

Summary
Type‐I interferons (IFN‐I) are used as common antiviral drugs for a range of viral diseases in clinic. However, the antiviral efficacy of IFN‐I is largely restricted by negative regulators of IFN‐I signaling in cells. Therefore, identification of intracellular inhibitors of IFN‐I signaling is important for developing novel targets to improve IFN‐I antiviral therapy. In this study, we report that the deubiquitinase ubiquitin‐specific protease 7 (USP7) negatively regulates IFN‐I‐mediated antiviral activity. USP7 physically interacts with suppressor of cytokine signaling 1 (SOCS1) and enhances SOCS1 protein stability by deubiquitination effects, which in turn restricts IFN‐I‐induced activation of Janus kinase–signal transducer and activator of transcription 1 signaling. Interestingly, viral infection up‐regulates USP7 and therefore facilitates viral immune evasion. Importantly, the USP7 small‐molecule inhibitors P5091 and P22077 inhibit SOCS1 expression and enhance IFN‐I antiviral efficacy. Our findings identify a novel regulator of IFN‐I antiviral activity and reveal that USP7 inhibitors could be potential enhancement agents for improving IFN‐I antiviral therapy.
Keywords: antiviral activity, interferon, small‐molecule inhibitor, suppressor of cytokine signaling 1, ubiquitin‐specific protease 7
The deubiquitinase USP7 inhibitors P5091 and P22077 enhance IFN‐I antiviral activity, so providing potential strategies for improving both host antiviral response and the efficacy of IFN‐I antiviral therapy.

Abbreviations
- CHX
cycloheximide
- DUBs
deubiquitinases
- IFN‐I
type‐I interferon
- ISGs
interferon‐stimulated genes
- ISRE
interferon‐stimulated response elements
- SeV
Sendai virus
- SOCS1
suppressor of cytokine signaling 1
- USP7
ubiquitin‐specific protease 7
- VSV
vesicular stomatitis virus
- VSV‐GFP
vesicular stomatitis virus with a green fluorescent protein gene
Introduction
Deubiquitinases (DUBs) have been widely studied and demonstrated to be important regulators for various biological functions, including tumor progression, apoptosis, cell cycle and anti‐microbial immunity. Recently, many human diseases have been linked to the dysfunction of DUBs,1, 2, 3 suggesting that DUBs could be potential therapeutic targets for human diseases. Some DUB inhibitors have entered clinical trials and proved to be potential candidates for the therapy of human tumors and other diseases.4
The ubiquitin‐specific protease (USP) family is the largest human DUB family and comprises more than 30 members. Among these USPs, ubiquitin‐specific protease 7 (USP7), also known as herpes virus‐associated ubiquitin‐specific protease (or HAUSP), is a well‐characterized deubiquitinase that plays essential roles in various biological functions, including tumor progression, DNA damage response, apoptosis, epigenetic regulation.5, 6, 7, 8 The roles of USP7 in tumorigenesis and tumor aggressiveness have attracted increasing attention. USP7 plays important roles in regulating the P53‐MDM2 complex.9 Also, USP7 can deubiquitinate phosphatase and tensin homolog (PTEN) to promote its nuclear exclusion and to induce tumor progression.10 Based on these important findings, specific anti‐USP7 inhibitors have been developed for the therapy of multiple human tumors.11, 12, 13
Interestingly, it has been reported that during herpes simplex virus (HSV) infection, USP7 can be recruited by HSV infected cell polypeptide 0 to attenuate nuclear factor‐κB‐ and c‐Jun N‐terminal kinase‐mediated inflammatory response.14 Kaposi's sarcoma herpesvirus recruits USP7 to the viral latency‐associated nuclear antigen 1 to modulate viral latent replication.15 These studies suggest that USP7 is involved in the regulation of virus infection. However, the detailed mechanisms by which USP7 regulates innate antiviral immunity remain to be illuminated. In particular, whether and how USP7 regulates interferon (IFN) ‐mediated antiviral defense remains unknown.
Type‐I interferon (IFN‐I, or IFN‐α/β), as a widely used antiviral drug in clinics, plays important roles in regulating antiviral innate immunity. Upon binding of IFN‐I to its receptors (IFNAR1 and IFNAR2), Janus kinase (JAK) family (JAK1 and Tyk2) will be activated. Then, activated JAK1 and Tyk2 result in tyrosine phosphorylation and activation of the signal transducers and activators of transcription STAT1 and STAT2. Activation of STAT1 largely determines the magnitude of the IFN signaling and antiviral activity. Subsequently, STAT1, STAT2 and interferon regulatory factor 9 (IRF9) form a complex, namely IFN‐stimulated gene factor 3 (ISGF3), which translocates into the nucleus to bind to the IFN‐stimulated response elements (ISRE) in the promoter of interferon‐stimulated genes (ISGs), and further induces transcription and expression of hundreds of antiviral ISGs. Our previous studies showed that the deubiquitinase USP2a positively regulates IFN‐I antiviral activity by maintaining the levels of activated STAT1.16 In addition, we revealed that the deubiquitinase Josephin Domain Containing 1 (JOSD1) negatively regulates IFN‐I antiviral activity by deubiquitinating and stabilizing SOCS1 at the early stage of viral infection.17 Interestingly, we noticed that SOCS1 proteins were gradually up‐regulated at the late stage of viral infection, whereas JOSD1 underwent down‐regulation at that stage, suggesting that SOCS1 protein levels could be regulated by other mechanisms. This interesting regulation needs to be further explored.
In this study, we revealed that USP7 physically interacts with SOCS1 and up‐regulates the level of SOCS1 protein. Moreover, USP7 is responsible for SOCS1 protein up‐regulation during long‐term viral infection. Interestingly, viral infection up‐regulates USP7 protein levels for viral immune evasion. Importantly, we further demonstrated that two USP7 small‐molecule inhibitors enhance IFN‐I‐induced STAT1 activation, so promoting IFN‐I signaling and antiviral activity. Our findings could provide new strategies for improving IFN‐based antiviral therapeutic efficacy.
Materials and methods
Cell culture and transfection
Human embryo kidney 293T cells, human fibrosarcoma 2fTGH cells, and human lung cancer A549 cells were cultured at 37° in humidified air containing 5% CO2. All culture media were supplemented with 10% fetal bovine serum (FBS; Gibco Life Technologies, Waltham, MA), 100 mg/ml streptomycin and 100 units/ml penicillin. Cells were transfected with either Polyfectine (PEI; Warrington, PA) or LongTrans (Ucallm, Wu Xi, China) according to the manufacturers' instructions.
Reagents and expression constructs
USP7 inhibitors (P5091 and P22077), Trypan Blue and PDTC ammonium (PDTC) were purchased from Selleck Chemicals (Houston, TX). Recombinant human IFN‐α was purchased from PBL Interferon Source (Piscataway, NJ, USA). Cycloheximide, and other chemicals were purchased from Sigma (St Louis, MO). Flag‐USP7 was a gift from Dr. J. Wade Harper (Harvard Medical School, Addgene Plasmids). HA‐Ub, Myc‐SOCS1, ISRE‐Luc and Renilla plasmids were gifts from Dr. Serge Y. Fuchs (University of Pennsylvania). The Flag‐USP7‐C223S mutant was generated by a QuickChange site‐directed mutagenesis kit (Stratagene, La Jolla, CA). Knockdown plasmids, including shUSP7 and shSOCS1, were purchased from GeneChem (Shanghai, China). All the plasmids were confirmed by DNA sequencing.
