

Abstract
Background:
The full spectrum of HIV-1 diversity can be found in Central Africa, including 2 divergent HIV-1 strains collected in 1983 and 1990 in Democratic Republic of Congo (DRC) that were preliminarily classified as group M subtype L. However, a third epidemiologically distinct subtype L genome must be identified to designate L as a true subtype.
Methods:
Specimen CG-0018a-01 was collected in 2001 in DRC as part of an HIV diversity study. Previous subgenomic HIV-1 sequences from this specimen branched closely with proposed subtype L references. Metagenomic next-generation sequencing (mNGS) and HIV-specific target-enriched (HIV-xGen) libraries were combined for NGS to extend genome coverage. mNGS reads were analyzed for the presence of other coinfections with the sequence-based ultrarapid pathogen identification bioinformatics pipeline.
Results:
A complete HIV-1 genome was generated with an average coverage depth of 47,783×. After bioinformatic analysis also identified hepatitis B virus reads, a complete hepatitis B virus genotype A genome was assembled with an average coverage depth of 73,830×. The CG-0018a-01 HIV-1 genome branched basal to the 2 previous putative subtype L strains with strong bootstrap support of 100. With no evidence of recombination present, the strain was classified as subtype L.
Conclusions:
The CG-0018a-01 HIV-1 genome establishes subtype L and confirms ongoing transmission in DRC as recently as 2001. Since CG-0018a-01 is more closely related to an ancestral strain than to isolates from 1983 to 1990, additional strains are likely circulating in DRC and possibly elsewhere.
Key Words: full-length genome, HIV-1 surveillance, subtype L, next-generation sequencing, xGen target enrichment, HIV diversity, phylogenetic analysis
INTRODUCTION
The origins of the HIV pandemic have been traced to the Democratic Republic of Congo (DRC), where estimates place the emergence of HIV in the 1920s.1,2 Consistent with an early expansion of HIV in this region, strains from DRC exhibit broad genetic diversity and include all the recognized subtypes, many circulating recombinant forms (CRFs), and an abundance of unique recombinant forms and unclassifiable sequences.3–8 Current HIV nomenclature guidelines specify that complete genome sequences from at least 3 nontransmission linked cases are required to establish a new subtype or CRF classification for HIV.9 Two unclassifiable complete genome sequences, 83CD003 (AF286236) and 90CD121E12 (AF457101), form a distinct branch from other subtypes and CRFs that is nearly equidistant from neighboring subtypes H and J, which led to a proposal that these sequences are members of a new subtype, L.7 The proposal was further supported by 13.2%–14.5% nucleotide divergence of each genome from all other group M subtypes.7 These strains were collected in 1983 and 1990 in DRC and have not been identified elsewhere. Since subtype L is not an official classification, these isolates are not typically included in reference sequence alignments used to classify HIV sequences. Therefore, it remains possible that other subtype L strains might be circulating, yet unclassified.
Specimen CG-0018a-01 was collected in 2001 at the Good Shepherd Hospital, located 12 kilometers from Kananga, Kasai-Occidental Province, in the DRC, and was previously classified as a putative subtype L based on the sequences of subgenomic polymerase chain reaction (PCR) fragments amplified from the gag, polymerase (pol) integrase, and envelope immunodominant (env IDR) regions.8 Previous efforts to obtain a full genome at that time10 were hampered by low viral load (<4 log10 copies/mL) and limited sample volume, which have now been overcome. Here, we describe the assembly and classification of the complete CG-0018a-01 genomic sequence obtained by application of the HIV-xGen11 method of target enrichment.
METHODS
Specimen
Plasma specimen CG-0018a-01 was collected as previously described8 and as approved by the University of Missouri—Kansas City Research Board with informed consent. The sample was identified as HIV reactive with the ARCHITECT HIV Combo Ag/Ab test (Abbott Diagnostics, Wiesbaden, Germany) with a signal to cutoff (S/CO) of 118.9. A viral load of 3.89 log10 copies/mL was determined by the HIV RealTime test (Abbott Molecular Diagnostics, Des Plaines, IL).
