

Abstract
A high allele burden of the KIT D816V mutation in peripheral blood or bone marrow aspirates indicates multi-lineage hematopoietic involvement and has been associated with an aggressive clinical course of systemic mastocytosis. Since mast cells are substantially underrepresented in these liquid specimens, their mutation burden likely underestimates the tumor burden of the disease. We used a novel previously validated digital polymerase chain reaction (PCR) method for KIT D816V analysis to systematically analyze the mutation burden in formalin-fixed, paraffin-embedded bone marrow tissue sections of 116 mastocytosis patients (91 with indolent and 25 with advanced systemic mastocytosis), and to evaluate for the first time the clinical value of the tissue mutation burden as a novel biomarker. The KIT D816V mutation burden in the tissue was significantly higher and correlated better with bone marrow mast cell infiltration (r=0.68 vs. 0.48) and serum tryptase levels (r=0.68 vs. 0.58) compared to that in liquid specimens. Furthermore, the KIT D816V tissue mutation burden was: (i) significantly higher in advanced than in indolent systemic mastocytosis (P=0.001); (ii) predicted survival of patients in multivariate analyses independently; and (iii) was significantly reduced after response to cytoreductive therapy. Finally, digital PCR was more sensitive in detecting KIT D816V in bone marrow sections of indolent systemic mastocytosis patients than melting curve analysis after peptide nucleic acid-mediated PCR clamping (97% vs. 89%; P<0.05). In summary, digital PCR-based measurement of KIT D816V mutation burden in the tissue represents a novel biomarker with independent prognostic significance that can also be employed for monitoring disease progression and treatment response in systemic mastocytosis.
Introduction
Systemic mastocytosis (SM) is a clonal hematopoietic disorder characterized by abnormal infiltration of mast cells (MC) in various organs, including the bone marrow (BM).1 The somatic KIT D816V mutation leads to growth factor-independent activation of the receptor tyrosine kinase KIT.2,3 Detection of the mutation in the BM, peripheral blood (PB), or another extra-cutaneous organ is a minor diagnostic criterion for SM.1,4 Although this typical driver mutation is present in a vast majority of all patients with SM, the clinical course in SM is highly variable.5 The World Health Organization (WHO) classification divides mastocytosis into cutaneous mastocytosis (CM), indolent SM (ISM), smoldering SM (SSM), aggressive SM (ASM), SM with an associated hematologic neoplasm (SM-AHN), and mast cell leukemia (MCL).1,4,6 These last three entities are collectively referred to as advanced mastocytosis, based on their increased risk of progression and death.4,7
Molecular diagnostics has become increasingly important in SM. Molecular techniques with a high analytical sensitivity, such as allele-specific quantitative polymerase chain reaction (qPCR) or digital PCR (dPCR), are required to reliably detect the KIT mutation in liquid specimens (PB or BM aspirate).8–13 Melting curve analysis after peptide nucleic acid (PNA)-mediated PCR clamping (clamp-PCR) is widely used for qualitative detection of the KIT mutation in formalin-fixed paraffin-embedded (FFPE) tissue biopsies despite its limited analytical sensitivity that requires micro-dissection of BM MC in a number of cases.14,15 We have recently shown that dPCR is suitable as a new sensitive method for KIT D816V testing in SM that also reliably quantifies the variant allele frequency (VAF).16 A high mutant allele burden in liquid specimens was indicative of multi-lineage involvement with KIT D816V and was associated with an aggressive clinical course and advanced forms of SM.16–20 However, in the majority of patients, only a small fraction of KIT D816V+ MC and/or MC precursors, if any, is found in liquid specimens.21 Therefore, quantification of KIT D816V VAF in liquid specimens typically only evaluates multi-lineage involvement in the non-MC compartment. In line with this observation, only a moderate correlation of KIT D816V VAF in liquid specimens with MC infiltration or serum tryptase as surrogate markers for disease burden in SM has been described.17–20 In contrast, the percentage of KIT D816V+ MC is typically much higher in BM tissue biopsies compared to BM aspirates.22,23 Therefore, molecular measurements in liquid specimens substantially underestimate the disease burden in SM, in particular in ISM. Moreover, while a reduction of the mutation burden in liquid specimens has been described in response to cytoreductive treatment in patients with advanced SM and multi-lineage involvement,18,24 its value as a follow-up parameter in ISM and advanced SM without multi-lineage involvement remains uncertain. Molecular quantification of KIT D816V disease burden in the tissue has the potential to overcome these limitations as a biomarker of disease burden in SM.
The quantification of KIT D816V VAF has not been assessed systematically in BM tissue sections of SM patients. Here we investigated the clinical value of dPCR-based KIT D816V tissue mutation burden quantification as a novel biomarker in SM.
