

Abstract
Histamine has been one of the most studied substances in medicine, playing a major role in diverse (patho)physiological processes. It elicits its multifaceted modulatory functions by activating four types of GPCRs, designated as H1–4. Despite the heterogeneity and the complexity of histamine receptor pharmacology, many discoveries over the past 100 years resulted in the development of H1 antihistamines and H2‐targeting ‘blockbuster’ therapeutics for the management of allergies and gastrointestinal disorders respectively. Recently, a first‐in‐class H3 inverse agonist was approved for the treatment of narcolepsy, whereas H4 antagonists are under clinical evaluation for their potential therapeutic exploitation in immune‐related diseases. This review critically presents the past successes and drawbacks in histamine research, complemented by the modern conceptual innovations in molecular and receptor pharmacology. It targets both young and experienced researchers in an ongoing effort to stimulate novel insights for the dissection of the translational potential of histamine pharmacology.
Linked Articles
This article is part of a themed section on New Uses for 21st Century. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v177.3/issuetoc
Abbreviations
- BBB
blood–brain barrier
- EHRS
European Histamine Research Society
- EMA
European Medicines Agency
- GORD
gastro‐oesophageal reflux disease
- HDC
histidine decarboxylase
- INSERM
Institut National de la Santé et de la Recherche Médicale
- JHRS
Japanese Histamine Research Society
- P‐gp
P‐glycoprotein
- Rα‐MeHA
(R)‐α‐methylhistamine
- TMN
tuberomamillary nucleus
All I ever promised was that I was sure I could develop a new pharmacological agent which might answer a physiological question. Any utility would be implicit in that answer.
Sir James W. Black – Biographical
Nobel Prize in Physiology or Medicine 1988
Honorary Member of the EHRS 1998
Introduction
The history of histamine effectively reflects the origins and the progress of the basic concepts that shaped modern pharmacology over the past 120 years (Figure 1). Most of the advancements in the field have been accomplished through the achievements of research groups led by outstanding scientists, including among others the Nobel Laureates Adolf Windaus (1928), Sir Henry H. Dale (1936), Daniel Bovet (1957) and Sir James W. Black (1988).
Figure 1.
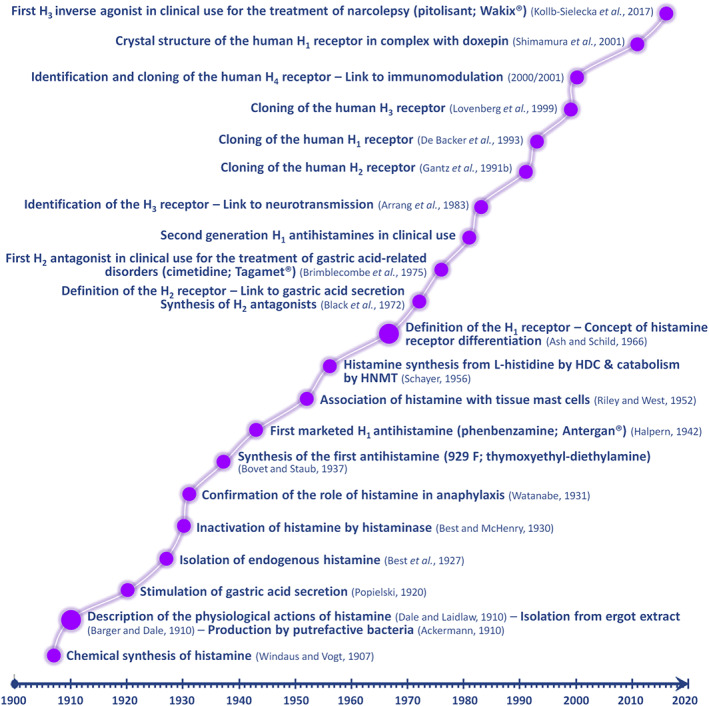
Timeline of the major achievements in histamine research and in the therapeutic exploitation of histamine receptor (H1–H4) ligands. HDC, histidine decarboxylase; HNMT, histamine N‐methyltransferase.
Histamine [2‐(4‐imidazolyl)‐ethylamine; β‐imidazolylethylamine] is an endogenous, short‐acting biogenic amine that is ubiquitously distributed in mammalian tissues at various levels (Shore et al., 1959). It is synthesized by decarboxylation of the semi‐essential amino acid L‐histidine via the catalytic activity of histidine decarboxylase (HDC, EC 4.1.1.22) (Schayer, 1956). Histamine is catabolized by ring methylation to N‐methylhistamine via the cytosolic enzyme histamine N‐methyltransferase (EC 2.1.1.8) or by oxidative deamination to imidazole acetic acid by the extracellular enzyme diamine oxidase (EC 1.4.3.22) (Best and McHenry, 1930; Schayer, 1956).
Being the first inflammatory biogenic amine to be characterized, histamine is one of the most studied substances in medicine. The relatively rapid progress in present‐day science and technology revealed that histamine elicits its pleiotropic functions in health and disease by activating four types of class A rhodopsin‐like GPCRs, which have been named chronologically in order of their discovery as histamine H1, H2, H3 and H4 receptors (Oda et al., 2000; Akdis and Simons, 2006; Parsons and Ganellin, 2006; Panula et al., 2015 ; Alexander et al., 2017a) (Figure 2). The H1 and H2 receptors have relatively low affinity for histamine (at the μM range) compared to their high affinity H3 and H4 counterparts (5–10 nM) (Figure 2) (Panula et al., 2015; Alexander et al., 2017a). Thus, the local tissue histamine concentration and functional expression of the different receptors seem to be important determinants of the biological response (Panula et al., 2015; Salem et al., 2017).
Figure 2.
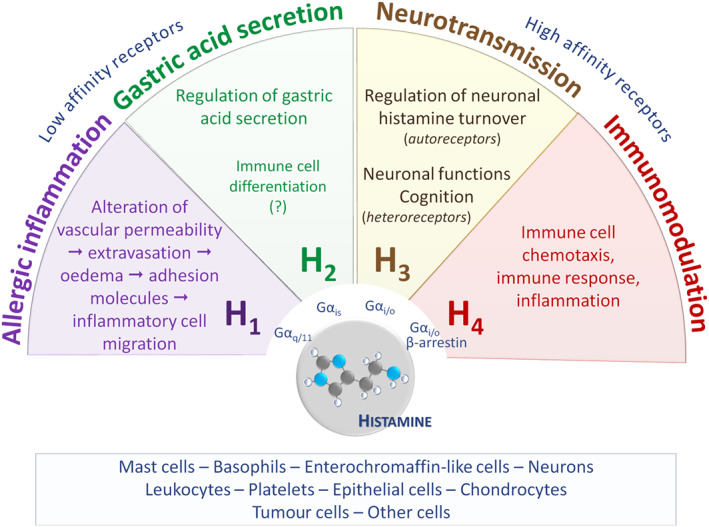
Overview of the main functions of the histamine receptors. Histamine is formed in various cell types (rectangular box) and orchestrates numerous actions via binding to four receptor types, designated as H1–H4. Gas, Gai/o, Gaq/11: G protein Ga subunits.
Notably, histamine receptor orthologues exhibit marked intra‐ and inter‐species variations that frequently hamper the translation of experimental pharmacological data into clinically useful therapies (Coruzzi et al., 2012; Strasser et al., 2013). Moreover, they exhibit pluridimensional efficacy leading to the recently described functional selectivity and biased signalling (Rosethorne and Charlton, 2011; Monczor and Fernandez, 2016; Riddy et al., 2017). These concepts have started to reshape GPCR ligand characteristics and drug discovery approaches (Kenakin, 2017). Thus, they contribute to the current tendency of redirecting biomedical experimentation from the subcellular and molecular levels back towards integrative and translational research approaches.
Despite the relatively recently exposed heterogeneity and complexity of histamine receptor pharmacology, a plethora of studies have related the H1, H2, H3 and H4 receptors to allergic inflammation (Lewis and Grant, 1924; Best et al., 1927), stimulation of gastric acid secretion (Black et al., 1972), neurotransmission (Arrang et al., 1983; Haas and Panula, 2003) and immune responses (Zampeli and Tiligada, 2009) respectively (Figure 2). These pleiotropic histamine actions provided the basis for the development and marketing of several ‘blockbuster’ drugs for the treatment of allergies and gastrointestinal disorders (Table 1) that antagonize the effects of histamine at the H1 (commonly referred to as H1 antihistamines) and H2 receptor respectively (Parsons and Ganellin, 2006). Pitolisant is the first H3 receptor‐targeting drug to reach the market (Table 1), being approved for the treatment of narcolepsy in 2016 (Kollb‐Sielecka et al., 2017). H4 receptor antagonists are currently under clinical evaluation (Table 1) for their potential therapeutic exploitation in inflammatory disorders such as atopic dermatitis (Werfel, 2018), as well as in vestibular disease (Attali et al., 2016).
Table 1.
Indicative, non‐exhaustive list of clinically important compounds targeting the histamine receptors
| INN (year approved) | ATC code | Ligand ID | Comments‐indicative brand names (see text for further details) |
|---|---|---|---|
| Drugs targeting the histamine H1 receptor (mainly indicated for the symptomatic treatment of allergic conditions) | |||
| First‐generation H1 antihistamines | |||
| • phenbenzamine (a) | a | a | Developed as RP 2339 at Rhône‐Poulenc in France and marketed as Antergan®; first antihistamine tested in humans in 1942 for the symptomatic relief of allergic manifestations. |
| • mepyramine (EURD 1969) | D04AA02c R06AC01d | 1227 | Also known as pyrilamine; substituted ethylene diamine introduced in 1944 by Rhône‐Poulenc in France and marketed as Neo‐Antergan®; sold among others as Anthisan®, Pyrlex®. |
| • diphenhydramine (FDA 1946) | D04AA32c D04AA33c R06AA02d R06AA52d , e | 1224 | Aminoalkyl ether; first FDA‐approved prescription antihistamine introduced by Parke, Davis and Co. and marketed as Benadryl®. Dimenhydrinate, the 8‐chlorotheophyllinate salt that is used for the management of nausea, vomiting and motion sickness was introduced by G.D. Searle & Co. in 1949 as Dramamine®; marketed among others as Gravol®, Vertirosan®, Vomex‐A®. Orphenadrine (ligand id 7251), an analogue of diphenhydramine developed by Parke, Davis and Co. in the late 1940s and acting, among others, on histamine H1 and on muscarinic ACh receptors is used as an anti‐Parkinson anticholinergic agent (N04AB02) and as a muscle relaxant (M03BC01, M03BC51e); sold as plain or combined preparations (e.g. Invagesic®, Norflex®, Norgesic®). |
| • tripelennamine (FDA 1948) | D04AA04c R06AC04d | 7318 | Substituted ethylene diamine introduced by Ciba as Pyribenzamine®; sold among others as Azaron®. |
| • promethazine (EURD 1948, FDA 1951) | D04AA10c R06AD02d R06AD52d , e | 7282 | Phenothiazine derivative developed in the 1940s by Rhône‐Poulenc as 3277 RP; marketed among others as Phenergan®; medically useful as antiemetic and sedative agent. |
| • doxylamine (EURD 1948, FDA 1948) | R06AA09d R06AA59d , e | 7171 | Aminoalkyl ether first approved as Decapryn® for Sanofi Aventis USA; has been used for the management of nausea and vomiting in pregnancy (Diclegis®, Diclectin®) and as a short‐term sedative (Unisom®). |
| • cyclizine (a) | R06AE03d R06AE53d , e | 7151 | Piperazine derivative developed by Burroughs Wellcome in the late 1940s and sold as Marezine®; used for the management of motion sickness, nausea and vomiting associated with narcotic analgesics, general anaesthetics and radiotherapy, and of vestibular disturbances; listed in the WHO Model List of Essential Medicines 2011–2017b; antiemetic chosen by the National Aeronautics and Space Administration (NASA) to be included in the medical kit of the first manned Apollo mission on October 11th, 1968 (Apollo 7). |
| • chlorphenamine (FDA 1950, EURD 1954) | R06AB04d R06AB54d , e | 6976 | Also known as chlorpheniramine; substituted alkylamine marketed as Chlor‐trimeton® by Schering and Bayer; sold among others as Phenetron®, Teldrin®; listed in the WHO Model List of Essential Medicines 1977–2011b; combined preparations for the relief of symptoms caused by common cold or influenza are available (e.g. Comtrex Cold ®); dexchlorpheniramine (R06AB02d) is the S‐enantiomer of chlorphenamine (e.g. Polaramine®). |
| • meclozine (EURD 1953, FDA 1957) | R06AE05d R06AE55d , e | 2757 | Also known as meclizine; piperazine derivative used for the management of motion sickness, vertigo and vestibular disturbances; marketed among others as Antivert®, Bonamine®, Dramamine II®, Postafen®, Sea‐Legs®. |
| • alimemazine (EURD 1956) | R06AD01d | 7237 | Also known as trimeprazine; phenothiazine derivative, used for the management of urticaria and pruritus; marketed among others as Nedeltran®, Temaril®, Theraligene®, Vallergan®. |
| • hydroxyzine (FDA 1956) | N05BB01f N05BB51e , f | 7199 | Diphenylmethane derivative synthesized by UCB and marketed by Pfizer in the USA as Atarax®; still widely used today; indicated for the management of anxiety and pruritus; restrictions to minimize cardiac risks released by the EMA in March 2015; also sold as Vistaril®. |
| • brompheniramine (FDA 1957) | R06AB01d R06AB51d , e | 7133 | Substituted alkylamine introduced by Wyeth as Dimetane®; combined preparations marketed among others as Bromfed®, Dimetapp®. |
| • dimetindene (EURD 1961) | D04AA13c R06AB01d | a | Substituted alkylamine developed by Ciba in the early 1960s as su‐6518 (dimethpyrindene, Forhistal®); marketed among others as Fenistil®, Foristal®. |
| • cyproheptadine (EURD 1961, FDA 1961) | R06AX02d | 277 | Also known as cyproheptidine; introduced by Merck as Periactin®; sold among others as Peritol®. |
| • clemastine (EURD 1966, FDA 1977) | D04AA14c R06AA04d R06AA54d , e | 6063 | Also known as meclastin; aminoalkyl ether introduced by Sandoz as HS 592; approved as Tavist® (Novartis); sold among others as Tavegil®. |
| • ketotifen (EURD 1977, FDA 1999) | R06AX17d S01GX08g | 7206 | Developed by Sandoz as HC 20511; inhibits PDE and inflammatory mediator release; marketed among others as Totifen®, Zaditen®, Zaditor®. |
| Second‐generation H1 antihistamines | |||
| • terfenadine (FDA 1985) withdrawn | R06AX12d | 2608 | First non‐sedating H1 antihistamine discovered in the early 1970s (RMI 9918) and introduced in the USA by Hoechst Marion Roussel as Seldane®; prodrug of fexofenadine; withdrawn in 1997 due to risk of QT interval prolongation and related potentially fatal ventricular arrhythmias; has also been marketed as Teldane®, Triludan®. |