Metagenomic Library Preparation and Target Enrichment
The previously described methods for nucleic acid extraction,10 library preparation, and HIV-xGen target enrichment11 were followed with modifications to accommodate enrichment of a single library. One-half (vol/vol) of the metagenomic next-generation sequencing (mNGS) Nextera library (∼250 ηg) was subjected to HIV-xGen hybridization. Hybridized targets were bound to streptavidin beads, washed to remove unbound DNA, and then amplified by 20 cycles of PCR. Postcapture DNA fragments were purified off the streptavidin beads and amplified by 37 cycles of PCR until a library was visible on the 2200 TapeStation (Agilent, Santa Clara, CA). PCR amplification after removing the library fragments from the streptavidin beads is essential to generating sufficient DNA for NGS. Both the mNGS and HIV-xGen target-enriched libraries were loaded onto the same MiSeq run.
Genome Assembly and Phylogenetic Analysis
The NGS raw data processing and sequence analysis workflow to build a complete viral genome has been described previously.8,10 After the 2 putative subtype L references and the CG-0018a-01 HIV genome were merged into a subset of the 2016 Los Alamos HIV Database full genome alignment (www.hiv.lanl.gov) containing HIV-1 subtypes A–K and at least one each of CRFs 1–88, neighbor-joining phylogenetic and recombinant analyses were completed as previously described.10 A simplified maximum likelihood tree was prepared for Figure 2A after removal of uninformative CRF references from the alignment using MEGA v6.06 and the general time reversible (GTR) + gamma distribution (G) + invariant (I) nucleotide substitution model with 500 bootstrap replicates.12 The trees were rooted to outgroup strain SIVcpz (X52154). Hepatitis B virus (HBV) phylogenetic analysis was completed with reference strains A–I as previously described,13 and HBV escape and resistance mutations were evaluated using Geno2Pheno hbv (2.0).14 Raw NGS data also were uploaded to the sequence-based ultrarapid pathogen identification pipeline15 for analysis and identification of any other human pathogens that might be present in the sample. The HIV and HBV genomes have been deposited in GenBank under accession numbers MN271384 and MN544634, respectively.
FIGURE 2.
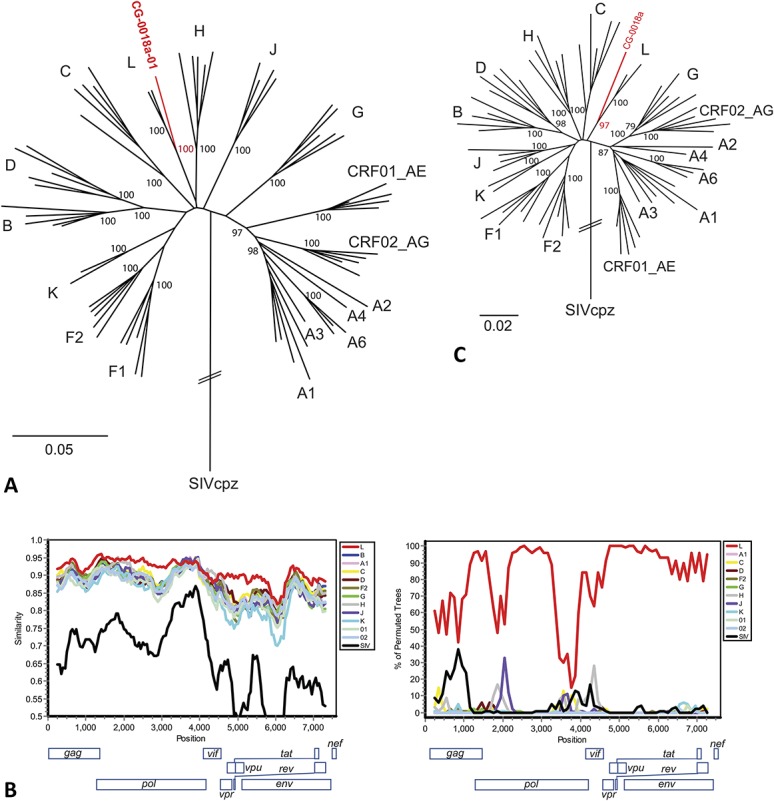
Sequence analysis of the CG-0018a-01 complete genome. A, HIV-1 group M maximum likelihood phylogenetic tree (7578 bp) shows the sequence of CG-0018a-01 groups with the 2 putative subtype L sequences with a bootstrap of 100. B, SimPlot (Window: 500 bp, Step: 50 bp) and Bootscan (Window: 500 bp, Step 100 bp) show % identity is highest to the putative subtype L reference sequences except in the well-conserved pol region (approximate positions 3500–4600). C, Results of neighbor-joining phylogenetic analysis of positions 3500–4600 show that the sequence of CG-0018a-01 continues to group with the putative subtype L sequences with a bootstrap of 97.