Methods
Patients
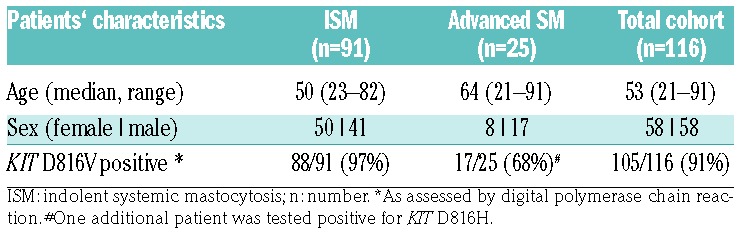
We examined 390 samples (211 FFPE BM sections, 106 BM aspirates, 73 PB) from 116 SM patients (58 females, 58 males), diagnosed between April 1988 and February 2016 and included in a local registry. PB and BM samples at diagnosis and during follow up were obtained after informed consent and the study was approved by the institutional review board (EK:1750/2017). According to WHO criteria,1,6 83 patients were diagnosed with ISM, 8 with SSM, 7 with ASM, 3 with MCL, and 15 with SM-AHN. Patients’ characteristics are shown in Table 1. During the course of disease, 36 patients (31%) received a cytoreductive treatment with a median of two different regiments (range 1-5) (Online Supplementary Table S1). Fifty-seven FFPE BM sections from lymphoma patients undergoing a staging biopsy, and in whom no BM infiltration was detected, were used as control material (Online Supplementary Table S2). Quantification of BM infiltration by MC, flow cytometry of MC, and measurement of serum tryptase is described in the Online Supplementary Methods.
Table 1.
Patients’ characteristics.
Molecular analysis of KIT D816V
Genomic DNA was extracted from (FFPE) BM sections as well as PB and/or aspirated BM cells as previously described and dPCR for KIT D816V was performed with the PrimePCR ddPCR mutation assay for KIT wild-type and the KIT D816V point mutation (Bio-Rad Laboratories, Munich, Germany) and analyzed on the QX-200 droplet-reader (Bio-Rad Laboratories), as described in detail in the Online Supplementary Methods.16 In addition, qualitative detection of KIT codon 816 mutations was performed using melting curve analysis after PNA-mediated PCR clamping as described.14–16
Statistical analysis
Statistical analysis was performed using R (version 3.4.2, Vienna, Austria)25 and GraphPad Prism (GraphPad Software, La Jolla, CA, USA). Applied tests are described in detail in the Online Supplementary Methods and P<0.05 was considered to be significant.
Results
The KIT D816V tissue allele burden is a novel biomarker in systemic mastocytosis
The KIT D816V tissue allele burden was studied by dPCR in 211 FFPE BM sections from 116 SM patients (Table 1). Median VAF was 1.9%, with a range between 0.027% and 60% indicating a substantial difference in the tissue mutation burden over various orders of magnitude between patients. When we compared the KIT D816V VAF in FFPE BM section with that in matched PB or BM aspirates samples, higher tissue allele burden levels were observed (Figure 1A), whereas a strong correlation was found between PB and BM aspirates (r=0.99) (Online Supplementary Figure S1). A comparison between the log transformed KIT D816V VAF in the tissue and liquid specimens (BM aspirate n=96, PB n=12 in cases for which no BM aspirate was available for molecular analysis) in 108 matched samples from 79 patients, revealed a direct correlation in non-parametric analysis (r=0.87) but a constant and proportional deviation was found in Passing Bablok regression analysis (intercept: 1.72, 95%CI: 1.53-1.91; slope: 0.59, 95%CI: 0.52-0.65) (Figure 1B). In addition, Bland-Altman plot displayed a deviation tendency to higher KIT D816V allele burden in FFPE BM sections, particularly in samples with low VAF (Figure 1C). In summary, these findings indicate that the KIT D816V VAF in FFPE BM sections was not interchangeable with allele burden measurements in liquid specimens and represents a new biological variable of disease burden in SM patients.
Figure 1.

Relation of KIT D816V allele burden in formalin-fixed paraffin-embedded (FFPE) bone marrow (BM) sections of systemic mastocytosis (SM) patients with that in liquid specimens. (A) Ternary plot of KIT D816V variant allele frequency (VAF) from 54 paired FFPE BM section (red axis), BM aspirate (green axis) and peripheral blood (PB) (blue axis) samples. (B) Comparison of KIT D816V VAF in 108 paired FFPE BM (VAF) tissue] and BM aspirate/PB samples (VAF liquid) showing a systematic constant and proportional deviation to higher VAF in the tissue (r=0.87; slope: 0.59 (95%CI: 0.52-0.65), intercept: 1.72 (95%CI: 1.53-1.91) for log transformed data). (C) Bland-Altman plot of KIT D816V VAF in the tissue and liquid specimens showing a skewing towards higher results in the tissue for samples with low average VAF.