| • cetirizine (EURD 1986, FDA 1995) | R06AE07d | 1222 | Piperazine derivative; active metabolite of hydroxyzine; registered in Belgium by UCB as Zyrtec®, reaching blockbuster drug status; also sold under various brand names (e.g. Reactine®). |
| • astemizole (FDA 1988) withdrawn | R06AX11d | 2603 | Developed in 1977 by Janssen Pharmaceutica (R43512); introduced in the UK and the USA in 1983 and 1988, respectively as Hismanal®; withdrawn in 1999 because of serious adverse cardiovascular adverse reactions. |
| • quifenadine (a) | R06AX31d | a | Developed by M.D. Mashkovsky (Phencarol®) in the late 1970s and marketed mainly in the former Soviet Republics. |
| • acrivastine (EURD 1990) | R06AX18d | a | Developed in the 1980s by Wellcome (BW 825C); available as plain preparations (e.g. Benadryl Allergy Relief®) or in combination with the nasal decongestant pseudoephedrine (e.g. Semprex‐D®). |
| • azelastine (EURD 1990, FDA 1996h) | R01AC03h R06AX19d S01GX07g | 7121 | Developed in the 1970s as Asta Pharma AG compound A 5610; nasal spray and eye drops formulations available; sold as plain products (e.g. Afluon®, Allergodil®, Astelin®, Optivar®, Rhinolast®) or in combination with corticosteroids (e.g. Dymista®). |
| • loratadine (EURD 1991, FDA 1993) | R06AX13d | 7216 | Developed in the late 1970s by Schering‐Plough (SCH 29851) and filed in 1981; marketed as Claritin®, achieving blockbuster drug status; representative H1 antihistamine in the WHO Model List of Essential Medicines 2013–2017b. |
| • levocabastine (EURD 1993, FDA 1993) | R01AC02h S01GX02g | 1586 | Discovered at Janssen Pharmaceutica in late 1970s; nasal and ophthalmic formulations marketed as Livostin®; also selective antagonist for the neurotensin receptor NTS2. |
| • mizolastine (EURD 1995) | R06AX25d | a | Developed by Synthelabo (SL 85.0324) and marketed as Mizollen®. |
| • ebastine (EURD 1996) | R06AX22d | a | Metabolized by CYP3A and CYP2J2 to the active metabolite carebastine; patented in the mid‐1980s and marketed by Almirall B. V as Kestine® in Europe; not approved for marketing in the US in 1999 because of cardiac safety concerns and significant pharmacokinetic interactions. |
| • fexofenadine (EURD 1996, FDA 1996) | R06AX26d | 4819 | Active metabolite of terfenadine generated primarily via CYP3A4 metabolism; identified by Sepracor and licensed to Hoechst Marion Roussel in 1993; first launched in the USA as Allegra®, achieving blockbuster drug status. |
| • olopatadine (FDA 1996, EURD 2002) | R01AC08h S01GX09g | 7249 | Developed by Kyowa Hakko, Japan (KW 4679); nasal and ophthalmic formulations available; sold as Patanol®, Pazeo®. |
| • emedastine (FDA 1997, EURD 1999) | S01GX06g | 7174 | First approved in the USA as an ophthalmic formulation marketed by Alcon laboratories as Emadine®. |
| • desloratadine (EURD & FDA 2001) | R06AX27d | 7157 | Active metabolite of loratadine produced primarily via CYP3A4 and CYP2D6 metabolism; identified by Sepracor and licensed to Schering‐Plough in 1997 (descarboethoxyloratadine, SCH 34117); brought to market as Clarinex® in the USA and as Aerius® in Europe; also sold as Neoclarityn® (MSD). |
| • levocetirizine (EMA 2001, FDA 2007) | R06AE09d | 1214 | Piperazine derivative; the (R)‐enantiomer of cetirizine licensed to UCB by Sepracor in 1999; first launched in Germany and marketed as Xusal® and Xyzal®. |
| • epinastine (EURD 2002, FDA 2003) | R06AX24d S01GX10g | 7176 | Developed by Boehringer Ingelheim (WAL 801 CL); launched in Japan; marketed as Alesion®, Elestat®. |
| • rupatadine (EURD 2005) | R06AX28d | a | Developed by J. Uriach y Cia (UR‐12592); possesses dual affinity for histamine H1 and PAF receptors; undergoes hepatic metabolism by CYP3A4; desloratadine is a metabolite; first launched in 2003 in Spain and marketed as Rupafin®; also sold as Rupall® and Urtimed®. |
| • bilastine (EMA 2010) | R06AX29d | a | Developed by FAES Farma in Spain and marketed as Bilaxten®; also sold among others as Bilaz®, Bilidren®, Blexten®, Ilaxten®. |
| • alcaftadine (FDA 2010) | S01GX11g | 7587 | Developed by Janssen Cilag (R89674); topical ophthalmic solution approved for Vistakon Pharmaceutical and marketed as Lastacaft®. |
| Drugs targeting the histamine H2 receptor (mainly indicated for the treatment of peptic ulcer and GORD) | |||
| • cimetidine (EURD 1976, FDA 1977) | A02BA01 A02BA51e | 1231 | Imidazole derivative synthesized in 1972 (SK&F 92334); first clinically used H2 receptor antagonist inhibiting gastric acid secretion; introduced in the UK as Tagamet® reaching blockbuster drug status; inhibits several CYP isoforms. |
| • ranitidine (FDA 1977; EURD 1981) | A02BA02 | 1234 | Substituted furan discovered at Glaxo‐Allenburys Research (AH 19065); first introduced in the UK in 1976 and marketed as Zantac®; listed under the antiulcer medicines in the WHO Model List of Essential Medicines 2002–2017b. |
| • famotidine (EURD 1984, FDA 1986) | A02BA03 A02BA53e | 7074 | Guanidinothiazole derivative developed by Yamanouchi Pharmaceutical Co. (YM‐11170), licensed by Merck & Co (MK‐208) and marketed as Pepcid®; also sold among others as Amfamox®, Famocid®, Famodil®, Gaster®, Peptan®. |
| • nizatidine (EURD 1985, FDA 1988) | A02BA04 | 7248 | Developed by Eli Lilly (LY 139037); marketed as Axid®, Tazac®; last new histamine H2 receptor antagonist approved for clinical use prior to omeprazole (A02BC01, Losec®), the first of the PPIs reaching the clinic (EURD 1987, FDA 1989). |
| • roxatidine (a) | A02BA06 | a | Introduced in 1980s (roxatine acetate; TZU 0460, Teikoku; HOE 760, Hoechst‐Roussel Pharmaceuticals) and marketed in a number of countries as Altat®, Rotane®. |
| • lafutidine (a) | A02BA08 | a | Introduced in the 1990s as FRG‐8813 by Fujirebio Inc., Japan; marketed in Japan by UCB since 2000 as Stogar®; also sold as Lafaxid®. |
| Drugs targeting the histamine H3 receptor | |||
| • pitolisant (EMA 2016) | N07XX11 | 8924 | First‐in‐class drug acting on histamine H3 receptors as antagonist/inverse agonist; N‐piperidyl derivative developed by the groups of Jean‐Charles Schwartz and Walter Schunack and introduced by Bioprojet, France as BF2.649; formerly designated tiprolisant by the WHO; orphan designation granted in EU in 2007 (EU/3/07/459) and by the FDA in 2010; first authorized in the EU for the treatment of narcolepsy with or without cataplexy marketed as Wakix®; Breakthrough Therapy and Fast Track designations granted by the FDA in 2018. |
| Compounds targeting the histamine H4 receptor (currently under clinical trials) | |||
| • seliforant (a) | a | a | Investigational histamine H4 receptor antagonist originally developed by Palau Pharma in Spain as UR‐63325; first in man histamine H4 receptor antagonist that entered clinical trials in 2010 for seasonal allergic rhinitis; subsequently licensed by the French biotech Sensorion as SENS‐111; INN granted in 2018; currently tested in Phase II clinical trials for the treatment of acute unilateral vestibulopathy, sponsored by Sensorion and registered in the EU and the USA. |
| • adriforant Proposed INN: List 199 (a) | a | 8985 | Investigational histamine H4 receptor antagonist originally developed by Pfizer UK as PF‐3893787 and subsequently licensed by the British biopharmaceutical firm Ziarco as ZPL‐389; currently tested in Phase II proof of concept clinical trials for the treatment of moderate to severe atopic dermatitis, sponsored by Novartis and registered in the EU and the USA. |
The substances are presented by their official international nonproprietary (INN) or generic name designated by the World Health Organization (WHO). The ATC code corresponds to the 5‐level hierarchical Anatomical Therapeutic Chemical (ATC) classification as defined by the WHO (WHO, 2018). Ligand ID refers to the classification of the IUPHAR/BPS Guide to PHARMACOLOGY developed jointly by the International Union of Basic and Clinical Pharmacology (IUPHAR) and the British Pharmacological Society (BPS) (Alexander et al., 2017a).
CYP, cytochrome P 450; CYP3A enzymes are of major importance in drug metabolism hence playing a significant role in drug interactions; EURD, European Union (EU) Reference Date maintained by the European Medicines Agency (EMA) corresponding to the date of the first marketing authorization in the EU or to the earliest known date of marketing authorization; FDA, U.S. Food and Drug Administration; MSD, Merck Sharp & Dohme; PPIs, proton pump inhibitors; UCB, Union Chimique Belge.
Not available.
Medicines satisfying the priority health care needs of the population and selected with due regard to public health relevance, evidence on efficacy and safety, and comparative cost‐effectiveness.
Dermatological preparation.
Antihistamine for systemic use.
Combinations.
Anxiolytic.
Ophthalmological preparation.
Nasal preparation.
Looking back at the major historical milestones, the phases of our understanding of histamine pharmacology span the 20th century and the beginning of the new millennium (Figure 1). Inspired predominantly by the work of the prominent members originally of the European (EHRS) and subsequently also of the Japanese (JHRS) Histamine Research Society, this review focuses on the critical and comprehensive description of the past and present developments and limitations in histamine pharmacology. The drug and molecular target nomenclature conforms to the Concise Guide to Pharmacology available in the British Journal of Pharmacology (Alexander et al., 2017a). The perspective on past work is complemented by the illustration of the current state‐of‐the‐art in the field in an effort to stimulate helpful insights for the translation of preclinical data into promising therapies for unmet medical needs.
The early years 1900–1950
The discovery of histamine and its physiological importance
Early on, histamine was referred to by its chemical name, β‐imidazolylethylamine (Dale and Laidlaw, 1910). Soon afterwards, the name ‘histamine’ was adopted to denote its derivation from histidine (from the Greek word for tissue ‘histos’, ιστός) (Dale and Richards, 1918). The chemical synthesis of histamine (Windaus and Vogt, 1907) and its production from histidine by putrefactive bacteria (Ackermann, 1910) were reported in the first decade of the 20th century, before the recognition of its biological significance. Shortly afterwards, along with its isolation from the extract of the rye fungus ergot (Claviceps purpurea; Ergotinum dialysatum of Wernich), Sir Henry H. Dale working with George Barger and Sir Patrick Laidlaw at the Wellcome laboratories in London, UK, pioneered the investigation of histamine physiology by conducting a series of biological assays in frogs, rodents, cats and dogs (Barger and Dale, 1910; Dale and Laidlaw, 1910). This early work of Sir Henry H. Dale represents one of the fundamental pillars in histamine research and is honoured during the annual meetings of the EHRS, where the participants sing the Society's unique Anthem at the farewell dinner (EHRS, 2018).
Consistent with the related literature available to date (Ennis et al., 1981; Coruzzi et al., 2012; Strasser et al., 2013), the actions of histamine reported over 100 years ago were described as ‘somewhat complicated’, exhibiting variations between the ‘different organs and in different species’ (Dale and Laidlaw, 1910). These included smooth muscle contraction, vasodilatation and depression of the CNS (Dale and Laidlaw, 1910). Interestingly, attention was drawn to the ability of histamine to mimic the immediate symptoms of hypersensitivity reactions (Dale and Laidlaw, 1910), which had been described as anaphylaxis (Portier and Richet, 1902) or allergy (von Pirquet and Schick, 1905) a few years earlier. After more than a decade, subcutaneous histamine administration was shown to elicit the triple response or wheal‐and‐flare reaction in the skin, characterized by vasodilatation, increased vascular permeability and axon reflex (Lewis and Grant, 1924).
