

Summary
Chloroquines are 4‐aminoquinoline‐based drugs mainly used to treat malaria. At pharmacological concentrations, they have significant effects on tissue homeostasis, targeting diverse signaling pathways in mammalian cells. A key target pathway is autophagy, which regulates macromolecule turnover in the cell. In addition to affecting cellular metabolism and bioenergetic flow equilibrium, autophagy plays a pivotal role at the interface between inflammation and cancer progression. Chloroquines consequently have critical effects in tissue metabolic activity and importantly, in key functions of the immune system. In this article, we will review the work addressing the role of chloroquines in the homeostasis of mammalian tissue, and the potential strengths and weaknesses concerning their use in cancer therapy.
Keywords: Autophagy, chloroquine, drug repurposing, inflammation, lysosome, neoplasm
Chloroquines are expected to have diverse effects on cancer progression. Inhibition of late autophagy can affect cell survival, as well as antigen processing and presentation in the immune system. Combinatorial study of these effects at several levels will help to enhance the capability to incorporate them in cancer treatment.
Abbreviations
- AMPK
adenosine monophosphate‐activated protein kinase
- ATG4C
autophagy related 4C cysteine peptidase
- ATM
ataxia telangiectasia mutated kinase
- BET
bromodomain and extra terminal domain
- CIC
cancer‐initiating cells
- CDK
cyclin‐dependent kinase
- CQ
chloroquine
- CSC
cancer stem cells
- CXCL
C‐X‐C motif chemokine
- CXCR
C‐X‐C chemokine receptor type
- DNA‐PK
DNA‐protein kinase
- GR
glucocorticoid receptor
- HCQ
hydroxychloroquine
- IDH
isocitrate dehydrogenase
- Ig
immunoglobulin
- IFN
interferon
- IκB
inhibitor of NF‐κB
- IL
interleukin
- MHC‐I
major histocompatibility complex class I
- NK
natural killer
- NF‐κB
nuclear factor κB
- NRF
nuclear factor erythroid‐related factor
- Par‐
prostate apoptosis response‐
- SIRT
sirtuin
- SQSTM
sequestosome
- STAT
signal transducer and activator of transcription
- TFEB
transcription factor EB
- Th
T‐helper
- TLR
Toll‐like receptor
- Treg
regulatory T cell
- TRAF
tumor necrosis factor receptor‐associated factor
- TRAIL
tumor necrosis factor‐related apoptosis‐inducing ligand
- UPS
ubiquitin‐proteasome system
Introduction
The chloroquine family of drugs (CQs), such as chloroquine (CQ) and hydroxychloroquine (HCQ), are 4‐aminoquinoline‐based compounds that have been used for the prevention and treatment of malaria. They have also been used as anti‐inflammatory agents for treating rheumatoid arthritis, and lupus erythematosus.1 CQ is one of the most prominent cases of drug repurposing in cancer2 and it was introduced as an auxiliary anticancer agent more than a decade ago.3 In preliminary research, CQ and HCQ have shown promising results in combination with other anticancer drugs.4, 5 Even though CQs have shown the potential to affect cell invasion, chemotaxis, trans‐differentiation, and clonogenicity in cancer research, they are mainly used as inhibitors of autophagy, which is a collective term for diverse mechanisms for intracellular degradation of macromolecules and organelles through lysosomes6 (Fig. 1a).
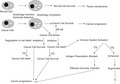
Figure 1.
(a) Depending on the cellular growth conditions, diverse macromolecules and organelles such as mitochondria and peroxisomes are selectively delivered to lysosomes to be degraded through autophagy. Autophagy enables cellular adaptation to increased mutagenic load or to changes in nutrient concentrations. Chloroquine blocks lysosome acidification and thereby inhibits degradation of macromolecules and organelles by lysosomes. (b) The process of autophagy commonly involves a complex series of molecular modifications that leads to the formation of the autophagosome. Docking of adaptor proteins such as p62 on the autophagosome is followed by fusion of the autophagosome with a lysosome. Depending on the identities of the molecules degraded by autophagy, the process may lead either to cell survival or cell death. These processes are targets of chloroquines (CQs), which thereby lead to cell death or cell survival by interfering with the functions of lysosomes
Autophagy is an intrinsic cellular mechanism by which cells degrade and recycle their dysfunctional components through lysosomes. Contrasting roles of autophagy have been reported in cancer cells.7 It may function as a tumor suppressor mechanism in the cells by degrading damaged proteins and organelles and therefore preventing their accumulation in the cells.8 However, it can also promote cell survival by recycling intracellular organelles and proteins in tumors.9 Hence, autophagy is considered a mechanism potentially leading to invasion, chemotaxis, trans‐differentiation, and clonogenicity of tumor cells.
Autophagy may be selective or non‐selective. In selective autophagy, substrates are selected for degradation by binding to specific adapter proteins, and the identity of the degraded molecules determines the impact of autophagy on cell fate.10 The outcome of autophagy, therefore, becomes an important variable and a criterion in the evaluation of the effects of CQ on cancer cells. Several clinical trials that have included CQ or its analogs in combination with other anticancer agents have been initiated and the results of a number of them have been evaluated, with inhibition of autophagy as one of the outcome measures11, 12 (updated information is available at https://clinicaltrials.gov/ct2/results?cond=cancer%26term=chloroquine%26cntry=%26state=%26city=%26dist=). It is important to note that the inhibition of autophagy is a crucial factor for the success of radiation therapy as well. It is known that radiotherapy leads to autophagy in the cells, which results in radioresistance, at least in glioblastoma.13 Therefore, CQ can also sensitize tumor cells to radiotherapy by inhibiting autophagy.14 This event was shown in various cancer cells including glioma, breast and lung cancers.15 In addition, CQ increased the survival time of glioblastoma patients.16 Overall, meta‐analysis shows that addition of CQs to chemotherapy or radiotherapy regimens increases overall survival and progression‐free survival.17
Overview of the strengths and weaknesses of CQ as a cancer drug
Chloroquine is on the World Health Organization's Essential Medicine list, and was a widely used medicine for decades without any major side effects. This, and its promising preclinical results, make CQ an outstanding candidate for repurposing in cancer treatment.1, 6, 18 The Repurposing Drugs in Oncology project that focuses on repurposing some well‐known and well‐characterized non‐cancer drugs for new uses in cancer treatment, has emphasized the importance of CQ and HCQ in sensitizing cancer cells to various cancer treatment protocols.12, 19 The easy synthesis of quinolines with selected chemical properties and their environmental sustainability20, 21, 22 are added bonuses for their use in research and clinics, if proven to be effective anticancer drugs.
A summary of past and ongoing clinical trials for CQs in cancer is given in Table 1. As expected of drug repurposing studies, the majority of trials are sponsored by hospitals/educational institutions. It is encouraging to note the increasing number of studies that combine CQ or HCQ with both established as well as novel methods of intervention, to explore concepts of synthetic lethality. Furthermore, it is understandable that a number of factors that include the legitimate concern for potential side effects may slow the pace of patient accrual. On the other hand, it would be desirable to see an increase in collaborative studies, and an improvement of the immunological approach of CQ use. In this direction, progress in drug delivery methodologies can help to focus the impact of CQ‐induced cellular stress on the components of the immune system that generate the fewest side effects. Increasingly sophisticated incorporation methods for CQ in protocols for cancer immunotherapy may benefit patients with co‐morbidities that cannot be included in trials without a better understanding of the immunological effects of CQs. The development of CQ‐incorporating immunotherapy schemes may require the design of parallel ex vivo research models to accompany clinical trials. Overall, clinical research for CQ delivery methods and their incorporation into cancer immunotherapy, by lowering the required CQ concentration, will help to address the side effects that occur due to the use of high CQ doses for prolonged time periods.
Table 1.
Clinical studies of chloroquines in cancer, registered with the database of the US National Library of Medicine at the National Institutes of Health. The safety and scientific validity of the listed studies has not been evaluated by the US Federal Government
| Registration Identifier | Recruitment Status as of 11/2019 | Aim | Responsible Party |
|---|---|---|---|
| 224978 | Completed | Chloroquine for Treatment of Glioblastoma Multiforme | National Institute of Neurology and Neurosurgery, Mexico |
| 568880 | Completed | Hydroxychloroquine and Bortezomib in Treating Patients With Relapsed or Refractory Multiple Myeloma | Abramson Cancer Center of the University of Pennsylvania |
| 771056 | Terminated | Hydroxychloroquine in Untreated B‐CLL Patients | Northwell Health |
| 969306 | Terminated (Poor accrual) | Chloroquine as an Anti‐Autophagy Drug in Stage IV Small Cell Lung Cancer Patients | Maastricht Radiation Oncology |
| 1023477 | Completed | Study of the Efficacy of Chloroquine in the Treatment of Ductal Carcinoma in Situ | Inova Health Care Services |
| 1227135 | Recruiting | Imatinib Mesylate With or Without Hydroxychloroquine in Treating Patients With Chronic Myeloid Leukemia | University of Glasgow |
| 1396200 | Completed | Cyclophosphamide and Pulse Dexamethasone With Rapamycin or Hydroxychloroquine for patients with relapsed/refractory multiple myeloma | Abramson Cancer Center of the University of Pennsylvania |
| 1438177 | Terminated | Chloroquine in Combination With VELCADE and Cyclophosphamide for Relapsed and Refractory Multiple Myeloma | NYU Langone Health |
| 1446016 | Completed | Chloroquine With Taxane Chemotherapy for Advanced or Metastatic Breast Cancer After Anthracycline Failure | The Methodist Hospital System |
| 1469455 | Completed | DNA Repair Inhibitor & Irradiation on Melanoma | DNA Therapeutics |
| 1575782 | Terminated (Poor accrual) | Chloroquine as an Anti‐autophagic Radiosensitizing Drug in Stage I–III Small Cell Lung Cancer | Maastricht Radiation Oncology |
| 1689987 | Completed | Hydroxychloroquine, Cyclophosphamide, Dexamethasone, and Sirolimus in Treating Patients With Relapsed or Refractory Multiple Myeloma | OHSU Knight Cancer Institute |
| 1727531 | Completed | IDO2 Genetic Status Informs the Neoadjuvant Efficacy of Chloroquine in Brain Metastasis Radiotherapy | Main Line Health |
| 1777477 | Completed | Adjuvant Effect of Chloroquine on Gemcitabine | University of Zurich |
| 1894633 | Terminated | Study of Whole‐brain Irradiation With Chloroquine for Brain Metastases | Instituto Nacional de Cancerologia de Mexico |
| 2071537 | Unknown | Chloroquine in Combination With Carboplatin/Gemcitabine in Advanced Solid Tumors | University of Cincinnati |
| 2333890 | Unknown | A Phase 2 Randomized, Double‐blind Trial Evaluating the Effects of Chloroquine in Breast Cancer | Ottawa Hospital Research Institute |
| 2366884 | Recruiting | Clinical Evaluation of a New Form of Cancer Therapy Based on the Principles of Atavistic Metamorphosis | Dr. Frank Arguello Cancer Clinic |
| 2378532 | Recruiting | The Addition of Chloroquine to Chemoradiation for Glioblastoma | Maastricht Radiation Oncology |
| 2432417 | Not yet recruiting | The Addition of Chloroquine to Chemoradiation for Glioblastoma | Maastricht Radiation Oncology |
| 2496741 | Unknown | Metformin And Chloroquine in IDH1/2‐mutated Solid Tumors | Universiteit van Amsterdam |
| 2631252 | Terminated (Inability to accrue) | Phase I Study of Mitoxantrone and Etoposide Combined With Hydroxychloroquine, for Relapsed Acute Myelogenous Leukemia | University of Pittsburgh |
| 2786589 | Recruiting | Plasmodium Immunotherapy for Lung Cancer | State Key Laboratory of Respiratory Disease |
| 3243461 | Recruiting | International Cooperative Phase III Trial of the HIT‐HGG Study Group | University of Göttingen |
| 3400865 | Not yet recruiting | Cabergoline Combined Hydroxychloroquine/Chloroquine to Treat Resistant Prolactinomas | Ruijin Hospital |
| 3979651 | Not yet recruiting | MEK and Autophagy Inhibition in Metastatic/Locally Advanced, Unresectable Neuroblastoma RAS Melanoma | Hospices Civils de Lyon |
| 4163107 | Not yet recruiting | Combined Carfilzomib and Hydroxychloroquine in Patients With Relapsed/Refractory Multiple Myeloma | Norwegian University of Science and Technology |
It is noteworthy that in addition to their antitumor effects, CQs also have some general clinical benefits. For example, HCQ shows a tendency to improve the metabolic, especially lipid, profile of diverse groups of patients.23, 24 Indeed, it was shown that the use of CQs improved lipid profiles of individuals with rheumatoid arthritis.25 Furthermore, CQ can protect mammalian tissues such as liver and bone marrow from the damaging effects of other drugs and irradiation.26 Consistently, CQ and HCQ can also protect skin cells against ultraviolet light damage.27 In further support of arguments for the study of innovation in drug repurposing, cancer is a global disease and the world's aging population will be in need of economically and environmentally sustainable cancer treatment options.28, 29
However, despite a wealth of preclinical experimental data on the beneficial impact of CQ on malignant tumors, clinical studies have not yet shown a substantial response of neoplastic diseases to CQs.6 There are two main potential reasons for this delay in the translation of experimental results into clinical results. The first is the inherent difficulty of CQ to pass through the cell membrane in the presence of an acidic extracellular microenvironment.30 The tumor microenvironment is often acidic, in contrast to normal tissue that, at least under homeostatic conditions, is mostly slightly alkaline.31 The acidity of the tumor microenvironment is mostly due to the ‘Warburg effect’ in cancer cells, which is the use of glycolysis rather than aerobic respiration for energy production.32 Compared with the normal cells, cancer cells use more glucose to sustain a high proliferation rate with an increase in the conversion of glucose to lactic acid with the release of protons, causing a decrease in the pH level that leads to elevated acidity. This problem can be overcome by using chemical or biological agents targeting pH regulatory mechanisms. An alternative approach may be the encapsulation of CQ with proper nanoparticles for efficient delivery into cancer cells in the acidic tumor microenvironment. The second reason is that CQ, as an inhibitor of lysosomal autophagy, interferes with several functions of tissue turnover and with key events in the immune response.33 This has been the subject of various studies, from which evidence is emerging that CQ can significantly strengthen the antineoplastic immune response, at least under certain conditions.34 Analysis of related data shows that a substantial part of the effects of CQ is due to its interference with cellular metabolism, gene regulation, and mechanisms that trigger innate immunity including signaling pathways through Toll‐like receptors (TLR) and tumor necrosis factor‐α(see Supplementary material, Appendix S1).35, 36 It is important to note that cell stress from antineoplastic treatment and CQs can activate some transcription factors, which may reprogram the cells and thereby enable tumor cells to escape from cell death induced by drug treatment or from the immune system.37, 38 It is important to track the regulatory mechanisms at the molecular level and design drug combinations accordingly to prevent tumor cell recovery.
Several recent findings have led to the expectation that CQs will be incorporated in cancer treatment schemes. However, supporting experimental data will be needed to close the gap between clinical drug repurposing trials and the related innovative basic and preclinical research, targeting to delineate the mechanisms of how to incorporate the repurposed drugs into treatment combination protocols.39, 40 At high concentrations, CQs can interact with several types of macromolecules, causing some effects such as inhibition of drug sequestration and degradation.41 However, due to the number and severity of characterized side effects of the systemic use of high CQ doses, it is imperative that translational study focuses on improving the antineoplastic effects of low micromolar drug concentrations.6, 42, 43 To this end, nanotechnology is expected to help to decrease the systemic exposure to CQs and their derivatives.44, 45, 46
Although we will show that CQ has diverse effects on tissue metabolism and homeostasis in general, the key scientific field that has the potential to enable CQ incorporation in anticancer drug combinations is the study of interference with immune functions. In the next paragraphs, we will attempt to explain why we consider this field as essential to the effective use of CQ in treatment combinations.
CQ inhibits lysosomal acidification
Theoretically, if a chemical base is weak enough, it will react selectively with the most acidic component within a given system. CQ is a weak base47 and the most significant effect of CQ as a cancer drug is its interference with lysosome acidification.6 Mechanistically, when CQ enters the lysosome, it becomes protonated due to the high internal acidity. Accumulation of protonated CQ in the lysosome increases lysosomal pH and consequently decreases lysosomal function.48 However, a recent study proposed that CQ can also inhibit autophagy by impairing autophagosome‐to‐lysosome fusion,49 an important observation that needs further investigation.
Some cell types rely on lysosomal degradation of macromolecules, especially when they depend on mitochondrial oxidative phosphorylation to generate energy. Damaged mitochondrial proteins are then gradually degraded in lysosomes.50 Likewise, various other conditions such as drug‐induced stress or mutations in the mitochondrial DNA may initiate mitochondrial degradation. Therefore, the cells under metabolic stress can target damaged mitochondria to lysosomes.51, 52 Moreover, some cancer cell types exhibit excessive acidity in their lysosomes, reaching pH values <4.53 In addition, mediators of apoptotic cell death can also be degraded in lysosomes, a process that gives a survival advantage to cancer cells.54, 55 CQ may also lead to a decrease in the quality and functions of mitochondria.56, 57 Therefore, the inhibitory effect of CQ on mitochondrial function and lysosomal acidification can be explored for a synergistic therapeutic benefit.
