

Abstract
There are currently seven approved immune checkpoint inhibitors (ICIs) for the treatment of various cancers. These drugs are associated with profound, durable responses in a subset of patients with advanced cancers. Unfortunately, in addition to individuals whose tumors show resistance, there is a minority subgroup treated with ICIs who demonstrate a paradoxical acceleration in the rate of growth or their tumors—hyperprogressive disease. Hyperprogressive disease is associated with significantly worse outcomes in these patients. This phenomenon, though still a matter of dispute, has been recognized by multiple groups of investigators across the globe and in diverse types of cancers. There are not yet consensus standardized criteria for defining hyperprogressive disease, but most commonly time to treatment failure less than 2 months and an increase in pace of progression of at least twofold between pre‐immunotherapy and on‐treatment imaging has been used. In some patients, the change in rate of progression can be especially dramatic—up to 35‐ to 40‐fold. MDM2 amplification and EGFR mutations have been suggested as genomic correlates of increased risk of hyperprogression, but these correlates require validation. The underlying mechanism for hyperprogression is not known but warrants urgent investigation.
Keywords: Hyperprogressive disease, Immunotherapy, Cancer clinical trials, Immune checkpoint inhibitors
Short abstract
The phenomenon of hyperprogression, the acceleration of cancer progression after immune checkpoint inhibitor therapy, is one of particular importance to all stakeholders in immunotherapy. This commentary serves as a call for increased awareness and investigation of these agents and hyperprogression.
Immunotherapy in the form of checkpoint blockade has resulted in impressive responses for several previously refractory tumor types. Indeed, the U.S. Food and Drug Administration (FDA) has now approved seven checkpoint inhibitors: pembrolizumab, nivolumab, durvalumab, avelumab, atezolizumab, cemiplimab, and ipilimumab 1, 2, 3, 4, 5, 6, 7. Immune checkpoint inhibitors mediate responses by reactivating the immune system. Reactivation occurs because these antibodies interfere with checkpoints such as programmed death‐ligand 1 (PD‐L1) and cytotoxic T‐lymphocyte–associated antigen 4 that have been exploited by tumor cells to evade the immune response, a necessity if the cancer is going to survive 8. The FDA approvals notwithstanding, there are now multiple groups that have reported that a minority of patients (albeit encompassing diverse cancers) experience a dramatic acceleration in the rate of tumor progression after starting checkpoint blockade—a phenomenon designated hyperprogressive disease (HPD; Table 1) 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20. Unfortunately, in the patients who are deemed to have HPD, their median overall survival is estimated to be roughly 3 months 21.
Table 1.
Criteria for and predictors of HPD according to different research groups
| Author | Criteria for HPD | Predictors of HPD |
|---|---|---|
| Peer‐reviewed manuscript | ||
| Champiat et al. 9 | RECIST progression after first evaluation and at least twofold increase of the TGR between pre‐immunotherapy imaging and on‐treatment | ≥65 years of age |
