

https://onlinelibrary.wiley.com/page/journal/23301619/homepage/mdc312892-sup-v001.htm
https://onlinelibrary.wiley.com/page/journal/23301619/homepage/mdc312892-sup-v002.htm
Classical atypical parkinsonian syndromes (APs) refers to a group of disorders including progressive supranuclear palsy (PSP), multiple system atrophy, corticobasal syndrome, and dementia with Lewy bodies.1 Clinical and pathological heterogeneity has been reported among APs.1 Additional features such as positive family history, “too‐long” stable course, and associated clinical/radiological findings may be “red flags” for APs and should direct clinicians toward alternative diagnosis and further investigations. This has led to the emerging concept of “atypical” APs. Among these, some genetic conditions might share some clinical features with the APs, and they are referred to as “AP‐look‐alikes.”2 Hirschbichler and colleagues1 reported that half of APs cases showed additional “atypical” features, and approximately 10% eventually received an alternative diagnosis.
Here we report a patient with adult, very late‐onset of Niemann Pick type C (NPC) disease with clinical presentation of PSP‐look‐alike disorder.
Case Report
The first symptoms (occasional impairment of nomination, dysarthria, and mild gait disturbances) were observed by the family members of a female patient aged 51 years. Soon after, sudden backward falls with preserved consciousness occurred, initially twice a day on average but became less frequent thereafter. She complained of speech difficulties, falls, and mild memory problems that did not interfere with activities of daily living. Therefore, she did not seek for medical advice until she has been admitted in our department 4 years after the disease onset. Personal history was unremarkable; her father and paternal grandmother had dementia after the age of 70.
Examination upon admission revealed very slow saccades in the vertical plane and vertical, especially downgaze, palsy with normal vestibulo‐ocular reflex that indicated supranuclear origin of vertical gaze palsy (VSGP), dysarthria, impersistence of tongue protrusion, mild generalized chorea, brisk muscle reflexes, and slightly wide‐based gait. Neuropsychological testing revealed executive dysfunction (verbal fluency and confrontational nomination), whereas other cognitive domains were preserved, including attention. Brain magnetic resonance imaging scans showed bilateral cortical encephalomalacia in the anterior parasagittal frontobasal regions and mild periventricular leukoencephalopathy. Brain 18‐F‐fluorodeoxyglucose–positron emission tomography detected hypometabolism in the frontobasal parasagittal regions (left > right) in both the thalami and less extensively in the mediotemporal regions (left > right).
Initially, her presentation resembled the PSP/Frontotemporal dementia (FTD) spectrum (VSGP, backward falls, and executive dysfunction), although with some “atypical” features such as absence of parkinsonism, a stable clinical course and presence of chorea. PSP/FTD look‐alike phenotype combined with chorea has been reported in patients with mutations in the C9orf72, FUS, and TARDBP genes.3, 4, 5
In addition to the neurological findings, we detected in our patient slight thrombocytopenia (140; normal range 150–400) and splenomegaly revealed by abdominal ultrasound examination (154 × 75 mm). Because isolated unexplained splenomegaly and the VSGP were strong predictors for the diagnosis of NPC disease,6 we conducted genetic analysis and found homozygous mutation in exon 19 of the NPC1 gene (c.2861C > T, p.S954L). This variant was previously described in the literature7, 8 but to the best of our knowledge not in the homozygous state.
We have clinically followed the patient for 6 years in 6‐month periods (miglustat has not been administered) and observed a surprisingly mild if any clinical and neuropsychological progression (independent in everyday tasks; Videos S1 and S2). However, repeated 18‐F‐fluorodeoxyglucose–positron emission tomography (3 years after the first) showed mild additional cerebellar hypometabolism besides the previously affected regions (Figs. 1 and 2).
Figure 1.
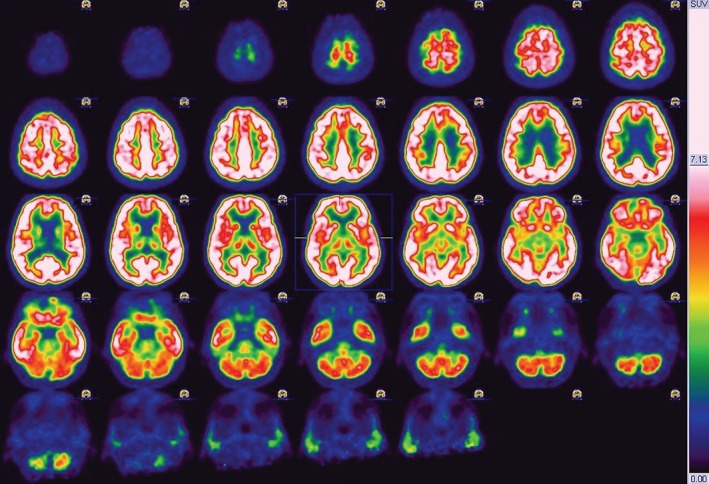
18‐F‐fluorodeoxyglucose–positron emission tomography showing hypometabolism in the frontobasal parasagittal regions (left > right) in both the thalami and less extensively in the medial temporal regions (left > right) and cerebellum. SUV, XXXXXXXXXX.