Immunoblotting and Western blot analysis
The detailed protocols for immunoblotting and Western blot analysis were described previously.16 Briefly, cells were harvested using lysis buffer containing 150 mm NaCl, 20 mm Tris–HCl (PH7·4), 1% Nonidet P‐40, 0·5 mm ethylenediamine tetraacetic acid, phenylmethylsulfonyl fluoride (50 μg/ml) and protease inhibitor mixtures (Sigma). Protein samples were resolved by sodium dodecyl sulfate—polyacrylamide gel electrophoresis. Immunoreactive bands were visualized with SuperSignal West Dura Extended kits (Thermo Scientific, Waltham, MA). The following antibodies were used: antibodies against USP7 (Santa Cruz Biotechnology, Dallas, TX; sc‐30164), SOCS1 (Millipore, Billierica, MA; 04‐002), JOSD1 (Santa Cruz; sc‐86154), Ubiquitin (Santa Cruz; sc‐8017), pY701‐STAT1 (Cell Signaling, Danvers, MA; #9167), p‐TYK2 (Cell Signaling; #9321), p‐JAK1 (Cell Signaling, #3331s) Viperin (Abcam, Cambridge, UK; #107359), Myc (Abmart; M20002), Flag (Sigma; F7425), HA (Abcam; ab9110), STAT1 (Cell Signalling; #8826) and Tubulin (Proteintech, Rosemont, IL; 66031‐1‐Ig).
Quantitative real‐time polymerase chain reaction
Total RNAs were isolated from 293T cells using a TRIzol reagent (Invitrogen, Carlsbad, CA). All cDNAs were synthesized by reverse transcription using oligo‐dT and subjected to quantitative real‐time polymerase chain reaction (PCR) with corresponding primers in the presence of SYBR Green Supermix (Bio‐Rad, Hercules, CA). The primer sequences are as follows.
IFIT1 (5'‐CACAAGCCATTTTCTTTGCT‐3' and 5'‐ACTTGGCTGCATATCGAAAG‐3'); ISG15 (5'‐GGGACCTGACGGTGAAGATG‐3' and 5'‐CGCCGATCTTCTGGGTGAT‐3'); ISG54 (5'‐CACCTCTGGACTGGCAATAGC‐3' and 5'‐GTCAGGATTCAGCCGAATGG‐3'); VSV (5'‐ACGGCGTACTTCCAGATGG‐3' and 5'‐CTCGGTTCAAGATCCAGGT‐3'); Sendai virus (5'‐GATGACGATGCCGCAGCAGTAG‐3' and 5'‐CCTCCGATGTCAGTTGGTTCACTC‐3'); H1N1 (5'‐TTCTAACCGAGGTCGAAACG‐3' and 5'‐ACAAAGCGTCTACGCTGCAG‐3'); USP7 (5'‐CCCTCCGTGTTTTGTGCGA‐3' and 5'‐AGACCATGACGTGGAATCAGA‐3'); β‐actin (5'‐ACCAACTGGGACGACATGGAGAAA‐3' and 5'‐ATAGCACAGCCTGGATAGCAACG‐3').
Reporter gene assay
To detect IFN‐I‐induced transcriptional activity, cells were transfected with the ISRE‐Luciferase and Renilla, together with either USP7 or shUSP7 constructs. Then, 36 hr after transfection, cells were treated with IFN‐α overnight and then collected. The Luciferase activities were measured using the Dual‐Luciferase Reporter Assay System (Promega, Madison, WI; #E1910). The activities were determined in three independent experiments and were shown as the mean ± standard deviation (SD).
Virus and viral infection
Vesicular stomatitis virus (VSV) was a gift from Dr. Chen Wang (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences). Vesicular stomatitis virus with a green fluorescent protein gene (VSV‐GFP) was a gift from Dr. Chunsheng Dong (Soochow University). Influenza A virus (H1N1, PR/8/34) was from Dr. Jianfeng Dai (Soochow University). The antiviral effect of IFN‐α was determined by pretreating cells overnight before infection with viruses. Briefly, cells were transfected with either Flag‐USP7 or shUSP7. After 48 hr, cells were pretreated with IFN‐α (50 IU/ml) overnight. After washing twice, cells were challenged with VSV or VSV‐GFP at a multiplicity of infection of either 1·0 or 0·5 for 1·5 hr. Then the infection medium was removed by washing twice. Cells were fed with fresh medium and incubated for 20 hr. Cells were analyzed by either immunofluorescence or reverse transcription PCR.
Stable cell lines of SOCS1‐knockdown by shSOCS1 in HeLa
293T cells were transfected with shCON or shSOCS1 plasmids, together with pMDL, VSVG and REV plasmids. Thirty‐six hours after transfection, the supernatant was collected and used to infect HeLa cells. The infected cells were cultured for 24 hr with polybrene (8 μg/ml). After removing the culture medium, the cells were cultured in 10% FBS without polybrene for 24 hr. Then cells were cultured in 10% FBS with puromycin (1·5 μg/ml) for 2 weeks of selection. The stable cell lines of SOCS1 knockdown were maintained in 10% FBS medium with puromycin (1·5 μg/ml) until 2 days before further experiments.
Cycloheximide chase assay
The half‐life of Myc‐SOCS1 was determined by cycloheximide (CHX) chase assay. For analysis of Myc‐SOCS1 levels, 293T cells were seeded in six‐well cell culture plates and were transfected with Myc‐SOCS1 and shUSP7. Sixy hours after transfection, cells were treated with dimethylsulfoxide (DMSO) or CHX (50 μg/ml) for the indicated time‐points. Cells were then collected and subjected to analysis by Western blotting.
Analysis of cell viability
Cell viability effects of P5091 and P22077 were evaluated by the trypan blue exclusion assay. HEK293T cells were treated with either P5091 or P22077 using different concentrations for 12 hr and cells were cultivated and collected. Next, both living and dead cells were quantified in a Neubauer chamber in the presence of Trypan blue (0·4%). The CC50 (50% cytotoxicity concentration) values were calculated.
Statistical analysis
Comparison between different groups was analyzed using Student's t‐test. All statistical data in this study are presented as mean ± SD. All differences were considered statistically significant when P < 0·05.
Results
Viral infection promotes expression of USP7 that up‐regulates SOCS1 protein levels at the late stage of viral infection
During our studies on regulation of IFN signaling proteins, we noticed that SOCS1 protein underwent strong up‐regulation at the late stage of viral infection (Fig. 1a). This up‐regulation cannot be due to the increase in JOSD1 protein that was reported to be a deubiquitinase of SOCS1 in our previous studies, as JOSD1 protein levels actually decreased to very low levels during that stage (Fig. 1a). Similarly, exogenously expressed Myc‐SOCS1 were also up‐regulated by viral infection (Fig. 1b). Virus‐mediated up‐regulation of both endogenous SOCS1 and exogenously expressed SOCS1 suggested that SOCS1 could be regulated at protein level. In line with the speculation, viral infection significantly lowered ubiquitination levels of SOCS1 protein (Fig. 1c). Hence, in this study we sought to determine which member of DUBs could contribute to SOCS1 protein up‐regulation at the late stage of viral infection. We noticed a deubiquitinase USP7 that can also promote SOCS1 protein stability (Fig. 1d), when we screened an expression library of DUBs. Importantly, knockdown of USP7 largely abolished virus‐mediated up‐regulation of SOCS1 at the late stage of viral infection (Fig. 1e). To confirm the role of the deubiquitinase USP7 in SOCS1 up‐regulation, we further used a USP7 inhibitor, P5091, to inhibit the deubiquitinase activity of USP7. The results showed that the USP7 inhibitor restricted SOCS1 protein up‐regulation induced by viral infection (Fig. 1f). These findings suggest that USP7 largely contributes to SOCS1 protein up‐regulation at the late stage of viral infection.
