

ABSTRACT
Background
Psychotic symptoms, such as delusions and hallucinations, are part of the clinical picture of several conditions presenting movement disorders. Phenomenology and epidemiology of psychosis in Parkinson's disease have received wide attention; however, the presence of psychosis in other movement disorders is, comparatively, less well known.
Objectives
To review psychotic symptoms present in different movement disorders.
Methods
A comprehensive and structured literature search was performed to identify and analyze data on patients with movement disorders and comorbid psychosis.
Results
In monogenic parkinsonisms, such as PARK‐GBA, PARK‐LRRK2, and PARK‐SNCA, visual hallucinations related to dopamine replacement therapy are frequent as well as are delusions in PARK‐LRRK2 and PARK‐SNCA, but not in PARK‐GBA. Different types of delusions and hallucinations are found in Huntington's disease and other choreic disorders. In Tourette's syndrome, paranoid delusions as well as visual, olfactory, and auditory hallucinations have been described, which usually develop after an average of 10 years of disease. Delusions in ataxias are more frequent in ATX‐TBP, ATX‐ATN1, and ATX‐ATXN3, whereas it is rare in Friedreich's ataxia. Psychosis is also a prominent and frequent clinical feature in Fahr's disease, Wilson's disease, neurodegeneration with brain iron accumulation, and some lysosomal storage disorders, whereas it is uncommon in atypical parkinsonisms and dystonia. Psychosis usually occurs at late disease stages, but may appear as onset symptoms of the disease, especially in Wilson's disease, Huntington's disease, late‐onset Tays‐Sachs, and Niemann‐Pick.
Conclusion
Psychosis is a frequent comorbidity in most hyper‐ and hypokinetic movement disorders. Appropriate recognition is relevant both in the early and late disease stages.
Keywords: psychosis, psychotic, delusions, hallucinations, psychiatry
During the past decade, more attention has been directed toward the nonmotor symptoms of movement disorders, a group of diseases and conditions classically recognized as mainly motor. The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition stresses the common cooccurrence of psychotic symptoms in neurocognitive disorders, and information on the neurobiology of hallucinations and delusions associated with neurological conditions has expanded rapidly over the past decade.1 Whereas an overwhelming number of studies have been published on psychosis in dementia with Lewy bodies (DLB) and on dopamine replacement therapy (DRT)‐associated psychosis in idiopathic Parkinson's disease (PD), phenomenology, epidemiology, and putative mechanisms of psychotic disorders in other movement disorders have received much less attention. The objective of the current educational review was to systematically search for and analyze clinical features of psychosis associated with movement disorders.
According to the American Psychiatric Association and the World Health Organization, current conceptualization of psychosis requires the presence of prominent hallucinations or delusions.1 The International Classification of Diseases, Tenth Revision (F06.0) describes Organic Hallucinosis as persistent or recurrent hallucinations occurring in clear consciousness, with or without preserved insight. Delusions may be present, but are less prominent. The Organic Delusional Disorder (F06.2) consists of persistent or recurrent delusions which dominate the clinical picture and may be accompanied by hallucinations or a thought disorder.
Hallucinations are defined as sensory perceptions in the absence of a corresponding external or somatic stimulus and are described according to the sensory domain in which it occurs. Hallucinations may occur with or without insight into their hallucinatory nature. Hallucinations are distinguished from illusions, which are misperceptions of a sensory stimulus. These perceptual phenomena occur in many neuropsychiatric conditions, including movement disorders.
Delusions are false beliefs based on false inferences about external reality or about oneself and maintained firmly despite conflicting evidence that contradicts the belief. Delusions are commonly present in patients with dementia and patients under DRT. Delusions may consist of different themes, such as persecutory, grandiose, erotomanic, nihilistic, and somatic content, or bizarre content, such as though control or withdrawal. Different and varied phenomenology may include the belief that one is being intentionally harmed, tricked, followed, spied on, poisoned or drugged, tormented, ridiculed, cheated, or conspired against (persecutory delusions); the conviction that one possesses special powers, talents, or abilities; or is famous or God, an angel, a devil, or a saint (grandiose delusions). Less frequent are beliefs that one's thoughts, feelings, or behaviors are being controlled by an external force (thought control) or that thoughts are being inserted into one's mind (thought insertion), that a person or group is removing or extracting one's thoughts (thought withdrawal), that one's mind can be or is being read by another person, the belief that a spouse or lover is unfaithful (delusion of infidelity), the belief that one is loved by another person of higher status (erotomania), the belief that one is infested with insects, spiders, worms, or other organisms (Ekbom syndrome), or the belief that one's body is abnormal, diseased, or changed in some manner.
Delusional misidentification includes various themes, such as the belief that a spouse, family member, or other familiar individual has been replaced by an impostor who is physically, but not psychologically, identical to the replaced person (Capgras delusional belief), or the belief that different people are in fact a single person (Fregoli delusion), or that a double of oneself exists (Doppelgänger), and the belief that one or more of one's organs or body parts are missing or no longer exist (Cotard delusion).
Methods
We conducted a comprehensive and structured search in PubMed following the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) guidelines (http://www.prisma-statement.org), using a list of keywords, namely: atypical parkinsonism, progressive supranuclear palsy, PSP, multiple system atrophy, MSA, corticobasal, CBS, CBD, genetic parkinsonism, SNCA, VPS35, LRRK2, Parkin, PINK1, PARK2, Kufor‐Rakeb, ATP13A2, DJ‐1, PARK9, GBA, glucocerebrosidase, Niemann‐Pick, chorea, Huntington disease, neuroacanthocytosis, Huntington‐like, Tourette syndrome, tic disorders, myoclonus, myoclonic, dystonia, tremor, ataxia, Wilson's disease, Fahr, PLA2G6, PANK2, NBIA, pallido‐pyramidal, hypokinetic, hyperkinetic, and basal ganglia in combination with psychosis, psychotic, hallucination, delusion, delusional, thought disorder, and illusion. Publications written in English and published up to December 31, 2018 were screened. Types of publications screened included case reports, case series or case‐controlled studies, literature reviews, and any other type of publication that could contain clinical information on psychosis in movement disorders. Abstracts and titles were screened and cross‐checked for relevance by two authors (M.R. and N.F.) working separately. Full texts containing clinical data of patients with movement disorders and comorbid psychosis were analyzed. Back‐searching of retrieved publication reference lists was conducted to identify gray literature. Publications based on DLB‐ or DRT‐associated psychosis in idiopathic PD were excluded because these entities were extensively reviewed in other publications. However, patients with mutations in PD‐related genes were included because traditionally they have received less attention and may present unique differential features, such as psychosis unrelated to DRT. In order to compare findings on monogenic parkinsonisms with idiopathic PD, data available on systematic reviews on psychosis in idiopathic PD were analyzed. Diseases classified as dementia, such as frontotemporal dementia and Creutzfeldt‐Jakob disease, or conditions eliciting movement disorders, such as myoclonus in the context of metabolic diseases, were excluded, given that other clinical features may predominate over movement disorders.
Results
Results of the systematic literature search are shown in Figure 1. In total, 11,635 publications were initially identified, of which 567 were included in qualitative and quantitative synthesis after excluding those publications without individual data or percentages of patients with movement disorders and psychotic symptoms. Frequency of psychotic patients reported in the selected movement disorders are shown in Table 1. The most relevant clinical features of each disorder are described below. In most cases, psychotic features were most frequent during the disease course or at advanced disease stages. However, in a few disorders, psychosis preceded all other clinical features and presented as a first symptom of the disease. Table 2 illustrates the phenomenology of psychosis in different movement disorders, as well as its relation to both the disease course and to DRT. Psychosis was considered as related to DRT if there was either a clear temporal association between onset or resolution of psychosis and starting or stopping of DRT, or if explicitly stated by the authors.
Figure 1.
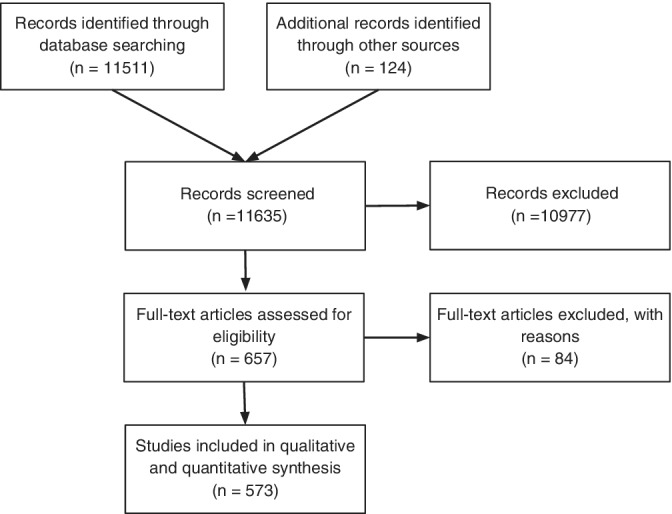
Flow diagram of the study selection process.
Table 1.
Frequency of psychotic patients reported in different movement disorders
| Movement Disorders | n = Patients/n = Articles |
|---|---|
| Monogenic parkinsonisms | At leasta 431/11S1–S56 b |
| Atypical parkinsonisms | At leasta 157/46S57–S103b |
| Choreas | At least 1,149/59S104–S163 |
| Tic disorders | 70/22S164–S180 |
| Ataxias | At least 63/29S181–S209 |
| Dystonia | 17/8S210–S217 |
| Myoclonus | 14/8S212, S218–224 |
| Basal ganglia calcification | At least 113/37S225–S260 |
| NBIA | 22/19S261–S278 |
| Others | |
| WD | At least 69/33S279–S310 |
| NP disease | At least 106/27S85, S311–S337 |
| LOTS | At least 40/15S338–S355 |
“At least” indicates that some minor inaccuracy exists given that some publications do not clearly report the total number of patients with psychosis for a specific condition.
“S” means Supporting Information references.
Table 2.
Phenomenology of psychosis in different movement disorders
| Movement Disorders | Psychosis at Disease Onset or in the Early Stages | Psychosis in Intermediate or Late Disease Stages | Psychosis Related to Dopamine‐Replacement Therapy |
|---|---|---|---|
| PD‐related genes | |||
| PARK‐GBA | – | +++ | ++ |
| PARK‐SNCA | + | +++ | +++ |
| PARK‐LRRK2 | + | +++ | +++ |
| PARK‐PRKN | + | ++ | + |
| PARK‐PINK1 | – | ++ | ++ |
| PARK‐DJ‐1 | – | ++ | ++ |
| 22qDS | + | ++ | NS |
| Atypical parkinsonisms | |||
| PARK‐ATP13A2 | + | +++ | +++ |
| PARK‐DNAJC6 | + | + | + |
| PARK‐FBXO7 | + | + | + |
| MSA | – | + | ++ |
| PSP | – | + | + |
| CBS | – | + | + |
| Choreas | |||
| HD | ++ | +++ | NS |
| Sydenham's chorea | – | ++ | NS |
| Benign hereditary chorea | – | + | NS |
| Chorea‐acanthocytosis | + | + | NS |
| McLeod syndrome | ++ | ++ | NS |
| ADCY5‐related dyskinesia | – | + | NS |
| Tic disorders | + | ++ | NS |
| Ataxias | + | ++ | NS |
| Dystonia | + | + | NS |
| Myoclonus | + | ++ | NS |
| Basal ganglia calcification | ++ | ++ | NS |
| NBIAs | ++ | ++ | NS |
| Others | |||
| WD | +++ | ++ | NS |
| NP disease | ++ | ++ | NS |
| LOTS | +++ | + | NS |
| CTX | – | + | NS |
: isolated cases (low frequency)
: some cases (intermediate frequency)
: many cases (high frequency)
: not reported. NS, not specified; 22qDS, 22q11.2 deletion syndrome; CBS, corticobasal syndrome; ADCY5, adenylyl cyclase 5; CTX, cerebrotendinous xanthomatosis.
