)-alpha-lipoic acid. Invest Ophthalmol Vis Sci, 2007. 48(1): p. 339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liu Z, et al. Hydroxytyrosol protects retinal pigment epithelial cells from acrolein-induced oxidative stress and mitochondrial dysfunction. J Neurochem, 2007. 103(6): p. 2690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dasgupta A, Zheng J, and Bizzozero OA, Protein carbonylation and aggregation precede neuronal apoptosis induced by partial glutathione depletion. ASN Neuro, 2012. 4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Dogan A, et al. Effects of MDL 72527, a specific inhibitor of polyamine oxidase, on brain edema, ischemic injury volume, and tissue polyamine levels in rats after temporary middle cerebral artery occlusion. J Neurochem, 1999. 72(2): p. 765–70. [DOI] [PubMed] [Google Scholar]
- 198.Hernandez C and Simo R, Somatostatin replacement: a new strategy for treating diabetic retinopathy. Curr Med Chem, 2013. 20(26): p. 3251–7. [DOI] [PubMed] [Google Scholar]
- 199.Dai H, et al. The polyamine oxidase inhibitor MDL-72,527 selectively induces apoptosis of transformed hematopoietic cells through lysosomotropic effects. Cancer Res, 1999. 59(19): p. 4944–54. [PubMed] [Google Scholar]
- 200.Duranton B, et al. Cytotoxic effects of the polyamine oxidase inactivator MDL 72527 to two human colon carcinoma cell lines SW480 and SW620. Cell Biol Toxicol, 2002. 18(6): p. 381–96. [DOI] [PubMed] [Google Scholar]
- 201.Agostinelli E, et al. Toxicity of enzymatic oxidation products of spermine to human melanoma cells (M14): sensitization by heat and MDL 72527. Biochim Biophys Acta, 2006. 1763(10): p. 1040–50. [DOI] [PubMed] [Google Scholar]
- 202.Agostinelli E, et al. Cytotoxicity of spermine oxidation products to multidrug resistant melanoma M14 ADR2 cells: sensitization by the MDL 72527 lysosomotropic compound. Int J Oncol, 2009. 35(3): p. 485–98. [DOI] [PubMed] [Google Scholar]
- 203.Seiler N, Duranton B, and Raul F, The polyamine oxidase inactivator MDL 72527. Prog Drug Res, 2002. 59: p. 1–40. [DOI] [PubMed] [Google Scholar]
- 204.Cervelli M, et al. Spermine oxidase (SMO) activity in breast tumor tissues and biochemical analysis of the anticancer spermine analogues BENSpm and CPENSpm. BMC Cancer, 2010. 10: p. 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Rubsam A, Parikh S, and Fort PE, Role of Inflammation in Diabetic Retinopathy. Int J Mol Sci, 2018. 19(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lois N, et al. Endothelial progenitor cells in diabetic retinopathy. Front Endocrinol (Lausanne), 2014. 5: p. 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Stitt AW, et al. The progress in understanding and treatment of diabetic retinopathy. Prog Retin Eye Res, 2016. 51: p. 156–86. [DOI] [PubMed] [Google Scholar]
- 208.Kowluru RA, et al. TIAM1-RAC1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia, 2014. 57(5): p. 1047–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Early Treatment Diabetic Retinopathy Study research group. Arch Ophthalmol, 1985. 103(12): p. 1796–806. [PubMed] [Google Scholar]
- 210.Ola MS, et al. Reduced levels of brain derived neurotrophic factor (BDNF) in the serum of diabetic retinopathy patients and in the retina of diabetic rats. Cell Mol Neurobiol, 2013. 33(3): p. 359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kaur C, Foulds WS, and Ling EA, Hypoxia-ischemia and retinal ganglion cell damage. Clin Ophthalmol, 2008. 2(4): p. 879–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Xiao C, et al. Physiological effects of superoxide dismutase on altered visual function of retinal ganglion cells in db/db mice. PLoS One, 2012. 7(1): p. e30343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fukumoto M, et al. Vulnerability of the retinal microvasculature to oxidative stress: ion channel-dependent mechanisms. Am J Physiol Cell Physiol, 2012. 302(9): p. C1413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wilkinson-Berka JL, Angiotensin and diabetic retinopathy. Int J Biochem Cell Biol, 2006. 38(5-6): p. 752–65. [DOI] [PubMed] [Google Scholar]
- 215.Frey RS, Ushio-Fukai M, and Malik AB, NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal, 2009. 11(4): p. 791–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Brownlee M, The pathobiology of diabetic complications: a unifying mechanism. Diabetes, 2005. 54(6): p. 1615–25. [DOI] [PubMed] [Google Scholar]
- 217.Narayanan SP, et al. Arginase in retinopathy. Prog Retin Eye Res, 2013. 36: p. 260–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Adamis AP, et al. Changes in retinal neovascularization after pegaptanib (Macugen) therapy in diabetic individuals. Ophthalmology, 2006. 113(1): p. 23–8. [DOI] [PubMed] [Google Scholar]
- 219.U.S. Food and Drug Adminstration. Drugs@FDA: FDA Approved Drug Products: Ranibizumab (Lucentis) 2017. [cited 2018 2 Oct]; available at https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=BasicSearch.process]. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=BasicSearch.process.
