

Summary
Genome‐wide association studies (GWAS) have identified many loci for systemic lupus erythematosus (SLE). However, identification of functionally relevant genes remains a challenge. The aim of this study was to highlight potential causal genes for SLE in the GWAS loci. By applying Mendelian randomization (MR) methods, such as summary data‐based MR (SMR), generalized SMR and MR pleiotropy residual sum and outlier, we identified DNA methylations in 15 loci and mRNA expression of 21 genes that were causally associated with SLE. The identified genes enriched in 14 specific KEGG pathways (e.g. SLE, viral carcinogenesis) and two GO terms (interferon‐γ‐mediated signaling pathway and innate immune response). Among the identified genes, UBE2L3 and BLK variants were significantly associated with UBE2L3 and BLK methylations and gene expressions, respectively. UBE2L3 was up‐regulated in SLE patients in several types of immune cells. Methylations (e.g. cg06850285) and mRNA expression of UBE2L3 were causally associated with SLE. Methylation site cg09528494 and mRNA expression of BLK were causally associated with SLE. BLK single nucleotide polymorphisms that were significantly associated with SLE were strongly associated with plasma cathepsin B level. Deep analysis identified that plasma cathepsin B level was causally associated with SLE. In summary, this study identified hundreds of DNA methylations and genes as potential risk factors for SLE. Genetic variants in UBE2L3 gene might affect SLE by influencing gene expression. Genetic variants in BLK gene might affect SLE by influencing BLK gene expression and plasma cathepsin B protein level.
Keywords: cathepsin B, genome‐wide association study, Mendelian randomization, methylation, systemic lupus erythematosus
Expressions of 21 genes and 152 methylations were causally associated with systemic lupus erythematosus (SLE). UBE2L3 and BLK variants were significantly associated with UBE2L3 and BLK methylations and gene expressions, respectively. Plasma cathepsin B level may be causally associated with SLE.

Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease in which the risk of disease is influenced by complex genetic and environmental contributions. During the past 30 years, many genetic association studies have been performed to identify genetic factors contributing to a susceptibility to SLE. Since 2008, genome‐wide association studies (GWAS) have been applied and represent a step forward in the evaluation of gene abnormalities in SLE. A variety of non‐HLA genes have emerged as possible candidates of genetic susceptibility to SLE. Among these genes, STAT4, IRF5, BLK and ITGAM were the first findings in GWAS1 and have been proven repeatedly.2, 3, 4, 5 As the sample size of GWAS has increased, more and more SLE susceptibility genes have been identified. Until now, a total of 794 genetic associations have been found for SLE by 33 GWAS according to the GWAS catalog (https://www.ebi.ac.uk/gwas/).
The GWAS exploits the linkage disequilibrium correlation structure of the genome so the identified associations always point to genomic regions that harbor many genes.6 A serious problem that arises is which gene or how many genes were functionally relevant to SLE. It is nearly impossible to prioritize among these genes to identify the most functionally relevant genes using GWAS data alone. Integration of GWAS data with data from DNA methylation and gene expression GWAS, which allowed identification of methylation and gene expression quantitative trait loci (meQTL and eQTL), respectively, is suggested to be a possible way to prioritize relevant genes in the GWAS‐identified regions.7, 8, 9
Mendelian randomization (MR) is a commonly used approach that can infer causality of an exposure for a complex disease outcome.10, 11 Genetic variants (e.g. QTLs) that are significantly associated with the exposure (e.g. gene expression and DNA methylation level) are used as genetic instruments to test for a causal effect on the outcome (e.g. SLE). If the exposure is causal, then variants affecting the exposure should affect the outcome proportionally. MR presents a number of advantages over observational epidemiology, including the ability to control for environmental confounders. Initial applications of MR used a single genetic variant as instrument and assessed the causal relationship of the modifiable intermediate phenotype on the outcome in a single sample. Recent two‐sample approaches based on GWAS summary‐level data used multiple genetic instruments to evaluate the impact of an exposure without necessitating the measurement of that exposure in the outcome group.12 These newly developed methods are much more efficient and powerful than the initial single instrument method.
Zhu et al.9 proposed a method called summary data‐based MR (SMR) that can integrate independent GWAS summary statistics data with QTL data to identify functionally relevant genes at the loci identified in GWAS. By applying this method, Zhu et al. have successfully identified novel trait‐associated genes for 33 complex traits.9, 13 However, this method has never been applied to SLE. In this study, we applied the MR methods on data from SLE GWAS,4 meQTL,14 eQTL15, 16, 17 and plasma protein level QTL (pQTL) studies18 to explore potential causal factors for SLE.
Materials and methods
Overview of the study design
This study was designed to identify potential causal factors (e.g. DNA methylations, gene expressions and plasma protein levels) for SLE. First, we conducted SMR analysis to identify DNA methylations that were causally associated with SLE by integrative analysis of data from one SLE GWAS4 and one meQTL study.14 Second, we conducted SMR analysis to identify gene expressions that were causally associated with SLE by integrative analysis of data from one SLE GWAS4 and three eQTL studies.15, 16, 17 Meanwhile, we tested the association between expression levels of the identified genes and SLE based on expression profile data from four studies available in the GEO database (http://www.ncbi.nlm.nih.gov/geo). Third, we looked for pQTLs within the identified causal genes using data from a large‐scale pQTL study.18 Finally, for proteins that were strongly affected by SNPs within the identified causal genes, we tested if the protein levels were causally associated with SLE using MR methods such as generalized SMR (GSMR) and MR pleiotropy residual sum and outlier (MR‐PRESSO).