The KIT D816V tissue allele burden correlates with mast cell infiltration and serum tryptase levels
In a next step, we correlated KIT D816V VAF with BM MC infiltration and serum tryptase levels as established surrogate parameters of disease burden in KIT D816V positive SM patients (n=105). The correlation of mutant allele burden with BM MC infiltration as determined by immunohistochemistry was higher for DNA isolated from BM FFPE sections (r=0.68) (Figure 2A) than from liquid specimens (r=0.48) (Figure 2B). Likewise, a higher correlation with serum tryptase was observed for tissue mutation burden (r=0.68) (Figure 2C) than for liquid mutation burden (r=0.58) (Figure 2D). When we stratified the samples into ISM and advanced SM, a higher correlation of BM MC infiltration with the mutation burden in the tissue (compared to liquid samples) was observed for both subgroups, with a generally higher correlation in ISM patients (Online Supplementary Figure S2A and B). In contrast, we found only a modest correlation of the mutation burden with serum tryptase for both specimens in advanced SM, while the correlation with tryptase substantially increased from liquid (r=0.55) to tissue VAF (r=0.70) in ISM (Online Supplementary Figure S2C and D). Together, the KIT D816V allele burden in FFPE BM sections reflects the burden of neoplastic cells in SM better than established mutant allele burden measurements in PB or BM aspirate.
Figure 2.

Association of KIT D816V allele burden with biomarkers of disease burden in systemic mastocytosis (SM). Correlation of KIT D816V mutation burden in 185 formalin-fixed paraffin-embedded (FFPE) bone marrow (BM) sections [variant allele frequency (VAF) tissue: A and C] and 108 BM aspirate/peripheral blood (PB) samples (VAF liquid: B and D) with immunohistologically determined BM mast cell (MC) infiltration (A and B) and serum tryptase (C and D). r: Spearman’s correlation coefficient.
The KIT D816V tissue allele burden is higher in advanced than in indolent systemic mastocytosis
Next, we compared surrogate markers of disease burden in SM patients between ISM (n=91) and advanced SM (n=25). The median BM MC infiltration was higher in advanced SM (20%) compared to ISM (7%) (P<0.01) (Figure 3A). Likewise, a tendency towards higher levels of serum tryptase levels were observed in advanced SM (median 140 ng/mL vs. 37 ng/mL; P=0.07) (Figure 3B). Within the KIT D816V positive patients, a significantly higher mutant allele burden was observed in advanced SM both in liquid specimens (21.31% vs. 0.34%; P<0.01) (Figure 3C) as well as in BM tissue samples (23.40% vs. 1.65%; P<0.001) (Figure 3D).
Figure 3.

Biomarkers of disease burden in indolent and advanced systemic mastocytosis (SM). Bone marrow (BM) mast cell (MC) infiltration (A), serum tryptase levels (B), and KIT D816V mutant allele burden in BM aspirate/peripheral blood samples [variant allele frequency (VAF) liquid: C] and formalin-fixed paraffin-embedded (FFPE) BM sections (VAF tissue; D) were assessed for indolent (ISM) (blue, n=91) and advanced (green, n=25) SM patients. Samples from smoldering SM patients within the ISM cohort are shown in orange. **P<0.01; ***P<0.001.
However, the observed difference was bigger in liquid specimens due to the higher KIT D816V allele burden in BM tissue (compared to liquid specimens) in our ISM patients. When we analyzed SSM patients within the ISM cohort (n=8), particularly high levels of MC infiltration (median: 35%, range: 20-80%), serum tryptase (median: 234 ng/mL, range: 188-545 ng/mL), liquid mutant allele burden (median: 35.40%, range: 10.60-39.10%) as well as tissue mutant allele burden (median: 10.94%, range: 3.90-36.90%) were found (Figure 3). In summary, our data indicate that tissue mutation burden is a promising novel bio-marker of disease burden in SM.
The KIT D816V tissue allele burden predicts survival in systemic mastocytosis and reflects response to cytoreductive treatment
To define the prognostic value of tissue mutation burden quantification, we associated KIT D816V tissue VAF results with the clinical end points overall survival (OS) and progression-free survival (PFS) in KIT D816V+ SM patients (n=103; no survival data available for 2 patients) in univariate and multivariate analyses including KIT D816V VAF in liquid specimens, age, sex, B-findings (BM MC infiltration, serum tryptase, organomegaly), and C-findings (anemia, thrombocytopenia, hypalbuminemia, weight loss, hepatomegaly with liver dysfunction, and increased alkaline phosphatase) as additional variables. In univariate analyses, PFS was adversely influenced by the majority of established risk factors including a high KIT D816V tissue allele burden with a hazard ratio (HR) of 15.82 (95%CI: 5.31-47.16) and a high KIT D816V liquid allele burden with a HR of 5.99 (95%CI: 2.41-14.88) (Table 2). In multivariate analysis including all molecular and clinical variables, only thrombocytopenia (HR: 21.26, 95%CI: 2.64-171.30; P=0.004) and a high KIT D816V allele burden in the tissue (HR: 50.71, 95%CI: 4.23-607.90; P=0.002) remained independent risk markers for PFS (Table 2). Similar results were obtained for OS with a HR of 12.79 (95%CI: 4.22-38.76) for a high KIT D816V tissue allele burden and 4.69 (95%CI: 1.84-11.98) for a high KIT D816V liquid allele burden in univariate analysis and a significant independent influence in multivariate analysis only for the tissue mutation burden (HR: 18.12, 95%CI: 1.98-165.57; P=0.01) (Online Supplementary Table S3). Using the maximum selected rank statistics method, 9% VAF in the tissue represents an optimal cut-off differentiating between surviving and non-surviving patients. In the group with <9% tissue mutant allele burden (n=79), the median PFS and OS was not reached, whereas in patients with a KIT D816V allele burden of ≥9% (n=24) the median PFS was 4.1 years (Figure 4A) and the median OS 4.6 years (Figure 4B). The observed differences in survival were highly significant both for OS and PFS (Log-rank test; P<0.0001 each).