Patients With Mutations in PD‐Related Genes
PARK‐GBA
Presence of visual hallucinations unrelated to DRT was reported in 45% (14 of 31) of PARK‐GBA patients.2 This frequency was surprisingly similar to the estimated 50% of lifetime prevalence of DRT‐related visual hallucinations found in idiopathic PD and is considered to be a consequence of Lewy body pathology spreading into the temporal lobe.2, 3 However, a high frequency (78%) of visual hallucinations developing under DRT was also reported in PARK‐GBA, a greater frequency than in PARK‐LRRK2 (38%) or in idiopathic PD (53%).4 Similar figures have been found in other studies, with rates of psychosis in PARK‐GBA patients receiving DRT ranging between 46%5 and 53%.6 Finally, a recent meta‐analysis found that PARK‐GBA was associated with a 1.83‐fold increased risk for psychosis (95% confidence interval: 1.23–2.74; P = 0.003).7 Visual hallucinations developed in PARK‐GBA after disease onset and were commonly associated with cognitive decline or dementia2, 6 as well as with the presence of deleterious or severe GBA variants causing Gaucher's disease types II and III.8, 9 To date, delusions have not been reported in PARK‐GBA patients.
PARK‐SNCA
Psychotic features in PARK‐SNCA have been reported among individuals with heterozygous mutations (e.g., p.A53T) and among those attributable to gene duplications, although the exact prevalence is not known. Psychotic symptoms, mostly consist of multimodal hallucinations (visual, auditory, and olfactory), often combined with paranoid delusions that may be persistent and refractory to treatment, eventually dominating the clinical picture.10, 11, 12 Psychotic symptoms presenting at disease onset are rare.12, 13 Takamura and colleagues reported on a single case with SNCA duplication that developed delusions and auditory hallucinations and was clinically diagnosed with schizophrenia 10 years before developing parkinsonian motor signs.13 Psychosis in PARK‐SNCA developed in most cases after several years of disease.10, 11, 12 Visual hallucinations were commonly related to DRT, cognitive decline, or dementia.10, 11, 12, 14, 15
PARK‐LRRK2
In a cohort of 23 PD patients carrying the G2019S mutation in the LRRK2 gene, delusions were found in 13% of cases and visual hallucinations in 26%, which was significantly more frequent than in noncarriers (odds ratio [OR] = 8.4).16 However, in a different sample of 27 PARK‐LRRK2 patients, no hallucinations or delusions were observed.14 Although psychotic symptoms of most PARK‐LRRK2 patients resemble those of patients with typical idiopathic PD given that they are induced by DRT, visual and auditory hallucinations as well as paranoid delusions unrelated to DRT have also been reported in PARK‐LRRK2.17, 18 In some cases, psychosis was associated with dementia and depression.17, 18
PARK‐PRKN
Psychosis has been rarely reported in PARK‐PRKN. Delusional jealousy, delusion of self‐persecution, paranoid delusions, and visual and third‐person auditory hallucinations have been reported in these patients, usually after several years of disease.19, 20 In only a few cases, psychosis occurred before motor symptom onset.19 Psychotic symptoms were usually related to DRT19, 20, 21 and persisted even after antiparkinsonian drug reduction and treatment with antipsychotics.21 Some cases were associated with depression, but not related to cognitive impairment.19
PARK‐PINK1
Psychosis in this condition was reported in isolated cases and included visual hallucinations,22, 23, 24, 25 paranoid and grandiose delusions, as well as hyper‐religiosity.26, 27, 28, 29, 30 Visual hallucinations were usually associated to DRT22, 23, 24, 25 and were also observed in the context of grandiose delusions and impulse control.31, 32 In a few cases, visual hallucinations were associated with dementia.23, 24, 29 Treatment with high doses of quetiapine or clozapine showed good efficacy in some cases.31, 32
PARK‐DJ‐1
Psychotic episodes in PD patients with the DJ‐1 gene mutation have been rarely described and include visual hallucinations and paranoid delusions. Some patients were on DRT, and psychosis developed most often after several years of disease.33, 34, 35, 36, 37
22q11.2 Deletion Syndrome
This multisystem condition is associated with an increased risk of early‐onset PD.38 The prevalence of psychosis in this condition ranged between 10% and 30% and developed usually during childhood or early adulthood.39, 40, 41 Psychotic symptoms included auditory and visual hallucinations, as well as paranoid delusions.39, 40, 42 Psychosis was frequently associated with mental retardation41, 42 as well as increased caudate head volume, principally on the left side.43 Psychotic symptoms sometimes improved with clozapine, quetiapine, and valproic acid.39, 44 Special attention should be paid to the risk of clozapine‐associated seizures.38
PARK‐VPS35
No reports of psychosis associated with mutations in this gene were found.
Idiopathic PD
Systematic reviews revealed that he most common psychotic symptom in idiopathic Parkinson's disease (IPD) is visual hallucination, with an estimated pooled prevalence of 28.2% (±19.1 to 39.5),45 followed by isolated delusions (mostly paranoid).46 DRT‐induced visual hallucinations are more frequent in IPD than in patients with mutations in PD‐related genes (except for PARK‐GBA that shows similar figures) and are also commonly associated with dementia.45, 47 In IPD, the content of hallucinations is often stereotyped with frequent descriptions of animals.48 Unlike genetic parkinsonisms, where multimodal hallucinations combined with delusions are the most frequent psychotic features, minor hallucinations, including passage, feeling of presence, or illusions, are one of the most common early psychiatric symptoms in IPD.49 Auditory hallucinations, like verbal hallucinations perceived as originating outside the head, are also more frequent in IPD (estimated pooled prevalence of 8.9%) compared to monogenic parkinsonisms, in which a few cases were reported.50 Psychosis, especially minor hallucinations, can affect around 10% to 42% of untreated IPD individuals at early disease stages,51 but is considerably more infrequent in monogenic parkinsonisms, in which psychosis is commonly associated with disease progression.
Atypical Parkinsonisms
PARK‐ATP13A2
Some cases with psychosis associated with this condition were reported, with patients presenting visual and auditory hallucinations as well as paranoid delusions.51, 52, 53, 54, 55 Visual hallucinations commonly occur several years after disease onset and are often associated with DRT and dementia.52, 54, 55, 56 Response to antipsychotic treatment has been only partial.47
PARK‐DNAJC6
Levodopa‐induced visual hallucinations have been reported in 3 patients with this genetic condition.57 One presented psychotic disturbances at disease onset at the age of 21 years. Another patient exhibited psychosis unrelated to DRT, with symptoms beginning at the age of 10 years, with vivid and terrifying visual hallucinations, which increased in frequency, followed within a few months by parkinsonism and cognitive decline.58
PARK‐FBXO7
In this type of genetic parkinsonism, DRT‐induced psychosis, characterized by delusions59 and hallucinations,60, 61 has been rarely reported.
PARK‐DCTN1 and PARK‐SYNJ1
No patients with psychosis have been reported on in these conditions.
MSA
Hallucinations related to DRT have been rarely reported in MSA.62 However, some patients with MSA did present psychotic features unrelated to DRT, such as religious, persecutory, and somatic delusions, as well as auditory and visual hallucinations (usually of small animals).63, 64, 65, 66 Psychotic manifestations were not related to dementia.63, 64, 66
PSP
Psychosis in PSP is rare. Visual and auditory hallucinations, as well as paranoid delusions, bizarre delusions, and an unusual schizophrenia‐like syndrome, have all been described.67, 68, 69, 70 In some cases, auditory hallucinations occurred during DRT; however, they were not considered related to treatment given that they persisted after l‐dopa discontinuation or, in other cases, hallucinations remitted spontaneously, before discontinuation of therapy.71 Unlike classical PSP, in the Guadeloupean population of PSP‐like parkinsonism, visual and auditory hallucinations unrelated to DRT have been reported in 59% of cases, some of them with comorbid delusions.72
Corticobasal Syndrome
Presence of psychotic symptoms is uncommon and consists of auditory and visual hallucinations as well as persecutory delusions.73, 74, 75 Psychosis, which has been usually associated with dementia, responds to antipsychotic treatment in some patients.73 Patients with alien limb syndrome may experience the delusion of external control.76
Choreas
Diagnosis of the underlying cause of chorea is challenging in cases in which psychosis develops before the onset of chorea. In fact, chorea may be misdiagnosed as tardive dyskinesia in psychotic patients receiving neuroleptics.77
Huntington's Disease
Patients with Huntington's disease (HD) frequently show depression, anxiety, irritability, aggression, apathy, and obsessive compulsive behaviors.78 Psychosis in HD disease is rare, and its prevalence is estimated to range between 3% and 30%, with most recent studies reporting a prevalence of 10%.78, 79, 80, 81, 82, 83, 84, 85, 86 It may manifest as a prodromal symptom, months or years preceding motor or cognitive dysfunction.84, 87, 88 Kirkwood and collaborators reported psychosis developing in 5.2% of the patients during the first year of disease and in 18.3% during the first 10 years of disease.80 More recently, a large prospective study failed to find psychosis in 34 premanifest mutation carriers, even late after motor‐based disease onset, whereas it was present in only 1 of 24 (4%) premanifest mutation carriers close to motor‐based disease onset and in 3 of 70 (4%) early‐symptomatic patients, suggesting that psychosis rarely occurs during the prodromal or early symptomatic HD stages.81 Psychosis in HD is characterized by different types of delusions, such as persecutory, grandiose, and nihilistic. However, other psychotic symptoms have also been reported, such as Othello syndrome (delusional jealousy), Ekbom syndrome (delusional parasitosis), folie à deux (a type of psychosis occurring simultaneously in 2 intimately related persons who share some elements of the illness), as well as somatic, auditory, and visual hallucinations.80, 82, 84 Psychosis is commonly associated with depression and cognitive decline, but the frequency of psychosis declines as cognitive impairment becomes more severe.78, 84, 85, 89 The association between psychosis and size of CAG expansion, age at disease onset, or sex remains inconclusive, with some studies failing to find a significant link78, 86, 90 and others finding lower number of CAG repeats and younger age at time of HD clinical diagnosis in individuals presenting with psychosis.85 Familial aggregation and predisposition to psychosis have been reported in some studies, suggesting that there may be modifying genes interacting with the HD gene to increase susceptibility to psychosis.88, 89 Psychosis in HD is one of the major predictors of nursing home placement.91 Severe or frequent visual and auditory hallucinations were more than twice as common in skilled nursing facility residents compared to patients living at home (4.9% vs. 2%; P = 0.007).91 Response to antipsychotics was variable, with partial response observed in several cases.92 Typical antipsychotics, such as haloperidol, may exert a dual effect, by controlling both psychotic symptoms and choreic movements; however, in more advanced stages of disease, atypical antipsychotics, such as risperidone, olanzapine, or quetiapine, are also effective and better tolerated than typical antipsychotics.92
