

Abstract
Sclerosing epithelioid fibrosarcoma (SEF) is an aggressive soft tissue sarcoma, characterized by a distinctive epithelioid phenotype in a densely sclerotic collagenous stroma, that shows frequent MUC4 immunoreactivity and recurrent gene fusions, often involving EWSR1 gene. A pathogenetic link with low grade fibromyxoid sarcoma (LGFMS) has been suggested, due to cases with hybrid morphology as well as overlapping genetic signature. However, a small subset of SEF is negative for MUC4 and lacks the canonical EWSR1/FUS gene rearrangements. Triggered by the identification of recurrent YAP1-KMT2A gene fusions by RNA sequencing in 3 index cases of MUC4-negative, EWSR1/FUS fusion-negative SEF, we further investigated a cohort of 14 similar SEF cases (MUC4-negative, EWSR1/FUS fusion-negative) by fluorescence in situ hybridization (FISH), reverse transcription polymerase chain reaction (RT-PCR), and/or DNA-based massively parallel sequencing (MSK-IMPACT) for abnormalities in these genes. Three additional SEFs with KMT2A gene rearrangements and one additional case with YAP1 gene rearrangements were identified by FISH. In addition, one case with YAP1-KMT2A and one with KMT2A-YAP1 fusion were detected by RT-PCR and MSK-IMPACT, respectively. As a control group, 24 fibromyxoid spindle cell tumors, diagnosed or suspected as fusion-negative LGFMS, were also tested for YAP1 and KMT2A abnormalities by FISH, but none were positive. The YAP1/KMT2A-rearranged SEF group affected patients ranging from 10–73 years old (average & median: 45) of both genders (4 females, 5 males). The tumors involved somatic soft tissues with a wide distribution, including extremities, trunk, pelvis, neck, and dura. Histologically, the tumors showed variable cellularity, with monotonous ovoid to epithelioid tumor cells and hyalinized collagenous background typical of SEF. More than half of the cases showed infiltrative borders, within fat or skeletal muscle. No LGFMS component was identified. All tumors were negative for MUC4 and had an otherwise non-specific immunophenotype. Of the 6 cases with available follow-up information, 2 had local recurrences, and 2 developed soft tissue and/or bone metastases, including 1 of them died of the disease.
Keywords: Sclerosing epithelioid fibrosarcoma, Low-grade fibromyxoid sarcoma, YAP1, KMT2A, MLL, MUC4
INTRODUCTION
Sclerosing epithelioid fibrosarcoma (SEF) is a highly aggressive soft tissue sarcoma closely related to low-grade fibromyxoid sarcoma (LGFMS).1,2 Some tumors display morphologic characteristics of both SEF and LGFMS, being classified as hybrid SEF/LGFMS. Furthermore, SEF and LGFMS share overexpression of MUC4 by immunohistochemistry. Although MUC4 immunohistochemistry represents a sensitive and specific marker for LGFMS,3 it is only present in up to 78% of SEFs.4 At the genetic level, a molecular dichotomy between these two tumors has emerged, with pure SEF harboring mostly EWSR1-CREB3L1 fusions, while LGFMS and hybrid SEF/LGFMS displaying FUS-CREB3L2 fusions.5,6 Nevertheless, significant morphologic, immunohistochemical and cytogenetic overlap exists in a minority of cases.
Triggered by a handful of cases with the morphologic appearance of SEF, but lacking MUC4 expression and the typical EWSR1/FUS gene rearrangements, we have explored their molecular abnormalities using a combined approach using targeted RNA sequencing, targeted DNA sequencing, fluorescence in situ hybridization (FISH), and/or reverse transcription polymerase chain reaction (RT-PCR) methodology. The recurrent genetic alterations identified were then screened in a larger cohort of cases.
MATERIALS AND METHODS
Index cases and cohort selection screening
Three index cases of soft tissue tumors with a histomorphology resembling SEF, but lacking MUC4 immunoreactivity and EWSR1/FUS gene rearrangements, were submitted to targeted RNA sequencing for fusion discovery. These patients were all in their forties, with tumors involving somatic soft tissues, including two in the lower extremities (thigh, lower leg) and one in the trunk (paraspinal) (Table 1). After identification of the fusion candidates, fluorescence in situ hybridization (FISH) was performed on all 3 cases, using a custom break-apart probe design of the respective fusion partner genes, in order to validate the gene rearrangements. Reverse transcription polymerase chain reaction (RT-PCR) was performed on case #3, using primers designed based on the fusion junction reads of RNA sequencing.
Table 1.
