

Abstract
Background and Purpose
Diacetyl‐bis(4‐methyl‐3‐thiosemicarbazonato)copperII (CuII(atsm)) ameliorates neurodegeneration and delays disease progression in mouse models of amyotrophic lateral sclerosis (ALS) and Parkinson's disease (PD), yet the mechanism of action remains uncertain. Promising results were recently reported for separate Phase 1 studies in ALS patients and PD patients. Affected tissue in these disorders shares features of elevated Fe, low glutathione and increased lipid peroxidation consistent with ferroptosis, a novel form of regulated cell death. We therefore evaluated the ability of CuII(atsm) to inhibit ferroptosis.
Experimental Approach
Ferroptosis was induced in neuronal cell models by inhibition of glutathione peroxidase‐4 activity with RSL3 or by blocking cystine uptake with erastin. Cell viability and lipid peroxidation were assessed and the efficacy of CuII(atsm) was compared to the known antiferroptotic compound liproxstatin‐1.
Key Results
CuII(atsm) protected against lipid peroxidation and ferroptotic lethality in primary and immortalised neuronal cell models (EC50: ≈130 nM, within an order of magnitude of liproxstatin‐1). NiII(atsm) also prevented ferroptosis with similar potency, whereas ionic CuII did not. In cell‐free systems, CuII(atsm) and NiII(atsm) inhibited FeII‐induced lipid peroxidation, consistent with these compounds quenching lipid radicals.
Conclusions and Implications
The antiferroptotic activity of CuII(atsm) could therefore be the disease‐modifying mechanism being tested in ALS and PD trials. With potency in vitro approaching that of liproxstatin‐1, CuII(atsm) possesses favourable properties such as oral bioavailability and entry into the brain that make it an attractive investigational product for clinical trials of ferroptosis‐related diseases.
Abbreviations
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- BCS
bathocuproinedisulfonic acid
- CuII(atsm)
diacetyl‐bis(4‐methyl‐3‐thiosemicarbazonato)copperII
- PD
Parkinson's disease
- RTA
radical trapping antioxidant
What is already known
CuII(atsm) benefits neurodegenerative diseases and their animal models through an uncertain mechanism of action.
Ferroptosis is a form of regulated cell death implicated in neurodegenerative diseases.
What this study adds
CuII(atsm) blocks ferroptotic neuronal death and lipid peroxidation in vitro by a radical trapping activity.
The antiferroptotic potency of CuII(atsm) approaches that of liproxstatin‐1, a potent antiferroptotic reference compound.
What is the clinical significance
Blocking ferroptosis may be the mechanism being tested in current efficacy trials of CuII(atsm).
Clinical testing of similarly potent antiferroptotic agents for neurodegenerative disease warrants further consideration.
1. INTRODUCTION
CuII(atsm) is a bis(thiosemicarbazone)copperII compound (Figure 1) that was recently reported to show promising preliminary results in both a Phase 1 trial of patients with amyotrophic lateral sclerosis (ALS; NCT02870634; Rowe et al., 2018) and a Phase 1 trial of patients with Parkinson's disease (PD; NCT03204929; Evans, Rowe, Lee, Noel, & Rosenfeld, 2019). In two Phase 0 clinical trials, CuII(atsm) crossed into the brain and concentrated in areas associated with disease activity (Ikawa et al., 2011; Ikawa et al., 2015). Furthermore, in preclinical studies CuII(atsm) ameliorated neurodegeneration in mutant SOD1 transgenic mouse models of ALS (Hilton et al., 2017; McAllum et al., 2013; Roberts et al., 2014; Soon et al., 2011; Vieira et al., 2017), transgenic and toxin‐induced mouse models of PD (Hung et al., 2012) and transient and permanent mouse models of ischaemic stroke (Huuskonen et al., 2017). However, the mechanism of neuroprotection of this molecule is uncertain. Proposed mechanisms of action have included controlled delivery of Cu ions (Hilton et al., 2017; Roberts et al., 2014). However, the affinity of CuII for the atsm scaffold is extremely high (Donnelly et al., 2008) and would not be released in physiological environments.
Figure 1.
