

Abstract
Selective modification of nucleobases with photolabile caging groups enables the study and control of processes and interactions of nucleic acids. Numerous positions on nucleobases have been targeted, but all involve formal substitution of a hydrogen atom with a photocaging group. Nature, however, also uses ring‐nitrogen methylation, such as m7G and m1A, to change the electronic structure and properties of RNA and control biomolecular interactions essential for translation and turnover. We report that aryl ketones such as benzophenone and α‐hydroxyalkyl ketone are photolabile caging groups if installed at the N7 position of guanosine or the N1 position of adenosine. Common photocaging groups derived from the ortho‐nitrobenzyl moiety were not suitable. Both chemical and enzymatic methods for site‐specific modification of N7G in nucleosides, dinucleotides, and RNA were developed, thereby opening the door to studying the molecular interactions of m7G and m1A with spatiotemporal control.
Keywords: benzophenone, N7G, photocaging, RNA, RNA modification
Light the way: A strategy was developed to chemically or enzymatically photocage the N7 position of nucleosides, dinucleotides, and RNA, which can be reversed upon irradiation at λ max=365 nm. The well‐known ortho‐nitrobenzyl photocaging groups were unsuitable, however aryl ketone derivatives such as benzophenone were identified as photocaging groups for purine imine positions.

Light is a versatile regulatory element because it is fully compatible with most cellular components, noninvasive, and controllable in timing and localization to tissues, cells, and even subcellular compartments.1 To gain optical control over nucleic acids and their respective biomolecular interactions, irreversible photo(de)activation (“uncaging”) has been applied to nucleosides, nucleotides, and oligonucleotides.1, 2 Various photolabile protecting groups have been placed either in the oligomer backbone or on the nucleobases, and their removal by light of a defined wavelength (≥365 nm) has been shown to be compatible with living cells and organisms.3 The ortho‐nitrobenzyl (ONB) group is the most widely used photocaging (PC) group, but numerous alternative scaffolds, derived from coumarins, quinolines, dibenzofuran, or the piperonyl group have been explored.1, 2
Typically, these PC groups are installed at exocyclic heteroatoms, such as O4 in thymidine or N4 of cytidine.4 In addition, the N3 position of thymidine was successfully used in optochemical biology.3b In purines, the exocyclic O6 of guanosine and N6 of adenosine are preferred sites for the installation of photocaging groups.5 In the case of guanosine, other positions like N1, C8, or the exocyclic N2 position have been explored albeit to a much lower extent.6 For chemoenzymatic approaches, where photocaged nucleoside triphosphates (NTPs) are synthesized and enzymatically introduced into DNA or RNA by polymerases, the 5 position of pyrimidines is particularly favorable.7 Recently, post‐enzymatic photocaging of DNA was also achieved by using methyltransferase (MTase)‐catalyzed transfer of PC groups to the N6 position of adenosines.8 In all of these cases, the PC group replaces a hydrogen atom and does not change the charge distribution of the nucleoside.
However, unlike the chemical modifications with PC groups developed to date, nature not only substitutes hydrogen atoms with modifications, but also uses methylation to change the electronic structure of the molecule. In naturally occurring m7G, m1A, and m3C, the modifications confer a positive charge on the nucleobase. Out of these, m7G deserves particular attention because it is 1) one of the most conserved modified nucleosides, 2) installed by numerous MTases in different organisms, and 3) found in rRNA and tRNA, as well as part of the 5′ cap in mRNA.9 In the latter case, m7G is crucial for coordinating mRNA translation and turnover by several interactions, such as binding to the translation initiation factor eIF4E or decapping scavenger enzymes DcpS.10 Due to its importance in biology, we chose the N7 position of guanosine for modification with functional groups with the goal of removing them upon irradiation with light and recover the free guanosine. In the 5′ cap of mRNAs, the G becomes remethylated in cells.11 To generate photocaged guanosines in different contexts, that is, from single nucleosides to long mRNAs, we devised both a chemical and an enzymatic preparative route (Scheme 1). PC groups for nucleosides that do not substitute a hydrogen atom but instead introduce a positive charge have not been reported to date to the best of our knowledge.