Sclerosing Epithelioid Fibrosarcoma with YAP1 and/or KMT2A rearrangements
| Age/Sex | Location (depth) | Size (cm) | Mitotic count (10HPF) | Genetics | Follow-up | |||
|---|---|---|---|---|---|---|---|---|
| RNAseq & RT-PCR (exon number) | YAP1 FISH | KMT2A FISH | ||||||
| 1 | 45/F | Paraspinal (deep) | 2.6 | P: 0 R1: 1 |
YAP1(5)-KMT2A(4)# | NEG (1X) | NEG | Recur (1yr) |
| 2 | 45/F | Thigh (subcutis) | 3.4 | P:17 Mets: 21 |
YAP1(4)-KMT2A(5)# | NEG (1X) | NEG | Mets to soft tissue (1.5yr) |
| 3 | 47/M | Lower leg (deep) | 2.0 | P: NA R1: 5 R2: 0 R3: 2 |
YAP1(5)-KMT2A(4)#† | POS | NEG | Recur (10, 17, 21 yr) |
| 4 | 73/F | Iliopsoas muscle (deep) | 10 | 8 | ND | NEG (1X) | POS | DOD (1yr), mets to contralateral pelvis & scalp |
| 5 | 35/M | Chest wall (deep) | 5.0 | 3 | ND | NEG | POS* | Recent |
| 6 | 10/F | Frontal dura (deep) | 12 | 9 | ND | NEG | POS | NED (2.5yr) |
| 7 | 86/M | Inguinal (NA) | NA | 11 | ND | POS | NEG | NA |
| 8 | 42/M | Thigh (subcutis) | 2.5 | 4 | YAP1(5)-KMT2A(4) † | NEG | NEG | Recent |
| 9 | 22/M | Neck/paraspinal (deep) | 8 | 4 | KMT2A(6)-YAP1(9)‡ | NEG (1X) | NEG (1X) | AWD (1yr) |
MAML2 FISH also positive for rearrangement
RNAseq
RT-PCR, (parenthesis are exonic breakpoint)
MSK-IMPACT; F, female; HPF, high power fields; M, male; mets, metastasis; NA, not available; ND, not done; NEG, negative; P, primary tumor; POS, positive; R1, first recurrence; R2, second recurrence; R3, third recurrence; RNAseq, RNA sequencing; RT-PCR, reverse transcription polymerase chain reaction; yr, year(s); 1X, only one copy of fused signals.
To further investigate the prevalence of the identified gene fusions in SEF and the closely related low-grade fibromyxoid sarcoma (LGFMS) and to better understand the clinicopathologic spectrum of such tumors, the consultation archive of the senior author (C.R.A.) was searched for cases diagnosed as or suspected to be SEF and LGFMS, which were negative for MUC4 immunostain and EWSR1/FUS gene rearrangements. The resultant screening cohorts of SEF (n=14) and LGFMS (n=24) were studied using break-apart FISH assays for YAP1 and KMT2A rearrangements. Cases with available material were also tested by RT-PCR for YAP1-KMT2A fusion. As part of the clinical work-up for diagnostic and therapeutic purposes, one SEF case was subjected to MSK-IMPACT (Memorial Sloan Kettering Cancer Center Integrated Mutation Profiling of Actionable Cancer Targets), a targeted DNA-based next generation sequencing using hybridization capture strategy as described previously.
Clinical follow-up information was obtained from the electronic medical records or the initial referral pathologists. The study was approved by the Institutional Review Board.
RNA sequencing and data analysis
Cases #1 and #2 were submitted to targeted RNA sequencing using TruSight RNA Fusion Panel (Illumina, San Diego, CA), while case #3 was studied using TruSeq RNA Exome (Illumina, San Diego, CA). RNA was extracted from formalin-fixed paraffin-embedded (FFPE) tissues, using Amsbio’s ExpressArt FFPE Clear RNA Ready kit (Amsbio LLC, Cambridge, MA) in cases #1 and #2, or RNeasy FFPE kit (Qiagen, USA) in case#3, according to the manufacturer’s instructions. Fragment length was assessed with an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). For cases #1–2, RNA sequencing libraries were prepared using 20–100 ng total RNA with the Trusight RNA Fusion Panel. Each sample was subjected to targeted RNA sequencing on an Illumina MiSeq at 8 samples per flow cell (approximately 3 million reads per sample). Sample#3 was ligated to adaptors for further amplification and exonic region enrichment using TruSeq RNA Exome, which focuses on capturing the coding regions of RNA. All reads were independently aligned with STAR (ver 2.3) and BowTie2 against the human reference genome (hg19) for Manta-Fusion or STAR-Fusion and TopHat-Fusion analysis, respectively.
Reverse transcription polymerase chain reaction (RT-PCR)
RT-PCR was performed on case #3 to validate the gene fusion and on one case (case #8) from the screening cohort. The primer sequences were designed based on the fusion junction read sequences from RNA sequencing (Supplementary Table 1). RNA after RNA sequencing was subjected to reverse transcription using SuperScript IV First-Strand Synthesis System (Invitrogen). PCR was performed by Phusion Green Hot Start II High-Fidelity DNA Polymerase (Thermo Scientific). The PCR products were then analyzed by gel electrophoresis and Sanger sequencing.
Fluorescence in situ hybridization (FISH)
FISH was performed on 4 μm-thick FFPE tissue sections. Custom probes were made by bacterial artificial chromosomes (BAC) clones (Supplementary Table 2) flanking YAP1, KMT2A, and MAML2, and flanking/covering VIM genes, according to UCSC genome browser (http://genome.ucsc.edu) and obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA; https://bacpacresources.org). Fusion FISH assays were also performed on selected cases. DNA from each BAC was isolated according to the manufacturer’s instructions. The BAC clones were labeled with fluorochromes by nick translation and validated on normal metaphase chromosomes. The slides were deparaffinized, pretreated, and hybridized with denatured probes. After overnight incubation, the slides were washed, stained with DAPI, mounted with an antifade solution, and then examined on a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany) controlled by Isis 5 software (Metasystems, Newton, MA).