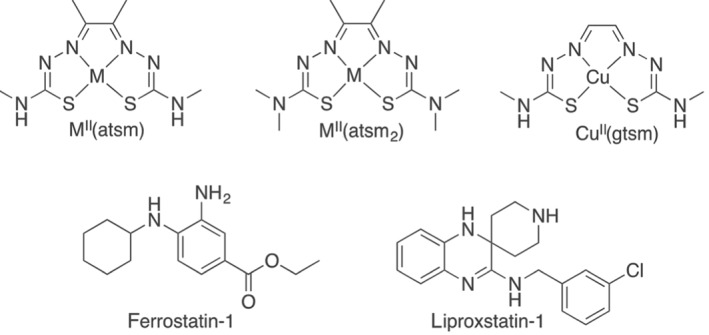
The chemical structures of MII(atsm) and MII(atsm2) (where M = either CuII or NiII), CuII(gtsm), ferrostatin‐1, and liproxstatin‐1
Recent work has implicated ferroptosis, a regulated cell death pathway characterised by Fe‐dependent lipid peroxidation in ALS, PD, Alzheimer's disease (AD) and stroke (Guiney, Adlard, Bush, Finkelstein, & Ayton, 2017; Masaldan, Bush, Devos, Rolland, & Moreau, 2018; Stockwell et al., 2017). Iron levels are elevated in affected brain regions in these neurological disorders (Ayton et al., 2015; Ayton et al., 2017; Ding et al., 2011; Grolez et al., 2016; Park et al., 2011; Pyatigorskaya et al., 2015; Raven, Lu, Tishler, Heydari, & Bartzokis, 2013; Tao, Wang, Rogers, & Wang, 2014; Tuo et al., 2017), as are lipid peroxidation products characteristic of ferroptosis (Cherubini, Ruggiero, Polidori, & Mecocci, 2005; Choi et al., 2015; Dexter et al., 1989; Jenner, Dexter, Sian, Schapira, & Marsden, 1992; Mandal, Saharan, Tripathi, & Murari, 2015; Reed, Pierce, Markesbery, & Butterfield, 2009; Sian et al., 1994; Simpson, Henry, Henkel, Smith, & Appel, 2004; Tohgi et al., 1999; Yu et al., 2016). The most potent known inhibitors of ferroptosis are the radical trapping antioxidants (RTAs), liproxstatin‐1 and ferrostatin‐1 (Dixon et al., 2012; Friedmann Angeli et al., 2014), which inhibit ferroptosis with nanomolar efficacy (Sheng et al., 2017; Zilka et al., 2017). Ferrostatin‐1 and liproxstatin‐1 have been reported to rescue ischaemia reperfusion injury and stroke models (Friedmann Angeli et al., 2014; Tuo et al., 2017) and PD (Do Van et al., 2016). Edaravone, an RTA approved for the treatment of acute ischaemic stroke and ALS, was recently reported to have antiferroptotic properties (Homma, Kobayashi, Sato, & Fujii, 2019).
Given the recent activity of CuII(atsm) in its first ALS and PD clinical trials, and that ferroptosis may play a role in neurodegenerative disorders, we hypothesised that CuII(atsm) has antiferroptosis properties. Utilising cellular and cell‐free systems, we found that CuII(atsm) possesses significant antiferroptotic activity by acting as a lipid RTA like ferrostatin‐1 and liproxstatin‐1.
2. METHODS
2.1. Bis(thiosemicarbazone) synthesis
H2(atsm), CuII(atsm) (Dearling, Lewis, Mullen, Welch, & Blower, 2002; Gingras, Suprunchuk, & Bayley, 1962), CuII (gtsm) (Beraldo, Boyd, & West, 1998) and CuII(atsm2) (Gingras et al., 1962; West et al., 1997) were synthesised as reported previously. 1H and 13C{1H} spectra were recorded using a Varian FT‐NMR 400 spectrometer (Agilent Technologies, Santa Clara, USA). All 1H NMR spectra were acquired at 400 MHz, and 13C{1H} spectra were acquired at 101 MHz. The reported peaks were all referenced to solvent peaks in the order of p.p.m. at 25°C. Microanalysis measurements were carried out by The Campbell Microanalytical Laboratory in the Department of Chemistry, University of Otago, Union Place, Dunedin, New Zealand. Analytical HPLC was performed on Agilent 1200 series HPLC system (Agilent Technologies) fitted with an Alltech Hypersil BDS–C18 column (4.6 × 150 nm, 5 μm). The mobile phase was a gradient consisting of Solvent A (0.1% TFA in H2O) and Solvent B (0.1% TFA in CH3CN) from 0% to 100% B over 25 min and UV detection at λ 220, 254, 275 and 350 nm. ESI‐QTOF MS was collected on an Exactive Plus Orbitrap Infusion mass spectrometer (Exactive Series, 2.8 Build 268801, ThermoFisher Scientific, Scoresby, Australia). Analysis was performed using Xcalibur 4.0.27.10 (ThermoFisher Scientific, RRID:SCR_014593).