Scheme 1.

Concept for chemical and enzymatic photocaging of the N7 of guanosine using a classical ortho‐nitrobenzyl (ONB) group or aryl ketones such as benzophenone (BP) to generate the respective nucleoside, 5′ cap, or RNA. N7‐BP‐modified guanosine is uncaged by subsequent irradiation with light (λmax=365 nm).
First, we explored the chemical photocaging of guanosine 1 using the well‐known ortho‐nitrobenzyl (ONB) group as well as para‐nitrobenzyl (PNB) as negative control. The N7 of guanosine is the most potent nucleophile in DNA and RNA.12 However, although N7G methylation was used in Maxam Gilbert sequencing,13 the chemical modification of N7 has not been exploited to manipulate and control interactions by orthogonal triggers such as light. Building on the good nucleophilicity of the N7 position of guanosine, we reacted guanosine with the PC bromides 2 a,b and obtained the expected N7‐ortho‐nitrobenzyl‐ (N7‐ONB) and N7‐para‐nitrobenzyl‐ (N7‐PNB) guanosines 3 a,b (Figures 1 A, Figure S3–S5 in the Supporting Information) in good yields (3 a: 53 % and 3 b: 56 %).14 Subsequent irradiation of 3 a, however, did not yield the desired photocleavage to release guanosine 1 but instead gave a different main product according to HPLC (Figure 1 B, Figure S6). Mass analysis revealed a mass of m/z=347.1475, indicating a mass loss of m/z=71.98 from 3 a, which would correspond to loss of CO and CO2 (Figure 1 C). After isolating the product, we identified the structure of 4, which contains a guanidine moiety, based on NMR and UHPLC‐MS/MS analyses (Figures S7, S54–59). Importantly, the control 3 b as well as unmodified 1 were stable when irradiated under the same conditions (Figures S8, S9). These data indicate that the N7 position of guanosine can be readily derivatized with the well‐known ONB group, however the photocleavage leads to an unusual product instead of guanosine, thus limiting its application in photocaging approaches.
Figure 1.

Chemical modification of the N7 position of guanosine derivatives with the common ONB group and subsequent irradiation. A) Concept. B) HPLC analysis before and after irradiation of 3 a at 365 nm. C) Mass analysis of 4 (calculated mass of [C15H19N6O4]+=347.1462 [M]+, found: 347.1475).
We therefore turned away from the classical PC groups and contemplated alternative light‐sensitive groups as potential position‐specific PC groups. Photoactivatable aryl ketone derivatives such as benzophenone (BP) are widely used biochemical probes.15 BP can be manipulated in ambient light and activated at λ≈360 nm. It is chemically stable and reacts preferentially with unreactive C−H bonds, which is widely used to study protein–protein as well as protein–RNA interactions.16 The substituents on BP can affect the photochemistry significantly and electron‐withdrawing groups increase the efficiency of H‐abstraction.15 We therefore reasoned that the positive charge at the N7 position might lead the radical to a different reaction pathway and therefore considered aryl ketone derivatives, such as BP or a 2‐hydroxy‐2‐methyl‐1‐phenylpropan‐1‐one group (HAK), as a potential PC groups for the N7 position of guanosine.
We chemically synthesized N7‐BP‐guanosine 3 c (Figures S3, S4) and tested its uncaging properties (Figure 2 A). After irradiation of 3 c with light (λ max=365 nm) in buffer containing EDTA and glycerol, we found free guanosine 1 and 4‐methylbenzophenone 5 as cleavage products, thus indicating that BP can be used as photocaging group for the N7 position (Figure S10 A–C). To learn more about the cleavage mechanism, we tested the components of the reaction buffer in more detail. We found that the cleavage reaction did not occur in plain water but when EDTA, glycerol, or especially cell lysate were present, thus suggesting that hydrogen abstraction occurs from these buffer components or other cell lysate components (Figure 2 B, Figure S11).