RESULTS
A recurrent YAP1-KMT2A fusion is identified in MUC4-negative SEFs
A YAP1-KMT2A fusion candidate was identified in all 3 index cases tested by 2 different RNA sequencing platforms. The fusions joined YAP1 exon 5 (NM_001130145) to KMT2A exon 4 (NM_001197104) in cases #1 and #3, and YAP1 exon 4 to KMT2A exon 5 in case #2 (Fig. 1). Both fusion transcript variants were predicted to be in-frame. In case #3, a variant YAP1-KMT2A fusion transcript was noted by RNA sequencing, showing an additional 12 bp of YAP1 intron 5 at the fusion juction, possibly due to alternative splicing. In addition, the reciprocal KMT2A-YAP1 fusion transcripts were present in all 3 cases, as KMT2A exon 6-YAP1 exon 9 (in-frame) in cases #1 and #3 and KMT2A exon 5-YAP1 exon 9 (breakpoints within exons, out-of-frame) in cases #2. RT-PCR confirmed the YAP1-KMT2A and KMT2A-YAP1 fusions in case #3 (Fig. 2A).
Figure 1. Diagrammatic representation of fusions involving KMT2A, YAP1, and MAML2 gene.

Upper panel illustrates chromosomal locations and orientations of KMT2A, YAP1, and MAML2 genes on chromosome 11. Middle and lower panels highlight the protein domains and exons contributed by each partner gene in the YAP1-KMT2A fusions, as well as the alignments of RNA sequences flanking the fusion junctions identified by RNA sequencing. YAP1 exon 5- KMT2A exon 4 fusion transcripts were more common (n=3) than YAP1 exon 4- KMT2A exon 5 (n=1). Both fusion variants were predicted to be in-frame.
Figure 2.

(A) RT-PCR and Sanger sequencing confirmed the fusion transcripts detected by targeted RNA sequencing in case #3. A 12-bp sequence represented by minor peaks immediately starting after the breakpoint (lower case letters in parenthesis) is derived from the 5’ end of YAP1 intron 5, likely the result of alternative splicing. (B-C) FISH detected rearrangements of YAP1 (case #3, unbalanced with 1 copy of unpaired green/telomeric signal)(B) or KMT2A (case #5)(C) in some cases.
In the screening cohort, one case (case #8) with available material was subjected to RT-PCR using the same primers and was shown to have the same YAP1 exon 5-KMT2A exon 4 fusion. No reciprocal KMT2A exon 6 -YAP1 exon 9 fusion transcript was found by RT-PCR. The case (case #9) studied by MSK-IMPACT showed fusion of KMT2A intron 6–7 (100bp) to YAP1 intron 8–9 (339bp). The predicted KMT2A exon 6-YAP1 exon 9 fusion transcript was identical to the reciprocal fusion transcripts found in cases #1 and #3 by RNA sequencing.
YAP1 and KMT2A rearrangements appear cryptic, not commonly detectable at the FISH resolution
FISH for YAP1 and KMT2A abnormalities were performed on the above-mentioned 5 cases with known YAP1 and KMT2A fusion sequences (cases #1–3, 8, 9). The presence of an YAP1 break-apart was observed only in case #3 (Fig. 2B), which showed one fused normal signal and one abnormal signal (single green, telomeric probe). In other cases, no YAP1 break-apart was detected by FISH, though cases #1, 2 and 9 had only 1 normal copy of YAP1. FISH for KMT2A rearrangements were negative in all 5 cases. Fusion FISH assay using telomeric probe of YAP1 (green) and centromeric probe of KMT2A was performed on case #3, but was negative for gene fusion. These findings suggest that YAP1 and KMT2A gene rearrangements in these tumors are often cryptic and beyond the FISH resolution. YAP1 and KMT2A genes are both located on chromosome 11q with the same direction of transcription, with a 16 Mb distance between these 2 genes. Based on the results of case# 3, it is likely that only a small fragment of the YAP1 locus is retained in the fusion event, being associated with a large deletion of the nearby locus (as demonstrated by the loss of the red, centromeric probe). Therefore, FISH studies may not represent the appropriate tool for screening, being associated with a high false-negative rate in these tumors.
Alternative fusion partners may occur in YAP1 or KMT2A-rearranged SEF
Four additional cases with either YAP1 or KMT2A gene abnormalities were identified by FISH in the remaining SEF screening cohort, 1 with YAP1 rearrangement and 3 with KMT2A rearrangements (Fig. 2C, Table 1). None of them showed concurrent YAP1 and KMT2A rearrangements by FISH. Fusion FISH assays of YAP1-KMT2A also showed negative results (cases #5–7). Case #4 showed a KMT2A gene rearrangement and only one normal copy of YAP1. To investigate other potential fusion gene partners, FISH for VIM and MAML2 gene rearrangements, two recently described fusion partners of YAP1 or KMT2A gene7–14, were performed on cases #4–7. Case #5 (KMT2A-rearranged) showed an additional MAML2 break-apart; a gene also located in chromosome 11q. Although FISH fusion assay using probes flanking MAML2 and centromeric to KMT2A did not show a fused signal between MAML2 and KMT2A, the possibility of a KMT2A-MAML2 fusion cannot be excluded considering the complex abnormalities and high false negative rates of FISH method in the index cases. Cases #4, 6 and 7 were negative for MAML2 rearrangements, and all 4 cases tested lacked abnormalities in VIM gene by FISH.