NiII(atsm) synthesis was adapted from a reported procedure (McCleverty & Jones, 1970). To a suspension of diacetyl‐bis(4‐methyl‐3‐thiosemicarbazone) (0.50 g, 1.9 mM) in ethanol (25 ml) was added Ni(OCOCH3)2·4H2O (0.48 g, 1.9 mM). The reaction mixture was heated at reflux for 5 hr and then allowed to cool to room temperature. The precipitate that formed was collected by filtration and washed with ethanol, water, followed by diethyl ether, to give a dark green powder (0.48 g, 1.5 mM, 79%). 1H NMR (400 MHz, DMSO‐d 6): δH/ppm 7.68 (br s, 2H, NH), 2.78 (d, J = 3.8 Hz, 6H, NH‐CH 3), 1.94 (s, 6H, CH3). 13C{1H} NMR (101 MHz, DMSO‐d 6): δC/ppm 176.08, 155.56, 32.15, 13.82. Analysis calculated for C8H14N6NiS2, C, 30.31; H, 4.45; N, 26.51; found: C, 30.49, H, 4.51, N, 26.85. MS (ESI/O‐TOF) (m/z): calculated for [C8H14N6NiS2 + H]+, 317.0148; found, 317.0148. RP‐HPLC: R t = 10.20 min.
2.2. Cell culture
The Immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology. Cell culture reagents were purchased from ThermoFisher Scientific unless otherwise specified, and all cells were cultured at 37°C with 5% CO2. N27 cells, derived from E12 rat mesencephalic tissue (Merck, Bayswater, Australia), were cultured in RPMI 1640 media supplemented with 10% fetal calf serum (Bovogen, Keilor East, Australia), penicillin, and streptomycin. Primary cortical neurons were isolated from E14 C57BL/6J mice (RRID:IMSR_JAX:000664) as previously described (Xu et al., 2016). Primary cells were maintained in neurobasal media supplemented with 2% B27, GlutaMAX, penicillin and streptomycin. RSL3 could not induce ferroptosis in these cells in the presence of B27, so after 3 days in vitro (DIV) the cells were transferred to RPMI 1640 media supplemented with 10% fetal calf serum, penicillin and streptomycin, with cytosine arabinoside (2 μM) to minimise astrocyte growth. Cells were used between DIV9 and DIV11. All cell‐based assays were conducted in RPMI 1640 media supplemented with 10% fetal calf serum, penicillin and streptomycin.
2.3. Cell viability
Cells in 96‐well plates were treated with candidate antiferroptotic compounds together with RSL3, erastin or Fe, for 24 hr as described in figure legends. C11‐BODIPY(581/591) was not present, and cell viability was determined colourimetrically using MTT as previously described (Xu et al., 2016). Absorbance was measured at 570 nm using a PowerWave XS microplate spectrophotometer (BioTek Instruments, Winooski, USA) and cell viability was expressed as a percentage of control cells.
2.4. Lipid peroxidation and FeII measurements
Lipid peroxidation was assessed with C11‐BODIPY(581/591), a ratiometric fluorophore that responds to lipid peroxidation with an increase in fluorescence emitted in the green range (excitation 484 nm; emission 530 nm) and a corresponding decrease in fluorescence emitted in the red range (excitation 581 nm; emission 600 nm; Pap et al., 1999). Fluorescence was measured with a FlexStation 3 multimode microplate reader (Molecular Devices, San Jose, USA). Lipid peroxidation was expressed as the ratio of green to red C11‐BODIPY(581/591) fluorescence.