Sydenham's Chorea
A large study examined the association of Sydenham's chorea and psychosis, and estimated OR was 13.8.93 Delusions of persecution and auditory hallucinations, mimicking schizophrenia, may develop93, 94, 95, 96, 97 and may frequently occur after chorea has subsided.94 Autopsy examination of 1 patient revealed nonspecific mineral deposits in the basal ganglia, similar to those found in schizophrenia and normal aging.94 Treatment usually shows good response to atypical antipsychotics.95, 96, 97, 98
TITF‐1‐Related Benign Hereditary Chorea
Psychosis in this condition was reported in isolated cases and included delusions and visual and auditory hallucinations,99 which were usually well controlled with atypical antipsychotics.100
Chorea‐Acanthocytosis (CHOR‐VPS13A1)
Delusions and hallucinations have been reported in some cases,77, 101, 102 of which a schizophrenia‐like syndrome was sometimes the first clinical manifestation, occurring months to years before neurological symptom onset.77, 102, 103 Psychosis in this condition was unrelated to seizures, albeit hallucinations, such as an epileptic aura, were occasionally reported.101 Response to antipsychotics was usually partial or ineffective.77, 102 Typical antipsychotics should be used with caution because of risk of severe parkinsonism.103
McLeod syndrome (CHOR‐XK)
Psychosis was reported in several patients,104, 105, 106 including a schizophrenia‐like presentation preceding onset of chorea by several years,107, 108 responding to both typical and atypical antipsychotics.105, 106
Adenylyl Cyclase 5–Related Dyskinesia
In this autosomal‐dominant disease, in which patients usually have chorea, including facial dyskinesia, and dystonia, psychosis (mainly auditory hallucinations and grandiose, religious, and persecutory delusions, thought insertion, and thought broadcasting) was reported in a few cases.109, 110
Tourette's Syndrome
Prevalence of psychosis in Tourette's syndrome ranged between 2.5% and 14.6%.111, 112, 113, 114 The most frequent psychotic symptoms were visual, olfactory, and auditory hallucinations and paranoid delusions, which usually developed after an average of 10 years of disease.115, 116, 117, 118 Even though some of the patients reported psychotic symptoms during childhood, many had already developed tics before onset of psychosis.113 Interestingly, some patients received antipsychotics as a treatment for tics before developing psychosis.113 Psychotic symptoms were effectively treated with haloperidol.113, 119
Ataxias
Psychosis has been reported in several conditions that include ataxia as a predominant or consistent clinical feature, such as dominant120, 121 recessive cerebellar ataxias.122, 123
Dominant Cerebellar Ataxias
In autosomal‐dominant cerebellar ataxias, psychosis was reported in different genetic subtypes, such as ATX‐ATXN2 (SCA2), ATX‐ATXN3 (SCA3), ATX‐ATXN7 (SCA7), ATX‐ATXN8 (SCA8), ATX‐ATXN10 (SCA10), ATX‐PPP2R2B (SCA12), ATX‐TBP (SCA17), and ATX‐ATN1 (dentatorubral‐pallidoluysian atrophy), most often after several years of disease course.120, 121, 124, 125 Among these genetic subtypes, psychosis is more frequent in ATX‐ATXN3, ATX‐TBP, and ATX‐ATN1 in comparison to other subtypes, such as ATX‐ATXN2, ATX‐ATXN8, ATX‐PPP2R2B, or ATX‐ATXN10, in which the presence of psychosis was reported in a few cases.120, 124, 125, 126, 127, 128, 129, 130 In ATX‐ATXN3 or Machado‐Joseph disease, the most common spinocerebellar ataxia worldwide, a frequency of psychosis of 4.5% was found in a large cohort of 112 patients.120 Patients with psychotic symptoms were older and presented later onset than those without psychosis.120 Anatomopathological studies in 5 patients with psychotic symptoms revealed severe loss of Purkinje cells and also loss of neurons in the dentate nucleus, inferior olives, and SN, but usually with preserved frontal, temporal, and parietal cortex. These findings are similar to those found in patients with autosomal‐dominant cerebellar ataxias without psychosis.120, 121 Patients with psychosis did not differ significantly in midbrain tyrosine hydroxylase activity staining.121 In fragile X tremor/ataxia syndrome, an X‐linked dominant ataxia, psychosis is uncommon.121
Recessive Cerebellar Ataxias
In Friedreich's ataxia, the most common autosomal‐recessive cerebellar ataxia worldwide, psychosis is rare, usually occurring after years or during the final stages of disease and responding in most cases to antipsychotics, such as risperidone, quetiapine, and aripiprazole.131, 132 In cerebrotendinous xanthomatosis (ATX‐CYP27A1), 7 patients developed delusions and hallucinations during the disease, responding partially to antipsychotics and chenodeoxycholic acid.133, 134, 135 In some patients, psychosis was associated with cognitive decline.133 Other recessive ataxias can show psychosis as part of a broader clinical picture, such as maple syrup urine disease (ATX‐BCKDHB), succinic semialdehyde dehydrogenase deficiency (ATX‐ALDH5A1), the hepatocerebral type of mitochondrial DNA depletion syndrome (ATX‐C10orf2), mitochondrial complex III deficiency nuclear type 2 (ATX‐TTC19), spastic paraplegia type 15 (HSP‐ZFYVE26), X‐linked mental retardation syndrome attributed to mutations in the MECP2 gene, and Hartnup disease (SLC6A19).123
In summary, psychosis in different neurodegenerative ataxias occur commonly after several years of disease and included auditory or visual and somatic hallucinations, paranoid delusions, and delusions of reference.120, 125, 126, 127, 128 Taking into consideration that psychosis as the initial symptom has rarely been reported, the presence of psychosis in early disease stages should point to ATX‐ATXN3, ATX‐TBP, ATX‐ATN1, or cerebrotendinous xanthomatosis. Dementia121, 136 or depression121, 125 have been occasionally associated with psychosis. Treatment with atypical antipsychotics is usually effective in controlling psychotic symptoms.125, 137
Dystonia
Psychosis was reported in some genetic dystonias, like myoclonus‐dystonia attributed to heterozygous mutations or deletions in the epsilon‐sarcoglycan gene (MYC/DYT‐SGCE). In this condition, both hallucinations and paranoid delusions have been reported, usually associated with other psychiatric conditions, such as depression, panic disorder, social phobia, obsessive‐compulsive disorder, and alcohol dependence or abuse, but not with cognitive decline.138, 139, 140, 141 In rapid‐onset dystonia‐parkinsonism caused by mutations in the ATP1A3 gene (DYT/PARK‐ATP1A3), psychotic symptoms may present before or concurrently with motor symptom onset142 or even in the absence of dystonia or parkinsonism.143 In other conditions, like secondary dystonia, dystonic features may be comorbid with the exacerbation or onset of psychotic symptoms.144
Myoclonus
Psychosis is extremely rare in conditions that feature myoclonus as a predominant, or long‐standing, clinical manifestation (myoclonus related to multisystemic general medical conditions, such as anti–N‐methyl‐D‐aspartate receptor encephalitis, has been excluded from this review). Psychosis has been reported in myoclonus‐dystonia attributed to heterozygous mutations or deletions in the epsilon‐sarcoglycan gene (MYC/DYT‐SGCE),139 in patients with myoclonic epilepsy of Lafora (MYC/ATX‐EPM2A and MYC/ATX‐NHLRC1),145 in which prolonged complex visual hallucinations are mostly of epileptic origin and may respond to antiepileptic drugs, rather than antipsychotics,145 and in some types of neuronal ceroid‐lipofuscinoses (NCLs) with myoclonus as a prominent and consistent associated movement disorder (MYC‐CLN6, MYC‐DNAJC5, MYC‐CLN3, and MYC/ATX‐KCTD7).146, 147 NCLs presents with psychosis in up to 20% of patients, although some of them combine psychosis with myoclonus or myoclonic epilepsy.148 Delusions, as well as visual and auditory hallucinations, have been reported.149, 150, 151 The association of psychosis with dementia is common,147, 149, 152 and a propensity toward neuroleptic malignant syndrome has been reported.153, 154
Tremor
Psychosis is absent in disorders that include tremor as a predominant or frequent clinical manifestation in the absence of other signs of parkinsonism.
Fahr's Disease or Idiopathic Basal Ganglia Calcification
Mutations in the SLC20A2, PDGFB, PDGFRB, and XPR1 genes have been identified in several idiopathic basal ganglia calcification (IBGC) families with autosomal‐dominant inheritance, some of which reported hallucinations and delusions.155, 156, 157, 158 Psychotic symptoms that were commonly reported include auditory and visual hallucinations as well as paranoid, reference, and grandiose delusions.159, 160, 161, 162 Age at presentation seems to influence the type of psychiatric symptom. Young individuals (aged <40 years) usually present psychosis without neurological features, whereas after the fifth decade of life, patients present movement disorders and dementia at disease onset.159, 160, 161, 163, 164 Imaging studies indicated that presence of psychosis was proportional to the extent of cerebral calcifications.163 Treatment with atypical antipsychotics is usually effective to alleviate psychotic symptoms158, 165; however, caution is advised given that patients with basal ganglia calcifications may be more vulnerable to developing parkinsonism.159
Neurodegeneration With Brain Iron Accumulation
Psychosis in neurodegeneration with brain iron accumulation (NBIA) has been reported in several cases, although the exact prevalence is unknown. Psychotic symptoms include visual and auditory hallucinations, as well as delusions.166, 167, 168, 169, 170 Psychosis has occasionally been associated with dementia or mental retardation166, 167, 168, 169, 170 and may frequently occur at disease presentation as the only symptom, especially in patients with PLA2G6 mutations, or years before the onset of neurological manifestations.171, 172, 173, 174, 175 In such cases, abnormal brain MRI findings, such as the “the eye of the tiger” sign can help avoid misdiagnosis of schizophrenia. Psychotic symptoms are generally unrelated to DRT,167, 169, 170, 176 although some DRT‐related psychosis cases have been reported.168 Psychotic symptoms in NBIA usually respond to neuroleptic treatment.166, 168, 171, 173 Use of typical antipsychotics requires special caution, given that these drugs may induce parkinsonism or dystonia.177 Though rare, increased sensitivity to neuroleptics (a paradoxical worsening of psychosis) has been reported.167