Figure 2.
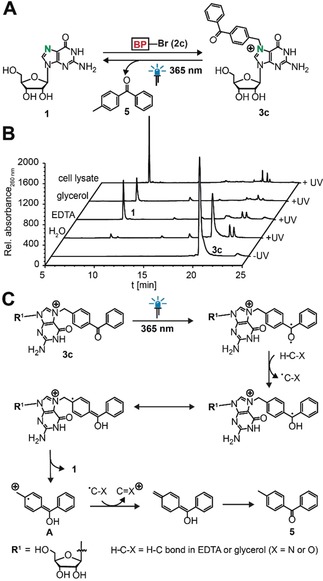
Chemical preparation of N7‐BP‐guanosine and subsequent irradiation. A) Concept. B) HPLC analysis before and after irradiation of 3 c at 365 nm in aqueous solution with different additives. C) Postulated mechanism for the photocleavage.
A possible cleavage mechanism is shown in Figure 2 C. Upon irradiation at λ max=365 nm, benzophenone generates a triplet ketone, which abstracts a hydrogen atom from a C−H bond of EDTA or glycerol.16 The ketyl radical thus formed does not engage in a C−C cross‐link, but the positive charge at the N7 position favors fragmentation to recover guanosine 1, along with the radical cation A. Single‐electron reduction of A by the EDTA‐ or glycerol‐derived radical and tautomerization eventually leads to 5.
To test whether this strategy can be applied to the other purine nucleosides, we synthesized N1‐benzophenone‐modified adenosine (7; Figure S4).17 The corresponding methylated nucleoside m1A has recently been identified in mRNA and its functional role is still under investigation.18 This compound also has a positive charge, thus suggesting that photo‐uncaging of aryl ketone derivatives might be possible in an analogous way. Indeed, we observed that irradiation of 7 with light of 365 nm led to complete formation of adenosine 6 and release of 5 similar to N7‐photocaged guanosine (Figure S12,S13).
To examine whether our strategy can be extended to more complex molecules, we chose the dinucleotide GpppA 8, which is the hallmark structure of the 5′ cap found in eukaryotic mRNAs. Since site‐specific chemical modification is not possible in this case, we devised an enzymatic approach, exploiting the site‐specificity of the cap N7‐methyltransferase Ecm1 together with its cosubstrate promiscuity.19 To this end, we synthesized analogues of the natural cosubstrate S‐adenosyl‐l‐methionine (AdoMet) that are suitable for transfer of the classical ONB‐based and novel BP‐based photocaging groups, that is, AdoONB (9 a), AdoPNB (9 b), AdoANB (9 c), and AdoBP (9 d; Figure S14).8, 19b These AdoMet analogues are well accepted by the highly promiscous Ecm1, resulting in efficient formation of the desired products N7‐PC‐GpppAs 10 a–d (Figure 3 A,B, Figure S15), according to HPLC and UHPLC‐MS analysis (Figure 3 C, Figures S16–S19). In line with the results obtained for the nucleoside, irradiation of N7‐ONB‐GpppA (10 a) did not yield the uncaged 8 but instead product 11 a with a mass loss of m/z=71.98 (Figures 3 C,D, Figure S20). Similarly, irradiation of N7‐ANB‐GpppA (10 c) revealed new product 11 c with a mass loss of m/z=71.98 (Figures S17, S19 E). More detailed analyses of 10 a and 11 a by enzymatic digestion into single nucleosides using snake venom phosphodiesterase revealed that the mass loss occurred exclusively at the N7‐ONB‐guanosine moiety of the dinucleotide (Figures S21–S23). The controls, PNB‐caged GpppA (10 b) and uncaged GpppA (8), remained stable when irradiated under these conditions, which is in line with our results from the guanosine (Figures S16, S24).