Nevertheless, degradation of intracellular components is not the only role of lysosomes. For example, macrophages and dendritic cells use lysosomes for microbe killing and antigen processing and presentation.58 Yet, another important role of lysosomes is the removal of dead cell debris to enable resolution of inflammation and tissue remodeling.59 Scavenging M2‐type macrophages are particularly effective in removing debris from tissues to help resolve inflammation; M2 macrophages have a highly acidic lysosomal pH (<5), whereas M1 macrophages that stimulate immune responses show lower lysosomal acidity (pH >5). An M2‐to‐M1 macrophage transition could be experimentally achieved by CQ at least partially through raising lysosomal pH.60 Moreover, CQ has the potential to alleviate pathological conditions associated with increased M2 activity, such as vascular disorder during lung carcinogenesis.61 Interestingly, it has also been shown that CQ attenuates lipopolysaccharide‐induced M1 macrophage activation.62 Dendritic cells also limit lysosomal acidification to optimize antigen processing and allow major histocompatibility complex class I (MHC‐I) dependent cross‐presentation.63 CQ may facilitate antigen cross‐presentation through late endosomes, and inhibit presentation from early endosomes, which is consistent with the inhibition of lysosomal acidification.64, 65
CQ is an inhibitor of late autophagy
In eukaryotic cells, autophagy is an evolutionarily conserved mechanism that plays important roles in degradation and recycling of intracellular components.66 This mechanism is a multi‐step complex process and involves the engulfment of targets by autophagosomes and their subsequent degradation by lysosomes.66, 67 Lysosomal degradation is the last step in the process of autophagy, which generates molecules to be used for the synthesis of macromolecules.67
Autophagy allows cells to degrade long‐lived proteins, aggregates and entire organelles.68 Processes of autophagy include mitophagy (for mitochondria), pexophagy (for peroxisomes), ER‐phagy (for endoplasmic reticulum), or ribophagy (for ribosomes), depending on which organelles are targeted for specific autophagic degradation.69 Hence, autophagy has the capacity to influence the entire spectrum of macromolecular turnover in a tissue. Under certain conditions, cells may undergo autophagic death, if intracellular survival pathways are blocked during autophagy.15, 70, 71 Pharmacological inhibitors of autophagy have been used extensively in various cellular conditions to delineate the molecular mechanism of autophagy and the effect of these inhibitors on autophagy. CQ is one such pharmacological inhibitor of autophagy that has been extensively used in research. It inhibits late stages of autophagy and may induce cell death even under conditions where inhibitors of early autophagy cannot72 (Fig. 1b). Furthermore, it impairs the late stages of acidic organelle fusion, especially autophagosome–lysosome fusion.49
Autophagy regulates cell and tissue homeostasis
Senescence is an important homeostatic process. It is triggered by various cell stress factors and arrests proliferation, which may lead to tumor cell survival and limit the therapeutic effects of pharmaceutical agents. Depending on the substrates targeted to the lysosomes, autophagy can prevent or accelerate cellular senescence.73 Specifically autophagy suppresses cellular senescence by removing damaged macromolecules or organelles, or promotes cellular senescence by facilitating the synthesis of senescence‐associated secretory proteins.73 This dual capacity of autophagy can therefore enable a tissue to either rescue or remove cells with malfunctioning organelles. This would preserve the integrity of tissue structure and function. At the organismal level, aging impairs regulation of autophagy, which may characteristically lead to a decreased protection of the heart, for example, from ischemia–reperfusion injury.74 For instance, fasting induces transcription factor EB (TFEB) which activates mitochondrial and lysosomal biogenesis, and enables autophagy, to allow removal and replacement of damaged organelles. TFEB protects the heart from cardiac injury during ischemia–reperfusion, which would otherwise cause cell death by hypoxia–reoxygenation.75 Drugs that interfere with the activation of TFEB, such as anthracyclines, impair autophagy and cause cardiomyopathy.76 The interference of anthracyclines with TFEB activity can also impair autophagy of peroxisomes, pexophagy, which may contribute to neurotoxicity, cognitive dysfunction, and accelerated brain aging in cancer patients and survivors.77
Autophagy has crucial roles in vertebrate development from the pre‐implantation stage to organogenesis.78, 79 Abnormalities in autophagy may lead to various developmental defects, as shown in several model organisms.79 For example, during chick embryogenesis, autophagy is essential in regulating the temporal expression and spatial localization of developmental genes and in coordinating epithelial‐to‐mesenchymal transitions that enable the establishment of the three germ layers.80 Consistently, developmental delay has been observed in chick embryos when autophagy was inhibited.80
CQ regulates tissue responses to metabolic and inflammatory stimuli
A tissue may regularly need cells to undergo autophagy as a means to remove damaged or misfolded proteins, conserve resources, and restore homeostasis. This is evident in a few experimental systems, where CQ exacerbates metabolite imbalance and causes tissue dysfunction and damage. For example, low micromolar CQ can exacerbate vascular calcification.81 Likewise, autophagy protects cardiomyocytes in mice fed a high‐fat diet, and this beneficial effect can be abrogated by CQ.82 However, HCQ may protect the myocardium in combination with phosphodiesterase inhibitors in type 2 diabetes.83 Autophagy in the heart is enhanced in type 1 diabetes but is suppressed in type 2 (insulin‐resistant) diabetes.84 Therefore CQ, being an inhibitor of late autophagy, might exacerbate cardiomyopathy in type 1 diabetes by causing myocardial hypertrophy and interstitial fibrosis.85 Also, in type 2 diabetes, inhibition of autophagy by CQ can be detrimental for circulation, especially in endothelial progenitor cells that are needed to preserve ischemic angiogenesis and blood perfusion.86
Autophagy is not always beneficial though; in excess, it can contribute to tissue damage, both in young and aged mammals.87, 88 Even in bone marrow, where autophagy provides essential maintenance function, excessive autophagy of mesenchymal stem cells is very likely the cause of cellular senescence during hyperglycemia‐induced marrow hematopoietic niche dysfunction.89 Treatment with the rapamycin can induce non‐coding RNA LCPAT1 and activate autophagy, to permit growth of lung tumors.90 It is interesting to note that increased lysosomal degradation can even suppress nuclear receptor signaling and can thereby blunt the glucocorticoid‐mediated inhibition of gene expression of inflammatory cytokines. Hence, CQ has the potential to modulate inflammation, in synergy with glucocorticoids.91 Viral infection, especially Epstein–Barr virus, can activate constitutive autophagy to support virus latency and permit lymphoproliferative disorders and lymphomas.92 Epstein–Barr virus‐induced autophagy is dependent on the expression of viral latent membrane proteins, which provide cells with an improved survival ability. In these cells, viruses inhibit lysosomal degradation in the maturation step of autophagy and use autophagic membranes for the formation and release of the viral particles. Therefore, inhibiting lysosomal degradation with CQ induces p53‐dependent cell death and prevents cancer in mouse models of lymphomagenesis.93 In fact, basal autophagy has been proposed to play a pivotal role in sustaining mitochondrial function in lymphoma, and low CQ concentration may cause apoptosis in susceptible lymphoma cells.94
In contrast, inhibition of autophagy by CQ may exacerbate diabetic neuropathy and impair neuronal function.95 Lysosomal proteolysis enables mitochondrial quality control in rat hippocampus, a process impaired by CQ.96 Likewise, the activity of autophagy is closely related to muscle diseases and inhibition of autophagy by HCQ may cause severe vacuolar myopathies in patients with Danon disease.97 CQ can also affect tissues such as kidney and pancreas. Similarly, CQ (10 mg/kg/day) can exacerbate renal ischemia/reperfusion injury in type 2 diabetes.98 In type 2 diabetes, low micromolar CQ could inhibit autophagy and cause apoptosis of podocytes and exacerbate nephropathy.99 In the pancreas of individuals with type 2 diabetes, cytokine interleukin‐6 (IL‐6) induces autophagy and protects pancreatic beta cells from apoptosis whereas CQ causes apoptosis and thereby elicits tissue damage.100
Failure of progenitor cells to regulate autophagy during tissue regeneration can contribute to carcinogenesis.37, 68 Likewise, cells can activate autophagic degradation of proteins as a compensatory mechanism under drug‐induced cytotoxic stress.101, 102 Through activation of cellular protein recycling during autophagy, lysosomes can help a cell survive endoplasmic reticulum stress that is often caused by anticancer drugs.37, 103 Endoplasmic reticulum stress otherwise kills the cell by inducing the unfolded protein response, mitochondrial membrane depolarization, and ensuing activation of caspases and degradation of DNA.104 Hence, autophagy could function as a rescue mechanism for malignant cells against treatment‐induced cell stress. In cancer, however, malignant cells might also acquire the capacity to induce autophagy of the mesenchymal cells in tumor stroma, to supply nutrients and generate a niche that fosters cancer cell survival and proliferation.32, 105
Chloroquine can also synergize with agents targeting the cAMP pathway, such as adenosine monophosphate‐activated protein kinase (AMPK). For example, in response to ATP deficiency, AMPK is activated and induces autophagy through inhibition of the mammalian target of rapamycin.106 AMPK activation thereby facilitates the restoration of the cellular energy status by switching on a catabolic pathway to generate ATP while simultaneously inhibiting ATP‐consuming processes such as cell proliferation and biosynthesis. However, when AMPK is aberrantly activated it can cause severe pathological manifestations associated with excessive glycolysis.107 Induction of autophagy by AMPK causes cell death when combined with inhibition of late autophagy by CQ.108 In some cases, the combination of CQ and AMPK activators, such as acetaminophen, can lead to side effects, such as liver toxicity.109 CQ‐AMPK synergy can also lead to cancer cell death.110 Examples include synergy of CQ with the two AMPK activators, the glucose analog 2‐deoxyglucose in killing prostate cancer cells, and with OSU‐53 in killing triple‐negative breast cancer cells.111, 112 Both of these AMPK activators also inhibit activation of transcription factor signal transducer and activator of transcription 3 (STAT3).111, 113 STAT3 can activate the expression of proteasome subunits such as β subunits of the 20 S proteasome core complex and immunoproteasome subunits latent membrane proteins7 and 2.114, 115 Thereby, STAT3 regulates proteostasis through the proteasome, a module that interacts with autophagy as we see next.116 STAT3 itself is a therapeutic target in cancer, and at least in some study systems, STAT3 blockers can be combined with autophagy inhibitors. In cancer cells, tyrosine kinase inhibitors can block STAT3 signaling, and thereby activate autophagy, making cells sensitive to death by CQ treatment.117, 118, 119 Hence, CQ regulates proteostasis, proteasome activity, and cell viability.
Proteostasis regulates cellular stress responses
In particular the Ubiquitin–Proteasome system (UPS) is a cellular mechanism degrading proteins that complements the activity of the lysosome.116 In general, proteins with a short half‐life undergo programmed degradation in the UPS after having completed their function.120 In addition, soluble misfolded and unfolded proteins can also be degraded by UPS.121 UPS is involved in vital cellular processes such as regulation of cell cycle progression, transcription, and DNA repair.122, 123, 124 The activities of UPS and autophagy are linked, and inhibition of the one causes activation of the other.116 Inhibitors of the proteasome and several anti‐inflammatory agents cause the redistribution of targeted proteins in organelles.125 Some protein aggregates inhibit proteasome function but trigger lysosomal protein degradation through a number of mechanisms.116, 126 The inhibition of proteasome induces transcription of p62 via transcription factor nuclear factor erythroid‐related factor 1 (NRF1).127 p62, also known as Sequestosome 1 (SQSTM1), is a ubiquitin‐binding adaptor protein that bridges the proteasome‐dependent degradation process to autophagy.128 It is a multifunctional protein, and its different domains are involved in both UPS and autophagy‐dependent degradation processes.128 Proteasome inhibition triggers autophagy by increased endoplasmic reticulum stress that releases NRF2 from Kelch‐like erythroid cell‐derived protein with CNC homology‐associated protein 1, leading to expression of NRF2 target genes that induce autophagy.129 Also, the transcription factor early growth response protein‐1 is a substrate of the proteasome and activates expression of genes within the autophagy pathway.130, 131 Conversely, RING (really interesting new gene)‐domain ubiquitin E3 ligases, which target proteins for proteasomal degradation, regulate autophagy and are themselves degraded by autophagy.132
The cellular proteolytic systems are therefore regulated in a coordinated fashion to enable adequate distribution of molecular resources according to changes in growth conditions. Practically this means that inhibition of one proteolytic system activates another proteolytic system. Inhibition of UPS by chemical agents leads to the activation of autophagy by increasing the expression levels of several autophagy‐related genes.133, 134 Consistently, the activity of UPS was increased when autophagy was inhibited by chemical agents or by small interfering RNAs targeting autophagy‐related genes.135, 136
There are several examples of this complementarity. When proteasome activity is impaired, its substrates may be imported into mitochondria to be degraded by mitophagy.137 Transcription factor NF‐κB is induced by inflammatory stimuli that activate proteasomal degradation of inhibitor of NF‐κBα (IκBα), which otherwise inhibits NF‐κB by sequestering it in the cytoplasm.138 After the degradation of IκBα, NF‐κB is translocated into the nucleus and interacts with nuclear hormone receptors, stress mediators, and tumor suppressors, to activate the expression of genes with different expression dynamics.139, 140 DNA damaging agents increase the activity of NF‐κB, both by increased recruitment to chromatin, as well as by increased interaction with protein complexes that recruit RNA polymerase II.141, 142, 143 As a mechanism of negative feedback regulation, the IκBα gene is expressed by activated NF‐κB.144, 145 To sustain NF‐κB activity in some cells, inhibition of the proteasome may induce proteolysis of IκBα by the lysosome, and inhibition of the lysosome can induce proteolysis of IκBα by calpain.146, 147 The degree of redundancy of these proteolytic systems in IκBα degradation depends on the cell type and the phase of the inflammatory cascade.148, 149, 150 In endothelial cells, inflammatory cytokines induce degradation of IκBα by autophagy, which leads to the expression of vascular cell adhesion molecule 1.151 By activating expression of vascular cell adhesion molecule 1, autophagy then enables the next step in the inflammatory cascade, which is the adhesion of lymphocytes to the endothelium and the recruitment of immune cells to the inflammation site.152
With regard to leukocytes, it was shown that 10 μm CQ caused M1 macrophage polarization through lysosomal calcium release and activation of protein kinase p38 and NF‐κB.60 Similarly, the proteasome inhibitor bortezomib induced autophagic degradation of IκBα leading to the activation of NF‐κB in diffuse large B‐cell lymphoma cells.153 Specifically, p62 recruits ubiquitinated proteins, including IκBα, to autophagosomes. Consequently, NF‐κB activation and degradation of p62 and IκBα was blocked by a higher CQ concentration (50 μm), which potentiated lymphoma cell death by bortezomib (Velcade). Furthermore, p62 can activate NF‐κB through diverse pathways.154 Autophagy can also activate NF‐κB by sequestering its inhibitor A20, thereby permitting expression of chemokines CXCL1 and CXCL2, recruiting neutrophils, and extending antimicrobial inflammatory responses.155 This partial redundancy may allow cell survival under inflammatory or toxic insult conditions.37 Under conditions of the physiological activity of the UPS, IκBα proteolysis permits NF‐κB to activate IκBα re‐synthesis, which in turn inhibits NF‐κB activity by sequestering NF‐κB in the cytoplasm. If NF‐κB was the only factor capable of inducing IκBα expression, then NF‐κB activation would function as a self‐limiting cycle, whereby NF‐κB activity gradually returned to basal levels.156
Expression of cell stress regulators affects cancer prognosis
In contrast to inflammatory genes, some endogenous anti‐inflammatory mediators, such as glucocorticoids, can activate IκBα and thereby maintain the inflammatory genes in a repressed state under normal homeostatic conditions.157 However, in triple‐negative breast cancer cells, the recognition of substrates by IκBα is dysregulated and therefore IκBα is not sufficient to block expression of NF‐κB‐driven inflammatory genes.158, 159 The expressed inflammatory genes inactivate natural killer (NK) cells and recruit neutrophils into both primary tumor and lung pre‐metastatic niche, enabling breast cancer cell metastasis.160 Can there be a prognostic impact of this dichotomy between triple‐negative breast cancer and the other breast cancer types with more efficient NF‐κB regulation by IκBα?
