

Abstract
The 2016 World Health Organization classification defines diffuse large B-cell lymphoma (DLBCL) subtypes based on Epstein-Barr virus (EBV) infection and oncogenic rearrangements of MYC/BCL2/BCL6 as drivers of lymphomagenesis. A subset of DLBCL, however, is characterized by activating mutations in MYD88/CD79B. We investigated whether MYD88/CD79B mutations could improve the classification and prognostication of DLBCL. In 250 primary DLBCL, MYD88/CD79B mutations were identified by allele-specific polymerase chain reaction or next-generation-sequencing, MYC/BCL2/BCL6 rearrangements were analyzed by fluorescence in situ hybridization, and EBV was studied by EBV-encoded RNA in situ hybridization. Associations of molecular features with clinicopathologic characteristics, outcome, and prognosis according to the International Prognostic Index (IPI) were investigated. MYD88 and CD79B mutations were identified in 29.6% and 12.3%, MYC, BCL2, and BCL6 rearrangements in 10.6%, 13.6%, and 20.3%, and EBV in 11.7% of DLBCL, respectively. Prominent mutual exclusivity between EBV positivity, rearrangements, and MYD88/CD79B mutations established the value of molecular markers for the recognition of biologically distinct DLBCL subtypes. MYD88-mutated DLBCL had a significantly inferior 5-year overall survival than wild-type MYD88 DLBCL (log-rank; P=0.019). DLBCL without any of the studied aberrations had superior overall survival compared to cases carrying ≥1 aberrancy (log-rank; P=0.010). MYD88 mutations retained their adverse prognostic impact upon adjustment for other genetic and clinical variables by multivariable analysis and improved the prognostic performance of the IPI. This study demonstrates the clinical utility of defining MYD88-mutated DLBCL as a distinct molecular subtype with adverse prognosis. Our data call for sequence analysis of MYD88 in routine diagnostics of DLBCL to optimize classification and prognostication, and to guide the development of improved treatment strategies.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is characterized by substantial heterogeneity in tumor biology and clinical behavior.1,2 Currently, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) is used as a ‘one-size-fits-all’ treatment. Unfortunately, a considerable percentage of patients will experience chemorefractory disease or relapse, resulting in a 5-year overall survival (OS) of approximately 60%.3 Particularly, patients with chemorefractory disease or an early relapse have a poor prognosis. For optimal counseling, DLBCL patients are categorized in risk groups according to the IPI.4 The IPI consists of clinical and biochemical parameters, but does not include tumor biological characteristics or provide any indication for precision medicine.5
The recently updated WHO classification of lymphoid neoplasms (2016) recognizes this heterogeneity by including selected drivers of lymphomagenesis for subclassification of DLBCL, i.e. the delineation of high-grade B-cell lymphomas (HGBL) with MYC and BCL2 and/or BCL6 rearrangements, and of Epstein-Barr virus-positive (EBV+) DLBCL.6 MYC, BCL2, and BCL6 rearrangements are found in respectively 4-14%, 20-30%, and ~20% of DLBCL.7–9 HGBL comprise approximately 5-10% of all DLBCL.9–11 It is thought that the combination of MYC-stimulated cell proliferation and anti-apoptotic effects of BCL2 in HGBL cause aggressive growth, relative resistance to therapy, and inferior OS.12 In addition, Asian studies showed a frequency of 1-14% EBV positivity in DLBCL and an association with inferior survival.13,14 EBV-associated viral proteins, such as latent membrane proteins (LMP)-1/2 and nuclear antigens, stimulate proliferation of B-cells via activation of nuclear factor-kappa-B (NFκB), regulate immune evasion, and inhibit apoptosis.13
In the search for additional oncogenic drivers and to discriminate different molecular DLBCL subtypes, large next-generation-sequencing (NGS) studies have revealed specific mutational profiles that reflect the dysregulation of distinct intracellular pathways, including epigenetic regulation and NF-κB, Toll-like receptor (TLR), and B-cell receptor (BCR) signalling.1,2,15,16 Recurrent ‘hotspot’ mutations in MYD88 (L265P) and CD79B (Y196) belong to the most prevalent sequence alterations in DLBCL. By altering the toll/interleukin-1 receptor domain of MYD88, the L265P increases interaction and consecutive phosphorylation of downstream targets, potentially without external stimuli from the TLR.17 The connection of MYD88 with BCR signalling within the so-called ‘My-T-BCR’ supercomplex facilitates activation of the NF-κB pathway via TLR9.2 Hotspot mutations, such as Y196, in the CD79B subunit of the BCR lead to increased BCR expression and inhibition of feedback in the BCR signalling pathway by attenuating downstream Lyn kinase. Therefore, CD79B mutations are thought to contribute to lymphomagenesis by enhancing chronic active BCR signalling.18
Both MYD88 and CD79B mutations are more prevalent in the so-called non-germinal center B-cell (GCB)-type DLBCL according to the cell-of-origin (COO) concept, originally developed on the basis of gene expression profiling.1,2,19 In addition, the prevalence of these mutations varies greatly among DLBCL originating at different anatomical sites. We recently described a high percentage of MYD88 L265P and CD79B Y196 mutations in intravascular large B-cell lymphomas (44% MYD88 and 26% CD79B).20 A high frequency of these mutations has also been found in other extranodal DLBCL, such as primary cutaneous DLBCL, leg type,21 orbita/vitreoretinal DLBCL,22–24 primary breast DLBCL,25 and DLBCL presenting at immune-privileged (IP) sites, i.e. primary testicular DLBCL (PTL)26 and primary central nervous system B-cell lymphoma (PCNSL).27–29 Several studies have shown that MYD88 mutations are associated with inferior OS in DLBCLs compared to wild-type MYD88.30, 31
Despite the increasing knowledge of the landscape of genetic drivers in DLBCL, the clinical implications of different oncogenic driver mutations remain unclear,32 and the R-CHOP regimen is used as a uniform treatment. Since patients with chemorefractory disease or relapses after R-CHOP have a poor outcome, the global 5-year OS in DLBCL is approximately 60%.3 While HGBL patients have been recognized as a particularly unfavorable subgroup, prognostication for the remaining DLBCL is based on clinical and biochemical parameters that define the IPI as well as primary extranodal manifestations.4,5 In contrast, the prognostic significance and interaction of mutations in MYD88 and CD79B with standard molecular aberrations (as designated by the WHO 2016) have not yet been conclusively elucidated. Therefore, the present study investigated whether the assessment of the mutational status of MYD88 and CD79B would improve the classification and prognostication of DLBCL.