- 220.U.S. Food and Drug Adminstration. Drugs@fda: Fda approved drug products: AFLIBERCEPT (Eylea). [cited 2018; available at https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=BasicSearch.process].
- 221.Arevalo JF, et al. Intravitreal bevacizumab (Avastin) for proliferative diabetic retinopathy: 6-months follow-up. Eye (Lond), 2009. 23(1): p. 117–23. [DOI] [PubMed] [Google Scholar]
- 222.Cho WB, et al. Panretinal photocoagulation combined with intravitreal bevacizumab in high-risk proliferative diabetic retinopathy. Retina, 2009. 29(4): p. 516–22. [DOI] [PubMed] [Google Scholar]
- 223.Wells JA, et al. Aflibercept, Bevacizumab, or Ranibizumab for Diabetic Macular Edema: Two-Year Results from a Comparative Effectiveness Randomized Clinical Trial. Ophthalmology, 2016. 123(6): p. 1351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rajendram R, et al. A 2-year prospective randomized controlled trial of intravitreal bevacizumab or laser therapy (BOLT) in the management of diabetic macular edema: 24-month data: report 3. Arch Ophthalmol, 2012. 130(8): p. 972–9. [DOI] [PubMed] [Google Scholar]
- 225.Nishijima K, et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol, 2007. 171(1): p. 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Price SA, et al. Gene transfer of an engineered transcription factor promoting expression of VEGF-A protects against experimental diabetic neuropathy. Diabetes, 2006. 55(6): p. 1847–54. [DOI] [PubMed] [Google Scholar]
- 227.Shin HJ, et al. Intravitreal Anti-Vascular Endothelial Growth Factor Therapy and Retinal Nerve Fiber Layer Loss in Eyes With Age-Related Macular Degeneration: A Meta-Analysis. Invest Ophthalmol Vis Sci, 2016. 57(4): p. 1798–806. [DOI] [PubMed] [Google Scholar]
- 228.Beck M, et al. Retinal Ganglion Cell Layer Change in Patients Treated With Anti-Vascular Endothelial Growth Factor for Neovascular Age-related Macular Degeneration. Am J Ophthalmol, 2016. 167: p. 10–7. [DOI] [PubMed] [Google Scholar]
- 229.Hombrebueno JR, et al. Sustained intraocular VEGF neutralization results in retinal neurodegeneration in the Ins2(Akita) diabetic mouse. Sci Rep, 2015. 5: p. 18316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Omay E, et al. The early effects of intravitreal anti vascular endothelial growth factor agents on intraocular pressure and central corneal thickness. Int Ophthalmol, 2016. 36(5): p. 665–70. [DOI] [PubMed] [Google Scholar]
- 231.Frenkel RE, et al. Intraocular pressure effects of pegaptanib (Macugen) injections in patients with and without glaucoma. Am J Ophthalmol, 2007. 143(6): p. 1034–5. [DOI] [PubMed] [Google Scholar]
- 232.Jalil A, Fenerty C, and Charles S, Intravitreal bevacizumab (Avastin) causing acute glaucoma: an unreported complication. Eye (Lond), 2007. 21(12): p. 1541. [DOI] [PubMed] [Google Scholar]
- 233.El Chehab H, et al. Intraocular Pressure Spikes after Aflibercept Intravitreal Injections. Ophthalmologica, 2016. 236(1): p. 43–7. [DOI] [PubMed] [Google Scholar]
- 234.Arevalo JF, et al. Tractional retinal detachment following intravitreal bevacizumab (Avastin) in patients with severe proliferative diabetic retinopathy. Br J Ophthalmol, 2008. 92(2): p. 213–6. [DOI] [PubMed] [Google Scholar]
- 235.Arevalo JF, et al. Retinal detachment after bevacizumab. Ophthalmology, 2011. 118(11): p. 2304.e3–7. [DOI] [PubMed] [Google Scholar]
- 236.Kuiper EJ, et al. The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS One, 2008. 3(7): p. e2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Jiao C, et al. APOPTOSIS AND ANGIOFIBROSIS IN DIABETIC TRACTIONAL MEMBRANES AFTER VASCULAR ENDOTHELIAL GROWTH FACTOR INHIBITION: Results of a Prospective Trial. Report No. 2. Retina, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Aggio FB, et al. Acute endophthalmitis following intravitreal bevacizumab (Avastin) injection. Eye (Lond), 2007. 21(3): p. 408–9. [DOI] [PubMed] [Google Scholar]