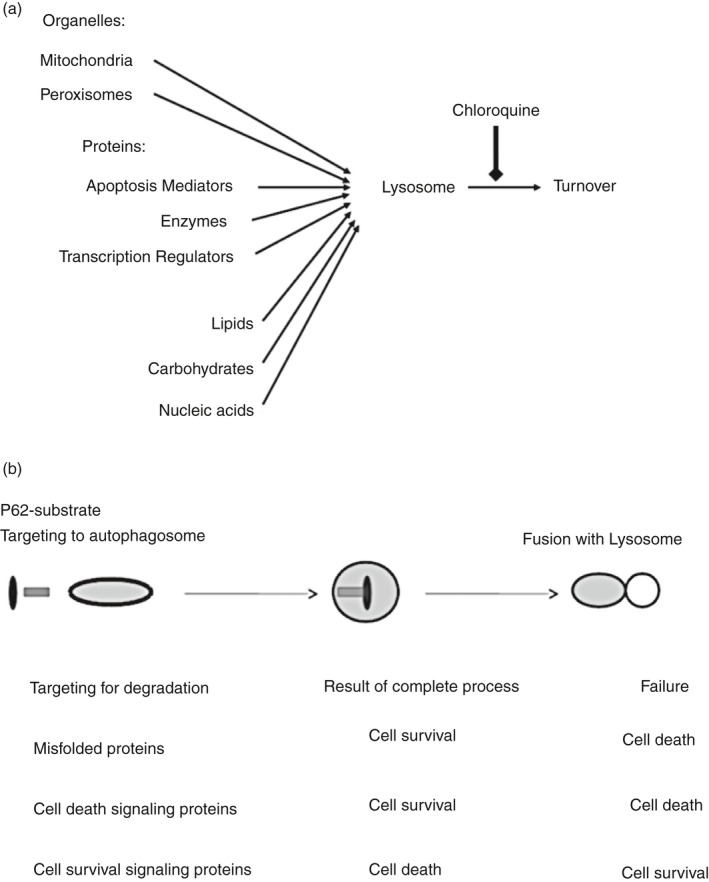
The fact that IκBα gene expression becomes less relevant in triple‐negative breast cancer cells can be illustrated by contrasting its effects on prognosis, when compared with the other types of breast cancer. In the case of triple‐negative breast cancer, neither higher IκBα expression nor a higher ratio of IκBα/ p62 gene expression offers survival benefit as seen in The Cancer Genome Atlas database (http://cancergenome.nih.gov/), whereas in other types of breast cancer, both higher IκBα as well as a higher ratio of IκBα/p62 gene expression has a significant positive effect on prognosis (Fig. 2a). The impact of changes in expression was calculated using the database proggenev2.161 Even higher prognostic divergence between triple‐negative and other types of breast cancers can be shown with the ratio between IκBα and Autophagy related 4C cysteine peptidase (ATG4C), which is required for stress‐induced autophagy, (such as under conditions of oxidative stress or prolonged starvation)162, 163 (Fig. 2b). Conversely, an eight‐gene signature composed of four autophagy‐related and four proteasome‐related genes offers a survival advantage in only triple‐negative breast cancer, while lacking this effect in the other types of breast cancer (Fig. 2c). This clearly shows a divergence in proteostatic effects for triple‐negative breast cancer and is in agreement with the hypothesis of a decreased impact of IκBα levels.
Figure 2.
Inhibitor of NF‐κBα (IκBα) is a key mediator at the interface between autophagy and the proteasome, and therefore the effect of its overexpression on cancer prognosis is an important factor. In triple‐negative breast cancer, the overexpression of proteasome and autophagic components (their gene products are also involved in IκBα degradation) is a positive prognostic factor, while in non‐triple‐negative breast cancer, it is IκBα that becomes a positive prognostic factor. This highlights the importance of proteostasis (proteasome and autophagy) in cancer prognosis. (a) Prognosis in triple‐negative breast cancer is not affected by expression of the gene encoding IκBα (NFkBIA) nor is prognosis affected by the ratio between the expression of NFkBIA and the p62 adaptor protein Sequestosome 1 (SQSTM1). In contrast, in other types of breast cancer, this ratio has a significant impact on survival. (b) The same divergence in prognosis between triple‐negative and the other types of breast cancer is true for the ratio between IκBα and ATG4C. (c) An eight‐gene signature (PSMD11, PSMG3, PSMB4, PSMC3, ATG4C, UVRAG, MAP1LC3A, MAP1LC3C) correlates with a survival advantage only in triple‐negative breast cancer (Black curve: high expression; grey: low expression)
The dichotomy in prognostic effects between ATG4C and IκBα is remarkable and shows that while a proper autophagic stress response protects from cancer, overexpression of a key component of this response mechanism in disproportion to the expression of anti‐inflammatory protein IκBα is tightly linked to negative prognosis. Furthermore, while both p62 and IκBα are targets of CQ, their expression has divergent results depending on the cancer subtype. As IκBα gene expression is a point of convergence of negative feedback mechanisms of inflammation, the dichotomy in prognostic effects demonstrates the importance of proteostasis in the regulation of genes that affect tissue integrity.
Cell stress regulators are partially redundant: impact on cancer progression
Nevertheless, NF‐κB activity may be inhibited at different levels; for example by marking of the upstream NF‐κB activating proteins tumor necrosis factor receptor‐associated factor 2 (TRAF2) and TRAF5 for degradation by the lysosome.164 This is important in the regulation of cell fate, as autophagic degradation of TRAF2 limits the capacity of a cell to undergo NF‐κB‐dependent epithelial to mesenchymal transitions.164 NF‐κB itself regulates genes that encode proteins operating both inside the cell, in its organelles, and in the extracellular space, and thereby controls all cellular functions, including internal homeostasis and communication with the microenvironment38, 147 (Fig. 3). Furthermore, NF‐κB also regulates the expression of autophagy genes to enable cellular adaptation and survival under adverse conditions.165
Figure 3.
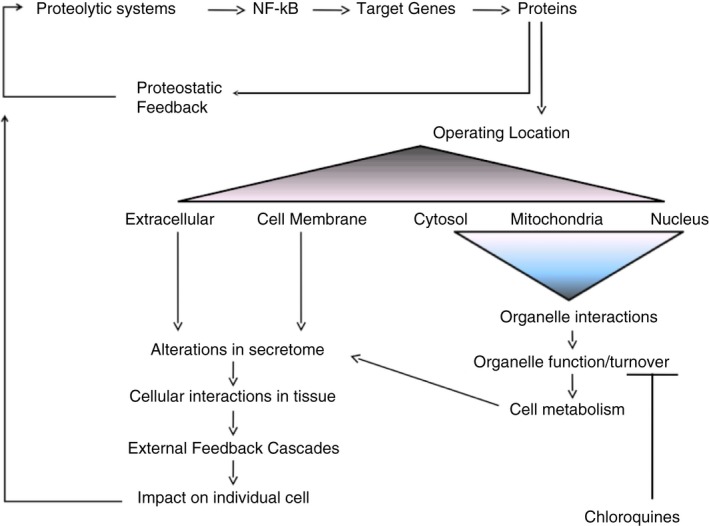
Proteostasis regulates gene expression and thereby controls the extent and duration of inflammation and cell stress. As an example, the transcription factor nuclear factor‐κB (NF‐κB) is induced by inflammatory stimuli and controls the expression of intracellular proteins and extracellular mediators. Proteolytic systems, primarily the proteasome and secondarily other systems such as lysosome, degrade the immediate NF‐κB inhibitor nhibitor of NF‐κBα (IκBα) to facilitate the pro‐inflammatory activity of NF‐κB, and expression of cell survival genes. Normally regulation of gene expression by NF‐κB ultimately includes cessation of transcriptional activity. A number of negative feedback mechanisms resolve inflammation by inhibiting pro‐inflammatory NF‐κB activity. Inhibition of lysosomal acidification by CQs disrupts the regulation of proteostasis, and a significant part of the linked molecular turnover
In cancer cells, adaptation mediated by the autophagy–proteostatic feedback is detrimental for the host, whereas in normal cells, this mechanism preserves essential tissue homeostasis. In melanoma cells, 25 μm CQ suffices to cause accumulation of the autophagy adaptor protein p62, inducing NF‐κB activity and thereby leading to expression of survival genes and p62 itself, culminating in cell resistance to apoptosis.166 Nevertheless, in normal cells, coordinated proteostatic feedback is beneficial for the host tissue. An example of such a beneficial function of the proteostatic feedback is seen when NF‐κB promotes mitophagy through induction of p62 expression in macrophages, thereby preventing the accumulation of damaged mitochondria and limiting excessive IL‐1β‐dependent inflammation.167
In the opposite direction, mechanisms that induce inflammation limit autophagy. NF‐κB‐target gene IL‐6 limits autophagy in healthy tissues.100 One can then ask, how do cells reprogram expression from inflammatory genes to autophagy genes if both share some of their transcriptional activators? The answer probably lies in the recruitment of chromatin regulators, whose activities are regulated through metabolic functions. Bromodomain and Extra Terminal domain (BET) family proteins are epigenetic readers that recognize histone acetylation and promote expression of inflammatory genes, whereas histone deacetylases such as sirtuin 1 (SIRT1) function as epigenetic erasers and promote expression of autophagy genes.168, 169 BET functions as a repressor for some genes such as TFEB, whereas activated AMPK allows SIRT1 to displace BET.170 This function could be expected to promote organelle restoration and cell survival under conditions of depleted nutrients and oxidative stress during inflammation. Indeed, negative feedback between autophagy and inflammation protects tissue integrity in the normal homeostatic state but fails in pathological states such as acute lung injury.171, 172, 173 Hence, intracellular IκBα proteostasis controls the immune system and tissue homeostasis through relocation of NF‐κB.
Disruption of tissue capacity to regulate autophagy can generate a suitable niche for senescent stromal cells that can either enhance tumor growth or provide a niche for metastatic cancer cells.174, 175 In fact, cancer cells may actively reprogram autophagy in stromal cells to promote cancer progression.176, 177 On the other hand, cancer cells treated with antimitotic drugs may escape mitotic arrest by entering interphase without proper chromosome segregation. This escape is linked to the induction of autophagy and leads to cellular senescence, migration, invasion and vascularization.178 Therefore, both impaired autophagy and excessive or dysregulated autophagy can disrupt tissue homeostasis and favor cancer progression (Fig. 4).
Figure 4.
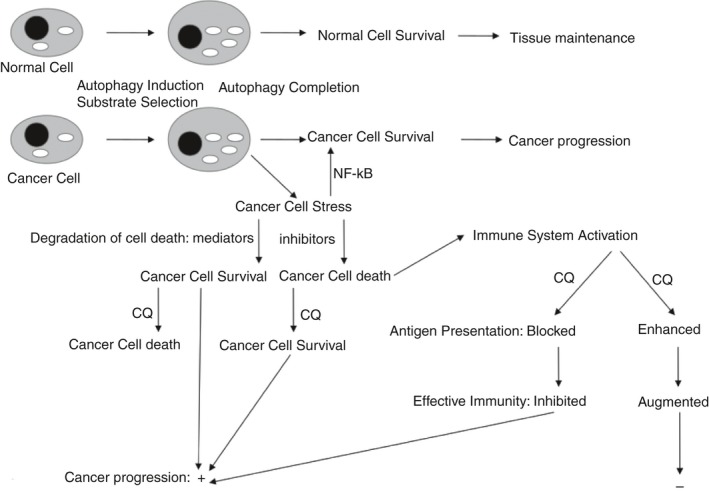
Chloroquines are expected to have diverse effects on cancer progression. Inhibition of late autophagy can affect cell survival, as well as antigen processing and presentation in the immune system. Combinatorial study of these effects at several levels will help enhance the capability to incorporate them in cancer treatment
CQ effects on cancer stem cells
Although conventional cancer treatment approaches reduce tumor mass/volume, they often fail to eradicate all tumor cells.179 The major reason for treatment failure is related to tumor heterogeneity.180 It has been proposed that cancer stem cells (CSC) or cancer‐initiating cells (CIC) with self‐renewal potential may be responsible for the cell heterogeneity in tumors.181 CSC or CIC are small sub‐populations of the cells that give rise to various cancer cell types and have the capacity of self‐regeneration within a tumor.182
Several properties of CSC affect the impact of CQ on tumors. CSC may derive from cells in a variety of differentiation stages, and autophagy bestows them with a critical degree of metabolic plasticity.183 During tumor growth, rapidly dividing cells that rely on glycolysis produce lactate. This, in turn, generates an acidic microenvironment that when combined with hypoxia leads to the development of quiescent cancer cell clones that share properties with stem cells.184 These quiescent cells rely on autophagy.185 Through autophagy, cancer cells and especially cancer stem‐like cell clones increase their capacity for material turnover and efflux of cancer drugs.186, 187 Therefore, autophagy has been considered as a major factor for the survival and chemotherapy resistance of CSCs.188 These protective effects of autophagy on CSC have been consistently shown in various cancers including colon and breast cancers and chronic myeloid leukemia.189, 190, 191 In addition, autophagy was proposed to enable neoplastic cells that have undergone epithelial‐to‐mesenchymal transitions to acquire CSC traits.192 Moreover, CSC markers and autophagy have been associated with poor prognosis in pancreatic cancer patients.193 Therefore, inhibition of autophagy is a promising approach to overcome chemotherapy resistance and consequently to eradicate tumor cells by reducing tumor heterogeneity.
In general, the capacity of CSC to maintain their carcinogenic potential is linked to their cell stress response mechanisms. As an example, CSC can increase their DNA double‐strand breaks, and thereafter induce the enzymatic activity of the ataxia telangiectasia mutated kinase (ATM), leading to activation of transcription factors NF‐κB and STAT3,194 two important transcription factors involved in tumorigenesis.195 Their deregulated activities were shown in various cancers including liver, lung, and prostate.196 In addition, STAT3 induces a number of DNA repair mechanisms, including ATM itself.197 ATM can further be induced by diverse conditions involving cell stress and organelle dysfunction.163 Moreover, alkylating agents such as temozolomide activate ATM, which induces cytoprotective autophagy to rescue tumor cells from chemotherapy, and even rescue cells from inhibition of NF‐κB and STAT3.198
Twenty micromolar CQ interferes specifically with ATM‐induced autophagy and stem cell‐like features in CSC.185 In a reported patient case with a follow up of 24 months, 150 mg/day CQ was well tolerated within an extensive ketogenic cancer treatment scheme that, in addition to temozolomide, also included AMPK activator metformin at 1 g/day.199 A clinical trial was initiated to determine metformin/CQ maximum tolerated doses for the treatment of solid tumors.200 Similar to other AMPK activators, mentioned herein, metformin inhibits activation of STAT3.201 Hence, it is becoming increasingly possible to evaluate the combination of autophagy activators with late‐stage autophagy inhibitors in the clinics.
The low micromolar concentration of CQ (1 or 5 μm) shows a striking capacity to decrease the expression of DNMT1, an important DNA methylation regulator that is highly expressed in various cancers and interferes with Janus‐activated kinase 2—STAT3 signaling in breast cancer CSC.202 In solid tumors, CQ interferes with STAT3, Hedgehog and CXCR4 signaling, as well as with autophagy in CSCs that are identified by the markers aldehyde dehydrogenase, CD44, and CD133.6, 203 In fact, aldehyde dehydrogenase sensitizes cancer cells to lysosomal autophagy inhibitors, including HCQ. Expression of helicase‐like transcription factor, a member of switch/sucrose non‐fermentable family proteins that regulate chromatin/nucleosome remodeling, promotes DNA damage repair and HCQ resistance, which indicates that HCQ could be combined with DNA repair inhibitors.204
Another class of inhibitors with which HCQ and CQ can be combined, are the inhibitors of the BET epigenetic readers, which drive the expression of genes promoting inflammation and cell survival.38, 170 What is encouraging is that a relatively low CQ/HCQ concentration (10 μm), in synergy with BET inhibition, is sufficient to cause apoptosis in CSC of both pancreatic cancer and acute myeloid leukemia.170, 205 CQ can also kill cancer cells in combination with cell cycle inhibitors.206, 207 A combination of CQ and proteasome inhibitor causes apoptosis in human liver tumors orthotopically or subcutaneously xenografted in mice.208 This demonstrates the importance of proteostasis for tumor cells.
Chloroquine and other inhibitors of autophagy have cytotoxic effects on diverse leukemia‐initiating cell types such as CD34‐positive and glucocorticoid‐resistant clones.205, 209 However, in leukemia patients, high doses of CQ are needed to inhibit autophagy, which limits their therapeutic efficacy.210 One potential explanation comes from the observation that CQ may kill leukemia cells independently from the early steps of the autophagy pathway, and this CQ effect can be neutralized by leukemia cells in vivo through exocytosis.211
There exists yet another mechanism that impairs the antineoplastic activity of CQ in a patients' tissue. Cancer cells commonly cause a localized acidosis that prevents the entry of CQ.30, 212 Nevertheless, CQ could still reach migrating cancer cells that have not yet entered or established a sufficiently acidic tumor niche to block CQ entry.213 Such a niche results from interactions between tumor and stromal cells.214 Hence, CQ can affect metastatic cells in transition and this can explain the statistically significant effect of CQ against metastatic cancer cells in patients.215 Furthermore, metastatic cells are preferentially vulnerable to lysosomal inhibition.216 The capacity of CQ to affect cancer cells in transition can also be linked to the interactions between transcription factors that steer cellular metabolism.217, 218 As an example, interactions between NF‐κB, STAT3, p53, and the glucocorticoid receptor (GR) guide dynamic transitions in the structures and functions of a cells' organelles and the resulting changes in the cells' phenotype.219, 220, 221, 222 Organelles, in turn, form intracellular networks that control autophagy and cell death.223, 224
Transcription factors such as NF‐κB, STAT3, p53, and GR interact at various levels. Their interactions impact the progress of the cell cycle, the response to nutritional changes, the induction of cell stress, organelle function, recycling and expression of cell surface molecules, and the secretion of diverse chemo‐attractants.225, 226, 227, 228 The importance of NF‐κB, STAT3, p53, and GR interactions is especially pronounced in malignant cells, and guides the cells' interactions with host tissue.147 CQ also impacts the dynamic interface between NF‐κB, STAT3, p53, and GR, and the phenomena these proteins control, including mitochondrial membrane potential229 and mitophagy.230 Although this interference can be associated with undesirable side effects of pharmacological CQ use at higher concentrations, it also forms a basis for the treatment of malignant tumors, especially to target drug‐resistant CSC.