Cells in 96‐well plates were incubated with candidate antiferroptotic compounds together with RSL3, erastin or Fe, as described in figure legends. C11‐BODIPY(581/591) (2.5 μM) was added immediately prior to the addition of the RSL3, erastin or Fe and was present for the duration of the treatment (24 hr). Media was then replaced with PBS for fluorescence measurements. Lipid peroxidation reported by C11‐BODIPY(581/591) was normalised to the maximum C11‐BODIPY(581/591) signal induced by RSL3, erastin or Fe, therefore adjusting for any potential antiferroptotic effect of the C11‐BODIPY(581/591) itself.
Cell‐free lipid peroxidation was examined using arachidonic acid in Locke's buffer (NaCl 154 mM; KCl 5.6 mM; HEPES 5 mM; glucose 5 mM; MgCl2 1 mM; CaCl2 2.3 mM; NaHCO3 3.6 mM; pH 7.4). Arachidonic acid was incubated with candidate antiferroptotic compounds together with FeII ((NH4)2Fe(SO4)2·6H2O) or FeIII ((NH4)5(Fe(C6H4O7)2)) as described in figure legends. C11‐BODIPY(581/591) (2.5 μM) was added immediately prior to the addition of the Fe and present for the duration of the treatment (30 min).
FeII levels were determined colourimetrically using Ferene‐S as previously described (Wong et al., 2014). Absorbance was measured at 590 nm using a PowerWave XS microplate spectrophotometer. FeII levels were calculated using an extinction coefficient of 36.6 mM−1 FeII cm−1.
2.5. Data and statistical analyses
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All quantitative data were analysed using Prism 8.1 (GraphPad Software, San Diego, USA, RRID:SCR_002798). As test compounds were coloured, the operator and data analysis were not blinded and samples were not randomised. Between two and four technical replicates were used to ensure the reliability of single values within each independent experiment. Results are expressed as means ± SEM from independent experiments. Cell viability and lipid peroxidation data were normalised to control samples to account for unwanted sources of variation between independent experiments. Comparisons between groups utilised nonparametric statistics and P values were calculated using either the Mann–Whitney test or the Kruskal–Wallis test corrected for multiple comparisons by controlling the false discovery rate with the Benjamini, Krieger and Yekutieli test, as described in the figure legends. The post hoc tests were conducted only if F achieved P < .05 and there was no significant variance inhomogeneity. P < .05 was deemed statistically significant. Non‐linear regression analysis with a variable slope model was used to fit a four‐parameter logistic curve to dose–response data to calculate EC50 with 95% CI.
2.6. Materials
Analytical reagents were purchased from Merck (Bayswater, Australia) unless otherwise specified. Erastin, RSL3 (Jomar Bioscience, Scoresby, Australia), liproxstatin‐1, clioquinol, C11‐BODIPY(581/591) (ThermoFisher Scientific), and bis(thiosemicarbazone) compounds were dissolved in DMSO. 3‐[4,5‐Dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide (MTT) was dissolved in PBS. CuSO4·5H2O, (NH4)2Fe(SO4)2·6H2O, (NH4)5(Fe(C6H4O7)2), Ferene‐S, and bathocuproinedisulfonic acid (BCS) were dissolved in highly purified water. Arachidonic acid was dissolved in ethanol.
3. RESULTS
The impact of CuII(atsm) on ferroptosis was evaluated in primary and immortalised neuronal models, with liproxstatin‐1 used as an antiferroptotic positive control (Figure 1). Primary cortical neurons were cultured with cytosine arabinoside to suppress astrocyte growth, rendering the culture predominantly neuronal (Figure S1). RSL3 was used to induce lipid peroxidation and ferroptotic cell death. Dose–response experiments revealed that CuII(atsm) (EC50:143 nM) could rescue RSL3‐induced death in primary neuronal cells with potency within an order of magnitude of liproxstatin‐1 (EC50: 32 nM; Figure 2a). Both CuII(atsm) and liproxstatin‐1 abolished RSL3‐induced lipid peroxidation detected by C11‐BODIPY(581/591) (Figure 2b), consistent with antiferroptotic activity. Inhibition of ferroptosis was next evaluated in immortalised neuronal cell lines, N27 (Figure 2c–f) and SN4741 (Figure S2), which, unlike primary neurons, were sensitive to ferroptosis induced by both RSL3 and erastin. CuII(atsm) exhibited a dose‐dependent rescue of cell death (measured by MTT assay) induced by RSL3 (Figure 2c) and erastin (Figure 2d) with EC50 values that were within an order of magnitude of those for liproxstatin‐1. Similar results were obtained when cell viability was measured by either PrestoBlue or LDH release (Figure S3). FeII‐induced cell death was ≈80% maximally rescued by CuII(atsm) and liproxstatin‐1 (Figure 2e), indicating that FeII induces some nonferroptotic toxicity. Both CuII(atsm) and liproxstatin‐1 suppressed lipid peroxidation in N27 cells induced by RSL3, erastin or Fe (Figure 2f). Similar results were seen when C11‐BODIPY(581/591) was added after the RSL3 treatment and detected by flow cytometry (Figure S4). CuII(atsm) and liproxstatin‐1 also prevented SN4741 cell death induced by RSL3 and erastin with EC50 values that were also within an order of magnitude of liproxstatin‐1 (Figure S2).