GWAS data set
The data set used in this study was from the SLE GWAS conducted by Bentham et al.,4 which comprised 7219 cases and 15 991 controls of European ancestry. It is the largest SLE GWAS of European ancestry so far. This data set was publically available in the ImmunoBase (http://www.immunobase.org/), a web‐based resource focused on the genetics and genomics of immunologically related human diseases. The summary data included the rs number, chromosome, position, allele information, odds ratio and corresponding confidence interval, and P‐values for nearly 8 million single nucleotide polymorphisms (SNPs). The requisite data (i.e. SNP rs number, allele 1, allele 2, frequency of allele 1, β, standard error, P‐value and sample size) were extracted from this GWAS data set and were organized to the specific format (the.ma file) for the SMR analysis softwares using the R language (https://www.r-project.org).
SMR analysis
We conducted an SMR analysis to identified DNA methylations and genes that were causally associated with SLE, by which we could prioritize functionally relevant genes in the GWAS loci. SMR applies the principles of MR to integrate summary‐level data from independent GWAS with data from eQTL studies to test for association between gene expression and a trait due to a shared variant at a locus. The SMR method implements a transcriptome‐wide association analysis in a formal statistical framework using summary data so that the statistical power is increased by using the GWAS and QTL data of very large sample size.
The meQTL summary data used in our analysis were from the study conducted by McRae et al.,14 which measured meQTL in the Brisbane Systems Genetics Study (n = 614) and the Lothian Birth Cohorts (n = 1366). The eQTL summary data from three studies were used in our SMR analysis. The first is the study conducted by Westra et al.,17 which is the largest eQTL meta‐analysis so far in peripheral blood samples of 5311 European healthy individuals. The second is the genetic architecture of gene expression study, which detected eQTLs in peripheral blood in 2765 European individuals.16 The third data set contains the cis‐eQTL summary data from the GTEx project (which only used data from whole blood).15
We ran SMR (version 0.712) with default parameters in a user‐friendly software tool according to the user manual (http://cnsgenomics.com/software/smr/). Genotype data of hapmap r23 CEU were used as a reference panel to calculate the linkage disequilibrium correlation for SMR analysis. All of the QTL summary data in SMR binary format were downloaded from http://cnsgenomics.com/software/smr/#DataResource, and all of the QTL data obtained were directly used in the analyses. The genome‐wide significance level for the SMR test is defined as 5·0 × 10−6. For probes with P SMR < 5·0 × 10−6, the heterogeneity in dependent instruments (HEIDI) test for heterogeneity in the resulting association statistics was conducted to test whether there is a single causal variant affecting SLE, methylation and gene expression. The HEIDI test is the procedure to test for pleiotropy, the fundamental assumption of MR analysis. Those probes with little evidence of heterogeneity (P HEIDI ≥ 0·05) were retained. The SMR locus plots for significant probes were generated by using the R code provided by Zhu et al.9 (http://cnsgenomics.com/software/smr/#SMRlocusplot19). The gene range list glist‐hg19 was used for plotting.
Differential expression analysis
We further found out if the expression levels of genes identified by SMR analysis were associated with SLE based on expression profile data from four studies. GSE10325 contained data of gene expression levels in CD19+ B cells (14 SLE cases and nine controls), CD33+ myeloid cells (11 SLE cases and 10 controls) and CD4+ T cells (13 SLE cases and nine controls).19 GSE13887 contained data of gene expression levels in CD4+ T cells from 10 SLE cases and nine controls.20 GSE30153 contained data of gene expression levels in B lymphocyte from 17 SLE cases and nine controls.21 GSE51997 contained data of gene expression levels in CD4+ T cells (six SLE cases and four controls), CD16− monocytes (four SLE cases and four controls) and CD16+ monocytes (four SLE cases and three controls).22 Differential expression was tested by comparing mean gene expression signals between cases and controls using t‐test. The significance level of P = 0·05 was used for the differential expression analysis.
GSMR analysis
To obtain additional supporting evidence, we took the advantage of the recently developed gsmr R‐package, which implements the GSMR method to test for a putative causal association between plasma protein levels and SLE.23 The summary GWAS data were from the SLE GWAS conducted by Bentham et al.,4 and the summary data of association between SNPs and plasma protein levels were obtained from a pQTL GWAS,18 which measured 509 946 SNPs for genome‐wide associations with 1124 protein levels measured in 1000 blood samples from the KORA study. The requisite data (i.e. SNP rs number, allele 1, allele 2, frequency of allele 1, β, standard error, P‐value and sample size) were extracted from each of the SLE GWAS and pQTL data sets and then organized (merged) to the specific format (a plain file) for the GSMR analysis softwares by using the R language. SNPs with P‐values < 5·0 × 10−6 in the pQTL study were used. Genotype data of hapmap r23 CEU were used to calculate the linkage disequilibrium correlation to construct the linkage disequilibrium correlation matrix for GSMR analysis. The HEIDI test is the procedure to test for pleiotropy. The parameters were left at the default setting in this analysis.
