

Abstract
Objective:
The purpose of this study was to evaluate a new synthesized multifunctional monomer, aminosilane functionalized methacrylate (ASMA), containing polymerizable methacrylate, tertiary amine, and methoxysilane functionalities in dental adhesive formulations, and to investigate the polymerization kinetics, leachates, thermal and mechanical properties of copolymers.
Methods:
Adhesive contained HEMA/BisGMA (45/55, w/w) was used as a control, and mixtures based on HEMA/BisGMA/ASMA at the mass ratio of 45/(55-x)/x were used as experimental adhesive. Adhesives were characterized with regard to water miscibility, photo-polymerization behavior (Fourier transform infrared spectroscopy, FTIR), leached co-monomers (high performance liquid chromatography, HPLC), thermal properties (modulated differential scanning calorimeter, MDSC), and mechanical properties (dynamic mechanical analyzer, DMA). Stress relaxation times and the corresponding moduli, obtained from stress relaxation tests, are used in a simulated linear loading case.
Results:
As compared to the control, ASMA-containing adhesives showed higher water miscibility, lower viscosity, improved monomer-to-polymer conversion, significantly greater Tg and rubbery modulus. HPLC results indicated a substantial reduction of leached HEMA (up to 85 wt%) and BisGMA (up to 55 wt%) in ethanol. The simulation reveals that the ASMA-containing adhesive becomes substantially stiffer than the control.
Significance:
ASMA monomer plays multiple roles, i.e. it serves as both a co-initiator and crosslinker while also providing autonomous strengthening and enhanced hydrolytic stability in the adhesive formulations. This multifunctional monomer offers significant promise for improving the durability of the adhesive at the composite/tooth interface.
Keywords: Co-initiator, dental adhesive, autonomous strengthening, multifunctional monomer, mechanical property, modulated DSC, hydrolytic stability, Prony series, stress relaxation
Graphical abstract
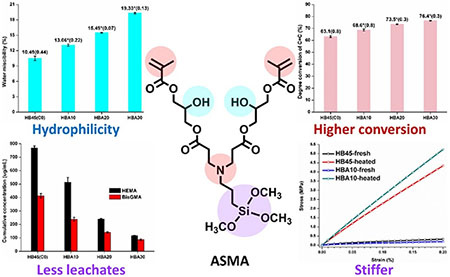
1. INTRODUCTION
Repeated dental-restoration replacement risks pulpal injury, increased tooth weakness, and eventually, tooth loss. The shift from amalgam to dental composite as the most popular material for the repair of lost or damaged tooth structure has increased these risks. Composite restorations fail at 2 to 3.5 times the rate of dental amalgam [1–4] and the average lifespan of composite restorations is about half the lifespan of amalgam restorations [5]. High caries-risk patients are particularly vulnerable to composite-restoration failure and the downward spiral associated with frequent replacements [3, 6].
The primary reason for replacing composite restorations is recurrent decay at the margins of existing restorations [7–9]. Unlike amalgam, composite lacks the inherent capability to seal gaps that develop at the interface between the restorative material and tooth structure. This interface is initially sealed by a low-viscosity adhesive but the adhesive seal to dentin is fragile, i.e. it is readily damaged by acids, enzymes and oral fluids [10–12]. Bacteria and bacterial by-products infiltrate the resulting marginal gaps. Bacterial acids decompose the tooth while bacterial enzymes erode the adhesive, leading to wider and deeper gaps [5] that provide an ideal environment for bacteria to proliferate.
Our research group has used complementary and synergistic approaches to address the issues associated with dental adhesive failure. We have designed, synthesized and systematically investigated various functional monomers, [13–20] e.g., amine co-monomers were explored as an approach for developing adhesives capable of buffering the microenvironment at the adhesive/dentin (a/d) interface [19, 21–23]. We developed alkoxysilane-containing adhesives that capitalize on free-radical polymerization and sol-gel reactions to provide self-strengthening polymers, i.e. polymers that achieve persistent, intrinsic network reinforcement [24–27].
While the beneficial effect of the self-strengthening strategy has been confirmed [24, 25, 27], our previous studies devoted limited attention to amphiphilicity. With wet bonding techniques, adhesive infiltration of the etched dentin (demineralized dentin substrate) was primarily determined by the amphiphilicity of adhesive monomers [28–31]. Inadequate adhesive infiltration could compromise both the integrity and durability of the a/d interface [28, 31–34].
The alkoxysilane monomers used in the self-strengthening adhesives [24, 25, 27] are relatively hydrophobic which could impede resin infiltration. To address this potential limitation, a balanced amphiphilic aminosilane functionalized methacrylate (ASMA) was designed and synthesized through aza-Michael addition. The monomer was introduced into dental adhesive formulations and properties such as, water miscibility, photopolymerization, monomer leaching, and viscoelastic behavior were investigated. The overall hypotheses of this investigation are: i) ASMA can act as a co-initator to replace ethyl-4-(dimethylamino) benzoate, EDMAB, ii) ASMA can act as a co-monomer to partially replace BisGMA and iii) the crosslinking density of the post-processed ASMA-containing polymer is significantly greater than the post-processed control.
2. MATERIALS & METHODS
2.1. Materials
2,2-Bis[4-(2-hydroxy-3-methacryloxypropoxy) phenyl]propane (BisGMA), 2-hydroxyethyl methacrylate (HEMA), 3-(acryloyloxy)-2-hydroxypropyl methacrylate (AHPMA), camphoroquinone (CQ), ethyl-4-(dimethylamino) benzoate (EDMAB), and diphenyliodonium hexafluorophosphate (DPIHP) were obtained from Sigma-Aldrich (St. Louis, MO) and used as received without further purification. 3-aminopropyl trimethoxysilane (APTMS, >95%) was purchased from Gelest Inc. (Morrisville, PA). Aminosilane functionalized methacrylate (ASMA) was synthesized in our laboratory and used as co-monomer. All the other chemicals were used as received without further purification.
2.2. Synthesis of ASMA
APTMS was reacted with stoichiometric amounts of AHPMA in dry methanol at 23±2 °C for 2 days. Briefly, to a 250-mL, round bottom flask, fitted with a magnetic stirrer and N2-purging, AHPMA (8.57 g, 0.04 mol) and methanol (50 mL) were added. Then, APTMS (3.59g, 0.02 mol) dissolved in methanol was added stepwise over 3 h. After the reaction was completed, the solvent, methanol, was removed by rotary evaporation under vacuum. As shown in Figure 1, the structure of the residual colorless liquid was confirmed using FTIR and 1H NMR spectroscopies (Bruker AVIIIHD 400 MHz Spectrometer, CDCl3 as solvent). The yield for ASMA synthesis was 97%.
Figure 1.

Reaction scheme, FTIR and 1H NMR spectra for the synthesized ASMA.
2.3. Preparation of adhesive formulations
Neat methacrylate resin made by mixing 45 wt% HEMA and 55 wt% BisGMA was used as the control adhesive (HB45) [35]. In the HB45 formulation, three components, e.g., CQ (0.5 wt%), EDMAB (0.5 wt%), and DPIHP (0.5 wt%) were used as the photoinitiator (PI) system [16, 36, 37]. For the experimental formulation containing ASMA, two components, e.g., CQ (0.5 wt%) and DPIHP (0.5 wt%) were used as the PI system. The monomer compositions of the control and experimental formulations are listed in Table 1. Mixtures of the monomers/PI were prepared in brown glass vials under amber light and thoroughly mixed for 24 h at 23±2 °C to promote complete dissolution and formation of a homogeneous resin.