Figure 3.
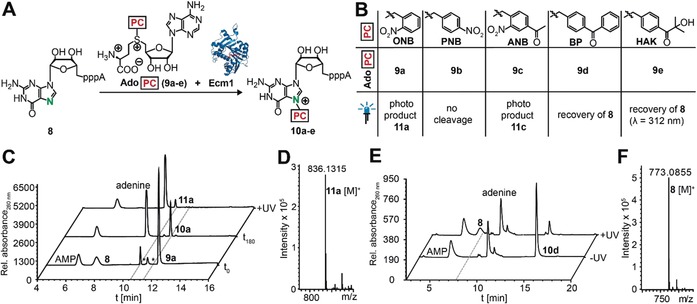
Enzymatic modification of the N7 position of the 5′ cap structure GpppA. A) Concept. B) Panel of PC groups tested and summary of irradiation results. B) HPLC analysis of enzymatic reaction of 8 to 10 a and subsequent irradiation at 365 nm. D) Mass analysis of 11 a (calculated mass of [C25H33N11O16P3]+=836.1314, found: 836.1315). E) HPLC analysis of enzymatic reaction of 8 to 10 d before and after irradiation at 365 nm in buffer. F) Mass analysis after irradiation, 8: Calculated mass of [C20H28N10O17P3]+=773.0841 [M+H]+, found: 773.0855. (*=impurities of 9 a).
The benzophenone group, however, was again suitable for photouncaging at the N7G of the 5′ cap. Specifically, 10 d was almost completely reacted (79 % decrease) when irradiated in buffer, and GpppA 8 was formed as the main product (Figure 3 E, Figure S18 A,B). As in the case of the nucleoside, addition of buffer was required to yield 8 and release 4‐methylbenzophenone 5 (Figure S25,S26). Importantly, the uncaged product 8 could be re‐modified enzymatically using the BP group, thus confirming that the free and functional GpppA (8) had been formed after photocleavage of the BP group (Figure S18 C).
To test whether other aryl ketone derivatives could also be suitable for photocaging, we used the α‐hydroxyalkyl ketone (HAK) substituent, which is known to normally react according to a Norrish type I mechanism to give the acylradical and ketylradical.20 We enzymatically produced N7‐HAK‐GpppA (10 e) from GpppA (8) using Ecm1 and AdoHAK (9 e; Figures S14, S15, S27). Irradiation of 10 e with light at 312 nm in the reaction mixture led to recovery of almost 60 % of 5 (Figure S28). UHPLC‐MS analysis verified the photocleavage to GpppA (8) and small amounts of side products, namely the N7 benzoic acid modified cap analogue 11 e,a and the corresponding N7‐benzaldehyde‐modified one 11 e,b (Figures S29, S30). These data show that the HAK group is also a photocaging group for the N7 position of guanosine nucleotides, thus suggesting that our approach can be extended to other aryl ketones.
Furthermore, we enzymatically modified plasmid DNA at the N6 position of deoxyadenosine using MTase TaqI and AdoBP 9 d, which worked efficiently (Figure S31). As expected, no uncaging and thus no enzymatic plasmid degradation were observed after irradiation with light at 365 nm in buffer (Figure S31 B). This showed that the positive charge at the N7 position is required for the photocleavage of aryl ketone derivatives such as BP.