Figure 2.
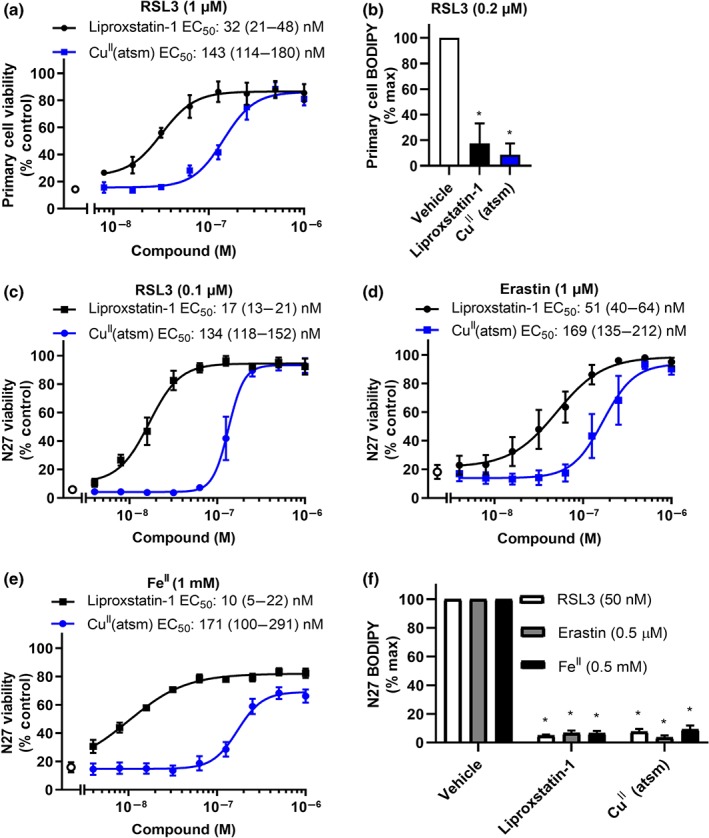
CuII(atsm) rescues ferroptosis and lipid peroxidation in cultured neurons. (a) Viability of cells treated with RSL3 (1 μM) ± liproxstatin‐1 or CuII(atsm) for 24 hr. Data are means ± SEM, n = 6 independent experiments. EC50 was determined by non‐linear regression, with 95% CI shown in parenthesis. (b) Lipid peroxidation measured by C11‐BODIPY(581/591) in cells treated with RSL3 (0.2 μM) ± liproxstatin‐1 or CuII(atsm) (1 μM) for 24 hr. Data are means ± SEM, n = 6 independent experiments. P values were calculated using the Kruskal–Wallis test corrected for multiple comparisons by controlling the false discovery rate with the Benjamini, Krieger, and Yekutieli test. *P < .05 compared to vehicle treated cells. (c–e) Viability of cells treated with RSL3 (0.1 μM, c), erastin (1 μM, d) or FeII (1 mM, e) ± liproxstatin‐1 or CuII(atsm) for 24 hr. Data are means ± SEM, n = 5 independent experiments. EC50 was determined by non‐linear regression, with 95% CI shown in parenthesis. (f) Lipid peroxidation measured by C11‐BODIPY(581/591) in cells treated with RSL3 (50 nM), erastin (0.5 μM), or FeII (0.5 mM) ± liproxstatin‐1 or CuII(atsm) (1 μM) for 24 hr. Data are means ± SEM, n = 5 independent experiments. P values were calculated using the Kruskal–Wallis test corrected for multiple comparisons by controlling the false discovery rate with the Benjamini, Krieger, and Yekutieli test. *P < .05 compared to vehicle treated cells
We evaluated whether a simple CuII salt could prevent ferroptosis in N27 cells (Figure 3). Neither RSL3‐ nor erastin‐induced cell death were prevented by addition of CuSO4 supplied at concentrations up to 300 μM (Figure 3a). CuII (gtsm) (Figure 3b) and clioquinol (Figure 3c), two compounds that promote cellular Cu uptake (Adlard et al., 2008; Crouch et al., 2009), were also unable to prevent ferroptosis. CuII (gtsm) is an analogous bis(thiosemicarbazone)CuII complex that has a higher CuII/I reduction potential than CuII(atsm) and is therefore more susceptible to intracellular reduction and release of the Cu ion from the ligand (Donnelly et al., 2008). Treatment with ≥100 nM of CuII (gtsm) resulted in significant reductions in cell viability suggesting the antiferroptotic effects of CuII(atsm) are unlikely to be mediated by merely increasing intracellular Cu concentrations.