| Kato et al. 10 | TTF <2 months, >50% increase in tumor burden compared with baseline pre‐immunotherapy imaging, and more than twofold increase in progression pace |
MDM2/MDM4 and EGFR alterations Poor TTF (defined as TTF <2 months) was not associated with age, tumor type, Royal Marsden or MD Anderson score, or type of checkpoint blockade DNMT3A alterations also significantly associated with poor TTF in multivariate analysis |
| Saada‐Bouzid et al. 11 |
TGKR calculated as ratio of the slope of tumor growth pre‐immunotherapy and the slope of tumor growth on‐treatment HPD was defined as a TGKR ≥ 2 |
HPD seen in 39% of patients with at least a locoregional recurrence and 9% of patients with exclusively distant metastases |
| Ferrara et al. 15 | Disease progression at the first evaluation with change in TGR exceeding 50% | More than two metastatic sites prior to immunotherapy |
| Kanjanapan et al. 16 | RECIST 1.1 [17] progression at the first on‐treatment scan and at least twofold increase in TGR between pre‐immunotherapy and on‐treatment | Female gender |
| Lo Russo et al. 18 |
TTF <2 months, increase ≥50% in the sum of target lesions major diameters, appearance of at least two new lesions in an organ already involved, spread of the disease to a new organ, ECOG performance status worse than ≥2 during the first 2 months HPD on the basis of three concomitant out of the five possible criteria |
Clustered macrophages with epithelioid morphology and colocalization of CD163, PD‐L1, and CD33 markers (defined as complete phenotype) in HPD cases |
| Kamada et al. 19 | TTF <2 months; >50% increase in tumor burden compared with pre‐immunotherapy imaging, and more than twofold increase in progression speed (same as per 10) |
PD‐1 blockade facilitated the proliferation of highly suppressive PD‐1+ effector (CD4+) T regulatory cells One of three patients with HPD had MDM2 amplification versus 0 of 18 patients without HPD |
| Kim et al. 20 | TTF <2 months or at least twofold increase of the TGR between pre‐immunotherapy and on‐treatment (same as 9) | HPD was associated with lower frequency of effector or memory (CCR7‐CD45RA‐) circulating CD8+ T cells, and higher frequency of severely exhausted (TIGIT+PD1+) circulating CD8+ T cells |
| Abstract only | ||
| Singavi et al. 12 | Progression at first restaging on‐treatment with increase in tumor size >50%, more than twofold increase in TGR | MDM2/MDM4, EGFR, amplifications on 11q13 (CCND1, FGF3, FGF4, FGF19) |
| Matos et al. 13 | TTF <2 months and minimum increase in measurable lesions of 10 mm plus (A) increase of ≥40% in target tumor burden compared with baseline or (B) increase ≥20% in target tumor burden plus multiple new lesions | HPD was not associated with age, tumor type, checkpoint inhibitor regimens, previous checkpoint inhibitor, or metastatic site |
| Kim et al. 14 | Defined by TGK pre‐immunotherapy versus on‐treatment (details not provided) | No associations found |
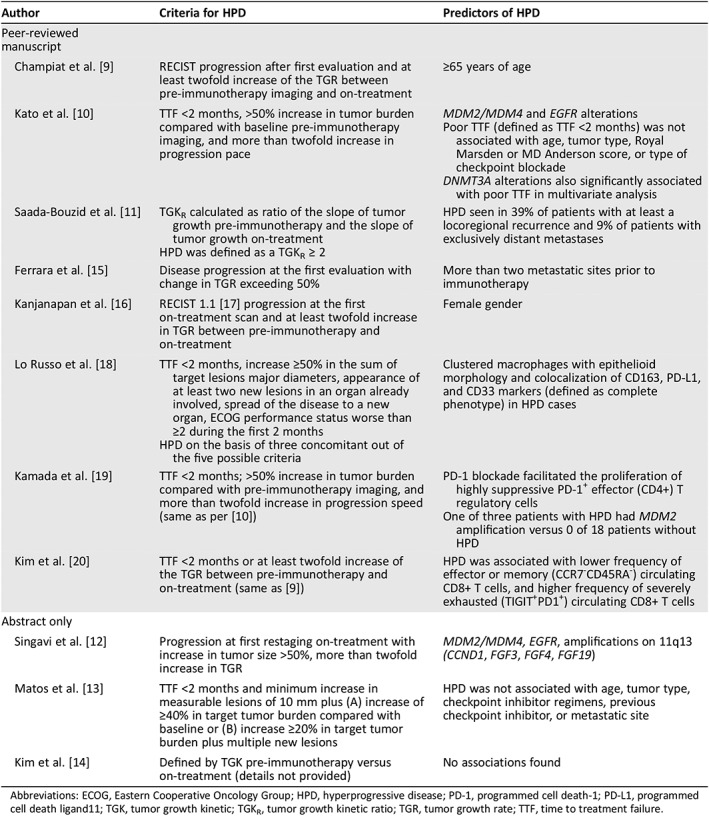
Abbreviations: ECOG, Eastern Cooperative Oncology Group; HPD, hyperprogressive disease; PD‐1, programmed cell death‐1; PD‐L1, programmed cell death ligand11; TGK, tumor growth kinetic; TGKR, tumor growth kinetic ratio; TGR, tumor growth rate; TTF, time to treatment failure.