Table 1.
Results of Water Miscibility and Polymerization Kinetics of Adhesive Formulations
| Run | HEMA (wt%) | BisGMA (wt%) | ASMA (wt%) | Wwm (wt%) | Viscosity (mPa·s) | DCa (%) | (1/s) |
|---|---|---|---|---|---|---|---|
| HB45 | 45 | 55 | / | 10.5 (0.4) | 171.0 (0.4) | 63.1 (0.8) | 18.5 (0.3) |
| HBA10 | 45 | 45 | 10 | 13.1*(0.2) | 134.8*(0.6) | 68.6*(0.8) | 20.3 (0.4) |
| HBA20 | 45 | 35 | 20 | 15.5*(0.1) | 103.9*(0.4) | 73.5*(0.3) | 17.7(1.2) |
| HBA30 | 45 | 25 | 30 | 19.3*(0.1) | 80.6* (0.2) | 76.4*(0.3) | 15.2*(1.3) |
degree of conversion of C=C double bond.
Significantly (p<0.05) different from the control (HB45). The value in the () is the standard deviation.
2.4. Water miscibility of adhesive formulations
Water miscibility of the adhesive formulations was determined according to the published protocol [15, 38]. In brief, ~0.5 g of each neat resin was weighed into a brown vial with water added in increments of approximately 0.005 g until the mixture was visually observed to be turbid. The percentage of water in the mixture was noted (w1). The mixture was then back-titrated using the neat resin until the turbidity disappeared, and the percentage of water in the mixture was noted (w2). The water miscibility (Wwm, %) of the liquid formulation was calculated as the average of w1 and w2. Three measurements of each formulation were recorded.
2.5. Viscosity measurement
The viscosity of the adhesive resins was measured using a Brookfield DV-II +Pro viscometer with a cone/plate set up (40 mm diameter and 2 °cone angle) at varying shear rate 37.5 s−1 to 195 s−1, with a step ramp 22.5 s−1. The measurements were made at 25.0 ± 0.2 °C and the sample volume was 0.5 mL. At each shear rate, shear was applied for 10 s before the viscosity measurement and eight viscosity measurements over the shear rate range were averaged.
2.6. Real-time C=C bond conversion and maximum polymerization rate
The degree of conversion (DC) of the reactive C=C bond and polymerization kinetics were determined by FTIR as reported [38], One drop of adhesive resin (5 μL) was placed on the crystal top plate of an attenuated total reflectance (ATR) accessory (PIKE Technologies Gladi-ATR, Madison, WI) and covered with a mylar film. After 30 IR spectra were collected, the adhesives were light-cured for 40 s at 23±2 °C using a commercial visible light lamp (Spectrum® 800, Dentsply, Milford, DE. Intensity is 550 mW/cm2). Real-time (in-situ) monitoring of the photopolymerization behavior of the adhesive formulations was performed using an infrared spectrometer (Spectrum 400, Perkin-Elmer, Waltham, MA) at a resolution of 4 cm−1. A time-based spectrum collector (Spectrum TimeBase, Perkin-Elmer) was used for continuous and automatic collection of spectra during polymerization. A minimum of three measurements were carried out for each adhesive formulation. Double bond conversion was monitored by the band ratio profile-1637 cm−1 (C=C)/1608 cm−1 (phenyl). The average of the last 30 values of the time-based spectra was reported as the DC value. The maximum polymerization rate () was determined using the maximum slope of the linear region of the DC vs. time plot [17, 39].
2.7. Preparation of polymer specimens
The adhesive specimens were prepared for mechanical and thermal property characterization according to published protocols [16]. The prepared resins were injected into a round glass-tubing mold (Wilmad LabGlass, P 1M-1.2M-0-914M, item #:1004-5014, Vineland, NJ) or a Tzero® Hermetic Lid (P/N: 900797.901 TA Instruments Waters LLC, New Castle, DE), covered with a mylar film and light-cured for 40 s at 23±2 °C with an LED light curing unit (LED Curebox, 100 mW/cm2 irradiance, Proto-tech, Portland, OR). The polymerized samples were stored in the dark at 23±2 °C for at least 48 h before testing. The resultant round beam specimens, 1mm diameter and ~15 mm length, were used to determine dynamic mechanical properties. Pellet samples, ~4 mm diameter and ~1.2 mm thick, were used for thermal property measurement and the leachable study.
2.8. Modulated differential scanning calorimetry (MDSC) test
The thermal behavior of polymer specimens was measured with a TA instruments Q200 (New Castle, DE). The pellet specimens (~20 mg) were purged with nitrogen with flow rate at 50 mL/min and heated from −20 to 200 °C at 3 °C/min, with a modulation period of 60 s and amplitude of ±2 °C. The second scan was consistent with the first scan. The Tg values were reported as the temperature of the inflection points in the reversing heat flow curves [27].
2.9. Dynamic mechanical analysis (DMA)
The dynamic mechanical analyses of methacrylate-based dental adhesive have been described [25, 39]. DMA tests of the round beam specimens were performed using a TA instruments Q800 DMA (TA Instruments, New Castle, USA) with a three-point bending clamp. For testing in dry condition, the following testing parameters were used: displacement amplitude of 15 μm, frequency of 1 Hz, and preload force of 0.001 N [40], The temperature was ramped at the rate of 3 °C /min from 20 to 200 °C. The testing parameters of the second and third scans were kept the same as the first scan. Glass transition temperature (Tg) measured by DMA is determined as the position of the maximum peak on tan δ versus temperature plot [41].
Stress relaxation tests of polymer specimens in dry condition were also performed with the three-point bending clamp. For the stress relaxation tests, 0.2% stain was applied for 24 h on a fresh or heated sample. The following parameters were used: preload force of 0.001 N, and frequency of 1Hz, temperature of 75 °C. The following samples were used: (i) Fresh sample: polymer specimen light irradiated for 40 seconds, 5 min post-cure and tested; (ii) Heated sample: polymerized specimen post-cured for 5 min and exposed to two-cycle heating by ramping temperature from 20 to 200°C at a rate of 3 °C/min. Following 5-minutes post-processing, the fresh HBA20/30 specimens were not stiff enough for stress relaxation test using the three-point bending clamp. Therefore, only the control and HBA10 specimens were tested.
Numerically, the stress-time relationship in the relaxation test is analyzed via nonlinear least squares data fitting in MATLAB platform (Mathworks®, Natick, MA, USA) to obtain relaxation times and moduli. The fitted Prony series is used to demonstrate the stiffening behavior by simulating behavior under monotonic loading [42, 43]. The resulting fitting coefficients of Prony series are listed in Table 2. E∞ is the rubbery modulus, and Ei and τi, are the coefficients and relaxation times in the series expansion.
Table 2.