Impact of CQ on antitumor immune responses
Antitumor immune response requires autophagy. Autophagy enhances immunogenic cell death and tumor antigen processing and presentation, thereby promoting adaptive antitumor immunity.231 In tumor cells, autophagy leads to immunogenic cell death by surface exposure of calreticulin (ecto‐CRT), secretion of ATP, and release of apoptotic proteins such as HMGB1. In antigen‐presenting cells, autophagy promotes antigen presentation by MHC.34 Lysosomal acidification in distinct phases of the pathways that lead to antigen presentation is needed and this need depends heavily on the nature of the antigen.64 MHC‐I‐driven presentation, which tends to be promoted by CQ, exposes cancer‐derived neoantigens to the immune system.232, 233
On the other hand, autophagy can also suppress immune responses. Autophagy, especially under hypoxia, permits activation of transcription factor STAT3, leading to the expression and secretion of cytokines such as IL‐10, and a number of other factors with immunosuppressive effects that help cancer cells escape multiple components of the immune system.34 This phenotype has often been observed in solid neoplasms and generally leads to more aggressive treatment‐resistant tumor phenotypes.9 Consequently, blocking the hypoxia‐induced autophagy in tumors restored cytotoxic T‐cell activity and promoted regression of melanoma xenografts in mice.234 Clearly, cancer is a complex condition, and difficult to describe immunologically. Therefore, the analysis of immune responses to antigens related to other diseases can aid in defining the immunological effect of CQs.
Do CQs have an immunosuppressive effect, especially at the antigen presentation level? In the case of malaria, an effective T helper type 1 (Th1) ‐dominated immune response requires TLR4, TLR9, interferon‐γ (IFN‐γ), and immunoglobulins IgG2a, IgG2b, and IgG3.235, 236 In malaria, a critical 3‐day time course is needed to mount an immune response before a 50 mg/kg dose of CQ can be added to the treatment regimen.237
The capacity of CQ to protect tumors from the mammalian immune system is evident by adding CQ to curcumin in immunocompetent mice. Namely, CQ treatment increases the cytotoxic effect of curcumin against epidermal growth factor receptor 2‐overexpressing breast cancer cells in nude mice while counteracting it in immune competent mice.238 Moreover, it was shown that CQ inhibits acidification of endolysosomes and consequently impairs antigen presentation to CD4+ T cells in HIV infection.239 Similarly, it was reported that HCQ inhibits intracellular TLR signaling in rheumatoid arthritis.239
Chloroquine has a beneficial immunosuppressive effect against IFN‐γ‐directed immune responses in autoimmunity and especially in lupus, where it balances the effect of autophagy in Th17 and regulatory T (Treg) cells, reducing IFN‐γ and inflammatory cytokines.240 Lupus also involves TLR9, IgG2a, and IgG2b, consistent with the beneficial immunosuppressive effect of CQ.241 In lectin‐stimulated human peripheral blood mononuclear cells, CQ inhibits soluble IL‐2R expression dose‐dependently at concentrations between 10 and 50 μm. It is interesting that under the same conditions, 10 μm CQ augments, while 50 μm CQ blocks neopterin expression, which suggests a potential of CQ to normalize IFN‐γ signaling in some tissues.242 Another potential beneficial effect of CQ against autoimmunity is evident in experimental autoimmune encephalomyelitis, a mouse model for multiple sclerosis. This condition is ameliorated by a CQ treatment protocol (5 mg/kg/day for 5 consecutive days) that promotes the expansion of Treg cells and decreases inflammatory cells.243 CQ‐treated mice showed a significant reduction in the number of IL‐17A‐ and IFN‐γ‐producing cells and a significant increase of IL‐10‐producing cells in the central nervous system.
On the other hand, CQs show a capacity to stimulate immune responses under certain conditions. T‐cell‐deficient mice depend on the activity of NK cells and on the tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) signaling, to mount antitumor immune responses.244 However, tumor cells may escape TRAIL and NK cells by activating autophagy.245, 246 This capacity is especially enhanced in tumors under hypoxia.247 CQ can be onco‐suppressive against tumor cells that activate autophagy to escape TRAIL‐induced apoptosis, or escape killing by NK cells.246, 248 In the case of human cancer xenografts in mice, 75 mg/kg CQ had an immunostimulatory effect, especially once the Treg cells were depleted by the addition of cyclophosphamide. Particularly, CQ resets the tumor‐associated macrophages from the immunosuppressive M2 toward a stimulatory M1 phenotype.60 CQ also prevents the clearance of apoptotic cells by macrophages.249 This effect could augment the immune response against tumors. In dendritic cells, autophagy that leads to MHC‐I depletion decreases their efficiency in stimulating CD8+ T cells against the influenza virus.250 In an experimental system of irradiated human bone marrow cells, 40 μm CQ and a stabilized derivative of cyclophosphamide were sufficient to stimulate elimination of contaminating B‐cell acute lymphoblastic leukemia cells by a selective B‐cell‐directed antibody coupled to an immunotoxin.251
However, in the presence of a fully functional immune system, low‐dose CQ (10 mg/kg) could trigger potent IFN‐γ‐associated immune responses against irradiated tumors, thereby protecting mice from further tumor challenge.252 Moreover, CQ has also shown the potential to prolong the survival of xenograft‐bearing mice by augmenting CD8+ T cells, and suppressing tumor‐associated macrophages, myeloid derived suppressor cells, and Treg cells in the tumor microenvironment.253 In parallel in vitro assays, CQ inhibited secretion of transforming growth factor‐β, while enhancing secretion of IFN‐γ. It can therefore be concluded that the balance of T‐cell types in the tumor microenvironment can determine the impact of CQs on the immune response. The capacity of CQ to stimulate immune responses can be extended to eliciting antiviral as well as antitumor activity in the clinical setting.254
What appears consistent between the lupus and cancer studies is the critical role played not only by macrophage polarizing conditions but also by the T‐helper phenotype. T cells are remarkably resistant to drugs that cause endoplasmic reticulum stress and autophagy.255 CQ can protect against autoimmune responses by decreasing expression of both Th1 and Th2 components.256 Suppression of Th1, albeit without the potential to inhibit Th17‐driven autoimmunity, was consistent also in cultured monocyte‐derived Langerhans‐like cells in response to IL‐1β, where 20 μm HCQ allowed them to prime CD4‐negative T cells for IL‐17 production, while suppressing IFN‐γ production.257 It would therefore be interesting to examine if in the immunocompetent mouse systems where CQs are immunosuppressive, this effect can be reversed by Treg cell depletion. In conclusion, even though lysosomes are an essential component of cells of the immune system, CQs can have beneficial effects, depending on the timing of administration and the cell types activated in the tumor microenvironment.
Clearly, other goals, such as the use of CQ to enhance antineoplastic drug retention, which requires high‐dose and simultaneous treatment with both drugs, are not consistent with the aim to avoid immunosuppression. This is because of the time needed for the immune response to commence. The choice of administering CQ after the antineoplastic drug is the first step for mutual compatibility, and the question remains, on how long the ideal pause of treatment before CQ administration is. The 3 days needed for the restoration of IFN‐γ response may be too long for cases where CQ is needed to act rapidly to enhance cell stress. One possible solution is to develop experimental treatment schemes that include both dosing with a 3‐day delay, as well as dosing with simultaneous administration of two drugs. During simultaneous administration with other drugs, CQ needs to be either at a smaller dose or in a specific nanocarrier that can escape scavenging by antigen‐presenting cells.
Feasibility of treating malignant tumors with CQ: strengths and weaknesses
Free CQ has limited utility as a prospective monotherapy. Individuals on long‐term exposure to HCQ or CQ are not at lower risk of cancer. However, there is statistical evidence that CQs may lower the risk of metastatic cancer and death.215 Therefore, CQ interferes with the capacity of malignant tumors to generate lethal metastases. Consistently, it was shown that CQ induces the secretion of tumor suppressor Par‐4 in a p53‐dependent manner.258 Secretion of Par‐4 triggered apoptosis and inhibited metastatic tumor growth.258 This effect of CQ would be consistent with autophagy inhibition, because autophagy bestows malignant cells a metabolic flexibility that is essential to survive in niches with limited nutrient availability. Hence CQ can be expected to inhibit homeostatic adaptation of malignant cells.37 Indeed, in triple‐negative breast cancer tumors, CQ kills CD44+/CD24−/low CSCs and diminishes the tumors' ability to metastasize in vitro and in mouse xenografts.259 It is important, however, to emphasize that CQs mainly affect tumor cells with stem‐like features.205 Therefore, their clinical application for cancer would be limited unless used in combination with drugs that kill cancer cells with more differentiated features.
Chloroquine cannot enter cells in an acidic microenvironment, which currently further limits its clinical application as explained in the introduction. The combination of CQ with biguanide metformin entered a clinical trial for patients with solid tumors bearing mutations in the IDH1 and IDH2 genes.200 IDH‐mutant gliomas might be suited for CQ treatment. These gliomas had shown a decreased capacity to generate an acidic extracellular microenvironment, an effect, however, that cannot be generalized.260 One potential solution to decrease tissue acidity is the use of inhibitors of glycolytic flux such as dichloroacetate, which might inhibit acidosis without compromising cancer treatment.261 Their effectiveness in pH regulation of the tumor microenvironment will still need to be empirically determined.
Given the low capacity of CQ to penetrate in cells in an acidic environment, it is imperative to develop methods that allow entry of CQ in malignant tumors in vivo. In addition to blocking entry of free CQ into cells, the acidic extracellular microenvironment triggers activation of the transcription factor TFEB that induces lysosomal biogenesis.30, 262 TFEB is one of the regulatory proteins that in cancer cells can become uncoupled from mechanisms that control their cytoplasmic retention. Deregulated TFEB leads to metabolic reprogramming of the cancer microenvironment.263 In vitro, cancer cell autophagy and TFEB activity can be modeled in three‐dimensional growth conditions.264 In many cell types, though, especially in normal macrophage cells, TFEB is mutually antagonistic to STAT3 activity, and would thereby limit the protection of a tumor by macrophages.265 It was shown that TFEB is activated and translocated to the nucleus via inhibition of mammalian target of rapamycin complex by CQ.266 Activation of TFEB by CQ can induce antitumor M1 macrophage activity.60 M1 macrophages could improve the effect of chemotherapy and lead to tumor growth regression.267 On the other hand, in cancer cells, active STAT3 could still facilitate lysosomal cell death by inhibiting TFEB in the nucleus.268 STAT3 is, however, also a factor critical in the M1‐to‐M2 activity transitions in the tumors.269 Therefore, an intervention that differentially targets malignant cells from macrophages could have an added value.
With respect to potential drug combinations, an example of a drug that targets CSCs, and in parallel induces cytoprotective autophagy, is polyether ionophore antibiotic salinomycin. On one hand, salinomycin was shown to interfere with tumor necrosis factor and IL‐6 signaling and to impair networking of transcription factors STAT1 and STAT3.270 On the other hand, it may target the Wnt/β‐catenin signaling pathway to promote differentiation and thus elimination of CSCs, while rescuing normal fibroblasts from CQ toxicity.271 Salinomycin is a drug that must be used at strictly limited concentrations, due to its potential neurotoxicity.272, 273 This can make its synergistic antineoplastic effects with other drugs beneficial, if they decrease systemic exposure and toxicity to non‐transformed cells. To this end, under the acidic conditions that inhibit CQ activity, salinomycin reaches the acidic core of multicellular tumor spheroids and impairs the autophagic flux, thereby killing malignant cells.274 This could allow a ‘first hit’ against a tumor and pave the way for a combination treatment. In line with the potential of CQ for synergy with inhibitors of DNA repair, the antineoplastic effect of salinomycin is neutralized by the induction of DNA repair via DNA–protein kinase (DNA‐PK) enzymes.275 Hence, theoretically both salinomycin and CQ can be combined with DNA repair inhibitors.
DNA‐PK enzymes are a group of interesting targets to inhibit, as they have the capacity to increase the ratio of IL‐10 to IL‐12, and thereby can inhibit Th1‐driven antitumor immune responses.276 Oncogene activation increases DNA replication stress, which can make at least a fraction of tumor cells highly dependent on DNA maintenance.277 In addition to impairing DNA maintenance, DNA‐PK inhibitors could affect immune responses at several levels, and therefore the kinetics and dynamics of adding them to a combination treatment scheme need to be studied.276, 278 The main difference between CQ and salinomycin is the capacity of salinomycin to both induce, and under some conditions inhibit the autophagic flux. Salinomycin can induce autophagy via AMPK, and in parallel inhibit autophagy at later stages: thereby salinomycin use exploits the imbalanced induction of autophagic pathway components that takes place in some cancer cell types.275, 279 Hence, the mechanisms through which salinomycin activates autophagy apparently make cancer cells prone to cell death, depending on the downstream signals it elicits. This is probably the main reason that salinomycin can sensitize some cells to CQ.
In fact, CQ can also inhibit topoisomerase II, although weakly, in addition to its lysosomotropic activity.280 Interestingly, it was reported that CQ preferentially enhances the death of Myc‐overexpressing cells, in a p53‐dependent but not an ATM‐dependent manner.93 Moreover, CQ can prevent genomic instability by inhibiting etoposide (Topoisomerase II inhibitor)‐induced centrosome amplification in a CDK2 inhibiting manner.14 Similarly, CQ can be combined with other drugs causing DNA damage, such as anthracyclines, especially when using nanocarriers that enable cell penetration, leading to inhibition of the growth of human prostate cancer xenografts in mice.45 Therefore, a combination with inhibitors of DNA repair can be studied, as under certain conditions it might improve Th1‐driven immune responses, and in parallel, provide additional cytotoxicity toward malignant cells.143 In human breast cancer cells, a mutant adenomatous polyposis coli gene induces STAT3, an activator of DNA repair genes, which renders cells resistant to anthracyclines.197, 281 In normal tissues, STAT3 would protect from excessive Th1 activity and inflammation.282 In normal and tumor tissue alike, the activity of STAT3 itself affects vascularity and pericyte interactions with their microenvironment, including myeloid cells.283, 284, 285, 286 However, unlike malignant tissue niches, normal tissue uses the intact regulatory network of STAT3 and NF‐κB as a common mechanism to recover from inflammation.147, 287, 288 Therefore, the activation of STAT3 by constitutive DNA damage in cancer cells could facilitate selective drug effects on the tumor nest through the circulation, while having discrete indirect effects on the local tissue microenvironment owing to the secretome of the tumor. It is therefore beneficial to dissect effects experimentally by cell‐selective agents. To some extent, cellular targeting by CQs can be achieved by conjugation with specific moieties, including sugar groups, and by the generation of hybrid molecules, such as CQ‐artemisinin.20, 289, 290
What is the technical maturity of the concepts of CQ use against tumors? Even though CQ is an established drug against certain conditions, its use in cancer is still in its infancy, as is evident by the clinical trials that have been publicized. The translational work of mechanistic CQ combination concepts on animal models is currently placed at an intermediate technology readiness level. Namely, as reviewed in previous sections, a number of concepts developed on the basis of induction of cell stress and under those conditions the experimental uses of CQ against tumors are gaining more attention.43, 68, 102, 291 Hopefully at least the most promising concepts and combination methods will ultimately reach the safety level required to enter clinical trials. On the other hand, in spite of the widely recognized dose‐limiting toxicity of CQs, the technology of molecular carriers for CQs or similar agents to reprogram immune responses, with cell‐selectivity and retaining direct antineoplastic activity at a low dose, is not yet close to clinical use.292, 293 In contrast to genomics, there does not seem to be a major change of research support in sight for high‐capacity molecular innovation.294, 295, 296 Furthermore, exploration of molecular concepts cannot be covered by biopharmaceutical industries, due to the fact that the concepts address essentially basic research. Research priorities, however, can change in the public sector.297, 298, 299 Grant calls that cover the topic of drug repurposing technology to reprogram immune responses with cell‐selectivity and parallel onco‐suppression will be beneficial to both basic and translational research.
Conclusion
Our understanding of cancer dynamics and metabolism has improved greatly. However, there are a lot of details in the field that need to be understood to develop effective treatment options for various cancers. Based on our knowledge of the molecular mechanisms of carcinogenesis and cancer cell survival, cancer treatment concepts are also evolving. The relationship between autophagy and cancer progression has been well documented. Therefore, an approach for cancer treatment can be formulated by targeting autophagic mechanisms.