- 239.Elman MJ, et al. Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology, 2010. 117(6): p. 1064–1077.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wu L, et al. Twelve-month safety of intravitreal injections of bevacizumab (Avastin): results of the Pan-American Collaborative Retina Study Group (PACORES). Graefes Arch Clin Exp Ophthalmol, 2008. 246(1): p. 81–7. [DOI] [PubMed] [Google Scholar]
- 241.Kumar B, et al. Current trends in the pharmacotherapy of diabetic retinopathy. J Postgrad Med, 2012. 58(2): p. 132–9. [DOI] [PubMed] [Google Scholar]
- 242.Ingber DE, Madri JA, and Folkman J, A possible mechanism for inhibition of angiogenesis by angiostatic steroids: induction of capillary basement membrane dissolution. Endocrinology, 1986. 119(4): p. 1768–75. [DOI] [PubMed] [Google Scholar]
- 243.Nauck M, et al. Corticosteroids inhibit the expression of the vascular endothelial growth factor gene in human vascular smooth muscle cells. Eur J Pharmacol, 1998. 341(2-3): p. 309–15. [DOI] [PubMed] [Google Scholar]
- 244.Googe J, et al. Randomized trial evaluating short-term effects of intravitreal ranibizumab or triamcinolone acetonide on macular edema after focal/grid laser for diabetic macular edema in eyes also receiving panretinal photocoagulation. Retina, 2011. 31(6): p. 1009–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Bressler SB, et al. Five-Year Outcomes of Ranibizumab With Prompt or Deferred Laser Versus Laser or Triamcinolone Plus Deferred Ranibizumab for Diabetic Macular Edema. Am J Ophthalmol, 2016. 164: p. 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Boyer DS, et al. Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology, 2014. 121(10): p. 1904–14. [DOI] [PubMed] [Google Scholar]
- 247.Campochiaro PA, et al. Sustained delivery fluocinolone acetonide vitreous inserts provide benefit for at least 3 years in patients with diabetic macular edema. Ophthalmology, 2012. 119(10): p. 2125–32. [DOI] [PubMed] [Google Scholar]
- 248.Zhang X, et al. Triamcinolone acetonide inhibits p38MAPK activation and neuronal apoptosis in early diabetic retinopathy. Curr Mol Med, 2013. 13(6): p. 946–58. [DOI] [PubMed] [Google Scholar]
- 249.Shen W, et al. Effect of glucocorticoids on neuronal and vascular pathology in a transgenic model of selective Muller cell ablation. Glia, 2014. 62(7): p. 1110–24. [DOI] [PubMed] [Google Scholar]
- 250.Prager SG, et al. Analysis of retinal layer thickness in diabetic macular oedema treated with ranibizumab or triamcinolone. Acta Ophthalmol, 2018. 96(2): p. e195–e200. [DOI] [PubMed] [Google Scholar]
- 251.Williams M, Hogg RE, and Chakravarthy U, Antioxidants and diabetic retinopathy. Curr Diab Rep, 2013. 13(4): p. 481–7. [DOI] [PubMed] [Google Scholar]
- 252.Rodriguez-Carrizalez AD, et al. The effect of ubiquinone and combined antioxidant therapy on oxidative stress markers in non-proliferative diabetic retinopathy: A phase IIa, randomized, double-blind, and placebo-controlled study. Redox Rep, 2016. 21(4): p. 155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lafuente M, et al. COMBINED INTRAVITREAL RANIBIZUMAB AND ORAL SUPPLEMENTATION WITH DOCOSAHEXAENOIC ACID AND ANTIOXIDANTS FOR DIABETIC MACULAR EDEMA: Two-Year Randomized Single-Blind Controlled Trial Results. Retina, 2017. 37(7): p. 1277–1286. [DOI] [PubMed] [Google Scholar]
- 254.Cao Y, et al. Effects of L-carnitine on high glucose-induced oxidative stress in retinal ganglion cells. Pharmacology, 2014. 94(3-4): p. 123–30. [DOI] [PubMed] [Google Scholar]