Summary of the fitted coefficients of Prony series
| HB45-fresh | HB45-heated | HBA10-fresh | HBAlO-heated | |
|---|---|---|---|---|
| E∞/MPa | 3.69E+01 | 8.31E+02 | 3.81E+01 | 1.41E+03 |
| E1/MPa | 2.29E+02 | 2.01E+02 | 1.86E+02 | 1.59E+02 |
| E2/MPa | 2.88E+02 | 4.05E+02 | 1.99E+02 | 3.22E+02 |
| E3/MPa | 7.91E+01 | 3.88E+02 | 4.92E+00 | 2.52E+02 |
| E4/MPa | 4.66E+01 | 8.97E+02 | 3.44E+01 | 8.66E+02 |
| E5/MPa | 1.00E−04 | 1.00E−04 | 8.58E−01 | 1.00E−04 |
| τ1/min | 1.01E−01 | 1.81E−01 | 1.07E−01 | 2.54E−01 |
| τ2/min | 1.06E+00 | 2.68E+00 | 1.11E+00 | 3.52E+00 |
| τ3/min | 1.00E+01 | 3.64E+01 | 1.00E+01 | 5.42E+01 |
| τ4/min | 1.31E+02 | 4.53E+02 | 1.05E+02 | 4.82E+02 |
| τ5/min | 1.01E+03 | 1.00E+03 | 1.00E+03 | 1.00E+03 |
| R2 | 0.999 | 0.999 | 0.999 | 0.999 |
2.10. Leachable study by high performance liquid chromatography (HPLC)
The polymerized pellets were submerged in 1 mL ethanol (200 proof) at 23±2 °C for up to 22 days. The storage solutions were collected at various time intervals, i.e. 1, 2, 4, 7, 10, 14 and 22 days, and quantified for concentration of leachates. Fresh ethanol was added to the pellet samples after each collection. The storage solutions were analyzed using high performance liquid chromatography (HPLC) on a system (Shimadzu® LC-2010C HT, software EZstart, version 7.4 SP2) equipped with a 250×4.6 mm column packed with 5μm C-18 silica (Luna®, Phenomenex Inc., Torrance, CA). The mobile phase was acetonitrile/water (70/30, v/v). The system was operated under the following conditions: 0.5 mL/min flow rate; detection at 208 nm; 20 μL sampling loop; 40 °C column temperature. The column was calibrated with known concentrations of the following compounds: HEMA (retention time 16.1 min), BisGMA (retention time 43.8 min), and ASMA (retention time 12.3 min), at concentrations of 5, 10, 20, 50, 100, 250 and 500 mg/L in ethanol. The HPLC chromatograms of each monomer are shown in SI Figure 2. The calibration curves with the linear fittings of BisGMA (5-100 mg/L, R2=0.99), HEMA (5-250 mg/L, R2=0.99), and ASMA (5-100 mg/L, R2=0.99) were used to calculate the concentration of these species in the extracts. The concentration was based on the intensity of the chromatographic peaks at the corresponding retention times. The HPLC analysis was performed using the extract of 5 specimens from each formulation.
2.11. Statistical analysis
The results (water miscibility, DC, rubbery moduli and glass transition temperature values obtained from DMA, and accumulative concentration of leachates determined by HPLC) were analyzed statistically using one-way analysis of variance (ANOVA) together with Tukey’s test at α = 0.05 (OriginPro Version 8.0, OriginLab Corporation, Northampton, MA) to identify significant differences in the means.
3. RESULTS
The water miscibility values and viscosities of the control (HB45) and the experimental formulations are shown in Table 1. The Wwm of the HB45 is 10.5±0.4 wt%. With the increase of ASMA content in the formulation from 10, 20, to 30 wt%, the Wwm increased significantly from 13.1±0.2, 15.5±0.1,to 19.3±0.1 wt% and the viscosities decreased from 134.8±0.6, 103.9±0.4, to 80.6±0.2 mPa·s. The viscosities of experimental formulations are significantly lower than that of the control, 171.0±0.4 mPa·s.
Figure 2 shows the real-time photopolymerization profiles of the control and experimental formulations. The experimental formulations exhibit significantly higher degree of conversion (DC) than that of the control (p<0.05). The maximum polymerization rates () of the experimental formulations were comparable to that of the control (p<0.05), with the exception of the HBA30 formulation.
Figure 2.

Real-time conversion of C=C bond (A) and maximum polymerization rates (B) of the control (HB45) and experimental formulations. (Light irradiation starts at approximately 33 seconds).
Figure 3 shows the reversing and nonreversing heat flow signals of the control and experimental polymer specimens. In the first heating cycle, two transitions are observed in the reversing heat flow signal. For the control (HB45), two transition temperatures are about 47 °C and 121 °C, respectively. For the experimental samples, with the increase in ASMA from 10, 20 to 30 wt%, the first transition temperatures are comparable at about 50 °C. However, the second transition temperatures decreased from 101 °C, 97 °C, and to 95 °C. Meanwhile, two exothermal peaks appeared in the nonreversing heat flow signal (one is about 60-65 °C and another is about 130-135 °C). For the control sample, the total peak area is 21.4 J/g. While for the experimental samples, the total peak areas are 16.1, 15.3, and 8.1 J/g, respectively. In the second heating cycle, just one transition, Tg (~122 °C), is observed in the reversing heat flow curves. With the increase of ASMA content, it is difficult to observe a clear step change due to the breadth of the transition, especially for the HBA20 and HBA30 formulations. Meantime, no exothermic peaks appeared in the nonreversing heat flow curves.
Figure 3.

Reversing heat flow (A and C) and nonreversing heat flow (B and D) versus temperature of the control and experimental polymer specimens in the first and second cycle.
Following 2-days post-processing, the HBA30 specimens could not be retrieved from the glass tubing without damage. Therefore, only the control and FIB A10/20 specimens were tested using DMA (Figure 4). With the increase in ASMA content, in the first testing cycle, the rubbery moduli of control and experimental samples are comparable (p<0.05), at about 30 MPa. However, the Tg of the samples obtained from the tan δ profiles are 124.5±0.9 °C (HB45), 113.7±0.1 °C (HBA10), and 102.6±0.7 °C (HBA20), respectively. The Tg of the experimental samples are significantly lower than that of the control (p<0.05).
Figure 4.

Representative storage modulus (Top) and tan δ (Bottom) vs. temperature curves of the control (A), HBA10 (B), and HBA20 (C) samples for three-cycle test in dry conditions.
The rubbery moduli (increasing from 29.6± 0.6 MPa to 31,0±0.6 MPa) and Tg of the control (increasing form 124.5±0.9 to 128.1±1.3 °C) showed a slight increase in the second and third testing cycle. In comparison, the HBA10/20 samples showed a significant increase in rubbery moduli and Tg in the second testing cycle. For HBA10, the rubbery modulus increased from 29.9±1.8 MPa to 43.1± 2.1 MPa, Tg increased from 113.7±0.1 °C to 135.1±0.6 °C. For HBA20, the rubbery modulus increased from 32.2±0.8 MPa to 65.1±0.1 MPa, and Tg increased from 102.6±0.7 °C to 138.2±0.5 °C. The rubbery moduli and Tg of the experimental polymers increased slightly in the third testing cycle. For the control, the intensity of tan δ increased from 0.66 (1st cycle) to 0.75 (2nd cycle). Meanwhile, the tan δ values for the experimental samples in the 2nd cycle decreased significantly. For the HBA10, the tan δ value decreased from 0.72 (1st cycle) to 0.55 (2nd cycle). For the HBA20, the value decreased from 0.72 (1st cycle) to 0.36 (2nd cycle).