YAP1 and/or KMT2A-rearranged SEFs show infiltrative borders and a deceptively hypocellular morphology
In total, 9 cases, including the 3 index cases and 6 additional from the screening cohort, were identified as harboring rearrangements in YAP1 and/or KMT2A genes (Table 1). The patients ranged from 10–73 years old (average & median: 45), with 6 (67%) patients diagnosed above the age of 40 years, in both genders (5 male and 4 female patients). The tumors involved mainly somatic soft tissues of the trunk and lower extremities, either superficial or deep-seated, including thigh, lower leg, inguinal area, iliopsoas muscle, paraspinal, and chest wall. Two tumors affected the head and neck region. One of them (case #6) presented as a brain tumor and involved frontal lobe dura, with secondary bone erosion, in a 10-year-old girl. The other was a large, unresectable cervical neck/paraspinal soft tissue mass of a 22-year-old (case #9) involving the brachial plexus. Tumor sizes ranged from 2.0 to 12.0 cm.
The morphology of the primary tumors was reviewed in all except 2 cases (#2 and #3), for which material was only available from the metastatic tumor and 3 recurrent tumors, respectively. Histologically, at least half of the cases showed infiltrative borders (5/9; 56%). Three tumors entrapped adipocytes from adjacent subcutaneous tissue (cases #4, 5 and 8) (Fig. 3A). Skeletal muscle infiltration was seen in case #1 and in the second recurrence of case #3 (Fig. 3B). Most tumors showed zonal variation in cellularity, ranging from low, intermediate, to high (Fig. 3C). The more cellular areas were often observed at the periphery (n=4), but occasionally were also noted intermixed with less cellular areas in the tumor (n=2) or as scattered small cellular foci in the center (n=1). The tumor cells ranged from rounded, ovoid, to epithelioid, with monotonous nuclei, fine chromatin, and small nucleoli (Fig. 3D–F). Short spindle cells arranged in a storiform growth were also seen in some cases as a focal pattern (Fig. 4A). Case #6 in addition showed more elongated spindle cells arranged in fascicles (Fig. 4B). Mitotic activity ranged widely, from 0 to 21 per 10 high power fields. The background stroma was hyalinized and sclerotic in all cases, including homogeneous fibrous background, thick collagenous bundles, and delicate dense collagen fibers enveloping individual tumor cells resembling osteoid matrix (Fig. 4C–E). Thin-walled dilated vessels were present in the tumor in about half of the cases (Fig. 3F, 4F). Intravascular tumor protrusion was seen in one case. Deceptively hypocellular fibroma-like areas composed of bland small ovoid cells in densely collagenous stroma were present in 5 cases and led to the initial diagnoses of benign lesions, including fibrous lesion, benign fibrous histiocytoma, and desmoid tumor (cases #1–3) (Fig. 4C, 4F). None of them showed areas of LGFMS morphology, lacking the typical alternating fibrous and myxoid components, with delicate spindle cells arranged in a swirling pattern. Recurrent tumors showed increased cellularity (case #1, third recurrence of case #3) (Fig. 4A), and osseous metaplasia was observed in the 3rd recurrence of case #3 (21 years after initial diagnosis).
Figure 3.
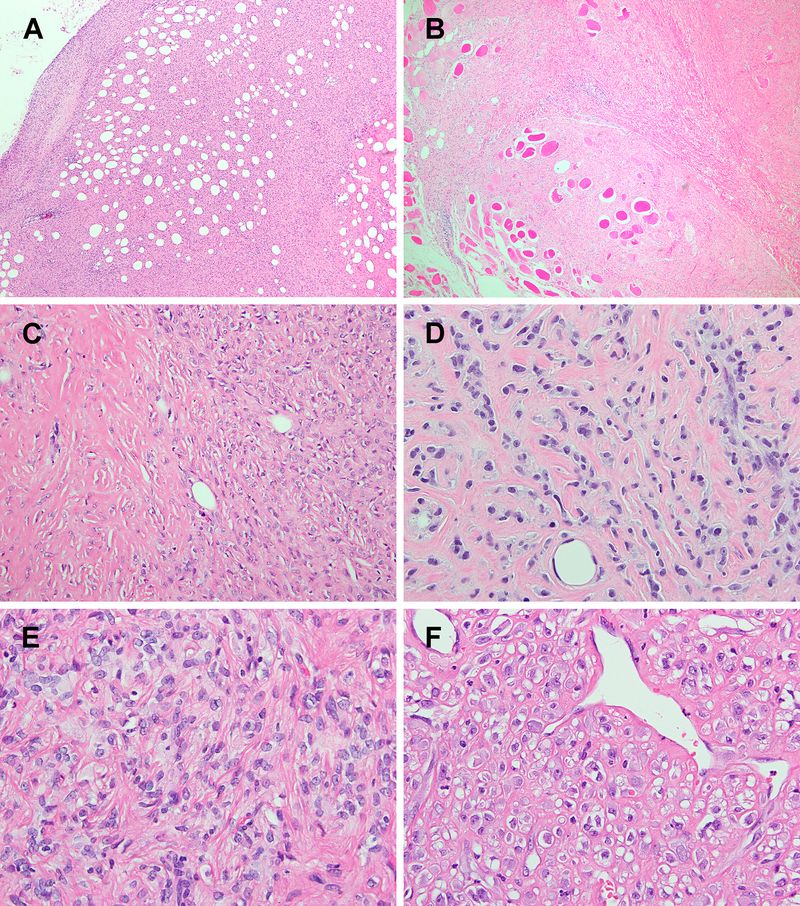
The tumors often show infiltrative borders within fat (A) or skeletal muscle (B). (C) zonal variation in cellularity is a striking feature, ranging from bland-looking hypocellular regions with marked stromal sclerosis (left) to back-to-back cellularity (right). (D-F) The tumor cytology ranged from ovoid to small epithelioid cells, arranged in a cord-like pattern, frequently admixed with thick collagen fibers, morphologically consistent with SEF. The tumor cells have uniform nuclei and fine chromatin, sometimes with small distinct nucleoli. Dilated thin-walled vessels are also occasionally observed (F).