The phenomenon of enhanced progression after checkpoint blockade has been described with different checkpoint blockade agents and in numerous tumor types including, but not limited to, non‐small cell lung, head and neck, breast, gastric, and genitourinary cancers 9, 10, 11, 12, 13, 14, 15, 16, 18, 19, 20, 21, 22, 23. The fact that various histologies that can be afflicted by HPD suggests that there could be common, histology‐agnostic biologic or molecular mechanism(s). A final reason for the controversy around the existence of HPD may be the reluctance of physicians and other stakeholders to acknowledge that therapies like checkpoint blockade could make some patients worse. Indeed, despite HPD, our impression remains that immune checkpoint inhibitors are some of the most effective drugs ever brought into the clinical cancer arena, with transformative activity in a broad range of lethal malignancies, including long‐term complete remissions in some individuals with highly refractory disease and heavy disease burden. For this reason, HPD should be considered “a toxicity,” or an immune‐related adverse event (irAE) similar to other potential side effects, and should not restrict the use of these important agents. Even so, there is an urgent imperative to inform patients of the risk of HPD, to determine the predictors of this phenomenon, and to unravel its underlying biology.
The frequency of HPD after immunotherapy varies depending on the publication, ranging from <5% to 29% (the latter reported in one study of head and neck squamous cell carcinoma) 9, 10, 11, 12, 13, 14, 15, 16, 21, 22, 23. One question that arises is whether or not HPD is unique to immunotherapy. One report suggests that HPD after chemotherapy can occur, albeit at a much lower rate of 5.1% (3/59) (vs. ∼14% after checkpoint blockade in that study) 15.
A key debate regarding the existence of HPD is whether or not the cancer was an aggressive one in the first place, with the thought being that rather than an induced immunologic effect, the aggressive growth is merely a lack of effective therapy (Fig. 1). In this regard, there are varying criteria that have been proposed to define HPD (Table 1). For instance, Champiat and colleagues 9 define HPD as RECIST progression after first evaluation and at least twofold increase of the tumor growth rate between the reference (prior) and the experimental periods; Kato et al. 10 defined HPD as >50% increase in tumor burden while on checkpoint blockade compared with pre‐immunotherapy, with a <2‐month time to treatment failure (TTF) and a more than twofold increase in progression pace 10. Importantly, the latter requires scans approximately 2 months before starting immunotherapy to be compared with pre‐immunotherapy scans, to exclude the possibility that the tumor had an aggressive pace of growth even before the start of immunotherapy. Virtually all other research groups have almost identical definitions for HPD (Table 1) except for Matos et al. 13 and Lo Russo et al. 18. The first group used the following definition: TTF <2 months and increase in measurable lesions of ≥10 mm plus the following: (A) increase of ≥40% in target tumor burden compared with pre‐immunotherapy or (B) increase of ≥20% in target tumor burden plus multiple new lesions. The second group used a similar definition, and patients with HPD had to fulfill at least three of the following clinical or radiological criteria: (A) TTF <2 months, (B) increase of ≥50% in the sum of target lesions’ major diameters between baseline and first radiologic evaluation, (C) appearance of at least two new lesions in an organ already involved between pre‐immunotherapy and first radiologic evaluation, (D) spread of the disease to a new organ between pre‐immunotherapy and first radiologic evaluation, and (E) clinical deterioration with decrease in Eastern Cooperative Oncology Group performance status ≥2 during the first 2 months of treatment. To avoid attributing rapid progression to immunotherapy when it simply reflects aggressive disease, some have argued that criteria that identifying HPD include a comparison with a prebaseline time period (perhaps ∼2 months) to demonstrate a substantial change in pace of tumor growth. Even this may not be valid, as patients are often on therapy during the period preceding initiation of immunotherapy. Furthermore, this strategy could be difficult to apply when immunotherapy is administered in the first line; therefore, validation of surrogate criteria that do not include a prebaseline scan will be an important future effort.