Figure 5 shows the stress relaxation behavior of the control (HB45) and experimental polymers (HBA10). For the fresh sample, it took ~30 s and ~20 s for HB45 and HBA10 to relax half of the stress, respectively (Fig. 5A). However, it took much longer for the heated samples to relax half of the stress. It took ~230 min and ~1000 min for HB45 and HBA10, respectively (Fig. 5B). Furthermore, the Young’s moduli of the heated samples are significantly higher than that of the fresh (Fig. 5C). The calculated Young’s moduli (based on the slopes of the stress-strain curves in Fig. 5C) of the fresh HB45 and HBA10 are 164 and 99 MPa, respectively. While the Young’s moduli of the heated HB45 and HBA10 are 2167 and 2613 MPa, respectively, reversing the trend.
Figure 5.

Representative stress relaxation behavior and fitted Prony series profiles of the control (HB45) and experimental adhesive (HBA10) at 75 °C: (A) fresh samples, (B) heated samples, and (C) simulated stress-strain curves under monotonic loading, with strain rate at 0.02% per min.
Figure 6 shows the results of cumulative amount of released monomer from the control and experimental polymers as a function of incubation time in ethanol at 23±2 °C. With the increase in ASMA content from 0, 10, 20, and 30 wt%, the cumulative release of HEMA (after 14 days) decreased from 769, 514, 239, to 117 μg/mL, and the percentage of leached HEMA based on the total HEMA in the corresponding formulation decreased from 8.0, 5.4, 2.5, to 1.2 wt%. The cumulative release of BisGMA (after 22 days) decreased from 413, 238, 140, to 87 pg/mL, and the percentage of leached BisGMA decreased from 3.5, 2.5, 1.9, to 1.6 wt%. Meanwhile, leached ASMA was detected in experimental polymer specimens, and the cumulative release of ASMA was 191, 251, and 256 μg/mL, respectively.
Figure 6.

Cumulative monomers release from the ASMA-containing polymers as a function of incubation time in ethanol: (A) HEMA, (B) BisGMA, and (C) ASMA.
4. DISCUSSION
Traditional dental adhesives contain mainly acrylic resin monomers such as HEMA, BisGMA, and triethylene glycol dimethacrylate (TEGDMA), photoinitiators, and organic solvents [44, 45], One advantage of this methacrylate chemistry is the high degree of conversion that can be achieved within 10-20 s at room temperature using visible-light irradiation. However, the ester bonds within methacrylate-based adhesives are inherently prone to hydrolysis—the hydrolysis can be accelerated at low pH and high temperature [46, 47]. Ester bond hydrolysis leads to adhesive degradation and deterioration of the adhesive seal at the composite/tooth interface [48]. Accumulation of the esterase-catalyzed degradation by-products promotes bacterial growth and up-regulates S. mutans virulence genes and proteins [49–51]. Thus, the failed adhesive provokes a cascade of events that lead ultimately to recurrent decay and composite-restoration failure.
Numerous strategies have been proposed to enhance the stability of dental adhesives. These strategies must consider a variety of physicochemical and mechanical parameters. As an example, balanced amphiphilicity of the adhesive monomers is one of the key parameters associated with adhesive infiltration of the wet, demineralized dentin substrate. Inadequate adhesive infiltration could compromise both the integrity and durability of the adhesive seal to dentin [28–31]. Strategies leading to self-strengthening offer promise for increasing the stability of adhesives, however earlier studies devoted limited attention to amphiphilicity [24, 25, 27]. To address this oversight, a multifunctional monomer was designed and synthesized to provide self-strengthening while also partially replacing hydrophobic components, e.g. relatively hydrophobic co-initiator, EDMAB, and co-monomer, BisGMA.
The Michael-addition reaction which is one of the most important and best studied reactions in organic chemistry [52] was explored as an approach for synthesizing the multifunctional dental monomer. The conjugate addition of nitrogen nucleophiles (aza-Michael addition) has been explored for a wide range of applications including biological and synthetic products [53–55]. Here, the aminosilane (APTMS) was used to act with acrylate monomer (AHPMA) to develop a new monomer, ASMA. ASMA contains dimethacrylate group (to provide crosslinking capabilities), tertiary amine group (to serve as a co-initiator), methoxysilane group (to provide autonomous strengthening), and hydroxyl group (to provide increased hydrophilicity). The FTIR and NMR results indicated that the new monomer was successfully synthesized.
Polymerization behavior, i.e. DC and maximum polymerization rate (), is a primary concern with the replacement of the commercial tertiary amine co-initiator, EDMAB. Our previous investigation [18] has confirmed that the DC and ) are significantly lower for the control formulation, HB45, without EDMAB (see SI Figure 1). In the current investigation, the significantly higher conversion and comparable polymerization rates (except HBA30) for the experimental resin formulated without EDMAB, indicates that ASMA can act simultaneously as a co-initiator and co-monomer. Therefore, the first hypothesis is accepted.
The DC of C=C bonds of polymerized adhesive is a critical parameter in the assessment of new resins. With the increase in ASMA concentration, the viscosity and the total C=C mole concentration decreased accordingly (see Table 1 and SI Table 1), which allows the propagation to continue for longer times and the autoacceleration was postponed [56, 57], Therefore, higher DC (Fig. 2) and converted C=C mole number (SI Table 1) were achieved in the experimental resins. This result can be viewed as indirect evidence of higher crosslinked methacrylate-based matrix obtained after light irradiation. Meanwhile, a shoulder peak appeared in the rate profiles, which has been reported in the literature [56–59]. Although the relationship between photopolymerization kinetics, resin composition, and curing parameters is complicated, the apparent shoulder was mainly attributed to the limited polymerization reaction in highly crosslinked networks, which significantly retarded the mobility of reactive species. In this study, the apparent shoulder observed in HBA30 could be attributed to the following: 1) more flexible backbone as compared to BisGMA, 2) by-products of the sol-gel reaction, and 3) multi-roles of ASMA as co-initiator and co-monomer. All of these factors could enhance the mobility of reactive species and contribute to the polymerization reactions. The observed lower maximum polymerization rate of HBA30 was attributed to the lower resin viscosity and indicated that the localized increase in viscosity (associated with the gel-effect) was less than that of the control.
Modulated DSC has been used to determine the glass transition temperature of polymers [60, 61] especially complex systems such as, reacting or curing polymers [62, 63] and dental adhesives [20, 64]. As shown in Table 1, the DC of the control and experimental specimens was about 63 to 76 %, which indicated that there are unpolymerized components trapped in the crosslinked network structure. The nonreversing signals exhibited a decreased total peak area (Fig. 3B) with the increase in ASMA content, which indicated the nonreversing signal in this study was related to the thermal curing of unpolymerized components [64]. After the specimens experienced the first heating cycle, most of the unpolymerized components have been polymerized therefore the first transition disappeared in the second reversing heat flow profiles (Fig. 3C). Meanwhile, the self-strengthening reaction can be prompted by the heating, [65] and the network of the experimental polymers was further crosslinked, which contributed to the increase in Tg of HBA10 from 101°C (first cycle) to 122 °C (second cycle). However, the breadth of the transition and lack of a clear step made it difficult to determine the Tg of HBA20 and HBA30.