The interest in histamine during the first half of the 20th century was intensified when the endogenous amine was isolated from normal tissues, thus documenting its physiological relevance (Best et al., 1927). Consequently, its mediator role in anaphylaxis was confirmed (Watanabe, 1931), and its contribution to the stimulation of hydrochloric acid secretion in the stomach was revealed (Popielski, 1920).
The emergence of antihistamines for the management of allergic disorders
The receptor theory of drug action was essentially formulated during the first three decades of the 20th century through the endeavours of Paul Ehrlich in Germany and John Newport Langley, Alfred Joseph Clark and Sir John Henry Gaddum in England (Clark, 1933; Rang, 2006). Towards this end, Paul Ehrlich characterized mast cells (Ehrlich, 1879) and introduced the term ‘receptor’ to refer to the diverse cellular ‘receptive substances’ that were assumed by J.N. Langley to dose‐dependently mediate the antagonistic effects of pharmacologically active compounds (Ehrlich and Morgenroth, 1900).
The search for antagonists to block the actions of histamine in anaphylaxis was undertaken at the Institut Pasteur in Paris, France, by Daniel Bovet using the bank of compounds of Ernest Fourneau (Fourneau and Bovet, 1933). The work on the pharmacology of cholinergic antagonists and antihistamines led by D. Bovet set the stage for the rational synthesis and testing of pharmacologically active agents (Bovet, 1950). The Nobel Prize in Physiology or Medicine was awarded to Daniel Bovet in 1957 ‘for his discoveries relating to synthetic compounds that inhibit the action of certain body substances, and especially their action on the vascular system and the skeletal muscles’.
The definition of the H1 receptor remained elusive until the mid‐1960s (Ash and Schild, 1966). However, the development of antihistamine drugs acting at a hitherto uncharacterized receptor marked the evolution of histamine pharmacology in the 1930s (Emanuel, 1999). The first substance with recognized antihistamine properties was the sympatholytic phenolic ether thymoxyethyl‐diethylamine (2‐isopropyl‐5‐methyl phenoxyethyl diethylamine; 929 F) that protected guinea pigs from histamine‐induced anaphylaxis (Fourneau and Bovet, 1933; Staub and Bovet, 1937). The toxicity of 929 F prevented its clinical use, but its thorough investigation in 1937 ascertained the existence of substances with antagonistic activity towards histamine (Staub and Bovet, 1937; Bovet, 1950). These discoveries paved the way for the development of phenbenzamine (N′‐benzyl‐N,N‐dimethyl‐N′‐phenylethane‐1,2‐diamine; RP 2339) that was patented by Rhône‐Poulenc in France and marketed as Antergan® (Bovet, 1950) (Table 1). Phenbenzamine was the first antihistamine successfully tested in humans in 1942 for the symptomatic relief of allergic manifestations (Halpern, 1942). Subsequently, mepyramine (pyrilamine; N‐p‐methoxybenzyl‐N‐dimethylaminoethyl‐α‐aminopyridine; RP 2786) was released in France as Neo‐Antergan® (Bovet et al., 1944) (Table 1).
In parallel, diphenhydramine (β‐dimethylaminoethyl benzhydryl ether) was developed by George Rieveschl, while he was a chemical engineering professor at the University of Cincinnati, Ohio in the USA and first marketed in 1946 as Benadryl® by Parke, Davis and Co. (Loew et al., 1945). Dimenhydrinate (Dramamine®, Gravol®, Vertirosan®, Vomex‐A®), the 8‐chlorotheophyllinate salt of diphenhydramine, was marketed in 1947 by G.D. Searle and Co. (Table 1) and is still in use for the prevention and relief of nausea and vomiting mostly associated with motion sickness (Parsons and Ganellin, 2006). At that time, many other new substances with antihistamine activity were developed for the management of conditions such as allergic rhinitis, conjunctivitis and urticaria (Table 1). These include promethazine (3277 RP; Phenergan®), hydroxyzine (Atarax®, Vistaril®), chlorpheniramine (Chlor‐trimeton®), dimethpyrindene (dimethindene; Fenistil®, Foristal®), and the list is continuing to expand (Table 1) (Emanuel, 1999; Church and Church, 2013). Several of the older antihistamines are still marketed as prescription or over‐the‐counter medicines, as exemplified by mepyramine cream preparations (Anthisan®).
The side effects of these ‘classical’ or ‘first‐generation’ H1 antihistamines were identified soon after their entry into the clinic in the 1940s. Sedation was a major limitation for their clinical use (Feinberg, 1947). Therefore, they are commonly referred to as ‘sedating’ H1 antihistamines (Timmerman, 1999; Church and Church, 2013). Additionally, drowsiness, dizziness, fatigue, interference with cognitive processes, headache, dry mouth and gastrointestinal disturbances were commonly observed, yet with variable incidence and severity among patients and drugs (Feinberg, 1947).
The central effects of H1 antihistamines have been attributed to their non‐polar and highly lipophilic structure that allows penetration through the blood–brain barrier (BBB) into brain, where they can potentially cross‐react with receptors of amine transmitters (Church and Church, 2013). Hence, they may elicit anti‐α‐adrenoceptor and anti‐5‐HT (serotonin) effects and, by occupying more than 50% of the histamine H1 receptors therein (Yanai et al., 2011), they interfere with the H1‐mediated excitatory histaminergic transmission and arousal in the circadian sleep/wake cycle (Church and Church, 2013). In addition, the sedative effect of the first‐generation H1 antihistamines seems to be related to their lack of interaction with P‐glycoprotein (P‐gp) that pumps drugs out of the BBB, contrary to most of the second‐generation H1 antihistamines (see below) (Hu et al., 2015). Moreover, considering the phylogenetic link of the H1 receptor to the muscarinic acetylcholine receptors (Leurs et al., 2000) and the evolution of the first‐generation H1 antihistamines from chemical compounds with cholinergic activity (Bovet, 1950), it comes as no surprise that these drugs have low H1 receptor selectivity and commonly elicit anti‐muscarinic effects (Church and Church, 2013).
The ‘refinement’ period 1950–1970
The heyday of histamine physiology and pharmacology in the first four decades of the 20th century (Dale, 1950) was followed by the considerable advances in histamine biochemistry (Schayer, 1956), as well as in the optimization mostly of fluorometric and radioenzymatic techniques for histamine quantification in biological samples (Shore et al., 1959; Snyder et al., 1966). Nevertheless, the discovery of the storage of histamine in mast cells (Riley and West, 1953), based mainly on studies conducted with the histamine liberators such as compound 48/80 (Paton, 1951), and the concept of histamine receptor differentiation into at least two classes (Ash and Schild, 1966) highlighted this period of histamine research.
Mast cells and basophils as professional cellular sources of histamine
An influential discovery in understanding the role of histamine in immunological processes was described by James F. Riley and Geoffrey B. West as the ‘intimate relationship’ of histamine with tissue mast cells (Riley and West, 1953). Actually, G.B. West was one of the founders of the EHRS and served as the Secretary‐General of the Society between 1978 and 1987 (Figure 3). Since 1992, the ‘GB West lecture’ is delivered during the annual EHRS meetings by a distinguished scientist who has contributed significantly to the field of histamine research (Table 2), in honour of the EHRS co‐founder and Late Honorary Member G.B. West (Figure 3) (EHRS, 2018).
Figure 3.
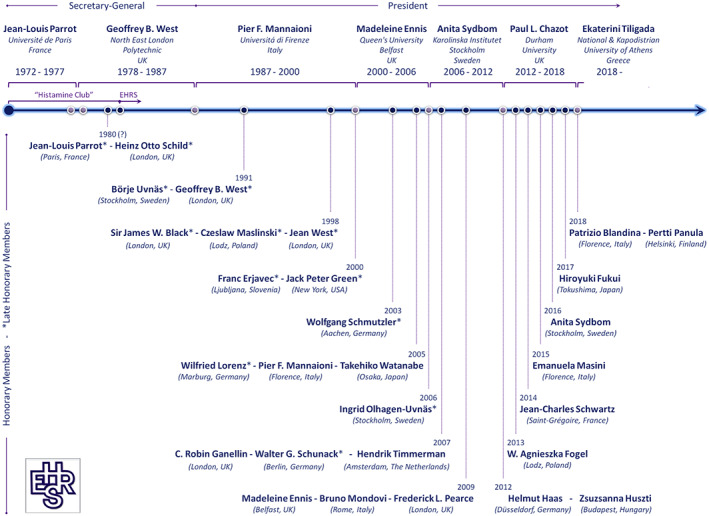
Historical features of the EHRS portraying the Honorary and Late Honorary Members and illustrating in chronological order the members who served as Secretary‐General and President of the Society since 1972. Honorary membership is the highest award of the EHRS, only given to very special people who contributed significantly to the Society and histamine research (EHRS, 2018). The members' place of work is shown in brackets. The EHRS logo is presented in the bottom left hand corner.
Table 2.
The ‘GB West lectures’ are delivered since 1992 during the Annual Meetings of the EHRS in honour of the EHRS co‐founder Geoffrey B. West (EHRS, 2018)
| Annual EHRS meeting | GB West lecture |
|---|---|
| Year, location (host) | Title, lecturer (affiliation) |
|
1992 Malaga, Spain (M. García‐Caballero) |
Highly potent and selective ligands for histamine receptor subtypes Walter Schunack (Freie Universität Berlin, Germany) |
|
1993 Cologne, Germany (E. Neugebauer) |
Histamine the heart of the matter Roberto Levi (Weill Cornell Medicine, NY, USA) |
|
1994 Budapest, Hungary (Z. Huszti) |
Histaminergic neuron system in brain: recent advances Takehito Watanabe (Tohoku University School of Medicine, Sendai, Japan) |
|
1995 Moscow, Russia (I.S. Gushchin) |
Design of H3‐receptor agonists and antagonists Walter Schunack (Freie Universität Berlin, Germany) |
|
1996 Antwerp, Belgium (F.J. van Overveld) |
The role of allergy in asthma – Implications for proper asthma management Michael A. Kaliner (Institute for Asthma and Allergy, Washington, DC, USA) |
|
1997 Seville, Spain (F. Lopez Elorza) |
Histamine and antihistamine drugs in septic shock Edmund Neugebauer (University of Cologne, Germany) |
|
1998 Lodz, Poland (A. Fogel) |
Mast cell heterogeneity Frederick L. Pearce (University College London, UK) |
|
1999 Lyon, France (P. Belon) |
Clinical significance of mast cell heterogeneity Madeleine Ennis (Queen's University Belfast, UK) |
|
2000 Nemi, Italy (B. Mondovi) |
Paradigms in histamine research: many controversies – many solutions Wilfried Lorenz (University of Marburg, Germany) |
|
2001 Turku, Finland (P. Panula) |
Histamine's wake Helmut L. Haas (Heinrich‐Heine‐Universität, Düsseldorf, Germany) |
|
2002 Eger, Hungary (A. Falus) |
Getting away from histamine C. Robin Ganellin (University College London, UK) |
|
2003 Noordwijkerhout, The Netherlands (H. Timmerman & R. Leurs) |
The role of chronic inflammation and remodelling in the pathogenesis of persistent asthma Stephen T. Holgate (University of Southampton, UK) |
|
2004 Bergisch‐Gladbach, Germany (H. Haas) |
The riddle of the mast cell receptors in the immune response Pier F. Mannaioni (University of Florence, Italy) |
|
2005 Bled, Slovenia (L. Stanovnik) |
My journey with histamine from the cardiovascular system to the brain Marija Čarman‐ Kržan (University of Ljubljana, Slovenia) |
|
2006 Delphi, Greece (E. Tiligada) |
Histamine and systems biology; genes and genomics beyond genes András Falus (Semmelweis University, Budapest, Hungary) |
|
2007 Florence, Italy (E. Masini) |
And then there were four … Rob Leurs (Vrije Universiteit Amsterdam, The Netherlands) |
|
2008 Stockholm, Sweden (S.‐E. Dahlén & A. Sydbom) |
Mast cells, nerves and IgE John Bienenstock (McMaster University, Hamilton, Ontario, Canada) |
|
2009 Fulda, Germany (F. Diel) |
Flexibilities within and around hematopoietic cell development Fritz Melchers (Max Planck Institute for Infection Biology, Berlin, Germany) |
|
2010 Durham, UK (P.L. Chazot) [Joint meeting of EHRS and COST Action BM0806] |
Clinical applications of pitolisant (BF2.649), an inverse agonist at the H3 receptor Jean‐Charles Schwartz (Bioprojet Biotech, Saint‐Grégoire, France) |
|
2011 Sochi, Russia (R. Khanferyan) |
The role of the histamine H4 receptor in allergy and inflammation Robin L. Thurmond (Johnson & Johnson Pharmaceutical R&D, San Diego, CA, USA) |
|
2012 Belfast, UK (M. Ennis) [Joint meeting of EHRS and COST Action BM0806] |
Interactions between mast cells and structural airway cells in the pathogenesis of asthma Peter Bradding (University of Leicester, UK) |
|
2013 Lodz, Poland (A. Fogel) |
Histamine synthesis and its functions in murine mast cells Satoshi Tanaka (Okayama University, Japan) |
|
2014 Lyon, France (J.‐S. Lin) |
Neuroimaging studies in humans on the histaminergic neuron system: past, present and future Katzuhiko Yanai (Tohoku University School of Medicine, Sendai, Japan) |
|
2015 Malaga, Spain (F.M. Sanchez Jimenez) |
Histamine and Relaxin: an intriguing connection Emanuela Masini (University of Florence, Italy) |
|
2016 Florence, Italy (E. Masini & B. Passani) |
The role of histamine in the memory of emotionally‐salient experiences Patricio Blandina (University of Florence, Italy) |
|
2017 Amsterdam, The Netherlands (R. Leurs) [1st joint meeting of the EHRS the Japanese Histamine Research Society (JHRS)] |
The functional significance of the histaminergic neuron system: Lessons from gene knockout mice and positron emission tomography Katzuhiko Yanai (Tohoku University School of Medicine, Sendai, Japan) |
|
2018 Dublin, Ireland (A. Sasse) |
Three out of four: A 25 year relationship with histamine receptors Nicholas Carruthers (Janssen R&D, San Diego, CA, USA) |
COST, European Cooperation in Science and Technology.