Figure 1.
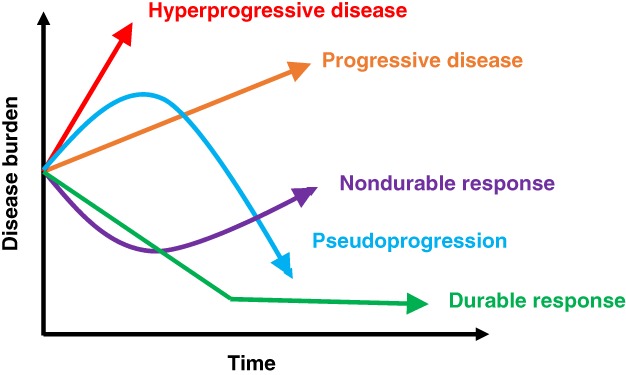
Potential outcomes after initiation of immunotherapy with immune checkpoint inhibitors for the treatment of various cancers over time. (Green): Durable response to treatment in which target lesions shrink on imaging and remain attenuated. (Purple): Nondurable response in which lesions initially response to therapy, but on subsequent surveillance imaging, lesions become resistant and increase in size. (Orange): Disease progression in which target lesions grow >20% from previous imaging. (Blue): Pseudoprogression in which tumors enlarge on imaging initially followed by decrease in size seen. (Red): Hyperprogressive disease in which rapid growth occurs after initiating immune checkpoint inhibitors.
Despite the controversy around the existence of HPD, unique response patterns after checkpoint blockade are not new 24, 25. For instance, a phenomenon termed pseudoprogression has been well established after checkpoint blockade, albeit in a small subgroup of patients 25, 26, 27. Pseudoprogression is defined by the appearance of progression on scans, probably because of immune infiltration, but the patient is asymptomatic or feels better (in contrast to hyperprogression, in which the patient, in our experience, feels worse; Fig. 1). Furthermore, with pseudoprogression, scans ultimately show tumor response. Forms of pseudoprogression have also been previously described, albeit rarely, with agents outside of immunotherapy, for example, after glioblastoma treatment and with some targeted therapeutics 28, 29, 30. The relatively unique response patterns after checkpoint blockade have resulted in development of modified RECIST criteria for immunotherapy—that is, iRECIST 31, 32. Importantly, with iRECIST, new lesions are assessed as per RECIST 1.1 17 but are recorded separately (and not included in the sum of target lesions identified at baseline). This type of evaluation results in a new category of unconfirmed progression (iUPD). Confirmed progression (iCPD) is only assigned if, at the next imaging, an increase in the size of new lesions is seen or additional new lesions appear.
Because of the urgency associated with the rapid progression that is the hallmark of HPD, it is crucial to differentiate between hyperprogression and pseudoprogression as early as possible, even before re‐imaging. With the former, checkpoint blockade should be immediately stopped; in contrast, with the latter, treatment should be continued. Liquid biopsies that interrogate serial blood‐derived circulating cell‐free DNA may be useful in this regard. It appears, at least based on one small study, that the genome instability number in cell‐free DNA rises precipitously with hyperprogression, but falls with pseudoprogression, when measured at about 3 to 6 weeks after starting immunotherapy 33.