Figure 1.
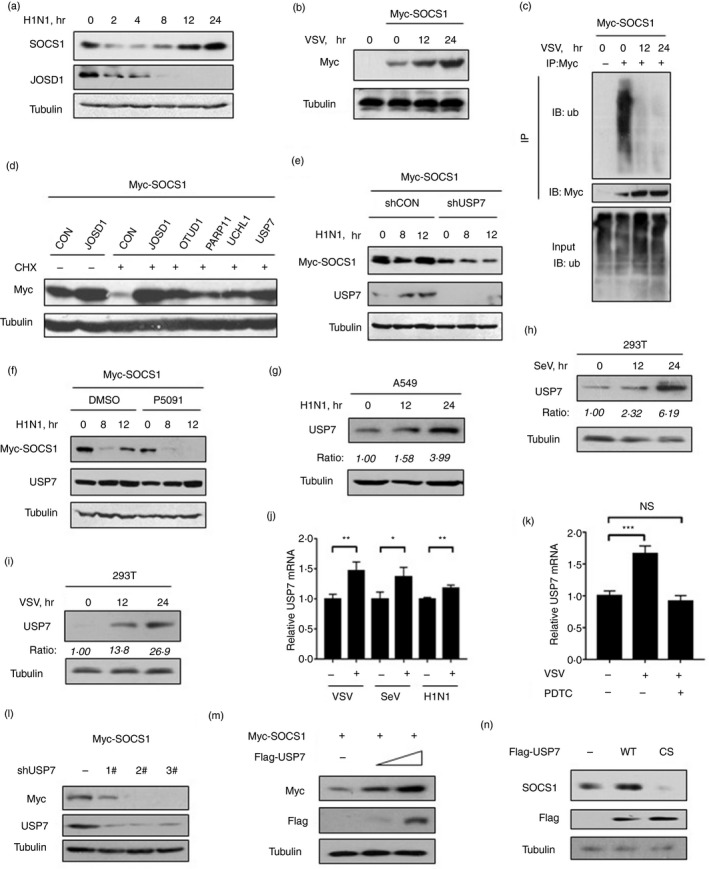
Viral infection promotes protein expression of USP7 that up‐regulates SOCS1 protein levels at the late stage of viral infection. (a) Western blot analysis of protein levels of SOCS1 and JOSD1 in A549 cells infected with H1N1 [multiplicity of infection (MOI) = 1·0] for the indicated times. (b) Western blot analysis of exogenously expressed Myc‐SOCS1 levels in HEK293T cells transfected with Myc‐SOCS1 and then infected with vesicular stomatitis virus (VSV) (MOI = 1·0). (c) Immunoprecipitation analysis of polyubiquitination of SOCS1 in HEK293T cells transfected with Myc‐SOCS1 and then infected with VSV (MOI = 1·0). (d) Western blot analysis of Myc‐SOCS1 protein levels in HEK293T cells transfected with Myc‐SOCS1 and different DUBs, followed by treatment with or without cycloheximide (50 μm) for 12 hr. (e) Western blot analysis of Myc‐SOCS1 protein levels in A549 cells co‐transfected with Myc‐SOCS1, control shRNAs (shCON) or shRNAs against human USP7 (shUSP7) as indicated and then infected with H1N1 (MOI = 1·0). (f) Western blot analysis of Myc‐SOCS1 protein levels in A549 cells transfected with Myc‐SOCS1 and then treated with dimethylsulfoxide or P5091 (100 nm) for 12 hr, followed by infection with VSV (MOI = 1·0). (g) Western blot analysis of USP7 protein levels in A549 cells infected with H1N1 (MOI = 1·0) for the indicated times. (h,i) Western blot analysis of USP7 protein levels in HEK293T cells infected with Sendai virus (SeV) (MOI = 1·0) (h) or VSV (MOI = 1·0) (i). The densities of protein bands were quantified with imageJ. (j) Quantitative PCR analysis of USP7 mRNA levels in HEK293T cells infected with VSV (MOI = 1·0), SeV (MOI = 1·0) or H1N1 (MOI = 1·0) for 24 hr. (k) Quantitative PCR analysis of USP7 mRNA levels in HEK293T cells pretreated with pyrrolidine dithiocarbamate ammonium (PDTC) (nuclear factor‐κB inhibitor, 10 μm) for 1 hr and then infected with VSV (MOI = 1·0) for 24 hr. The data were shown as fold change normalized to that in uninfected control cells. (l) Western blot analysis of Myc‐SOCS1 protein levels in A549 cells co‐transfected with Myc‐SOCS1, together with control shRNAs (–) or three shUSP7 as indicated. (m) Western blot analysis of Myc‐SOCS1 protein levels in A549 cells co‐transfected with Myc‐SOCS1 and increasing amounts of Flag‐USP7. (n) Western blot analysis of SOCS1 protein levels in A549 cells co‐transfected with Flag‐USP7 (WT or inactive C223S mutant) as indicated
Consistent with SOCS1 protein up‐regulation, we observed that USP7 protein levels were gradually up‐regulated in cells infected with influenza A virus (H1N1, PR/8/34) (Fig. 1g), Sendai virus (Fig. 1h) and VSV (Fig. 1i). By reverse transcription quantitative PCR analysis, we found that all these viruses can promote USP7 mRNA expression (Fig 1j). Furthermore, we found that PDTC, which is a nuclear factor‐κB inhibitor, strongly inhibited virus‐induced up‐regulation of USP7 mRNA transcriptional expression (Fig. 1k), suggesting that USP7 mRNA up‐regulation during viral infection is through activation of NF‐κB.
Next, we found that knockdown of USP7 by two specific short hairpin RNAs obviously down‐regulated Myc‐SOCS1 protein levels (Fig. 1l). Conversely, USP7 overexpression up‐regulated Myc‐SOCS1 protein levels in a dose‐dependent manner (Fig. 1m). Moreover, we found that USP7‐mediated up‐regulation of SOCS1 protein was dependent on the deubiquitinase activity of USP7, as the deubiquitinase inactive USP7‐C223S (CS) mutant lost the ability to enhance SOCS1 protein expression (Fig. 1n). Taken together, these findings suggest that viral infection up‐regulates USP7, which results in increase of SOCS1 protein at the late stage of viral infection.