Mast cells had been characterized 70 years earlier by Paul Ehrlich (Ehrlich, 1879) and they were linked to anaphylactic shock in the 1940s (Jacques and Water, 1941). Interestingly, although a strong positive correlation was reported between histamine and mast cells in various normal and pathological tissues, it was suggested that other cells may also store histamine (Riley and West, 1953). In fact, further to mast cells, basophils (Graham et al., 1955), enterochromaffin‐like cells in the stomach (Rubin and Schwartz, 1979) and histaminergic neurons in the CNS (Watanabe et al., 1984) are considered as typical cellular sources of histamine (Figure 2).
Numerous subsequent studies have shown the presence of histamine in a range of other immune and non‐immune cells and tissues of various species, under physiological and pathological conditions (Shore et al., 1959; Zimmermann et al., 2011). A non‐exhaustive list of additional histamine sources comprises platelets (Mannaioni et al., 1995), white blood cells such as polymorphonuclear neutrophils, monocytes and lymphocytes (Zwadlo‐Klarwasser et al., 1994), chondrocytes (Tetlow and Woolley, 2004), vascular smooth muscle and endothelial cells (Tippens and Gruetter, 2004), as well as epithelial cells including dermal (Gutowska‐Owsiak et al., 2014) and oral (Salem et al., 2017) keratinocytes (Figure 2).
However, the pluripotent mast cell and the basophil represent the primary histamine source in the immune system (Borriello et al., 2017; Varricchi et al., 2018). Thus, they are considered as ‘professional’ histamine‐synthesizing cells. Notably, these cells display considerable morphological, structural, biochemical and pharmacological heterogeneity not only between them but also across different tissue locations and species (Ennis and Pearce, 1980; Varricchi et al., 2018). Yet one of the hallmarks of these cells is that they store histamine in cytoplasmic granules, where it is associated with anionic proteoglycans and released by exocytosis upon cellular activation by a plethora of immune and non‐immune stimuli (Borriello et al., 2017). These include allergens, drugs, mechanical stimuli, cold, UV radiation, endogenous components such as the complement anaphylatoxin C5a and substance P, as well as different bacterial products that bind to the Toll‐like receptors (Cross et al., 1996; Borriello et al., 2017; Olivera et al., 2018). Other preformed and newly formed mediators are differentially secreted from the professional histamine‐synthesizing cells, including heparin, proteases, leukotrienes, prostaglandins and a wide repertoire of cytokines and chemokines (Ennis et al., 1984; Varricchi et al., 2018). Moreover, evidence argues for the metabolic interplay between histamine and other biogenic amines, such as 5‐HT (serotonin) and polyamines in mast cells (Acosta‐Andrade et al., 2016). These complex processes provide examples of how the micro‐environment may govern the involvement of histamine in driving the inflammatory process, including adaptive, innate and autoimmune responses (Forsythe et al., 1999; Zampeli et al., 2009a; Papathanassiou et al., 2011; Borriello et al., 2017; Olivera et al., 2018).
By studying its actions 70 years ago, Sir Henry H. Dale suggested that extrinsic tissue histamine diffuses before exerting its effects, whereas intrinsic histamine – secreted by antigen–antibody reactions – may act even on the cells that it is released from, which had not been identified at the time (Dale, 1948). More recent research challenges the hypothesis that histamine release may be regulated by autocrine negative feedback pathways that are mediated via H1 receptors (Levi‐Schaffer and Eliashar, 2009) and H4 receptors (Hofstra et al., 2003) in mast cells, and via H2 receptors in basophils (Lichtenstein and Gillespie, 1973; Novak et al., 2012). Furthermore, pharmacological evidence implicates the H2 receptors in the autocrine, negative modulation of cardiac anaphylaxis, whereas presynaptic H3 receptors are likely to serve a modulatory role in cardiac function (Bani et al., 2006). Interestingly, H3 and H4 receptors seem to exert differential automodulating actions on tissue histamine levels in health and disease, with or without the involvement of professional histamine‐synthesizing cells (Zampeli et al., 2009b; Kyriakidis et al., 2015; Salem et al., 2017).
The designation of the histamine H1 receptor
In 1960, Ullrich Trendelenberg first used the pA2 criterion defined by Heinz Otto Schild (Rang, 2006) to suggest that the cardiac histamine receptors differ from those antagonized by antihistamines in the smooth muscle (Trendelenburg, 1960). Six years later, A.S. Ash and H.O. Schild postulated that the actions of a series of histamine analogues in gastric acid secretion, in the rat uterus and in the guinea pig ileum were likely to be mediated by at least two distinct types of histamine receptors (Ash and Schild, 1966). Hence, the receptors that were specifically antagonized by low concentrations of antihistamine drugs, such as mepyramine, were designated as H1 (Ash and Schild, 1966). Following this notation, the histamine receptors have since been classified as H1–4 (Alexander et al., 2017a) (Figure 2).
The ‘rises and falls’ 1970–2000
The prototype Η2 receptor antagonist and the treatment of peptic ulcers
In addition to the clinical exploitation of the classical H1 antihistamines for the management of allergic diseases, the interest of the pharmaceutical industry was expanded to the investigation of the putative histamine H2 receptor as a drug target for controlling gastric acid secretion in peptic ulcers and gastro‐oesophageal reflux disease (GORD) (Ganellin, 2011).
Following the development of the β‐adrenoceptor antagonist propranolol (ICI 45520; Inderal®) that changed the face of cardiovascular medicine, the EHRS Late Honorary Member Sir James W. Black moved from the Imperial Chemical Industries' pharmaceutical research centre in Cheshire, UK, to the SmithKline and French laboratory in Hertfordshire, UK. He was joined by the pharmacologist Michael Parsons and the chemists Graham Durant, John Emmett and Robin Ganellin, an EHRS Honorary Membership since 2007 (Figure 3). Almost 20 years before the cloning of the human H2 receptor (Gantz et al., 1991a), this interdisciplinary team not only placed histamine in the forefront of the control of gastric acid secretion but also contributed greatly to the rational approach of drug design and development (Ganellin, 2011). In a series of bioassays – by comparing the histamine responses antagonized by drugs such as mepyramine to those that were refractory to the antihistamines available –, the development of the thiourea derivative burimamide confirmed the heterogeneity of histamine receptors and represented a new class of drugs that served to define the second type of histamine receptors (Black et al., 1972; Parsons and Ganellin, 2006; Ganellin, 2011) (Figure 2). Interestingly, after the characterization of the H3 and the H4 receptors, burimamide was found to block both H2 and H3 receptors (Parsons and Ganellin, 2006) and to be an agonist at the human H4 receptor (Lim et al., 2005). The Nobel Prize in Physiology or Medicine 1988 was awarded jointly to Sir James W. Black, Gertrude B. Elion and George H. Hitchings ‘for their discoveries of important principles for drug treatment’.
The efforts to improve the potency of burimamide when given orally led to metiamide, which was withdrawn during early clinical trials due to the occurrence of reversible granulocytopenia (Ganellin, 2011). Subsequently, the new imidazole derivative cimetidine (SK&F 92334) inhibited the stimulated and basal acid secretion by antagonizing the H2 receptor and showed clinical efficacy in gastric acid‐related diseases (Brimblecombe et al., 1975; Ganellin, 2011). Cimetidine was introduced in the UK under the brand name Tagamet® (derived from anTAGonist and ciMETidine) in November 1976 and in the USA less than a year later (Table 1). Tagamet was the world's first ‘blockbuster’ drug that generated $14 billion during its years of patent protection.
By revolutionizing the treatment of peptic ulcer and GORD, cimetidine guided a new era of histamine pharmacology. The original breakthrough was rapidly followed by the discovery of the substituted furan ranitidine (AH 19065; Zantac®), an H2 receptor antagonist with improved pharmacokinetic properties and fewer side effects and drug interactions (Bradshaw et al., 1979). Ranitidine was launched in 1981in the UK and in Italy and became world's best‐selling drug by 1986, leaving cimetidine behind. The addition of famotidine (MK‐208, YM‐11170, Pepcid®) and nizatidine (Axid®, Tazac®) complete the armamentarium of the major clinically exploited H2 antagonists (Table 1). Although, these drugs are still in use, the introduction of the proton pump inhibitors in 1988 and their mainstay in the management of acid‐related disorders ended the glory of the therapeutic exploitation of H2 antagonists in gastroenterology (Strand et al., 2017). However, the role of distinct histamine receptor subtypes in gastrointestinal pathologies other than acid‐related disorders remains a largely unexplored research area, mostly associated with the complex and species‐specific effects of histamine on smooth muscle (Poli et al., 2006).
The development of second‐generation H1 antihistamines
The efficacy of H1 antihistamines in managing many allergic disorders stimulated the search for compounds with less adverse effects (Simons and Simons, 2011; Church and Church, 2013). Structural changes in the molecules of classical antihistamines led to the emergence of the more hydrophilic and longer acting ‘second‐generation’ Η1 antihistamines, the mainstay of treatment for urticaria (Simons and Simons, 2011; Church and Church, 2013). These drugs were characterized by low interaction with other biogenic amine systems and by limited penetration of the BBB, mainly due to their high affinity for the P‐gp efflux transporter (Timmerman, 1999; Hu et al., 2015). It should be noted that, although some of them have the potential to cause various degrees of somnolence, they are commonly referred to as ‘non‐sedating’ rather than ‘minimally sedating’ H1 antihistamines (Hu et al., 2015).
Among the first second‐generation H1 antihistamines marketed were terfenadine (RMI 9918; Seldane®, Triludan®, Teldane®) and astemizole (R43512; Hismanal®) that were launched in the 1980s (Table 1) (Simons and Simons, 2011). They were followed by a growing list of compounds that exhibit differences mainly in their pharmacokinetic profile (Church and Church, 2013). The second‐generation H1 antihistamines are available in various formulations and under a variety of brand names around the world (Table 1). They include the hydroxyzine metabolite cetirizine (Zirtek®, Zyrtec®) and its l‐enantiomer levocetirizine (Xozal®, Xyzal®), loratadine (Claritin®), ebastine (Ebastel®, Kestine®), terfenadine's metabolite fexofenadine (Allegra®), the dual‐acting rupatadine (Rupafin®) that antagonizes the effects of both histamine and PAF (platelet‐activating factor), the metabolite of loratadine and rupatadine desloratadine (Clarinex®, Aerius®, NeoClarityn®) and bilastine (Bilaxten®, Bilargen®, Bilaz®, Ilaxten®).
Despite their clinical success, terfenadine and astemizole were withdrawn by their manufacturers in 1997 and 1999, respectively, due to their association with the occurrence of potentially fatal cardiac arrhythmias (Leurs et al., 2002). Following this drawback, experimental, clinical and epidemiological evidence revealed that some of the older H1 antihistamines may also exhibit cardiotoxicity (Taglialatela et al., 2000). In fact, in March 2015, the European Medicines Agency (EMA) released restrictions to minimize the risks of effects on heart rhythm with hydroxyzine‐containing medicines that are indicated, among others, for the treatment of anxiety and sleep disorders and for pruritus relief in various countries (EMA, 2015). Subsequently, similar warnings were released around the world. Scepticism about the cardiotoxicity of H1 antihistamines inspired a number of investigations. It is now acknowledged that cardiac toxicity is not a class effect and does not occur through the histamine H1 receptor (Church and Church, 2013). Actually, structure–activity studies suggested that the QT‐prolonging drugs have at least one aromatic ring in their structure, which interacts with the voltage‐dependent human ether‐à‐go‐go related (hERG) gene potassium channels that are essential for the cardiac electrical activity (Taglialatela et al., 2000; Leurs et al., 2002).
Moreover, the advances in molecular biology in the 1990s led to the concept of constitutive receptor activity that directed the reclassification of GPCR antagonists as inverse agonists that can reduce constitutive activity, or neutral antagonists, which do not affect the basal receptor activity but interfere with agonist binding (Leurs et al., 2002). Since H1 antihistamines may be inverse agonists or neutral antagonists, this therapeutic drug class is commonly referred to by the suggested term ‘H1 antihistamines’ rather than ‘H1 antagonists’ (Leurs et al., 2002; Church and Church, 2013).