Another key question in HPD is whether there are clinical or molecular features that are associated with an increased risk of accelerated growth after checkpoint blockade. Predictors of HPD have included age ≥ 65 years, female gender, regional recurrence of disease, having more than two sites of metastases, low baseline highly differentiated CD4+ T cells or effector memory CD8+ T cells, high severely exhausted T cells or proliferating T regulatory cells, clustered CD163+ PD‐L1+ CD33+ macrophages with epithelioid morphology, and genomic markers (mainly MDM2/MDM4 alterations and EGFR alterations; Table 1) 9, 10, 11, 12, 13, 14, 15, 16, 18, 19, 20, 22. There are inconsistencies between studies in that some have not shown age or sex to be predictors. Furthermore, although it has been described by several groups including ours 10, 12, 19, 34, the putative genomic correlates (e.g., MDM2/MDM4 and EGFR alterations) require larger sample size validation and an understanding of potential mechanisms by which these alterations could mediate or facilitate accelerated tumor growth after checkpoint blockade.
Despite the current uncertainty regarding molecular markers such as MDM2 amplification and EGFR alterations that may predict HPD 9, 12, 34, the use of genomic aberrations as biomarkers for immunotherapy response pattern has been previously established 35, 36, 37, 38, 39, 40, 41. Indeed, although genomics and immunotherapy are often considered as separate fields, in reality, they are tightly linked 42. There are various genomic aberrations that correlate with immunotherapy response, including (but not limited to) (A) mismatch repair gene defects that result in high microsatellite instability (MSI), (B) high tumor mutational burden (TMB), (C) PBRM1 and CDK12 mutations, and (D) PD‐L1 amplification 35, 36, 37, 38, 39, 40, 41. Other biomarkers include high PD‐L1 protein expression, gut microbiome, and POLE 43, ATM 44 (TMB‐mediated), ATR 45 (TMB‐mediated), and CDK12 46 mutations, which have been shown to predict response to immunotherapy 33, 47, 48, 49. Of interest in this regard, pembrolizumab was granted the first tissue‐agnostic approval by the FDA in patients with mismatch repair gene‐altered/MSI‐high solid tumors of any type, based on response rates of ∼40% 1. The reasons that high MSI and high TMB predict response to immunotherapy are probably related, because high MSI almost inevitably leads to a high TMB 50. High TMB means that there are likely many neoantigens produced by the tumor mutational genome and, hence, a greater chance that the reactivated immune system after checkpoint blockade will be able to differentiate the neoplasm from normal tissue elements and target it for eradication 51, 52. In certain tumor types, such as clear cell renal cell carcinoma, PBRM1 mutations have been associated with response to immunotherapy 35, 40. PBRM1 encodes a subunit of the PBAF switch‐sucrose nonfermentable chromatin remodeling complex, which regulates how tightly DNA is packaged in cells; its loss may increase expression of T‐cell cytotoxicity 35, 40. Similarly, PD‐L1 amplification in Hodgkin lymphoma and various solid tumors also associates with immunotherapy benefit 39, 53, 54. There are also several markers of tumor resistance, again of genomic origin: (A) STK11 and KRAS co‐mutations in lung cancer 55; (B) loss‐of‐function mutations in the genes encoding interferon‐receptor–associated Janus kinase 1 or Janus kinase 2, concurrent with deletion of the wild‐type allele 56; and (C) truncating mutations in the gene encoding the antigen‐presenting protein beta‐2‐microglobulin (which leads to loss of surface expression of major histocompatibility complex class I resulting in attenuated neoantigen presentation) 56. These observations suggest that genomic markers can predict response pattern after checkpoint blockade and that their mechanism of action is not always fully understood, at least initially.