Figure 4.
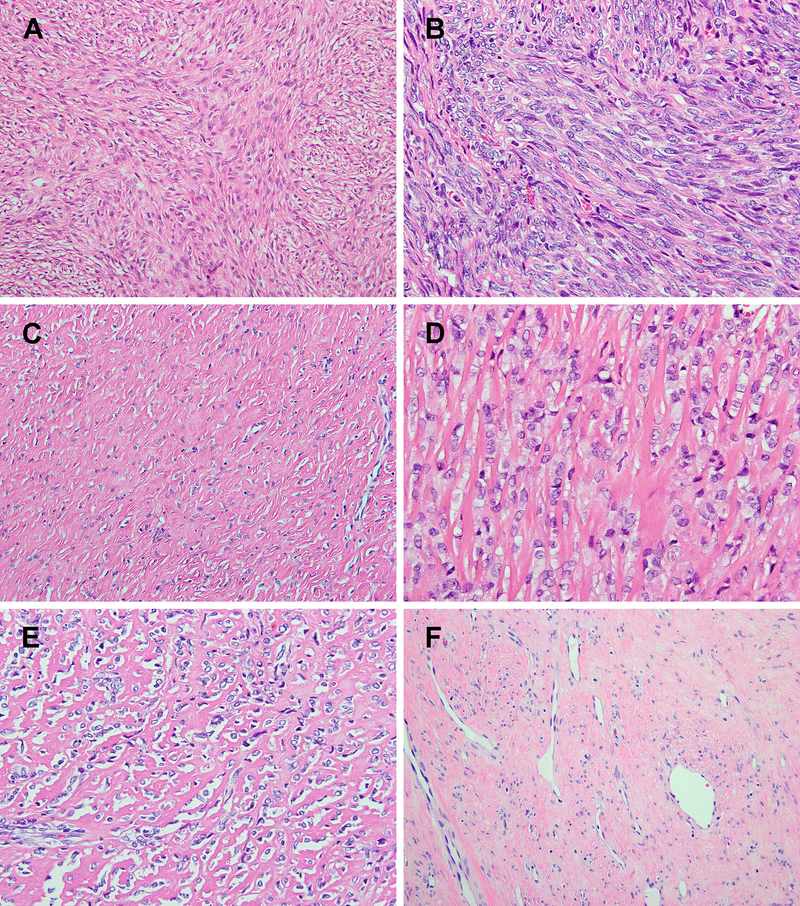
Storiform and fascicular pattern of more elongated spindle cells was focally noted in some cases (A & B). All cases displayed large areas of sclerotic stroma, including a homogeneous fibrotic stroma (C), thick collagen fibers (D), and confluent collagen deposition resembling osteoid matrix (E). Deceptively hypocellular, ‘fibroma’-like areas were observed in most cases (C & F), and may lead to an erroneous diagnosis of benign lesions.
All tumors were negative for MUC4 immunostaining. Other immunostains were negative or non-specific. S100 stain showed patchy staining in about half of the cases tested (3/7), but they were negative for SOX10 in 5 cases tested. Multifocal weak EMA, focal CD34, ERG, and focal desmin expressions were seen in the 5/7, 3/7, 1/3, and 1/7 cases tested, respectively. Variable intensity of BCOR (4/4; weak in 2, moderate in 1, strong in 1) and strong cyclin D1 (2/2) staining were also seen. CD31 (n=3), beta-catenin (n=3), and SMA (n=5) stains were negative.
Of the 6 cases with available follow-up information, local recurrence and distant metastasis were observed in 2 patients each. Case #1 underwent a piecemeal excision of the paraspinal tumor and had local recurrence 1 year later. The recurrent tumor was treated with neoadjuvant radiotherapy and resection. The recurrent specimen showed increased cellularity compared to the primary tumor. No further follow-up was available after the resection. Case #2 had a subcutaneous thigh mass which was excised with free margins and developed soft tissue metastasis 1.5 years later. Case #3 presented as a 2.0 cm intramuscular tumor of the lower leg which was treated with surgery alone and subsequently developed multiple late recurrences 10, 17, and 21 years after the diagnosis. No metastasis was observed. Case #4, a deep-seated tumor involving the iliopsoas muscle and adjacent bone in a 73-year-old, developed bone and soft tissue metastases to the contralateral pelvis and scalp and died of the disease 1 year after diagnosis. Case #6, a dural-based tumor of a 10-year-old, showed no evidence of disease 2.5 years after the surgery. Lastly, case#9 was unresectable and the patient received chemotherapy with some decrease in tumor size, and is alive with disease 1 year later.
MUC4-negative low-grade fibromyxoid sarcomas lacked YAP1 or KMT2A gene alterations
Considering the overlapping clinicopathologic and genetic characters of SEF and LGFMS, a screening cohort of 24 MUC4-negative fibromyxoid spindle cell tumors, diagnosed or suspected as fusion-negative LGFMS, were also examined by FISH for YAP1 and KMT2A abnormalities. All these cases showed no evidence of YAP1 and KMT2A gene rearrangements. However, due to high false negative rate by FISH in detecting this fusion, further studies using other methodologies are needed to clarify if this genetic group is truly only observed in SEF but not in LGFMS cases, unlike the EWSR1/FUS gene rearrangements.