Critical information on the selectivity of H1 antihistamines was obtained when the crystal structure of the human H1 receptor complexed with its inverse agonist doxepin, a first‐generation H1 antihistamine, was determined in 2011 by the Human Receptor Crystallography Project ERATO in Kyoto, Japan (Shimamura et al., 2011). The findings revealed that the H1 receptor is structurally most similar to that of the β2‐ and β1‐adrenoceptors and the dopamine D3 receptor, while it shows larger deviations from the adenosine A2A receptor and the chemokine receptor CXCR4 (Shimamura et al., 2011). The low selectivity of the first‐generation H1 antihistamines for the H1 receptor is likely attributed to their interaction with residues deep inside the ligand‐binding pocket that are highly conserved among amine receptors. On the other hand, the high selectivity of the second‐generation H1 antihistamines stems from the interaction of their carboxyl group with the anion‐binding site near the extracellular part of the receptor, which consists of residues that are specific for the H1 receptor (Shimamura et al., 2011).
The localization of histaminergic neurons in the brain
In the 1960s, the enormous interest of the neuroscientists in o‐phtalaldehyde histochemistry enabled the visualization of biogenic amine systems and their involvement in major neuropsychiatric diseases (Carlsson et al., 1961). Unfortunately, the cross‐reaction of histamine with spermidine held back the investigation of histamine localization in the brain during that time (Haas et al., 2008). Nevertheless, the relatively scarce reports published before the more systematic investigation of brain histamine in the 1970s and 1980s were suggestive of the putative neurotransmitter role of histamine in the CNS (Green, 1970) and its involvement in the regulation of wakefulness (Monnier et al., 1967). In the preceding decades, besides the sedative side effects of the first‐generation H1 antihistamines (Staub and Bovet, 1937), the demonstration of the presence of histamine in the brain of various species, including man, supported the idea of histaminergic nerves (Kwiatkowski, 1943). Systemically applied histamine does not cross the BBB (Parsons and Ganellin, 2006). Yet studies on its metabolism in the brain showed that histamine formation was high in the hypothalamus (White, 1959). The subsequent biochemical advances revealed two cellular pools of histamine in the brain, one of putative neuronal origin exhibiting a rapid turnover and a second showing very slow turnover most probably of mast cell origin (Schwartz, 1975).
In the mid‐1970s, the existence of histaminergic neurons and the novel putative role of histamine as a cerebral neurotransmitter were predicted by lesion studies (Schwartz, 1975), contrary to the widespread belief in neurosciences that restricted the role of neurotransmitters to acetylcholine and the catecholamines (Schwartz, 2011). Indeed, the localization of histamine‐containing neurons was confirmed almost 10 years later. The EHRS Honorary Members (Figure 3) Takehiko Watanabe at Osaka University, Japan and Pertti Panula in Washington DC in the USA identified the histaminergic neurons in the rat brain by histochemical approaches using anti‐HDC antibodies (Watanabe et al., 1983, 1984) and an antiserum against histamine (Panula et al., 1984) respectively. Cell bodies of histaminergic neurons were shown to be localized in the tuberomamillary nucleus (TMN) of the posterior hypothalamus and to send projections to essentially all areas of the CNS, actually contributing to the modulation of alertness, sleep/wakefulness, feeding, endocrine, memory and other processes (Haas and Panula, 2003). It is noteworthy that a prominent transient histamine‐containing neuronal system in the raphe nuclei that projects to forebrain and hindbrain areas and is anatomically distinct from the mature hypothalamic TMN system implies the developmental role of histamine in the brain (Schwartz, 1975; Bessinis et al., 2012; Panula et al., 2014).
It is now established that histamine is produced in the brain of almost all animal species, although substantial inter‐species variations are detected in both the number of histaminergic neurons and the content of neuronal and/or of non‐neuronal histamine (Schwartz, 1975; Haas and Panula, 2003; Nuutinen and Panula, 2010). For example, the histamine‐containing neurons are ~4000 in the rat and ~64 000 in the human TMN (Nuutinen and Panula, 2010). The current knowledge of the histaminergic system and its interactions with other systems in the brain derives predominately from extensive studies in rodents, humans and the zebrafish, as summarized in a number of excellent reviews that are available in the literature (Haas and Panula, 2003; Haas et al., 2008; Nuutinen and Panula, 2010; Sundvik and Panula, 2015; Nieto‐Alamilla et al., 2016; Provensi et al., 2018). In particular, the zebrafish has recently been shown to be a valuable tool for dissecting the role of histaminergic and histaminoceptive neurons in the brain by employing cutting‐edge methodologies, such as genome modification using the CRISPR/Cas9 editing tool (Haas, 2018).
The characterization of H3 receptors
Three years after the report on the H2 receptors by Sir James W. Black (Black et al., 1972), Jean‐Charles Schwartz at Institut National de la Santé et de la Recherche Médicale (INSERM) in Paris, France, suggested that ‘specific receptors to this amine (histamine), as evidenced by electrophysiological and behavioural studies, as well as by the activation of cyclic AMP formation, are present in brain’ (Schwartz, 1975). At a time when the interest of neuropharmacology focused on the characterization of catecholamine autoreceptors, and only months before the report on the visualization of the histaminergic neurons (Watanabe et al., 1983, 1984; Panula et al., 1984), J.‐C. Schwartz working with Jean‐Michel Arrang and Monique Garbarg proposed that presynaptic autoreceptors were able to inhibit histamine release from depolarized slices of rat cerebral cortex (Arrang et al., 1983). These receptors were pharmacologically distinct from the H1 and H2 receptors and they were designated as histamine H3 receptors (Arrang et al., 1983) (Figure 2). EHRS Honorary Membership was awarded to Jean‐Charles Schwartz in 2014 (Figure 3). In later studies, evidence pointed to the presence of H3 postsynaptic receptors or heteroreceptors in different neuronal populations, controlling the release of various neurotransmitters, including histamine, glutamate, γ‐aminobutyric acid (GABA), dopamine, noradrenaline and acetylcholine (Gbahou et al., 2012), thus being implicated in various (patho)physiological conditions (Tiligada et al., 2011; Sundvik and Panula, 2015; Nieto‐Alamilla et al., 2016; Provensi et al., 2018).
The collaborative research of J.‐C. Schwartz at INSERM and Jeanne‐Marie Lecomte at the young research company Bioprojet in Paris with Max Robba at the University of Caen, France and the EHRS Late Honorary Member Walter Schunack (Figure 3) at the University of Berlin, Germany led to the development of H3 receptor ligands that were imperative to explore the function of the novel H3 autoreceptors. These included (R)‐α‐methylhistamine (Rα‐MeHA) and thioperamide, a potent agonist and antagonist, respectively, with high selectivity for the H3 compared to the H1 and H2 receptors (Arrang et al., 1987). Interestingly, following the identification of the H4 receptor in the 2000s (see below), thioperamide was shown to also act as an inverse agonist and Rα‐MeHA as an agonist at the H4 receptor (Lim et al., 2005). The continuing collaborative effort to develop H3 ligands involving additional groups from academia and the pharmaceutical industry in Europe and in the USA did not manage to get a novel drug to the clinic at that point (Parsons and Ganellin, 2006; Schwartz, 2011). However, it contributed greatly to the current understanding of the role of histamine in the sleep–wake cycle and in neuropsychiatric (patho)physiology (Haas et al., 2008; Nuutinen and Panula, 2010; Schwartz, 2011; Sundvik and Panula, 2015; Nieto‐Alamilla et al., 2016; Provensi et al., 2018) and laid the foundation for the prospective exploitation of the histamine H3 receptor as a novel drug target (Kollb‐Sielecka et al., 2017).
In fact, after the deorphanization of the human H3 receptor in 1999 (Lovenberg et al., 1999) and the discovery that inverse agonists were able to reverse the constitutive activity of the H3 receptor, thus triggering the release of endogenous histamine, pitolisant (BF2.649; formerly designated tiprolisant) was selected for further development (Schwartz, 2011). Pitolisant showed promising results in a preclinical model of narcolepsy, a rare disorder characterized, among others, by excessive daytime sleepiness and caused by deficient orexin neurotransmission that affects histaminergic neurons, which are essential for its waking actions (Schwartz, 2011; Sundvik and Panula, 2015). Pitolisant (Wakix®, Bioprojet Pharma) received an orphan designation from the Committee for Orphan Medicinal Products of the EMA in 2007, and it was approved by the EMA in 2016 as a new first‐in‐class H3 inverse agonist for the treatment of narcolepsy with or without cataplexy in adults (Kollb‐Sielecka et al., 2017). Thus, pitolisant became the first Η3 receptor‐targeting compound in clinical use (Table 1).
Cloning of the histamine receptors
The vast body of physiological and pharmacological data that accumulated over the years offered strong support for the (patho)physiological properties of histamine and for the continuous efforts to improve the selectivity and specificity of both clinically and experimentally relevant ligands targeting the H1–H3 receptors (Hill et al., 1997; Parsons and Ganellin, 2006; Seifert et al., 2013). The development of selective radioligands, such as [3H]mepyramine, [125I]iodoaminopotentidine and [3H]Rα‐MeHA significantly aided the studies on the distribution of the histamine H1, H2 and H3 receptors, respectively, in different tissues, whereas a series of biochemical investigations greatly contributed to the elucidation of the signal transduction pathways associated with receptor activation (Hill, 1990; Hill et al., 1997).
However, the structure of the three known types of histamine receptors remained elusive until the last decade of the 2nd millennium, when GPCR cloning had already become a regular exercise in many molecular pharmacology and biochemistry laboratories worldwide (Hill et al., 1997; Hill, 2006; Lefkowitz, 2013). The canine H2 receptor was the first histamine receptor to be cloned (Gantz et al., 1991b), rapidly followed by the human H2 receptor (Gantz et al., 1991b). Within a few years, both the H1 and H2 receptors were cloned from several species (Hill et al., 1997), including the human H1 receptor in 1993 (De Backer et al., 1993). It was shown that the amino acid sequence of the H1 and H2 receptors has the conserved seven transmembrane domain organization of the class A rhodopsin‐like GPCRs and possesses N‐terminal glycosylation sites (Hill et al., 1997; Lefkowitz, 2013).
Following numerous attempts, the H3 receptor was selected from a human thalamic cDNA library and cloned in 1999 at the R.W. Johnson Pharmaceutical Research Institute in San Diego, CA in the USA (Lovenberg et al., 1999). The H3 receptor is also a member of the class A GPCR family, yet it shows a greater amino acid homology between humans and rodents (>92%), compared to the H1 and H2 receptors (72–78% and 85–86%, respectively) (Liu et al., 2001b). In addition, the gene encoding the human H3 receptor is not phylogenetically closely related to those encoding the human H1 or H2 receptors (Leurs et al., 2000). Moreover, the H3 receptor exhibits very low overall amino acid homology with other GPCRs, being 31% with the α2A‐ and α2C‐adrenoceptors and the muscarinic M1 receptor, and only 22% and 20% with the H1 and H2 receptors respectively (Leurs et al., 2000). Subsequent studies confirmed the existence of H3 receptor isoforms generated by alternative mRNA splicing that is consistent with the pharmacological heterogeneity of the H3 receptor (Gbahou et al., 2012; Riddy et al., 2017).
The ‘resurgence’ at the beginning of the 3rd millennium
Discovery of the H4 receptor through GPCR deorphanization
The H3 was the last histamine receptor to be discovered using traditional pharmacological approaches (Arrang et al., 1983). A year after its cloning (Lovenberg et al., 1999), the discovery of the novel histamine H4 receptor using genomics‐based reverse pharmacological approaches for screening orphan GPCRs reflected the impact of ‘molecular screening’ on histamine pharmacology. The description of the new histamine receptor was reported almost simultaneously, yet independently, by a number of groups working in Japan, the USA and Canada (Liu et al., 2001a,b; Morse et al., 2001; Nakamura et al., 2000; Nguyen et al., 2001; Oda et al., 2000; Zhu et al., 2001).
The first reported cloning and functional characterization of the novel human histamine receptor was achieved by obtaining an orphan GPCR named GPRv53 through a search of the human genomic DNA database at the Helix Research Institute, Japan (Oda et al., 2000). After only a month, a group at the Banyu Pharmaceutical Company, also in Japan, reported that a new histamine receptor, homologous to the H3, was cloned from the same gene (named BG26 in this study) using human leukocyte cDNA (Nakamura et al., 2000). This novel receptor was identified as the new subtype of human histamine H4 receptor (Nakamura et al., 2000) (Figure 2). Within the next 3 months, four more reports confirmed the existence of the histamine H4 receptor (Morse et al., 2001; Zhu et al., 2001; Liu et al., 2001a; Nguyen et al., 2001). The fourth histamine receptor was shown to exhibit rapid internalization upon agonist exposure (Nguyen et al., 2001) and to share higher sequence similarity with the H3 receptor (~40%) compared to ~20% with the H1 or H2 and to <30% with other amine GPCRs (Morse et al., 2001; Zhu et al., 2001; Nguyen et al., 2001).
Using various H1–3 receptor ligands for the functional characterization of the novel GPCR, the data indicated that, while the new receptor was structurally similar to the H3, it possessed a unique pharmacological profile. The H4 receptor exhibited high affinity for histamine, the equilibrium K D being at the lower nM range (Oda et al., 2000; Liu et al., 2001a). No or, occasionally, weak binding of known H1 and H2 receptor ligands was observed (Nakamura et al., 2000; Oda et al., 2000; Morse et al., 2001; Liu et al., 2001a; Nguyen et al., 2001), yet modest affinity was shown for the H2 agonist dimaprit (Liu et al., 2001a). Interestingly, the new receptor was able to bind H3 agonists, including immepip, imetit, Rα‐MeHA and N‐α‐methylhistamine, and H3 antagonists such as clobenpropit, thioperamide and burimamide, albeit with different affinity and/or potency compared to the H3 receptor (Liu et al., 2001a,b; Morse et al., 2001; Nakamura et al., 2000; Nguyen et al., 2001; Oda et al., 2000; Zhu et al., 2001). Moreover, clobenpropit behaved as a partial agonist, rather than as an antagonist (Liu et al., 2001a,b; Oda et al., 2000), whereas clozapine, an atypical antipsychotic drug with considerable affinity for numerous GPCRs that acts as an H1 and H2 antagonist, also exerted agonistic activity (Oda et al., 2000; Liu et al., 2001a).