Methods
Patient cohort
This retrospective study investigated a cohort of 250 primary DLBCL. DLBCL patients were diagnosed between 2000-2016 at the Amsterdam University Medical Center (AUMC), the Leiden University Medical Center (LUMC), and their affiliated hospitals. In all cases, diagnosis was centrally revised following the WHO classification 2008. A subset of this cohort was previously published without survival analysis.28,29 As our academic hospitals are tertiary referral centers, this cohort is enriched for IP locations. Formalin-fixed and paraffin-embedded (FFPE) tissue samples were obtained during standard diagnostic procedures. The study was performed in accordance with the Dutch Code for Proper Secondary Use of Human Tissue in accordance with the local institutional board requirements and the revised Declaration of Helsinki 2008 and was approved by the medical ethics committees of both the AUMC (W15_213#15.0253) and the LUMC (B16.048). Patients were eligible in case tissue was available and MYD88 mutational analysis was successful.
Histopathologic and molecular characterization
In the majority, immunohistochemistry was performed for CD20, CD10, BCL6, MUM1, and BCL2. The Hans’ algorithm was used for the COO classification.33 The EBV status was assessed by EBER-ISH. MYC, BCL2, and BCL6 rearrangements were analyzed by FISH using break-apart probes. Antibodies and probes are depicted in the Online Supplementary Table S1.20,29 In the AUMC, DNA was isolated using the QIAamp DNA Micro kit (Qiagen) and mutational status of MYD88 and CD79B was established by allele-specific PCR, followed by mutation-specific primers and confirmed by Sanger sequencing, as described before.28, 29 In the LUMC, DNA isolation was automatically performed with the TPS robot (Siemens Healthcare Diagnostics), as presented previously.34 The Ampliseq Cancer Hotspot Panel V.2-V.4 (Thermo Fisher Scientific) was used for the detection of variants in MYD88 (exons 3&5) and CD79B (exons 5&6). The minimum coverage threshold was 100 on-target reads with a minimum variant allele frequency of ≥10% of the reads. Variants were analyzed using Geneticist Assistant NGS Interpretative Workbench (v.1.4.15, SoftGenetics, State College). As described, identified variants were classified into five classes based on potential pathogenicity and only class 4 (possibly pathogenic) and class 5 variants (pathogenic) were reported.35
Statistical analysis
The correlation between the clinicopathologic parameters and biological aberrations was examined with the Chi-square test or ANOVA. The Kaplan-Meier method was applied to estimate 5-year OS and progression free survival (PFS). The starting point for time-to-event analysis was the date of the histological diagnosis. An event for OS was defined as death by any cause. An event for PFS was determined as relapse, disease progression, or death by any cause (whatever came first). If patients received palliative treatment and no remission evaluation was performed during the follow-up, an event for progression was defined at three weeks before patients succumbed to their disease. Observational intervals of patients without any event at the time of the last follow up or at 5 years after diagnosis were censored. The median follow up time for the whole cohort was determined by the use of reverse Kaplan-Meier.36 The log-rank test was performed to compare risk groups. The Cox proportional-hazards model was used to estimate hazard ratios (HR) including 95% confidence intervals (95%-CI). Adjusted HR were obtained in a multivariable Cox model. Competing risks analysis was used to estimate the cumulative incidences of relapse/progression, with non-relapse mortality considered as competing risk. Gray’s test was performed to compare cumulative incidences, whereas a cause-specific Cox proportional-hazards model was used to estimate the impact of risk factors on them.37 The incremental prognostic value of MYD88 and/or CD79B was assessed by comparing Harrell’s cross-validated C statistic for Cox models with and without MYD88 and/or CD79B.38 All statistical analyses were performed using SPSS software (version 23, IBM SPSS statistics) and RStudio (version 1.1442, RStudio, Inc. packages survival, prodlim, dynpred and cmprsk). P-values were two-sided and P<0.05 was considered statistically significant.