USP7 physically interacts with SOCS1 and enhances SOCS1 protein stability
To further address how USP7 up‐regulates SOCS1, we first tested whether USP7 interacts with SOCS1. Immunoprecipitation analysis indicated a clear interaction between Flag‐USP7 and Myc‐SOCS1 (Fig. 2a,b). Furthermore, we observed that endogenous USP7 can interact with endogenous SOCS1 in cells (Fig. 2c,d). These findings suggest that USP7 physically interacts with SOCS1. Given that USP7‐mediated up‐regulation of SOCS1 protein levels is dependent on its deubiquitinase activity (Fig. 1l), we asked whether USP7 could regulate SOCS1 protein stability. To answer this question, a CHX pulse‐chase assay was performed. CHX is a protein synthesis inhibitor and can be used for measurement of the stability of cellular proteins. To this end, HEK293T cells transfected with Myc‐SOCS1 and shUSP7 were treated with DMSO or CHX (50 μg/ml) for 3 and 6 hr. Cells were then collected and subjected to Western blot analysis. This result showed that when endogenous USP7 was knocked down, the degradation rate of Myc‐SOCS1 protein was markedly accelerated under conditions of CHX treatment (Fig. 2e), indicating that SOCS1 protein stability is regulated by cellular USP7. Based on these findings, we next questioned whether the effect of USP7 on SOCS1 protein levels is mediated by the ubiquitin–proteasome system. We found that the proteasome inhibitor MG132 was able to block the down‐regulation of SOCS1 proteins induced by USP7 knockdown (Fig. 2f), suggesting that USP7 could regulate SOCS1 in a ubiquitin‐proteasome‐dependent manner. Hence, we demonstrated that USP7 interacts with SOCS1 and enhances protein stability of SOCS1.
Figure 2.
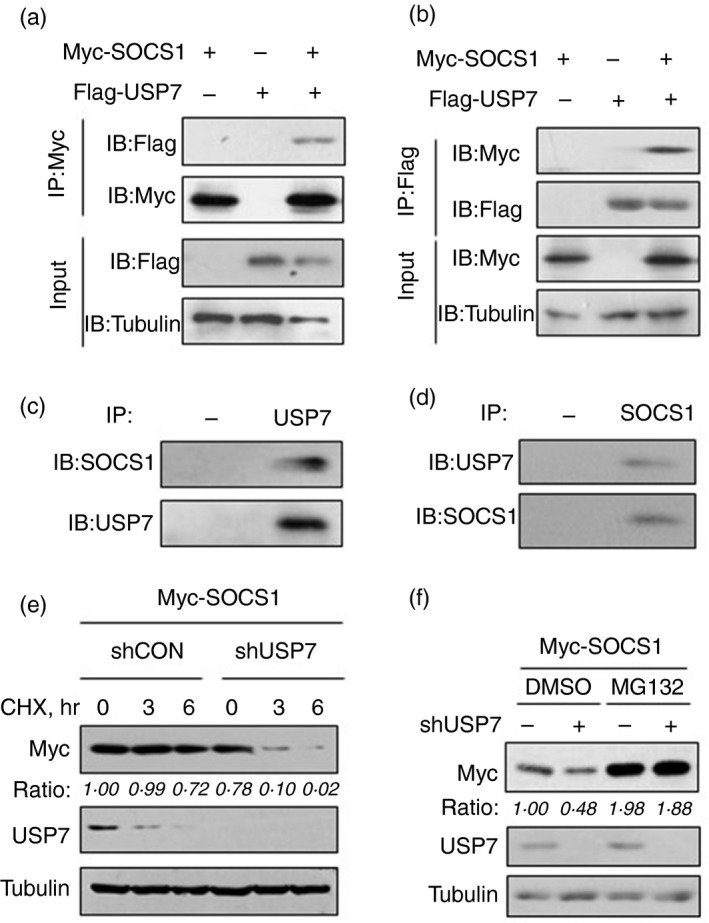
USP7 interacts with and stabilizes SOCS1 protein. (a,b) Immunoprecipitation analysis of the interaction between Flag‐USP7 and Myc‐SOCS1 in A549 cells co‐transfected with Myc‐SOCS1 and Flag‐USP7 using anti‐Myc (a) or anti‐Flag (b) antibodies for immunoprecipitation. (c) Immunoprecipitation analysis of the interaction between endogenous USP7 and SOCS1 in A549 cells using either the isotype IgG (−) or anti‐USP7 antibodies (USP7) for immunoprecipitation. (d) Immunoprecipitation analysis of the interaction between endogenous USP7 and SOCS1 in A549 cells using either the isotype IgG (−) or anti‐SOCS1 antibodies (SOCS1) for immunoprecipitation. (e) Western blot analysis of Myc‐SOCS1 protein levels in HEK293T cells transfected with Myc‐SOCS1, together with shCON or shUSP7, followed by treatment with cycloheximide (50 μm) as indicated. (f) Western blot analysis of Myc‐SOCS1 protein levels in HEK293T cells transfected with Myc‐SOCS1, together with shCON (−) or shUSP7, followed by treatment with dimethylsulfoxide or MG132 (10 μm) for 12 hr
USP7 removes polyubiquitination of SOCS1 protein
The above findings suggest that USP7 could regulate ubiquitination levels of SOCS1 as a deubiquitinase. To address this possibility, we analyzed SOCS1 protein ubiquitination in cells. The result showed that overexpression of USP7 significantly reduced polyubiquitination of Myc‐SOCS1 (Fig. 3a), which was dependent on the dosage of USP7 (Fig. 3b). Furthermore, we determined the effect of USP7 on endogenous SOCS1 ubiquitination. Consistent with USP7‐mediated regulation of exogenously expressed Myc‐SOCS1, overexpression of USP7 dramatically inhibited ubiquitination of endogenous SOCS1 (Fig. 3c), suggesting that USP7 could deubiquitinate SOCS1 in cells. Conversely, when endogenous USP7 in cells was knocked down, ubiquitination levels of Myc‐SOCS1 significantly increased (Fig. 3d). Additionally, as compared with wild‐type USP7, the deubiquitinase inactive USP7‐C223S (CS) cannot deubiquitinate Myc‐SOCS1 (Fig. 3e), confirming the role of USP7 in regulating SOCS1 ubiquitination as a deubiquitinase. Hence, we concluded that USP7 deubiquitinates SOCS1 protein in cells.
Figure 3.