Chloroquine and its analogues are valuable tools in cancer research. They will become consolidated as antineoplastic agents as soon as our progress in cancer research helps elucidate their molecular mode of action. Research shows that under certain circumstances cancer cells develop a dependency on autophagy, either of their own contents, or on autophagy activity of stromal cells. In such cases, an organelle‐selective or cell‐selective and intense exposure to CQs or their derivatives could provide researchers with the opportunity to sensitize tumors to a number of antineoplastic drugs. Development of CQ‐based cancer treatment options is expected to advance significantly within the next decade. Through this development, the bench‐based research has the capacity to provide translational research with several interesting targets, which in the long run will certainly improve our concepts of renewing the antineoplastic armamentarium.
The best known effect of CQs is the inhibition of autophagy by increasing lysosomal pH. However, another mechanism to inhibit autophagy, via preventing autophagosome–lysosome fusion, has recently been proposed. This new mechanism should be further investigated due to its direct influence on immune response, inflammation and carcinogenesis. Concordantly, the study of uncharacterized effects of CQs may lead to an unexpected discovery of modulating factors for immunological activity, a crucial parameter for both carcinogenesis and cancer treatment.
Disclosures
All authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Informed consent
For this type of study, formal consent is not required.
Supporting information
Appendix S1. Genomic targets of chloroquine pharmacological activity. Shown is a snapshot from the query of the database DRUGSURV (http://www.bioprofiling.de/cgi-bin/GEO/DRUGSURV/start_DRUG.pl) for chloroquine.
References
- 1. Al‐Bari MA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother 2015; 70:1608–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baker NC, Ekins S, Williams AJ, Tropsha A. A bibliometric review of drug repurposing. Drug Discovery Today 2018; 23:661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Briceno E, Calderon A, Sotelo J. Institutional experience with chloroquine as an adjuvant to the therapy for glioblastoma multiforme. Surg Neurol 2007; 67:388–91. [DOI] [PubMed] [Google Scholar]
- 4. Amann VC, Dreier J, Ignatova D, Kamarashev J, Kerl K, Kempf W et al Disseminated primary cutaneous CD8+ small/medium‐sized pleomorphic T‐cell lymphoma responding to hydroxychloroquine. Acta dermato‐venereologica 2015; 95:602–3. [DOI] [PubMed] [Google Scholar]
- 5. Samaras P, Tusup M, Nguyen‐Kim TDL, Seifert B, Bachmann H, von Moos R et al Phase I study of a chloroquine‐gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother Pharmacol 2017; 80:1005–12. [DOI] [PubMed] [Google Scholar]
- 6. Vlahopoulos S, Critselis E, Voutsas IF, Perez SA, Moschovi M, Baxevanis CN et al New use for old drugs? Prospective targets of chloroquines in cancer therapy. Curr Drug Targets 2014; 15:843–51. [DOI] [PubMed] [Google Scholar]
- 7. Manic G, Obrist F, Kroemer G, Vitale I, Galluzzi L. Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol 2014; 1:e29911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H et al Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402(6762):672–6. [DOI] [PubMed] [Google Scholar]
- 9. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G et al Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu Z, Yang L, Xu S, Zhang Z, Cao Y. The receptor proteins: pivotal roles in selective autophagy. Acta Biochim Biophys Sin 2015; 47:571–80. [DOI] [PubMed] [Google Scholar]
- 11. Chude CI, Amaravadi RK. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci 2017; 18:E1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Verbaanderd C, Maes H, Schaaf MB, Sukhatme VP, Pantziarka P, Sukhatme V et al Repurposing Drugs in Oncology (ReDO)‐chloroquine and hydroxychloroquine as anti‐cancer agents. Ecancermedicalscience 2017; 11:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koukourakis MI, Mitrakas AG, Giatromanolaki A. Therapeutic interactions of autophagy with radiation and temozolomide in glioblastoma: evidence and issues to resolve. Br J Cancer 2016; 114:485–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen TY, Syu JS, Lin TC, Cheng HL, Lu FL, Wang CY. Chloroquine alleviates etoposide‐induced centrosome amplification by inhibiting CDK2 in adrenocortical tumor cells. Oncogenesis 2015; 4:e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Levy JM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther 2011; 131:130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S et al A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014; 10:1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu R, Ji Z, Xu C, Zhu J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: a systematic review and meta‐analysis. Medicine 2018; 97:e12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Das AK. Anticancer effect of antimalarial artemisinin compounds. Ann Med Health Sci Res 2015; 5:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP, Vikas P. The Repurposing Drugs in Oncology (ReDO) Project. Ecancermedicalscience 2014; 8:442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bharate JB, Vishwakarma RA, Bharate SB. Metal‐free domino one‐pot protocols for quinoline synthesis. RSC Adv 2015; 5:42020–53. [Google Scholar]
- 21. Gattu R, Bagdi PR, Basha RS, Khan AT. Camphorsulfonic acid catalyzed one‐pot three‐component reaction for the synthesis of fused quinoline and benzoquinoline derivatives. J Org Chem 2017; 82:12416–29. [DOI] [PubMed] [Google Scholar]
- 22. Zheng J, Li Z, Huang L, Wu W, Li J, Jiang H. Palladium‐catalyzed intermolecular aerobic annulation of o‐alkenylanilines and alkynes for quinoline synthesis. Org Lett 2016; 18:3514–7. [DOI] [PubMed] [Google Scholar]
- 23. Pareek A, Chandurkar N, Thomas N, Viswanathan V, Deshpande A, Gupta OP et al Efficacy and safety of hydroxychloroquine in the treatment of type 2 diabetes mellitus: a double blind, randomized comparison with pioglitazone. Curr Med Res Opin 2014; 30:1257–66. [DOI] [PubMed] [Google Scholar]
- 24. Rempenault C, Combe B, Barnetche T, Gaujoux‐Viala C, Lukas C, Morel J et al Metabolic and cardiovascular benefits of hydroxychloroquine in patients with rheumatoid arthritis: a systematic review and meta‐analysis. Ann Rheum Dis 2018; 77:98–103. [DOI] [PubMed] [Google Scholar]
- 25. Restrepo JF, Del Rincon I, Molina E, Battafarano DF, Escalante A. Use of hydroxychloroquine is associated with improved lipid profile in rheumatoid arthritis patients. J Clin Rheumatol: Pract Rep Rheumatic Musculoskeletal Dis 2017; 23:144–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lim Y, Hedayati M, Merchant AA, Zhang Y, Yu HH, Kastan MB et al Chloroquine improves survival and hematopoietic recovery after lethal low‐dose‐rate radiation. Int J Radiat Oncol Biol Phys 2012; 84:800–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wozniacka A, Lesiak A, Boncela J, Smolarczyk K, McCauliffe DP, Sysa‐Jedrzejowska A. The influence of antimalarial treatment on IL‐1β, IL‐6 and TNF‐α mRNA expression on UVB‐irradiated skin in systemic lupus erythematosus. Br J Dermatol 2008; 159:1124–30. [DOI] [PubMed] [Google Scholar]
- 28. Ettinger WH Jr. Forces of change in the health care system. Implications for cancer care in the 1990s. Cancer 1991; 67(6 Suppl):1728–31. [DOI] [PubMed] [Google Scholar]
- 29. Soto‐Perez‐de‐Celis E, de Glas NA, Hsu T, Kanesvaran R, Steer C, Navarrete‐Reyes AP et al Global geriatric oncology: achievements and challenges. J Geriatric Oncol 2017; 8:374–86. [DOI] [PubMed] [Google Scholar]
- 30. Pellegrini P, Strambi A, Zipoli C, Hagg‐Olofsson M, Buoncervello M, Linder S et al Acidic extracellular pH neutralizes the autophagy‐inhibiting activity of chloroquine: implications for cancer therapies. Autophagy 2014; 10:562–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shan L. Hyperpolarized 13C‐labeled bicarbonate (H13CO3–) for in vivo pH measurement with 13C magnetic resonance spectroscopy In: Molecular Imaging and Contrast Agent Database (MICAD). Bethesda, MD: National Center for Biotechnology Information, 2004. [PubMed] [Google Scholar]
- 32. Gonzalez CD, Alvarez S, Ropolo A, Rosenzvit C, Bagnes MF, Vaccaro MI. Autophagy, Warburg, and Warburg reverse effects in human cancer. Biomed Res Int 2014; 2014:926729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhong Z, Sanchez‐Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell 2016; 166:288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Viry E, Noman MZ, Arakelian T, Lequeux A, Chouaib S, Berchem G et al Hijacker of the antitumor immune response: autophagy is showing its worst facet. Front Oncol 2016; 6:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Antonov AV. BioProfiling.de: analytical web portal for high‐throughput cell biology. Nucleic Acids Res 2011; 39(suppl):W323–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Antonov AV, Krestyaninova M, Knight RA, Rodchenkov I, Melino G, Barlev NA. PPISURV: a novel bioinformatics tool for uncovering the hidden role of specific genes in cancer survival outcome. Oncogene 2014; 33:1621–8. [DOI] [PubMed] [Google Scholar]
- 37. Moschovi M, Critselis E, Cen O, Adamaki M, Lambrou GI, Chrousos GP et al Drugs acting on homeostasis: challenging cancer cell adaptation. Expert Rev Anticancer Ther 2015; 15:1405–17. [DOI] [PubMed] [Google Scholar]
- 38. Vlahopoulos SA. Aberrant control of NF‐κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: molecular mode. Cancer Biol Med 2017; 14:254–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bertolini F, Sukhatme VP, Bouche G. Drug repurposing in oncology – patient and health systems opportunities. Nat Rev Clin Oncol 2015; 12:732–42. [DOI] [PubMed] [Google Scholar]
- 40. Hernandez JJ, Pryszlak M, Smith L, Yanchus C, Kurji N, Shahani VM et al Giving drugs a second chance: overcoming regulatory and financial hurdles in repurposing approved drugs as cancer therapeutics. Front Oncol 2017; 7:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol 2009; 625:220–33. [DOI] [PubMed] [Google Scholar]
- 42. Angelakis E, Million M, Kankoe S, Lagier JC, Armougom F, Giorgi R et al Abnormal weight gain and gut microbiota modifications are side effects of long‐term doxycycline and hydroxychloroquine treatment. Antimicrob Agents Chemother 2014; 58:3342–7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43. Mei L, Chen Y, Wang Z, Wang J, Wan J, Yu C et al Synergistic anti‐tumour effects of tetrandrine and chloroquine combination therapy in human cancer: a potential antagonistic role for p21. Br J Pharmacol 2015; 172:2232–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gao M, Xu Y, Qiu L. Sensitization of multidrug‐resistant malignant cells by liposomes co‐encapsulating doxorubicin and chloroquine through autophagic inhibition. J Liposome Res 2017; 27:151–60. [DOI] [PubMed] [Google Scholar]
- 45. Panagiotaki KN, Sideratou Z, Vlahopoulos SA, Paravatou‐Petsotas M, Zachariadis M, Khoury N et al A triphenylphosphonium‐functionalized mitochondriotropic nanocarrier for efficient co‐delivery of doxorubicin and chloroquine and enhanced antineoplastic activity. Pharmaceuticals 2017; 10:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Y, Shi K, Zhang L, Hu G, Wan J, Tang J et al Significantly enhanced tumor cellular and lysosomal hydroxychloroquine delivery by smart liposomes for optimal autophagy inhibition and improved antitumor efficiency with liposomal doxorubicin. Autophagy 2016; 12:949–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Natarajan JK, Alumasa JN, Yearick K, Ekoue‐Kovi KA, Casabianca LB, de Dios AC et al 4‐N‐, 4‐S‐, and 4‐O‐chloroquine analogues: influence of side chain length and quinolyl nitrogen pKa on activity vs chloroquine resistant malaria. J Med Chem 2008; 51:3466–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double‐edged sword of autophagy. Can Res 2013; 73:3–7. [DOI] [PubMed] [Google Scholar]
- 49. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ et al Chloroquine inhibits autophagic flux by decreasing autophagosome‐lysosome fusion. Autophagy 2018; 14:1435–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yoo SM, Jung YK. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells 2018; 41:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Biel TG, Rao VA. Mitochondrial dysfunction activates lysosomal‐dependent mitophagy selectively in cancer cells. Oncotarget 2018; 9:995–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik‐Zainal S, Ramakrishna M et al Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014; 3:e02935 10.7554/eLife.02935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen X, Chen Z, Hu B, Cai P, Wang S, Xiao S et al Synergistic lysosomal activatable polymeric nanoprobe encapsulating pH sensitive imidazole derivative for tumor diagnosis. Small 2018; 14:1703164. [DOI] [PubMed] [Google Scholar]
- 54. Chen S, Zhang Y, Zhou L, Leng Y, Lin H, Kmieciak M et al A Bim‐targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood 2014; 124:2687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guan JJ, Zhang XD, Sun W, Qi L, Wu JC, Qin ZH. DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis 2015; 6:e1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chaanine AH, Gordon RE, Nonnenmacher M, Kohlbrenner E, Benard L, Hajjar RJ. High‐dose chloroquine is metabolically cardiotoxic by inducing lysosomes and mitochondria dysfunction in a rat model of pressure overload hypertrophy. Physiol Rep 2015; 3:e12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Redmann M, Benavides GA, Berryhill TF, Wani WY, Ouyang X, Johnson MS et al Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol 2017; 11:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hipolito VEB, Ospina‐Escobar E, Botelho RJ. Lysosome remodelling and adaptation during phagocyte activation. Cell Microbiol 2018; 20:e12824. [DOI] [PubMed] [Google Scholar]
- 59. Lim JJ, Grinstein S, Roth Z. Diversity and versatility of phagocytosis: roles in innate immunity, tissue remodeling, and homeostasis. Front Cell Infect Microbiol 2017; 7:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J et al Chloroquine modulates antitumor immune response by resetting tumor‐associated macrophages toward M1 phenotype. Nat Commun 2018; 9:873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li GG, Guo ZZ, Ma XF, Cao N, Geng SN, Zheng YQ et al The M2 macrophages induce autophagic vascular disorder and promote mouse sensitivity to urethane‐related lung carcinogenesis. Dev Comp Immunol 2016; 59:89–98. [DOI] [PubMed] [Google Scholar]
- 62. Long Y, Liu X, Wang N, Zhou H, Zheng J. Chloroquine attenuates LPS‐mediated macrophage activation through miR‐669n‐regulated SENP6 protein translation. Am J Transl Res 2015; 7:2335–45. [PMC free article] [PubMed] [Google Scholar]
- 63. Alloatti A, Kotsias F, Magalhaes JG, Amigorena S. Dendritic cell maturation and cross‐presentation: timing matters! Immunol Rev 2016; 272:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Belizaire R, Unanue ER. Targeting proteins to distinct subcellular compartments reveals unique requirements for MHC class I and II presentation. Proc Natl Acad Sci USA 2009; 106:17463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vieira OV, Botelho RJ, Grinstein S. Phagosome maturation: aging gracefully. Biochem J 2002; 366(Pt 3):689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330(6009):1344–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728–41. [DOI] [PubMed] [Google Scholar]
- 68. Dash S, Chava S, Chandra PK, Aydin Y, Balart LA, Wu T. Autophagy in hepatocellular carcinomas: from pathophysiology to therapeutic response. Hepatic Med: Evid Resarch 2016; 8:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Coto‐Montes A, Boga JA, Rosales‐Corral S, Fuentes‐Broto L, Tan DX, Reiter RJ. Role of melatonin in the regulation of autophagy and mitophagy: a review. Mol Cell Endocrinol 2012; 361:12–23. [DOI] [PubMed] [Google Scholar]
- 70. He M, Luo M, Liu Q, Chen J, Li K, Zheng M et al Combination treatment with fasudil and clioquinol produces synergistic anti‐tumor effects in U87 glioblastoma cells by activating apoptosis and autophagy. J Neurooncol 2016; 127:261–70. [DOI] [PubMed] [Google Scholar]
- 71. Zhang Z, Liu T, Yu M, Li K, Li W. The plant alkaloid tetrandrine inhibits metastasis via autophagy‐dependent Wnt/β‐catenin and metastatic tumor antigen 1 signaling in human liver cancer cells. J Exp Clin Cancer Res: CR 2018; 37:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li C, Liu Y, Liu H, Zhang W, Shen C, Cho K et al Impact of autophagy inhibition at different stages on cytotoxic effect of autophagy inducer in glioblastoma cells. Cell Physiol Biochem: Int J Exp Cell Physiol Biochem Pharmacol 2015; 35:1303–16. [DOI] [PubMed] [Google Scholar]