Figure 2.
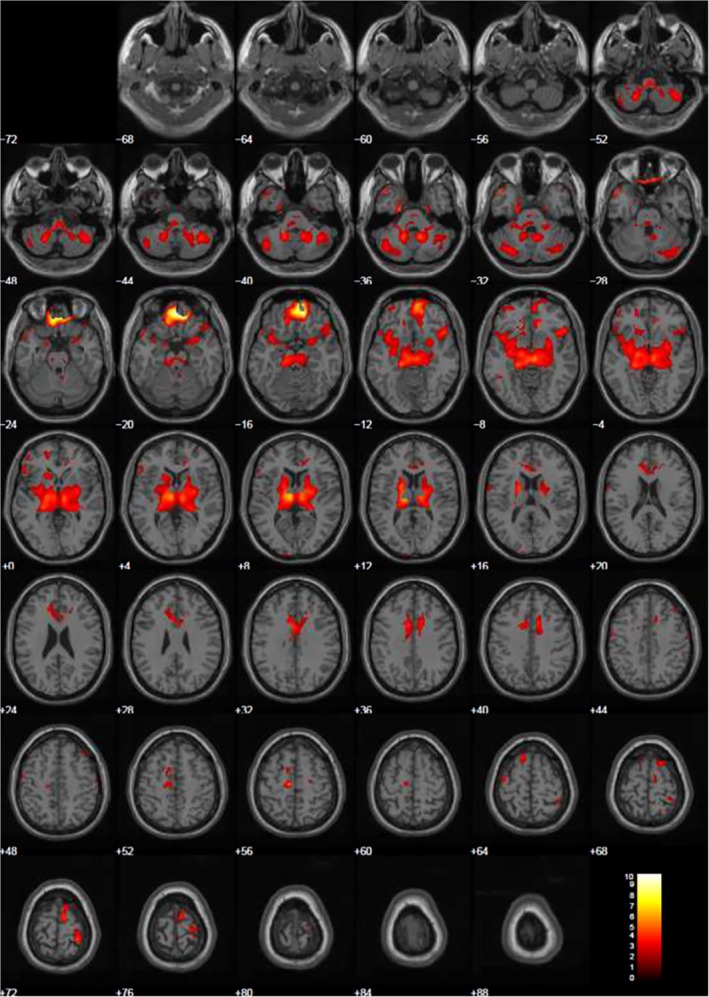
Statistical parametric mapping analysis‐t‐maps of the same abovementioned regions of hypometabolism overlaid on magnetic resonance imaging.
Discussion
NPC is a rare progressive autosomal recessive lysosomal storage disorder caused by mutations in either the NPC1 or NPC2 genes. These mutations affect the late endosomal–lysosomal trafficking of cellular cholesterol and other lipids.9 Clinical presentation of NPC depends on the age of onset. Early infantile form (<2 years) is neurovisceral with almost invariably the presence of hepatosplenomegaly and/or a neonatal prolonged jaundice, hypotonia, and delay in developmental motor milestones. The juvenile form (6–15 years) manifests as cognitive impairment, frequent falls, progressive ataxia, and dystonia and VSGP. In the adult‐onset group (>15 years), visceral symptoms are less common and typically there is the co‐occurrence of cognitive impairment, psychiatric symptoms, VSGP and progressive ataxia, or movement disorders.7, 9 Movement disorders are common in adult‐onset NPC; dystonia is the most frequent, followed by chorea, myoclonus, and parkinsonism.7, 10 When present in NPC, parkinsonism is mild, consisting in bradykinesia, axial rigidity, hypomimia, or isolated rest tremor.7 Gelastic cataplexy is sensitive for NPC diagnosis, but is present in only 16% of these patients.6 One may assume that the falls in our patient were the result of cataplexy, but we have not witnessed them.