Our studies have confirmed the autonomous strengthening characteristic in alkoxysilane-containing dental adhesive formulations [24, 25]. Due to the highly crosslinked structure of polymethacrylate matrix and relatively slow rate of the sol-gel reaction, a considerable amount of time was required to observe the self-strengthening effect under wet conditions. In the current study, three-cycle DMA under dry conditions was used to determine the mechanical properties of the control and experimental samples. Following 2-days post-processing, the HBA30 specimens could not be retrieved from the glass tubing without damage. Therefore, only the control and HBA10/20 specimens were tested using DMA.
For the control and experimental samples in the 1st DMA test cycle, it can be observed that the rubbery moduli are comparable (~30-33 MPa). These results indicated that the crosslinking densities (crosslinked by C-C bonds) were similar. However, the Tg of the experimental samples were significantly lower than that of the control. There were two main reasons. First, the more flexible backbone of ASMA as compared to BisGMA. Second, the by-product of sol-gel reaction, methanol, plays a plasticizer effect. Therefore, the initial mechanical properties of ASMA-containing samples showed poor performance than that of the control. However, both the rubbery modulus and Tg of the ASMA-containing samples, showed a substantial increase after the first and second heating cycles. In comparison, for the control polymer, the rubbery moduli of each test were comparable at about 30 MPa. Tg of the control polymer increased slightly from 124.5±0.9 °C (1st cycle) to 129.7±0.1 °C (3rd cycle). The very limited change in rubbery modulus and Tg for the control formulation indicated that the network structure (crosslink density) does not change significantly after the 1st and 2nd heating cycles.
With the increase in ASMA content from 10 to 20 wt%, the rubbery modulus increased from about 30 MPa (1st cycle) to ~43 MPa (HBA10, 2nd cycle) and ~65MPa (HBA20, 2nd cycle), respectively. The inverse ratio (ζ) of the rubbery modulus to the temperature has been used to present the relative crosslinking density of polymers [22] and the results have been shown in SI Table 3. For the control, the ζ values of three-cycle tests (at about 1.26-1.34 Pa−1•K) are not significantly different at level 0.05, which indicated the crosslinking densities were comparable. In contrast, for the experimental samples, the significantly lower ζ values suggested higher crosslinking densities were achieved after heating and the significantly lower ζ values of the experimental sample indicated that its crosslinking density was higher than the control. Meanwhile, the Tg observed from the tan δ peak also increased, over 20 °C (HBA10) and 30 °C (HBA20), respectively. The significant increase in Tg in the heated experimental samples is difficult to observe using modulated DSC. In comparison, the DMA test is substantially more sensitive for measuring the glass transition temperature in the highly crosslinked materials.
Tan δ is a measure of the energy dissipation of a material, and the intensity of maximum tan δ peak usually reflects the mobility of the chain segments at the transition temperature [40, 66, 67]. In the first cycle, the tan δ values of the experimental samples were higher than that of the control, which indicated that the chain mobility of experimental samples was higher than that of the control. The tan δ values of the HBA10 or HBA20 decreased significantly in the second cycle, and the values were much lower than that of the control. These differences are attributed to further crosslinking by the sol-gel reaction when the experimental samples were heated [65]. These results indicate a substantial increase in crosslink density in the experimental polymer as compared to the control. Based on the polymerization kinetics and DMA results, the second hypothesis is accepted. The ASMA can act as a co-monomer to partially replace BisGMA.
Stress relaxation tests were performed to characterize the viscoelastic behavior of fresh and 2-cycle heated control and HBA10 formulations. The results showed significant differences between the fresh and heated samples (Fig. 5A and 5B). For the fresh samples, the crosslinking densities were comparable (SI Table 3), and the monomer-to-polymer conversion of HB45 (control) and HBA10 (experimental) was about 60% (Fig. 2). These results indicate that the network includes covalent crosslinked structure and unpolymerized components, such as HEMA, BisGMA, and ASMA. When stress was applied to fresh samples, both the crosslink chains and unpolymerized components (e.g., monomers and oligomers [18, 68, 69]) were mobile. Due to the faster migration of the unpolymerized components [70], the stress for fresh samples decreased to half within ~30 s. The relaxation behavior of HB45 and HBA10 was comparable.
After two-cycle heating, the post-curing reactions are complete and the mechanical properties reach equilibrium (this relationship was supported by the DMA results, Fig. 4). Accordingly, in the heated samples, only crosslinked chains migrate to relax the applied stress, and it took much longer to relax half of the stress (~460 times for heated HB45 and ~3000 times for heated HBA10). The relaxation time for the heated HBA10 was ~5 times longer than that of heated HB45, which indicates the limited mobility of the chains in the heated HBA10 specimens. These results further support the significantly increased crosslinking density in experimental samples as compared to HB45, which is consistent with the DMA data (Fig. 4) and relative crosslinking density (SI Table 3).
The stress relaxation data alone cannot clearly show how the material strengthens at different time scales. Since obtaining the relaxation time constants and the associated moduli is not straightforward due to the time-history effects present in viscoelastic materials [71], the constitutive behavior of the material was modeled using generalized Maxwell model or Weichert model represented as Prony series [42, 72]. Stress relaxation test data were broken down into relaxation times and corresponding moduli over a wide range of time. A linear loading case was simulated incorporating the obtained relaxation times and corresponding moduli for each specimen (Fig 5C). The simulation based on Prony-series-fitting provides information about the self-strengthening phenomenon which is not accessible through the raw stress relaxation data. Based on the DMA and simulation results, the third hypothesis is accepted. The mechanical properties, including the crosslinking density, of the post-processed ASMA-containing polymers showed significant improvement due to the self-strengthening reaction.
Leaching of unreacted components is inevitable since the conversion of methacrylate monomers in the adhesive formulations is incomplete. The leachates may provoke toxicity concerns [73–75] and degradation of the adhesive [76–78]. In the present study, with the increase in ASMA content, the amount of leached HEMA and BisGMA decreased dramatically. The leached percentage of HEMA decreased from 8 wt% (HB45) to 1.2 wt% (HBA30), BisGMA decreased from 3.5 wt% (HB45) to 1.6 wt% (HBA30) (see SI Table 4). The decrease in leached HEMA and BisGMA is attributed to the higher DC of C=C bond during light-curing and the crosslinking reactions between the formed silanol and hydroxyl groups of HEMA/BisGMA [24–26].
ASMA was leached from the experimental polymers. This unexpected result may be attributed, in part, to the relatively slow rate of the sol-gel reaction as compared to free-radical polymerization [25, 26]. When the liquid resin formulation is irradiated by visible-light, a polymethacrylate-based crosslinked network is formed by free-radical polymerization of methacrylate monomers and limited hydrolysis and condensation of the silane. The limited condensation reaction of the silane in the highly crosslinked polymethacrylate-based network [26] translates to ASMA that is trapped in the matrix and thus, available for leaching. The effectiveness of ASMA as both a co-monomer and co-initiator may also be a factor, i.e. ASMA may act more efficiently as a co-monomer.
It must be noted that ethanol is not a clinically relevant solvent for the HPLC study. Ethanol was used in this in vitro investigation to accelerate the leaching. HPLC data determined in ethanol would be expected to have a higher cumulative leachate concentration as compared to the clinical conditions, and the results should be interpreted with caution.