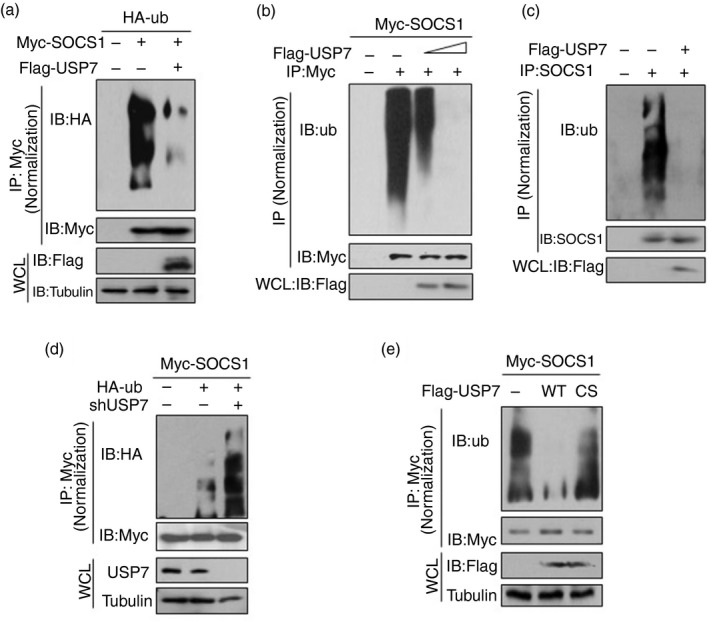
USP7 removes polyubiquitination of SOCS1 dependently on its deubiquitinase activity. (a) Immunoprecipitation analysis of polyubiquitination of Myc‐SOCS1 in HEK293T cells co‐transfected with Myc‐SOCS1, HA‐ub and Flag‐USP7 as indicated. (b) Immunoprecipitation analysis of polyubiquitination of Myc‐SOCS1 in A549 cells co‐transfected with Myc‐SOCS1 and increasing amounts of Flag‐USP7. (c) Immunoprecipitation analysis of polyubiquitination of endogenous SOCS1 in HEK293T cells transfected with empty vectors (−) or Flag‐USP7. (d) Immunoprecipitation analysis of polyubiquitination of SOCS1 in HEK293T cells co‐transfected with Myc‐SOCS1, HA‐ub and shUSP7 as indicated. (e) Immunoprecipitation analysis of polyubiquitination of SOCS1 in HEK293T cells co‐transfected with Myc‐SOCS1 and Flag‐USP7 (WT or inactive C223S mutant) as indicated
USP7 regulates IFN‐I‐induced JAK‐STAT1 activation through SOCS1
SOCS1 is an important negative regulator of IFN‐I signaling. It has been demonstrated that the inhibitory effect of SOCS1 on IFN signaling is through inhibition of tyrosine phosphorylation of JAK family proteins,18 leading to inhibition of STAT1 activation. Hence, we further analyzed whether USP7 regulates JAK–STAT1 phosphorylation and activation. We found that knockdown of USP7 by all three shUSP7 constructs up‐regulated IFN‐α‐induced STAT1 phosphorylation at tyrosine 701 site, which is a marker of STAT1 activation (Fig. 4a). Next, we observed that USP7 knockdown enhanced IFN‐α‐induced STAT1 phosphorylation in a time‐dependent manner (Fig. 4b). Conversely, overexpression of USP7 obviously lowered the levels of STAT1 phosphorylation under IFN‐α treatment in a time‐dependent manner (Fig. 4c), as well as in a dose‐dependent manner (Fig. 4d). Moreover, USP7‐mediated inhibition of STAT1 activation was dependent on the deubiquitinase activity of USP7, because the USP7‐C223C inactive mutant lost the ability to restrict STAT1 phosphorylation (Fig. 4e). Furthermore, we determined IFN‐induced tyrosine phosphorylation and activation of JAK family members, including JAK1 and Tyk2. We found that overexpression of USP7 robustly inhibited tyrosine phosphorylation of both Tyk2 and JAK1 (Fig. 4f). To further address the importance of SOCS1 in USP7‐mediated regulation of IFN‐induced JAK–STAT1 signaling, two short hairpin RNAs against human SOCS1 were used to establish stable cell lines with SOCS1 knockdown (Fig. 4g). We found that USP7‐mediated inhibition of STAT1 activation in IFN signaling was largely abolished in stable cells with SOCS1 knockdown (Fig. 4h). Collectively, these findings demonstrated that USP7 can regulate IFN‐I‐induced JAK–STAT1 signaling through SOCS1.
Figure 4.
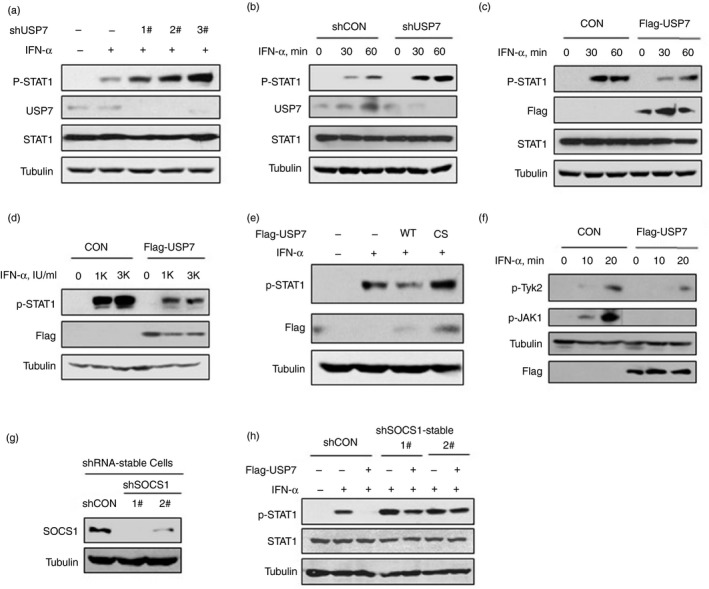
USP7 regulates interferon type I (IFN‐I) ‐induced Janus kinase–signal transducer and activator of transcription (JAK–STAT1) activation through SOCS1. (a) Western blot analysis of STAT1 phosphorylation on tyrosine 701 (p‐STAT1) in HEK293T cells transfected with control shRNA (−) or three shUSP7 and then treated with IFN‐α (1000 IU/ml) for 30 min. (b) Western blot analysis of p‐STAT1 in HEK293T cells transfected with shCON or shUSP7 and then treated with IFN‐α (1000 IU/ml) as indicated. (c) Western blot analysis of p‐STAT1 in HEK293T cells transfected with empty vectors (CON) or Flag‐USP7 and then treated with IFN‐α (1000 IU/ml) as indicated. (d) Western blot analysis of p‐STAT1 in HEK293T cells transfected with Flag‐USP7 and then treated with IFN‐α (0, 1000, 3000 IU/ml) for 30 min. (e) Western blot analysis of p‐STAT1 in HEK293T cells transfected with empty vectors (−) or Flag‐USP7 (WT or inactive C223S mutant) and then treated with IFN‐α (1000 IU/ml) as indicated. (f) Western blot analysis of phosphorylated Tyk2 on tyrosine 1054/1055 (p‐Tyk2) and phosphorylated JAK1 on serine 1022/1023 (p‐JAK1) in HEK293T cells transfected with Flag‐USP7 and then treated with IFN‐α (1000 IU/ml) as indicated. (g) Western blot analysis of endogenous SOCS1 protein levels in stable SOCS1‐knockdown HeLa cells (shSOCS1: 1# and 2#) or stable control‐shRNA HeLa cells (shCON). (h) Western blot analysis of p‐STAT1 levels in stable SOCS1‐knockdown HeLa cells transfected with Flag‐USP7 and then treated with IFN‐α (1000 IU/ml) for 30 min
USP7 regulates the strength of IFN‐I signaling
JAK–STAT1 activation largely determines the strength of IFN antiviral signaling, so we further analyzed the effect of USP7 on the IFN‐I signaling pathway. ISRE is commonly used to effectively evaluate the promoter activities of ISGs. We found that knockdown of USP7 by all three shUSP7 constructs obviously enhanced IFN‐I‐induced ISRE activity (Fig. 5a). Conversely, overexpression of USP7 significantly inhibited ISRE activity in IFN‐I signaling (Fig. 5b). Next, we analyzed the effect of USP7 deubiquitinase activity on ISRE promoter activity. The result showed that USP7‐WT, but not USP7‐CS, inhibited IFN‐α‐induced ISRE activity (Fig. 5c). These findings suggest that USP7 restricts the promoter activity of ISGs in IFN‐I signaling in a deubiquitinase activity‐dependent manner. To further assess the strength of IFN signaling, we next analyzed IFN‐I‐induced transcriptional expression of ISGs. We found that USP7 knockdown remarkably up‐regulated mRNA levels of several representative ISGs, including IFIT1, ISG15 and ISG54, induced by IFN‐α (Fig. 5d). Overexpression of USP7 significantly inhibited IFN‐I‐induced transcriptional expression of these representative ISGs (Fig. 5e). Moreover, the deubiquitinase inactive USP7‐CS lost the ability to inhibit ISG expression (Fig. 5e). Together, these findings demonstrated that USP7 is a negative regulator of IFN‐I signaling pathway.