Table 2.
Parameters for progression-free survival (PFS) in systemic mastocytosis.

Figure 4.

KIT D816V tissue mutation burden for prognostication and therapy response monitoring in systemic mastocytosis (SM). (A and B) Kaplan-Meier plot for progression-free survival (PFS) (A) and overall survival (OS) (B) of SM patients with a KIT D816V variant allele frequency (VAF) <9% or ≥9% in the bone marrow (BM) tissue. (C-E) Follow up of KIT D816V tissue mutation burden (blue) and serum tryptase (black) in three patients with SM who received cytoreductive treatment is shown: a patient initially diagnosed as indolent SM (ISM) with disease progression to SM with an associated hematologic neoplasm (SM-AHN) [aggressive SM (ASM)-chronic myelomonocytic leukemia (CMML)] and response to cytoreductive therapy with cladribine (2-CDA). Later on, the patient progressed to acute myeloid leukemia (AML) (C). A patient diagnosed with mast cell leukemia who responded to alternating treatment with the tyrosine kinase inhibitor midostaurin and 2-CDA (D). A patient diagnosed with ASM who responded to treatment with midostaurin, 2-CDA and achieved complete remission after hematopoietic stem cell transplantation (HSCT) (E).
Moreover, the KIT D816V tissue allele burden could be followed in 26 patients over a median observation time of 42 months (range 2-172 months). In patients with stable disease and no cytoreductive therapy, no substantial increase or decrease in the KIT D816V allele burden was observed. In contrast, marked changes in KIT D816V tissue VAF were found in patients with advanced SM receiving cytoreductive therapy. In particular, six patients who showed a response to cytoreductive treatment (cladribine n=3, midostaurin and cladribine n=3) showed a significant decrease in the KIT D816V tissue burden (92% median reduction comparing post- with pre-therapeutic samples; P<0.01). Representative examples of two patients are shown in Figure 4C and D. In addition, Figure 4E shows a patient with complete remission after allogeneic stem cell transplantation, in whom low minimal residual disease (MRD) was detected by dPCR at an early time point (day 93) after transplantation. In total, our data show that dPCR-based tissue mutation burden measurement is feasible for monitoring treatment responses and to assess MRD levels in patients with SM.
Digital polymerase chain reaction is a sensitive diagnostic test to detect KIT D816V in formalin-fixed paraffin-embedded bone marrow sections
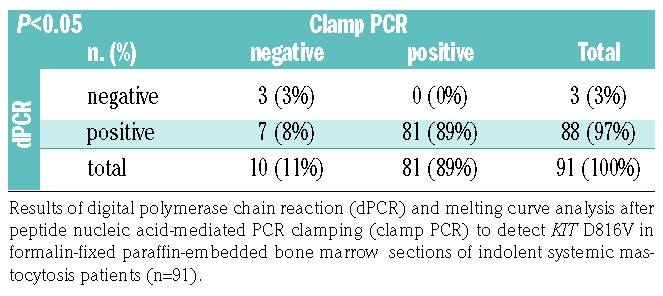
Finally, we assessed the performance of dPCR as a diagnostic test to detect the KIT D816V mutation in FFPE BM section compared to melting curve analysis after PNA-mediated PCR clamping on diagnostic BM section from 116 SM patients (Table 1). In the total cohort, 105 patients (91%) were tested positive for KIT D816V by dPCR whereas the mutation was detected in 98 patients (84%) by melting curve analysis. When analyzing SM subgroups, the mutation was detected with equal sensitivity in 17 patients with advanced SM (68%) by both methods. In contrast, 88 ISM patients (97%) were tested positive by dPCR compared to 81 (89%) by clamp PCR (Table 3). The observed difference in sensitivity was statistically significant (P<0.05) in favor of dPCR. To confirm the specificity of the results, we analyzed FFPE BM sections of control subjects (n=57). No KIT codon 816 mutation was detected by either method, indicating 100% specificity.
Table 3.
Sensitivity of molecular techniques to detect the KIT D816V in the tissue.
When we further characterized patients that were tested ‘false-negative’ by melting curve analysis after PNA-mediated PCR clamping (n=7), a relatively low VAF for KIT D816V was observed (median 0.7%, range 0.027-2.1%). While some of these patients showed a mutant allele burden clearly below the limit of detection established for this method,15 others were found to have a low amount of total KIT copies (<1000) reflecting impaired quality or quantity of these specimens (Online Supplementary Figure S3D). However, importantly, constrained validity of the analysis was not recognizable by melting curve analysis after PNA-mediated PCR clamping. In this regard, quantitative dPCR results allow for an additional quality control of the specimen within the same assay. Altogether, dPCR was superior over clamp PCR to detect the KIT D816V mutation in FFPE BM sections of patients with SM.