The results suggest that ASMA may act more efficiently as a co-monomer as opposed to a co-initiator. During light irradiation, a photo-induced electron transfer occurred between the electron acceptor (CQ) and electron donor (EDMAB or ASMA) [18, 79, 80]. It has been observed that the polymerization rate of ASMA-containing formulations with DPIHP was significantly faster than without DPIHP (see SI Figure 1). This result indicated that the phenyl free radical generated from the electron transfer products and DPIHP played pivotal role in the polymerization. At the same time, it is noted that the DC and polymerization rate of HB45 without EDMAB was relatively low (27%, see SI Figure 1). These results indicated that limited free radicals are generated by the reaction between excited CQ and DPIHP. Considering the chemical structure and basicity of EDMAB and ASMA, it can be surmised that the ASMA is less efficient than EDMAB in two-component photo-initiator system. Additional investigations are required to unravel these complex relationships.
In the oral environment, the poorly polymerized hydrophilic-rich phase will be degraded rapidly [47, 81]. Due to the reduced hydrophobicity of ASMA as compared to BisGMA, the inclusion of ASMA may result in more ASMA present in the hydrophilic-rich phase following physical phase separation of the adhesive under wet bonding conditions. While further research is required to confirm the benefit of ASMA under degradative conditions, it is likely that the higher DC, significantly reduced leachates, and higher crosslinking density noted with the ASMA-containing adhesives will have a positive impact on the hydrophilic-rich phase.
5. Conclusion
Novel dental adhesives with capabilities of higher water miscibility, higher degree of conversion, and autonomous strengthening have been developed by introducing a balanced amphiphilic multifunctional monomer ASMA in the HEMA/BisGMA formulation. ASMA can act as amine-based co-initiator without using EDMAB, simplifying the adhesive formulation. The lower viscosity and higher hydrophilicity of the ASMA-containing formulations may promote adhesive infiltration of the wet, demineralized dentin substrate. The relaxation behavior and the fitted Prony series indicated the stiffening properties for ASMA-containing adhesives. The ability of ASMA to act simultaneously as co-monomer and co-initiator offers promise for developing a simplified adhesive that achieves enhanced durability as a result of intrinsic self-strengthening properties. While the results are promising, future studies are required to determine the quality and durability of the seal at the interface with dentin using ASMA containing adhesive.
Supplementary Material
Highlights:
ASMA monomer serves multiple roles, i.e. acts simultaneously as co-initiator and co-monomer and provides self-strengthening.
ASMA-containing adhesive shows a substantial reduction of leached of HEMA and BisGMA.
Simulation-based on Prony-series-fitting provides information about the self-strengthening phenomenon.
Acknowledgements
This investigation was supported by research grant R01DE025476 from the National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, Maryland. The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Afrashtehfar KI, Emami E, Ahmadi M, Eilayyan O, Abi-Nader S, Tamimi F, Failure rate of single-unit restorations on posterior vital teeth: A systematic review, J Prosthet Dent, 117 (2017) 345–353 e348. [DOI] [PubMed] [Google Scholar]
- [2].Ferracane JL, Resin-based composite performance: are there some things we can’t predict?, Dent Mater, 29 (2013) 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schwendicke F, Gostemeyer G, Blunck U, Paris S, Hsu LY, Tu YK, Directly Placed Restorative Materials: Review and Network Meta-analysis, J Dent Res, 95 (2016) 613–622. [DOI] [PubMed] [Google Scholar]
- [4].Makvandi P, Jamaledin R, Jabbari M, Nikfarjam N, Borzacchiello A, Antibacterial quaternary ammonium compounds in dental materials: A systematic review, Dent Mater, 34 (2018) 851–867. [DOI] [PubMed] [Google Scholar]
- [5].Stewart CA, Finer Y, Biostable, antidegradative and antimicrobial restorative systems based on host-biomaterials and microbial interactions, Dent. Mater, 35 (2019) 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kopperud SE, Espelid I, Tveit AB, Skudutyte-Rysstad R, Risk factors for caries development on tooth surfaces adjacent to newly placed class II composites--a pragmatic, practice based study, J Dent, 43 (2015) 1323–1329. [DOI] [PubMed] [Google Scholar]
- [7].Ferracane JL, Models of Caries Formation around Dental Composite Restorations, J Dent Res, 96 (2017) 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kuper NK, Opdam NJM, Bronkhorst EM, Ruben JL, Huysmans MCDNJM, Hydrodynamic Flow through Loading and in vitro Secondary Caries Development, Journal of Dental Research, 92 (2013) 383–387. [DOI] [PubMed] [Google Scholar]
- [9].Chisini LA, Collares K, Cademartori MG, de Oliveira LJC, Conde MCM, Demarco FF, Correa MB, Restorations in primary teeth: a systematic review on survival and reasons for failures, Int J Paediatr Dent, 28 (2018) 123–139. [DOI] [PubMed] [Google Scholar]
- [10].Brackett MG, Li N, Brackett WW, Sword RJ, Qi YP, Niu LN, Pucci CR, Dib A, Pashley DH, Tay FR, The critical barrier to progress in dentine bonding with the etch-and-rinse technique, J. Dent, 39 (2011) 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bourbia M, Ma D, Cvitkovitch DG, Santerre JP, Finer Y, Cariogenic Bacteria Degrade Dental Resin Composites and Adhesives, J. Dent. Res, 92 (2013) 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Spencer P, Ye Q, Misra A, Goncalves SEP, Laurence JS, Proteins, Pathogens, and Failure at the Composite-Tooth Interface, J. Dent. Res, 93 (2014) 1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Park JG, Ye Q, Topp EM, Kostoryz EL, Wang Y, Kieweg SL, Spencer P, Preparation and properties of novel dentin adhesives with esterase resistance, J. Appl. Polym. Sci, 107 (2008) 3588–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Park JG, Ye Q, Topp EM, Spencer P, Enzyme-Catalyzed Hydrolysis of Dentin Adhesives Containing a New Urethane-Based Trimethacrylate Monomer, J. Biomed. Mater. Res. Part B, 91B (2009) 562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Park J, Ye Q, Singh V, Kieweg SL, Misra A, Spencer P, Synthesis and evaluation of novel dental monomer with branched aromatic carboxylic acid group, J. Biomed. Mater. Res. Part B, 100B (2012) 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Song