Shortly after these notable findings, the evaluation of the interaction of the human H4 receptor with various known histamine receptor ligands revealed, among others, that the standard H3 receptor inverse agonist thioperamide (Arrang et al., 1987) also acts as an inverse agonist at the H4 receptor and that the H3 agonists immepip, imetit and Rα‐MeHA (Arrang et al., 1987) are also H4 receptor agonists (Lim et al., 2005). Furthermore, the H2 agonist/H3 antagonist dimaprit exerts partial H4 agonistic activity; the H2/H3 antagonist burimamide and the H3 antagonist clobenpropit displays agonism at the human H4 receptor, whereas 4‐methylhistamine was identified as the first selective H4 receptor agonist (Lim et al., 2005).
Breakthroughs and drawbacks in the expanding function of histamine in immunomodulation
The preferential expression of the H4 receptor in the immune system and its pharmacological profile (Nakamura et al., 2000; Oda et al., 2000; Morse et al., 2001; Zhu et al., 2001; Liu et al., 2001a; Nguyen et al., 2001) were in good agreement with the previously hypothesized existence of a novel histamine receptor mediating calcium mobilization in eosinophils (Raible et al., 1994). Subsequent studies provided evidence for the functional role of the H4 receptor not only in eosinophil biology (Ling et al., 2004; Grosicki et al., 2016) but also in the chemotaxis, differentiation and crosstalk of other immune cell types (Akdis and Simons, 2006; Zampeli and Tiligada, 2009; O'Mahony et al., 2011).
The discovery of the H4 receptor coincided with a turning point in pharmacology, illustrated by major advances in our understanding of GPCR properties (Lefkowitz, 2013; Kenakin, 2017), as well as by the ongoing scientific and biotechnological innovations in immunopharmacology (Tiligada et al., 2015). The timely characterization of a histamine receptor with putative immunomodulating properties inspired novel attractive perspectives in histamine research at the turn of the millennium (Akdis and Simons, 2006). Likewise, it raised hopes for the translational exploitation of the histamine H4 receptor as a new therapeutic target for unmet medical needs, particularly with respect to allergy, asthma, autoimmune diseases, host defence, neuropathic pain and even cancer (Zampeli and Tiligada, 2009; Panula et al., 2015; Thurmond et al., 2017).
The investigation of the contribution of histamine in acute and chronic inflammation was led by the development of the first highly selective H4 receptor antagonist JNJ 7777120 {1‐[(5‐chloro‐1H‐indol‐2‐yl)carbonyl]‐4‐methylpiperazine} (Jablonowski et al., 2003; Thurmond et al., 2004), followed by the reevaluation and the synthesis of numerous histamine receptor‐targeting compounds (Lim et al., 2005; Tiligada et al., 2009; Walter and Stark, 2012; Seifert et al., 2013; Panula et al., 2015; Wifling et al., 2015). Moreover, the investigation of the pharmacology and the (patho)physiological significance of the H4 receptor was intensified by the highly successful 4 years long Action BM0806 that was funded in 2009 by the European Cooperation in Science and Technology (COST). The Action specifically promoted the networking of several EHRS members and prominent groups interested in histamine research across the world. The outcomes of this multidisciplinary effort are elegantly summarized in a book published at the end of the Action (Stark, 2013).
The translational potential of the extensive efforts that revived the interest in histamine research was hampered, however, by several drawbacks that emerged over the past two decades (Thurmond et al., 2017). Illustrative examples include the recurrent concerns as regards the specificity of the available antibodies used in receptor localization studies (Seifert et al., 2013), as well as the receptor variations, not only relating to the species and strain differences (Liu et al., 2001b; Poli et al., 2006; Coruzzi et al., 2012; Neumann et al., 2013; Seifert et al., 2013) but also to a multitude of pharmacologically important parameters, including constitutive activity, ligand binding, intrinsic activity and functional selectivity (Nijmeijer et al., 2013; Wifling et al., 2015). Importantly, while JNJ 7777120 has been used as a prototype experimental tool, its short half‐life in vivo and its toxicity prevented its clinical development (Thurmond et al., 2017). Following the description of agonist‐biased signalling in various systems, JNJ 7777120 was demonstrated to be a biased agonist at the human H4 receptor (Rosethorne and Charlton, 2011) the year before the Nobel Prize in Chemistry 2012 was awarded to Robert Lefkowitz and Brian Kobilka ‘for studies of G‐protein‐coupled receptors’ (Lefkowitz, 2013).
Thus, although JNJ 7777120 acts as an antagonist with regard to G protein‐dependent signalling, it was shown to be an agonist in a non‐G protein‐dependent manner to recruit β‐arrestin to the receptor (Rosethorne and Charlton, 2011). Subsequent studies showed that JNJ 7777120 acts as a partial inverse agonist at the human H4 receptor, but as a partial agonist at the rat and mouse H4 receptors (Wifling et al., 2015), which possess lower constitutive activity than their human counterparts (Morse et al., 2001; Seifert et al., 2013; Strasser et al., 2013). Consequently, these properties of JNJ 7777120 may, at least in part, account for the frequently generated controversies and even misleading conclusions of studies conducted, especially in vivo, in a variety of experimental models (Zampeli et al., 2009b; Neumann et al., 2013; Seifert et al., 2013). Therefore, the experimental findings on the role of the H4 receptor need to be interpreted and/or to be extrapolated with caution.
The timing that the H4 receptor actually triggered research into the role of histamine in the regulation of immune (patho)physiology seems to be both a ‘wish’ and a ‘curse’. The current era of dynamic conceptual innovations in GPCR signalling clearly complicates further drug discovery efforts (Lefkowitz, 2013; Kenakin, 2017). Yet it provides the potential for the rational design of specific biased ligands with fewer unwanted effects (Nijmeijer et al., 2013; Riddy et al., 2017). Nevertheless, personalized or precision medicine is rapidly gaining ground in immunopharmacology and clinical immunology and challenges the therapeutic exploitation of ‘traditional’ therapies for unmet medical needs (Tiligada et al., 2015; Riccardi et al., 2018; Werfel, 2018). However, in addition to the H4 receptor, the vast body of accumulating evidence confers yet elusive immunomodulatory properties to the H2 receptor (O'Mahony et al., 2011; Tiligada, 2012; Monczor and Fernandez, 2016; Werfel, 2018), thus being invaluable for the dissection of the functions of histamine in the immune system and beyond.
Challenges and perspectives
Histamine research has been at the forefront of pharmacological advancements for more than a century. The investigations are largely supported to date by the members of the long standing EHRS that was founded in Paris in 1972 as the ‘Histamine Club’ led by a Secretary‐General and National Secretaries (Figure 3). After the annual meeting in Hannover in 1981, the ‘Histamine Club’ became the EHRS, a non‐profit making association of scientists and physicians interested in histamine. According to the statutes adopted at the meeting in Rome in 2000, the EHRS is led by a President (Figure 3) and a Council elected by the General Assembly during an annual meeting traditionally held in May (EHRS, 2018). Today, the EHRS is a leading global multidisciplinary network of senior experts and young investigators that promotes basic and translational histamine research in collaboration with a number of national societies, including the JHRS initiated in 1979 by the late Professors Kenji Tasaka of Okayama University Medical School and Hiroshi Wada of Osaka University, Japan (EHRS, 2018).
Although, GPCRs constitute the largest family of proteins targeted by approved drugs, small molecules, such as histamine receptor ligands, have to compete with cutting‐edge targeted therapeutics for the development of more effective and safer options for a number of debilitating pathologies with high economic and societal impact. Histamine remains one of the richest sources of therapeutic tools (Table 1). The development of new and the re‐evaluation of old histamine receptor ligands is an ongoing process. Building on its past, the elucidation of the functional complexity of histamine networks in human (patho) physiology requires defragmentation of the current research based on accurate hypotheses of scientific and translational relevance. The standardization of the methodological tools is an imperative task that challenges cross‐disciplinary efforts, spanning a broad spectrum of both traditional and modern drug discovery approaches and conceptual innovations in receptor pharmacology.
In the inaugural lecture on ‘The multiple facets of histamine research’ at the 9th EHRS meeting in Visegrad, Hungary in 1980, the EHRS Late Honorary Member Heinz Otto Schild (Figure 3) concluded by ‘I wanted to emphasize that the decarboxylated son of histidine is not resting. I feel confident that histamine is preparing many more interesting surprises …’. Twenty one years later, during the 30th EHRS meeting in Turku, Finland in 2001, the EHRS Honorary Member Hendrik Timmerman (Figure 3) summed up his lecture on ‘Histamine receptors, a versatile subject for studying molecular aspects of receptors and receptor mechanisms’ ‘… as being about turning fantasies into hypotheses and then into research’.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
Conflict of interest
E.T. is currently president and M.E. is past president of the EHRS.
Acknowledgements
We would like to express our sincere gratitude to Dr Anita Sydbom and Dr Paul Chazot for sharing their information on the history of the EHRS.
Tiligada E., and Ennis M. (2020) Histamine pharmacology: from Sir Henry Dale to the 21st century, British Journal of Pharmacology, 177, 469–489, doi: 10.1111/bph.14524.
References
- Ackermann D (1910). Über den bakteriellen Abbau des Histidin. Hoppe‐Seylers. Z Physiol Chem 65: 504–510. [Google Scholar]
- Acosta‐Andrade C, Lambertos A, Urdiales JL, Sánchez‐Jiménez F, Peñafiel R, Fajardo I (2016). A novel role for antizyme inhibitor 2 as a regulator of serotonin and histamine biosynthesis and content in mouse mast cells. Amino Acids 48: 2411–2421. [DOI] [PubMed] [Google Scholar]
- Akdis CA, Simons FE (2006). Histamine receptors are hot in immunopharmacology. Eur J Pharmacol 533: 69–76. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Lancelot JC, Lecomte JM, Pollard H, Robba M et al (1987). Highly potent and selective ligands for histamine H3‐receptors. Nature 327: 117–123. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Schwartz JC (1983). Auto‐inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 302: 832–837. [DOI] [PubMed] [Google Scholar]
- Ash AS, Schild HO (1966). Receptors mediating some actions of histamine. Br J Pharmacol Chemother 27: 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attali P, Gomeni R, Wersinger E, Poli S, Venail F (2016). The effects of SENS‐111, a new H4R antagonist, on vertigo induced by caloric test in healthy volunteers (HV) is related to plasma concentrations. Clin Ther 38 (10S): e4. [DOI] [PubMed] [Google Scholar]
- Bani D, Nistri S, Mannaioni PF, Masini E (2006). Cardiac anaphylaxis: pathophysiology and therapeutic perspectives. Curr Allergy Asthma Rep 6: 14–19. [DOI] [PubMed] [Google Scholar]
- Barger G, Dale HH (1910). Chemical structure and sympathomimetic action of amines. J Physiol 41: 19–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessinis DP, Dalla C, Daifoti ZP, Tiligada E (2012). Histamine involvement in visual development and adaptation. Invest Ophthalmol Vis Sci 53: 7498–7503. [DOI] [PubMed] [Google Scholar]
- Best CH, Dale HH, Dudley HW, Thorpe WV (1927). The nature of the vaso‐dilator constituents of certain tissue extracts. J Physiol 62: 397–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best CH, McHenry EW (1930). The inactivation of histamine. J Physiol 70: 349–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JW, Duncan WAM, Durant CJ, Ganellin CR, Parsons EM (1972). Definition and antagonism of histamine H2‐receptors. Nature 236: 385–390. [DOI] [PubMed] [Google Scholar]
- Borriello F, Iannone R, Marone G (2017). Histamine release from mast cells and basophils. Handb Exp Pharmacol 241: 121–139. [DOI] [PubMed] [Google Scholar]
- Bovet D (1950). Introduction to antihistamine agents and antergan derivative. Ann N Y Acad Sci 50: 1089–1126. [DOI] [PubMed] [Google Scholar]
- Bovet D, Horclois R, Fournel J (1944). L'antagonisme exercé par la Np‐méthoxybenzyl‐N‐diméthylamino‐éthyl‐α‐amino‐pyridine sur l'hypotension et la vasodilatation histaminiques. C R Seanc Soc Biol (Paris) 138: 185. [Google Scholar]
- Bradshaw J, Brittain RT, Clitherow JW, Daly MJ, Jack D, Price BJ et al (1979). Ranitidine (AH 19065): a new potent, selective histamine H2‐receptor antagonist. Br J Pharmacol 66 464P. [PMC free article] [PubMed] [Google Scholar]
- Brimblecombe RW, Duncan WAM, Durant GJ, Ganellin CR, Parsons ME, Black JW (1975). The pharmacology of cimetidine, a new histamine H2‐receptor antagonist. Br J Pharmacol 53: 435–436. [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Falck B, Hillarp NA, Thieme G, Torp A (1961). A new histochemical method for visualization of tissue catechol amines. Med Exp Int J Exp Med 4: 123–125. [DOI] [PubMed] [Google Scholar]
- Church MK, Church DS (2013). Pharmacology of antihistamines. Indian J Dermatol 58: 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AJ (1933). The Mode of Action of Drugs on Cells. E. Arnold: London. [Google Scholar]
- Coruzzi G, Pozzoli C, Adami M, Grandi D, Guido N, Smits R et al (2012). Strain‐dependent effects of the histamine H₄ receptor antagonist JNJ7777120 in a murine model of acute skin inflammation. Exp Dermatol 2: 32–37. [DOI] [PubMed] [Google Scholar]
- Cross LJ, Heaney LG, Ennis M (1996). Further characterisation of substance P induced histamine release from human bronchoalveolar lavage mast cells. Inflamm Res 45 (Suppl. 1): S11–S12. [DOI] [PubMed] [Google Scholar]
- Dale H (1948). Antihistamine substances. Br Med J 2: 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale H (1950). The pharmacology of histamine: with a brief survey of evidence for its occurrence, liberation, and participation in natural reactions. Ann N Y Acad Sci 50: 1017–1028. [DOI] [PubMed] [Google Scholar]
- Dale HH, Laidlaw PP (1910). The physiological action of β‐imidazolylethylamine. J Physiol 41: 318–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale HH, Richards AN (1918). The vasodilator action of histamine and of some other substances. J Physiol 52: 110–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Backer MD, Gommeren W, Moereels H, Nobels G, Van Gompel P, Leysen JE et al (1993). Genomic cloning, heterologous expression and pharmacological characterization of a human histamine H1 receptor. Biochem Biophys Res Commun 197: 1601–1608. [DOI] [PubMed] [Google Scholar]
- Ehrlich P (1879). Beitrage Zur Kenntniss der Granulirten Bindegewebszellen und der Eosinophilen Leukocythen. Arch Anat Physiol Physiol 6: 166–169. [Google Scholar]
- Ehrlich P, Morgenroth J (1900). Ueber Haemolysine: dritte Mittheilung. Berlin Klin Wochschr 37: 453–458. [Google Scholar]
- Emanuel MB (1999). Histamine and the antiallergic antihistamines: a history of their discoveries. Clin Exp Allergy 29 (Suppl. 3): 1–11. [DOI] [PubMed] [Google Scholar]
- Ennis M, Barrow SE, Blair IA (1984). Prostaglandin and histamine release from stimulated rat peritoneal mast cells. Agents Actions 14: 397–400. [DOI] [PubMed] [Google Scholar]
- Ennis M, Pearce FL (1980). Differential reactivity of isolated mast cells from the rat and guinea pig. Eur J Pharmacol 66: 339–345. [DOI] [PubMed] [Google Scholar]
- Ennis M, Truneh A, White JR, Pearce FL (1981). Inhibition of histamine secretion from mast cells. Nature 289: 186–187. [DOI] [PubMed] [Google Scholar]
- European Histamine Research Society (2018). Available at: https://www.ehrs.org.uk/ (accessed 10/12/2018).