Discussion
Although it is generally appreciated that the quantification of the total burden of KIT-mutated neoplastic cells in SM is an important prognostic parameter, quantification of the KIT mutant allele burden has so far been limited to liquid specimens (PB or BM aspirate). This is a critical point, as neoplastic MC and their progenitors are not easily aspirable from BM and are very rarely circulating in the PB; they are thus substantially underrepresented in liquid specimens compared to BM biopsy material. We had previously investigated the analytical validity of dPCR for KIT D816V in detail according to laboratory standards before applying the technology here.16 In the current study, we used dPCR for mutant allele burden measurement in FFPE BM sections of SM patients. To the best of our knowledge, this is the first study that comprehensively assesses the tissue mutation burden as a novel molecular biomarker in SM.
Our results suggest that the KIT D816V allele burden in BM tissue was not interchangeable with that in liquid specimens (PB or BM aspirate). In particular, a number of ISM patients showed substantially higher levels in FFPE tissue. In SM patients with multi-lineage involvement, the KIT D816V mutation is also present in CD34+ hematopoietic stem/precursor cells, eosinophils, basophils, monocytes or neutrophils.26–28 These cells are abundant in the liquid specimens and represent the main source of KIT D816V in PB of ISM patients with high mutant allele burden.19 In contrast, a MC infiltration of >10% is commonly found in the BM tissue of ISM patients.22,23 The mutation burden in FFPE BM sections, therefore, reflects both the ‘infiltration burden’ of KIT D816V+ MC in the tissue and the multi-lineage involvement of the mutation in non-MC. Accordingly, the mutation burden in BM tissue correlated better with the established markers of disease burden in SM (serum tryptase levels and BM MC infiltration) than molecular parameters performed on liquid specimens.
A high MC burden in the BM biopsy (>30% infiltration of cellularity) and high serum total tryptase (>200 ng/mL) also represent B-findings (‘burden of disease’) for definition of SSM.4 Although the clinical course in SSM is often stable for many years, progression to advanced SM can occur. Therefore, SSM represents a rare high-risk subcategory compared to ISM.29 Quantification of KIT D816V allele burden in FFPE might be useful as an additional molecular marker of disease burden in SSM since it includes all KIT D816V positive cells in the tissue. In our study, a particularly high tissue allele burden was found in SSM patients. However, the numbers of samples tested were too small to allow us to draw a final conclusion as to the value of KIT D816V allele burden measurement in FFPE BM sections as an additional criterion of SSM. Multi-center studies with larger patient cohorts are currently being prepared to investigate the definitive value of measurement of tissue mutation burden and its applicability as a B-finding in SSM.
The current definition of advanced SM relies largely on the presence of C-findings (‘cytoreduction-requiring’) as markers of organopathy in ASM.1,4 For these patients, cytoreductive treatment is required.4,30 With the development of tyrosine kinase inhibitor (TKI) treatment in SM as a more specific therapeutic option,31,32 additional subgroups of patients might benefit from treatment and are the subject of ongoing clinical trials.33–35 Thus, prognostic biomarkers are warranted to define patients at risk that do not meet the current ASM criteria. We and others have shown that multi-lineage involvement of KIT D816V indicated by a high mutant allele burden in PB was associated with an aggressive clinical course.17–20 In liquid specimens, we have previously used a 2% VAF cut-off to stratify OS in SM patients.18 Jara-Acevedo et al. used a 6% VAF cut-off in PB to discriminate between MC-restricted versus multilineage SM.19 In this study, we used a higher cut-off of 9% VAF in BM sections to take into account the higher ISM mutational burden in the tissue. Using this cut-off, highly significant differences in both PFS and OS were observed and the tissue mutation burden remained an independent poor risk marker in multivariate analysis when B- and C-findings were considered. This is an important and novel finding as all previous studies assessing the mutation burden in liquid samples (PB and BM aspirate samples) found a significant effect on survival only in univariate but not in multivariate analyses.17,18,20,36,37 This difference might be explained by the close association of multi-lineage KIT D816V involvement (indicated by a high liquid mutation burden) with advanced SM (indicated by the presence of C-findings). Our observations have a clear clinical impact and strongly argue for inclusion of assessment of the KIT D816V mutation burden in prognostic scoring systems for mastocytosis. A potential limitation of our study is that different cytoreductive treatment modalities were applied in this retrospective analysis with a relatively high proportion of interferon-α or cladribine. In fact, more effective treatment regimens may improve OS in the future.31 However, the vast majority of patients with a low tissue mutation burden experienced no events despite the lack of any cytoreductive treatment, suggesting mutation burden analysis is important irrespective of treatment.