Results
Patient characteristics
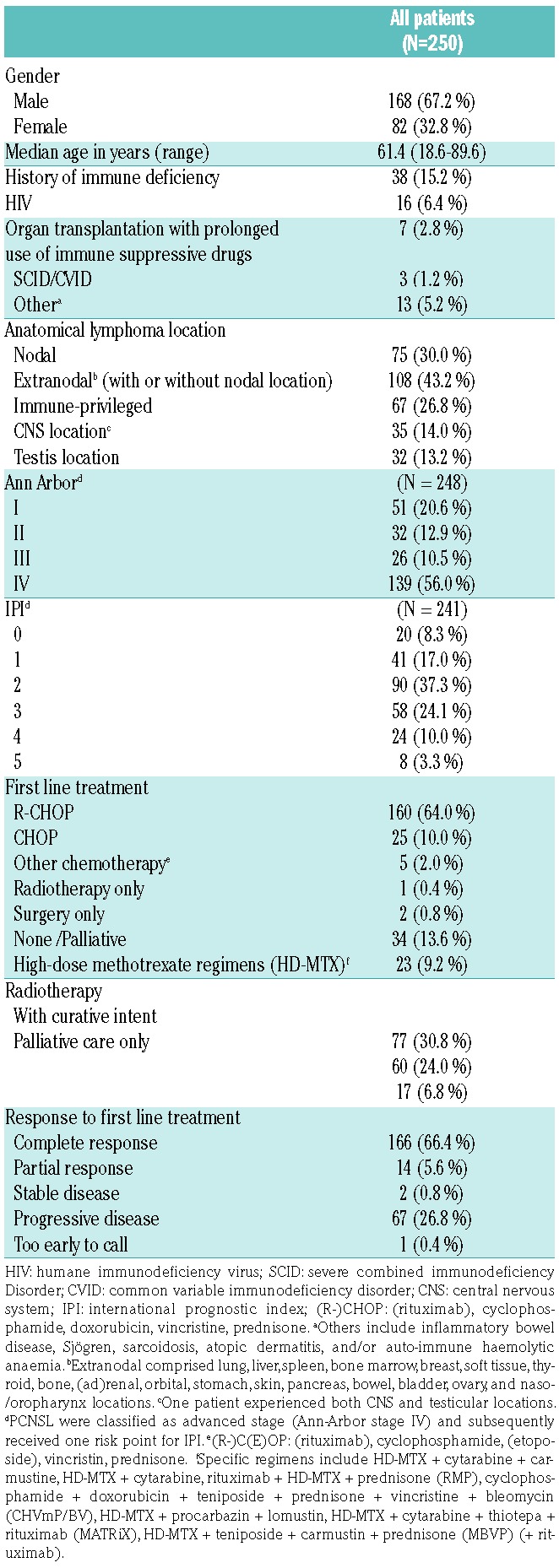
Table 1 depicts the baseline characteristics of the 250 DLBCL patients (AUMC N=224 patients and LUMC N=26 cases). The median age at diagnosis was 61.4 years (range 18.6-89.6). A total of 38 DLBCL patients were immune-compromised, due to inherited conditions (severe combined immunodeficiency disorder, common variable immunodeficiency disorder), human immunodeficiency virus (HIV) infection, or extended use of therapeutic immunosuppression necessitated by organ transplantation or auto-immune disorders. Based on anatomical locations, 75 patients (30.0%) had strictly nodal DLBCL and in 67 patients (26.8%) the lymphoma presented in IP sites: 33 patients with PTL and 35 patients with PCNSL of whom one patient had testicular and CNS locations synchronously. The remaining 108 patients (43.2%) had extranodal disease in non-IP sites (with or without nodal involvement). With respect to staging, PCNSL was considered as advanced disease equivalent to Ann Arbor Stage IV for assignment of the IPI and subsequent statistical analyses. With this definition, 83 patients (33.5%) were categorized as having regional disease (Ann Arbor stage I-II) and 165 patients (66.5%) had advanced disease (stage III-IV). Sixty-one patients (25.3%) had an IPI risk score of 0/1, 148 patients (61.4%) an IPI of 2-3, and 32 patients (13.3%) an IPI of 4-5. The IPI of nine patients was unknown. The majority of (extra)nodal and testicular DLBCL patients were treated with R-CHOP (N=160), CHOP (N=25), or (R)CHOP-like treatments (N=5) with curative intent. Curative treatment regimens incorporating high-dose methotrexate were initiated for 23 patients with PCNSL. Because of older age, poor clinical Eastern Cooperative Oncology Group Performance Status (ECOG-PS), or patients’ refusal of treatment, 34 patients received palliative care only, mainly with steroids or (local) radiotherapy. The median follow up time was 6.6 years (range 0.1-15.7).36
Table 1.
Patient characteristics at time of diagnosis.
Molecular characterization: mutated MYD88 discriminates a distinct DLBCL subgroup
According to the Hans’ algorithm, DLBCL were classified as GCB (N=100, 40.0%), non-GCB (N=130, 52.0%), or unclassifiable (N=20, 8.0%), with no statistical difference between nodal, extranodal, and IP locations (P=0.228)(Table 2).33
Table 2.
Hans’ algorithm and molecular analysis at time of diagnosis.

In 198 patients (79.2%), molecular analysis for MYD88 and CD79B mutations, MYC, BCL2, and BCL6 rearrangements, and EBV infection was complete, whereas in 52 patients, partial data sets were available (Figure 1; Table 2). MYD88 mutations were identified in 74 cases (29.6%), of whom 67 harbored the hotspot L265P mutation. The other MYD88 variants were S219C (N=5) and S243N (N=2). In line with a published meta-analysis,30 mutated MYD88 was significantly correlated with older age (≥65 years), anatomical lymphoma location, and non-GCB subtype (P=0.006; P<0.001; P=0.042, respectively). CD79B mutations were detected in 29 patients (12.3%), including the hotspot Y196 mutation (N=28) and the L188 mutation (N=2, one patient had both mutations). MYC, BCL2, and BCL6 were rearranged in 23 (10.6%), 30 (13.6%), and 44 (20.3%) DLBCL, respectively, with a total of nine HGBL patients (4.1%).
Figure 1.

Oncoprint plot of the molecular analysis of 250 cases with diffuse large B-cell lymphoma (DLBCL). EBV: Epstein-Barr virus; GCB: germinal center B-cell; IP: immune-privileged. Of 52 cases, molecular analysis was not complete due to results that were ambiguous to interpret or no FFPE material was left for subsequent analysis.
As suggested by previous reports and other studies, MYD88 and CD79B mutations were significantly more common in IP-DLBCL (67.2% resp. 25.8%) compared to nodal (17.3% resp. 4.1%) and other extranodal sites (14.8% resp. 9.3%)(P<0.001 and P<0.001).26,29,39 In contrast, BCL2 rearrangements were more prevalent in nodal and extranodal DBLCL (P=0.001), whereas MYC and BCL6 rearrangements were evenly distributed across the anatomical sites. EBV was positive in 28 patients (11.7%) and was not associated with an anatomical location (P=0.091).