DISCUSSION
Sclerosing epithelioid fibrosarcoma is a rare soft tissue tumor exhibiting a characteristic morphologic pattern of epithelioid cells arranged in strands, cords, nests and sheets embedded in a sclerosed and hyalinized stroma.1,2 A subset of SEF shares morphologic overlap with low grade fibromyxoid sarcoma, that features a deceptively benign histology of bland appearing spindle cells in an alternating fibrous and myxoid stroma.15,16. SEF tends to have an aggressive clinical course with high rates of local recurrence (50%), distant metastasis (40–80%) and mortality (25–57%).1,2 Although LGFMS have been initially described as following a protracted clinical course with local recurrences and late metastasis,15,16 in a more recent comprehensive study of 33 LGFMS cases with long-term follow-up (mean of 14 years), half of the patients developed metastases and 42% died of disease.17
MUC4 was found to be among the top upregulated genes in LGFMS and thus detectable at the protein level;18 with subsequent comprehensive studies demonstrating that MUC4 immunohistochemistry represents a sensitive and specific marker for this entity.3 Expression of MUC4 was also demonstrated in the majority but not all SEF cases (up to 78%),4 suggesting a certain heterogeneity in this group of tumors. Moreover, in our prior study, only one EWSR1-rearranged pure SEF was negative for MUC4 staining, while all the SEF/LGFMS hybrid tumors tested expressed MUC4.6 Additionally, a FUS-CREB3L2 translocation positive, MUC4-negative LGFMS was also recently published.19
At the genetic level, most pure SEF cases showed EWSR1-CREB3L1 fusions, while the overwhelming majority of hybrid SEF/LGFMS lesions are characterized by FUS-CREB3L2.5,6 With few exceptions,6,19 EWSR1 and FUS-rearranged SEF cases are often MUC4 positive,4,6 while the molecular pathogenesis of MUC4-negative SEF remains unknown. In this study, we demonstrated that at least half (9/17, including index cases and screening cohort) of MUC4-negative SEF harbors YAP1 and/or KMT2A gene rearrangements.
YAP1-KMT2A fusions have been reported in rare sarcoma cases.7,20 A chest wall tumor diagnosed as ‘sclerosing fibrosarcoma’ in a 35-year-old female was found to have YAP1-KMT2A fusion by RNA sequencing in a large-scale study focused on small round cell sarcomas.20 Although the histomorphology and immunoprofile of this case are not available, it is likely that it also falls into the morphologic spectrum of SEF as our cases. More recently, two sarcoma cases with KMT2A rearrangements, fused with YAP1 and VIM, respectively, were identified by RNA sequencing.7 The first case harboring a YAP1-KMT2A fusion presented in a 20-year-old female with a deep thigh tumor (8.5 cm) showing similar histomorphology to our cases, with infiltrative borders, epithelioid to round cells embedded in a sclerotic stroma, and focal storiform growth of short spindle cells. Immunohistochemically, this case showed focal EMA, CD31, CD34, and ERG staining, and was negative for MUC4. Some of these markers were also focally positive in some of our cases, however none of our cases showed CD31 positivity (tested in 3 cases). Similar to our cases (#1, 3, and 8), the exon composition of their case included a YAP1 exon 5- KMT2A exon 4 fusion transcript. Although the reciprocal KMT2A-YAP1 fusion transcript was present in the majority of these tumors reported to date and the number of reciprocal fusion reads exceeded YAP1-KMT2A reads in some of our cases, the immunoreactivity to the N-terminal YAP1 but not C-terminal YAP1 is in keeping with a YAP1-KMT2A being the main functional transcript.7
Their second case, harboring a VIM-KMT2A fusion, presented in a 30 year-old woman with a femoral bone lesion,7 displayed a somewhat distinct morphology from our cases. It showed a hypercellular round to spindle cell morphology, with a sclerotic stroma being more prominent in the resection specimen after chemotherapy. We examined our KMT2A rearranged cases with unknown fusion partners for VIM gene abnormalities by FISH, but did not identify any positive cases. In addition to focal EMA positivity, the VIM-KMT2A case also showed immunoreactivity to BCOR (diffuse strong), WT1, and NKX2.2. BCOR staining was also observed in all 4 cases tested in our study, but more commonly with a non-specific weak to moderate intensity. By RNA sequencing data, our cases #1–3 did not show significant up-regulation of WT1 and NKX2.2. Both of their patients developed pulmonary and/or bone metastases and died of disease.7
The present study expands the clinicopathologic spectrum of sarcomas harboring YAP1-KMT2A fusions. Our patients ranged from 10 to 86 years old. In addition to chest wall and lower extremities, some cases involved the paraspinal soft tissue, neck, and dura. Some of our patients presented with large and deep-seated tumors, like most SEF with EWSR1/FUS fusions and the previously reported YAP1-KMT2A positive sarcomas; while others were smaller in size and involved superficial subcutaneous tissues only. Our results also showed a more variable clinical course, with some patients developing early metastases and others having prolonged multiple recurrence or being disease free. Further larger studies are needed to better determine their biologic potential and prognostic significance of different fusion genotypes in SEF.