Reports of very late adult‐onset NPC (after age 50) are rare.7, 11 NPC should be considered in the differential diagnosis of late‐onset vertical gaze palsy among more common disorders (such as PSP) to minimize diagnostic and therapy delay.11 In the presented case, the presence of chorea and the absence of parkinsonism are clinical findings against a PSP diagnosis. However, according to the recently published Movement Disorder Society criteria,12 our patient had core clinical PSP features (repeated unprovoked falls within 3 years and VSGP) and therefore fulfilled the clinical criteria for probable PSP. Biochemical or genetic confirmation of NPC is the exclusion criteria for the diagnosis of PSP. These tests, among others, have been recommended in patients with PSP phenotype if aged <45 years.12 However, based on our limited experience, in some patients with the PSP‐look‐alike phenotype, further etiological investigations should not be restricted by the age of disease onset, especially if NPC is suspected, which is a treatable disorder.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
N.K.: 1A, 1B, 1C, 3A
G.M.‐S.: 1A, 1B, 3B
V.D.: 1B, 1C
I.P.: 1A, 1B, 1C, 3B
L.B.: 1B, 1C
E.S.: 1B, 1C
M.S.: 1A, 3B
V.K.: 1A, 1B, 3B
Disclosures
Ethical Compliance Statement
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Written consent to publish all shown materials was obtained from the patient in Serbian.
Funding Sources and Conflicts of Interest
This study was supported by the Ministry of Education, Science, and Technological Development of the Republic of Serbia (project no. 175090 to V.K.). The authors report no conflicts of interest.
Financial Disclosures for Previous 12 Months
Nikola Kresojević, Gorana Mandić‐Stojmenović, Valerija Dobričić, Igor Petrović, Leposava Brajković and Elka Stefanova: report no sources of funding and no conflicts of interest. Marina Svetel has received speaker's honoraria from Actavis. Vladimir Kostić has received research grants from the Ministry of Education, Science, and Technological Development, Republic of Serbia and the Serbian Academy of Science and Arts; and speaker honoraria from Actavis and Salveo.
Supporting information
Video S1. Patient was 55 years old when this video was recorded. Video shows dyshartria and mild chorea, vertical supranuclear downgaze palsy, normal finger‐to‐nose test, no hypo/bradykinesia, difficulty in repeating Luria sequence, slightly wide‐based gait, and postural instability.
Video S2. Patient was 58 years old when this video was recorded. Video shows vertical supranuclear downgaze palsy, slow saccades in vertical plane, “round the houses” sign, impersistence of tongue protrusion, mild chorea, no stimulus sensitivity and bradykinesia, normal finger‐to‐nose test, and brisk patellar reflex.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Hirschbichler ST, Erro R, Ganos C, et al. “Atypical” atypical parkinsonism: critical appraisal of a cohort. Parkinsonism Relat Disord 2017;37:36–42. [DOI] [PubMed] [Google Scholar]
- 2. Stamelou M, Quinn NP, Bhatia KP. “Atypical” atypical parkinsonism: new genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy‐a diagnostic guide. Mov Disord 2013;28(9):1184–1199. [DOI] [PubMed] [Google Scholar]
- 3. Kovacs GG, Murrell JR, Horvath S, et al. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord 2009;24(12):1843–1847. [DOI] [PubMed] [Google Scholar]
- 4. Siuda J, Fujioka S, Wszolek ZK. Parkinsonian syndrome in familial frontotemporal dementia. Parkinsonism Relat Disord 2014;20(9):957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hensman Moss DJ, Poulter M, Beck J, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology 2014;82(4):292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wijburg FA, Sedel F, Pineda M, et al. Development of a suspicion index to aid diagnosis of Niemann‐Pick disease type C. Neurology 2012;78(20):1560–1567. [DOI] [PubMed] [Google Scholar]
- 7. Sévin M, Lesca G, Baumann N, et al. The adult form of Niemann‐Pick disease type C. Brain 2007;130(Pt 1):120–133. [DOI] [PubMed] [Google Scholar]
- 8. Reunert J, Fobker M, Kannenberg F, et al. Rapid diagnosis of 83 patients with niemann pick type C disease and related cholesterol transport disorders by cholestantriol screening. EBioMedicine 2015;4:170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geberhiwot T, Moro A, Dardis A, et al. Consensus clinical management guidelines for Niemann‐Pick disease type C. Orphanet J Rare Dis 2018;13(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nadjar Y, Hütter‐Moncada AL, Latour P, et al. Adult Niemann‐Pick disease type C in France: clinical phenotypes and long‐term miglustat treatment effect. Orphanet J Rare Dis 2018;13(1):175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumar N, Rizek P, Mohammad Y, et al. Pearls & Oy‐sters: Niemann‐Pick disease type C in a 65‐year‐old patient. Neurology 2016;87(8):e79–e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord 2017;32(6):853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Patient was 55 years old when this video was recorded. Video shows dyshartria and mild chorea, vertical supranuclear downgaze palsy, normal finger‐to‐nose test, no hypo/bradykinesia, difficulty in repeating Luria sequence, slightly wide‐based gait, and postural instability.
Video S2. Patient was 58 years old when this video was recorded. Video shows vertical supranuclear downgaze palsy, slow saccades in vertical plane, “round the houses” sign, impersistence of tongue protrusion, mild chorea, no stimulus sensitivity and bradykinesia, normal finger‐to‐nose test, and brisk patellar reflex.