MR‐PRESSO analysis
We also applied the MR‐PRESSO method to test for causal association between plasma protein levels and SLE followed by retesting for heterogeneity.24 This test identifies possible bias from horizontal pleiotropy. In the case of evidence of horizontal pleiotropy, the test compares the expected and observed distributions of individual variants to identify outlier variants. We used an implementation of MR‐PRESSO in R (https://github.com/rondolab/MR-PRESSO) to detect horizontal pleiotropy, which is the MR‐PRESSO global test; the outlier‐corrected causal estimate; and the MR‐PRESSO distortion, which estimates whether the causal estimate is significantly different (at P < 0·05) after adjustment for outliers. The outlier test in MR‐PRESSO is the procedure to test for the assumption of no pleiotropy. Data used in the MR‐PRESSO analysis were the same as the GSMR analysis. The requisite data (i.e. SNP rs number, β, standard error and P‐value) were extracted from each of the SLE GWAS and pQTL data sets and then organized (merged) to the specific format (a plain file) for the MR‐PRESSO analysis by using the R language. SNPs with P‐value < 5·0 × 10−6 in the pQTL study were used. The default values were used for all of the MR‐PRESSO analysis parameters.
Results
SLE‐associated methylation sites and genes
We first performed an SMR analysis to determine if DNA methylations were causally associated with SLE. By integrating data from a large‐scale SLE GWAS and meQTL study, we found 152 methylation sites in 15 loci (73 genes), which were associated with SLE (P SMR < 5 × 10–6, P HEIDI > 0·05) (Table 1). There are CpG islands in all of the loci except 2q21.2 (MGAT5) (Table 1; see Supplementary material, Table S1). The detail information about these 152 SLE‐associated methylation sites was presented in the Supplementary material (Table S1. P HEIDI > 0·05 indicates that there was no significant heterogeneity underlying the meQTL signals. Among the 15 loci, 10 have been reported in GWAS and five have never been reported.
Table 1.
Summary of the systemic lupus erythematosus‐associated methylations identified by Mendelian randomization analysis
| Loci | Genes | Reported genes | CpG sites 1 | CpG islands 2 |
|---|---|---|---|---|
| 2q21·2 | MGAT5 | No | 4 | 0 |
| 2q32·2 | NAB1 | Yes | 1 | 1 |
| 3p14·3 | PXK, PDHB | Yes | 2 | 2 |
| 4p16·1 | PPP2R2C | No | 1 | 1 |
| 4q21·21 | PRDM8 | No | 1 | 7 |
| 5q31·1 | TCF7 | Yes | 1 | 1 |
| 6p22·2 | BTN2A1, HIST1H2BJ, ZSCAN23 | Yes | 3 | 2 |
| 6p22·1 | ZNF391, HIST1H2BL, HIST1H2AI, HIST1H2BN, OR2B2, ZNF165, ZSCAN12L1, ZSCAN16, ZNF389, ZSCAN9, NKAPL, ZNF323, ZSCAN12, ZKSCAN3, PGBD1, HIST1H4K, TRIM27, ZSCAN31, OR12D3, GABBR1, MOG, ZFP57, HCG4, HLA‐A, TRIM31, HLA‐L | Yes | 66 | 22 |
| 6p21·3 | FLOT1, DDAH2, CLIC1, NEU1, SLC44A4, STK19, HLA‐DQB2, HLA‐DPB1, COL11A2, RXRB | Yes | 11 | 7 |
| 8p23·1 | BLK | Yes | 1 | 1 |
| 11p15·5 | HRAS, MIR210, PHRF1, IRF7, CDHR5, SCT, DRD4, EPS8L2 | Yes | 32 | 18 |
| 12q24·33 | SLC15A4 | Yes | 2 | 2 |
| 17p13·1 | NEURL4, ACAP1, KCTD11 | No | 16 | 5 |
| 19q13·2 | ITPKC | No | 1 | 1 |
| 22q11·21 | UBE2L3, YDJC, CCDC116 | Yes | 10 | 3 |
Number of methylation sites that were significantly associated with systemic lupus erythematosus in the gene regions.
Number of CpG islands in the gene regions according to UCSC genome browser.
We found that expressions of 21 genes in six loci were significantly associated with SLE (P SMR < 5 × 10–6), and there was no significant heterogeneity underlying the eQTL signals (P HEIDI > 0·05) (Table 2). After literature searching, we found that among these 21 genes, GLS and ACAP1 have not been reported to be associated with SLE. All of these 21 genes passed HEIDI tests (P HEIDI > 0·05), suggesting that there was no heterogeneity, and the expressions of these genes and SLE were affected by the same variant, suggesting that the same causal SNPs contributed to both SLE risk and gene expression. We compared mRNA expression signals in gene expression studies for the 21 genes and found that 15 of them were differentially expressed (P < 0·05) (Table 2). For the 73 genes identified in meQTL SMR analysis (Table 1), the eQTL SMR tests identified that ZSCAN16, GABBR1, NEU1, BLK, DRD4, ACAP1 and UBE2L3 were significantly associated with SLE.