In the 198 cases with complete molecular analysis, hardly any overlap between the presence of oncogenic rearrangements, EBV positivity, or MYD88 and/or CD79B mutations was observed (Figure 2A), suggesting that they represent distinct DLBCL subgroups with different drivers of lymphomagenesis. CD79B mutations co-occurred with MYD88 mutations in 18 of 23 cases (78.2%). In contrast, MYD88 mutations co-occurred with any rearrangement in only 7 of 60 patients (11.7%) and with EBV positivity in only one case (1.7%). EBV infection was detected in only 3 of 71 cases (4.2%) with a rearrangement. In 51 patients (25,8%) with full molecular characterization, no aberrancy was detected.
Figure 2.

Molecular characterization discriminates distinct DLBCL subgroups with prognostic impact. (A) Venn diagram demonstrating the overlap of aberrations for 198 fully analysed DLBCL. (B) DLBCL without detected aberrations showed a superior overall survival compared to DLBCL with ≥1 affected aberrations (for cases with complete aberration analysis), identifying a novel good-risk group. (C) PFS of the novel identified risk group (for cases with complete driver analysis). (D) Cumulative incidences of novel identified risk group (for cases with complete driver analysis). CRS: competing risk; PFS: progression free survival.
Mutated MYD88 predicts inferior survival
All outcomes are reported at the 5-year survival. For the entire cohort, the OS was 61.0% (95%-CI 55.1-67.5) and the PFS was 52.6% (95%-CI 46.6-59.3). Cumulative incidences of relapse/progression and non-relapse mortality were 37.2% (95%-CI 31.2-43.3) and 10.1% (95%-CI 6.4- 13.9), respectively. Figure 3 shows the survival outcomes presented for the anatomical location, IPI-score, and MYD88 status. The survival outcomes of COO and the other aberrations are outlined in the Online Supplementary Figure S2 (none of these factors had a significant impact).
Figure 3.

Prognostic significance of anatomical location, IPI Score and MYD88 in DLBCL. OS, PFS, and cumulative incidence of relapse/progression compared to NRM (1st row: Location, 2nd row: IPI Score, 3rd row: MYD88). CRS: competing risk; OS: overall survival; PFS: progression free survival; NRM: non-relapsed mortality.
The IPI clearly predicted OS (Figure 3): patients with IPI scores of 0/1, 2/3, and 4/5 had an OS of 84.9% (95%-CI 76.3-94.5), 58.0% (95%-CI 50.3-66.8), and 34.4% (95%-CI 21.3-55.5), respectively. IPI also showed a significant difference in cumulative incidences of relapses (Gray’s; P=0.025) and non-relapse mortality (Gray’s; P=0.006). In addition to the IPI, DLBCL with IP locations had inferior outcomes (OS 47.1%, 95%-CI 36.5-60.9; PFS 41.0%, 95%-CI 30.7-54.9) compared to nodal (OS 71.2%, 95%-CI 61.4-82.4; PFS 55.7%, 95%-CI 45.3-68.6) and other extranodal sites (OS 62.6%, 95%-CI 53.9-72.7; PFS 58.1%, 95%-CI 49.4-68.2) (log-rank; P=0.004 and P=0.024). This unfavorable prognosis was particularly associated with the CNS location. Within the IP group, patients with CNS location had a significantly inferior 5-year OS of 29.9% (95%-CI 17.7-50.5) compared to 65.5% (95%-CI 50.9-84.3%) for PTL (log-rank; P=0.003).
With respect to the molecular markers, patients without any detected aberrancy demonstrated a good-risk profile with a superior OS (78.0%, 95%-CI 67.2-90.4, versus 56.3%, 95%-CI:48.6-65.2; Figure 2B) (log-rank; P=0.010) and PFS (65.4%, 95%-CI 53.2-80.3, versus 48.2%, 95%-CI 40.6-57.3; Figure 2C) (log-rank; P=0.031) compared to patients who had one or more aberration(s). The cumulative incidence of relapse/progression for this good-risk profile was 28.6% (95%-CI 15.8-41.4) compared to 39.3% (95%-CI 31.2-47.4) (Gray’s; P=0.155). This good risk profile included patients with a lower ECOG-PS, age<60 years, and more GCB subtypes (Chi square; P=0.012, P=0.001, and P=0.006, respectively) compared to patients with one or more aberrations. Patients in the good risk category seem to be susceptible for immune-chemotherapy with enduring responses, however, the molecular background of this subgroup remains unknown. In IP-DLBCL, a total of 93.8% of the patients were classified in the risk group with ≥1 aberrations.
MYD88-mutated DLBCLs had a significantly inferior 5-year OS compared to DLBCL with wild-type MYD88 (log-rank; P=0.019; HR 1.64, 95%-CI 1.08-2.48) and significantly inferior 5-year PFS (log-rank; P=0.049; HR 1.46, 95%-CI 1.00-2.14). Employing competing risk analysis, MYD88-mutated DLBCL revealed significantly higher relapse rates (46.6%, 95%-CI 35.1-58.1) than cases with wild-type MYD88 (33.3%, 95%-CI 26.2-40.4)(Gray’s; P=0.029; CSH 1.62, 95%-CI 1.06-2.48), while non-relapse mortality showed no significant difference (Gray’s; P=0.832). Mutated CD79B showed higher cumulative incidence for relapse/progression (56.3%, 95%-CI 37.9-74.8) versus wild-type CD79B (35.1%, 95%-CI 28.5- 41.8)(Gray’s; P=0.019, CSH: 1.82, 95%-CI 1.06-3.14), whereas no significant difference was found for OS (HR 1.43, 95%-CI 0.81-2.53).