Therapy response criteria in advanced SM mainly rely on resolution of C-findings as markers of SM-mediated organopathy to define major response.38 In addition, reduction of MC infiltration and/or of serum tryptase levels is used to define complete remission, incomplete remission, and pure clinical response.38 The TKI midostau-rin showed high efficacy in advanced SM with 45% major response and marked decreases in BM MC burden and serum tryptase.31 Both measurements are valuable surrogate parameters of disease burden in SM, but they do have some limitations.39 While the basal level of total tryptase is well established as being quite a stable parameter in SM, single time-point measurements might be substantially influenced by MC activation or allergic reactions.40–42 On the other hand, quantification of MC burden is a rather robust parameter of disease burden, but relies on experienced hematopathologists.43 In this regard, measurement of KIT D816V allele burden might be useful as an additional objective response parameter. Recently, a ≥25% reduction in expressed KIT D816V allele burden in PB was described as an independent ‘on treatment’ marker for improved OS in midostaurin-treated patients with advanced SM.24 Based on the results of individual patients before and after cytoreductive treatment, FFPE-based allele burden measurement as a more direct marker of the number of all KIT D816V positive cells in the tissue might be an interesting additional follow-up parameter for treatment response. This might be of particular relevance for ISM patients undergoing TKI treatment in the future, since the mutation burden in liquid specimens substantially underestimates the disease burden in ISM. Furthermore, the high sensitivity of the assay makes it applicable for KIT D816V-based MRD measurement in patients that achieve complete remission in the histopathological assessment. However, further multi-centric studies are needed to definitively establish the tissue mutation burden as a parameter for therapy response in SM.
A potential limitation of dPCR and any other molecular test detecting specifically KIT D816V is that neither rare non-D816V mutations of KIT nor somatic mutations in other genes can be monitored. In particular, mutations of SRSF2, ASXL1 and RUNX1 are found in aggressive forms of mastocytosis and TET2 mutations have been described to precede the acquisition of KIT D816V in some patients.37,44 KIT D816V-negative clonal cells in advanced SM might be overlooked by dPCR while sequencing of a gene panel would identify them. Thus, additional next generation sequencing (NGS)-based monitoring of the disease might be warranted, particularly when there is clinical suspicion of disease progression despite a low KIT D816V allele burden. Here we show a higher clinical sensitivity of dPCR to detect KIT D816V in BM sections of SM patients compared to the clamp PCR that is widely used for tissue analysis. This is partly due to the higher analytical sensitivity of dPCR, which allows low mutant alleles to be detected.16 The analytical sensitivity of clamp PCR can be increased by analysis of micro-dissected neoplastic MC.14 Micro-dissection of MC has been used to detect KIT D816V in SM patients but requires time-consuming sample processing and is not widely available for routine diagnostics. In this regard, dPCR seems sufficiently sensitive, as it allows the detection of KIT D816V without micro-dissection in >95% of all ISM patients. However, micro-dissection of MC is still valuable to detect non-D816V mutations in KIT by clamp PCR or sequencing. Thus, both molecular techniques for KIT D816V detection are necessary and helpful, but the dPCR technique may have some advantages and should be considered in the future. One advantage of dPCR is the quantification of KIT wild-type alleles in the sample as quality control. Samples with borderline quality/quantity of DNA isolated from FFPE material are easily identified. In a number of these samples, clamp PCR could have shown false negative results if no additional (quantitative) quality control measurements for amplifiable DNA had been applied. Beside these assays, a number of other molecular tests, such as qPCR or ultra-sensitive NGS, have been described for quantification of KIT D816V in PB or BM aspirate.13,17,45 We have previously shown an excellent correlation of dPCR and qPCR in these liquid specimens with easier inter-laboratory standardization and lack of amplification bias in highly fragmented DNA as potential advantages of dPCR.16 For diagnostic assessment, the high analytical sensitivity of digital PCR is sufficient to use not only trephines but also BM aspirates.16 In addition, qPCR and dPCR detect the mutation in the PB of the majority of patients,16,46,47 arguing for an early molecular assessment during the diagnostic workup of SM.9
In summary, dPCR for KIT D816V is a valuable diagnostic test to detect and quantify the mutation in FFPE BM sections of SM patients. The clinical sensitivity to detect the mutation is superior to clamp PCR without micro-dissection of MC. The KIT D816V mutant allele burden in FFPE BM sections correlates with BM MC infiltration and serum tryptase levels and represents a novel molecular parameter which differs from the mutant allele burden in PB or BM aspirate. Importantly, dPCR-based measurement of KIT D816V mutation burden in the tissue represents a novel biomarker with independent prognostic significance that can also be employed for follow-up analyses in SM. We therefore propose to include the measurement of tissue mutation burden in future studies for prognosis, SSM definition, and monitoring of disease progression and treatment response in SM patients.
Supplementary Material
Acknowledgments
The authors thank Jana Strasakova (Department of Laboratory Medicine, Medical University of Vienna) for technical support.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/105/2/366
Funding
This study was supported by the Austria Science Fund (FWF) project P26079-B13, the SFB projects F4701-B20 and F4704-B20, the Medical-Scientific Fund of the Mayor of Vienna and a research grant of the Austrian Society of Laboratory Medicine and Clinical Chemistry (ÖGLMKC).