Table 2.
The systemic lupus erythematosus‐associated mRNA expressions identified by Mendelian randomization analysis
| Gene | CHR | SMR analysis | eQTL study | DEG | |||
|---|---|---|---|---|---|---|---|
| β | SE | P SMR | P HEIDI | ||||
| GLS | 2 | 0·8039 | 0·1657 | 1·23E‐06 | 0·18 | Westra | Yes |
| ZSCAN16 | 6 | 1·6606 | 0·2708 | 8·71E‐10 | 0·91 | CAGE | Yes |
| GABBR1 | 6 | 1·1092 | 0·2226 | 6·26E‐07 | 0·90 | GTEx | Yes |
| RPP21 | 6 | −3·2586 | 0·6150 | 1·17E‐07 | 0·05 | CAGE | Yes |
| HCP5 | 6 | −2·6344 | 0·4136 | 1·90E‐10 | 0·38 | CAGE | Yes |
| C6orf48 | 6 | 2·0189 | 0·3789 | 9·88E‐08 | 0·73 | GTEx | Yes |
| NEU1 | 6 | −3·4882 | 0·6370 | 4·36E‐08 | 0·95 | CAGE | Yes |
| NELFE | 6 | 0·7827 | 0·1594 | 9·05E‐07 | 0·06 | CAGE | No |
| SKIV2L | 6 | −0·6047 | 0·0887 | 9·49E‐12 | 0·12 | GTEx | No |
| C4A | 6 | −0·9127 | 0·1106 | 1·54E‐16 | 0·07 | GTEx | Yes |
| C4B | 6 | 0·8036 | 0·0962 | 6·64E‐17 | 0·13 | GTEx | Yes |
| CYP21A2 | 6 | 1·2416 | 0·2254 | 3·61E‐08 | 0·67 | GTEx | No |
| RNF5 | 6 | 1·9483 | 0·2168 | 2·56E‐19 | 0·07 | CAGE | Yes |
| C8ORF13 | 8 | 0·4747 | 0·0520 | 7·50E‐20 | 0·23 | Westra | No |
| FAM167A | 8 | 0·3658 | 0·0481 | 2·84E‐14 | 0·21 | CAGE | Yes |
| FAM167A | 8 | 0·3369 | 0·0521 | 1·01E‐10 | 0·41 | GTEx | Yes |
| BLK | 8 | −0·9493 | 0·1602 | 3·13E‐09 | 0·35 | GTEx | Yes |
| BLK | 8 | −0·5048 | 0·0625 | 6·88E‐16 | 0·11 | Westra | Yes |
| BLK | 8 | −0·3890 | 0·0515 | 4·35E‐14 | 0·33 | CAGE | Yes |
| ANO9 | 11 | −0·7016 | 0·1274 | 3·62E‐08 | 0·07 | CAGE | No |
| IRF7 | 11 | 0·4601 | 0·0846 | 5·33E‐08 | 0·81 | CAGE | Yes |
| DRD4 | 11 | 1·0887 | 0·2296 | 2·12E‐06 | 0·12 | Westra | No |
| ACAP1 | 17 | −0·3421 | 0·0610 | 2·06E‐08 | 0·32 | CAGE | Yes |
| ACAP1 | 17 | −0·3222 | 0·0557 | 7·07E‐09 | 0·10 | Westra | Yes |
| UBE2L3 | 22 | 1·3527 | 0·2462 | 3·93E‐08 | 0·06 | GTEx | Yes |
| UBE2L3 | 22 | 0·2671 | 0·0326 | 2·62E‐16 | 0·08 | CAGE | Yes |
CHR, chromosome; DEG, differentially expressed genes; eQTL, expression quantitative trait locus; HEIDI, heterogeneity in dependent instruments; SMR, summary data‐based Mendelian randomization.
The identified genes were found to enrich in 14 specific KEGG pathways (including SLE and viral carcinogenesis) and two GO biological process terms (interferon‐γ‐mediated signaling pathway and innate immune response) (see Supplementary material, Table S2).
QTL analysis
According to the SMR analysis, SNPs in the identified genes are strongly associated with DNA methylation and gene expression levels. We found eQTLs that significantly associated with SLE (P < 5·0 × 10−8) for ZSCAN16, GABBR1, NEU1, BLK and UBE2L3 (see Supplementary material, Table S1). We further looked for pQTLs in ZSCAN16, GABBR1, NEU1, BLK, DRD4, ACAP1 and UBE2L3, because they showed evidence in both eQTL and meQTL SMR analysis. In the UCSC database, we obtained 5835 SNPs for these seven genes. Plasma protein level QTLs that significantly associated with SLE (P < 5·0 × 10−8) were found for BLK (Table 3) and UBE2L3 (see Supplementary material, Table S3).
Table 3.