To study whether our strategy is applicable to photocaging of oligonucleotides, we used a panel of short 5–21 nucleotide RNAs with internal guanosine sites (Figure S33 A). These RNA oligonucleotides were incubated with BP bromide 2 c, resulting in N7G but not N1A modification according to UHPLC‐MS analysis after digestion and dephosphorylation to single nucleosides. The modified RNAs were then irradiated with light of 365 nm in buffer containing EDTA, and single nucleosides were analyzed by UHPLC‐MS. The data showed that BP was removed from N7‐BP‐guanosine after irradiation, thus suggesting that photocaging and uncaging is also possible in the context of RNA oligonucleotides (Figures S10 D,E, S32–S39). Neither reaction with 2 c nor irradiation at λ max=365 nm led to RNA degradation, as shown in denaturing PAGE analysis (Figure S38). Furthermore, the N7‐modified caps were successfully used for in vitro transcription to produce long reporter mRNAs (>1000 nt) and these also remained intact after irradiation under the same conditions (Figure 4 B).
Figure 4.

Photocaging of N7 of guanosines blocks cap binding proteins and can be used to generate long RNAs. A) Binding assay of N7‐BP‐GpppA was performed with DcpS (H277N) and eIF4E before and after photouncaging and remethylation. B) N7‐BP‐modified cap was used to produce long mRNAs. These were stable under irradiation, if no H‐donor was added. C, D) Binding of 10 d to eIF4E and DcpS (H277N) is restored by irradiation and remethylation.
Finally, we tested whether photocaging the N7 position of guanosine can be used to block a biological function (Figure 4 A). The 5′ cap is involved in several interactions, most notably with eIF4E, for translation, and decapping enzymes, such as DcpS, for RNA turnover.10 These interactions require N7 methylation, and unmodified caps have been show to become remethylated in the cytoplasm.11 We measured the K d values of recombinantly produced eIF4E and the inactive variant DcpS (H277N) for the native and modified cap and found that these proteins do not bind to N7‐BP‐modified‐GpppA (10d), whereas m7GpppA is bound, showing a K d value in the sub‐micromolar range, which is in line with reported values (Figures S40, S41, Table S1).10d, 21 After irradiation and enzymatic remethylation to mimic cellular remethylation, binding to both proteins was fully restored (Figure 4 C,D). These data show that benzophenone can be used to block and release biologically relevant functions.
In summary, we have developed a strategy for photocaging purine nucleosides via the formation of purine iminium ions. We present aryl ketone derivatives as new class of photolabile groups for these purine imine positions, as exemplified for N7G and N1A, and show that the common ONB group is not suitable for N7G. We developed both a chemical and an enzymatic strategy to photocage and release N7G, which to the best of our knowledge is the first reported photocaged nucleoside that strictly affects the Hoogsteen recognition site of G. Hoogsteen interactions are important in RNA biology, for example, in riboswitches, ribozymes, or the formation of G quadruplexes. Furthermore, due to the biological importance of m7G in the 5′ cap and the enzymatic remethylation process in nature, our approach significantly expands the chemical biology toolbox and opens the door to control the functions of the 5′ cap with spatiotemporal control in a biological context. The photocaging of N7G and N1A could be further improved by testing additional aryl ketone derivatives that are excited at longer wavelengths and exploiting the two‐photon excitation properties of BP in the future.22
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Dr. W. Dörner, A.‐M. Dörner, S. Hüwel and S. Wulff for experimental assistance and Dr. M. Teders for helpful discussion (all WWU). We thank the Glorius group, Dr. F. Höhn, and the technical workshop for providing and building custom‐made LED boxes (all WWU Münster). This work was supported by the Deutsche Forschungsgemeinschaft [RE2796/6‐1], the European Research Council (77280)and the Fonds der Chemischen Industrie (doctoral fellowship to L.A. and Dozentenpreis to A.R.). We thank Prof. Dr. C. Lima (Sloan Kettering Institute) for a plasmid coding for DcpS (H277N).
L. Anhäuser, N. Klöcker, F. Muttach, F. Mäsing, P. Špaček, A. Studer, A. Rentmeister, Angew. Chem. Int. Ed. 2020, 59, 3161.