Despite relatively high HR, none of the other molecular aberrations was a significantly adverse prognostic factor for OS (Table 3), which can be explained by the lack of power due to the low incidence of these aberrations. For these molecular data, univariate cause-specific hazards for relapse/progression showed similar results. The nine HGBL had an OS of 50.0% (95%-CI 24.1-100) compared to 63.6% (95%-CI 57.3-70.6) (log-rank; P=0.628) for non-HGBL.
Table 3A.
Prognostic impact of molecular aberrations and IPI risk factors on overall survival: univariable and multivariable analysis.

Prognostic significance of MYD88 mutations in multivariable analysis
To evaluate the prognostic impact of mutated MYD88 on survival outcomes in addition to other molecular aberrations and the IPI, the initial multivariable Cox regression model included the standard individual IPI risk factors (Model 1, Table 3A/3B). In the second model, the current WHO 2016 molecular aberrations (EBV and oncogenic rearrangements) were added. In the third model, also MYD88 and CD79B mutations were included. MYD88 mutations showed prognostic significance for OS (HR 1.87, 95%-CI 1.10-3.20) in addition to ECOG-PS (≥2) (HR 8.16, 95%-CI 4.90-13.59) and Ann Arbor stage (III/IV) (HR 1.84, 95%-CI 1.04-3.25). In this third model, oncogenic rearrangements, mutated CD79B, elevated LDH, and age (>65 years) did not have a significant impact. The performance of the IPI prognostic model was improved by adding all molecular aberrations and mutated MYD88 and CD79B as risk factors, as indicated by an increase in cross-validated C-index (CVC) from 0.67 to 0.70. MYD88 did not have a significant impact on the cause-specific survival (HR 1.42, 95%-CI 0.85-2.37), whilst ECOG-PS, Ann Arbor stage, and extranodal location were prognostic in this model.
Table 3B.
Prognostic impact of molecular aberrations and IPI risk factors on relapse/progression: univariable and multivariable analysis.

Further multivariable analyses were performed to evaluate the prognostic significance of MYD88 mutational status in comparison to the COO subtype or anatomical lymphoma location. The COO subtype did not improve the performance of models 2 and 3 (results not shown). However, the prognostic impact of model 2 was improved by adding the anatomical lymphoma location (CVC index = 0.71, model 4, presented in the Online Supplementary Table S1) and outperforms model 2 (Table 3A, CVC index = 0.69, including the IPI factors and molecular aberrations of WHO 2016). Model 4 demonstrated a nearly identical prognostic performance when compared to model 3 (CVC index = 0.70, including the IPI factors, molecular aberrations of WHO 2016 and the mutational status of MYD88 and CD79B). When adding the mutational status of MYD88 and CD79B to model 4, the performance of this model 5 was not improved (CVC index 0.71, Online Supplementry Table S1). As such, the prognostic impact of the MYD88 mutational status on mortality was not superior to the anatomical lymphoma location.
Next, we explored whether mutated MYD88 could improve the prognostic performance of the currently used IPI risk model (Table 4). The inclusion of the IPI as a continuous variable (0-5 points) and the MYD88 status in the multivariable analysis demonstrated an independent and similar impact of mutated MYD88 (HR 1.83, 95%-CI 1.19-2.80) and IPI (HR 1.77, 95%-CI 1.47-2.13) on OS. Similar effects were observed for cause-specific survival (Table 4). For the models, OS and relapse/progression, an increase in CVC-index was observed from 0.57 to 0.61 and 0.53 to 0.57, respectively. Altogether, these multivariable survival analyses demonstrated the significant prognostic importance of mutated MYD88, next to (genetic) aberrations and clinical/biochemical variables, and the improvement of adding mutated MYD88 to the prognostic performance of the IPI.
Table 4.
Mutated MYD88 improved the prognostic performance of the IPI.

To evaluate possible confounding of the impact of mutated MYD88 and with the outcomes by anatomical lymphoma location, we performed a sensitivity analysis for OS on the cohort stratified by the anatomical lymphoma location, including CNS involvement. For patients with CNS involvement (N=35), MYD88 had an unadjusted HR of 1.94 (95%-CI 0.77-4.90) in the univariable analysis. For patients without CNS involvement (N=215), MYD88 did not have a significant impact on OS with an adjusted HR of 1.81 (95%-CI 0.96-3.42), when applying the multivariable analysis as described for model 3 (Table 3B). Although not statistically significant, the adjusted HR for this subgroup was similar to the original HR for the entire cohort.
Discussion
To the best of our knowledge, this is the first study evaluating the clinical significance of mutated MYD88 and CD79B in DLBCL, in addition to the oncogenic drivers that are currently included in the WHO classification 2016 (EBV status and MYC, BCL2, and BCL6 rearrangements), the IPI risk factors, and well-defined anatomical locations.
The strength of this study is the large number of patients with good clinical annotation and complete molecular analysis (N=198). In addition, our study shows that the incorporation of the mutational status of MYD88 into a clinical/biochemical risk score as the IPI is feasible. An increase in the predictive performance of the IPI risk model as is illustrated by an increase in the CVC-index, suggests that this model can be improved by the introduction of molecular aberrations. However, while interpreting the results, we have encountered several limitations. MYD88-mutated DLBCL more often had extranodal location, older age (and thus a high IPI), and non-GCB subtype. Therefore, these patients were more frequently subjected to palliative care. Possibly the interaction between treatment and mutated MYD88 has not been tested as more data is needed. We present an average effect over different treatment modalities. Since the reported frequencies and survival outcomes are similar to previous reports in the literature, our cohort appears to be representative for the target population.3,7–9,13 To investigate the prognostic significance of mutated MYD88 adjusted for the IPI for the entire cohort, we considered PCNSL as advanced disease stage, although it is not common practice to apply the IPI in PCNSL patients. Additionally, our cohort is enriched for IP locations. Therefore, a sensitivity analysis was performed excluding PCNSL patients, demonstrating that the adjusted HR of MYD88 for OS was similar to the entire cohort. This indicates that our results are not affected by confounding by CNS localisation. Hence, we believe that our data corroborate the clinical relevance of mutant MYD88 for the diagnostic classification and prognostication of DLBCL and support implementation of MYD88 mutational analysis in routine diagnostics. The simplicity and accessibility to examine MYD88 mutations and associated low costs permit an efficient timely implementation. In addition, CD79B mutations were prognostic in the univariate analysis, but when adjusted for other aberrations in the multivariable analysis the prognostic importance disappeared. This finding may be explained by the prominent overlap between MYD88 and CD79B mutations, as 78.2% of mutated CD79B had co-occurring MYD88 mutations.