Association between BLK single nucleotide polymorphisms and systemic lupus erythematosus and plasma cathepsin B level
| SNP | Position (GRCh37.p13) | Minor allele | Major allele | MAF 1 | SLE association | Cathepsin B level association | eQTL | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| β | SE | P | β | SE | P | ||||||
| rs2736345 | 11352485 | G | A | 0·3058 | 0·2311 | 0·0292 | 1·49E‐13 | ‐0·1316 | 0·0486 | 6·84E‐03 | – |
| rs2618476 | 11352541 | C | T | 0·2688 | 0·2390 | 0·0289 | 1·58E‐14 | ‐0·1674 | 0·0507 | 1·00E‐03 | – |
| rs998682 | 11353052 | A | G | 0·1566 | −0·1278 | 0·0423 | 1·20E‐03 | ‐0·1571 | 0·0589 | 7·83E‐03 | – |
| rs1478895 | 11353335 | G | C | 0·1566 | −0·1278 | 0·0423 | 1·06E‐03 | ‐0·1551 | 0·0587 | 8·41E‐03 | – |
| rs2618479 | 11355821 | A | G | 0·1680 | −0·1054 | 0·0352 | 9·27E‐03 | ‐0·1658 | 0·0582 | 4·45E‐03 | – |
| rs1600249 | 11359638 | T | G | 0·2398 | 0·1906 | 0·0304 | 5·45E‐09 | ‐0·1366 | 0·0528 | 9·76E‐03 | cis, trans |
| rs2618466 | 11362698 | C | T | 0·0142 | 0·1989 | 0·0914 | 2·98E‐02 | 0·1394 | 0·1544 | 3·67E‐01 | – |
| rs2736354 | 11368731 | T | C | 0·1589 | ‐0·0943 | 0·0348 | 2·09E‐02 | ‐0·1390 | 0·0588 | 1·83E‐02 | – |
| rs12674768 | 11371547 | T | C | 0·2563 | 0·1484 | 0·0271 | 3·23E‐06 | ‐0·1411 | 0·0511 | 5·87E‐03 | – |
| rs4841548 | 11374779 | C | T | 0·3622 | −0·0943 | 0·0288 | 4·33E‐03 | 0·0877 | 0·0472 | 6·34E‐02 | – |
| rs2729940 | 11382367 | A | G | 0·4294 | 0·1133 | 0·0329 | 7·24E‐05 | ‐0·2005 | 0·0440 | 5·73E‐06 | cis, trans |
| rs2252797 | 11382659 | G | C | 0·4021 | −0·1054 | 0·0352 | 2·10E‐04 | 0·0784 | 0·0444 | 7·77E‐02 | cis, trans |
| rs2618443 | 11384556 | T | C | 0·4556 | 0·1310 | 0·0323 | 2·80E‐06 | ‐0·1703 | 0·0433 | 8·88E‐05 | trans |
| rs2252534 | 11384713 | C | A | 0·2483 | −0·1054 | 0·0352 | 1·31E‐03 | 0·0484 | 0·0490 | 3·23E‐01 | – |
| rs12677843 | 11387189 | T | C | 0·2899 | 0·1823 | 0·0307 | 3·27E‐08 | ‐0·1284 | 0·0487 | 8·48E‐03 | cis, trans |
| rs2248932 | 11391650 | A | G | 0·3468 | 0·1740 | 0·0309 | 1·48E‐08 | ‐0·1497 | 0·0469 | 1·47E‐03 | cis, trans |
| rs2248325 | 11396874 | A | G | 0·4977 | 0·1054 | 0·0276 | 6·92E‐05 | 0·1882 | 0·0438 | 1·92E‐05 | – |
eQTL, expression quantitative trait locus; MAF, minor allele frequency; SLE, systemic lupus erythematosus; SNP, single nucleotide polymorphism.
Minor allele frequency in the European populations.
The relationship between UBE2L3 and SLE
Single nucleotide polymorphisms in the UBE2L3 gene were strongly associated with SLE (P < 5·0 × 10−8)4 (Fig. 1a). We also found that UBE2L3 was differentially expressed between SLE cases and controls in CD33+ myeloid cells, CD19+ B cells, CD4+ T cells, CD16− monocytes and CD16+ monocytes (P < 0·05; Fig. 1b). A total of 87 SNPs that were significantly associated with SLE (P < 5·0 × 10−8) were associated with UBE2L3 gene expression according to the haploreg database. We noticed that UBE2L3 gene expression was directly associated with SLE according to the SMR analysis based on eQTL data from the CAGE study (P SMR = 2·62 × 10–16) and GTEx project (P SMR = 3·93 × 10–8l Fig. 1c,d, respectively). These data showed that SNPs in UBE2L3 gene might affect SLE by influencing gene expression. We did not detect a causal association between the plasma ubiquitin‐conjugating enzyme E2 L3 (UBE2L3) level and SLE in GSMR or MR‐PRESSO analyses.
Figure 1.
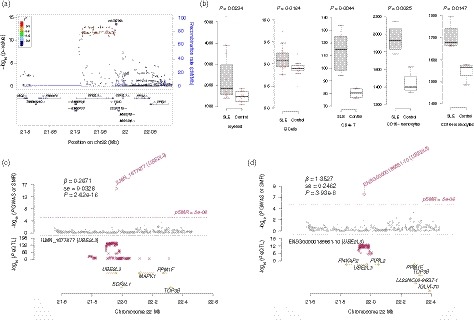
The relationship between UBE2L3 and systemic lupus erythematosus (SLE). (a) Regional association plots of the UBE2L3 locus. The regional plots were plotted based on the published data of the SLE genome‐wide association studies (GWAS)4 using the locuszoom website tool (http://locuszoom.sph.umich.edu//). (b) UBE2L3 was differentially expressed between SLE cases and controls according to gene expression profile data from the GEO database. (c,d) UBE2L3 gene expression was causally associated with SLE according to the SMR analysis.