Our study also pointed out that this intra-chromosomal 11 translocation resulting in a YAP1-KMT2A fusion is likely complex and unbalanced, rather than a simple translocation or interstitial deletion, as it is often not detected by either break-apart or fusion FISH assays. YAP1 (11q22.1–22.2) and KMT2A (11q23.3) are both located on the long arm of chromosome 11, 16 Mb apart, with the same direction of transcription. These findings may explain the high false-negative rate of the FISH assays, as none of our 3 index cases with YAP1-KMT2A fusions by RNA sequencing revealed KMT2A rearrangements and only one case showed YAP1 split signals. Therefore, the prevalence of this fusion among MUC4-negative SEF is expected to be higher.
Awareness of the existence of MUC4-negative SEF and its potential aggressive behavior is crucial. Some of the tumors in our cohort showed deceptively bland hypocellular fibrous morphology, similar to the previously described fibroma-like area in SEF,2 and only focally the more cellular areas with round to epithelioid tumor cells in a sclerotic stroma, and therefore could be misdiagnosed as benign tumors or tumors with intermediate behavior, such as fibrous histiocytoma, desmoid tumor, solitary fibrous tumor, etc. In addition, although hypocellular areas were observed in 5 of 9 cases, none of them showed fibromyxoid stroma typical of LGFMS.
KMT2A (MLL), a transcriptional coactivator with H3K4 methyltransferase activity, is a well-known gene involved in genetic rearrangements in leukemias, including infantile, pediatric, adult and therapy-induced leukemias, and are associated with poor outcome.21 In KMT2A-rearranged leukemia, KMT2A is the 5’ fusion partner gene and most commonly fused to AF4, AF9, AF10, and ENL, among many other fusion partners. The most prevalent breakpoints of KMT2A in KMT2A-rearranged leukemia are between exon 9 and intron 11, resulting in retained N-terminal DNA binding domains but losing the C-terminal SET domain with H3K4 methyltransferase activity in the chimeric protein.21 In SEF cases, KMT2A is presumably the 3’ fusion partner gene and the most common breakpoints were observed in exons 4–5, resulting in a partially disrupted or totally lost DNA binding domain and intact SET domain. In our index cases, RNA sequencing showed no significant up-regulation of YAP1 or KMT2A, while the KMT2A expression levels were even slightly lower than control (data not shown). We also did not observe up-regulation of the target genes of KMT2A, such as HOXA9 and MEIS, which are up-regulated in KMT2A-rearranged leukemia.22 Nevertheless, in the previously reported sarcoma with VIM-KMT2A fusion, up-regulation of KMT2A and HOXA genes were noted.7 The pathogenic mechanism of KMT2A-rearranged sarcoma remains to be explored.
One of our cases (case #5) showed rearrangements of both KMT2A and MAML2 genes by FISH. MAML2 (11q21) is also located on chromosome 11, centromeric to YAP1 and KMT2A and in the opposite transcriptional direction. It has been reported as rare fusion partners of KMT2A in leukemias.8–10 Since 3 of our cases had only KMT2A or YAP1 rearrangements by FISH, other alternative fusion partner genes likely exist.
Our case #6 stood out in this study cohort with regard to the particularly young age of 10 years, the unusual dura location, and the round to elongated spindle cell morphology. Areas more typical of SEF with dense sclerotic collagen fibers enveloping ovoid to rounded tumor cells were present and alternating with the fascicular spindle cell areas. Interestingly, a dura-based spindle cell tumor with MN1-KMT2A fusion has been reported recently in a 22-year-old female.23 However, the morphology of that case is composed of more plump spindle cells arranged in hypercellular sheets and fascicles, lacking the sclerotic stroma as identified in our case, and expresses diffuse and strong S-100 and PR staining. FISH for MN1 gene rearrangement was negative in our case #6, as was S100 protein staining.
YAP1, a downstream effector of the Hippo signaling pathway, is also known to be involved in recurrent gene fusions in a subset of epithelioid hemangioendothelioma (YAP1-TFE3) and ependymoma (YAP1- MAMLD1 and FAM118B).24,25 In YAP1-KMT2A fusions, the N-terminal TEAD binding domain of YAP1 is retained, while the C-terminal transcriptional activation domain is lost. Interestingly, YAP1-MAML2 fusions through chromosomal inversions have also been described in poroma (88.5%), porocarcinoma (63.6%), and rare cases of various carcinoma types.11–14 Our case #7 also showed an inversion pattern of YAP1 rearrangement by FISH, but was negative for MAML2 rearrangements by FISH.
In summary, we report a group of MUC4-negative SEF with YAP1 and/or KMT2A gene rearrangements. While many of the clinicopathologic features overlap with MUC4-positive SEF, these tumors in particular can present as superficial and small tumors with infiltrative borders and could be deceptively fibrotic and hypocellular, mimicking benign lesions. Due to the lack of specific immunohistochemical markers at this point and the high false negative rate by FISH, a high index of suspicion and incorporating different diagnostic methodology, including next generation sequencing, are needed to avoid misdiagnosis.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Chung-Hsi Wang, Dr. Cheng-Hsiang Hsiao, and Dr. Chin-Cheng Lee for contribution of cases and providing the follow-up information.