Figure 5.
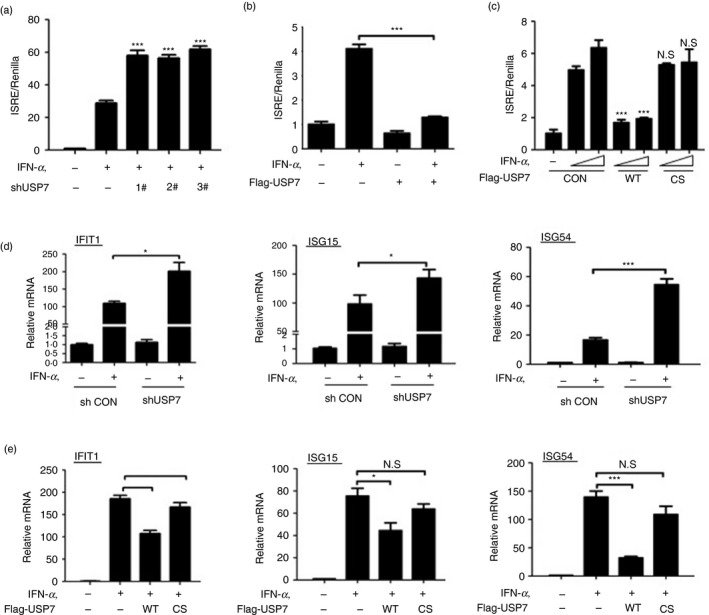
USP7 regulates the strength of interferon type I (IFN‐I) signaling. (a) The ISRE‐luciferase activity was measured in HEK293T cells transfected with control shRNA (−) or three shUSP7, together with ISRE‐luciferase and Renilla, and then treated with IFN‐α (500 IU/ml) for 18 hr. (b) The ISRE‐luciferase activity was measured in HEK293T cells transfected with Flag‐USP7, together with ISRE‐luciferase and Renilla, and then treated with IFN‐α (500 IU/ml) for 18 hr. (c) The ISRE‐luciferase activity was measured in HEK293T cells transfected with Flag‐USP7 (WT or inactive C223S mutant), together with ISRE‐luciferase and Renilla, and then treated with IFN‐α (200, 500 IU/ml) for 18 hr. (d) Quantitative‐PCR analysis of IFIT1, ISG15 and ISG54 mRNA levels in HEK293T cells transfected with shCON or shUSP7 and then treated with IFN‐α (1000 IU/ml) for 8 hr. (e) Quantitative‐PCR analysis of IFIT1, ISG15 and ISG54 mRNA levels in HEK293T cells transfected with Flag‐USP7 (WT or inactive C223S mutant) and then treated with IFN‐α (1000 IU/ml) for 8 hr. NS, not significant, *P < 0·05, ***P < 0·001 (two‐tailed unpaired Student's t‐test). All data are shown as mean ± SD of three independent replicates
USP7 regulates IFN‐I‐mediated antiviral activity
To explore the role of USP7 in IFN‐I antiviral function, we used VSV and H1N1, which have been widely used as sensitive viral models, to assess IFN‐I antiviral activity. Using immunofluorescence analysis, we observed that IFN‐α treatment efficiently inhibited infection by a VSV‐GFP virus, which is a VSV virus with a GFP gene (Fig. 6a), whereas knockdown of USP7 significantly promoted IFN‐I‐mediated antiviral activity (Fig. 6a). Consistent with the antiviral activity, knockdown of USP7 markedly up‐regulated ISG expression during viral infection (Fig. 6b). Furthermore, we found that overexpression of USP7 remarkably restricted IFN‐α‐mediated antiviral activity, as shown by increased levels of VSV viral RNA (Fig. 6c). Consistently, when H1N1 viruses were employed, we found that USP7 overexpression promoted H1N1 viral infection in a dose‐dependent manner (Fig. 6d). Importantly, the deubiquitinase activity of USP7 was required for USP7‐mediated inhibition of cellular antiviral activity, as the inactive USP7‐CS cannot promote viral infection (Fig. 6e). Hence, we demonstrated that USP7 inhibits IFN‐I‐mediated antiviral activity.
Figure 6.
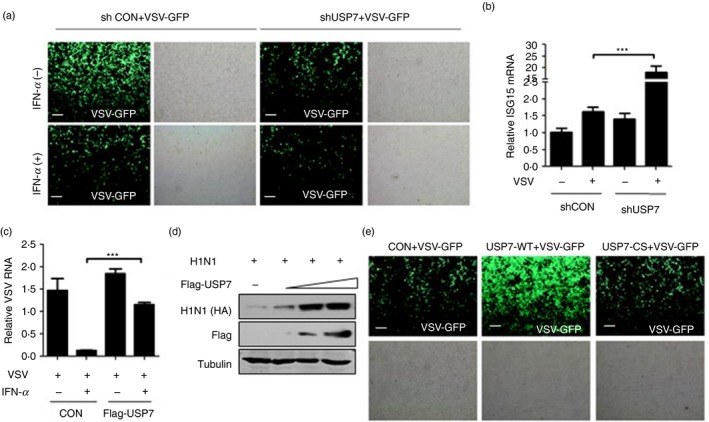
USP7 regulates interferon type I (IFN‐I) ‐mediated antiviral activity. (a) Fluorescence microscopy of vesicular stomatitis virus (VSV) with a green fluorescent protein gene (GFP) in HEK293T cells transfected with shCON or shUSP7 and then pretreated with or with IFN‐α (50 IU/ml) for 20 hr, followed by infection with VSV‐GFP [multiplicity of infection (MOI) = 0·5] for 20 hr. Scale bars, 100 µm. (b) Quantitative PCR analysis of mRNA levels of a representative ISG (ISG15) in HEK293T cells transfected with shCON or shUSP7 and then infected with VSV (MOI = 1·0) for 20 hr. (c) Quantitative PCR analysis of VSV viral RNA in HEK293T cells transfected with empty vectors (CON) or Flag‐USP7 and then pretreated with or without IFN‐α (50 IU/ml) for 20 hr, followed by infection with VSV (MOI = 1·0) for 20 hr. (d) Western blot analysis of H1N1‐encoded HA protein levels in HEK293T cells transfected with increasing amounts of Flag‐USP7 and then infected with H1N1 (MOI = 1·0) for 20 hr. (e) Fluorescence microscopy of VSV‐GFP viruses in HEK293T cells transfected with Flag‐USP7 (WT or inactive C223S mutant) and then infected with VSV‐GFP (MOI = 0·5) for 20 hr. Scale bars, 100 µm. ***P < 0·001 (two‐tailed unpaired Student's t‐test). Data are shown as mean ± SD of three independent replicates (b,c)
USP7 small‐molecule inhibitors enhance IFN‐I signaling and antiviral efficacy
Given that the deubiquitinase activity of USP7 is necessary for the inhibition of IFN‐I antiviral activity, we sought to develop possible strategies to enhance IFN‐I antiviral therapeutic efficacy. Recently, two potent inhibitors of USP7 deubiquitinase activity, P5091 and P22077, have been reported. We first observed the effects of these two inhibitors on SOCS1 ubiquitination and protein levels. We found that both P5091 and P22077 significantly up‐regulated ubiquitination levels of Myc‐SOCS1 (Fig. 7a). Consistently, two USP7 inhibitors markedly down‐regulated protein levels of endogenous SOCS1 (Fig. 7b), which depended on proteasome‐dependent degradation because MG132 abolished Myc‐SOCS1 protein down‐regulation mediated by USP7 inhibitors (Fig. 7c). Furthermore, we analyzed the effects of two USP7 inhibitors on STAT1 activation in IFN‐I signaling. The results showed that both P22077 and P5091 promoted IFN‐I‐induced activation of STAT1 (Fig. 7d). Next, when cells were pretreated with either P5091 or P22077, IFN‐I‐induced ISRE promoter activities were obviously up‐regulated by these two inhibitors (Fig. 7e). Consistently, USP7 inhibitors remarkably enhanced expression of ISGs, including IFIT1, ISG15 and ISG54, in IFN‐I signaling (Fig. 7f). These findings suggest that USP7 inhibitors promote activation of IFN‐I signaling.