- 255.Al-Dosary DI, Alhomida AS, and Ola MS, Protective Effects of Dietary Flavonoids in Diabetic Induced Retinal Neurodegeneration. Curr Drug Targets, 2017. 18(13): p. 1468–1476. [DOI] [PubMed] [Google Scholar]
- 256.Chen M, et al. Protective effect of mitochondriatargeted peptide MTP131 against oxidative stressinduced apoptosis in RGC5 cells. Mol Med Rep, 2017. 15(4): p. 2179–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kan E, et al. Effects of alpha-lipoic acid on retinal ganglion cells, retinal thicknesses, and VEGF production in an experimental model of diabetes. Int Ophthalmol, 2017. 37(6): p. 1269–1278. [DOI] [PubMed] [Google Scholar]
- 258.Santos AR, et al. Functional and Structural Findings of Neurodegeneration in Early Stages of Diabetic Retinopathy: Cross-sectional Analyses of Baseline Data of the EUROCONDOR Project. Diabetes, 2017. 66(9): p. 2503–2510. [DOI] [PubMed] [Google Scholar]
- 259.Simo R and Hernandez C, Novel approaches for treating diabetic retinopathy based on recent pathogenic evidence. Prog Retin Eye Res, 2015. 48: p. 160–80. [DOI] [PubMed] [Google Scholar]
- 260.Mohan N, et al. Imbalanced levels of angiogenic and angiostatic factors in vitreous, plasma and postmortem retinal tissue of patients with proliferative diabetic retinopathy. J Diabetes Complications, 2012. 26(5): p. 435–41. [DOI] [PubMed] [Google Scholar]
- 261.Yoshida Y, et al. Protective role of pigment epithelium-derived factor (PEDF) in early phase of experimental diabetic retinopathy. Diabetes Metab Res Rev, 2009. 25(7): p. 678–86. [DOI] [PubMed] [Google Scholar]
- 262.Liu Y, et al. Pigment epithelium-derived factor (PEDF) peptide eye drops reduce inflammation, cell death and vascular leakage in diabetic retinopathy in Ins2(Akita) mice. Mol Med, 2012. 18: p. 1387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Calado SM, Diaz-Corrales F, and Silva GA, pEPito-driven PEDF Expression Ameliorates Diabetic Retinopathy Hallmarks. Hum Gene Ther Methods, 2016. 27(2): p. 79–86. [DOI] [PubMed] [Google Scholar]
- 264.Carrasco E, et al. Lower somatostatin expression is an early event in diabetic retinopathy and is associated with retinal neurodegeneration. Diabetes Care, 2007. 30(11): p. 2902–8. [DOI] [PubMed] [Google Scholar]
- 265.Hernandez C, et al. Topical administration of somatostatin prevents retinal neurodegeneration in experimental diabetes. Diabetes, 2013. 62(7): p. 2569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Arroba AI, et al. Somatostatin protects photoreceptor cells against high glucose-induced apoptosis. Mol Vis, 2016. 22: p. 1522–1531. [PMC free article] [PubMed] [Google Scholar]
- 267.Shah SM, et al. A randomized, double-masked controlled clinical trial of Sandostatin long-acting release depot in patients with postsurgical cystoid macular edema. Retina, 2010. 30(1): p. 160–6. [DOI] [PubMed] [Google Scholar]
- 268.53rdEASD Annual Meeting of the European Association for the Study of Diabetes. Diabetologia, 2017. 60(1): p. 1–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Mohamed R and El-Remessy AB, Imbalance of the Nerve Growth Factor and Its Precursor: Implication in Diabetic Retinopathy. J Clin Exp Ophthalmol, 2015. 6(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Mantelli F, et al. NGF and VEGF effects on retinal ganglion cell fate: new evidence from an animal model of diabetes. Eur J Ophthalmol, 2014. 24(2): p. 247–53. [DOI] [PubMed] [Google Scholar]
- 271.Zhang P and Zhou Z, Combination of Bevacizumab and NGF Reduces the Risk of Diabetic Retinopathy. Cell Biochem Biophys, 2015. 73(1): p. 79–85. [DOI] [PubMed] [Google Scholar]