- 73. Kwon Y, Kim JW, Jeoung JA, Kim MS, Kang C. Autophagy is pro‐senescence when seen in close‐up, but anti‐senescence in long‐shot. Mol Cells 2017; 40:607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ma L, Zhu J, Gao Q, Rebecchi MJ, Wang Q, Liu L. Restoring pharmacologic preconditioning in the aging heart: role of mitophagy/autophagy. J Gerontol Ser A, Biol Sci Med Sci 2017; 72:489–98. [DOI] [PubMed] [Google Scholar]
- 75. Godar RJ, Ma X, Liu H, Murphy JT, Weinheimer CJ, Kovacs A et al Repetitive stimulation of autophagy‐lysosome machinery by intermittent fasting preconditions the myocardium to ischemia‐reperfusion injury. Autophagy 2015; 11:1537–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bartlett JJ, Trivedi PC, Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol Cell Cardiol 2017; 104:1–8. [DOI] [PubMed] [Google Scholar]
- 77. Moruno‐Manchon JF, Uzor NE, Kesler SR, Wefel JS, Townley DM, Nagaraja AS et al Peroxisomes contribute to oxidative stress in neurons during doxorubicin‐based chemotherapy. Mol Cell Neurosci 2018; 86:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Aburto MR, Hurle JM, Varela‐Nieto I, Magarinos M. Autophagy during vertebrate development. Cells 2012; 1:428–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lu WH, Wang G, Li Y, Li S, Song XY, Wang XY et al Autophagy functions on EMT in gastrulation of avian embryo. Cell Cycle 2014; 13:2752–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cai Y, Wang XL, Flores AM, Lin T, Guzman RJ. Inhibition of endo‐lysosomal function exacerbates vascular calcification. Sci Rep 2018; 8:3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yu Y, Wang L, Delguste F, Durand A, Guilbaud A, Rousselin C et al Advanced glycation end products receptor RAGE controls myocardial dysfunction and oxidative stress in high‐fat fed mice by sustaining mitochondrial dynamics and autophagy‐lysosome pathway. Free Radic Biol Med 2017; 112:397–410. [DOI] [PubMed] [Google Scholar]
- 83. Wang R, Xi L, Kukreja RC. PDE5 inhibitor tadalafil and hydroxychloroquine cotreatment provides synergistic protection against type 2 diabetes and myocardial infarction in mice. J Pharmacol Exp Ther 2017; 361:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A et al Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 2015; 11:1146–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wei H, Qu H, Wang H, Ji B, Ding Y, Liu D et al 1,25‐Dihydroxyvitamin‐D3 prevents the development of diabetic cardiomyopathy in type 1 diabetic rats by enhancing autophagy via inhibiting the β‐catenin/TCF4/GSK‐3β/mTOR pathway. J Steroid Biochem Mol Biol 2017; 168:71–90. [DOI] [PubMed] [Google Scholar]
- 86. Dai X, Zeng J, Yan X, Lin Q, Wang K, Chen J et al Sitagliptin‐mediated preservation of endothelial progenitor cell function via augmenting autophagy enhances ischaemic angiogenesis in diabetes. J Cell Mol Med 2018; 22:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li X, Wu Z, Zhang Y, Xu Y, Han G, Zhao P. Activation of autophagy contributes to sevoflurane‐induced neurotoxicity in fetal rats. Front Mol Neurosci 2017; 10:432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhang X, Zhou Y, Xu M, Chen G. Autophagy is involved in the sevoflurane anesthesia‐induced cognitive dysfunction of aged rats. PLoS ONE 2016; 11:e0153505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chang TC, Hsu MF, Wu KK. High glucose induces bone marrow‐derived mesenchymal stem cell senescence by upregulating autophagy. PLoS ONE 2015; 10:e0126537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yu X, Ye X, Lin H, Feng N, Gao S, Zhang X et al Knockdown of long non‐coding RNA LCPAT1 inhibits autophagy in lung cancer. Cancer Biol Med 2018; 15:228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. He Y, Xu Y, Zhang C, Gao X, Dykema KJ, Martin KR, et al Identification of a lysosomal pathway that modulates glucocorticoid signaling and the inflammatory response. Sci Signal 2011; 4:ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pujals A, Favre L, Pioche‐Durieu C, Robert A, Meurice G, Le Gentil M et al Constitutive autophagy contributes to resistance to TP53‐mediated apoptosis in Epstein–Barr virus‐positive latency III B‐cell lymphoproliferations. Autophagy 2015; 11:2275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53‐dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Investig 2008; 118:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Birkenmeier K, Moll K, Newrzela S, Hartmann S, Drose S, Hansmann ML. Basal autophagy is pivotal for Hodgkin and Reed–Sternberg cells' survival and growth revealing a new strategy for Hodgkin lymphoma treatment. Oncotarget 2016; 7:46579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li XC, Hu QK, Chen L, Liu SY, Su S, Tao H et al HSPB8 promotes the fusion of autophagosome and lysosome during autophagy in diabetic neurons. Int J Med Sci 2017; 14:1335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Luo L, Dai JR, Guo SS, Lu AM, Gao XF, Gu YR et al Lysosomal proteolysis is associated with exercise‐induced improvement of mitochondrial quality control in aged hippocampus. J Gerontol Ser A, Biol Sci Med Sci 2017; 72:1342–51. [DOI] [PubMed] [Google Scholar]
- 97. Bolanos‐Meade J, Zhou L, Hoke A, Corse A, Vogelsang G, Wagner KR. Hydroxychloroquine causes severe vacuolar myopathy in a patient with chronic graft‐versus‐host disease. Am J Hematol 2005; 78:306–9. [DOI] [PubMed] [Google Scholar]
- 98. Muratsubaki S, Kuno A, Tanno M, Miki T, Yano T, Sugawara H et al Suppressed autophagic response underlies augmentation of renal ischemia/reperfusion injury by type 2 diabetes. Sci Rep 2017; 7:5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jiang XS, Chen XM, Wan JM, Gui HB, Ruan XZ, Du XG. Autophagy protects against palmitic acid‐induced apoptosis in podocytes in vitro . Sci Rep 2017; 7:42764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Linnemann AK, Blumer J, Marasco MR, Battiola TJ, Umhoefer HM, Han JY et al Interleukin 6 protects pancreatic beta cells from apoptosis by stimulation of autophagy. FASEB J 2017; 31:4140–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dimitrakis P, Romay‐Ogando MI, Timolati F, Suter TM, Zuppinger C. Effects of doxorubicin cancer therapy on autophagy and the ubiquitin‐proteasome system in long‐term cultured adult rat cardiomyocytes. Cell Tissue Res 2012; 350:361–72. [DOI] [PubMed] [Google Scholar]
- 102. Lee YJ, Lee SH. Pro‐oxidant activity of sulforaphane and cisplatin potentiates apoptosis and simultaneously promotes autophagy in malignant mesothelioma cells. Mol Med Rep 2017; 16:2133–41. [DOI] [PubMed] [Google Scholar]
- 103. Koubkova L, Vyzula R, Karban J, Pinkas J, Ondrouskova E, Vojtesek B et al Evaluation of cytotoxic activity of titanocene difluorides and determination of their mechanism of action in ovarian cancer cells. Invest New Drugs 2015; 33:1123–32. [DOI] [PubMed] [Google Scholar]
- 104. Szebeni GJ, Balazs A, Madarasz I, Pocz G, Ayaydin F, Kanizsai I et al Achiral mannich‐base curcumin analogs induce unfolded protein response and mitochondrial membrane depolarization in PANC‐1 cells. Int J Mol Sci 2017; 18:2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I et al Warburg meets autophagy: cancer‐associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal 2012; 16:1264–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ke R, Xu Q, Li C, Luo L, Huang D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol Int 2018; 42:384–92. [DOI] [PubMed] [Google Scholar]
- 107. Lin DS, Kao SH, Ho CS, Wei YH, Hung PL, Hsu MH et al Inflexibility of AMPK‐mediated metabolic reprogramming in mitochondrial disease. Oncotarget 2017; 8:73627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Singh K, Matsuyama S, Drazba JA, Almasan A. Autophagy‐dependent senescence in response to DNA damage and chronic apoptotic stress. Autophagy 2012; 8:236–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zai W, Chen W, Luan J, Fan J, Zhang X, Wu Z et al Dihydroquercetin ameliorated acetaminophen‐induced hepatic cytotoxicity via activating JAK2/STAT3 pathway and autophagy. Appl Microbiol Biotechnol 2018; 102:1443–53. [DOI] [PubMed] [Google Scholar]
- 110. Xie WY, Zhou XD, Yang J, Chen LX, Ran DH. Inhibition of autophagy enhances heat‐induced apoptosis in human non‐small cell lung cancer cells through ER stress pathways. Arch Biochem Biophys 2016; 607:55–66. [DOI] [PubMed] [Google Scholar]
- 111. Lee KH, Hsu EC, Guh JH, Yang HC, Wang D, Kulp SK et al Targeting energy metabolic and oncogenic signaling pathways in triple‐negative breast cancer by a novel adenosine monophosphate‐activated protein kinase (AMPK) activator. J Biol Chem 2011; 286:39247–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wang L, Wang J, Xiong H, Wu F, Lan T, Zhang Y et al Co‐targeting hexokinase 2‐mediated Warburg effect and ULK1‐dependent autophagy suppresses tumor growth of PTEN‐ and TP53‐deficiency‐driven castration‐resistant prostate cancer. EBioMedicine 2016; 7:50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hu K, Yang Y, Lin L, Ai Q, Dai J, Fan K et al Caloric restriction mimetic 2‐deoxyglucose alleviated inflammatory lung injury via suppressing nuclear pyruvate kinase M2‐signal transducer and activator of transcription 3 pathway. Front Immunol 2018; 9:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Vangala JR, Dudem S, Jain N, Kalivendi SV. Regulation of PSMB5 protein and beta subunits of mammalian proteasome by constitutively activated signal transducer and activator of transcription 3 (STAT3): potential role in bortezomib‐mediated anticancer therapy. J Biol Chem 2014; 289:12612–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhang XD, Baladandayuthapani V, Lin H, Mulligan G, Li B, Esseltine DW et al Tight junction protein 1 modulates proteasome capacity and proteasome inhibitor sensitivity in multiple myeloma via EGFR/JAK1/STAT3 signaling. Cancer Cell 2016; 29:639–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ji CH, Kwon YT. Crosstalk and Interplay between the ubiquitin‐proteasome system and autophagy. Mol Cells 2017; 40:441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Fraser J, Cabodevilla AG, Simpson J, Gammoh N. Interplay of autophagy, receptor tyrosine kinase signalling and endocytic trafficking. Essays Biochem 2017; 61:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Liu X, Sun K, Wang H, Dai Y. Inhibition of autophagy by chloroquine enhances the antitumor efficacy of sorafenib in glioblastoma. Cell Mol Neurobiol 2016; 36:1197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. You L, Shou J, Deng D, Jiang L, Jing Z, Yao J et al Crizotinib induces autophagy through inhibition of the STAT3 pathway in multiple lung cancer cell lines. Oncotarget 2015; 6:40268–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lambrou GI, Papadimitriou L, Chrousos GP, Vlahopoulos SA. Glucocorticoid and proteasome inhibitor impact on the leukemic lymphoblast: multiple, diverse signals converging on a few key downstream regulators. Mol Cell Endocrinol 2012; 351:142–51. [DOI] [PubMed] [Google Scholar]
- 121. Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature 2003; 426(6968):895–99. [DOI] [PubMed] [Google Scholar]
- 122. Bader M, Steller H. Regulation of cell death by the ubiquitin‐proteasome system. Curr Opin Cell Biol 2009; 21:878–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Muratani M, Tansey WP. How the ubiquitin‐proteasome system controls transcription. Nat Rev Mol Cell Biol 2003; 4:192–201. [DOI] [PubMed] [Google Scholar]
- 124. Peters JM. The anaphase‐promoting complex: proteolysis in mitosis and beyond. Mol Cell 2002; 9:931–43. [DOI] [PubMed] [Google Scholar]
- 125. Latonen L, Moore HM, Bai B, Jaamaa S, Laiho M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 2011; 30:790–805. [DOI] [PubMed] [Google Scholar]
- 126. Ciechanover A, Kwon YT. Protein quality control by molecular chaperones in neurodegeneration. Front Neurosci 2017; 11:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell 2010; 38:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL et al p62 links the autophagy pathway and the ubiqutin‐proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett 2016; 21:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Jiang T, Harder B, Rojo de la Vega M, Wong PK, Chapman E, Zhang DD. p62 links autophagy and Nrf2 signaling. Free Radic Biol Med 2015; 88(Pt B):199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Bae MH, Jeong CH, Kim SH, Bae MK, Jeong JW, Ahn MY et al Regulation of Egr‐1 by association with the proteasome component C8. Biochem Biophys Acta 2002; 1592:163–7. [DOI] [PubMed] [Google Scholar]
- 131. Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali‐Bostwick C, Stolz DB et al Egr‐1 regulates autophagy in cigarette smoke‐induced chronic obstructive pulmonary disease. PLoS ONE 2008; 3:e3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kuang E, Qi J, Ronai Z. Emerging roles of E3 ubiquitin ligases in autophagy. Trends Biochem Sci 2013; 38:453–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ge PF, Zhang JZ, Wang XF, Meng FK, Li WC, Luan YX et al Inhibition of autophagy induced by proteasome inhibition increases cell death in human SHG‐44 glioma cells. Acta Pharmacol Sin 2009; 30:1046–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhu K, Dunner K Jr, McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010; 29:451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM et al Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation 2008; 117:3070–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wang XJ, Yu J, Wong SH, Cheng AS, Chan FK, Ng SS et al A novel crosstalk between two major protein degradation systems: regulation of proteasomal activity by autophagy. Autophagy 2013; 9:1500–8. [DOI] [PubMed] [Google Scholar]
- 137. Ruan L, Zhou C, Jin E, Kucharavy A, Zhang Y, Wen Z et al Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 2017; 543(7645):443–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Vlahopoulos S, Boldogh I, Casola A, Brasier AR. Nuclear factor‐κB‐dependent induction of interleukin‐8 gene expression by tumor necrosis factor alpha: evidence for an antioxidant sensitive activating pathway distinct from nuclear translocation. Blood 1999; 94:1878–89. [PubMed] [Google Scholar]
- 139. Copland JA, Sheffield‐Moore M, Koldzic‐Zivanovic N, Gentry S, Lamprou G, Tzortzatou‐Stathopoulou F et al Sex steroid receptors in skeletal differentiation and epithelial neoplasia: is tissue‐specific intervention possible? BioEssays: News Rev Mol Cell Dev Biol 2009; 31:629–41. [DOI] [PubMed] [Google Scholar]
- 140. Kolovos P, Georgomanolis T, Koeferle A, Larkin JD, Brant L, Nikolicc M et al Binding of nuclear factor βB to noncanonical consensus sites reveals its multimodal role during the early inflammatory response. Genome Res 2016; 26:1478–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Pan L, Zhu B, Hao W, Zeng X, Vlahopoulos SA, Hazra TK et al Oxidized guanine base lesions function in 8‐oxoguanine DNA glycosylase‐1‐mediated epigenetic regulation of nuclear factor κB‐driven gene expression. J Biol Chem 2016; 291:25553–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Vlahopoulos S, Adamaki M, Khoury N, Zoumpourlis V, Boldogh I. Roles of DNA repair enzyme OGG1 in innate immunity and its significance for lung cancer. Pharmacol Ther 2019; 194:59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Zaffini R, Gotte G, Menegazzi M. Asthma and poly(ADP‐ribose) polymerase inhibition: a new therapeutic approach. Drug Des Dev Ther 2018; 12:281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Amici C, Rossi A, Costanzo A, Ciafre S, Marinari B, Balsamo M et al Herpes simplex virus disrupts NF‐κB regulation by blocking its recruitment on the IkappaBalpha promoter and directing the factor on viral genes. J Biol Chem 2006; 281:7110–17. [DOI] [PubMed] [Google Scholar]
- 145. Algarte M, Kwon H, Genin P, Hiscott J. Identification by in vivo genomic footprinting of a transcriptional switch containing NF‐κB and Sp1 that regulates the IκBα promoter. Mol Cell Biol 1999; 19:6140–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Lee KH, Jeong J, Yoo CG. Long‐term incubation with proteasome inhibitors (PIs) induces IkappaBalpha degradation via the lysosomal pathway in an IκB kinase (IKK)‐dependent and IKK‐independent manner. J Biol Chem 2013; 288:32777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Vlahopoulos SA, Cen O, Hengen N, Agan J, Moschovi M, Critselis E et al Dynamic aberrant NF‐κB spurs tumorigenesis: a new model encompassing the microenvironment. Cytokine Growth Factor Rev 2015; 26:389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Colleran A, Ryan A, O'Gorman A, Mureau C, Liptrot C, Dockery P et al Autophagosomal IκBα degradation plays a role in the long term control of tumor necrosis factor‐α‐induced nuclear factor‐κB (NF‐κB) activity. J Biol Chem 2011; 286:22886–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Han Y, Weinman S, Boldogh I, Walker RK, Brasier AR. Tumor necrosis factor‐α‐inducible IκBα proteolysis mediated by cytosolic m‐calpain. A mechanism parallel to the ubiquitin‐proteasome pathway for nuclear factor‐κb activation. J Biol Chem 1999; 274:787–94. [DOI] [PubMed] [Google Scholar]