The relationship between BLK‐CTSB and SLE
We detected significant associations between methylations and gene expressions of BLK and SLE (Fig. 2), which provided strong evidence for the relationship between BLK and SLE. SNPs in the BLK gene were strongly associated with SLE (P < 5·0 × 10−8)4 (Fig. 3a), which has been confirmed by several GWAS. We also found that BLK was differentially expressed between SLE cases and controls in CD4+ T cells (P = 0·0286). A total of 24 SNPs that were significantly associated with SLE (P < 5·0 × 10−8) were associated with BLK gene expression according to eQTL analysis.
Figure 2.
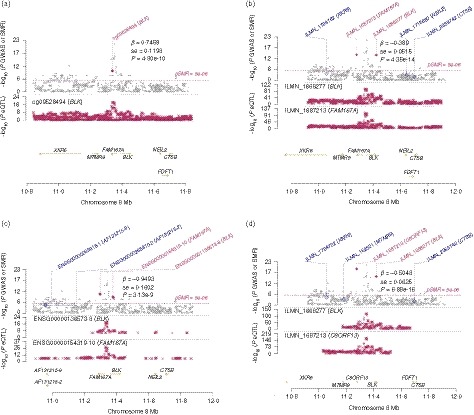
The association between BLK and systemic lupus erythematosus (SLE). The methylation level of cg09528494 in BLK and BLK gene expression were directly associated with SLE according to the summary data‐based Mendelian randomization (SMR) analysis based on methylation quantitative trait loci (meQTL) data from the study conducted by McRae et al.14 (a) and expression quantitative trait loci (eQTL) data from the CAGE study (b), GTEx project (c) and the study conducted by Westra et al.17 (d), respectively.
Figure 3.
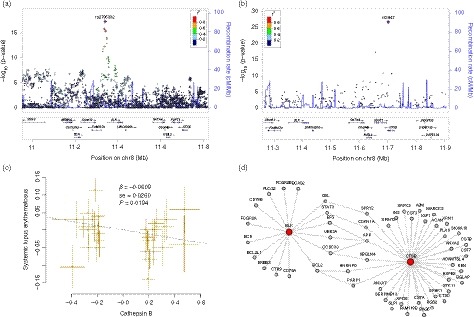
The relationship between BLK, CTSB and systemic lupus erythematosus (SLE). (a) Regional association plots of the BLK‐CTSB locus. The regional plots were plotted using published data of the SLE genome‐wide association studies (GWAS)4 in the locuszoom website tool (http://locuszoom.sph.umich.edu//). The BLK single nucleotide polymorphisms (SNPs) were significantly associated with SLE, whereas SNPs in the CTSB gene were not. (b) The CTSB SNPs were significantly associated with plasma cathepsin B level, and BLK SNPs also had suggested association evidence. (c) Mendelian randomization analysis showed the causal relationship between plasma cathepsin B level and SLE. (d) Protein–protein interaction analysis showed that BLK might interact with CTSB through several interactors.
On the other hand, we found that several SLE‐associated SNPs within BLK were strongly associated with plasma cathepsin B level according to pQTL analysis (Table 3, Fig. 3b). Hence, we further performed GSMR and MR‐PRESSO analyses to examine if there was a causal relationship between plasma cathepsin B level and SLE. In GSMR analysis, we found that plasma cathepsin B level may be causally associated with SLE (P = 0·0194; Fig. 3c). In MR‐PRESSO analysis, the causal association between plasma cathepsin B level and SLE was also significant (β = −0·2301, SE = 0·0355, P = 2·73 × 10−7), even after removing outliers (β = −0·2193, SE = 0·0340, P = 3·43 × 10−7).
Additionally, in protein–protein interaction analysis by using the LENS website tool (https://hagrid.dbmi.pitt.edu/LENS/), we found that BLK might interact with CTSB through several interactors (Fig. 3d). Some of these interactors have been reported to be associated with SLE, including CBL, STAT3, BCL2, CDKN1A and PARP1.
Discussion
The current study represented the first effort to identify potential causal genes for SLE by integrating GWAS, QTL and other data using the SMR method. This study leveraged results of several large‐scale GWAS initiatives on DNA methylation, gene expression, plasma protein levels and SLE risk. Many important methylation sites and genes (e.g. BLK and UBE2L3) were identified to be causally associated with SLE using our strategy.