An important result of our study is the recognition of the prominent mutual exclusivity between the presence of mutations in MYD88 and/or CD79B, MYC, BCL2, and BCL6 rearrangements, and EBV infection, indicating that MYD88 and/or CD79B-mutated tumors present a distinct DLBCL subcategory. In accordance with a large meta-analysis and two other studies,30,40,41 MYD88 L265P mutations were preferentially found in specific anatomical sites (e.g. testis and CNS) and were significantly associated with non-GCB subtypes, older age, and poor OS. However, the published literature has studied neither explicitly analysed IP sites, nor evaluated the interaction of MYD88 mutations with EBV status or oncogenic rearrangements in multivariable analysis. Other NGS studies have recently demonstrated high frequencies of mutated MYD88 (15-18%) in large cohorts of DLBCL.1,2,15,42–44 Besides a certain association of mutated MYD88 with poor OS (e.g. in non-GCB DLBCL), cluster analysis of multiple genes indicated distinct DLBCL subentities, including mutated MYD88 as an important classifier for NF-κB pathway activation. Again, these NGS studies did not take into account specific anatomical sites or investigated the interaction and prognostic significance of mutated MYD88 in correlation with the EBV status or MYC, BCL2, and BCL6 rearrangements.
In this context, our study adds important new knowledge by demonstrating MYD88 mutations as an adverse prognostic factor for OS and relapse/progression in a multivariate analysis that takes all major known clinical and WHO classification-defined risk factors into account. This insight does not only show that the incorporation of the mutational status of MYD88 into a clinical/biochemical risk score as the IPI is feasible, but also highlights the importance of assessing MYD88 at the time of diagnosis for an optimal classification and patient counselling. An increase in the predictive performance of the IPI risk model, as is illustrated by an increase in the CVC-index, formally suggests that this model can be improved by the introduction of molecular aberrations. However, the prognostic impact of the MYD88 mutational status on the presented multivariable models was not superior to anatomical lymphoma location. Whether the MYD88 mutational status outperforms the predictive performance of anatomical lymphoma location in the described prognostic models needs further validation in an external cohort. Of note, no difference was found for non-relapse mortality, indicating that mutated MYD88 is a lymphoma-specific poor prognostic factor. Routine diagnostic assessment of MYD88 mutations is likely to gain decisive importance for DLBCL since several approaches may therapeutically target MYD88.45,46 Several studies have indicated that DLBCL with mutated MYD88 and/or CD79B are more sensitive to Bruton’s Tyrosine Kinase (BTK)-inhibitors.46–48 As such, the objective analysis of MYD88 mutations will not only improve diagnostic classification and prognostication, but might also enable patient selection for precision medicine such as treatment with BTK-inhibitors. However, the predictive significance of mutated MYD88 with or without CD79B mutations needs to be validated in upcoming clinical trials, including precision medicine targeting the BCR and TLR cascades.
Finally, as a corollary of this study, we identified a novel good risk DLBCL group characterized by the absence of detected genetic aberrations. These DLBCL appeared to be highly sensitive to standard immune-chemotherapy as a first-line treatment. Future studies, employing a larger NGS targeted gene panel, may elucidate the genetic drivers in this group. We anticipate that there might be a parallel with the study of Chapuy et al.,15 which identified a good-risk DLBCL group harbouring mainly aberrations in epigenetic pathways.
Studies by Rossi et al. and Kurtz et al.,49,50 have analysed liquid biopsies in DLBCL demonstrating that the mutational load in circulating-free tumor DNA obtained by NGS technologies reliably mirror the mutational profiles of DLBCL tissues, including mutated MYD88. Additionally, digital droplet PCR techniques enable the quantification of low amounts of mutated MYD88 in any physiological fluid.51 Further investigation is needed to determine whether the analysis of mutated MYD88 in liquid biopsies prior to and during therapy will be significantly predictive for the treatment response and to establish its specificity and sensitivity.
Conclusion
The present study demonstrates that the presence of MYD88 and CD79B mutations is almost mutually exclusive with EBV infection and MYC, BCL2, and BCL6 rearrangements, indicating distinctive molecular DLBCL subgroups that can be readily appreciated in clinical practice. Mutant MYD88 showed its prognostic importance for inferior survival outcomes, even next to other genetic and clinical prognosticators and IPI. Additionally, patients lacking all analysed abberrancies represented a novel risk group with superior survival outcomes. Taken together and after validation in an independent cohort, these results provide a rationale for including MYD88 mutational analysis in the routine diagnostics of DLBCL, to improve classification and prognostication, as well as to guide future treatment strategies.
Supplementary Material
Acknowledgments
The authors would like to thank the involved research technicians, data managers and physicians for their contributions to this manuscript.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/105/2/424
Funding
This study was supported in part by research funding from ‘Egbers Stichting AMC Foundation’, ‘Stichting Fonds Oncologie Holland’, and Lymph&Co (JSV, SFS, RAG, AHC, WK, MS, MJK, and STP).