Disclosures: Supported in part by: P50 CA 140146-01 (CRA), P50 CA217694 (CRA), P30 CA008748, Cycle for Survival (CRA), Kristin Ann Carr Foundation (CRA), St Baldrick Foundation (CRA)
REFERENCES
- 1.Meis-Kindblom JM, Kindblom LG, Enzinger FM. Sclerosing epithelioid fibrosarcoma. A variant of fibrosarcoma simulating carcinoma. Am J Surg Pathol. 1995;19:979–993. [DOI] [PubMed] [Google Scholar]
- 2.Antonescu CR, Rosenblum MK, Pereira P, et al. Sclerosing epithelioid fibrosarcoma: a study of 16 cases and confirmation of a clinicopathologically distinct tumor. Am J Surg Pathol. 2001;25:699–709. [DOI] [PubMed] [Google Scholar]
- 3.Doyle LA, Moller E, Dal Cin P, et al. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am J Surg Pathol. 2011;35:733–741. [DOI] [PubMed] [Google Scholar]
- 4.Doyle LA, Wang WL, Dal Cin P, et al. MUC4 is a sensitive and extremely useful marker for sclerosing epithelioid fibrosarcoma: association with FUS gene rearrangement. Am J Surg Pathol. 2012;36:1444–1451. [DOI] [PubMed] [Google Scholar]
- 5.Arbajian E, Puls F, Magnusson L, et al. Recurrent EWSR1-CREB3L1 gene fusions in sclerosing epithelioid fibrosarcoma. Am J Surg Pathol. 2014;38:801–808. [DOI] [PubMed] [Google Scholar]
- 6.Prieto-Granada C, Zhang L, Chen HW, et al. A genetic dichotomy between pure sclerosing epithelioid fibrosarcoma (SEF) and hybrid SEF/low-grade fibromyxoid sarcoma: a pathologic and molecular study of 18 cases. Genes Chromosomes Cancer. 2015;54:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida A, Arai Y, Tanzawa Y, et al. KMT2A (MLL) fusions in aggressive sarcomas in young adults. Histopathology. 2019. [DOI] [PubMed] [Google Scholar]
- 8.Menu E, Beaufils N, Usseglio F, et al. First case of B ALL with KMT2A-MAML2 rearrangement: a case report. BMC Cancer. 2017;17:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metzler M, Staege MS, Harder L, et al. Inv(11)(q21q23) fuses MLL to the Notch co-activator mastermind-like 2 in secondary T-cell acute lymphoblastic leukemia. Leukemia. 2008;22:1807–1811. [DOI] [PubMed] [Google Scholar]
- 10.Nemoto N, Suzukawa K, Shimizu S, et al. Identification of a novel fusion gene MLL-MAML2 in secondary acute myelogenous leukemia and myelodysplastic syndrome with inv(11)(q21q23). Genes Chromosomes Cancer. 2007;46:813–819. [DOI] [PubMed] [Google Scholar]
- 11.Sekine S, Kiyono T, Ryo E, et al. Recurrent YAP1-MAML2 and YAP1-NUTM1 fusions in poroma and porocarcinoma. J Clin Invest. 2019;130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valouev A, Weng Z, Sweeney RT, et al. Discovery of recurrent structural variants in nasopharyngeal carcinoma. Genome Res. 2014;24:300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papp E, Hallberg D, Konecny GE, et al. Integrated Genomic, Epigenomic, and Expression Analyses of Ovarian Cancer Cell Lines. Cell Rep. 2018;25:2617–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Picco G, Chen ED, Alonso LG, et al. Functional linkage of gene fusions to cancer cell fitness assessed by pharmacological and CRISPR-Cas9 screening. Nat Commun. 2019;10:2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans HL. Low-grade fibromyxoid sarcoma. A report of two metastasizing neoplasms having a deceptively benign appearance. Am J Clin Pathol. 1987;88:615–619. [DOI] [PubMed] [Google Scholar]
- 16.Evans HL. Low-grade fibromyxoid sarcoma. A report of 12 cases. Am J Surg Pathol. 1993;17:595–600. [DOI] [PubMed] [Google Scholar]
- 17.Evans HL. Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol. 2011;35:1450–1462. [DOI] [PubMed] [Google Scholar]
- 18.Moller E, Hornick JL, Magnusson L, et al. FUS-CREB3L2/L1-positive sarcomas show a specific gene expression profile with upregulation of CD24 and FOXL1. Clin Cancer Res. 2011;17:2646–2656. [DOI] [PubMed] [Google Scholar]
- 19.Linos K, Bridge JA, Edgar MA. MUC 4-negative FUS-CREB3L2 rearranged low-grade fibromyxoid sarcoma. Histopathology. 2014;65:722–724. [DOI] [PubMed] [Google Scholar]
- 20.Watson S, Perrin V, Guillemot D, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol. 2018;245:29–40. [DOI] [PubMed] [Google Scholar]
- 21.Meyer C, Burmeister T, Groger D, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawrence HJ, Rozenfeld S, Cruz C, et al. Frequent co-expression of the HOXA9 and MEIS1 homeobox genes in human myeloid leukemias. Leukemia. 1999;13:1993–1999. [DOI] [PubMed] [Google Scholar]
- 23.Chen S, Dickson BC, Mohammed S, et al. A dural-based spindle cell neoplasm characterized by a novel MN1-KMT2A fusion gene. Neuro Oncol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antonescu CR, Le Loarer F, Mosquera JM, et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer. 2013;52:775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malgulwar PB, Nambirajan A, Pathak P, et al. C11orf95-RELA fusions and upregulated NF-KB signalling characterise a subset of aggressive supratentorial ependymomas that express L1CAM and nestin. J Neurooncol. 2018;138:29–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.