LY, Ye Q, Ge XP, Misra A, Laurence JS, Berrie CL, Spencer P, Synthesis and evaluation of novel dental monomer with branched carboxyl acid group, J. Biomed. Mater. Res. Part B, 102 (2014) 1473–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ge XP, Ye Q, Song LY, Misra A, Spencer P, Synthesis and evaluation of novel siloxane-methacrylate monomers used as dentin adhesives, Dent. Mater, 30 (2014) 1073–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ge XP, Ye Q, Song LY, Laurence JS, Spencer P, Synthesis and Evaluation of a Novel Co-Initiator for Dentin Adhesives: Polymerization Kinetics and Leachables Study, JOM, 67 (2015) 796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ge XP, Ye Q, Song LY, Laurence JS, Misra A, Spencer P, Probing the dual function of a novel tertiary amine compound in dentin adhesive formulations, Dent. Mater, 32 (2016) 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Song LY, Ye Q, Ge XP, Singh V, Misra A, Laurence JS, Berrie CL, Spencer P, Development of methacrylate/silorane hybrid monomer system: Relationship between photopolymerization behavior and dynamic mechanical properties, J. Biomed. Mater. Res. Part B, 104 (2016) 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ge XP, Ye Q, Song LY, Spencer P, Laurence JS, Effect of crosslinking density of polymers and chemical structure of amine-containing monomers on the neutralization capacity of dentin adhesives, Dent. Mater, 31 (2015) 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Song LY, Ye Q, Ge XP, Spencer P, Compositional design and optimization of dentin adhesive with neutralization capability, J. Dent, 43 (2015) 1132–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Song LY, Ye Q, Ge XP, Misra A, Tamerler C, Spencer P, Probing the neutralization behavior of zwitterionic monomer-containing dental adhesive, Dent. Mater, 33 (2017) 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Song LY, Ye Q, Ge XP, Misra A, Spencer P, Mimicking nature: Self-strengthening properties in a dental adhesive, Acta Biomater., 35 (2016) 138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Song LY, Ye Q, Ge XP, Misra A, Tamerler C, Spencer P, Self-strengthening hybrid dental adhesive via visible-light irradiation triple polymerization, RSC Adv., 6 (2016) 52434–52447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Song LY, Ye Q, Ge XP, Misra A, Tamerler C, Spencer P, Fabrication of hybrid crosslinked network with buffering capabilities and autonomous strengthening characteristics for dental adhesives, Acta Biomater., 67 (2018) 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Song LY, Ye Q, Ge XP, Misra A, Tamerler C, Spencer P, New silyl-functionalized BisGMA provides autonomous strengthening without leaching for dental adhesives, Acta Biomater, 83 (2019) 130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Spencer P, Wang Y, Adhesive phase separation at the dentin interface under wet bonding conditions, J. Biomed. Mater. Res, 62 (2002) 447–456. [DOI] [PubMed] [Google Scholar]
- [29].Nishitani Y, Yoshiyama M, Donnelly AM, Agee KA, Sword J, Tay FR, Pashley DH, Effects of resin hydrophilicity on dentin bond strength, J. Dent. Res, 85 (2006) 1016–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Abedin F, Ye Q, Good HJ, Parthasarathy R, Spencer P, Polymerization- and solvent-induced phase separation in hydrophilic-rich dentin adhesive mimic, Acta Biomater, 10 (2014) 3038–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Breschi L, Maravic T, Cunha SR, Comba A, Cadenaro M, Tjaderhane L, Pashley DH, Tay FR, Mazzoni A, Dentin bonding systems: From dentin collagen structure to bond preservation and clinical applications, Dent. Mater, 34 (2018) 78–96. [DOI] [PubMed] [Google Scholar]
- [32].Wang Y, Spencer P, Overestimating hybrid layer quality in polished adhesive/dentin interfaces, J. Biomed. Mater. Res. Part A, 68A (2004) 735–746. [DOI] [PubMed] [Google Scholar]
- [33].Spencer P, Wang Y, Bohaty B, Interfacial chemistry of moisture-aged class II composite restorations, J. Biomed. Mater. Res. Part B, 77B (2006) 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Guo X, Spencer P, Wang Y, Ye Q, Yao X, Williams K, Effects of a solubility enhancer on penetration of hydrophobic component in model adhesives into wet demineralized dentin, Dent. Mater, 23 (2007) 1473–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kostoryz EL, Dharmala K, Ye Q, Wang Y, Huber J, Park JG, Snider G, Katz JL, Spencer P, Enzymatic Biodegradation of HEMA/BisGMA Adhesives Formulated With Different Water Content, J. Biomed. Mater. Res. Part B, 88B (2009) 394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Guo X, Wang Y, Spencer P, Ye Q, Yao X, Effects of water content and initiator composition on photopolymerization of a model BisGMA/HEMA resin, Dent. Mater, 24 (2008) 824–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ye Q, Park J, Topp E, Spencer P, Effect of photoinitiators on the in vitro performance of a dentin adhesive exposed to simulated oral environment, Dent. Mater, 25 (2009) 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Parthasarathy R, Misra A, Park J, Ye Q, Spencer P, Diffusion coefficients of water and leachables in methacrylate-based crosslinked polymers using absorption experiments, J. Mater. Sci.-Mater. Med, 23 (2012) 1157–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Park J, Ye Q, Topp EM, Misra A, Kieweg SL, Spencer P, Effect of photoinitiator system and water content on dynamic mechanical properties of a light-cured bisGMA/HEMA dental resin, J. Biomed. Mater. Res. Part A, 93A (2010) 1245–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Park JG, Ye Q, Topp EM, Lee CH, Kostoryz EL, Misra A, Spencer P, Dynamic Mechanical Analysis and Esterase Degradation of Dentin Adhesives Containing a Branched Methacrylate, J. Biomed. Mater. Res. Part B, 91B (2009) 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ge XP, Ye Q, Song LY, Misra A, Spencer P, The influence of water on visible-light initiated free-radical/cationic ring-opening hybrid polymerization of methacrylate/epoxy: polymerization kinetics, crosslinking structure and dynamic mechanical properties, RSC Adv., 5 (2015) 77791–77802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Park SW, Schapery RA, Methods of interconversion between linear viscoelastic material functions. Part I - a numerical method based on Prony series, Int J Solids Struct, 36 (1999) 1653–1675. [Google Scholar]
- [43].Chen T, Determining a Prony series for a viscoelastic material from time varying strain data, 2000.