References
- 1.Horny H-P, Metcalfe DD, Akin C, et al. Mastocytosis. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue. Revised 4th ed. Lyon: International Agency for Research on Cancer (IARC), 2017:62–69. [Google Scholar]
- 2.Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A. 1995;92(23):10560–10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Longley BJ, Tyrrell L, Lu SZ, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12(3):312–314. [DOI] [PubMed] [Google Scholar]
- 4.Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010;3(4):497–516. [DOI] [PubMed] [Google Scholar]
- 6.Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25(7):603–625. [DOI] [PubMed] [Google Scholar]
- 7.Valent P, Akin C, Hartmann K, et al. Advances in the Classification and Treatment of Mastocytosis: Current Status and Outlook toward the Future. Cancer Res. 2017;77(6):1261–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37(6):435–453. [DOI] [PubMed] [Google Scholar]
- 9.Arock M, Sotlar K, Akin C, et al. KIT mutation analysis in mast cell neoplasms: recommendations of the European Competence Network on Mastocytosis. Leukemia. 2015; 29(6):1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valent P, Escribano L, Broesby-Olsen S, et al. Proposed diagnostic algorithm for patients with suspected mastocytosis: a proposal of the European Competence Network on Mastocytosis. Allergy. 2014;69(10):1267–1274. [DOI] [PubMed] [Google Scholar]
- 11.Kristensen T, Vestergaard H, Bindslev-Jensen C, Moller MB, Broesby-Olsen S, Mastocytosis Centre OUH. Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am J Hematol. 2014;89(5):493–498. [DOI] [PubMed] [Google Scholar]
- 12.Kristensen T, Vestergaard H, Bindslev-Jensen C, et al. Prospective evaluation of the diagnostic value of sensitive KIT D816V mutation analysis of blood in adults with suspected systemic mastocytosis. Allergy. 2017;72(11):1737–1743. [DOI] [PubMed] [Google Scholar]
- 13.Kristensen T, Vestergaard H, Moller MB. Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time qPCR assay. J Mol Diagn. 2011;13(2):180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sotlar K. c-kit mutational analysis in paraffin material. Methods Mol Biol. 2013;999:59–78. [DOI] [PubMed] [Google Scholar]
- 15.Sotlar K, Escribano L, Landt O, et al. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol. 2003;162(3):737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greiner G, Gurbisz M, Ratzinger F, et al. Digital PCR: A Sensitive and Precise Method for KIT D816V Quantification in Mastocytosis. Clin Chem. 2018;64(3):547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erben P, Schwaab J, Metzgeroth G, et al. The KIT D816V expressed allele burden for diagnosis and disease monitoring of systemic mastocytosis. Ann Hematol. 2014;93(1):81–88. [DOI] [PubMed] [Google Scholar]
- 18.Hoermann G, Gleixner KV, Dinu GE, et al. The KIT D816V allele burden predicts survival in patients with mastocytosis and correlates with the WHO type of the disease. Allergy. 2014;69(6):810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jara-Acevedo M, Teodosio C, Sanchez-Munoz L, et al. Detection of the KIT D816V mutation in peripheral blood of systemic mastocytosis: diagnostic implications. Mod Pathol. 2015;28(8):1138–1149. [DOI] [PubMed] [Google Scholar]
- 20.Broesby-Olsen S, Kristensen T, Vestergaard H, et al. KIT D816V mutation burden does not correlate to clinical manifestations of indolent systemic mastocytosis. J Allergy Clin Immunol. 2013;132(3):723–728. [DOI] [PubMed] [Google Scholar]
- 21.Mayado A, Teodosio C, Dasilva-Freire N, et al. Characterization of CD34(+) hematopoietic cells in systemic mastocytosis: Potential role in disease dissemination. Allergy. 2018; 73(6):1294–1304. [DOI] [PubMed] [Google Scholar]
- 22.Horny HP, Parwaresch MR, Lennert K. Bone marrow findings in systemic mastocytosis. Hum Pathol. 1985;16(8):808–814. [DOI] [PubMed] [Google Scholar]
- 23.Krokowski M, Sotlar K, Krauth MT, Fodinger M, Valent P, Horny HP. Delineation of patterns of bone marrow mast cell infiltration in systemic mastocytosis: value of CD25, correlation with subvariants of the disease, and separation from mast cell hyperplasia. Am J Clin Pathol. 2005;124(4): 560–568. [DOI] [PubMed] [Google Scholar]
- 24.Jawhar M, Schwaab J, Naumann N, et al. Response and progression on midostaurin in advanced systemic mastocytosis: KIT D816V and other molecular markers. Blood. 2017;130(2):137–145. [DOI] [PubMed] [Google Scholar]
- 25.R: Development Core Team. A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: ISBN 3-900051-07-0 2005. [Google Scholar]