In summary, despite the numerous research teams that have documented HPD 9, 10, 11, 12, 13, 14, 15, 16, 18, 20, 21, 22, the existence of this phenomenon remains a matter of dispute. Indeed, an analysis of the OAK trial 57, which was a randomized study of checkpoint blockade versus chemotherapy in lung cancer, did not show a difference in the number of “fast progressors” between the arms. However, this trial had no pre‐immunotherapy evaluation to demonstrate whether or not the pace of progression had increased. Patients with rapid progression who do not have pre‐immunotherapy imaging available may be currently difficult to designate as having HPD. It is important to note, however, that using pre‐immunotherapy imaging may not always be feasible for all treatment settings; for example, in the first‐line setting not all cancer patients have pre‐immunotherapy scans available. Therefore, some groups (Table 1) 13, 18 have suggested criteria for HPD that do not require pre‐ immunotherapy scans; these criteria will need to be validated in patients with existing pre‐immunotherapy scans. Recent data have shown that HPD can be recapitulated in preclinical models 18. As physicians make immunotherapy a mainstay of treatment in more cancer types, it will be imperative to develop predictive markers for HPD and to understand the biology that underlies this devastating irAE.
Disclosures
Shumei Kato: Foundation Medicine (C/A); Roberto Ferrara: Merck Sharp and Dohme (C/A); Giuseppe Lo Russo: Merck Sharp and Dohme (SAB), Merck Sharp and Dohme, Bristol‐Myers Squibb, Roche, AstraZenea (C/A); Razelle Kurzrock: IDbyDNA, CureMatch, Inc., Soluventis (OI), Gaido, LOXO, X‐Biotech, Actuate Therapeutics, Roche, NeoMed, Soluventis (C/A), Incyte, Genentech, Merck Serono, Pfizer, Sequenom, Foundation Medicine, Guardant Health, Grifols, Konica Minolta, OmniSeq (RF—institutional), Roche (other—speaker's fee). Jacob J. Adashek indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Acknowledgments
R.K. is funded in part by the Joan and Irwin Jacobs Fund and NIH P30 CA023100.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Marcus L, Lemery SJ, Keegan P et al. FDA approval summary: Pembrolizumab for the treatment of microsatellite instability‐high solid tumors. Clin Cancer Res 2019;25:3753–3758. [DOI] [PubMed] [Google Scholar]
- 2. Nivolumab approved for lung cancer . Cancer Discov 2015;5:OF1. [DOI] [PubMed] [Google Scholar]
- 3. Syed YY. Durvalumab: First global approval. Drugs 2017;77:1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim ES. Avelumab: First global approval. Drugs 2017;77:929–937. [DOI] [PubMed] [Google Scholar]
- 5. Weinstock C, Khozin S, Suzman D et al. U.S. Food and Drug Administration approval summary: Atezolizumab for metastatic non‐small cell lung cancer. Clin Cancer Res 2017;23:4534–4539. [DOI] [PubMed] [Google Scholar]
- 6. Cemiplimab approved for treatment of CSCC . Cancer Discov 2018;8:OF2. [DOI] [PubMed] [Google Scholar]
- 7. Culver ME, Gatesman ML, Mancl EE et al. Ipilimumab: A novel treatment for metastatic melanoma. Ann Pharmacother 2011;45:510–519. [DOI] [PubMed] [Google Scholar]
- 8. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359:1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Champiat S, Dercle L, Ammari S et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti‐PD‐1/PD‐L1. Clin Cancer Res 2017;23:1920–1928. [DOI] [PubMed] [Google Scholar]
- 10. Kato S, Goodman A, Walavalkar V et al. Hyperprogressors after immunotherapy: Analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res 2017;23:4242–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saada‐Bouzid E, Defaucheux C, Karabajakian A et al. Hyperprogression during anti‐PD‐1/PD‐L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol 2017;28:1605–1611. [DOI] [PubMed] [Google Scholar]
- 12. Singavi AK, Menon S, Kilari D et al. Predictive biomarkers for hyper‐progression (HP) in response to immune checkpoint inhibitors (ICI) – Analysis of somatic alterations (SAs). Ann Oncol 2017;28(suppl 5):1140PDA. [Google Scholar]