Hundreds of genetic variants associated with SLE have been confirmed using GWAS. However, elucidating the causal genes underlying GWAS hits remains challenging given the complexities of a typical genome‐wide significant locus and the regulatory process. Laboratory‐based evaluation of all of the genes in the associated regions is costly and hard to achieve at this stage. On the other hand, although traditional case–control studies have identified DNA methylations25 and gene expressions19, 20, 21, 22 implicated in the pathogenesis of SLE, the power is very limited because of the small sample size. Moreover, traditional observational studies are subject to confounding and reverse causation and have not been able to disentangle which genetic, epigenetic and other factors directly influence SLE. MR‐based studies can circumvent these limitations by use of genetic proxies of putative risk factors when evaluating their associations with disease risk, as they are not subject to reverse causation.10, 11 More importantly, the SMR method allows the evaluation of the association between methylation and expression levels and SLE risk in very large samples by using data from large‐scale GWAS. In other words, the MR method not only circumvents the limitations of observational studies, but also increases power. As we showed in this study, MR analysis provided robust evidence on the important roles of hundreds of DNA methylations and genes in SLE.
DNA methylation, an important epigenetic modification in embryonic development, cell differentiation and diseases, has been implicated in the pathogenesis of SLE.25, 26, 27 Our study identified hundreds of DNA methylations that were associated with SLE. We have noticed that there are CpG islands in most of the identified gene regions, which implied a regulatory potential of DNA methylation on the gene expression. The findings raised the possibility that DNA methylation affected gene expression and then caused SLE. For example, in the UBE2L3 gene region we found six methylation sites to be causally associated with SLE. Five of these methylation sites locate in CpG island 85 (chr22: 21921937–21922715). And among them, cg06850285 has been identified as associated with SLE (discovery P = 2·8 × 10−105, replication P = 8·6 × 10−29) in the latest large case–control study.26 These methylations were supposed to affect UBE2L3 expression (needs to be proven). As we have shown that UBE2L3 was up‐regulated in SLE patients in several types of immune cells, and UBE2L3 expression level was causally associated with SLE, which was identified by SMR analysis. The UBE2L3 gene encodes the ubiquitin‐conjugating enzyme E2. It should be a key gene for SLE because the association between this gene and SLE has been proven by many studies in different populations.2, 4, 5, 28, 29 Further studies are suggested to elucidate the mechanisms.
Another arresting example was the B lymphoid tyrosine kinase (BLK) gene. This is a well‐known SLE‐associated gene that was first reported in the first SLE GWAS and has been proven by many subsequent studies. However, how BLK affects SLE is still unclear. In the BLK gene region, we found one methylation site cg09528494 (chr8: 11338675) to be significantly associated with SLE. The association between BLK methylation and SLE has not been reported. But DNA methylation levels have been showed to be related to BLK expression.30 BLK expression level was also found to be causally associated with SLE in our analysis. So it is possible that DNA methylation may affect BLK gene expression and then cause SLE. We also found that several BLK SNPs that significantly associated with SLE were strongly associated with plasma cathepsin B level, and plasma cathepsin B level was causally associated with SLE. This finding suggested that BLK SNPs may affect SLE through influencing the cathepsin B level. Cathepsin B was encoded by the CTSB gene, which was involved in the antigen processing and presentation KEGG pathway. The associations between cathepsin S and cathepsin K and SLE have been reported.31, 32 But to our knowledge, the association between Cathepsin B and SLE was first reported in the present study. How BLK interacts with CTSB is unknown. These genes were both involved in the innate immune response GO biological process. As suggested by the protein–protein interaction analysis, these two genes might interact with each other via other SLE‐related genes such as CBL, STAT3, BCL2, CDKN1A and PARP1. The interaction between these genes may point to a pathway for SLE.
In summary, the MR method not only circumvents the limitations of observational studies, but also increases power. We applied the SMR method in this study to provide robust evidence on the important roles of DNA methylations and gene expressions of BLK and UBE2L3 as important risk factors of SLE. This study provided novel evidence of an important role of circulating cathepsin B in SLE etiology. However, MR studies rest on strong assumptions and the causal relationship identified by MR analysis is always conditional and requires close scrutiny. Further research is needed to fully elucidate the important relationship between these factors and SLE.
Disclosures
The authors declare no conflict of interests.
Supporting information
Table S1 . The systemic lupus erythematosus‐associated methylations identified by Mendelian randomization analysis.
Table S2 . Functional enrichments of the systemic lupus erythematosus‐associated genes identified by Mendelian randomization analysis.
Table S3 . Association between UBE2L3 single nucleotide polymorphisms and systemic lupus erythematosus and plasma protein levels.