References
- 1.Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171(2):481–94 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. 2018;560(7718):387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cunningham D, Hawkes EA, Jack A, Qian W, Smith P, Mouncey P, et al. Rituximab plus cyclophosphamide, doxorubicin, vin-cristine, and prednisolone in patients with newly diagnosed diffuse large B-cell non-Hodgkin lymphoma: a phase 3 comparison of dose intensification with 14-day versus 21-day cycles. Lancet (London, England). 2013;381(9880):1817–1826. [DOI] [PubMed] [Google Scholar]
- 4.A predictive model for aggressive non-Hodgkin’s lymphoma. N Engl J Med. 1993; 329(14):987–994. [DOI] [PubMed] [Google Scholar]
- 5.Wight JC, Chong G, Grigg AP, Hawkes EA. Prognostication of diffuse large B-cell lymphoma in the molecular era: moving beyond the IPI. Blood Rev. 2018;32(5):400–415. [DOI] [PubMed] [Google Scholar]
- 6.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenthal A, Younes A. High grade B-cell lymphoma with rearrangements of MYC and BCL2 and/or BCL6: Double hit and triple hit lymphomas and double expressing lymphoma. Blood Rev. 2017;31(2):37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shustik J, Han G, Farinha P, Johnson NA, Ben Neriah S, Connors JM, et al. Correlations between BCL6 rearrangement and outcome in patients with diffuse large B-cell lymphoma treated with CHOP or R-CHOP. Haematologica. 2010;95(1):96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt-Hansen M, Berendse S, Marafioti T, McNamara C. Does cell-of-origin or MYC, BCL2 or BCL6 translocation status provide prognostic information beyond the International Prognostic Index score in patients with diffuse large B-cell lymphoma treated with rituximab and chemotherapy? A systematic review. Leukemia & lymphoma. 2017;58(10):2403–2418. [DOI] [PubMed] [Google Scholar]
- 10.Akyurek N, Uner A, Benekli M, Barista I. Prognostic significance of MYC, BCL2, and BCL6 rearrangements in patients with diffuse large B-cell lymphoma treated with cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab. Cancer. 2012;118(17):417347–83. [DOI] [PubMed] [Google Scholar]
- 11.McPhail ED, Maurer MJ, Macon WR, Feldman AL, Kurtin PJ, Ketterling RP, et al. Inferior survival in high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements is not associated with MYC/IG gene rearrangements. Haematologica. 2018;103(11):1899–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leskov I, Pallasch CP, Drake A, Iliopoulou BP, Souza A, Shen CH, et al. Rapid generation of human B-cell lymphomas via com bined expression of Myc and Bcl2 and their use as a preclinical model for biological therapies. Oncogene. 2013;32(8):1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao X, Li J, Wang Y, Liu S, Yue B. Clinical characteristics and prognostic significance of EBER positivity in diffuse large B-cell lymphoma: A meta-analysis. PloS one. 2018;13(6):e0199398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu TX, Liang JH, Miao Y, Fan L, Wang L, Qu XY, et al. Epstein-Barr virus positive diffuse large B-cell lymphoma predict poor outcome, regardless of the age. Scientific reports. 2015;5:12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nature medicine. 2018;24(5):679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. New Engl J Med. 2018; 378(15):1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ngo VNYRM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;(3)470:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. [DOI] [PubMed] [Google Scholar]
- 20.Schrader AMR, Jansen PM, Willemze R, Vermeer MH, Cleton-Jansen AM, Somers SF, et al. High prevalence of MYD88 and CD79B mutations in intravascular large B-cell lymphoma. Blood. 2018;131(18):2086–2089. [DOI] [PubMed] [Google Scholar]
- 21.Zhou XA, Louissaint A, Jr, Wenzel A, Yang J, Martinez-Escala ME, Moy AP, et al. Genomic analyses identify recurrent alterations in immune evasion genes in diffuse large B cell lymphoma, leg type. J Invest Dermatol. 2018;138(11):2365–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonzheim I, Giese S, Deuter C, Susskind D, Zierhut M, Waizel M, et al. High frequency of MYD88 mutations in vitreoretinal B-cell lymphoma: a valuable tool to improve diagnostic yield of vitreous aspirates. Blood. 2015;126(1):76–79. [DOI] [PubMed] [Google Scholar]
- 23.Cani AK, Soliman M, Hovelson DH, Liu CJ, McDaniel AS, Haller MJ, et al. Comprehensive genomic profiling of orbital and ocular adnexal lymphomas identifies frequent alterations in MYD88 and chromatin modifiers: new routes to targeted therapies. Mod Pathol. 2016; 29(7):685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raja H, Salomao DR, Viswanatha DS, Pulido JS. Prevalence of Myd88 L265p mutation in histologically proven, diffuse large B-cell vtreoretinal lymphoma. Retina. 2016;36(3):624–628. [DOI] [PubMed] [Google Scholar]
- 25.Taniguchi K, Takata K, Chuang SS, Miyata-Takata T, Sato Y, Satou A, et al. Frequent MYD88 L265P and CD79B mutations in primary breast diffuse large B-cell lymphoma. Am J Surg Pathol. 2016; 40(3):324–334. [DOI] [PubMed] [Google Scholar]
- 26.Kraan W, van Keimpema M, Horlings HM, Schilder-Tol EJ, Oud ME, Noorduyn LA, et al. High prevalence of oncogenic MYD88 and CD79B mutations in primary testicular diffuse large B-cell lymphoma. Leukemia. 2014; 28(3):719–720. [DOI] [PubMed] [Google Scholar]