- 13. Matos I, Martin‐Liberal J, Hierro C et al. Incidence and clinical implications of a new definition of hyperprogression (HPD) with immune checkpoint inhibitors (ICIs) in patients treated in phase 1 (Ph1) trials. J Clin Oncol 2018;36(suppl 15):3032A. [Google Scholar]
- 14. Kim Y, Kim CH, Kim HS et al. Hyperprogression after immunotherapy: Clinical implication and genomic alterations in advanced non‐small cell lung cancer patients (NSCLC). J Clin Oncol 2018;36(suppl 15):9075A. [Google Scholar]
- 15. Ferrara R, Mezquita L, Texier M et al. Hyperprogressive disease in patients with advanced non‐small cell lung cancer treated with PD‐1/PD‐L1 inhibitors or with single‐agent chemotherapy. JAMA Oncol 2018;4:1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanjanapan Y, Day D, Wang L et al. Hyperprogressive disease in early‐phase immunotherapy trials: Clinical predictors and association with immune‐related toxicities. Cancer 2019;125:1341–1349. [DOI] [PubMed] [Google Scholar]
- 17. Schwartz LH, Litiere S, de Vries E et al. RECIST 1.1‐Update and clarification: From the RECIST committee. Eur J Cancer 2016;62:132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lo Russo G, Moro M, Sommariva M et al. Antibody‐Fc/FcR Interaction on macrophages as a mechanism for hyperprogressive disease in non‐small cell lung cancer subsequent to PD‐1/PD‐L1 blockade. Clin Cancer Res 2019;25:989–999. [DOI] [PubMed] [Google Scholar]
- 19. Kamada T, Togashi Y, Tay C et al. PD‐1(+) regulatory T cells amplified by PD‐1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci USA 2019;116:9999–10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim CG, Kim KH, Pyo KH et al. Hyperprogressive disease during PD‐1/PD‐L1 blockade in patients with non‐small‐cell lung cancer. Ann Oncol 2019;30:1104–1113. [DOI] [PubMed] [Google Scholar]
- 21. Champiat S, Ferrara R, Massard C et al. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat Rev Clin Oncol 2018;15:748–762. [DOI] [PubMed] [Google Scholar]
- 22. Zuazo‐Ibarra M, Arasanz H, Fernandez‐Hinojal G et al. Highly differentiated CD4 T cells unequivocally identify primary resistance and risk of hyperprogression to PD‐L1/PD‐1 immune checkpoint blockade in lung cancer. Ann Oncol 2018;29:viii14–viii57. [Google Scholar]
- 23. Matos I GA, Martin‐Liberal J, Hierro C et al. Refining criteria of hyperprogression (HPD) with immune checkpoint inhibitors (ICIs) to improve clinical applicability. Presented at: European Society for Medical Oncology 2018 Congress; October 20, 2018; Munich, Germany.
- 24. Popat S. Hyperprogression with immunotherapy: Is it real? Cancer 2019;125:1218–1220. [DOI] [PubMed] [Google Scholar]
- 25. Borcoman E, Kanjanapan Y, Champiat S et al. Novel patterns of response under immunotherapy. Ann Oncol 2019;30:385–396. [DOI] [PubMed] [Google Scholar]
- 26. Kurra V, Sullivan RJ, Gainor JF et al. Pseudoprogression in cancer immunotherapy: Rates, time course and patient outcomes. J Clin Oncol 2016;34(suppl 15):6580A. [Google Scholar]
- 27. Soria F, Beleni AI, D'Andrea D et al. Pseudoprogression and hyperprogression during immune checkpoint inhibitor therapy for urothelial and kidney cancer. World J Urol 2018;36:1703–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Balana C, Capellades J, Pineda E et al. Pseudoprogression as an adverse event of glioblastoma therapy. Cancer Med 2017;6:2858–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benjamin RS, Choi H, Macapinlac HA et al. We should desist using RECIST, at least in GIST. J Clin Oncol 2007;25:1760–1764. [DOI] [PubMed] [Google Scholar]
- 30. Kurzrock R, Atkins J, Wheler J et al. Tumor marker and measurement fluctuations may not reflect treatment efficacy in patients with medullary thyroid carcinoma on long‐term RET inhibitor therapy. Ann Oncol 2013;24:2256–2261. [DOI] [PubMed] [Google Scholar]
- 31. Beer L, Hochmair M, Prosch H. Pitfalls in the radiological response assessment of immunotherapy. Memo 2018;11:138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seymour L, Bogaerts J, Perrone A et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol 2017;18:e143–e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jensen TJ, Goodman AM, Kato S et al. Genome‐wide sequencing of cell‐free DNA identifies copy‐number alterations that can be used for monitoring response to immunotherapy in cancer patients. Mol Cancer Ther 2019;18:448–458. [DOI] [PubMed] [Google Scholar]