Wilson's Disease
In the first description of the disease, S.A. Kinnier Wilson described psychotic symptoms in 2 of 12 patients.178 Even though psychiatric disturbances have been reported in 50% to 70% of Wilson's disease (WD) patients,179, 180 < 10% showed psychosis, suggesting that this manifestation is not a hallmark of the disease.179, 181, 182, 183 On the other hand, psychotic symptoms at disease onset may be present in 35% of patients,180, 184 with an interval between onset of psychosis and diagnosis of WD of 2.4 years.185 This poses an important differential diagnosis with schizophrenia. In most cases in which psychotic symptoms were the first manifestation of WD, these were paranoid delusions and hallucinations.180, 186, 187 Psychotic symptoms during the course of WD are similar to those appearing at disease onset.188, 189 Psychosis may be related to discontinuation of penicillamine treatment,190, 191 which usually reverts after treatment is restored or antipsychotics prescribed192 or after liver transplantation.193 Psychotic symptoms may also be related to an ineffective zinc treatment.194 Treatment of psychotic symptoms in WD includes penicillamine monotherapy, which resolves psychosis indirectly by improving copper metabolism. However, redistribution of copper from the liver to other organs, including the brain, after penicillamine treatment may paradoxically cause or aggravate psychosis in some patients.195 Most studies show poor or lack of response to antipsychotics, except for high doses of clozapine or risperidone.189, 196 In some cases of psychosis refractory to antipsychotics, electroconvulsive therapy reverted hallucinations and delusions.197, 198
Niemann‐Pick Disease
Psychotic manifestations are prominent and frequent in Niemann‐Pick disease (NP) types C, A, and B. A recent systematic review of psychiatric signs in NP type C found a prevalence of psychosis of 62%.199 Psychosis may present as paranoid delusions, thought disorder, and visual, auditory, or somatic hallucinations.200, 201, 202, 203 Psychosis as the initial presentation was reported in several cases and may be the only symptom for several years, resulting in misdiagnosis of schizophrenia.174, 200, 201, 202, 204, 205 In these cases, diagnosis of NP disease may be delayed, especially when patients develop dyskinesias after neuroleptic use, which can be mistakenly interpreted as a drug‐induced movement disorder rather than clinical manifestation of NP disease.201, 206 The presence of neurological manifestations, (e.g., vertical supranuclear gaze palsy, gelastic cataplexia, ataxia, dystonia, and seizures), treatment‐resistant psychosis, or paradoxical worsening of psychosis with neuroleptics suggest an organic cause like NP disease.205, 207, 208 Psychosis is frequently associated with other psychiatric symptoms, such as depression203, 204, 208 and cognitive decline.200, 202, 205, 208 Postictal psychosis was also reported in NP disease type C, generally limited to a psychotic disorder that follows complex partial or generalized seizure activity, or a cluster of seizures.209 Frontal lobe atrophy may be prominent in NP disease type C and might be associated with psychosis.174 Psychosis usually responds to antipsychotic medications, but paradoxical worsening and resistance to antipsychotics can be observed, which might be ameliorated in some cases by using miglustat, a drug that inhibits glycosphingolipid synthesis and the only approved targeted therapy for the disease.210, 211 Antipsychotics should be used carefully because of the high frequency of dystonia, which worsens with these types of medications. Risk of a neuroleptic‐induced lipidosis has also been reported.212
Late‐onset Tay‐Sachs or Chronic GM2 Gangliosidosis
Hexosaminidase A deficiency in adults or late‐onset Tay‐Sachs (LOTS) is a common cause of recurrent psychotic syndrome.213 The prevalence of psychosis ranges from 30% to 50% among adult‐onset cases, and many patients are misdiagnosed with paranoid schizophrenia.214 Other psychotic features include visual and auditory hallucinations and Capgras syndrome.215, 216, 217 Psychosis precedes all other clinical features in most cases.213, 217, 218, 219 Nevertheless, in some patients, psychosis develops late after many years of neurological symptoms.213 Psychosis is frequently associated with depression,213, 215, 217 catatonia,216,218 and, in some cases, with dementia.213 Postpartum psychosis, with affective and bizarre delusions, was also reported in LOTS and may resolve with lithium.220 Treatment of psychotic patients with LOTS should not include phenothiazines or tricyclic antidepressants given that they may worsen neurological manifestations by inhibiting the activity of lysosomal enzymes.214, 221 Psychosis refractory to neuroleptics as well as neuroleptic malignant syndrome116,218 have been described in several cases.215,216,218 Lithium and electroconvulsive therapy have frequently proven successful to treat psychotic symptoms in this condition.215, 216, 219
Neurobiology of Psychosis in Movement Disorders
The pathophysiology of psychosis and the role of motor circuits in the development of psychosis is complex and will be summarized here only briefly, given that it is beyond the scope of this review. It has been shown that lesions of the right lateral prefrontal cortex or its efferent projections, such as the basal ganglia and limbic system, are associated with delusions.222 Likewise, frontostriatal circuitry disruption after loss of neurons, as in caudate atrophy observed in patients with HD, may alter relevant processing of striatal‐limbic information and favor the development of psychosis.223 Cerebellar dysfunction has also been described in both schizophrenia and in populations at risk for psychosis, suggesting that the cerebellum may play a role in the development of psychosis.224 In addition to structural alterations, faulty dopamine signaling, including altered dopamine receptor modulation, has been proposed as a possible pathway for the genesis of delusions.225 [18F] Fluoro‐2‐deoxy‐d‐glucose hypometabolism of the striatal and temporal lobes was found in some IBGC patients with psychosis,161, 226 suggesting a disruption of cortico‐subcortical neural circuits. In tauopathies, the abnormal tissue burden distribution, more than the disease type, may trigger psychosis.74 In summary, brain mechanisms underlying delusional symptoms may be similar in both primary and secondary psychosis, but this requires further study.
Conclusion
Psychosis is a common symptom in many conditions in which the primary manifestation is a movement disorder, making it important to evaluate the presence of psychosis during patient monitoring. The most frequent types of psychotic manifestations reported among patients with movement disorders include visual hallucinations and delusions, which occur most often after several years of disease duration and are sometimes associated with cognitive decline or depression. In cases in which psychosis is the presenting clinical manifestation, the diagnosis of secondary psychosis may often be delayed.
Comprehensive neuropsychiatric assessment of psychosis in movement disorder conditions, as well as the association with cognitive or other psychiatric manifestations, is lacking in most reports. Although typical and atypical antipsychotics may be useful to treat psychosis among patients with movement disorders, no structured or class‐I evidence studies have been published to date. DRT discontinuation and use of neuroleptics have been the main treatment strategies reported in the literature. Psychosocial interventions, which are also important tools for the treatment of patients with primary psychosis, were not mentioned in any of the high‐quality studies analyzed for this review.
Psychotic manifestations in the absence of signs and symptoms of a movement disorder should motivate clinicians to a close follow‐up of these patients. Treatment with neuroleptics may confuse the clinical picture because of the development, in some cases, of secondary movement disorders, stressing the challenge to identify a specific movement disorder as a comorbid condition of psychosis in clinical practice. We hope this review will provide the clinician with useful guidelines to improve awareness about the comorbidity of psychosis with different movement disorders.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
M.R.: 1A, 1B, 1C, 2A
N.F.: 1A, 1B, 1C, 2A
S.E.S.: 1A, 1B, 2B
M.M.: 1A, 1B, 1C, 2B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. The authors confirm that the approval of an institutional review board and informed consent was not required for this work.
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: The authors declare that there are no disclosures to report.
Supporting information
Appendix S1: Supporting Information
Dr. Rossi and Farcy contributed equally to this work.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Lawrie SM, O'Donovan MC, Saks E, Burns T, Lieberman JA. Improving classification of psychoses. Lancet Psychiatry 2016;3:367–374. [DOI] [PubMed] [Google Scholar]
- 2. Neumann J, Bras J, Deas E, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain 2009;132:1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Williams DR,Lees AJ. Visual hallucinations in the diagnosis of idiopathic Parkinson's disease: a retrospective autopsy study. Lancet Neurol 2005;4:605–610. [DOI] [PubMed] [Google Scholar]
- 4. Wang C, Cai Y, Gu Z, et al. Clinical profiles of Parkinson's disease associated with common leucine‐rich repeat kinase 2 and glucocerebrosidase genetic variants in Chinese individuals. Neurobiol Aging 2014;35:725.e1–e6. [DOI] [PubMed] [Google Scholar]
- 5. Yahalom G, Greenbaum L, Israeli‐Korn S, et al. Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson's disease: risk estimates and genotype‐phenotype correlations. Parkinsonism Relat Disord 2019;62:179–184. [DOI] [PubMed] [Google Scholar]
- 6. Oeda T, Umemura A, Mori Y, et al. Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson's disease. Neurobiol Aging 2015;36:3306–3313. [DOI] [PubMed] [Google Scholar]
- 7. Creese B, Bell E, Johar I, Francis P, Ballard C, Aarsland D. Glucocerebrosidase mutations and neuropsychiatric phenotypes in Parkinson's disease and Lewy body dementias: review and meta‐analyses. Am J Med Genet B Neuropsychiatr Genet 2018;177:232–241. [DOI] [PubMed] [Google Scholar]
- 8. Barrett MJ, Shanker VL, Severt WL, et al. Cognitive and antipsychotic medication use in monoallelic GBA‐related Parkinson disease. JIMD Rep 2014;16:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jesus S, Huertas I, Bernal‐Bernal I, et al. GBA variants influence motor and non‐motor features of Parkinson's disease. PLoS One 2016;11:e0167749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kasten M, Klein C. The many faces of alpha‐synuclein mutations. Mov Disord 2013;28:697–701. [DOI] [PubMed] [Google Scholar]
- 11. Kiely AP, Ling H, Asi YT, et al. Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener 2015;10:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Konno T, Ross OA, Puschmann A, Dickson DW,Wszolek ZK. Autosomal dominant Parkinson's disease caused by SNCA duplications. Parkinsonism Relat Disord 2016;22(Suppl. 1):S1–S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takamura S, Ikeda A, Nishioka K, et al. Schizophrenia as a prodromal symptom in a patient harboring SNCA duplication. Parkinsonism Relat Disord 2016;25:108–109. [DOI] [PubMed] [Google Scholar]
- 14. Somme JH, Gomez‐Esteban JC, Molano A, Tijero B, Lezcano E,Zarranz JJ. Initial neuropsychological impairments in patients with the E46K mutation of the alpha‐synuclein gene (PARK 1). J Neurol Sci 2011;310:86–89. [DOI] [PubMed] [Google Scholar]
- 15. Zarranz JJ, Alegre J, Gomez‐Esteban JC, et al. The new mutation, E46K, of alpha‐synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004;55:164–173. [DOI] [PubMed] [Google Scholar]
- 16. Belarbi S, Hecham N, Lesage S, et al. LRRK2 G2019S mutation in Parkinson's disease: a neuropsychological and neuropsychiatric study in a large Algerian cohort. Parkinsonism Relat Disord 2010;16:676–679. [DOI] [PubMed] [Google Scholar]
- 17. Kasten M, Marras C, Klein C. Nonmotor signs in genetic forms of Parkinson's disease. Int Rev Neurobiol 2017;133:129–178. [DOI] [PubMed] [Google Scholar]
- 18. Tomiyama H, Li Y, Funayama M, et al. Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson's disease patients from 18 countries. Mov Disord 2006;21:1102–1108. [DOI] [PubMed] [Google Scholar]
- 19. Khan NL, Graham E, Critchley P, et al. Parkin disease: a phenotypic study of a large case series. Brain 2003;126:1279–1292. [DOI] [PubMed] [Google Scholar]
- 20. Kim TJ, Kim TJ, Lee H, Kim YE,Jeon BS. A case of Parkin disease (PARK2) with schizophrenia: evidence of regional selectivity. Clin Neurol Neurosurg 2014;126:35–37. [DOI] [PubMed] [Google Scholar]
- 21. Yamamura Y, Hattori N, Matsumine H, Kuzuhara S, Mizuno Y. Autosomal recessive early‐onset parkinsonism with diurnal fluctuation: clinicopathologic characteristics and molecular genetic identification. Brain Dev 2000;22(Suppl. 1):S87–S91. [DOI] [PubMed] [Google Scholar]
- 22. Hatano Y, Sato K, Elibol B, et al. PARK6‐linked autosomal recessive early‐onset parkinsonism in Asian populations. Neurology 2004;63:1482–1485. [DOI] [PubMed] [Google Scholar]
- 23. Healy DG, Abou‐Sleiman PM, Gibson JM, et al. PINK1 (PARK6) associated Parkinson disease in Ireland. Neurology 2004;63:1486–1488. [DOI] [PubMed] [Google Scholar]
- 24. Li Y, Tomiyama H, Sato K, et al. Clinicogenetic study of PINK1 mutations in autosomal recessive early‐onset parkinsonism. Neurology 2005;64:1955–1957. [DOI] [PubMed] [Google Scholar]
- 25. Marongiu R, Brancati F, Antonini A, et al. Whole gene deletion and splicing mutations expand the PINK1 genotypic spectrum. Hum Mutat 2007;28:98. [DOI] [PubMed] [Google Scholar]
- 26. Hedrich K, Hagenah J, Djarmati A, et al. Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease: role of a single hit? Arch Neurol 2006;63:833–838. [DOI] [PubMed] [Google Scholar]
- 27. Reetz K, Lencer R, Steinlechner S, et al. Limbic and frontal cortical degeneration is associated with psychiatric symptoms in PINK1 mutation carriers. Biol Psychiatry 2008;64:241–247. [DOI] [PubMed] [Google Scholar]
- 28. Samaranch L, Lorenzo‐Betancor O, Arbelo JM, et al. PINK1‐linked parkinsonism is associated with Lewy body pathology. Brain 2010;133:1128–1142. [DOI] [PubMed] [Google Scholar]
- 29. Savettieri G, Annesi G, Civitelli D, et al. Identification of the novel D297fsX318 PINK1 mutation and phenotype variation in a family with early‐onset Parkinson's disease. Parkinsonism Relat Disord 2008;14:509–512. [DOI] [PubMed] [Google Scholar]
- 30. Steinlechner S, Stahlberg J, Volkel B, et al. Co‐occurrence of affective and schizophrenia spectrum disorders with PINK1 mutations. J Neurol Neurosurg Psychiatry 2007;78:532–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ephraty L, Porat O, Israeli D, et al. Neuropsychiatric and cognitive features in autosomal‐recessive early parkinsonism due to PINK1 mutations. Mov Disord 2007;22:566–569. [DOI] [PubMed] [Google Scholar]
- 32. Paviour DC,Marion MH. PINK1: pumps, paraesthesia, punding and psychosis. J Neurol 2012;259:1241–1242. [DOI] [PubMed] [Google Scholar]
- 33. Annesi G, Savettieri G, Pugliese P, et al. DJ‐1 mutations and parkinsonism‐dementia‐amyotrophic lateral sclerosis complex. Ann Neurol 2005;58:803–807. [DOI] [PubMed] [Google Scholar]
- 34. Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ‐1 gene associated with autosomal recessive early‐onset parkinsonism. Science 2003;299:256–259. [DOI] [PubMed] [Google Scholar]
- 35. Hanagasi HA, Giri A, Kartal E, et al. A novel homozygous DJ1 mutation causes parkinsonism and ALS in a Turkish family. Parkinsonism Relat Disord 2016;29:117–120. [DOI] [PubMed] [Google Scholar]
- 36. Taghavi S, Chaouni R, Tafakhori A, et al. A clinical and molecular genetic study of 50 families with autosomal recessive parkinsonism revealed known and novel gene mutations. Mol Neurobiol 2018;55:3477–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Duijn CM, Dekker MC, Bonifati V, et al. Park7, a novel locus for autosomal recessive early‐onset parkinsonism, on chromosome 1p36. Am J Hum Genet 2001;69:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boot E, Bassett AS, Marras C. 22q11.2 deletion syndrome‐associated Parkinson's disease. Mov Disord Clin Pract 2019;6:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krahn LE, Maraganore DM, Michels VV. Childhood‐onset schizophrenia associated with parkinsonism in a patient with a microdeletion of chromosome 22. Mayo Clin Proc 1998;73:956–959. [DOI] [PubMed] [Google Scholar]
- 40. Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo‐cardio‐facial syndrome. Arch Gen Psychiatry 1999;56:940–945. [DOI] [PubMed] [Google Scholar]
- 41. Shprintzen RJ, Goldberg R, Golding‐Kushner KJ, Marion RW. Late‐onset psychosis in the velo‐cardio‐facial syndrome. Am J Med Genet 1992;42:141–142. [DOI] [PubMed] [Google Scholar]
- 42. Chow EW, Bassett AS, Weksberg R. Velo‐cardio‐facial syndrome and psychotic disorders: implications for psychiatric genetics. Am J Med Genet 1994;54:107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eliez S, Barnea‐Goraly N, Schmitt JE, Liu Y, Reiss AL. Increased basal ganglia volumes in velo‐cardio‐facial syndrome (deletion 22q11.2). Biol Psychiatry 2002;52:68–70. [DOI] [PubMed] [Google Scholar]
- 44. Verhoeven WM, Egger JI. Atypical antipsychotics and relapsing psychoses in 22q11.2 deletion syndrome: a long‐term evaluation of 28 patients. Pharmacopsychiatry 2015;48:104–110. [DOI] [PubMed] [Google Scholar]
- 45. Alzahrani H, Venneri A. Cognitive and neuroanatomical correlates of neuropsychiatric symptoms in Parkinson's disease: a systematic review. J Neurol Sci 2015;356:32–44. [DOI] [PubMed] [Google Scholar]
- 46. Warren N, O'Gorman C, Hume Z, Kisely S, Siskind D. Delusions in Parkinson's disease: a systematic review of published cases. Neuropsychol Rev 2018;28:310–316. [DOI] [PubMed] [Google Scholar]
- 47. Swann P, O'Brien JT. Management of visual hallucinations in dementia and Parkinson's disease. Int Psychogeriatr 2019;31:815–836. [DOI] [PubMed] [Google Scholar]
- 48. Barnes J, David AS. Visual hallucinations in Parkinson's disease: a review and phenomenological survey. J Neurol Neurosurg Psychiatry 2001;70:727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Factor SA, McDonald WM, Goldstein FC. The role of neurotransmitters in the development of Parkinson's disease‐related psychosis. Eur J Neurol 2017;24:1244–1254. [DOI] [PubMed] [Google Scholar]
- 50. Eversfield CL, Orton LD. Auditory and visual hallucination prevalence in Parkinson's disease and dementia with Lewy bodies: a systematic review and meta‐analysis. Psychol Med 2019;49:2342–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weintraub D, Mamikonyan E. The neuropsychiatry of Parkinson disease: a perfect storm. Am J Geriatr Psychiatry 2019;27:998–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.52.Behrens MI, Bruggemann N, Chana P, et al. Clinical spectrum of Kufor‐Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord 2010;25:1929–1937. [DOI] [PubMed] [Google Scholar]
- 53. Di Fonzo A, Chien HF, Socal M, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 2007;68:1557‐1562. [DOI] [PubMed] [Google Scholar]
- 54. Eiberg H, Hansen L, Korbo L, et al. Novel mutation in ATP13A2 widens the spectrum of Kufor‐Rakeb syndrome (PARK9). Clin Genet 2012;82:256–263. [DOI] [PubMed] [Google Scholar]
- 55. Ramirez A, Heimbach A, Grundemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P‐type ATPase. Nat Genet 2006;38:1184–1191. [DOI] [PubMed] [Google Scholar]
- 56. Williams DR, Hadeed A, al‐Din AS, Wreikat AL, Lees AJ. Kufor Rakeb disease: autosomal recessive, levodopa‐responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov Disord 2005;20:1264–1271. [DOI] [PubMed] [Google Scholar]
- 57. Olgiati S, Quadri M, Fang M, et al. DNAJC6 mutations associated with early‐onset Parkinson's disease. Ann Neurol 2016;79:244–256. [DOI] [PubMed] [Google Scholar]
- 58. Elsayed LE, Drouet V, Usenko T, et al. A novel nonsense mutation in DNAJC6 expands the phenotype of autosomal‐recessive juvenile‐onset Parkinson's disease. Ann Neurol 2016;79:335–337. [DOI] [PubMed] [Google Scholar]
- 59. Yalcin‐Cakmakli G, Olgiati S, Quadri M, et al. A new Turkish family with homozygous FBXO7 truncating mutation and juvenile atypical parkinsonism. Parkinsonism Relat Disord 2014;20:1248–1252. [DOI] [PubMed] [Google Scholar]
- 60. Di Fonzo A, Dekker MC, Montagna P, et al. FBXO7 mutations cause autosomal recessive, early‐onset parkinsonian‐pyramidal syndrome. Neurology 2009;72:240–245. [DOI] [PubMed] [Google Scholar]
- 61. Lohmann E, Coquel AS, Honore A, et al. A new F‐box protein 7 gene mutation causing typical Parkinson's disease. Mov Disord 2015;30:1130–1133. [DOI] [PubMed] [Google Scholar]
- 62. Miki Y, Foti SC, Asi YT, et al. Improving diagnostic accuracy of multiple system atrophy: a clinicopathological study. Brain 2019;142:2813–2827. [DOI] [PubMed] [Google Scholar]
- 63. Khan S, Moore J, Ealing J, Kobylecki C. Psychosis in a patient with probable multiple system atrophy of cerebellar type. J Neurol Sci 2015;358:501–502. [DOI] [PubMed] [Google Scholar]