References
- 1. Liu Q., Deiters A., Acc. Chem. Res. 2014, 47, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brieke C., Rohrbach F., Gottschalk A., Mayer G., Heckel A., Angew. Chem. Int. Ed. 2012, 51, 8446–8476; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8572–8604. [Google Scholar]
- 3.
- 3a. Ando H., Furuta T., Tsien R. Y., Okamoto H., Nat. Genet. 2001, 28, 317–325; [DOI] [PubMed] [Google Scholar]
- 3b. Hemphill J., Govan J., Uprety R., Tsang M., Deiters A., J. Am. Chem. Soc. 2014, 136, 7152–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Heckel A., Mayer G., J. Am. Chem. Soc. 2005, 127, 822–823; [DOI] [PubMed] [Google Scholar]
- 4b. Schäfer F., Joshi K. B., Fichte M. A., Mack T., Wachtveitl J., Heckel A., Org. Lett. 2011, 13, 1450–1453. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Menge C., Heckel A., Org. Lett. 2011, 13, 4620–4623; [DOI] [PubMed] [Google Scholar]
- 5b. Mikat V., Heckel A., RNA 2007, 13, 2341–2347; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Buff M. C., Schäfer F., Wulffen B., Müller J., Pötzsch B., Heckel A., Mayer G., Nucleic Acids Res. 2010, 38, 2111–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Lusic H., Lively M. O., Deiters A., Mol. Biosyst. 2008, 4, 508–511; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Ogasawara S., ChemBioChem 2014, 15, 2652–2655; [DOI] [PubMed] [Google Scholar]
- 6c. Ogasawara S., ACS Synth. Biol. 2018, 7, 2507–2513; [DOI] [PubMed] [Google Scholar]
- 6d. Ogasawara S., ACS Chem. Biol. 2017, 12, 351–356. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Vaníková Z., Hocek M., Angew. Chem. Int. Ed. 2014, 53, 6734–6737; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6852–6855; [Google Scholar]
- 7b. Boháčová S., Ludvíková L., Poštová Slavětínská L., Vaniková Z., Klán P., Hocek M., Org. Biomol. Chem. 2018, 16, 1527–1535; [DOI] [PubMed] [Google Scholar]
- 7c. Boháčová S., Vaníková Z., Poštová Slavětínská L., Hocek M., Org. Biomol. Chem. 2018, 16, 5427–5432; [DOI] [PubMed] [Google Scholar]
- 7d. Vaníková Z., Janoušková M., Kambová M., Krásná L., Hocek M., Chem. Sci. 2019, 10, 3937–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Anhäuser L., Muttach F., Rentmeister A., Chem. Commun. 2018, 54, 449–451. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Tomikawa C., Int. J. Mol. Sci. 2018, 19, 4080; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. White J., Li Z., Sardana R., Bujnicki J. M., Marcotte E. M. and Johnson A. W., Mol. Cell. Biol. 2008, 28, 3151–3161; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Ramanathan A., Robb G. B. and Chan S. H., Nucleic Acids Res. 2016, 44, 7511–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Liu H., Rodgers N. D., Jiao X., Kiledjian M., EMBO J. 2002, 21, 4699–4708; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Liu S. W., Jiao X., Liu H., Gu M., Lima C. D., Kiledjian M., RNA 2004, 10, 1412–1422; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Gu M., Fabrega C., Liu S. W., Liu H., Kiledjian M., Lima C. D., Mol. Cell 2004, 14, 67–80; [DOI] [PubMed] [Google Scholar]
- 10d. Niedzwiecka A., Marcotrigiano J., Stepinski J., Jankowska-Anyszka M., Wyslouch-Cieszynska A., Dadlez M., Gingras A. C., Mak P., Darzynkiewicz E., Sonenberg N., Burley S. K., Stolarski R., J. Mol. Biol. 2002, 319, 615–635; [DOI] [PubMed] [Google Scholar]