- 26.Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108(7): 2366–2372. [DOI] [PubMed] [Google Scholar]
- 27.Kocabas CN, Yavuz AS, Lipsky PE, Metcalfe DD, Akin C. Analysis of the lineage relationship between mast cells and basophils using the c-kit D816V mutation as a biologic signature. J Allergy Clin Immunol. 2005;115(6):1155–1161. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Montero AC, Jara-Acevedo M, Alvarez-Twose I, et al. KIT D816V-mutated bone marrow mesenchymal stem cells in indolent systemic mastocytosis are associated with disease progression. Blood. 2016;127(6):761–768. [DOI] [PubMed] [Google Scholar]
- 29.Pardanani A, Lim KH, Lasho TL, et al. WHO subvariants of indolent mastocytosis: clinical details and prognostic evaluation in 159 consecutive adults. Blood. 2010;115(1):150–151. [DOI] [PubMed] [Google Scholar]
- 30.Ustun C, Arock M, Kluin-Nelemans HC, et al. Advanced systemic mastocytosis: from molecular and genetic progress to clinical practice. Haematologica. 2016;101(10):1133–1143. [DOI] [PubMed] [Google Scholar]
- 31.Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N Engl J Med. 2016;374(26):2530–2541. [DOI] [PubMed] [Google Scholar]
- 32.Bibi S, Arock M. Tyrosine Kinase Inhibition in Mastocytosis: KIT and Beyond KIT. Immunol Allergy Clin North Am. 2018;38(3):527–543. [DOI] [PubMed] [Google Scholar]
- 33.Lortholary O, Chandesris MO, Bulai Livideanu C, et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo-controlled, phase 3 study. Lancet. 2017; 389(10069):612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Anrooij B, Oude Elberink JNG, Span LF, et al. Midostaurin in indolent systemic mas-tocytosis patients: an open-label phase 2 trial. J Allergy Clin Immunol. 2018; 142(3):1006–1008. [DOI] [PubMed] [Google Scholar]
- 35.DeAngelo DJ, Quiery AT, Radia D, et al. Clinical Activity in a Phase 1 Study of Blu-285, a Potent, Highly-Selective Inhibitor of KIT D816V in Advanced Systemic Mastocytosis (AdvSM). Blood. 2017;130(Suppl 1):2–2.28684445 [Google Scholar]
- 36.Jawhar M, Schwaab J, Hausmann D, et al. Splenomegaly, elevated alkaline phosphatase and mutations in the SRSF2/ASXL1/RUNX1 gene panel are strong adverse prognostic markers in patients with systemic mastocytosis. Leukemia. 2016;30(12):2342–2350. [DOI] [PubMed] [Google Scholar]
- 37.Jawhar M, Schwaab J, Schnittger S, et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia. 2016; 30(1):136–143. [DOI] [PubMed] [Google Scholar]
- 38.Valent P, Akin C, Sperr WR, et al. Aggressive systemic mastocytosis and related mast cell disorders: current treatment options and proposed response criteria. Leuk Res. 2003; 27(7):635–641. [DOI] [PubMed] [Google Scholar]
- 39.Sperr WR, Jordan JH, Fiegl M, et al. Serum tryptase levels in patients with mastocytosis: correlation with mast cell burden and implication for defining the category of disease. Int Arch Allergy Immunol. 2002;128(2):136–141. [DOI] [PubMed] [Google Scholar]
- 40.Kabashima K, Nakashima C, Nonomura Y, et al. Biomarkers for evaluation of mast cell and basophil activation. Immunol Rev. 2018;282(1):114–120. [DOI] [PubMed] [Google Scholar]
- 41.Theoharides TC, Valent P, Akin C. Mast Cells, Mastocytosis, and Related Disorders. N Engl J Med. 2015;373(2):163–172. [DOI] [PubMed] [Google Scholar]
- 42.Valent P, Sperr WR, Sotlar K, et al. The serum tryptase test: an emerging robust bio-marker in clinical hematology. Expert Rev Hematol. 2014;7(5):683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jawhar M, Schwaab J, Horny HP, et al. Impact of centralized evaluation of bone marrow histology in systemic mastocytosis. Eur J Clin Invest. 2016;46(5):392–397. [DOI] [PubMed] [Google Scholar]
- 44.Jawhar M, Schwaab J, Schnittger S, et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia. 2015;29(5):1115–1122. [DOI] [PubMed] [Google Scholar]
- 45.Kristensen T, Broesby-Olsen S, Vestergaard H, Bindslev-Jensen C, Moller MB. Targeted ultradeep next-generation sequencing as a method for KIT D816V mutation analysis in mastocytosis. Eur J Haematol. 2016;96(4): 381–388. [DOI] [PubMed] [Google Scholar]
- 46.Kristensen T, Vestergaard H, Bindslev-Jensen C, et al. Prospective evaluation of the diagnostic value of sensitive KIT D816V mutation analysis of blood in adults with suspected systemic mastocytosis. Allergy. 2017;72(11):1737–1743. [DOI] [PubMed] [Google Scholar]
- 47.Broesby-Olsen S, Oropeza AR, Bindslev-Jensen C, et al. Recognizing mastocytosis in patients with anaphylaxis: value of KIT D816V mutation analysis of peripheral blood. J Allergy Clin Immunol. 2015;135(1): 262–264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.