Figure 7.
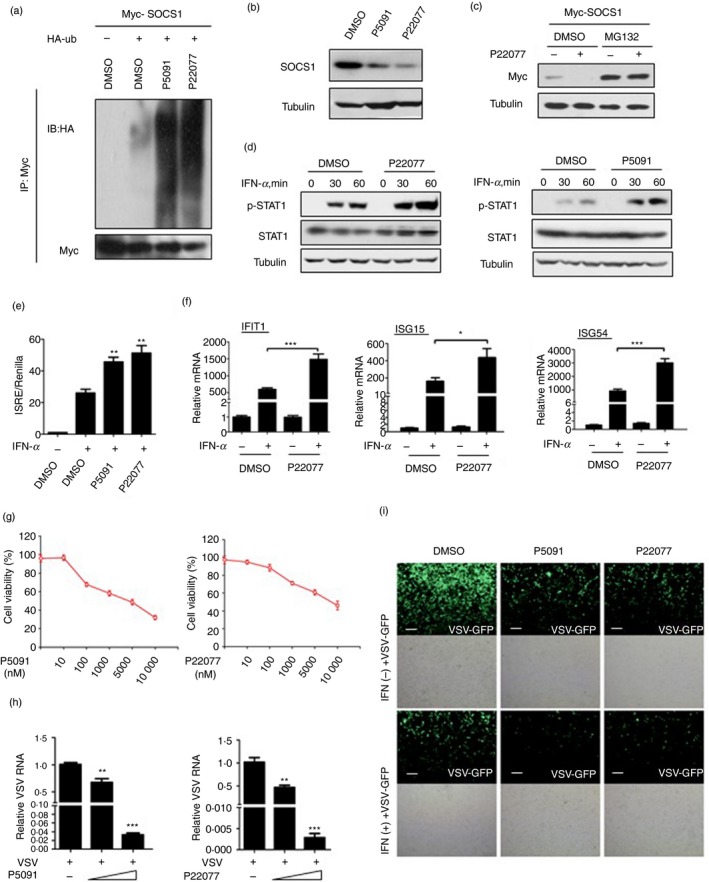
USP7 Small‐molecule inhibitors enhance interferon type I (IFN‐I) signaling and antiviral efficacy. (a) Immunoprecipitation analysis of polyubiquitination of SOCS1 in HEK293T cells co‐transfected with Myc‐SOCS1 and HA‐ub and then treated with either P5091 (100 nm) or P22077 (100 nm) for 12 hr. (b) Western blot analysis of endogenous SOCS1 protein levels in A549 cells treated with P5091 (100 nm) or P22077 (100 nm) for 12 hr. (c) Western blot analysis of Myc‐SOCS1 protein levels in HEK293T cells transfected with Myc‐SOCS1 and then pretreated with MG132 (10 μm) for 12 hr, followed by treatment with P22077 (100 nm) for 12 hr. (d) Western blot analysis of p‐STAT1 in HEK293T cells pretreated with P22077 or P5091 (100 nm) for 12 hr and then treated with IFN‐α (1000 IU/ml) as indicated. (e) The ISRE‐luciferase activity was measured in HEK293T cells co‐transfected with ISRE‐luciferase and Renilla and then treated with P5091 (100 nm) or P22077 (100 nm) for 12 hr, followed by treatment with IFN‐α (500 IU/ml) for 18 hr. (f) Quantitative PCR analysis of mRNA levels of IFIT1, ISG15 and ISG54 in HEK293T cells pretreated with dimethylsulfoxide or P22077 (100 nm) for 12 hr, and then treated with or without IFN‐α (1000 IU/ml) for 6 hr. (g) Cell viability was measured in HEK293T cells treated with either P5091 or P22077 using different concentrations as indicated. (h) Quantitative PCR analysis of vesicular stomatitis virus (VSV) viral RNA in HEK293T cells pretreated with P5091 or P22077 (20 and 100 nm) for 12 hr and then challenged with VSV [multiplicity of infection (MOI) = 1·0] for 24 hr. (i) Fluorescence microscopy of VSV‐GFP viruses in HEK293T cells pretreated with DMSO, P5091 and P22077 (100 nm) for 12 hr and then treatment with or without IFN‐α (50 IU/ml) for 20 hr, followed by infection with VSV‐GFP (MOI = 0·5) for 24 hr. Scale bars, 100 µm. *P < 0·05, **P < 0·01 and ***P < 0·001 (two‐tailed unpaired Student's t‐test). Data are shown as mean ± SD. of three independent replicates (e,f,h)
To further analyze the effects of USP7 inhibitors on IFN‐I antiviral activity in cells, we first analyzed the cytotoxicity effect of these two inhibitors on HEK293T cells. The results showed that the 50% of cellular viability (CC50) value of P5091 in HEK293T cells was 5·0 μm and the CC50 value of P22077 was 10·9 μm (Fig. 7g). Next, we pretreated cells with increasing amounts (20 nm and 100 nm) of either P5091 or P22077 and then infected with VSV. We found that two USP7 inhibitors robustly inhibited viral infection in a dose‐dependent manner (Fig. 7h). Moreover, IFN‐I‐mediated antiviral efficacy was significantly enhanced by two USP7 inhibitors (Fig. 7i). Taken all together, these findings clearly demonstrated that two USP7 inhibitors, P5091 and P22077, up‐regulate SOCS1 protein levels by deubiquitination effects, promote activation of IFN‐I signaling, and improve IFN‐mediated antiviral activity.
Discussion
Recent studies have demonstrated that USP7 is essential for regulating diverse signaling pathways related to human diseases. In particular, USP7 plays critical roles in tumorigenesis and tumor aggressiveness. It has been reported that USP7 can regulate the P53–MDM2 complex9 and also deubiquitinates PTEN to induce tumor progression.10 However, whether or how USP7 plays roles in host antiviral innate immunity remains largely unexplored. Here, we reported a novel biological function of USP7, uncovering that USP7 is an important negative regulator of IFN‐I antiviral activity during viral infection.