- 10e. Marcotrigiano J., Gingras A. C., Sonenberg N., Burley S. K., Cell 1997, 89, 951–961. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Otsuka Y., Kedersha N. L., Schoenberg D. R., Mol. Cell. Biol. 2009, 29, 2155–2167; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Mukherjee C., Bakthavachalu B., Schoenberg D. R., PLoS Biol. 2014, 12, e1001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gillingham D., Geigle S., Anatole von Lilienfeld O., Chem. Soc. Rev. 2016, 45, 2637–2655. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Maxam A. M., Gilbert W., Proc. Natl. Acad. Sci. USA 1977, 74, 560–564; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Sauter B., Gillingham D., ChemBioChem 2018, 19, 1638–1642. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. La D. K., Upton P. B., Swenberg J. A. in 14.05—Carcinogenic Alkylating Agents (Ed.: C. A. McQueen), Elsevier, Oxford, 2010, pp. 63–83; [Google Scholar]
- 14b. Pautus S., Sehr P., Lewis J., Fortune A., Wolkerstorfer A., Szolar O., Guilligay D., Lunardi T., Decout J. L., Cusack S., J. Med. Chem. 2013, 56, 8915–8930. [DOI] [PubMed] [Google Scholar]
- 15. Dorman G., Prestwich G. D., Biochemistry 1994, 33, 5661–5673. [DOI] [PubMed] [Google Scholar]
- 16. Preston G. W., Wilson A. J., Chem. Soc. Rev. 2013, 42, 3289–3301. [DOI] [PubMed] [Google Scholar]
- 17. Vik A., Hedner E., Charnock C., Tangen L. W., Samuelsen Ø., Larsson R., Bohlin L., Gundersen L. L., Bioorg. Med. Chem. 2007, 15, 4016–4037. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Dominissini D., Nachtergaele S., Moshitch-Moshkovitz S., Peer E., Kol N., Ben-Haim M. S., Dai Q., Di Segni A., Salmon-Divon M., Clark W. C., Zheng G., Pan T., Solomon O., Eyal E., Hershkovitz V., Han D., Dore L. C., Amariglio N., Rechavi G., He C., Nature 2016, 530, 441–446; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Li X., Xiong X., Wang K., Wang L., Shu X., Ma S., Yi C., Nat. Chem. Biol. 2016, 12, 311–316; [DOI] [PubMed] [Google Scholar]
- 18c. Safra M., Sas-Chen A., Nir R., Winkler R., Nachshon A., Bar-Yaacov D., Erlacher M., Rossmanith W., Stern-Ginossar N., Schwartz S., Nature 2017, 551, 251–255; [DOI] [PubMed] [Google Scholar]
- 18d. Schwartz S., RNA 2018, 24, 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Holstein J. M., Anhäuser L., Rentmeister A., Angew. Chem. Int. Ed. 2016, 55, 10899–10903; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11059–11063; [Google Scholar]
- 19b. Muttach F., Mäsing F., Studer A., Rentmeister A., Chem. Eur. J. 2017, 23, 5988–5993; [DOI] [PubMed] [Google Scholar]
- 19c. Muttach F., Muthmann N., Reichert D., Anhäuser L., Rentmeister A., Chem. Sci. 2017, 8, 7947–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mäsing F., Mardyukov A., Doerenkamp C., Eckert H., Malkus U., Nüsse H., Klingauf J., Studer A., Angew. Chem. Int. Ed. 2015, 54, 12612–12617; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12803–12808. [Google Scholar]
- 21. Niedzwiecka A., Stepinski J., Darzynkiewicz E., Sonenberg N., Stolarski R., Biochemistry 2002, 41, 12140–12148. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. McGimpsey W. C., Scaiano J. C., Chem. Phys. Lett. 1987, 138, 13–17; [Google Scholar]
- 22b. Cai X., Sakamoto M., Yamaji M., Fujitsuka M., Majima T., J. Phys. Chem. A 2005, 109, 5989–5994. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary