

Abstract
Development of photocatalysts (PCs) with diverse properties has been essential in advancement of organocatalyzed atom transfer radical polymerization (O-ATRP). In this work, dimethyl-dihydroacridines are presented as a new family of organic PCs, for the first time enabling controlled polymerization of challenging acrylate monomers via O-ATRP. Structure-property relationships for seven PCs are established, demonstrating tunable photochemical and electrochemical properties and accessing a strongly oxidizing 2PC●+ intermediate for efficient deactivation. In O-ATRP, a combination of PC choice, implementation of continuous-flow reactors, and promotion of deactivation through addition of LiBr are critical to producing well-defined acrylate polymers with dispersities as low as 1.12. The utility of this approach is established through demonstration of the oxygen tolerance of the system and application to diverse acrylate monomers, including the synthesis of well-defined di- and triblock copolymers.
Keywords: photoredox catalysis, polymerization, monomers, microreactors
Graphical Abstract
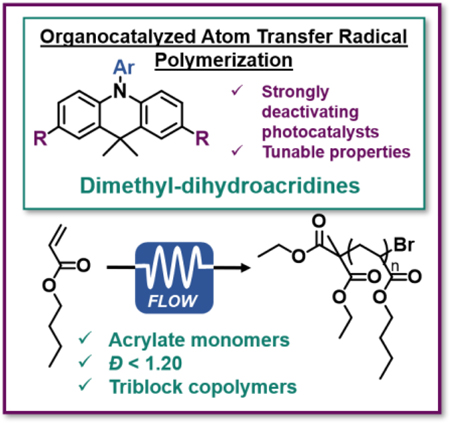
Organic photoredox catalysis is applied to the controlled synthesis of poly(acrylates). This advancement is enabled by development of new organic photoredox catalysts with unique properties, as well as through application to continuous flow reactors.
Introduction
The ability of photoredox catalysis to manipulate electron or energy transfer reactivity has revolutionized small molecule and macromolecular chemistry, presenting opportunities to develop new chemical transformations under mild and energy efficient reaction conditions.[1] Recently, photoredox catalysis has been applied in controlled radical polymerization (CRP) approaches for light-regulated synthesis of well-defined polymers, most commonly in atom transfer radical polymerization (ATRP) and reversible addition-fragmentation transfer (RAFT).[2] ATRP, the most widely studied CRP methodology, is used to access polymers with controlled properties, higher-order architectures, and consequently diverse applications.[3] Traditionally, ATRP is operated through activation of a Cu(I) catalyst by heat to promote an inner-sphere electron transfer to generate a propagating radical species. However, in recent advances, new light-driven ATRP processes have been reported using photocatalysts (PCs) derived from copper, ruthenium, or iridium.[4]
Organocatalyzed atom transfer radical polymerization (O-ATRP) is a metal-free variant of photoredox-catalyzed ATRP which eliminates the concern of trace metal contamination in the polymer product and is advantageous in electronic and biomedical applications, while also enabling opportunities for “greener” reaction design in polymer synthesis.[5] Induced by light, O-ATRP relies on a strongly reducing organic PC to mediate an oxidative quenching catalytic cycle (Figure 1a). O-ATRP processes following a reductive quenching pathway have also been reported but rely on the presence of stoichiometric quantities of sacrificial electron donors, which can also induce undesirable side reactions.[6] The proposed O-ATRP mechanism proceeds through four central steps.[7] Photoexcitation of a ground-state PC generates 1PC*, which can undergo intersystem crossing to produce a long-lived 3PC*. Either 1PC* or 3PC* then directly reduces an alkyl halide initiator through outer-sphere electron transfer to produce a propagating radical species, as well as the ion-pair 2PC●+X-. Deactivation of the propagating chain-end occurs through reinstallation of the halide, generating the PC and a dormant polymer. Central to success in O-ATRP, as determined by control over polymer molecular weight (MW) and dispersity (Đ) close to 1.0, is the presence of a dynamic equilibrium between the activation and deactivation steps, where the rate of deactivation (with rate constant kd) must be higher than the rates of propagation (kp) and activation (ka), limiting radical concentrations and undesirable termination events via radical quenching.
Figure 1.

(a) Proposed mechanism of O-ATRP. b) Previously reported PCs developed for O-ATRP (left) with the new dimethyl-dihydroacridine PC family investigated in this work (right).
To date, advances in O-ATRP have been enabled through development of strongly reducing organic PCs, which are capable of directly reducing an activated alkyl bromide ATRP initiator or dormant polymer chain-end (~−0.8 V vs. SCE).[8] In 2014, perylene and N-phenyl phenothiazine were reported as strongly-reducing PCs for the polymerization of methacrylate monomers via O-ATRP.[9] Since then, other organic PCs derived from N,N-diaryl dihydrophenazine[10] and N-aryl phenoxazine[11] families, among others[12], have been developed (Figure 1b). N-aryl phenothiazine PCs have been applied in diverse contexts[13] and have also been used for mechanistic analysis[14]. Recently, PC structure-property relationships have been studied using N,N-diaryl dihydrophenazine and N-aryl phenoxazine PCs.[15] Empirically, these studies have determined key PC design principles for effective catalytic performance in O-ATRP, among which are the ability of the PC to exhibit intramolecular charge transfer (CT) excited states, redox reversibility, and sufficient thermodynamic driving forces (redox potentials) to mediate the oxidative quenching O-ATRP cycle.[16] N,N-diaryl dihydrophenazine and N-aryl phenoxazine PCs have also been studied in diverse polymerization-related contexts, including the effects of light intensity and solvent, adaptation of O-ATRP to continuous-flow reactors, synthesis of star polymers, and demonstration of oxygen tolerance.[17a-e] In addition, these organic PCs were also applied in small molecule reactions, including trifluoromethylation, C-N and C-S cross couplings via dual catalytic approach with Ni(II) salts, and the reduction of carbon dioxide to methane using sunlight for solar fuel generation.[17f-g]
Despite advances in PC design, O-ATRP has largely been limited to polymerization of methacrylate monomers, but the controlled polymerization of other monomers is highly desired.[18] Poly(acrylates) possess disparate thermal and mechanical properties, enabling widespread industrial and academic use, including drug delivery, superabsorbent materials, coatings, adhesives and additive manufacturing.[19] As such, we sought to leverage current understanding of organic PC design to target the O-ATRP of acrylate monomers. The CRP of acrylates is inherently challenging due to high kp, with values ranging from 15,000 to 24,000 L mol−1s−1,[20] an order of magnitude larger than methacrylates. Furthermore, acrylate chain-end groups containing bromides are more difficult to reduce compared to the corresponding methacrylates, emphasizing the need for efficient PCs for activation.[x]
Photoinduced copper-catalyzed ATRP processes have been reported for successful CRP of acrylate monomers.[21] Additionally, a photoredox-catalyzed ATRP approach of acrylates was also performed using a precious metal-based fac-Ir(ppy)3 PC following an oxidative quenching catalytic cycle, accessing control over MW growth to produce polymers with moderate Đ.[22] To access this polymerization with an organic PC, we hypothesized that a PC candidate must possess both a sufficiently oxidizing 2PC●+ with a correspondingly high E1/2 (2PC●+/1PC) potential to promote fast deactivation (high kd) to compensate for high kp, but also maintain a strongly reducing 3PC* with sufficiently negative E0(2PC●+/3PC*) value for efficient alkyl bromide activation.
Using density functional theory (DFT) calculations to guide organic PC design, herein dimethyl-dihydroacridines are reported as a new class of organic PCs adept at controlled polymerizations of acrylate monomers via O-ATRP. Due to structural similarity to previously employed PCs in O-ATRP and tunable donor-acceptor motifs, we sought to investigate this class of molecules for use in photoredox-catalyzed processes, accessing tailored photo- and electrochemical properties. In this approach, well-defined acrylate polymers with controlled molecular weights and low dispersities (Đ ≤ 1.20) were synthesized using a 365 nm LED in a continuous-flow reactor in conjuction with LiBr salt additives, which are hypothesized to promote efficient deactivation.
Results and Discussion
1. Photocatalyst Development
Dimethyl-dihydroacridines have previously been applied in the development of organic LEDs as thermally activated delayed fluorescence emitters. Tunable absorption profiles, small S1 and T1 energy gaps (ΔEST < 0.4 eV), as well as CT characteristics were reported,[23] making this a promising structural motif for application in photoredox catalysis. Initial DFT calculations for 9,9-dimethyl-10-(naphthalen-1-yl)-9,10-dihydroacridine (1a) predicted an E0ox (2PC●+/1PC) value of 0.72 V vs. SCE. Corroborating DFT prediction, 1a was experimentally determined to have an Ep/2 value of 0.82 V vs. SCE (Figure 2a). Although 1a displayed a non-reversible cyclic voltammogram, this relatively high Ep/2 value encouraged further exploration of dimethyl-dihydroacridines as potential PC candidates, as current successful strongly-reducing PCs in O-ATRP only have E1/2 up to ~ 0.7 V vs. SCE.[16]
Figure 2.

(a) Cyclic voltammogram of PC 1a and PC 1. (b) UV-vis spectrum of PCs 1a, 1, 2, and 3. (c) Electrochemical series of experimentally-measured excited state redox potentials E0*T1, exp. = E0(2PC●+/3PC*) and oxidation potentials E1/2 (2PC●+/1PC) of PCs investigated in this study. High- and low-lying SOMO for PCs with electronically neutral N-aryl groups, overlays of the absorption profiles (purple) and emission profiles (teal) with the experimentally determined Stokes shifts for each PC, and photographs of the PCs dissolved in solvents with increasing polarity with λmax,emission for each solvent for PC 1 (d), 4 (e), and 5 (f). All CV, UV-vis, and emission data collected in N,N-dimethylformamide. See SI for full experimental and computational details.
Notably, installation of biphenyl groups at the 2 and 7 positions of 1a imparted redox reversibility, a key requirement for catalyst turnover (Figure 2a). Furthermore, relative to 1a, increasing the conjugation of the dimethyl-dihydroacridine molecules also red-shifted the absorption profile by ~80 nm, presenting the possible use of milder irradiation conditions (Figure 2b). These results motivated us to synthesize a library of core-substituted derivatives and evaluate their catalytic potential within the previously established design framework for O-ATRP PCs, including absorption properties, excited-state characteristics, and redox properties.[7,15a] Seven dimethyl-dihydroacridine PCs with diverse electron-poor (cyanophenyl), electron-rich (methoxyphenyl), and highly conjugated (biphenyl or naphthalene) groups on both core and N-aryl positions were synthesized (Figure 2c). UV-vis spectroscopy was used to probe the ability to tune the frontier orbitals with various substitutions, and as such tune the absorption profiles of the PC (Figure 2b and Figures S41-S47).
Installation of electron-donating or -withdrawing groups onto the core-substituent was found to strongly influence λmax,abs. For example, installation of 4-cyanophenyl groups (PC 3, 382 nm) red-shifted λmax,abs by 42 nm compared to 4-methoxyphenyl groups (PC 2, 340 nm) (Figure 2b). In a comparison of N-aryl modifications with biphenyl core substituents, decreasing N-aryl conjugation from a 1-naphthyl group (PCs 1 and 4) to a phenyl group (PC 5) led to a decrease in molar absorptivity (ε), (difference of ~18,000 M−1cm−1) but similar λmax,abs at 361 nm and 360 nm, respectively (Table 1, entries 1 and 4). Despite increasing pi-system conjugation through core-substitution, most PCs did not absorb beyond 400 nm. As an exception, the presence of 4-cyanophenyl groups at either core or N-aryl positions resulted in greater red-shifting of λmax,abs, yielding some visible-light absorption. Notably, all the core-modified dimethyl-dihydroacridine PC candidates showed strong light absorption compared to non-core-modified 1a, with ε ranging from 31,500 M−1cm−1 to 50,140 M−1cm-1. This high degree of efficiency in photoexcitation is also corroborated through computationally predicted oscillator strengths (f values ranging from 1.211 to 1.749) indicating high π- π* transition probabilities (Table S2).
Table 1.
Summary of photophysical and electrochemical measurements for PCs 1–7.
| PC | λmax,abs .(nm)[a] |
ε (M−1cm−1) |
λmax,S1 em. (nm)[b] |
Φf (%) |
ES1,exp. (eV)[c] |
ET1,calc.
(eV)[d] |
ET1,77 K (eV)[c,e] |
E1/2 (2PC●+/1PC) (V vs. SCE)[f] |
E0*S1,exp. (2PC●+/1PC*) (V vs. SCE)[g] |
E0*T1,exp. 77 K (2PC●+/3PC*) (V vs. SCE)[e,g] |
E0*T1,calc. (2PC●+/3PC*) (V vs. SCE)[d] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 361 | 46,300 | 487 | 0.3 | 2.55 | 2.41 | 2.34 | 0.76 | −1.79 | −1.58 | −1.86 |
| 2 | 340 | 38,600 | 509 | 0.1 | 2.44 | 2.36 | 2.33 | 0.71 | −1.73 | −1.62 | −1.94 |
| 3 | 382 | 44,300 | 458 | 83 | 2.71 | 2.43 | 2.39 | 0.90 | −1.81 | −1.49 | −1.69 |
| 4 | 360 | 49,800 | 453 | 3.8 | 2.74 | 2.30 | 2.34 | 0.77 | −1.98 | −1.57 | −1.77 |
| 5 | 361 | 31,500 | 443 | 27 | 2.80 | 2.29 | 2.35 | 0.76 | −2.04 | −1.59 | −1.75 |
| 6 | 363 | 43,100 | 444 | 70 | 2.79 | 2.28 | 2.34 | 0.75 | −2.04 | −1.59 | −1.78 |
| 7 | 355 | 50,100 | 535 | 8.2 | 2.32 | 2.39 | 2.37 | 0.82 | −1.50 | −1.55 | −1.78 |
Absorption wavelength measured using ultraviolet-visible spectroscopy in DMF.
Emission wavelength measured using steady-state fluorescence spectroscopy in DMF.
Energies were calculated using the maximum wavelength of emission.
DFT calculations performed at uM06/6–311+Gdp/uM06/6–31+Gdp level of theory with CPCM-described solvation in aqueous solvent.
Spectral emission measured at 77 K after 1 ms gate-delay.
All measurements were performed in a 3-compartment electrochemical cell with an Ag/AgNO3 reference electrode in MeCN (0.01 M) and 0.1 M NBu4PF6 electrolyte solution with DMF analyte solution. Platinum was used at the working and counter electrodes.
Excited-state redox potentials were calculated using energies estimated from the maximum wavelength of singlet or triplet emission and the experimentally measured E1/2; E0*S1,exp.= E1/2 − ES1,exp. and E0*T1, exp. 77K = E1/2 − ET1,77K.
To study the nature of PC*, which governs alkyl bromide activation, a combination of DFT calculations and experimental studies were used to evaluate the ability of these PCs to access CT excited states, which have been empirically shown beneficial for good O-ATRP performance in structurally similar N,N-diaryl dihydrophenazine and N-aryl phenoxazine PCs.[24,15a] In these systems, electron density is transferred from the electron rich core to either the N-aryl or core-substituent, generating a shift in charge density within the molecule in its excited state, which is dictated by the electron accepting ability of the aryl “acceptor” as well as the environment surrounding the “donor” tricyclic core. The connection between PC CT and superior O-ATRP performance were investigated but with dissimilar conclusions. For N-aryl phenoxazines, increasing CT character has been shown to augment triplet yields, positing that higher concentrations of 3PC* promotes fast activation.[26] Conversely, studies with N,N-diaryl dihydrophenazine PCs suggests that CT lowers *PC reduction potentials, slowing down activation, reducing radical concentrations, and minimizing termination.[15c]
Computationally, CT characteristics can be predicted through the presence of charge-separated singly occupied molecular orbitals (SOMOs) for 3PC*. Of the PCs evaluated in this study, PCs 1, 2, and 7, which possess either electron withdrawing groups or an extended π system as the N-aryl moiety, were predicted to have localization of the higher-lying SOMO onto the N-aryl substituent. PCs 4, 5, and 6 all showed a higher-lying SOMO localization onto one core substituent, while PC 3 showed a higher-lying SOMO distributed across both core substituents (Figure S1).
Experimentally, CT character can be observed through a large Stokes shift and visualized through solvatochromism, where the polar 1PC* is progressively stabilized by increasing solvent polarity, resulting in lower-energy emission and a corresponding red-shift in λmax,em. Evaluation of CT from 1PC* can estimate the CT character of 3PC*, as CT singlet and triplet excited states are expected to be energetically degenerate, with low Δ EST.[23] A high fluorescence quantum yield (Φf) can indicate a lack of CT, as CT states have been shown to minimize fluorescence and increase triplet yields.[26] Consistent with previous observations in N,N-diaryl dihydrophenazine and N-aryl phenoxazine studies, for dimethyl-dihydroacridines subtle N-aryl substitution differences were found to be significant in influencing the nature of experimentally-observed CT character. Of these candidates, PCs 1 (Figure 2d), 2, and 7 displayed the largest degree of CT through the largest measured Stokes shifts (ranging from 126 to 180 nm) paired with low Φf (0.1% to 8.7%), and the most dramatic solvatochromism spanning blue to yellow wavelengths of emission (Figure 2d and S1). By the same analysis, PCs 4 and 5 (Figure 2e and f) displayed a moderate degree of CT, while PCs 3 and 6 displayed the least amount of CT character (Figure S1). Notably, Φf values ranging from 0.1% (PC 2) to 83% (PC 3) can be obtained by modulating core-substitution.
Evaluation of excited-state redox potentials of these PCs was performed by DFT calculations to predict E0*T1,calc. (2PC●+/3PC*) values (Table 1). Experimentally, E0*S1,exp. (2PC●+/1PC*) was determined by a modified Rehm-Weller equation: E0*S1,exp.= E1/2 – ES1,exp., where ES1,exp. was measured from the maximum wavelength of steady-state emission at room temperature. Experimental triplet energies (ET1,exp.) were measured from PC phosphorescence at 77 K with a 1 ms gate-delay using time-resolved spectroscopy. ES1,exp. was also evaluated at 77 K with no gate delay, finding significant shifts in emission profiles. Interestingly, the PCs with the highest Stokes shifts, PCs 2 and 7 presented the lowest ΔEST values at 77 K (0.39 and 0.34 eV) (Table S4). Furthermore, computational ET1 predictions corresponded well with experimental values, with differences less than 0.07 eV.
Like absorption, the electronics of core-substitution strongly influenced E0*T1,exp., where the PC with the lowest energy absorption (PC 3) also possessed the lowest E0*T1,exp. (−1.49 V vs. SCE). Correspondingly, PC 2, with a relatively high energy absorption, was predicted to have the most reducing E0*T1,exp. of −1.62 V vs SCE. Despite diverse absorption and CT properties, systematic study of various withdrawing, donating, and neutral N-aryl groups showed minimal influence on reduction potential, with E0*T1,exp. ranging from −1.59 to −1.55 V vs. SCE.
The stability of the deactivating species 2PC●+ was measured by CV to determine E1/2 (2PC●+/1PC), finding similar influence of the electronics of the core subsituent as was observed for both measured absorption characteristics and E0*T1,exp. values. The presence of donating groups (PC 2) resulted in stabilization of 2PC●+ (E1/2 = 0.71 V vs SCE). Withdrawing groups (PC 3) destabilized 2PC●+ and accessed a strongly oxidizing 2PC●+ (E1/2 = 0.90 V vs. SCE). As in evaluation of E0*T1,exp., the nature of the N-aryl group has minimal influence on E1/2, with a range of 0.75 to 0.82 V vs. SCE. Computationally, the oxidizing ability of 2PC●+ (E0ox) was predicted by DFT, with values systematically ~0.25 V lower than the experimentally measured E1/2, justifying the difference in magnitude between E0*T1,exp. and E0*T1,calc.. In sum, the characterization of these dimethyl-dihydroacridine PC candidates shows that photophysical and electrochemical properties can be tuned in an analagous fashion to other established organic PCs with similar donor-acceptor motifs, while accessing more strongly oxidizing 2PC●+ characteristics and offering opportunities for previously unaccessable reactivity.
2. Application of Dimethyl-dihydroacridines to O-ATRP of Acrylate Monomers
To test the ability of these dimethyl-dihydroacridine PC candidates to catalyze O-ATRP of challenging acrylate monomers, initial polymerizations were conducted using n-butyl acrylate (BA) monomer, Diethyl 2-bromo-2-methylmalonate (DBMM) initiator, in N,N-dimethylacetamide (DMAc) solvent under 365 nm LED irradiation in batch reactor conditions. For comparison to dimethyl-dihydroacridine PCs, other well-studied organic PCs with diverse redox properties, including 3,7-di(4-biphenyl) 1-naphthalene-10-phenoxazine (PhenO), 5,10-di-(2-naphthyl)-5–10-dihydrophenazine (PhenN), and 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4Cz-IPN) were applied in the O-ATRP of BA under the same reaction conditions. PhenO and PhenN both possess strongly reducing E0*T1,calc (−1.70 and −2.12 V vs. SCE) and somewhat stabilized E1/2 (0.65 and 0.21 V vs. SCE) while 4Cz-IPN is a moderate reductant and strong oxidant (−1.06 and 1.50 V vs. SCE, respectively).[17f,7] In all cases these polymerizations were uncontrolled. For 4Cz-IPN, Mn decreased over the course of polymerization from 170 kDa to 63 kDa, with Đ > 2.0 (Figure S93). PhenN and PhenO showed some characteristics of an O-ATRP process, but suffered from non-linear growth of MW and high Đ (Figures S91 and S92).
Initial application of PCs 1–7 to O-ATRP of BA supported that CT was required for good O-ATRP performance, similar to what was observed in N,N-diaryl dihydrophenazines.[24] For the PCs with the least amount of CT character (PCs 3 and 6), the polymerization was uncontrolled, with MWs decreasing with increasing monomer conversion and Đ > 2.0. Excitingly, control over polymer MW growth, an indication of efficient deactivation processes, was realized for PCs 1, 2, 4, 5, and 7, with all I* values near 100% (Table 2, entries 1–7), where I*=(Mn,GPC/Mn, theo.) x 100. PC 2 provided the best results with I* = 96% at 77% conversion (Figure 4a), but produced a polymer with high Đ at 1.53. For PCs 1, 2, 4, 5, and 7, bimodal gel permeation chromatography (GPC) traces revealed an accompanying high MW p(BA) species present in low quantities. Control experiments with BA in DMAc under 365 nm irradiation revealed significant monomer autopolymerization (63% conversion at 2.5 hours) (Table S3) and thus likely contributed to the formation of this undesired high MW species.
Table 2:
Summary of results of O-ATRP of BA performed in batch and flow using PCs 1–7[a]
| Entry | PC | Reactor | Conv. (%)[b] |
Mn,calc,
(kDa)[c] |
Đ
(Mw/Mn)[c] |
I*
(%)[d] |
|---|---|---|---|---|---|---|
| 1 | 1 | Batch | 65 | 9.3 | 1.64 | 92 |
| 2 | 2 | Batch | 77 | 10.6 | 1.53 | 96 |
| 3 | 3 | Batch | 42 | 31.4 | 4.93 | 35 |
| 4 | 4 | Batch | 68 | 9.6 | 1.62 | 93 |
| 5 | 5 | Batch | 59 | 8.7 | 1.70 | 90 |
| 6 | 6 | Batch | 72 | 25.8 | 3.52 | 37 |
| 7 | 7 | Batch | 76 | 10.9 | 1.89 | 92 |
| 8 | 1 | Flow | 67 | 8.9 | 1.59 | 100 |
| 9 | 2 | Flow | 81 | 11.0 | 1.35 | 97 |
| 10 | 3 | Flow | 71 | 13.1 | 4.57 | 72 |
| 11 | 4 | Flow | 81 | 11.1 | 1.48 | 96 |
| 12 | 5 | Flow | 79 | 10.2 | 1.48 | 102 |
| 13 | 6 | Flow | 82 | 11.4 | 3.58 | 94 |
| 14 | 7 | Flow | 73 | 9.6 | 1.54 | 100 |
Conditions are [1000]:[10]:[1] of [BA]:[DBMM]:[PC] with 1.0 eq. DMAc to BA by volume and irradiated by 365 nm LEDs. Batch reactions are conducted under ambient temperatures and flow reactions at 22 ºC.
Determined by 1H NMR.
Measured using GPC.
Calculated by (Conv. × [Mon]/[RX] × Mw,Mon)/1000.
Initiator efficiency (I*) calculated by (Theo. Mn/Calc. Mn) × 100.
Figure 4.

(a) Plots of Mn vs. conversion (blue) and Đ vs. conversion (red) for O-ATRP of BA plotted against the theoretical Mn (dashed line) using PC 2 conducted in batch reactor, (b) flow reactor, and (c) flow reactor with 30 eq. LiBr relative to PC. (d) First order kinetic plot of O-ATRP of BA with varying LiBr eq. relative to PC. Conditions are [1000]:[10]:[1]:[x] of [BA]:[DBMM]:[PC 2]:[LiBr] with 1.0 eq. by volume of DMAc for (a) and 1.5 eq. for (b) and (c), and irradiated by 365 nm LEDs.
As the best performing PC in these conditions (PC 2) possessed the highest E0*T1,exp. but also the lowest E1/2, we hypothesized that promoting efficient activation could outcompete autopolymerization side-reactivity, leading to lower Đ while maintaining control over MW growth. Notably, acrylate alkyl halide chain-end groups are more difficult to reduce compared to methacrylates, which can be observed by CV measurements of representative alkyl bromide initiators with methacrylate and acrylate functionalities. An acrylate analogue, methyl 2-bromopropionate, was found to undergo one-electron reduction at Ep/2 = −0.96 V vs SCE, while a methacrylate model, methyl α-bromoisobutyrate, was reduced at Ep/2 = −0.80 V vs. SCE, or a difference of 0.16 V (Figure S67), highlighting the need for high E0*T1,exp. for efficient activation of acrylate-derived chain-end groups.
One alternative to batch reactor systems are continuous-flow reactors, which provide more uniform reaction irradiation and have been shown to improve photoinduced CRP systems through enhancing PC photoexcitation, thus promoting efficient alkyl bromide activation.[27] In this approach, a commercially available temperature-controlled Hepatochem PhotoRedOx box, an 18 W EvoluChem 365 nm LED, and a 2 mL PFA flow reactor was employed. Importantly, control experiments with BA in DMAc under these conditions did not produce any undesired autopolymerization (Table S14). PCs 1–7 were then evaluated for the O-ATRP of BA in continuous-flow, showing similar trends in performance as batch conditions, albeit with consistently lower Đ (Table 2, entries 8–14). PC 2 again proved superior, however with initial high Đ > 1.5 at conversions < 50%, but lowered to 1.35 with I* = 97% at 81% conversion.
Using PC 2, further reaction optimization analyzing the effects of initiator, PC loading, and reaction concentration were conducted (Figures S124-S131). Increasing the DMF:BA ratio (1:1 to 1.5:1 v/v) significantly improved polymerization results, with Đ < 1.5 at all monomer conversions (Figure 4b). To further improve the system, the addition of various bromide salts was investigated, as this has been shown to promote deactivation in aqueous copper-catalyzed ATRP systems and in some photoredox-catalyzed ATRP systems.[28] Analysis of LiBr, NaBr, KBr, and tetrabutylammonium bromide salts showed some decrease in Đ, with LiBr providing the greatest effect (Đ ≤ 1.23) (Table S20). O-ATRP of BA using lithium salts with diverse anions (LiCl, LiI, LiPF6) was also performed. A complete loss of MW control and Đ > 2.0 in all cases was observed (Table S20), illustrating the significance of the bromide anion in deactivation. Analysis of relative LiBr concentrations showed the conditions producing polymers with lowest Đ (30 eq. LiBr relative to PC, or 83.3 mM) to also have the slowest overall reaction rate (Figure 4c and d), which paired with lower Đ, suggests LiBr concentration plays a key role in deactivation.
Next, a combination of DFT calculations and experimental studies were employed to explore the role of the LiBr salt in this O-ATRP system, probing its potential influence on both activation and deactivation mechanistic steps. One possibility is a Lewis-acid driven activation by Li+ of the alkyl bromide ester polymer chain-end, facilitating rapid activation. However, CV revealed no change in reduction Ep/2 (RX/RX●-) of methyl-2-bromopropionate upon addition of LiBr (Ep/2 = −0.92 V vs. SCE) (Figure S61). To evaluate potential effects of LiBr on PC photophysical behavior, PC 2 absorption and emission characterization was performed in the presence of LiBr, finding no changes in these properties (Figure S83 and S84).
Formally, the deactivation step is a termolecular process requiring low concentrations of Pn●, 2PC●+, and X- to collide, which is entropically unfavorable. Previous studies with dihydrophenazines have shown that decreasing solvent polarity promotes ion-pairing of 2PC●+X-, with a subsequent decrease in polymerization rate and improvement in performance.[24] As such, we propose that efficient deactivation requires oxidation of Pn● by a 2PC●+X- ion-pair, which shifts a formal three-body collisional event to a more favorable pseudo-two-body event.11,22a In addition, we hypothesize a concerted mechanism for the oxidation of Pn● by a 2PC●+X- ion-pair, where the formation of Pn-X is tied to the nuclear coordinate of X- thus avoiding thermodynamically unfavorable carbocation Pn+ intermediary species. Using DFT, the association of 2PC●+ + X- → 2PC●+X- using PC 2 was predicted to be slightly endergonic (∆G0complex = 0.6 kcal/mol). As such, we postulate that the presence of excess bromide ions through LiBr addition increases the population of the proposed 2PC●+X- deactivator species, promoting rapid deactivation of the propagating chain-end and explaining the observed decrease in polymerization rates and lower Đ.
To further elucidate the relative importance of PC choice and the optimized reaction conditions in successful O-ATRP of acrylates, polymerizations were also conducted using 4-CzIPN, PhenO, and PhenN in continuous-flow with LiBr additives. For 4Cz-IPN, no polymerization was observed in the presence of LiBr, while O-ATRP without LiBr yielded an uncontrolled polymerization with Mn > 40 kDa and Đ > 2.0. (Table S21). The O-ATRP of BA using PhenO and PhenN was moderately improved under the optimized conditions, showing some control over molecular weight growth and with Đ < 1.5 at higher monomer conversions. However, both polymerizations displayed bimodal MW distributions by GPC, with I* ranging from 31–109%.
Using these optimized conditions, MW control in the O-ATRP of BA catalyzed by PC 2 was demonstrated through adjustment of reaction stoichiometry, accessing MWs ranging from 2 to 26 kDa with Đ < 1.36 (Table 2, entries 1–5). However, targeting high MWs proved challenging, with I* > 100%, perhaps from a confluence of increased termination events and viscosity changes within the reactor. The O-ATRP of other alkyl and glycol acrylate monomers was performed using PC 2 with excellent control over MW and Đ (Table 2, entries 7–11). O-ATRP of methyl methacrylate (MMA) was also accomplished, realizing low Đ but with slightly bimodal GPC traces (Table 2, entry 12). As expected due to lower kp, the O-ATRP of MMA was significantly slower than BA requiring 600 minutes of residence time for 72% monomer conversion, as compared to ~100 minutes for acrylates.
In batch conditions, no monomer conversion was observed using PC 2 under ambient atmosphere (Table S5). Recently, N-aryl phenoxazines were found to perform a well-controlled O-ATRP under ambient conditions when no vial headspace was present.[17e] To further test the oxygen-tolerance of dimethyl-dihydroacridines, the O-ATRP of BA was performed in optimized flow conditions using reagents and solvents previously exposed proceeded efficiently with no observed induction period, with Đ ≤ 1.24 and I* close to 100% (Table 3, entry 6).
Table 3:
Summary of results of O-ATRP of acrylate monomers performed using PC 2.[a]
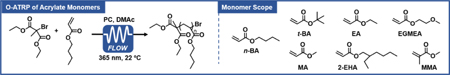 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Monomer | [Monomer]:[DBMM] | Res. Time (min.) |
Conv. (%) |
Mn,calc,
(kDa) |
Mw,calc,
(kDa) |
Đ
(Mw/Mn) |
I*
(%) |
| 1 | n-BA | [1000]:[2.5] | 90 | 89 | 26.4 | 45.7 | 1.35 | 173 |
| 2 | n-BA | [1000]:[5] | 120 | 88 | 15.2 | 22.8 | 1.32 | 150 |
| 3 | n-BA | [1000]:[10] | 120 | 73 | 9.4 | 9.7 | 1.20 | 103 |
| 4 | n-BA | [1000]:[20] | 180 | 80 | 7.3 | 5.4 | 1.19 | 74 |
| 5 | n-BA | [1000]:[40] | 180 | 67 | 5.4 | 2.4 | 1.18 | 44 |
| 6[b] | n-BA | [1000]:[10] | 120 | 85 | 10.3 | 11.1 | 1.24 | 108 |
| 7 | t-BA | [1000]:[10] | 120 | 92 | 13.8 | 12.1 | 1.23 | 88 |
| 8 | MA | [1000]:[10] | 90 | 92 | 10.0 | 8.1 | 1.30 | 81 |
| 9 | EA | [1000]:[10] | 90 | 76 | 7.5 | 7.8 | 1.19 | 105 |
| 10 | 2-EHA | [1000]:[10] | 90 | 90 | 16.3 | 16.8 | 1.53 | 104 |
| 11 | EGMEA | [1000]:[10] | 60 | 92 | 10.5 | 12.3 | 1.37 | 117 |
| 12 | MMA | [1000]:[10] | 600 | 72 | 8.8 | 7.4 | 1.17 | 85 |
Conditions are [1000]:[×]:[1]:[30] of [monomer]:[DBMM]:[PC 2]:[LiBr] with 1.5 mL DMAc relative to 1 mL monomer performed in continuous-flow and irradiated by 18 W 365 nm LED at 22 ºC.
Reaction components sparged with air for 30 minutes before polymerization. See SI for full experimental details.
To demonstrate the temporal control characteristic of an O-ATRP process, pulsed irradiation polymerization experiments were performed in batch conditions, showing no monomer conversion during dark periods with no irradiation (Figure S115). Analysis of bromide chain-end group retention of p(BA) was performed through MALDI mass spectrometry, revealing the presence of H-terminated and bimolecular radical termination products (Figure S161). Notably, no significant differences in chain-end groups between polymers synthesized with and without LiBr were observed (Figure S162).
To validate the chain-end group fidelity of the system and demonstrate the ability of this system to produce complex polymeric materials, chain-extensions were performed in continous-flow of an isolated p(n-BA) macroinitiator (Mn = 4.6 kDa, Đ = 1.26) with ethyl acrylate (EA) to produce a block-copolymer p(n-BA)-b-p(EA) with Đ = 1.16 (12 kDa). This polymer was then again reintroduced as a macroinitiator and further extended with tert-butyl acrylate to produce a well-defined triblock copolymer (Mn = 20 kDa, Đ = 1.44) (Figure 5). An 1H NMR spectrum for the p(n-BA) macroinitiator with detailed assignments (Figure S158) also shows the presence of α- and ω- chain-end groups.
Figure 5:

Results of sequential chain-extension experiments with corresponding GPC traces to produce a p(n-BA)-b-p(EA)-b-p(t-BA) triblock copolymer.
Conclusion
In summary, we have reported the first successful O-ATRP of acrylate monomers with controlled MW and low Đ, enabled by rational design and development of a new family of dimethyl-dihydroacridine photocatalysts. Relationships between PC properties and catalytic performance are discussed, finding an interplay of photophysical and electrochemical properties is necessary to achieve the desired reactivity, as is supported by the best performing PC having the highest E0*T1,exp. but the lowest E1/2. A combination of reactor choice and LiBr additives is found to be significant in achieving a well-controlled system, which are proposed to promote the respective activation and deactivation steps of the O-ATRP cycle. We envision that dimethyl-dihydroacridines, with distinct properties and new reactivity, will further enable the advancement of organic photoredox catalysis in both small molecule and macromolecular syntheses, aiding in the replacement of precious-metal catalysts for sustainable photoredox processes.
Supplementary Material
Acknowledgements
This work was supported by Colorado State University and the NIH (R35GM119702). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant ACI-1548562. C.-H. L. acknowledges a National Institutes of Health F32 Postdoctoral Fellowship (F32GM122392). The authors thank Dr. Matthew Ryan, Dr. Dianfeng Chen, and Max Kudisch for technical assistance and helpful discussions.
References
- [1].a) Romero NA, Nicewicz DA, Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]; b) Prier CK, Rankic DA, MacMillan DWC, Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Narayanam JMR, Stephenson CRJ, Chem. Soc. Rev. 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; d) Buzzetti L, Crisenza GEM, Melchiorre P, Angew. Chem. Int. Ed. 2019, 58, 3730–3747. [DOI] [PubMed] [Google Scholar]; e) Corrigan N, Shanmugam S, Xu J, Boyer C, Chem. Soc. Rev. 2016, 45, 6165–6212. [DOI] [PubMed] [Google Scholar]
- [2].a) Leibfarth FA, Mattson KM, Fors BP, Collins HA, Hawker CJ, Angew.Chem. Int. Ed. 2012, 52, 199–210. [DOI] [PubMed] [Google Scholar]; b) Chen M, Zhong M, Johnson JA, Chem. Rev. 2016, 116, 10167–10211. [DOI] [PubMed] [Google Scholar]; c) Corrigan N, Yeow J, Judzewitsch P, Xu J, Boyer C, Angew. Chem. Int. Ed. 2019, 131, 5224–5243. [DOI] [PubMed] [Google Scholar]
- [3].a) Matyjaszewski K, Adv. Mater. 2018, 30, 1706441. [DOI] [PubMed] [Google Scholar]; b) Ribelli TG, Lorandi F, Fantin M, Matyjaszewski K, Macromol. Rapid Commun, 2019, 40, 1800616. [DOI] [PubMed] [Google Scholar]; c) Ouchi M, Terashima T, Sawamoto M, Chem. Rev. 2009, 109, 4963–5050. [DOI] [PubMed] [Google Scholar]
- [4].a) Yang Q, Dumur F, Morlet-Savary F, Poly J, Lalevee J, Macromolecules 2015, 48, 1972–1980. [Google Scholar]; b) Zhang G, Song IY, Ahn KH, Park T, Choi W, Macromolecules, 2011, 44, 7594–7599. [Google Scholar]; c) Fors BP, Hawker CJ, Angewandte Chemie 2012, 51, 8850–8853. [DOI] [PubMed] [Google Scholar]
- [5].Shanmugam S, Boyer C, Science 2016, 352, 1053–1054 [DOI] [PubMed] [Google Scholar]
- [6].Yilmaz G, Yagci Y, Polym. Chem. 2018, 9, 1757–1762. [Google Scholar]
- [7].Theriot JC, McCarthy BG, Lim C-H, Miyake GM, Macromol. Rapid Commun. 2017, 38, 1700040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Roth HG, Romero NA, Nicewicz DA, Synlett 2016, 27, 714–723. [Google Scholar]
- [9].a) Miyake GM, Theriot JC, Macromolecules 2014, 47, 8255–8261. [Google Scholar]; b) Treat NJ, Sprafke H, Kramer JW, Clark PG, Barton BE, Alaniz JR, Fors BP, Hawker CJ, J. Am. Chem. Soc. 2014, 136, 16090–166101 [DOI] [PubMed] [Google Scholar]
- [10].a) Theriot JC, Lim C-H, Yang H, Ryan MD, Musgrave CB, Miyake GM, Science 2016, 352, 1082–1086. [DOI] [PubMed] [Google Scholar]