We clearly demonstrated that USP7 physically interacts with SOCS1 and enhances SOCS1 protein stability by deubiquitinating SOCS1. It is well known that SOCS1 is a negative regulator of IFN‐induced JAK–STAT signaling. Consistently, we found that USP7 can inhibit JAK–STAT1 signaling activation and restrict IFN‐I‐induced ISG expression. Importantly, USP7‐mediated inhibition of JAK–STAT1 signaling is dependent on SOCS1, suggesting that USP7 affects the IFN signaling pathway through SOCS1. We further demonstrated that USP7 significantly inhibits IFN‐I antiviral activity. Hence, our findings revealed a negative regulatory mechanism of IFN‐I antiviral signaling mediated by the USP7–SOCS1 axis.
An interesting observation is that viral infection promotes USP7 protein expression. Given that we have demonstrated that USP7 is an important inhibitor of IFN‐I signaling, we believe that virus‐induced up‐regulation of USP7 inhibits the host IFN‐I response and promotes viral infection. We also found that USP7 is responsible for SOCS1 protein up‐regulation at the late stage of viral infection, which suggests that viruses use host deubiquitinase USP7 to improve cellular SOCS1 protein levels for efficient viral evasion. Hence, this finding suggests a novel viral evasion mechanism exploited by viruses.
Interferon‐I plays important roles in host defense against viral infection through up‐regulation of hundreds of antiviral ISGs. Given that we demonstrated that USP7 is an important negative regulator of IFN‐I signaling and that the deubiquitinase activity of USP7 is required for USP7‐mediated inhibition of IFN‐I signaling and antiviral activity, we speculated that inhibition of USP7 could be a potential strategy for enhancing IFN‐I antiviral activity. In line with our speculation, we found that two USP7 inhibitors, P5091 and P22077, up‐regulated SOCS1 ubiquitination, inhibited SOCS1 protein expression, promoted IFN‐I signaling and enhanced IFN‐I antiviral efficacy. Hence, our findings revealed that USP7 inhibitors could be effective enhancement agents for improving IFN‐I antiviral therapeutic efficacy.
In summary, we identified USP7 as an important negative regulator of the IFN‐I signaling pathway and antiviral activity, revealed the underlying mechanisms, and demonstrated that USP7 inhibitors enhance IFN‐I antiviral efficacy. These findings uncovered a novel biological function of USP7 and provided evidence for USP7 inhibitors as potential enhancement agents for improving IFN‐based antiviral therapy.
Author contributions
HZ and HL conceived and designed the projects. YY, YM and CZ performed most of the experiments and data analysis. JL, XC, LQ and XW contributed to cellular experiments and plasmid purification. QF, ZY, JW, GQ and QF provided technical support and literature retrieval. HZ and YY wrote the manuscript.
Disclosures
The authors declare no competing interests.
Acknowledgements
We thank Prof. Serge Y. Fuchs for plasmids and thank Prof. Chen Wang, Prof. Chunsheng Dong, Prof. Jianfeng Dai for important viruses. We also thank the laboratory members for helpful discussions and suggestions. This work was supported by the National Key R&D Program of China (2018YFC1705500): 2018YFC1705505, the National Natural Science Foundation of China (81870365, 31770177, 31970846, and 81971477), the Key Project of University Natural Science Foundation of Jiangsu Province (18KJA180010) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Contributor Information
Haitao Lv, Email: haitaosz@163.com.
Hui Zheng, Email: huizheng@suda.edu.cn.
References
- 1. D'Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M et al Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med 2011; 17:1636–40. [DOI] [PubMed] [Google Scholar]
- 2. Pal A, Young MA, Donato NJ. Emerging potential of therapeutic targeting of ubiquitin‐specific proteases in the treatment of cancer. Cancer Res 2014; 74:4955–66. [DOI] [PubMed] [Google Scholar]
- 3. Mattern MR, Wu J, Nicholson B. Ubiquitin‐based anticancer therapy: carpet bombing with proteasome inhibitors vs surgical strikes with E1, E2, E3, or DUB inhibitors. Biochim Biophys Acta 2012; 1823:2014–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lopez‐Castejon G, Edelmann MJ. Deubiquitinases: Novel Therapeutic Targets in Immune Surveillance? Mediators Inflamm 2016; 2016:3481371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu J, Kumar S, Wang F, Wang H, Chen L, Arsenault P et al Chemical Approaches to Intervening in Ubiquitin Specific Protease 7 (USP7) Function for Oncology and Immune Oncology Therapies. J Med Chem 2018; 61:422–43. [DOI] [PubMed] [Google Scholar]
- 6. Wang Q, Ma S, Song N, Li X, Liu L, Yang S et al Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J Clin Invest 2016; 126:2205–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lecona E, Narendra V, Reinberg D. USP7 cooperates with SCML2 to regulate the activity of PRC1. Mol Cell Biol 2015; 35:1157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alonso‐de Vega I, Martin Y, Smits VA. USP7 controls Chk1 protein stability by direct deubiquitination. Cell Cycle 2014; 13:3921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mungamuri SK, Qiao RF, Yao S, Manfredi JJ, Gu W, Aaronson SA. USP7 Enforces Heterochromatinization of p53 Target Promoters by Protecting SUV39H1 from MDM2‐Mediated Degradation. Cell Rep 2016; 14:2528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Song MS, Salmena L, Carracedo A, Egia A, Lo‐Coco F, Teruya‐Feldstein J et al The deubiquitinylation and localization of PTEN are regulated by a HAUSP‐PML network. Nature 2008; 455:813–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R et al A small molecule inhibitor of ubiquitin‐specific protease‐7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012; 22:345–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gavory G, O'Dowd CR, Helm MD, Flasz J, Arkoudis E, Dossang A et al Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat Chem Biol 2018; 14:118–25. [DOI] [PubMed] [Google Scholar]
- 13. Turnbull AP, Ioannidis S, Krajewski WW, Pinto‐Fernandez A, Heride C, Martin ACL et al Molecular basis of USP7 inhibition by selective small‐molecule inhibitors. Nature 2017; 550:481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Daubeuf S, Singh D, Tan Y, Liu H, Federoff HJ, Bowers WJ et al HSV ICP0 recruits USP7 to modulate TLR‐mediated innate response. Blood 2009; 113:3264–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jager W, Santag S, Weidner‐Glunde M, Gellermann E, Kati S, Pietrek M et al The ubiquitin‐specific protease USP7 modulates the replication of Kaposi's sarcoma‐associated herpesvirus latent episomal DNA. J Virol 2012; 86:6745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ren Y, Zhao P, Liu J, Yuan Y, Cheng Q, Zuo Y et al Deubiquitinase USP2a Sustains Interferons Antiviral Activity by Restricting Ubiquitination of Activated STAT1 in the Nucleus. PLoS Pathog 2016; 12:e1005764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X, Zhang L, Zhang Y, Zhao P, Qian L, Yuan Y et al JOSD1 Negatively Regulates Type‐I Interferon Antiviral Activity by Deubiquitinating and Stabilizing SOCS1. Viral Immunol 2017; 30:342–9. [DOI] [PubMed] [Google Scholar]
- 18. Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K et al A new protein containing an SH2 domain that inhibits JAK kinases. Nature 1997; 387:921–4. [DOI] [PubMed] [Google Scholar]