- [44].Van Landuyt KL, Snauwaert J, De Munck J, Peurnans M, Yoshida Y, Poitevin A, Coutinho E, Suzuki K, Lambrechtsa P, Van Meerbeek B, Systematic review of the chemical composition of contemporary dental adhesives, Biomaterials, 28 (2007) 3757–3785. [DOI] [PubMed] [Google Scholar]
- [45].Ikemura K, Endo T, A review of our development of dental adhesives - Effects of radical polymerization initiators and adhesive monomers on adhesion, Dent. Mater. J, 29 (2010) 109–121. [DOI] [PubMed] [Google Scholar]
- [46].Gopferich A, Mechanisms of polymer degradation and erosion, Biomaterials, 17 (1996) 103–114. [DOI] [PubMed] [Google Scholar]
- [47].Delaviz Y, Finer Y, Santerre JP, Biodegradation of resin composites and adhesives by oral bacteria and saliva: A rationale for new material designs that consider the clinical environment and treatment challenges, Dent. Mater, 30 (2014) 16–32. [DOI] [PubMed] [Google Scholar]
- [48].Huang B, Cuitizouitch DG, Santerre JP, Finer Y, Biodegradation of resin-dentin interfaces is dependent on the restorative material, mode of adhesion, esterase or MMP inhibition, Dent. Mater, 34 (2018)1253–1262. [DOI] [PubMed] [Google Scholar]
- [49].Singh J, Khalichi P, Cvitkovitch DG, Santerre JP, Composite resin degradation products from BisGMA monomer modulate the expression of genes associated with biofilm formation and other virulence factors in Streptococcus mutans, J. Biomed. Mater. Res. Part A, 88a (2009) 551–560. [DOI] [PubMed] [Google Scholar]
- [50].Khalichi P, Singh J, Cvitkovitch DG, Santerre JP, The influence of triethylene glycol derived from dental composite resins on the regulation of Streptococcus mutans gene expression, Biomaterials, 30 (2009) 452–459. [DOI] [PubMed] [Google Scholar]
- [51].Sadeghinejad L, Cvitkovitch DG, Siqueira WL, Merritt J, Santerre JP, Finer Y, Mechanistic, genomic and proteomic study on the effects of BisGMA-derived biodegradation product on cariogenic bacteria, Dent. Mater, 33 (2017) 175–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Perlmutter P, Conjugate addition reactions in organic synthesis, 1st ed., Pergamon, Oxford; ; New York, 1992. [Google Scholar]
- [53].Enders D, Wang C, Liebich JX, Organocatalytic Asymmetric Aza-Michael Additions, Chem.-Eur. J, 15(2009)11058–11076. [DOI] [PubMed] [Google Scholar]
- [54].Wang J, Li PF, Choy PY, Chan ASC, Kwong FY, Advances and Applications in Organocatalytic Asymmetric aza-Michael Addition, Chemcatchem, 4 (2012) 917–925. [Google Scholar]
- [55].Jung B, Theato P, Chemical Strategies for the Synthesis of Protein-Polymer Conjugates, Bio-Synthetic Polymer Conjugates, 253 (2013) 37–70. [Google Scholar]
- [56].Ye Q, Wang Y, Williams K, Spencer P, Characterization of photopolymerization of dentin adhesives as a function of light source and irradiance, J. Biomed. Mater. Res. Part B, 80B (2007) 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dickens SH, Stansbury JW, Choi KM, Floyd CJE, Photopolymerization kinetics of methacrylate dental resins, Macromolecules, 36 (2003) 6043–6053. [Google Scholar]
- [58].Horie K, Otagawa A, Muraoka M, Mita I, Calorimetric Investigation of Polymerization Reactions .5. Crosslinked Copolymerization of Methyl-Methacrylate with Ethylene Dimethacrylate, J. Polym. Sci. Pol. Chem, 13 (1975) 445–454. [Google Scholar]
- [59].Kloosterboer JG, Network Formation by Chain Crosslinking Photopolymerization and Its Applications in Electronics, Adv Polym Sci, 84 (1988) 1–61. [Google Scholar]
- [60].Hourston DJ, Song M, Schafer FU, Pollock HM, Hammiche A, Modulated-temperature differential scanning calorimetry: 15. Crosslinking in polyurethane-poly(ethyl methacrylate) interpenetrating polymer networks, Polymer, 40 (1999) 4769–4775. [Google Scholar]
- [61].Hempel E, Beiner M, Huth H, Donth E, Temperature modulated DSC for the multiple glass transition in poly(n-alkyl methacrylates), Thermochim. Acta, 391 (2002) 219–225. [Google Scholar]
- [62].Van Mele B, Van Assche G, Van Hemelrijck A, Modulated differential scanning calorimetry to study reacting polymer systems, J. Reinf. Plast. Compos, 18 (1999) 885–894. [Google Scholar]
- [63].Jenninger W, Schawe JEK, Alig I, Calorimetric studies of isothermal curing of phase separating epoxy networks, Polymer, 41 (2000) 1577–1588. [Google Scholar]
- [64].Ye Q, Spencer P, Wang Y, Misra A, Relationship of solvent to the photopolymerization process, properties, and structure in model dentin adhesives, J. Biomed. Mater. Res. Part A, 80A (2007) 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Issa AA, Luyt AS, Kinetics of Alkoxysilanes and Organoalkoxysilanes Polymerization: A Review, Polymers-Basel, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Menard KP, Dynamic mechanical analysis : a practical introduction, CRC Press, Boca Raton, FL, 2008. [Google Scholar]
- [67].Menczel JD, Prime RB, Thermal analysis of polymers : fundamentals and applications, John Wiley, Hoboken, N.J., 2009. [Google Scholar]
- [68].Van Landuyt KL, Nawrot T, Geebelen B, De Munck J, Snauwaert J, Yoshihara K, Scheers H, Godderis L, Hoet P, Van Meerbeek B, How much do resin-based dental materials release? A meta-analytical approach, Dent. Mater, 27 (2011) 723–747. [DOI] [PubMed] [Google Scholar]
- [69].Kloukos D, Pandis N, Eliades T, Bisphenol-A and residual monomer leaching from orthodontic adhesive resins and polycarbonate brackets: A systematic review, Am. J. Orthod. Dentofac. Orthop, 143 (2013) S104-+. [DOI] [PubMed] [Google Scholar]
- [70].Zhao XH, Huebsch N, Mooney DJ, Suo ZG, Stress-relaxation behavior in gels with ionic and covalent crosslinks, J Appl Phys, 107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].O’Brien DJ, Mather PT, White SR, Viscoelastic properties of an epoxy resin during cure, J. Compos Mater, 35 (2001) 883–904. [Google Scholar]
- [72].Kwok K, Pellegrino S, Folding, Stowage, and Deployment of Viscoelastic Tape Springs, Aiaa J, 51 (2013) 1908–1918. [Google Scholar]
- [73].Liso PA, Vazquez B, Rebuelta M, Hernaez ML, Rotger R, SanRoman J, Analysis of the leaching and toxicity of new amine activators for the curing of acrylic bone cements and composites, Biomaterials, 18 (1997) 15–20. [DOI] [PubMed] [Google Scholar]
- [74].Geurtsen W, Biocompatibility of resin-modified filling materials, Crit Rev Oral Biol M, 11 (2000) 333–355. [DOI] [PubMed] [Google Scholar]
- [75].Teti G, Mazzotti G, Zago M, Ortolani M, Breschi L, Pelotti S, Ruggeri A, Falconi M, HEMA down-regulates procollagen alpha 1 type I in human gingival fibroblasts, J. Biomed. Mater. Res. Part A, 90a (2009) 256–262. [DOI] [PubMed] [Google Scholar]
- [76].Lee SY, Huang HM, Lin CY, Shih YH, Leached components from dental composites in oral simulating fluids and the resultant composite strengths, J. Oral Rehabil, 25 (1998) 575–588. [DOI] [PubMed] [Google Scholar]
- [77].Breschi L, Mazzoni A, Ruggeri A, Cadenaro M, Di Lenarda R, Dorigo ED, Dental adhesion review: Aging and stability of the bonded interface, Dent. Mater, 24 (2008) 90–101. [DOI] [PubMed] [Google Scholar]
- [78].Manuja N, Nagpal R, Pandit IK, Dental Adhesion: Mechanism, Techniques and Durability, J. Clin. Pediatr. Dent, 36 (2012) 223–234. [PubMed] [Google Scholar]
- [79].Fouassier JP, Allonas X, Lalevee J, Visconti M, Radical polymerization activity and mechanistic approach in a new three-component photoinitiating system, J. Polym. Sci. Pol. Chem, 38 (2000) 4531–4541. [Google Scholar]
- [80].Fouassier JP, Lalevee J, Three-component photoinitiating systems: towards innovative tailor made high performance combinations, RSC Adv., 2 (2012) 2621–2629. [Google Scholar]
- [81].Frassetto A, Breschi L, Turco G, Marchesi G, Di Lenarda R, Tay FR, Pashley DH, Cadenaro M, Mechanisms of degradation of the hybrid layer in adhesive dentistry and therapeutic agents to improve bond durability-A literature review, Dent. Mater, 32 (2016) E41–E53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.