- [11].a) Pearson RM, Lim C-H, McCarthy BG, Musgrave CB, Miyake GM, J. Am. Chem. Soc. 2016, 138, 11399–11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Singh VK, Yu C, Badgujar S, Kim Y, Kwon Y, Kim D, Lee J, Akhter T, Thangavel G, Park LS, Lee J, Nandajan PC, Wannemacher R, Milian-Medina B, Luer L, Kim KS, Gierschner J, Kwon MS, Nature Catalysis, 2018, 1, 794–804. [Google Scholar]; b) Discekici EH, Anastasaki A, Read de Alaniz J, Hawker CJ, Macromolecules 2018. 51, 7421–7434. [Google Scholar]
- [13].a) Narupai B, Page ZA, Treat NJ, McGrath AJ, Pester CW, Discekici EH, Dolinski ND, Meyers GF, Read de Alaniz J, Hawker CJ Angewandte Chemie 2018, 57, 13433–13438. [DOI] [PubMed] [Google Scholar]; b) Discekici EH, Pester CW, Treat NJ, Lawrence J, Mattson KM, Narupai B, Toumayan EP, Luo Y, McGrath AJ, Clark PG, Read de Alaniz J, Hawker CJ ACS Macro Lett. 2016, 5, 258–262. [DOI] [PubMed] [Google Scholar]
- [14].Pan X, Fang C, Fantin M, Malhotra N, So WY, Peteanu LA, Isse AA, Gennaro A, Liu P, Matyjaszewski K, J. Am. Chem. Soc. 2016, 138, 2411–2425. [DOI] [PubMed] [Google Scholar]
- [15].a) McCarthy BG, Pearson RM, Lim C-H, Sartor SM, Damrauer NH, Miyake GM, J. Am. Chem. Soc. 2018, 140, 5088–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cole JP, Federico CR, Lim C-H, Miyake GM, Macromolecules 2019, 52, 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Koyama D, Dale HJA, Orr-Ewing AJ, J. Am. Chem. Soc. 2018, 140, 1285–1293. [DOI] [PubMed] [Google Scholar]
- [16].a) Corbin DA, Lim C-H, Miyake GM, Aldrichimica Acta. 2019, 52, 7–21. [PMC free article] [PubMed] [Google Scholar]
- [17].a) Ryan MD, Pearson RM, French TA. Miyake GM, Macromolecules, 2017, 50, 4616–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ryan MD, Theriot JC, Lim C-H, Yang H, Lockwood AG, Garrison NG, Lincoln SR, Musgrave CB, Miyake GM, J. Polym. Sci. Part A: Polym. Chem. 2017, 55, 3017–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ramsey BL, Pearson RM, Beck LR, Miyake GM, Macromolecules, 2017, 50, 28–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Buss BL, Beck LR, Miyake GM, Polym. Chem. 2018, 9, 1658–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) McCarthy B, Miyake GM, ACS Macro. Lett. 2018, 7, 1016–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Du Y, Pearson RM, Lim C-H, Sartor SM, Ryan MD, Yang H, Damrauer NH, Miyake GM, Chem. Euro. J. 2017, 23, 10962–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Rao H, Lim C-H, Bonin J, Miyake GM, Robert M, J. Am. Chem. Soc. 2018, 140, 17830–17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Pan X, Lamson M, Yan J, Matyjaszewski K, ACS Macro Lett. 2015, 4, 192–196. [DOI] [PubMed] [Google Scholar]; b) Chen D-F, Boyle BM, McCarthy BG, Lim CH, Miyake GM, J. Am. Chem. Soc. 2019, 141, 13268–13277. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ramakers G, Krivkov A, Trouillet V, Welle A, Mobius H, Junkers T, Macromol. Rapid Commun. 2017, 38, 1700423. [DOI] [PubMed] [Google Scholar]
- [19].Destarac M, Polym. Chem. 2018, 9, 4947–4967. [Google Scholar]
- [20].Beuermann S, Buback M, Prog. Polym. Sci. 2002, 27, 191–254. [Google Scholar]
- [21].Anastasaki A, Nikolaou V, Zhang Q, Burns J, Samanta SR, Waldron C, Haddleton AJ, McHale R, Fox D, Percec V, Wilson P, Haddleton DM, J. Am. Chem. Soc. 2014, 136, 1141–1149. [DOI] [PubMed] [Google Scholar]
- [22].Treat NJ, Fors BP, Kramer JW, Christianson M, Chiu C-Y, Read de Alaniz J, Hawker CJ, ACS Macro. Lett. 2014, 3, 580–584. [DOI] [PubMed] [Google Scholar]
- [23].Wong MY, Zysman-Colman E, Advanced Materials 2017, 29, 1605444. [DOI] [PubMed] [Google Scholar]; Li W, Li B, Cai X, Gan L, Xu Z, Li W, Liu K, Chen D, Su S-J, Angew. Chem. Int. Ed. 2019, 58, 11301–11305. [DOI] [PubMed] [Google Scholar]
- [24].Lim C-H, Ryan MD, McCarthy BG, Theriot JC, Sartor SM, Damrauer NH, Musgrave CB, Miyake GM, J. Am. Chem. Soc. 2017, 139, 348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Arias-Rotondo DM, McCusker JK, Chem. Soc. Rev. 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- [26].Sartor SM, McCarthy BG, Pearson RM, Miyake JM, Damrauer NH, J. Am. Chem. Soc. 2018, 140, 4778–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Hartman RL, McMullen JP, Jenson KF, Angew. Chem. Int. Ed. 2011, 50, 7502–7519. [DOI] [PubMed] [Google Scholar]; b) Buss BL, Miyake GM, Chem. Mater. 2018, 30, 3931–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Junkers T, Wenn B, React. Chem. Eng. 2016, 1, 60. [Google Scholar]; d) Tucker JW, Zhang Y, Jamison TF, Stephenson CRJ, Angew. Chem. Int. Ed. 2012, 51, 4144–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Simakova A, Averick SE, Konkolewicz D, Matyjaszewski K, Macromolecules 2012, 45, 6371–6379. [Google Scholar]; b) Fantin M, Isse AA, Gennaro A, Matyjaszewski K, Macromolecules, 2015, 48, 6862–6875. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.