- 150. Iguchi‐Hashimoto M, Usui T, Yoshifuji H, Shimizu M, Kobayashi S, Ito Y et al Overexpression of a minimal domain of calpastatin suppresses IL‐6 production and Th17 development via reduced NF‐κB and increased STAT5 signals. PLoS ONE 2011; 6:e27020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Chu LY, Hsueh YC, Cheng HL, Wu KK. Cytokine‐induced autophagy promotes long‐term VCAM‐1 but not ICAM‐1 expression by degrading late‐phase IκBα . Sci Rep 2017; 7:12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Zapolska‐Downar D, Siennicka A, Kaczmarczyk M, Kolodziej B, Naruszewicz M. Butyrate inhibits cytokine‐induced VCAM‐1 and ICAM‐1 expression in cultured endothelial cells: the role of NF‐κB and PPARα . J Nutr Biochem 2004; 15:220–8. [DOI] [PubMed] [Google Scholar]
- 153. Jia L, Gopinathan G, Sukumar JT, Gribben JG. Blocking autophagy prevents bortezomib‐induced NF‐κB activation by reducing I‐κBα degradation in lymphoma cells. PLoS ONE 2012; 7:e32584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Islam MA, Sooro MA, Zhang P. Autophagic regulation of p62 is critical for cancer therapy. Int J Mol Sci 2018; 19:1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Kanayama M, Inoue M, Danzaki K, Hammer G, He YW, Shinohara ML. Autophagy enhances NFκB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun 2015; 6:5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kourdis PD, Palasantza AG, Goussis DA. Algorithmic asymptotic analysis of the NF‐κB signaling system. Comput Math Appl 2013; 65:1516–34. [Google Scholar]
- 157. Deroo BJ, Archer TK. Glucocorticoid receptor activation of the IκBα promoter within chromatin. Mol Biol Cell 2001; 12:3365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Fusella F, Secli L, Busso E, Krepelova A, Moiso E, Rocca S et al The IKK/NF‐κB signaling pathway requires Morgana to drive breast cancer metastasis. Nat Commun 2017; 8:1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Uddin MM, Zou Y, Sharma T, Gatla HR, Vancurova I. Proteasome inhibition induces IKK‐dependent interleukin‐8 expression in triple negative breast cancer cells: opportunity for combination therapy. PLoS ONE 2018; 13:e0201858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Fusella F, Secli L, Brancaccio M. Escaping NK cells and recruiting neutrophils: how Morgana/NF‐κB signaling promotes metastasis. Mol Cell Oncol 2018; 5:e1432258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Goswami CP, Nakshatri H. PROGgeneV2: enhancements on the existing database. BMC Cancer 2014; 14:970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Marino G, Salvador‐Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez‐Otin C. Tissue‐specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin‐3. J Biol Chem 2007; 282:18573–83. [DOI] [PubMed] [Google Scholar]
- 163. Stagni V, Cirotti C, Barila D. Ataxia‐telangiectasia mutated kinase in the control of oxidative stress, mitochondria, and autophagy in cancer: a maestro with a large orchestra. Front Oncol 2018; 8:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Choi JM, Devkota S, Sung YH, Lee HW. EI24 regulates epithelial‐to‐mesenchymal transition and tumor progression by suppressing TRAF2‐mediated NF‐κB activity. Oncotarget 2013; 4:2383–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Wang HL, Fan SS, Pang M, Liu YH, Guo M, Liang JB et al The ankyrin repeat domain 49 (ANKRD49) augments autophagy of serum‐starved GC‐1 cells through the NF‐κB pathway. PLoS ONE 2015; 10:e0128551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Yang S, Qiang L, Sample A, Shah P, He YY. NF‐κB signaling activation induced by chloroquine requires autophagosome, p62 protein, and c‐Jun N‐terminal kinase (JNK) signaling and promotes tumor cell resistance. J Biol Chem 2017; 292:3379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Zhong Z, Umemura A, Sanchez‐Lopez E, Liang S, Shalapour S, Wong J et al NF‐κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016; 164:896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kokkola T, Suuronen T, Pesonen M, Filippakopoulos P, Salminen A, Jarho EM et al BET inhibition upregulates SIRT1 and alleviates inflammatory responses. Chembiochem: Eur J Chem Biol 2015; 16:1997–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Ou X, Lee MR, Huang X, Messina‐Graham S, Broxmeyer HE. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014; 32:1183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Sakamaki J‐I, Wilkinson S, Hahn M, Tasdemir N, O'Prey J, Clark W, et al Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Molecular Cell 2017; 66:517–32.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Chen ZH, Wu YF, Wang PL, Wu YP, Li ZY, Zhao Y et al Autophagy is essential for ultrafine particle‐induced inflammation and mucus hyperproduction in airway epithelium. Autophagy 2016; 12:297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Hu Y, Lou J, Mao YY, Lai TW, Liu LY, Zhu C et al Activation of MTOR in pulmonary epithelium promotes LPS‐induced acute lung injury. Autophagy 2016; 12:2286–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Pan H, Zhang Y, Luo Z, Li P, Liu L, Wang C et al Autophagy mediates avian influenza H5N1 pseudotyped particle‐induced lung inflammation through NF‐κB and p38 MAPK signaling pathways. Am J Physiol Lung Cell Mol Physiol 2014; 306:L183–95. [DOI] [PubMed] [Google Scholar]
- 174. Cai L, Xu S, Piao C, Qiu S, Li H, Du J. Adiponectin induces CXCL1 secretion from cancer cells and promotes tumor angiogenesis by inducing stromal fibroblast senescence. Mol Carcinog 2016; 55:1796–806. [DOI] [PubMed] [Google Scholar]
- 175. Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T. The senescent bystander effect is caused by ROS‐activated NF‐κB signalling. Mech Ageing Dev 2018; 170:30–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Gouirand V, Guillaumond F, Vasseur S. Influence of the tumor microenvironment on cancer cells metabolic reprogramming. Front Oncol 2018; 8:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Yang A, Herter‐Sprie G, Zhang H, Lin EY, Biancur D, Wang X et al Autophagy sustains pancreatic cancer growth through both cell‐autonomous and nonautonomous mechanisms. Cancer Discov 2018; 8:276–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Jakhar R, Luijten MNH, Wong AXF, Cheng B, Guo K, Neo SP et al Autophagy governs protumorigenic effects of mitotic slippage‐induced senescence. Mol Cancer Research: MCR 2018; 16:1625–40. [DOI] [PubMed] [Google Scholar]
- 179. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646–74. [DOI] [PubMed] [Google Scholar]
- 180. Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer 2013; 108:479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 2009; 138:822–9. [DOI] [PubMed] [Google Scholar]
- 182. Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells – old concepts, new insights. Cell Death Differ 2008; 15:947–58. [DOI] [PubMed] [Google Scholar]
- 183. Carnero A, Garcia‐Mayea Y, Mir C, Lorente J, Rubio IT, LLeonart ME. The cancer stem‐cell signaling network and resistance to therapy. Cancer Treat Rev 2016; 49:25–36. [DOI] [PubMed] [Google Scholar]
- 184. Wang L, Shang Z, Zhou Y, Hu X, Chen Y, Fan Y et al Autophagy mediates glucose starvation‐induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis 2018; 9:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Antonelli M, Strappazzon F, Arisi I, Brandi R, D'Onofrio M, Sambucci M et al ATM kinase sustains breast cancer stem‐like cells by promoting ATG4C expression and autophagy. Oncotarget 2017; 8:21692–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Alakhova DY, Zhao Y, Li S, Kabanov AV. Effect of doxorubicin/pluronic SP1049C on tumorigenicity, aggressiveness, DNA methylation and stem cell markers in murine leukemia. PLoS ONE 2013; 8:e72238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Klohs WD, Steinkampf RW. The effect of lysosomotropic agents and secretory inhibitors on anthracycline retention and activity in multiple drug‐resistant cells. Mol Pharmacol 1988; 34:180–5. [PubMed] [Google Scholar]
- 188. Ojha R, Bhattacharyya S, Singh SK. Autophagy in cancer stem cells: a potential link between chemoresistance, recurrence, and metastasis. BioResearch Open Access 2015; 4:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M et al Targeting autophagy potentiates tyrosine kinase inhibitor‐induced cell death in Philadelphia chromosome‐positive cells, including primary CML stem cells. J Clin Investig 2009; 119:1109–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Gong C, Bauvy C, Tonelli G, Yue W, Deloménie C, Nicolas V, et al Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem‐like/progenitor cells. Oncogene 2013; 32:2261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Kantara C, O'Connell M, Sarkar S, Moya S, Ullrich R, Singh P. Curcumin promotes autophagic survival of a subset of colon cancer stem cells, which are ablated by DCLK1‐siRNA. Can Res 2014; 74:2487–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Marcucci F, Ghezzi P, Rumio C. The role of autophagy in the cross‐talk between epithelial‐mesenchymal transitioned tumor cells and cancer stem‐like cells. Mol Cancer 2017; 16:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Valle S, Martin‐Hijano L, Alcala S, Alonso‐Nocelo M, Sainz B Jr. The ever‐evolving concept of the cancer stem cell in pancreatic cancer. Cancers 2018; 10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Liu X, Li F, Huang Q, Zhang Z, Zhou L, Deng Y et al Self‐inflicted DNA double‐strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res 2017; 27:764–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Fan Y, Mao R, Yang J. NF‐κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein & Cell 2013; 4:176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF‐κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 2010; 21:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Barry SP, Townsend PA, Knight RA, Scarabelli TM, Latchman DS, Stephanou A. STAT3 modulates the DNA damage response pathway. Int J Exp Pathol 2010; 91:506–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Zanotto‐Filho A, Braganhol E, Klafke K, Figueiro F, Terra SR, Paludo FJ et al Autophagy inhibition improves the efficacy of curcumin/temozolomide combination therapy in glioblastomas. Cancer Lett 2015; 358:220–31. [DOI] [PubMed] [Google Scholar]
- 199. Elsakka AMA, Bary MA, Abdelzaher E, Elnaggar M, Kalamian M, Mukherjee P et al Management of glioblastoma multiforme in a patient treated with ketogenic metabolic therapy and modified standard of care: a 24‐month follow‐up. Front Nutr 2018; 5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Molenaar RJ, Coelen RJS, Khurshed M, Roos E, Caan MWA, van Linde ME et al Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with IDH1‐mutated or IDH2‐mutated solid tumours. BMJ Open 2017; 7:e014961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Nerstedt A, Johansson A, Andersson CX, Cansby E, Smith U, Mahlapuu M. AMP‐activated protein kinase inhibits IL‐6‐stimulated inflammatory response in human liver cells by suppressing phosphorylation of signal transducer and activator of transcription 3 (STAT3). Diabetologia 2010; 53:2406–16. [DOI] [PubMed] [Google Scholar]
- 202. Choi DS, Blanco E, Kim YS, Rodriguez AA, Zhao H, Huang TH et al Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells 2014; 32:2309–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Balic A, Sorensen MD, Trabulo SM, Sainz B Jr, Cioffi M, Vieira CR et al Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther 2014; 13:1758–71. [DOI] [PubMed] [Google Scholar]
- 204. Piao S, Ojha R, Rebecca VW, Samanta A, Ma XH, McAfee Q et al ALDH1A1 and HLTF modulate the activity of lysosomal autophagy inhibitors in cancer cells. Autophagy 2017; 13:2056–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Jang JE, Eom JI, Jeung HK, Cheong JW, Lee JY, Kim JS et al AMPK‐ULK1‐mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin Cancer Res 2017; 23:2781–94. [DOI] [PubMed] [Google Scholar]
- 206. Okada Y, Kato S, Sakamoto Y, Oishi T, Ishioka C. Synthetic lethal interaction of CDK inhibition and autophagy inhibition in human solid cancer cell lines. Oncol Rep 2017; 38:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Zhu J, Zheng Y, Zhang H, Zhu J, Sun H. Low concentration of chloroquine enhanced efficacy of cisplatin in the treatment of human ovarian cancer dependent on autophagy. Am J Transl Res 2017; 9:4046–58. [PMC free article] [PubMed] [Google Scholar]
- 208. Hui B, Shi YH, Ding ZB, Zhou J, Gu CY, Peng YF et al Proteasome inhibitor interacts synergistically with autophagy inhibitor to suppress proliferation and induce apoptosis in hepatocellular carcinoma. Cancer 2012; 118:5560–71. [DOI] [PubMed] [Google Scholar]
- 209. Jiang L, Xu L, Xie J, Li S, Guan Y, Zhang Y et al Inhibition of autophagy overcomes glucocorticoid resistance in lymphoid malignant cells. Cancer Biol Ther 2015; 16:466–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Sehgal AR, Konig H, Johnson DE, Tang D, Amaravadi RK, Boyiadzis M et al You eat what you are: autophagy inhibition as a therapeutic strategy in leukemia. Leukemia 2015; 29:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Chen X, Clark J, Wunderlich M, Fan C, Davis A, Chen S et al Autophagy is dispensable for Kmt2a/Mll‐Mllt3/Af9 AML maintenance and anti‐leukemic effect of chloroquine. Autophagy 2017; 13:955–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Di Pompo G, Lemma S, Canti L, Rucci N, Ponzetti M, Errani C et al Intratumoral acidosis fosters cancer‐induced bone pain through the activation of the mesenchymal tumor‐associated stroma in bone metastasis from breast carcinoma. Oncotarget 2017; 8:54478–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Li Q, Yuan DM, Ma LH, Ma CH, Liu YF, Lv TF et al Chloroquine inhibits tumor growth and angiogenesis in malignant pleural effusion. Tumour Biol: J Int Soc Oncodev Biol Med 2016; 37:16249–58. [DOI] [PubMed] [Google Scholar]
- 214. Wu Y, Dong Y, Atefi M, Liu Y, Elshimali Y, Vadgama JV. Lactate, a neglected factor for diabetes and cancer interaction. Med Inflamm 2016; 2016:6456018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Fardet L, Nazareth I, Petersen I. Effects of chronic exposure of hydroxychloroquine/chloroquine on the risk of cancer, metastasis, and death: a population‐based cohort study on patients with connective tissue diseases. Clin Epidemiol 2017; 9:545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Morgan MJ, Fitzwalter BE, Owens CR, Powers RK, Sottnik JL, Gamez G et al Metastatic cells are preferentially vulnerable to lysosomal inhibition. Proc Natl Acad Sci USA 2018; 115:E8479–E8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Wang H, Wang L, Cao L, Zhang Q, Song Q, Meng Z et al Inhibition of autophagy potentiates the anti‐metastasis effect of phenethyl isothiocyanate through JAK2/STAT3 pathway in lung cancer cells. Mol Carcinog 2018; 57:522–35. [DOI] [PubMed] [Google Scholar]
- 218. Wang J, Zou K, Feng X, Chen M, Li C, Tang R et al Downregulation of NMI promotes tumor growth and predicts poor prognosis in human lung adenocarcinomas. Mol Cancer 2017; 16:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Psarra AM, Hermann S, Panayotou G, Spyrou G. Interaction of mitochondrial thioredoxin with glucocorticoid receptor and NF‐κB modulates glucocorticoid receptor and NF‐κB signalling in HEK‐293 cells. Biochem J 2009; 422:521–31. [DOI] [PubMed] [Google Scholar]
- 220. Psarra AM, Sekeris CE. Glucocorticoid receptors and other nuclear transcription factors in mitochondria and possible functions. Biochem Biophys Acta 2009; 1787:431–6. [DOI] [PubMed] [Google Scholar]
- 221. Szczepanek K, Lesnefsky EJ, Larner AC. Multi‐tasking: nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol 2012; 22:429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Zouein FA, Duhe RJ, Arany I, Shirey K, Hosler JP, Liu H et al Loss of STAT3 in mouse embryonic fibroblasts reveals its Janus‐like actions on mitochondrial function and cell viability. Cytokine 2014; 66:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Patergnani S, Missiroli S, Marchi S, Giorgi C. Mitochondria‐associated endoplasmic reticulum membranes microenvironment: targeting autophagic and apoptotic pathways in cancer therapy. Front Oncol 2015; 5:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Sionov RV, Vlahopoulos SA, Granot Z. Regulation of bim in health and disease. Oncotarget 2015; 6:23058–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Guo AK, Hou YY, Hirata H, Yamauchi S, Yip AK, Chiam KH et al Loss of p53 enhances NF‐κB‐dependent lamellipodia formation. J Cell Physiol 2014; 229:696–704. [DOI] [PubMed] [Google Scholar]
- 226. Kfir‐Erenfeld S, Yefenof E. Non‐genomic events determining the sensitivity of hemopoietic malignancies to glucocorticoid‐induced apoptosis. Cancer Immunol Immunother: CII 2014; 63:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Makhov P, Naito S, Haifler M, Kutikov A, Boumber Y, Uzzo RG et al The convergent roles of NF‐κB and ER stress in sunitinib‐mediated expression of pro‐tumorigenic cytokines and refractory phenotype in renal cell carcinoma. Cell Death Dis 2018; 9:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Meier JA, Larner AC. Toward a new STATe: the role of STATs in mitochondrial function. Semin Immunol 2014; 26:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Vessoni AT, Quinet A, de Andrade‐Lima LC, Martins DJ, Garcia CC, Rocha CR et al Chloroquine‐induced glioma cells death is associated with mitochondrial membrane potential loss, but not oxidative stress. Free Radic Biol Med 2016; 90:91–100. [DOI] [PubMed] [Google Scholar]
- 230. Hoshino A, Matoba S, Iwai‐Kanai E, Nakamura H, Kimata M, Nakaoka M et al p53‐TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol 2012; 52:175–84. [DOI] [PubMed] [Google Scholar]
- 231. You L, Jin S, Zhu L, Qian W. Autophagy, autophagy‐associated adaptive immune responses and its role in hematologic malignancies. Oncotarget 2017; 8:12374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Accapezzato D, Visco V, Francavilla V, Molette C, Donato T, Paroli M et al Chloroquine enhances human CD8+ T cell responses against soluble antigens in vivo. J Exp Med 2005; 202:817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Marty R, de Prisco N, Carter H, Font‐Burgada J. MHC‐I genotype drives early immune selection of oncogenic mutations. Mol Cell Oncol 2018; 5:e1409863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P et al Blocking hypoxia‐induced autophagy in tumors restores cytotoxic T‐cell activity and promotes regression. Can Res 2011; 71:5976–86. [DOI] [PubMed] [Google Scholar]
- 235. Ishih A, Kawakami C, Todoroki A, Hirai H, Ohori K, Kobayashi F. Outcome of primary lethal and nonlethal Plasmodium yoelii malaria infection in BALB/c and IFN‐γ receptor‐deficient mice following chloroquine treatment. Parasitol Res 2013; 112:773–80. [DOI] [PubMed] [Google Scholar]
- 236. Zhang Y, Zhu X, Feng Y, Pang W, Qi Z, Cui L et al TLR4 and TLR9 signals stimulate protective immunity against blood‐stage Plasmodium yoelii infection in mice. Exp Parasitol 2016; 170:73–81. [DOI] [PubMed] [Google Scholar]
- 237. Qin X, Chen G, Feng Y, Zhu X, Du Y, Pang W et al Early treatment with chloroquine inhibits the immune response against Plasmodium yoelii infection in mice. Tohoku J Exp Med 2014; 234:271–80. [DOI] [PubMed] [Google Scholar]
- 238. Masuelli L, Granato M, Benvenuto M, Mattera R, Bernardini R, Mattei M et al Chloroquine supplementation increases the cytotoxic effect of curcumin against Her2/neu overexpressing breast cancer cells in vitro and in vivo in nude mice while counteracts it in immune competent mice. Oncoimmunology 2017; 6:e1356151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Kyburz D, Brentano F, Gay S. Mode of action of hydroxychloroquine in RA‐evidence of an inhibitory effect on toll‐like receptor signaling. Nat Clin Pract Rheumatol 2006; 2:458–9. [DOI] [PubMed] [Google Scholar]
- 240. An N, Chen Y, Wang C, Yang C, Wu ZH, Xue J et al Chloroquine autophagic inhibition rebalances Th17/treg‐mediated immunity and ameliorates systemic lupus erythematosus. Cell Physiol Biochem: Int J Exp Cell Physiol Biochem Pharmacol 2017; 44:412–22. [DOI] [PubMed] [Google Scholar]
- 241. Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 2006; 203:553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Gostner JM, Schrocksnadel S, Becker K, Jenny M, Schennach H, Uberall F et al Antimalarial drug chloroquine counteracts activation of indoleamine (2,3)‐dioxygenase activity in human PBMC. FEBS Open Bio 2012; 2:241–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Thome R, Moraes AS, Bombeiro AL, Farias Ados S, Francelin C, da Costa TA et al Chloroquine treatment enhances regulatory T cells and reduces the severity of experimental autoimmune encephalomyelitis. PLoS ONE 2013; 8:e65913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Wennerberg E, Sarhan D, Carlsten M, Kaminskyy VO, D'Arcy P, Zhivotovsky B et al Doxorubicin sensitizes human tumor cells to NK cell‐ and T‐cell‐mediated killing by augmented TRAIL receptor signaling. Int J Cancer 2013; 133:1643–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Messai Y, Noman MZ, Hasmim M, Janji B, Tittarelli A, Boutet M et al ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Can Res 2014; 74:6820–32. [DOI] [PubMed] [Google Scholar]
- 246. Singh K, Sharma A, Mir MC, Drazba JA, Heston WD, Magi‐Galluzzi C et al Autophagic flux determines cell death and survival in response to Apo2L/TRAIL (dulanermin). Mol Cancer 2014; 13:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K et al Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer‐mediated lysis under hypoxia. Proc Natl Acad Sci USA 2013; 110:17450–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E et al Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5‐dependent manner. Proc Natl Acad Sci USA 2017; 114:E9271–E9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Nyati MK, Feng FY, Kanade VD, Nayak R. Chloroquine treatment increases detection of 5‐fluorouracil‐induced apoptosis index in vivo. Mol Imaging 2006; 5:148–52. [PubMed] [Google Scholar]
- 250. Loi M, Muller A, Steinbach K, Niven J, Barreira da Silva R, Paul P et al Macroautophagy proteins control MHC class I levels on dendritic cells and shape anti‐viral CD8+ T cell responses. Cell Rep 2016; 15:1076–87. [DOI] [PubMed] [Google Scholar]
- 251. Uckun FM, Ramakrishnan S, Houston LL. Increased efficiency in selective elimination of leukemia cells by a combination of a stable derivative of cyclophosphamide and a human B‐cell‐specific immunotoxin containing pokeweed antiviral protein. Can Res 1985; 45:69–75. [PubMed] [Google Scholar]
- 252. Ratikan JA, Sayre JW, Schaue D. Chloroquine engages the immune system to eradicate irradiated breast tumors in mice. Int J Radiat Oncol Biol Phys 2013; 87:761–8. [DOI] [PubMed] [Google Scholar]
- 253. Zhang Y, Cao Y, Sun X, Feng Y, Du Y, Liu F et al Chloroquine (CQ) exerts anti‐breast cancer through modulating microenvironment and inducing apoptosis. Int Immunopharmacol 2017; 42:100–7. [DOI] [PubMed] [Google Scholar]
- 254. Moschovi M, Adamaki M, Vlahopoulos SA. Progress in treatment of viral infections in children with acute lymphoblastic leukemia. Oncol Rev 2016; 10:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Sato A. The human immunodeficiency virus protease inhibitor ritonavir is potentially active against urological malignancies. OncoTargets Ther 2015; 8:761–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Ouyang Q, Huang Z, Wang Z, Chen X, Ni J, Lin L. Effects of pristane alone or combined with chloroquine on macrophage activation, oxidative stress, and TH1/TH2 skewness. J Immunol Res 2014; 2014:613136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Said A, Bock S, Lajqi T, Muller G, Weindl G. Chloroquine promotes IL‐17 production by CD4+ T cells via p38‐dependent IL‐23 release by monocyte‐derived Langerhans‐like cells. J Immunol 2014; 193:6135–43. [DOI] [PubMed] [Google Scholar]
- 258. Burikhanov R, Hebbar N, Noothi SK, Shukla N, Sledziona J, Araujo N et al Chloroquine‐inducible Par‐4 secretion is essential for tumor cell apoptosis and inhibition of metastasis. Cell Rep 2017; 18:508–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Liang DH, Choi DS, Ensor JE, Kaipparettu BA, Bass BL, Chang JC. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett 2016; 376:249–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Emir UE, Larkin SJ, de Pennington N, Voets N, Plaha P, Stacey R et al Noninvasive quantification of 2‐hydroxyglutarate in human gliomas with IDH1 and IDH2 mutations. Can Res 2016; 76:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Galgamuwa R, Hardy K, Dahlstrom JE, Blackburn AC, Wium E, Rooke M et al Dichloroacetate prevents cisplatin‐induced nephrotoxicity without compromising cisplatin anticancer properties. J Am Soc Nephrol: JASN 2016; 27:3331–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Giatromanolaki A, Sivridis E, Kalamida D, Koukourakis MI. Transcription factor EB expression in early breast cancer relates to lysosomal/autophagosomal markers and prognosis. Clin Breast Cancer 2017; 17:e119–e125. [DOI] [PubMed] [Google Scholar]
- 263. Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M et al Transcriptional control of autophagy‐lysosome function drives pancreatic cancer metabolism. Nature 2015; 524:361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Bingel C, Koeneke E, Ridinger J, Bittmann A, Sill M, Peterziel H et al Three‐dimensional tumor cell growth stimulates autophagic flux and recapitulates chemotherapy resistance. Cell Death Dis 2017; 8:e3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Fang L, Hodge J, Saaoud F, Wang J, Iwanowycz S, Wang Y et al Transcriptional factor EB regulates macrophage polarization in the tumor microenvironment. Oncoimmunology 2017; 6:e1312042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Roczniak‐Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, et al The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012; 5:ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Jarosz‐Biej M, Kaminska N, Matuszczak S, Cichon T, Pamula‐Pilat J, Czapla J et al M1‐like macrophages change tumor blood vessels and microenvironment in murine melanoma. PLoS ONE 2018; 13:e0191012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Li L, Sun B, Gao Y, Niu H, Yuan H, Lou H. STAT3 contributes to lysosomal‐mediated cell death in a novel derivative of riccardin D‐treated breast cancer cells in association with TFEB. Biochem Pharmacol 2018; 150:267–79. [DOI] [PubMed] [Google Scholar]
- 269. Szebeni GJ, Vizler C, Kitajka K, Puskas LG. Inflammation and cancer: extra‐ and intracellular determinants of tumor‐associated macrophages as tumor promoters. Med Inflamm 2017; 2017:9294018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Chung SS, Adekoya D, Enenmoh I, Clarke O, Wang P, Sarkyssian M et al Salinomycin abolished STAT3 and STAT1 interactions and reduced telomerase activity in colorectal cancer cells. Anticancer Res 2017; 37:445–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Jangamreddy JR, Ghavami S, Grabarek J, Kratz G, Wiechec E, Fredriksson BA et al Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: differences between primary and cancer cells. Biochem Biophys Acta 2013; 1833(9):2057–69. [DOI] [PubMed] [Google Scholar]
- 272. Omidi A, Aslani MR, Movassaghi AR, Mohri M, Dadfar M. Accidental salinomycin intoxication in calves. Can Vet J = La revue veterinaire canadienne 2010; 51:1143–5. [PMC free article] [PubMed] [Google Scholar]
- 273. van der Linde‐Sipman JS, van den Ingh TSGAM, van Nes JJ, Verhagen H, Kersten JGTM, Beynen AC, et al Salinomycin‐induced polyneuropathy in cats: morphologic and epidemiologic data. Vet Pathol 1999; 36:152–6. [DOI] [PubMed] [Google Scholar]
- 274. Pellegrini P, Dyczynski M, Sbrana FV, Karlgren M, Buoncervello M, Hagg‐Olofsson M et al Tumor acidosis enhances cytotoxic effects and autophagy inhibition by salinomycin on cancer cell lines and cancer stem cells. Oncotarget 2016; 7:35703–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Jiang J, Li H, Qaed E, Zhang J, Song Y, Wu R et al Salinomycin, as an autophagy modulator – a new avenue to anticancer: a review. J Exp Clin Cancer Res: CR 2018; 37:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Yotsumoto S, Saegusa K, Aramaki Y. Endosomal translocation of CpG‐oligodeoxynucleotides inhibits DNA‐PKcs‐dependent IL‐10 production in macrophages. J Immunol 2008; 180:809–16. [DOI] [PubMed] [Google Scholar]
- 277. Pawlowska E, Szczepanska J, Blasiak J. DNA2 – An important player in DNA damage response or just another DNA maintenance protein? Int J Mol Sci 2017; 18:1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Ma C, Spies NP, Gong T, Jones CX, Chu WM. Involvement of DNA‐PKcs in the type I IFN response to CpG‐ODNs in conventional dendritic cells in TLR9‐dependent or ‐independent manners. PLoS ONE 2015; 10:e0121371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Zhu LQ, Zhen YF, Zhang Y, Guo ZX, Dai J, Wang XD. Salinomycin activates AMP‐activated protein kinase‐dependent autophagy in cultured osteoblastoma cells: a negative regulator against cell apoptosis. PLoS ONE 2013; 8:e84175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Jensen PB, Sorensen BS, Sehested M, Grue P, Demant EJ, Hansen HH. Targeting the cytotoxicity of topoisomerase II‐directed epipodophyllotoxins to tumor cells in acidic environments. Can Res 1994; 54:2959–63. [PubMed] [Google Scholar]
- 281. VanKlompenberg MK, Leyden E, Arnason AH, Zhang JT, Stefanski CD, Prosperi JR. APC loss in breast cancer leads to doxorubicin resistance via STAT3 activation. Oncotarget 2017; 8:102868–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Alabbas SY, Begun J, Florin TH, Oancea I. The role of IL‐22 in the resolution of sterile and nonsterile inflammation. Clin Transl Immunol 2018; 7:e1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Carlsson R, Ozen I, Barbariga M, Gaceb A, Roth M, Paul G. STAT3 precedes HIF1α transcriptional responses to oxygen and oxygen and glucose deprivation in human brain pericytes. PLoS ONE 2018; 13:e0194146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Li Y, Zhang X, Cui L, Chen R, Zhang Y, Zhang C et al Salvianolic acids enhance cerebral angiogenesis and neurological recovery by activating JAK2/STAT3 signaling pathway after ischemic stroke in mice. J Neurochem 2017; 143:87–99. [DOI] [PubMed] [Google Scholar]
- 285. Matsumoto J, Dohgu S, Takata F, Machida T, Bolukbasi Hatip FF, Hatip‐Al‐Khatib I et al TNF‐α‐sensitive brain pericytes activate microglia by releasing IL‐6 through cooperation between IκB‐NFκB and JAK‐STAT3 pathways. Brain Res 2018; 1692:34–44. [DOI] [PubMed] [Google Scholar]
- 286. Riu F, Slater SC, Garcia EJ, Rodriguez‐Arabaolaza I, Alvino V, Avolio E et al The adipokine leptin modulates adventitial pericyte functions by autocrine and paracrine signalling. Sci Rep 2017; 7:5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Adamaki M, Tsotra M, Vlahopoulos S, Zampogiannis A, Papavassiliou AG, Moschovi M. STAT transcript levels in childhood acute lymphoblastic leukemia: STAT1 and STAT3 transcript correlations. Leuk Res 2015; 39:1285–91. [DOI] [PubMed] [Google Scholar]
- 288. Stephanou A, Latchman DS. Opposing actions of STAT‐1 and STAT‐3. Growth Factors 2005; 23:177–82. [DOI] [PubMed] [Google Scholar]
- 289. Li X, Zhou Y, Liu Y, Zhang X, Chen T, Chen K et al Preclinical efficacy and safety assessment of artemisinin‐chemotherapeutic agent conjugates for ovarian cancer. EBioMedicine 2016; 14:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Srivastava V, Lee H. Chloroquine‐based hybrid molecules as promising novel chemotherapeutic agents. Eur J Pharmacol 2015; 762:472–86. [DOI] [PubMed] [Google Scholar]
- 291. Wang Y, Tai X, Zhang L, Liu Y, Gao H, Chen J et al A novel antitumour strategy using bidirectional autophagic vesicles accumulation via initiative induction and the terminal restraint of autophagic flux. J Controlled Release 2015; 199:17–28. [DOI] [PubMed] [Google Scholar]
- 292. Ha J, Kim J. Novel pharmacological modulators of autophagy: an updated patent review (2012–2015). Expert Opin Ther Pat 2016; 26:1273–89. [DOI] [PubMed] [Google Scholar]
- 293. Njaria PM, Okombo J, Njuguna NM, Chibale K. Chloroquine‐containing compounds: a patent review (2010–2014). Expert Opin Ther Pat 2015; 25:1003–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Chen PC, Liu X, Lin Y. Drug repurposing in anticancer reagent development. Comb Chem High Throughput Screening 2017; 20:395–402. [DOI] [PubMed] [Google Scholar]
- 295. Doble B, Schofield DJ, Roscioli T, Mattick JS. Prioritising the application of genomic medicine. NPJ Genomic Med 2017; 2:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Neagu M, Albulescu R, Tanase C. Biotechnology landscape in cancer drug discovery. Fut Sci OA 2015; 1:FSO12 10.4155/FSO.15.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Cardoso F, Harbeck N, Barrios CH, Bergh J, Cortes J, El Saghir N et al Research needs in breast cancer. Ann Oncol 2017; 28:208–17. [DOI] [PubMed] [Google Scholar]
- 298. Engert A, Balduini C, Brand A, Coiffier B, Cordonnier C, Dohner H, et al The European Hematology Association Roadmap for European Hematology Research: a consensus document. Haematologica 2016; 101:115–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Smith AB, Chisolm S, Deal A, Spangler A, Quale DZ, Bangs R et al Patient‐centered prioritization of bladder cancer research. Cancer 2018; 124:3136–44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Genomic targets of chloroquine pharmacological activity. Shown is a snapshot from the query of the database DRUGSURV (http://www.bioprofiling.de/cgi-bin/GEO/DRUGSURV/start_DRUG.pl) for chloroquine.