RESULTS
To obtain a complete genome from sample CG-0018a-01, a target enrichment (HIV-xGen) method was applied to a cDNA library (mNGS) followed by sequencing of both metagenomic and HIV-xGen target-enriched libraries with a single barcode. Iterative refinement of the consensus sequence identified 4,363,031 of 11,046,542 total reads (39.5%) which mapped to the final 9681 bp complete genome sequence of CG-0018a-01 at an average coverage depth of 47,783× (Fig. 1).
FIGURE 1.
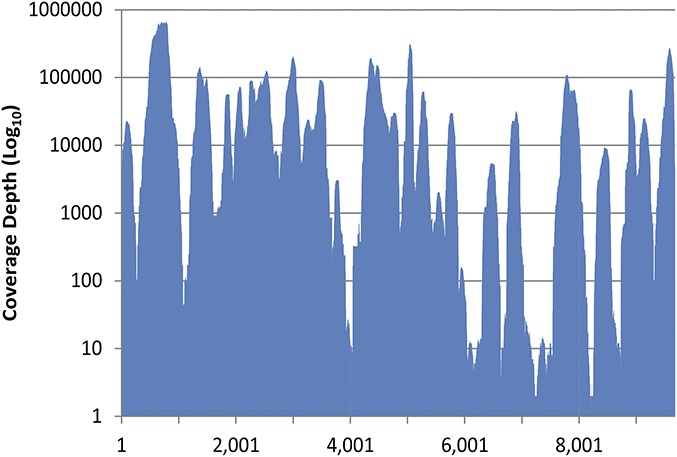
Coverage plot of NGS data for the CG-0018a-01 HIV-xGen library illustrates the average coverage depth of 47,783 across the length of the genome.
A basic local alignment search tool query of the CG-0018a-01 sequence to the NCBI nt database retrieved 90CD121E12, a putative subtype L sequence,7 as the top hit (92% identity, e-value 0.0). Phylogenetic analysis indicates CG-0018a-01 branches with L-83CD003 and L-90CD121E12 with a bootstrap value of 100 (Fig. 2A). Consistent with our previous evaluation of subgenomic sequences,8 full-length CG-0018a-01 branched basal to L-83CD003 and L-90CD121E12, suggesting it may be ancestral to these strains or that it represents a recombinant sequence. SimPlot analysis shows percent identity remains highest to the putative subtype L sequences across the entire genome, except in the well-conserved pol region where percent identity is 90%–95% among all group M subtypes (Fig. 2B). Bootscanning confirmed the absence of a recombination event (Fig. 2B), and an individual tree of the pol region (positions 3500–4600 in the gap-stripped alignment) demonstrates that CG-0018a-01 still branches with the putative subtype L isolates with a bootstrap of 97 (Fig. 2C). Therefore, we classify the sequence of CG-0018a-01 as the third nontransmission linked genome of HIV-1 group M subtype L.
To identify any additional viruses present in the CG-0018a-01 specimen, all NGS reads were processed by the sequence-based ultrarapid pathogen identification bioinformatics tool.15 Unexpectedly, HBV reads were also present, comprising 23.4% (N = 2,588,714) of the NGS reads and indicating that patient CG-0018a-01 was coinfected with HIV and HBV. A complete HBV genotype A sequence was assembled with an average coverage depth of 73,830×. There are no HBV probes in the HIV-xGen probe set, and only 96 reads were mapped from our positive control library spiked with 4 log10 HBV copies/mL, suggesting the HBV viral load in CG-0018a-01 is high (>>5 log 10), although quantitation could not be performed due to limited specimen volume. The HBV surface antigen (HBsAg) “a” determinant region did not encode any known escape mutations, and resistance mutations were absent from the reverse transcriptase region of the pol gene.
DISCUSSION
The complete genome sequence of CG-0018a-01 from the DRC has been assembled from NGS of metagenomic and HIV-xGen target-enriched libraries. The HIV-xGen method has been described previously for target enrichment of a pool of barcoded libraries,11 but the work described here also shows the method can be successful for a single library if additional postcapture amplification is performed. By loading both the mNGS and the HIV-xGen target-enriched libraries on 1 MiSeq run, we were able to obtain complete genome coverage despite divergent viral sequences which may have posed a challenge to probe capture. The mNGS approach also enabled identification of an HBV coinfection and the assembly of a complete HBV genome with comparable deep read coverage.