- 27.Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood. 2016;127(7):869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kersten MJ, Kraan W, Doorduijn J, Bromberg J, Lam K, Kluin PM, et al. Diffuse large B cell lymphomas relapsing in the CNS lack oncogenic MYD88 and CD79B mutations. Blood Cancer J. 2014;12;4: e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraan W, Horlings HM, van Keimpema M, Schilder-Tol EJ, Oud ME, Scheepstra C, et al. High prevalence of oncogenic MYD88 and CD79B mutations in diffuse large B-cell lymphomas presenting at immune-privileged sites. Blood Cancer J. 2013; 3:e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JH, Jeong H, Choi JW, Oh H, Kim YS. Clinicopathologic significance of MYD88 L265P mutation in diffuse large B-cell lymphoma: a meta-analysis. Scientific reports. 2017;7(1):1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu S, Luo H, Pan M, Palomino LA, Song X, Wu P, et al. High frequency and prognostic value of MYD88 L265P mutation in diffuse large B-cell lymphoma with R-CHOP treatment. Oncology Lett. 2018;15(2):1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vermaat JS, Pals ST, Younes A, Dreyling M, Federico M, Aurer I, et al. Precision medicine in diffuse large B-cell lymphoma: hitting the target. Haematologica. 2015; 100(8):989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103(1):275–282. [DOI] [PubMed] [Google Scholar]
- 34.van Eijk R, Stevens L, Morreau H, van Wezel T. Assessment of a fully automated high-throughput DNA extraction method from formalin-fixed, paraffin-embedded tissue for KRAS, and BRAF somatic mutation analysis. Exp Mol Pathol. 2013; 94(1):121–125. [DOI] [PubMed] [Google Scholar]
- 35.Sibinga Mulder BG, Mieog JS, Handgraaf HJ, Farina Sarasqueta A, Vasen HF, Potjer TP, et al. Targeted next-generation sequencing of FNA-derived DNA in pancreatic cancer. J Clin Pathol. 2017;70(2):174–178. [DOI] [PubMed] [Google Scholar]
- 36.Schemper M, Smith TL. A note on quantifying follow-up in studies of failure time. Control Clin Trials. 1996;17(4):343–346. [DOI] [PubMed] [Google Scholar]
- 37.Kim HT. Cumulative incidence in competing risks data and competing risks regression analysis. Clin Cancer Res. 2007;15;13(2 Pt 1): 559–565. [DOI] [PubMed] [Google Scholar]
- 38.Houwelingen HCPH. Dynamic Prediction in Clinical Survival Analysis. Chapman & Hall, 2012. [Google Scholar]
- 39.Zheng M, Perry AM, Bierman P, Loberiza F, Jr, Nasr MR, Szwajcer D, et al. Frequency of MYD88 and CD79B mutations, and MGMT methylation in primary central nervous system diffuse large B-cell lymphoma. Neuropathology. 2017;37(6):509–516. [DOI] [PubMed] [Google Scholar]
- 40.Rovira J, Karube K, Valera A, Colomer D, Enjuanes A, Colomo L, et al. MYD88 L265P mutations, but no other variants, identify a subpopulation of DLBLC patients of activated B-cell origin, extranodal involvement, and poor outcome. Clin Cancer Res. 2016;22(11):10. [DOI] [PubMed] [Google Scholar]
- 41.Dubois S, Viailly PJ, Bohers E, Bertrand P, Ruminy P, Marchand V, et al. Biological and clinical relevance of associated genomic alterations in MYD88 L265P and non-L265P-mutated diffuse large B-cell lymphoma: analysis of 361 cases. Clin Cancer Res. 2017;23(9):2232–2244. [DOI] [PubMed] [Google Scholar]
- 42.Intlekofer AM, Joffe E, Batlevi CL, Hilden P, He J, Seshan VE, et al. Integrated DNA/RNA targeted genomic profiling of diffuse large B-cell lymphoma using a clinical assay. Blood Cancer J. 2018;12;8(6):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karube K, Enjuanes A, Dlouhy I, Jares P, Martin-Garcia D, Nadeu F, et al. Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets. Leukemia. 2018;32(3):675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu PP, Zhong HJ, Huang YH, Gao XD, Zhao X, Shen Y, et al. B-cell function gene mutations in diffuse large B-cell lymphoma: a retrospective cohort study. EBioMedicine. 2017;16:106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu X, Li W, Deng Q, Li L, Hsi ED, Young KH, et al. MYD88 L265P Mutation in lymphoid malignancies. Cancer Res. 2018; 78(10):2457–2462. [DOI] [PubMed] [Google Scholar]
- 46.de Groen RAL, Schrader AMR, Kersten MJ, Pals ST, Vermaat JSP. MYD88 in the driver’s seat of B-cell lymphomagenesis: from molecular mechanisms to clinical implications. Haematologica. 2019;104(12):2337–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grommes C, Pastore A, Palaskas N, Tang SS, Campos C, Schartz D, et al. Ibrutinib unmasks critical role of Bruton tyrosine kinase in primary CNS lymphoma. Cancer Discov. 2017;7(9):1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, et al. Inhibition of B cell receptor signaling by ibrutinib in primary CNS lymphoma. Cancer Cell. 2017;31(6):833–43 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med 2015; 21(8):922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurtz DM, Scherer F, Jin MC, Soo J, Craig AFM, Esfahani MS, et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J Clin Oncol. 2018; 36(28):2845–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossi D, Diop F, Spaccarotella E, Monti S, Zanni M, Rasi S, et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood. 2017;129(14):1947–1957. [DOI] [PubMed] [Google Scholar]
- 52.Hiemcke-Jiwa LS, Minnema MC, Radersma-van Loon JH, Jiwa NM, de Boer M, Leguit RJ, et al. The use of droplet digital PCR in liquid biopsies: a highly sensitive technique for MYD88 p.(L265P) detection in cerebrospinal fluid. Hematol Oncol. 2018;36(2):429–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.