Acknowledgements
The study was supported by the Natural Science Foundation of China (81773508), the Key Research Project (Social Development Plan) of Jiangsu Province (BE2016667), the Startup Fund from Soochow University (Q413900313, Q413900412), and a Project of the Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
- 1. Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S et al Association of systemic lupus erythematosus with C8orf13‐BLK and ITGAM‐ITGAX. N Engl J Med 2008; 358:900–9. [DOI] [PubMed] [Google Scholar]
- 2. Yang W, Tang H, Zhang Y, Tang X, Zhang J, Sun L et al Meta‐analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am J Hum Genet 2013; 92:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y, Yang J, Zhang J, Sun L, Hirankarn N, Pan HF et al Genome‐wide search followed by replication reveals genetic interaction of CD80 and ALOX5AP associated with systemic lupus erythematosus in Asian populations. Ann Rheum Dis 2016; 75:891–8. [DOI] [PubMed] [Google Scholar]
- 4. Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW et al Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet 2015; 47:1457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Langefeld CD, Ainsworth HC, Cunninghame Graham DS, Kelly JA, Comeau ME, Marion MC et al Transancestral mapping and genetic load in systemic lupus erythematosus. Nat Commun 2017; 8:16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet 2012; 90:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW et al Integrative approaches for large‐scale transcriptome‐wide association studies. Nat Genet 2016; 48:245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino‐Michaels K, Carroll RJ et al A gene‐based association method for mapping traits using reference transcriptome data. Nat Genet 2015; 47:1091–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE et al Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016; 48:481–7. [DOI] [PubMed] [Google Scholar]
- 10. Smith GD, Ebrahim S. 'Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003; 32:1–22. [DOI] [PubMed] [Google Scholar]
- 11. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 2014; 23:R89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burgess S, Timpson NJ, Ebrahim S, Davey Smith G. Mendelian randomization: where are we now and where are we going? Int J Epidemiol 2015; 44:379–88. [DOI] [PubMed] [Google Scholar]
- 13. Pavlides JM, Zhu Z, Gratten J, McRae AF, Wray NR, Yang J. Predicting gene targets from integrative analyses of summary data from GWAS and eQTL studies for 28 human complex traits. Genome Med 2016; 8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McRae AF, Marioni RE, Shah S, Yang J, Powell JE, Harris SE et al Identification of 55,000 Replicated DNA Methylation QTL. Sci Rep 2018; 8:17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature 2017; 550:204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lloyd‐Jones LR, Holloway A, McRae A, Yang J, Small K, Zhao J et al The genetic architecture of gene expression in peripheral blood. Am J Hum Genet 2017; 100:228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J et al Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 2013; 45:1238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suhre K, Arnold M, Bhagwat AM, Cotton RJ, Engelke R, Raffler J et al Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun 2017; 8:14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK 3rd, Wu T, Li QZ et al Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity 2008; 28:206–17. [DOI] [PubMed] [Google Scholar]
- 20. Fernandez DR, Telarico T, Bonilla E, Li Q, Banerjee S, Middleton FA et al Activation of mammalian target of rapamycin controls the loss of TCRζ in lupus T cells through HRES‐1/Rab4‐regulated lysosomal degradation. J Immunol 2009; 182:2063–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garaud JC, Schickel JN, Blaison G, Knapp AM, Dembele D, Ruer‐Laventie J et al B cell signature during inactive systemic lupus is heterogeneous: toward a biological dissection of lupus. PLoS ONE One 2011; 6:e23900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kyogoku C, Smiljanovic B, Grun JR, Biesen R, Schulte‐Wrede U, Haupl T et al Cell‐specific type I IFN signatures in autoimmunity and viral infection: what makes the difference? PLoS ONE One 2013; 8:e83776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu Z, Zheng Z, Zhang F, Wu Y, Trzaskowski M, Maier R et al Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun 2018; 9:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018; 50:693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao M, Wang J, Liao W, Li D, Li M, Wu H et al Increased 5‐hydroxymethylcytosine in CD4+ T cells in systemic lupus erythematosus. J Autoimmun 2016; 69:64–73. [DOI] [PubMed] [Google Scholar]
- 26. Imgenberg‐Kreuz J, Carlsson Almlof J, Leonard D, Alexsson A, Nordmark G, Eloranta ML et al DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann Rheum Dis 2018; 77:736–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joseph S, George NI, Green‐Knox B, Treadwell EL, Word B, Yim S et al Epigenome‐wide association study of peripheral blood mononuclear cells in systemic lupus erythematosus: Identifying DNA methylation signatures associated with interferon‐related genes based on ethnicity and SLEDAI. J Autoimmun 2019; 96:147–57. [DOI] [PubMed] [Google Scholar]
- 28. Chung SA, Taylor KE, Graham RR, Nititham J, Lee AT, Ortmann WA et al Differential genetic associations for systemic lupus erythematosus based on anti‐dsDNA autoantibody production. PLoS Genet 2011; 7:e1001323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z et al Genome‐wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 2009; 41:1234–7. [DOI] [PubMed] [Google Scholar]
- 30. Zhu H, Wu LF, Mo XB, Lu X, Tang H, Zhu XW et al Rheumatoid arthritis‐associated DNA methylation sites in peripheral blood mononuclear cells. Ann Rheum Dis 2019; 78:36–42. [DOI] [PubMed] [Google Scholar]
- 31. Zhou Y, Chen H, Liu L, Yu X, Sukhova GK, Yang M et al Cathepsin K deficiency ameliorates systemic lupus erythematosus‐like manifestations in Fas(lpr) mice. J Immunol 2017; 198:1846–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rupanagudi KV, Kulkarni OP, Lichtnekert J, Darisipudi MN, Mulay SR, Schott B et al Cathepsin S inhibition suppresses systemic lupus erythematosus and lupus nephritis because cathepsin S is essential for MHC class II‐mediated CD4 T cell and B cell priming. Ann Rheum Dis 2015; 74:452–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 . The systemic lupus erythematosus‐associated methylations identified by Mendelian randomization analysis.
Table S2 . Functional enrichments of the systemic lupus erythematosus‐associated genes identified by Mendelian randomization analysis.
Table S3 . Association between UBE2L3 single nucleotide polymorphisms and systemic lupus erythematosus and plasma protein levels.