- European Medicines Agency (2015). Hydroxyzine. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/referrals/Hydroxyzine/human_referral_prac_000043.jsp&mid=WC0b01ac05805c516f (accessed 8/15/2018).
- Feinberg SM (1947). The antihistaminic drugs pharmacology and therapeutic effects. Am J Med 3: 560–570. [DOI] [PubMed] [Google Scholar]
- Forsythe P, McGarvey LP, Heaney LG, MacMahon J, Ennis M (1999). Adenosine induces histamine release from human bronchoalveolar lavage mast cells. Clin Sci (Lond) 96: 349–355. [PubMed] [Google Scholar]
- Fourneau E, Bovet D (1933). Recherches sur l'action sympathicolytique d'un nouveau dérivé du dioxane. Arch Int Pharmacodyn Thér 46: 178–191. [Google Scholar]
- Ganellin CR (2011). Personal reflections on Sir James Black (1924‐2010) and histamine. Inflamm Res 60: 103–110. [DOI] [PubMed] [Google Scholar]
- Gantz I, Munzert G, Tashiro T, Schäffer M, Wang L, DelValle J et al (1991a). Molecular cloning of the human histamine H2 receptor. Biochem Biophys Res Commun 178: 1386–1392. [DOI] [PubMed] [Google Scholar]
- Gantz I, Schäffer M, DelValle J, Logsdon C, Campbell V, Uhler M et al (1991b). Molecular cloning of a gene encoding the histamine H2 receptor. Proc Natl Acad Sci U S A 88: 429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gbahou F, Rouleau A, Arrang JM (2012). The histamine autoreceptor is a short isoform of the H₃ receptor. Br J Pharmacol 166: 1860–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham HT, Lowry OH, Wheelwright F, Lenz MA, Parish HH Jr (1955). Distribution of histamine among leukocytes and platelets. Blood 10: 467–481. [PubMed] [Google Scholar]
- Green JP (1970). Histamine In: Lajtha A. (ed). Handbook of Neurochemistry, Vol. IV Plenum Press: New‐York, pp. 221–250. [Google Scholar]
- Grosicki M, Wójcik T, Chlopicki S, Kieć‐Kononowicz K (2016). In vitro study of histamine and histamine receptor ligands influence on the adhesion of purified human eosinophils to endothelium. Eur J Pharmacol 777: 49–59. [DOI] [PubMed] [Google Scholar]
- Gutowska‐Owsiak D, Greenwald L, Watson C, Selvakumar TA, Wang X, Ogg GS (2014). The histamine‐synthesizing enzyme histidine decarboxylase is upregulated by keratinocytes in atopic skin. Br J Dermatol 171: 771–778. [DOI] [PubMed] [Google Scholar]
- Haas H, Panula P (2003). The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci 4: 121–130. [DOI] [PubMed] [Google Scholar]
- Haas HL (2018). Fishing for histamine H3 receptor functions. Acta Physiol (Oxf) 222 (3). [DOI] [PubMed] [Google Scholar]
- Haas HL, Sergeeva OA, Selbach O (2008). Histamine in the nervous system. Physiol Rev 88: 1183–1241. [DOI] [PubMed] [Google Scholar]
- Halpern BN (1942). Les antihistaminiques de synthèse. Essai de chimiothérapie des états allergiques. Arch Int Pharmacodyn Ther 68: 339–408. [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ (1990). Distribution, properties and functional characteristics of three classes of histamine receptor. Pharmacol Rev 42: 45–83. [PubMed] [Google Scholar]
- Hill SJ (2006). G‐protein‐coupled receptors: past, present and future. Br J Pharmacol 147: S27–S37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ, Ganellin CR, Timmerman H, Schwartz JC, Shankley NP, Young JM et al (1997). International Union of Pharmacology. XIII Classification of histamine receptors. Pharmacol Rev 49: 253–278. [PubMed] [Google Scholar]
- Hofstra CL, Desai PJ, Thurmond RL, Fung‐Leung WP (2003). Histamine H4 receptor mediates chemotaxis and calcium mobilization of mast cells. J Pharmacol Exp Ther 305: 1212–1221. [DOI] [PubMed] [Google Scholar]
- Hu Y, Sieck DE, Hsu WH (2015). Why are second‐generation H1‐antihistamines minimally sedating? Eur J Pharmacol 765: 100–106. [DOI] [PubMed] [Google Scholar]
- Jablonowski JA, Grice CA, Chai W, Dvorak CA, Venable JD, Kwok AK et al (2003). The first potent and selective non‐imidazole human histamine H4 receptor antagonists. J Med Chem 46: 3957–3960. [DOI] [PubMed] [Google Scholar]
- Jacques LB, Water ET (1941). The identity and origin of the anticoagulant of anaphylactic shock in the dog. J Physiol (London) 99: 454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T (2017). Signaling bias in drug discovery. Expert Opin Drug Discov 12: 321–333. [DOI] [PubMed] [Google Scholar]
- Kollb‐Sielecka M, Demolis P, Emmerich J, Markey G, Salmonson T, Haas M (2017). The European Medicines Agency review of pitolisant for treatment of narcolepsy: summary of the scientific assessment by the Committee for Medicinal Products for Human Use. Sleep Med 33: 125–129. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski H (1943). Histamine in nervous tissue. J Physiol 102: 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakidis K, Zampeli E, Palaiologou M, Tiniakos D, Tiligada E (2015). Histamine H3 and H4 receptor ligands modify vascular histamine levels in normal and arthritic large blood vessels in vivo . Inflammation 38: 949–958. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ (2013). A brief history of G‐protein coupled receptors (Nobel Lecture). Angew Chem Int Ed Engl 52: 6366–6378. [DOI] [PubMed] [Google Scholar]
- Leurs R, Church MK, Taglialatela M (2002). H1‐antihistamines: inverse agonism, anti‐inflammatory actions and cardiac effects. Clin Exp Allergy 32: 489–498. [DOI] [PubMed] [Google Scholar]
- Leurs R, Hoffmann M, Wieland K, Timmerman H (2000). H3 receptor gene is cloned at last. Trends Pharmacol Sci 21: 11–12. [DOI] [PubMed] [Google Scholar]
- Levi‐Schaffer F, Eliashar R (2009). Mast cell stabilizing properties of antihistamines. J Invest Dermatol 129: 2549–2551. [DOI] [PubMed] [Google Scholar]
- Lewis T, Grant RT (1924). Vascular reactions of the skin to injury. Part II. The liberation of histamine‐like substance in the injured skin, the underlying cause of factitious urticaria and of wheals produced by burning: and observations upon the nervous control of certain skin reactions. Heart 11: 209–265. [Google Scholar]
- Lichtenstein LM, Gillespie E (1973). Inhibition of histamine release by histamine controlled by H2 receptor. Nature 244: 287–288. [DOI] [PubMed] [Google Scholar]
- Lim HD, van Rijn RM, Ling P, Bakker RA, Thurmond RL, Leurs R (2005). Evaluation of histamine H1‐, H2‐, and H3‐receptor ligands at the human histamine H4 receptor: identification of 4‐methylhistamine as the first potent and selective H4 receptor agonist. J Pharmacol Exp Ther 314: 1310–1321. [DOI] [PubMed] [Google Scholar]
- Ling P, Ngo K, Nguyen S, Thurmond RL, Edwards JP, Karlsson L et al (2004). Histamine H4 receptor mediates eosinophil chemotaxis with cell shape change and adhesion molecule upregulation. Br J Pharmacol 142: 161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Ma X, Jiang X, Wilson SJ, Hofstra CL, Blevitt J et al (2001a). Cloning and pharmacological characterization of a fourth histamine receptor (H4) expressed in bone marrow. Mol Pharmacol 59: 420–426. [DOI] [PubMed] [Google Scholar]
- Liu C, Wilson SJ, Kuei C, Lovenberg TW (2001b). Comparison of human, mouse, rat, and guinea pig histamine H4 receptors reveals substantial pharmacological species variation. J Pharmacol Exp Ther 299: 121–130. [PubMed] [Google Scholar]
- Loew ER, Kaiser ME, Moore V (1945). Synthetic benzhydryl alkamine ethers effective in preventing fatal experimental asthma in guinea pigs exposed to atomized histamine. J Pharmacol Exp Ther 83: 120–129. [Google Scholar]
- Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A et al (1999). Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol 55: 1101–1107. [PubMed] [Google Scholar]
- Mannaioni PF, Di Bello MG, Raspanti S, Romano V, Bani Sacchi T, Cappugi P et al (1995). Storage and release of histamine in human platelets. Inflamm Res 44 (Suppl. 1): S16–S17. [DOI] [PubMed] [Google Scholar]
- Monczor F, Fernandez N (2016). Current knowledge and perspectives on histamine H1 and H2 receptor pharmacology: functional selectivity, receptor crosstalk, and repositioning of classic histaminergic ligands. Mol Pharmacol 90: 640–648. [DOI] [PubMed] [Google Scholar]
- Monnier M, Fallert M, Battacharya IC (1967). The waking action of histamine. Experientia 23: 21–22. [DOI] [PubMed] [Google Scholar]
- Morse KL, Behan J, Laz TM, West RE Jr, Greenfeder SA, Anthes JC et al (2001). Cloning and characterization of a novel human histamine receptor. J Pharmacol Exp Ther 296: 1058–1066. [PubMed] [Google Scholar]
- Nakamura T, Itadani H, Hidaka Y, Ohta M, Tanaka K (2000). Molecular cloning and characterization of a new human histamine receptor, HH4R. Biochem Biophys Res Commun 279: 615–620. [DOI] [PubMed] [Google Scholar]
- Neumann D, Beermann S, Burhenne H, Glage S, Hartwig C, Seifert R (2013). The dual H3/4R antagonist thioperamide does not fully mimic the effects of the ‘standard’ H4R antagonist JNJ 7777120 in experimental murine asthma. Naunyn Schmiedebergs Arch Pharmacol 386: 983–990. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Shapiro DA, George SR, Setola V, Lee DK, Cheng R et al (2001). Discovery of a novel member of the histamine receptor family. Mol Pharmacol 59: 427–433. [DOI] [PubMed] [Google Scholar]
- Nieto‐Alamilla G, Márquez‐Gómez R, García‐Gálvez AM, Morales‐Figueroa GE, Arias‐Montaño JA (2016). The histamine H3 receptor: structure, pharmacology, and function. Mol Pharmacol 90: 649–673. [DOI] [PubMed] [Google Scholar]
- Nijmeijer S, Vischer HF, Sirci F, Schultes S, Engelhardt H, de Graaf C et al (2013). Detailed analysis of biased histamine H4 receptor signalling by JNJ 7777120 analogues. Br J Pharmacol 170: 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak N, Mete N, Bussmann C, Maintz L, Bieber T, Akdis M et al (2012). Early suppression of basophil activation during allergen‐specific immunotherapy by histamine receptor 2. J Allergy Clin Immunol 130: 1153–1158.e2. [DOI] [PubMed] [Google Scholar]
- Nuutinen S, Panula P (2010). Histamine in neurotransmission and brain diseases. Adv Exp Med Biol 709: 95–107. [DOI] [PubMed] [Google Scholar]
- Oda T, Morikawa N, Saito Y, Masuho Y, Matsumoto S (2000). Molecular cloning and characterization of a novel type of histamine receptor preferentially expressed in leukocytes. J Biol Chem 275: 36781–36786. [DOI] [PubMed] [Google Scholar]
- Olivera A, Beaven MA, Metcalfe DD (2018). Mast cells signal their importance in health and disease. J Allergy Clin Immunol 142: 381–393. [DOI] [PubMed] [Google Scholar]
- O'Mahony L, Akdis M, Akdis CA (2011). Regulation of the immune response and inflammation by histamine and histamine receptors. J Allergy Clin Immunol 128: 1153–1162. [DOI] [PubMed] [Google Scholar]
- Panula P, Chazot PL, Cowart M, Gutzmer R, Leurs R, Liu WL et al (2015). International Union of Basic and Clinical Pharmacology. XCVIII. Histamine receptors. Pharmacol Rev 67: 601–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panula P, Sundvik M, Karlstedt K (2014). Developmental roles of brain histamine. Trends Neurosci 37: 159–168. [DOI] [PubMed] [Google Scholar]