- 64. Koga S, Aoki N, Uitti RJ, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology 2015;85:404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Malone D, Dennis MS. Multiple system atrophy and hallucinations—a short report. Int J Geriatr Psychiatry 2005;20:699–700. [DOI] [PubMed] [Google Scholar]
- 66. Papapetropoulos S, Tuchman A, Laufer D, Mash DC. Hallucinations in multiple system atrophy. Parkinsonism Relat Disord 2007;13:193–194. [DOI] [PubMed] [Google Scholar]
- 67. Compta Y, Marti MJ, Rey MJ, Ezquerra M. Parkinsonism, dysautonomia, REM behaviour disorder and visual hallucinations mimicking synucleinopathy in a patient with progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 2009;80:578–579. [DOI] [PubMed] [Google Scholar]
- 68. Lee S. The neuropsychiatric evolution of a case of progressive supranuclear palsy. Br J Psychiatry 1991;158:273–275. [DOI] [PubMed] [Google Scholar]
- 69. Ovsiew F, Schneider J. Schizophrenia and atypical motor features in a case of progressive supranuclear palsy (the Steele‐Richardson‐Olszewski syndrome). Behav Neurol 1993;6:243–247. [DOI] [PubMed] [Google Scholar]
- 70. Perkin GD, Lees AJ, Stern GM, Kocen RS. Problems in the diagnosis of progressive supranuclear palsy. (Steele–Richardson–Olszewski syndrome). Can J Neurol Sci 1978;5:168–173. [PubMed] [Google Scholar]
- 71. Papapetropoulos S, Mash DC. Visual hallucinations in progressive supranuclear palsy. Eur Neurol 2005;54:217–219. [DOI] [PubMed] [Google Scholar]
- 72. Lannuzel A, Ruberg M, Michel PP. Atypical parkinsonism in the Caribbean island of Guadeloupe: etiological role of the mitochondrial complex I inhibitor annonacin. Mov Disord 2008;23:2122–2128. [DOI] [PubMed] [Google Scholar]
- 73. Bensaidane MR, Fortin MP, Damasse G, et al. Clinical utility of amyloid imaging in a complex case of corticobasal syndrome presenting with psychiatric symptoms. J Neurol Disord 2014;2:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Diederich NJ, Leurgans S, Fan W, Chmura TA, Goetz CG. Visual hallucinations and symptoms of REM sleep behavior disorder in Parkinsonian tauopathies. Int J Geriatr Psychiatry 2008;23:598–603. [DOI] [PubMed] [Google Scholar]
- 75. Ikeda C, Yokota O, Nagao S, et al. Corticobasal degeneration initially developing motor versus non‐motor symptoms: a comparative clinicopathological study. Psychogeriatrics 2014;14:152–164. [DOI] [PubMed] [Google Scholar]
- 76. Litvan I, Cummings JL, Mega M. Neuropsychiatric features of corticobasal degeneration. J Neurol Neurosurg Psychiatry 1998;65:717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bruneau MA, Lesperance P, Chouinard S. Schizophrenia‐like presentation of neuroacanthocytosis. J Neuropsychiatry Clin Neurosci 2003;15:378–380. [DOI] [PubMed] [Google Scholar]
- 78. van Duijn E, Craufurd D, Hubers AA, et al. Neuropsychiatric symptoms in a European Huntington's disease cohort (REGISTRY). J Neurol Neurosurg Psychiatry 2014;85:1411–1418. [DOI] [PubMed] [Google Scholar]
- 79. Jensen P, Sorensen SA, Fenger K, Bolwig TG. A study of psychiatric morbidity in patients with Huntington's disease, their relatives, and controls. Admissions to psychiatric hospitals in Denmark from 1969 to 1991. Br J Psychiatry 1993;163:790–797. [DOI] [PubMed] [Google Scholar]
- 80. Kirkwood SC, Su JL, Conneally P, Foroud T. Progression of symptoms in the early and middle stages of Huntington disease. Arch Neurol 2001;58:273–278. [DOI] [PubMed] [Google Scholar]
- 81. Martinez‐Horta S, Perez‐Perez J, van Duijn E, et al. Neuropsychiatric symptoms are very common in premanifest and early stage Huntington's disease. Parkinsonism Relat Disord 2016;25:58–64. [DOI] [PubMed] [Google Scholar]
- 82. Pflanz S, Besson JA, Ebmeier KP, Simpson S. The clinical manifestation of mental disorder in Huntington's disease: a retrospective case record study of disease progression. Acta Psychiatr Scand 1991;83:53–60. [DOI] [PubMed] [Google Scholar]
- 83. Watt DC, Seller A. A clinico‐genetic study of psychiatric disorder in Huntington's chorea. Psychol Med 1993;Suppl. 23:1–46. [DOI] [PubMed] [Google Scholar]
- 84. Weigell‐Weber M, Schmid W, Spiegel R. Psychiatric symptoms and CAG expansion in Huntington's disease. Am J Med Genet 1996;67:53–57. [DOI] [PubMed] [Google Scholar]
- 85. Rocha NP, Mwangi B, Gutierrez Candano CA, Sampaio C, Furr Stimming E, Teixeira AL. The clinical picture of psychosis in manifest Huntington's disease: a comprehensive analysis of the Enroll‐HD Database. Front Neurol 2018;9:930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Saft C, Andrich JE, Brune N, et al. Apolipoprotein E genotypes do not influence the age of onset in Huntington's disease. J Neurol Neurosurg Psychiatry 2004;75:1692–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gomez‐Esteban JC, Lezcano E, Zarranz JJ, et al. Monozygotic twins suffering from Huntington's disease show different cognitive and behavioural symptoms. Eur Neurol 2007;57:26–30. [DOI] [PubMed] [Google Scholar]
- 88. Lovestone S, Hodgson S, Sham P, Differ AM, Levy R. Familial psychiatric presentation of Huntington's disease. J Med Genet 1996;33:128–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tsuang D, Almqvist EW, Lipe H, et al. Familial aggregation of psychotic symptoms in Huntington's disease. Am J Psychiatry 2000;157:1955–1959. [DOI] [PubMed] [Google Scholar]
- 90. Tsuang D, DiGiacomo L, Lipe H, Bird TD. Familial aggregation of schizophrenia‐like symptoms in Huntington's disease. Am J Med Genet 1998;81:323–327. [PubMed] [Google Scholar]
- 91. Wheelock VL, Tempkin T, Marder K, et al. Predictors of nursing home placement in Huntington disease. Neurology 2003;60:998–1001. [DOI] [PubMed] [Google Scholar]
- 92. Bonelli RM, Hofmann P. A review of the treatment options for Huntington's disease. Expert Opin Pharmacother 2004;5:767–776. [DOI] [PubMed] [Google Scholar]
- 93. Wilcox JA, Nasrallah HA. Sydenham's chorea and psychosis. Neuropsychobiology 1986;15:13–14. [DOI] [PubMed] [Google Scholar]
- 94. Casanova MF, Crapanzano KA, Mannheim G, Kruesi M. Sydenham's chorea and schizophrenia: a case report. Schizophr Res 1995;16:73–76. [DOI] [PubMed] [Google Scholar]
- 95. Teixeira AL, Jr. , Maia DP, Cardoso F. Psychosis following acute Sydenham's chorea. Eur Child Adolesc Psychiatry 2007;16:67–69. [DOI] [PubMed] [Google Scholar]
- 96. Hu MT, Butterworth R, Giovannoni G, Church A, Logsdail S. Chorea. Clin Med (Lond) 2009;9:188–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Umene W, Yoshimura R, Hori H, et al. Blood levels of catecholamine metabolites and brain‐derived neurotrophic factor in a case of Sydenham's chorea. World J Biol Psychiatry 2009;10:248–251. [DOI] [PubMed] [Google Scholar]
- 98. Wilcox JA, Nasrallah H. Sydenham's chorea and psychopathology. Neuropsychobiology 1988;19:6–8. [DOI] [PubMed] [Google Scholar]
- 99. Ferrara JM, Adam OR, Kirwin SM, et al. Brain‐lung‐thyroid disease: clinical features of a kindred with a novel thyroid transcription factor 1 mutation. J Child Neurol 2012;27:68–73. [DOI] [PubMed] [Google Scholar]
- 100. Glik A, Vuillaume I, Devos D, Inzelberg R. Psychosis, short stature in benign hereditary chorea: a novel thyroid transcription factor‐1 mutation. Mov Disord 2008;23:1744–1747. [DOI] [PubMed] [Google Scholar]
- 101. Al‐Asmi A, Jansen AC, Badhwar A, et al. Familial temporal lobe epilepsy as a presenting feature of choreoacanthocytosis. Epilepsia 2005;46:1256–1263. [DOI] [PubMed] [Google Scholar]
- 102. Yamada H, Ohji T, Sakurai S, et al. Chorea‐acanthocytosis presenting with schizophrenia symptoms as first symptoms. Psychiatry Clin Neurosci 2009;63:253–254. [DOI] [PubMed] [Google Scholar]
- 103. Muller‐Vahl KR, Berding G, Emrich HM, Peschel T. Chorea‐acanthocytosis in monozygotic twins: clinical findings and neuropathological changes as detected by diffusion tensor imaging, FDG‐PET and (123)I‐beta‐CIT‐SPECT. J Neurol 2007;254:1081–1088. [DOI] [PubMed] [Google Scholar]
- 104. Danek A, Rubio JP, Rampoldi L, et al. McLeod neuroacanthocytosis: genotype and phenotype. Ann Neurol 2001;50:755–764. [DOI] [PubMed] [Google Scholar]
- 105. Jung HH, Haker H. Schizophrenia as a manifestation of X‐linked Mcleod‐Neuroacanthocytosis syndrome. J Clin Psychiatry 2004;65:722–723. [DOI] [PubMed] [Google Scholar]
- 106. Miranda M, Castiglioni C, Frey BM, Hergersberg M, Danek A, Jung HH. Phenotypic variability of a distinct deletion in McLeod syndrome. Mov Disord 2007;22:1358–1361. [DOI] [PubMed] [Google Scholar]
- 107. Chen PY, Lai SC, Yang CC, et al. A novel XK gene mutation in a Taiwanese family with McLeod syndrome. J Neurol Sci 2014;340:221–224. [DOI] [PubMed] [Google Scholar]
- 108. Hewer E, Danek A, Schoser BG, et al. McLeod myopathy revisited: more neurogenic and less benign. Brain 2007;130:3285–3296. [DOI] [PubMed] [Google Scholar]
- 109. Chen DH, Meneret A, Friedman JR, et al. ADCY5‐related dyskinesia: broader spectrum and genotype‐phenotype correlations. Neurology 2015;85:2026–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vijiaratnam N, Newby R, Kempster PA. Depression and psychosis in ADCY5‐related dyskinesia‐part of the phenotypic spectrum? J Clin Neurosci 2018;57:167–168. [DOI] [PubMed] [Google Scholar]
- 111. Bruun RD, Budman CL. Hallucinations in nonpsychotic children. J Am Acad Child Adolesc Psychiatry 1999;38:1328–1329. [DOI] [PubMed] [Google Scholar]
- 112. Comings DE, Comings BG. Tourette syndrome: clinical and psychological aspects of 250 cases. Am J Hum Genet 1985;37:435–450. [PMC free article] [PubMed] [Google Scholar]
- 113. Kerbeshian J, Peng CZ, Burd L. Tourette syndrome and comorbid early‐onset schizophrenia. J Psychosom Res 2009;67:515–523. [DOI] [PubMed] [Google Scholar]
- 114. Shapiro AK, Shapiro E, Wayne H, Clarkin J. The psychopathology of Gilles de la Tourette's syndrome. Am J Psychiatry 1972;129:427–434. [DOI] [PubMed] [Google Scholar]
- 115. Duggal HS. Cognitive affective psychosis syndrome in a patient with sporadic olivopontocerebellar atrophy. J Neuropsychiatry Clin Neurosci 2005;17:260–262.15939990 [Google Scholar]
- 116. Fennig S, Fennig S, Pomeroy J, Calev A. Developmental disorder, Tourette disorder and schizophrenia: a case study. Isr J Psychiatry Relat Sci 1997;34:239–243. [PubMed] [Google Scholar]
- 117. Sandyk R, Bamford CR. Gilles de la Tourette's syndrome associated with chronic schizophrenia. Int J Neurosci 1988;41:83–86. [DOI] [PubMed] [Google Scholar]
- 118. Takeuchi K, Yamashita M, Morikiyo M, et al. Gilles de la Tourette's syndrome and schizophrenia. J Nerv Ment Dis 1986;174:247–248. [DOI] [PubMed] [Google Scholar]
- 119. Fuse‐Nagase Y, Boku M. Psychotic symptoms and a diagnosis of schizophrenia follow an initial diagnosis of tic disorder. J Psychiatry Neurosci 1996;21:346–348. [PMC free article] [PubMed] [Google Scholar]