We conclude that the epidemiologically unlinked isolates CG-0018a-01, 83CD003, and 90CD121E12 may now be classified as HIV-1 group M subtype L. This is the first new subtype classification identified since the nomenclature guidelines were established in 2000.9 Despite being the most recently sequenced subtype L strain, CG-0018a-01 branched basal to the 2 older strains from 1990 and 1983 (Fig. 2A), consistent with CG-0018a-01 being more closely related to the ancestral subtype L strain than the other 2 isolates. Therefore, the CG-0018a-01 sequence will be important for determining the origins and age of subtype L. Furthermore, our identification of CG-0018a-01 decades after the first subtype L strain was collected also suggests that ongoing transmission of subtype L is likely, albeit poorly sampled. Although CG-0018a-01 was one of 172 specimens sequenced in this study,8 we expect the prevalence of subtype L is much lower than was found in this small cohort. Although subtype L is currently restricted to the DRC, it remains possible that future sequence analyses that include this clade as a reference may identify more subtype L infections in DRC or elsewhere. Continued molecular surveillance will be essential to determining the true prevalence of subtype L and other rare or emerging strains of HIV.
Footnotes
The study was funded by Abbott Laboratories.
J.Y., A.V., G.A.C., M.G.B., and M.A.R. are employees and shareholders of Abbott Laboratories. The remaining authors have no conflicts of interest to disclose.
REFERENCES
- 1.Worobey M, Gemmel M, Teuwen DE, et al. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature. 2008;455:661–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faria NR, Rambaut A, Suchard MA, et al. HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science. 2014;346:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao F, Trask SA, Hui H, et al. Molecular characterization of a highly divergent HIV type 1 isolate obtained early in the AIDS epidemic from the Democratic Republic of Congo. AIDS Res Hum Retroviruses. 2001;17:1217–1222. [DOI] [PubMed] [Google Scholar]
- 4.Mokili JL, Wade CM, Burns SM, et al. Genetic heterogeneity of HIV type 1 subtypes in Kimpese, rural Democratic Republic of Congo. AIDS Res Hum Retroviruses. 1999;15:655–664. [DOI] [PubMed] [Google Scholar]
- 5.Vidal N, Peeters M, Mulanga-Kabeya C, et al. Unprecedented degree of human immunodeficiency virus type 1 (HIV-1) group M genetic diversity in the Democratic Republic of Congo suggests that the HIV-1 pandemic originated in Central Africa. J Virol. 2000;74:10498–10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tongo M, Dorfman JR, Martin DP. High degree of HIV-1 group M (HIV-1M) genetic diversity within circulating recombinant forms: insight into the early events of HIV-1M evolution. J Virol. 2015;90:2221–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mokili JL, Rogers M, Carr JK, et al. Identification of a novel clade of human immunodeficiency virus type 1 in Democratic Republic of Congo. AIDS Res Hum Retroviruses. 2002;18:817–823. [DOI] [PubMed] [Google Scholar]
- 8.Rodgers MA, Wilkinson E, Vallari A, et al. Sensitive next-generation sequencing method reveals deep genetic diversity of HIV-1 in the democratic republic of the Congo. J Virol. 2017;91:pii: e01841–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robertson DL, Anderson JP, Bradac JA, et al. HIV-1 nomenclature proposal. Science. 2000;288:55–56. [DOI] [PubMed] [Google Scholar]
- 10.Berg MG, Yamaguchi J, Alessandri-Gradt E, et al. A pan-HIV strategy for complete genome sequencing. J Clin Microbiol. 2015;54:868–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi J, Olivo A, Laeyendecker O, et al. Universal target capture of HIV sequences from NGS libraries. Front Microbiol. 2018;9:2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodgers MA, Vallari AS, Harris B, et al. Identification of rare HIV-1 Group N, HBV AE, and HTLV-3 strains in rural South Cameroon. Virology. 2017;504:141–151. [DOI] [PubMed] [Google Scholar]
- 14.Informatik MPI. Geno2pheno (hbv) v2.0. Available at: http://hbv.geno2pheno.org. Accessed June, 2016. [Google Scholar]
- 15.Naccache SN, Federman S, Veeraraghavan N, et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014;24:1180–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]