- 34. Kato S, Ross JS, Gay L et al. Analysis of MDM2 amplification: Next‐generation sequencing of patients with diverse malignancies. JCO Precis Oncol 2018. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bratslavsky G, Gay LM, Sokol E et al. PBRM1 mutation and immunotherapy efficacy: A comprehensive genomic profiling (CGP) assessment. J Clin Oncol 2018;36(suppl 15):12091A. [Google Scholar]
- 36. Goodman A, Patel SP, Kurzrock R. PD‐1‐PD‐L1 immune‐checkpoint blockade in B‐cell lymphomas. Nat Rev Clin Oncol 2017;14:203–220. [DOI] [PubMed] [Google Scholar]
- 37. Goodman AM, Kato S, Bazhenova L et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 2017;16:2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chan TA, Jaffee E, Yarchoan M et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann Oncol 2018;30:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goodman AM, Piccioni D, Kato S et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol 2018;4:1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miao D, Margolis CA, Gao W et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018;359:801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Le DT, Durham JN, Smith KN et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 2017;357:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Subbiah V, Kurzrock R. The marriage between genomics and immunotherapy: Mismatch meets its match. The Oncologist 2019;24:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mehnert JM, Panda A, Zhong H et al. Immune activation and response to pembrolizumab in POLE‐mutant endometrial cancer. J Clin Invest 2016;126:2334–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: Therapeutic implications. Mol Cancer Ther 2016;15:1781–1791. [DOI] [PubMed] [Google Scholar]
- 45. Mouw KW, Goldberg MS, Konstantinopoulos PA et al. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov 2017;7:675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu YM, Cieslik M, Lonigro RJ et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018;173:1770–1782 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Patel SP, Kurzrock R. PD‐L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 2015;14:847–856. [DOI] [PubMed] [Google Scholar]
- 48. Topalian SL, Hodi FS, Brahmer JR et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012;366:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sears CL, Pardoll DM. The intestinal microbiome influences checkpoint blockade. Nat Med 2018;24:254–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chalmers ZR, Connelly CF, Fabrizio D et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yi M, Qin S, Zhao W et al. The role of neoantigen in immune checkpoint blockade therapy. Exp Hematol Oncol 2018;7:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gubin MM, Artyomov MN, Mardis ER et al. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J Clin Invest 2015;125:3413–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roemer MG, Advani RH, Ligon AH et al. PD‐L1 and PD‐L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ansell SM, Lesokhin AM, Borrello I et al. PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015;372:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Skoulidis F, Goldberg ME, Greenawalt DM et al. STK11/LKB1 mutations and PD‐1 inhibitor resistance in KRAS‐mutant lung adenocarcinoma. Cancer Discov 2018;8:822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zaretsky JM, Garcia‐Diaz A, Shin DS et al. Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med 2016;375:819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gandara DR, Reck M, Cardona A et al. Fast progression in patients treated with a checkpoint inhibitor (cpi) vs chemotherapy in OAK, a phase III trial of atezolizumab (atezo) vs docetaxel (doc) in 2L+ NSCLC. Ann Oncol 2018;29(suppl 10):LBA1A. [Google Scholar]