- 120. Braga‐Neto P, Pedroso JL, Gadelha A, et al. Psychosis in Machado‐Joseph disease: clinical correlates, pathophysiological discussion, and functional brain imaging. Expanding the cerebellar cognitive affective syndrome. Cerebellum 2016;15:483–490. [DOI] [PubMed] [Google Scholar]
- 121. Turk KW, Flanagan ME, Josephson S, Keene CD, Jayadev S, Bird TD. Psychosis in spinocerebellar ataxias: a case series and study of tyrosine hydroxylase in substantia nigra. Cerebellum 2018;17:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Mignarri A, Tessa A, Carluccio MA, et al. Cerebellum and neuropsychiatric disorders: insights from ARSACS. Neurol Sci 2014;35:95–97. [DOI] [PubMed] [Google Scholar]
- 123. Rossi M, Anheim M, Durr A, et al. The genetic nomenclature of recessive cerebellar ataxias. Mov Disord 2018;33:1056–1076. [DOI] [PubMed] [Google Scholar]
- 124. Hering S, Achmuller C, Kohler A, Poewe W, Schneider R, Boesch SM. Phenotype variability in spinocerebellar ataxia type 2: a longitudinal family survey and a case featuring an unusual benign course of disease. Mov Disord 2009;24:774–777. [DOI] [PubMed] [Google Scholar]
- 125. Trikamji B, Singh P, Mishra S. Spinocerebellar ataxia‐10 with paranoid schizophrenia. Ann Indian Acad Neurol 2015;18:93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Day JW, Schut LJ, Moseley ML, Durand AC, Ranum LP. Spinocerebellar ataxia type 8: clinical features in a large family. Neurology 2000;55:649–657. [DOI] [PubMed] [Google Scholar]
- 127. O'Hearn E, Holmes SE, Calvert PC, Ross CA,Margolis RL. SCA‐12: Tremor with cerebellar and cortical atrophy is associated with a CAG repeat expansion. Neurology 2001;56:299–303. [DOI] [PubMed] [Google Scholar]
- 128. Wexler E, Fogel BL. New‐onset psychosis in a patient with spinocerebellar ataxia type 10. Am J Psychiatry 2011;168:1339–1340. [DOI] [PubMed] [Google Scholar]
- 129. Bruni AC, Takahashi‐Fujigasaki J, Maltecca F, et al. Behavioral disorder, dementia, ataxia, and rigidity in a large family with TATA box‐binding protein mutation. Arch Neurol 2004;61:1314–1320. [DOI] [PubMed] [Google Scholar]
- 130. Fujigasaki H, Martin JJ, De Deyn PP, et al. CAG repeat expansion in the TATA box‐binding protein gene causes autosomal dominant cerebellar ataxia. Brain 2001;124:1939–1947. [DOI] [PubMed] [Google Scholar]
- 131. Chan YC. Aripiprazole treatment for psychosis associated with Friedreich's ataxia. Gen Hosp Psychiatry 2005;27:372. [DOI] [PubMed] [Google Scholar]
- 132. Salbenblatt MJ, Buzan RD, Dubovsky SL. Risperidone treatment for psychosis in end‐stage Friedreich's ataxia. Am J Psychiatry 2000;157:303. [DOI] [PubMed] [Google Scholar]
- 133. Berginer VM, Foster NL, Sadowsky M, Townsend JA III, Siegel GJ, Salen G. Psychiatric disorders in patients with cerebrotendinous xanthomatosis. Am J Psychiatry 1988;145:354–357. [DOI] [PubMed] [Google Scholar]
- 134. Dotti MT, Rufa A, Federico A. Cerebrotendinous xanthomatosis: heterogeneity of clinical phenotype with evidence of previously undescribed ophthalmological findings. J Inherit Metab Dis 2001;24:696–706. [DOI] [PubMed] [Google Scholar]
- 135. Fraidakis MJ. Psychiatric manifestations in cerebrotendinous xanthomatosis. Transl Psychiatry 2013;3:e302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chen KH, Lin CH, Wu RM. Psychotic‐affective symptoms and multiple system atrophy expand phenotypes of spinocerebellar ataxia type 2. BMJ Case Rep 2012;2012:bcr1020115061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Demily C, Duwime C, Poisson A, Boddaert N, Munnich A. Low dose clozapine controls adult‐onset psychosis associated with the neurogenic ataxia‐retinitis pigmentosa (NARP) mutation. Mol Genet Metab Rep 2017;10:20–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Dale RC, Nasti JJ, Peters GB. Familial 7q21.3 microdeletion involving epsilon‐sarcoglycan causing myoclonus dystonia, cognitive impairment, and psychosis. Mov Disord 2011;26:1774–1775. [DOI] [PubMed] [Google Scholar]
- 139. Doheny DO, Brin MF, Morrison CE, et al. Phenotypic features of myoclonus‐dystonia in three kindreds. Neurology 2002;59:1187–1196. [DOI] [PubMed] [Google Scholar]
- 140. Peall KJ, Smith DJ, Kurian MA, et al. SGCE mutations cause psychiatric disorders: clinical and genetic characterization. Brain 2013;136:294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Wong SH, Steiger MJ, Larner AJ, Fletcher NA. Hereditary myoclonus dystonia (DYT11): a novel SGCE gene mutation with intrafamilial phenotypic heterogeneity. Mov Disord 2010;25:956–957. [DOI] [PubMed] [Google Scholar]
- 142. Brashear A, Cook JF, Hill DF, et al. Psychiatric disorders in rapid‐onset dystonia‐parkinsonism. Neurology 2012;79:1168–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Smedemark‐Margulies N, Brownstein CA, Vargas S, et al. A novel de novo mutation in ATP1A3 and childhood‐onset schizophrenia. Cold Spring Harb Mol Case Stud 2016;2:a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Thornton A, McKenna PJ. Acute dystonic reactions complicated by psychotic phenomena. Br J Psychiatry 1994;164:115–118. [DOI] [PubMed] [Google Scholar]
- 145. Andrade DM, del Campo JM, Moro E, Minassian BA, Wennberg RA. Nonepileptic visual hallucinations in Lafora disease. Neurology 2005;64:1311–1312. [DOI] [PubMed] [Google Scholar]
- 146. van der Veen S, Zutt R, Klein C, et al. Nomenclature of genetically determined myoclonus syndromes: recommendations of the International Parkinson and Movement Disorder Society Task Force. Mov Disord 2019;34:1602–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Berkovic SF, Oliver KL, Canafoglia L, et al. Kufs disease due to mutation of CLN6: clinical, pathological and molecular genetic features. Brain 2019;142:59–69. [DOI] [PubMed] [Google Scholar]
- 148. Walterfang M, Bonnot O, Mocellin R, Velakoulis D. The neuropsychiatry of inborn errors of metabolism. J Inherit Metab Dis 2013;36:687–702. [DOI] [PubMed] [Google Scholar]
- 149. Jarrett P, Easton A, Rockwood K, et al. Evidence for cholinergic dysfunction in autosomal dominant Kufs disease. Can J Neurol Sci 2018;45:150–157. [DOI] [PubMed] [Google Scholar]
- 150. Backman ML, Aberg LE, Aronen ET, Santavuori PR. New antidepressive and antipsychotic drugs in juvenile neuronal ceroid lipofuscinoses—a pilot study. Eur J Paediatr Neurol 2001;5(Suppl A):163–166. [DOI] [PubMed] [Google Scholar]
- 151. Iannetti P, Messa C, Spalice A, Lucignani G, Fazio F. Positron emission tomography in neuronal ceroid lipofuscinosis (Jansky‐Bielschowsky disease): a case report. Brain Dev 1994;16:459–462. [DOI] [PubMed] [Google Scholar]
- 152. Nijssen PC, Brekelmans GJ, Roos RA. Electroencephalography in autosomal dominant adult neuronal ceroid lipofuscinosis. Clin Neurophysiol 2009;120:1782–1786. [DOI] [PubMed] [Google Scholar]
- 153. Vercammen L, Buyse GM, Proost JE, Van Hove JL. Neuroleptic malignant syndrome in juvenile neuronal ceroid lipofuscinosis associated with low‐dose risperidone therapy. J Inherit Metab Dis 2003;26:611–612. [DOI] [PubMed] [Google Scholar]
- 154. Reif A, Schneider MF, Hoyer A, et al. Neuroleptic malignant syndrome in Kufs' disease. J Neurol Neurosurg Psychiatry 2003;74:385–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hayashi T, Legati A, Nishikawa T, Coppola G. First Japanese family with primary familial brain calcification due to a mutation in the PDGFB gene: an exome analysis study. Psychiatry Clin Neurosci 2015;69:77–83. [DOI] [PubMed] [Google Scholar]
- 156. Kasuga K, Konno T, Saito K, Ishihara A, Nishizawa M, Ikeuchi T. A Japanese family with idiopathic basal ganglia calcification with novel SLC20A2 mutation presenting with late‐onset hallucination and delusion. J Neurol 2014;261:242–244. [DOI] [PubMed] [Google Scholar]
- 157. Legati A, Giovannini D, Nicolas G, et al. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet 2015;47:579–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Nicolas G, Pottier C, Charbonnier C, et al. Phenotypic spectrum of probable and genetically‐confirmed idiopathic basal ganglia calcification. Brain 2013;136:3395–3407. [DOI] [PubMed] [Google Scholar]
- 159. Chiu HF, Lam LC, Shum PP, Li KW. Idiopathic calcification of the basal ganglia. Postgrad Med J 1993;69:68–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Lauterbach EC, Spears TE, Prewett MJ, Price ST, Jackson JG, Kirsh AD. Neuropsychiatric disorders, myoclonus, and dystonia in calcification of basal ganglia pathways. Biol Psychiatry 1994;35:345–351. [DOI] [PubMed] [Google Scholar]
- 161. Le Ber I, Marie RM, Chabot B, Lalevee C, Defer GL. Neuropsychological and 18FDG‐PET studies in a family with idiopathic basal ganglia calcifications. J Neurol Sci 2007;258:115–122. [DOI] [PubMed] [Google Scholar]
- 162. Shouyama M, Kitabata Y, Kaku T, Shinosaki K. Evaluation of regional cerebral blood flow in Fahr disease with schizophrenia‐like psychosis: a case report. AJNR Am J Neuroradiol 2005;26:2527–2529. [PMC free article] [PubMed] [Google Scholar]
- 163. Chabot B, Roulland C, Dollfus S. Schizophrenia and familial idiopathic basal ganglia calcification: a case report. Psychol Med 2001;31:741–747. [DOI] [PubMed] [Google Scholar]
- 164. Cummings JL, Gosenfeld LF, Houlihan JP, McCaffrey T. Neuropsychiatric disturbances associated with idiopathic calcification of the basal ganglia. Biol Psychiatry 1983;18:591–601. [PubMed] [Google Scholar]
- 165. Johnson JM, Legesse B, Camprodon JA, Murray E, Price BH. The clinical significance of bilateral basal ganglia calcification presenting with mania and delusions. J Neuropsychiatry Clin Neurosci 2013;25:68–71. [DOI] [PubMed] [Google Scholar]
- 166. Maciel P, Cruz VT, Constante M, et al. Neuroferritinopathy: missense mutation in FTL causing early‐onset bilateral pallidal involvement. Neurology 2005;65:603–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Neumann M, Adler S, Schluter O, Kremmer E, Benecke R, Kretzschmar HA. Alpha‐synuclein accumulation in a case of neurodegeneration with brain iron accumulation type 1 (NBIA‐1, formerly Hallervorden‐Spatz syndrome) with widespread cortical and brainstem‐type Lewy bodies. Acta Neuropathol 2000;100:568–574. [DOI] [PubMed] [Google Scholar]
- 168. Wirth T, Weibel S, Montaut S, et al. Severe early‐onset impulsive compulsive behavior and psychosis in PLA2G6‐related juvenile Parkinson's disease. Parkinsonism Relat Disord 2017;41:127–129. [DOI] [PubMed] [Google Scholar]
- 169. Yoshino H, Tomiyama H, Tachibana N, et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14‐linked parkinsonism. Neurology 2010;75:1356–1361. [DOI] [PubMed] [Google Scholar]
- 170. Yousaf M, Ramakrishnaiah RH, Kaushik C, Kumar M, Shah CC. Pantothenate kinase 2 deficiency: a neurodegeneration with brain iron accumulation. Radiol Case Rep 2009;4:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Attademo L, Paolini E, Bernardini F, Quartesan R, Moretti P. Adult‐onset case of undiagnosed neurodegeneration with brain iron accumulation with psychotic symptoms. Case Rep Psychiatry 2014;2014:742042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. del Valle‐Lopez P, Perez‐Garcia R, Sanguino‐Andres R, Gonzalez‐Pablos E. Adult onset Hallervorden‐Spatz disease with psychotic symptoms. Actas Esp Psiquiatr 2011;39:260–262. [PubMed] [Google Scholar]