- Panula P, Yang HY, Costa E (1984). Histamine‐containing neurons in the rat hypothalamus. Proc Natl Acad Sci U S A 81: 2572–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papathanassiou M, Giannoulaki V, Zampeli E, Tiligada E (2011). Effect of aminoguanidine on the conjunctival histamine and nitrite levels in experimental conjunctivitis. J Ocul Pharmacol Ther 27: 137–142. [DOI] [PubMed] [Google Scholar]
- Parsons ME, Ganellin CR (2006). Histamine and its receptors. Br J Pharmacol 147: S127–S135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton WD (1951). Compound 48/80: a potent histamine liberator. Br J Pharmacol Chemother 6: 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli E, Pozzoli C, Regina G, Menozzi A, Roncoroni L, Coruzzi G (2006). Dualistic H1‐mediated effect of histamine on the human isolated intestine. 5. Histamine in cardiovascular and gastrointestinal systems. Inflamm Res 55 (S1): S65–S66. [DOI] [PubMed] [Google Scholar]
- Popielski L (1920). β‐Imidazolylathylamin und die Organextrakte Erster Teil: b‐Imidazolylathylamin als mechtiger Errezer der Magendrucken. Pfluegers Arch 178: 214–236. [Google Scholar]
- Portier P, Richet CR (1902). De l'action anaphylactique de certains venins. C R Seanc Soc Biol 54: 170–172. [Google Scholar]
- Provensi G, Passani MB, Costa A, Izquierdo I, Blandina P (2018). Neuronal histamine and the memory of emotionally‐salient events. Br J Pharmacol 10.1111/bph.14476 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raible DG, Lenahan T, Fayvilevich Y, Kosinski R, Schulman ES (1994). Pharmacologic characterization of a novel histamine receptor on human eosinophils. Am J Respir Crit Care Med 149: 1506–1511. [DOI] [PubMed] [Google Scholar]
- Rang HP (2006). The receptor concept: pharmacology's big idea. Br J Pharmacol 147 (Suppl. 1): S9–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi C, Levi‐Schaffer F, Tiligada E (2018). Immunopharmacology and Inflammation. Springer International Publishing: Cham, Switzerland. [Google Scholar]
- Riddy DM, Cook AE, Diepenhorst NA, Bosnyak S, Brady R, Mannoury la Cour C et al (2017). Isoform‐specific biased agonism of histamine H3 receptor agonists. Mol Pharmacol 91: 87–99. [DOI] [PubMed] [Google Scholar]
- Riley JF, West GB (1953). The presence of histamine in tissue mast cells. J Physiol 120: 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosethorne EM, Charlton SJ (2011). Agonist‐biased signaling at the histamine H4 receptor: JNJ7777120 recruits β‐arrestin without activating G proteins. Mol Pharmacol 79: 749–757. [DOI] [PubMed] [Google Scholar]
- Rubin W, Schwartz B (1979). Electron microscopic radioautographic identification of the ECL cell as the histamine‐synthesizing endocrine cell in the rat stomach. Gastroenterology 77: 458–467. [PubMed] [Google Scholar]
- Salem A, Rozov S, Al‐Samadi A, Stegajev V, Listyarifah D, Kouri VP et al (2017). Histamine metabolism and transport are deranged in human keratinocytes in oral lichen planus. Br J Dermatol 176: 1213–1223. [DOI] [PubMed] [Google Scholar]
- Schayer RW (1956). The origin and fate of histamine in the body In: O'Connor CM. (ed). Wolstenholme GEW. Ciba Foundation Symposium on Histamine. J. and A. Churchill Ltd: London, pp. 183–188. [Google Scholar]
- Schwartz JC (1975). Histamine as a transmitter in brain. Life Sci 17: 503–517. [DOI] [PubMed] [Google Scholar]
- Schwartz JC (2011). The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br J Pharmacol 163: 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R, Strasser A, Schneider EH, Neumann D, Dove S, Buschauer A (2013). Molecular and cellular analysis of human histamine receptor subtypes. Trends Pharmacol Sci 34: 33–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V et al (2011). Structure of the human histamine H1 receptor complex with doxepin. Nature 475: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore PA, Burkhalter A, Cohn VH Jr (1959). A method for the fluorometric assay of histamine in tissues. J Pharmacol Exp Ther 127: 182–186. [PubMed] [Google Scholar]
- Simons FE, Simons KJ (2011). Histamine and H1‐antihistamines: celebrating a century of progress. J Allergy Clin Immunol 128: 1139–1150.e4. [DOI] [PubMed] [Google Scholar]
- Snyder SH, Baldessarini RJ, Axelrod J (1966). A sensitive and specific enzymatic isotopic assay for tissue histamine. J Pharmacol Exp Ther 153: 544–549. [PubMed] [Google Scholar]
- Stark H (2013). Histamine H4 Receptor: A Novel Drug Target in Immunoregulation and Inflammation. Versita Ltd: Warsaw. [Google Scholar]
- Staub AM, Bovet D (1937). Action de la thymoxyethyldiethylamine (929F) et des ethers pehnoliques sur le choc anaphylactique. C R Seances Soc Bio 125: 818–821. [Google Scholar]
- Strand DS, Kim D, Peura DA (2017). 25 Years of proton pump inhibitors: a comprehensive review. Gut Liver 11: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Wittmann HJ, Buschauer A, Schneider EH, Seifert R (2013). Species‐dependent activities of G‐protein‐coupled receptor ligands: lessons from histamine receptor orthologs. Trends Pharmacol Sci 34: 13–32. [DOI] [PubMed] [Google Scholar]
- Sundvik M, Panula P (2015). Interactions of the orexin/hypocretin neurones and the histaminergic system. Acta Physiol 213: 321–333. [DOI] [PubMed] [Google Scholar]
- Taglialatela M, Timmerman H, Annunziato L (2000). Cardiotoxic potential and CNS effects of first‐generation antihistamines. Trends Pharmacol Sci 21: 52–56. [DOI] [PubMed] [Google Scholar]
- Tetlow LC, Woolley DE (2004). Immunolocalisation of histamine and histidine decarboxylase in chondrocytes of arthritic cartilage. Inflamm Res 53 (Suppl. 1): S21–S22. [DOI] [PubMed] [Google Scholar]
- Thurmond RL, Desai PJ, Dunford PJ, Fung‐Leung WP, Hofstra CL, Jiang W et al (2004). A potent and selective histamine H4 receptor antagonist with anti‐inflammatory properties. J Pharmacol Exp Ther 309: 404–413. [DOI] [PubMed] [Google Scholar]
- Thurmond RL, Venable J, Savall B, La D, Snook S, Dunford PJ et al (2017). Clinical development of histamine H4 receptor antagonists. Handb Exp Pharmacol 241: 301–320. [DOI] [PubMed] [Google Scholar]
- Tiligada E (2012). Editorial: is histamine the missing link in chronic inflammation? J Leukoc Biol 92: 4–6. [DOI] [PubMed] [Google Scholar]
- Tiligada E, Ishii M, Riccardi C, Spedding M, Simon HU, Teixeira MM et al (2015). The expanding role of immunopharmacology: IUPHAR Review 16. Br J Pharmacol 172: 4217–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiligada E, Kyriakidis K, Chazot PL, Passani MB (2011). Histamine pharmacology and new CNS drug targets. CNS Neurosci Ther 17: 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiligada E, Zampeli E, Sander K, Stark H (2009). Histamine H3 and H4 receptors as novel drug targets. Expert Opin Investig Drugs 18: 1519–1531. [DOI] [PubMed] [Google Scholar]
- Timmerman H (1999). Why are non‐sedating antihistamines non‐sedating? Clin ExpvAllergy 29 (Suppl. 3): 13–18. [PubMed] [Google Scholar]
- Tippens AS, Gruetter CA (2004). Detection of histidine decarboxylase mRNA in human vascular smooth muscle and endothelial cells. Inflamm Res 53: 215–216. [DOI] [PubMed] [Google Scholar]
- Trendelenburg U (1960). The action of histamine and 5‐hydroxytryptamine on isolated mammalian atria. J Pharm Exp Ther 130: 450–460. [PubMed] [Google Scholar]
- Varricchi G, Raap U, Rivellese F, Marone G, Gibbs BF (2018). Human mast cells and basophils—How are they similar how are they different? Immunol Rev 282: 8–34. [DOI] [PubMed] [Google Scholar]
- von Pirquet CP, Schick B (1905). Die Serumkrankeit. F. Deuticke: Wien. [Google Scholar]
- Walter M, Stark H (2012). Histamine receptor subtypes: a century of rational drug design. Front Biosci (Schol Ed) 2012 4: 461–488. [DOI] [PubMed] [Google Scholar]
- Watanabe KL (1931). Quantitative Untersuchungen über den gehalt an darkmontrahierenden Stoffen von Lunge und Leber bei Meerschweinchen in Stadium der Eiweissensibilisierung und im anaphylaktischen Shock. Ztschr Immunitats Forsch Exper Ther 72: 50–56. [Google Scholar]
- Watanabe T, Taguchi Y, Hayashi H, Tanaka J, Shiosaka S, Tohyama M et al (1983). Evidence for the presence of a histaminergic neuron system in the rat brain: an immunohistochemical analysis. Neurosci Lett 39: 249–254. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Taguchi Y, Shiosaka S, Tanaka J, Kubota H, Terano Y et al (1984). Distribution of the histaminergic neuron system in the central nervous system of rats; a fluorescent immunohistochemical analysis with histidine decarboxylase as a marker. Brain Res 295: 13–25. [DOI] [PubMed] [Google Scholar]
- Werfel T (2018). Novel systemic drugs in treatment of atopic dermatitis: results from phase II and phase III studies published in 2017/2018. Curr Opin Allergy Clin Immunol 18: 432–437. [DOI] [PubMed] [Google Scholar]
- White T (1959). Formation and catabolism of histamine in brain tissue in vitro. J Physiol 149: 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2018). ATC/DDD Index 2018. Available at: https://www.whocc.no/atc_ddd_index/ (accessed 9/11/2018).
- Wifling D, Löffel K, Nordemann U, Strasser A, Bernhardt G, Dove S et al (2015). Molecular determinants for the high constitutive activity of the human histamine H4 receptor: functional studies on orthologues and mutants. Br J Pharmacol 172: 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windaus A, Vogt W (1907). Synthese des Imidazolyläthylamins. Ber Dtsch Chem Ges 40: 3691–3695. [Google Scholar]
- Yanai K, Zhang D, Tashiro M, Yoshikawa T, Naganuma F, Harada R et al (2011). Positron emission tomography evaluation of sedative properties of antihistamines. Expert Opin Drug Saf 10: 613–622. [DOI] [PubMed] [Google Scholar]
- Zampeli E, Pitychoutis PM, Papadopoulou‐Daifoti Z, Tiligada E (2009a). Systemic challenge with lipopolysaccharide increases histamine levels in the conjunctiva and cartilage, but not hypothalamus of Sprague Dawley rats. Inflamm Res 58 (Suppl. 1): 49–50. [DOI] [PubMed] [Google Scholar]
- Zampeli E, Thurmond RL, Tiligada E (2009b). The histamine H4 receptor antagonist JNJ7777120 induces increases in the histamine content of the rat conjunctiva. Inflamm Res 58: 285–291. [DOI] [PubMed] [Google Scholar]
- Zampeli E, Tiligada E (2009). The role of histamine H4 receptor in immune and inflammatory disorders. Br J Pharmacol 157: 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Michalovich D, Wu H, Tan KB, Dytko GM, Mannan IJ et al (2001). Cloning, expression, and pharmacological characterization of a novel human histamine receptor. Mol Pharmacol 59: 434–441. [DOI] [PubMed] [Google Scholar]
- Zimmermann AS, Burhenne H, Kaever V, Seifert R, Neumann D (2011). Systematic analysis of histamine and N‐methylhistamine concentrations in organs from two common laboratory mouse strains: C57Bl/6 and Balb/c. Inflamm Res 60: 1153–1159. [DOI] [PubMed] [Google Scholar]
- Zwadlo‐Klarwasser G, Braam U, Mühl‐Zürbes P, Schmutzler W (1994). Macrophages and lymphocytes: alternative sources of histamine. Agents Actions 41 Spec No 41: C99–C100. [DOI] [PubMed] [Google Scholar]