- 173. Panas M, Spengos K, Koutsis G, et al. Psychosis as presenting symptom in adult‐onset Hallervorden‐Spatz syndrome. Acta Neuropsychiatr 2007;19:122–124. [DOI] [PubMed] [Google Scholar]
- 174. Walterfang M, Fietz M, Fahey M, et al. The neuropsychiatry of Niemann‐Pick type C disease in adulthood. J Neuropsychiatry Clin Neurosci 2006;18:158–170. [DOI] [PubMed] [Google Scholar]
- 175. Yazar MS, Fistikci N, Balaban OD, Eradamlar N, Alpkan L. Psychotic disorder in neurodegeneration with brain iron accumulation. Clin Schizophr Relat Psychoses 2013. Sep 18. 10.3371/CSRP.YAFI.091313. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 176. Oner O, Oner P, Deda G, Icagasioglu D. Psychotic disorder in a case with Hallervorden‐Spatz disease. Acta Psychiatr Scand 2003;108:394–397; discussion, 397–398. [DOI] [PubMed] [Google Scholar]
- 177. Huang MH, Chiu YC, Tsai CF. Aripiprazole in a patient of PLA2G6‐associated neurodegeneration with psychosis. Clin Neuropharmacol 2018;41:136–137. [DOI] [PubMed] [Google Scholar]
- 178. Compston A. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver, by S. A. Kinnier Wilson, (From the National Hospital, and the Laboratory of the National Hospital, Queen Square, London) Brain 1912: 34; 295‐509. Brain 2009;132 (Pt. 8):1997–2001. [DOI] [PubMed] [Google Scholar]
- 179. Kumar R, Datta S, Jayaseelan L, Gnanmuthu C, Kuruvilla K. The psychiatric aspects of Wilson's disease—a study from a neurology unit. Indian J Psychiatry 1996;38:208–211. [PMC free article] [PubMed] [Google Scholar]
- 180. Zimbrean PC, Schilsky ML. The spectrum of psychiatric symptoms in Wilson's disease: treatment and prognostic considerations. Am J Psychiatry 2015;172:1068–1072. [DOI] [PubMed] [Google Scholar]
- 181. Akil M, Schwartz JA, Dutchak D, Yuzbasiyan‐Gurkan V, Brewer GJ. The psychiatric presentations of Wilson's disease. J Neuropsychiatry Clin Neurosci 1991;3:377–382. [DOI] [PubMed] [Google Scholar]
- 182. Demily C, Sedel F. Psychiatric manifestations of treatable hereditary metabolic disorders in adults. Ann Gen Psychiatry 2014;13:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Piga M, Murru A, Satta L, et al. Brain MRI and SPECT in the diagnosis of early neurological involvement in Wilson's disease. Eur J Nucl Med Mol Imaging 2008;35:716–724. [DOI] [PubMed] [Google Scholar]
- 184. Srinivas K, Sinha S, Taly AB, et al. Dominant psychiatric manifestations in Wilson's disease: a diagnostic and therapeutic challenge! J Neurol Sci 2008;266:104–108. [DOI] [PubMed] [Google Scholar]
- 185. Biswas S, Paul N, Das SK. Nonmotor manifestations of Wilson's disease. Int Rev Neurobiol 2017;134:1443–1459. [DOI] [PubMed] [Google Scholar]
- 186. Czlonkowska A, Rodo M. Late onset of Wilson's disease. Report of a family. Arch Neurol 1981;38:729–730. [DOI] [PubMed] [Google Scholar]
- 187. Elyasi F. Misidentification of Wilson disease as schizophrenia (1998–2013): case report and review. Indian J Psychol Med 2017;39:675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Huang CC, Chu NS. Psychosis and epileptic seizures in Wilson's disease with predominantly white matter lesions in the frontal lobe. Parkinsonism Relat Disord 1995;1:53–58. [DOI] [PubMed] [Google Scholar]
- 189. Raveh Y, Khoury T, Lachish M, Safadi R, Kohn Y. Acute psychosis and movement disorders as first presentations of Wilson's disease. Isr Med Assoc J 2018;20:788–789. [PubMed] [Google Scholar]
- 190. Lappas AS, Jan F. Unusual psychotic presentation after discontinuation of treatment in a patient with Wilson's disease: a case report. Clin Schizophr Relat Psychoses 2016. Dec 20. 10.3371/CSRP.LAJA.112316. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 191. Modai I, Karp L, Liberman UA, Munitz H. Penicillamine therapy for schizophreniform psychosis in Wilson's disease. J Nerv Ment Dis 1985;173:698–701. [DOI] [PubMed] [Google Scholar]
- 192. Grover S, Sarkar S, Jhanda S, Chawla Y. Psychosis in an adolescent with Wilson's disease: a case report and review of the literature. Indian J Psychiatry 2014;56:395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Sorbello O, Riccio D, Sini M, Carta M, Demelia L. Resolved psychosis after liver transplantation in a patient with Wilson's disease. Clin Pract Epidemiol Ment Health 2011;7:182–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Wichowicz HM, Cubala WJ, Slawek J. Wilson's disease associated with delusional disorder. Psychiatry Clin Neurosci 2006;60:758–760. [DOI] [PubMed] [Google Scholar]
- 195. McDonald LV, Lake CR. Psychosis in an adolescent patient with Wilson's disease: effects of chelation therapy. Psychosom Med 1995;57:202–204. [DOI] [PubMed] [Google Scholar]
- 196. Dudani K, Solanki RK, Sharma P, Singh D. Adolescent onset Wilson's disease misdiagnosed as psychosis. Indian J Psychiatry 2016;58:480–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Rodrigues AC, Dalgalarrondo P, Banzato CE. Successful ECT in a patient with a psychiatric presentation of Wilson's disease. J ECT 2004;20:55. [DOI] [PubMed] [Google Scholar]
- 198. Sahoo MK, Avasthi A, Sahoo M, Modi M, Biswas P. Psychiatric manifestations of Wilson's disease and treatment with electroconvulsive therapy. Indian J Psychiatry 2010;52:66–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Bonnot O, Klunemann HH, Velten C, Torres Martin JV, Walterfang M. Systematic review of psychiatric signs in Niemann‐Pick disease type C. World J Biol Psychiatry 2019;20:320–332. [DOI] [PubMed] [Google Scholar]
- 200. Campo JV, Stowe R, Slomka G, Byler D, Gracious B. Psychosis as a presentation of physical disease in adolescence: a case of Niemann‐Pick disease, type C. Dev Med Child Neurol 1998;40:126–129. [DOI] [PubMed] [Google Scholar]
- 201. Imrie J, Vijayaraghaven S, Whitehouse C, et al. Niemann‐Pick disease type C in adults. J Inherit Metab Dis 2002;25:491–500. [DOI] [PubMed] [Google Scholar]
- 202. Koens LH, Kuiper A, Coenen MA, et al. Ataxia, dystonia and myoclonus in adult patients with Niemann‐Pick type C. Orphanet J Rare Dis 2016;11:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Nadjar Y, Hutter‐Moncada AL, Latour P, et al. Adult Niemann‐Pick disease type C in France: clinical phenotypes and long‐term miglustat treatment effect. Orphanet J Rare Dis 2018;13:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Bonnot O, Gama CS, Mengel E, et al. Psychiatric and neurological symptoms in patients with Niemann‐Pick disease type C (NP‐C): findings from the International NPC Registry. World J Biol Psychiatry 2019;20:310–319. [DOI] [PubMed] [Google Scholar]
- 205. Sevin M, Lesca G, Baumann N, et al. The adult form of Niemann‐Pick disease type C. Brain 2007;130:120–133. [DOI] [PubMed] [Google Scholar]
- 206. Josephs KA, Van Gerpen MW, Van Gerpen JA. Adult onset Niemann‐Pick disease type C presenting with psychosis. J Neurol Neurosurg Psychiatry 2003;74:528–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Wijburg FA, Sedel F, Pineda M, et al. Development of a suspicion index to aid diagnosis of Niemann‐Pick disease type C. Neurology 2012;78:1560–1567. [DOI] [PubMed] [Google Scholar]
- 208. Wraith JE, Sedel F, Pineda M, et al. Niemann‐Pick type C Suspicion Index tool: analyses by age and association of manifestations. J Inherit Metab Dis 2014;37:93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Walterfang M, Kornberg A, Adams S, Fietz M, Velakoulis D. Post‐ictal psychosis in adolescent Niemann‐Pick disease type C. J Inherit Metab Dis 2010;33(Suppl. 3):S63–S65. [DOI] [PubMed] [Google Scholar]
- 210. Kawazoe T, Yamamoto T, Narita A, et al. Phenotypic variability of Niemann‐Pick disease type C including a case with clinically pure schizophrenia: a case report. BMC Neurol 2018;18:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Szakszon K, Szegedi I, Magyar A, et al. Complete recovery from psychosis upon miglustat treatment in a juvenile Niemann‐Pick C patient. Eur J Paediatr Neurol 2014;18:75–78. [DOI] [PubMed] [Google Scholar]
- 212. Turpin JC, Masson M, Baumann N. Clinical aspects of Niemann‐Pick type C disease in the adult. Dev Neurosci 1991;13:304–306. [DOI] [PubMed] [Google Scholar]
- 213. Navon R, Argov Z, Frisch A. Hexosaminidase A deficiency in adults. Am J Med Genet 1986;24:179–196. [DOI] [PubMed] [Google Scholar]
- 214. MacQueen GM, Rosebush PI, Mazurek MF. Neuropsychiatric aspects of the adult variant of Tay‐Sachs disease. J Neuropsychiatry Clin Neurosci 1998;10:10–19. [DOI] [PubMed] [Google Scholar]
- 215. Hamner MB. Recurrent psychotic depression associated with GM2 gangliosidosis. Psychosomatics 1998;39:446–448. [DOI] [PubMed] [Google Scholar]
- 216. Manor I, Hermesh H, Munitz H, Weizman A. Neuroleptic malignant syndrome with gangliosidosis type II. Biol Psychiatry 1997;41:1222–1224. [DOI] [PubMed] [Google Scholar]
- 217. Neudorfer O, Kolodny EH. Late‐onset Tay‐Sachs disease. Isr Med Assoc J 2004;6:107–111. [PubMed] [Google Scholar]
- 218. Rosebush PI, MacQueen GM, Clarke JT, Callahan JW, Strasberg PM, Mazurek MF. Late‐onset Tay‐Sachs disease presenting as catatonic schizophrenia: diagnostic and treatment issues. J Clin Psychiatry 1995;56:347–353. [PubMed] [Google Scholar]
- 219. Stendel C, Gallenmuller C, Peters K, et al. Paranoid delusion as lead symptom in two siblings with late‐onset Tay‐Sachs disease and a novel mutation in the HEXA gene. J Neurol 2015;262:1072–1073. [DOI] [PubMed] [Google Scholar]
- 220. Lichtenberg P, Navon R, Wertman E, Dasberg H, Lerer B. Post‐partum psychosis in adult GM2 gangliosidosis. A case report. Br J Psychiatry 1988;153:387–389. [DOI] [PubMed] [Google Scholar]
- 221. Federico A, Palmeri S, Malandrini A, Fabrizi G, Mondelli M, Guazzi GC. The clinical aspects of adult hexosaminidase deficiencies. Dev Neurosci 1991;13:280–287. [DOI] [PubMed] [Google Scholar]
- 222. Joyce EM. Organic psychosis: the pathobiology and treatment of delusions. CNS Neurosci Ther 2018;24:598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Walterfang M, Evans A, Looi JC, et al. The neuropsychiatry of neuroacanthocytosis syndromes. Neurosci Biobehav Rev 2011;35:1275–1283. [DOI] [PubMed] [Google Scholar]
- 224. Picard H, Amado I, Mouchet‐Mages S, Olie JP, Krebs MO. The role of the cerebellum in schizophrenia: an update of clinical, cognitive, and functional evidences. Schizophr Bull 2008;34:155–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Quintana C, Beaulieu JM. A fresh look at cortical dopamine D2 receptor expressing neurons. Pharmacol Res 2019;139:440–445. [DOI] [PubMed] [Google Scholar]
- 226. Benke T, Karner E, Seppi K, Delazer M, Marksteiner J, Donnemiller E. Subacute dementia and imaging correlates in a case of Fahr's disease. J Neurol Neurosurg Psychiatry 2004;75:1163–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information