Figure 3.
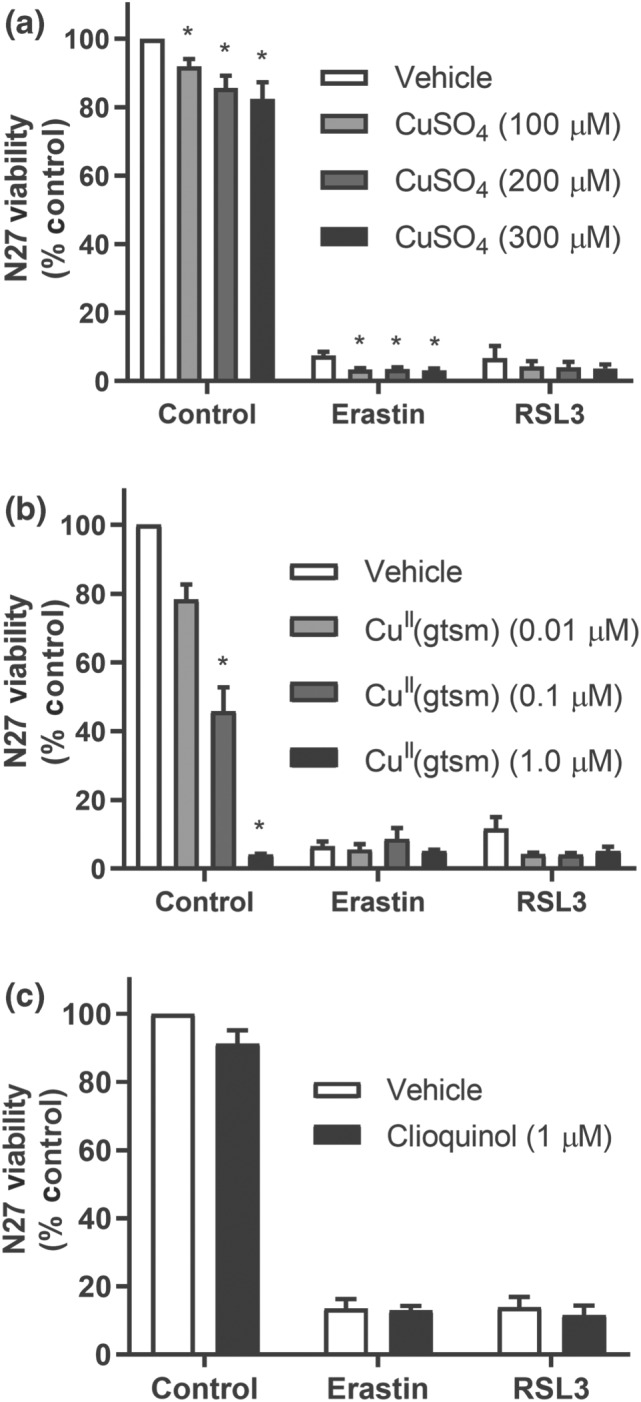
Cu does not prevent ferroptosis in N27 cells. (a–c) Viability of cells treated with RSL3 (0.1 μM) or erastin (1 μM) ± CuSO4 (a), CuII (gtsm) (b), or clioquinol (c) for 24 hr. Data are means ± SEM, n = 5 independent experiments. (a, b) P values were calculated using the Kruskal–Wallis test corrected for multiple comparisons by controlling the false discovery rate with the Benjamini, Krieger, and Yekutieli test. (c) P values were calculated using the Mann–Whitney test. *P < .05 compared to vehicle
To probe the role of the Cu ion in the antiferroptotic activity of CuII(atsm) we investigated the activity of the analogous NiII complex, NiII(atsm), and the metal‐free ligand H2(atsm) (Figure 1). H2(atsm) and NiII(atsm) both prevented ferroptosis in N27 cells (Figure 4a). It is likely that the activity of H2(atsm) is a consequence of the ligand forming CuII(atsm) by complexing Cu ions present in the cell media. BCS, a selective copper chelator (Xiao et al., 2011), abolished the antiferroptotic activity of H2(atsm), consistent with the premise that the activity of H2(atsm) is due to the ligand coordinating to CuII in the culture media to form CuII(atsm). NiII(atsm) (EC50 = 178 nM) prevented RSL3‐induced ferroptosis in N27 cells similarly to CuII(atsm) (EC50 = 131 nM; Figure 4b). Analysis of cellular metal concentrations by inductively coupled plasma MS indicated that CuII(atsm) and NiII(atsm) increased Cu and Ni levels, respectively (Figure S5). As expected, cellular Fe levels were not altered by addition of any atsm compounds. Cellular levels of GPX4, a lipid peroxide scavenging peroxidase, were unaffected by treatment with either liproxstatin‐1 or CuII(atsm) (Figure S6).
Figure 4.
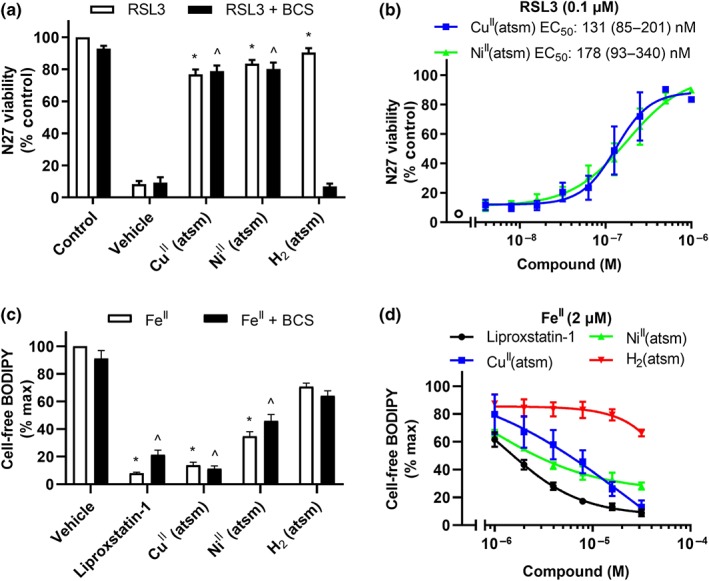
NiII(atsm) prevents ferroptosis in N27 cells and lipid peroxidation induced by FeII. (a, b) Viability in cells treated with RSL3 (0.1 μM) for 24 hr. (a) Cells treated with RSL3 ± CuII(atsm), NiII(atsm) or H2(atsm) (1 μM) for 24 hr in the presence or absence of the Cu chelator BCS (100 μM). Data are means ± SEM, n = 6 independent experiments. P values were calculated using the Kruskal–Wallis test corrected for multiple comparisons by controlling the false discovery rate with the Benjamini, Krieger, and Yekutieli test. *P < .05 compared to vehicle with RSL3, ^P < .05 compared to vehicle with RSL3 and BCS. (b) Cells treated with RSL3 ± CuII(atsm) or NiII(atsm) for 24 hr. Data are means ± SEM, n = 5 independent experiments. EC50 was determined by non‐linear regression, with 95% CI shown in parenthesis. (c, d) Lipid peroxidation measured by C11‐BODIPY(581/591) in Locke's buffer with arachidonic acid (50 μM) treated with FeII (2 μM) for 30 min. (c) Arachidonic acid treated with FeII ± liproxstatin‐1, CuII(atsm), NiII(atsm) or H2(atsm) (1 μM) in the presence or absence of the Cu chelator BCS (12.5 μM). Data are means ± SEM, n = 6 independent experiments. P values were calculated using the Kruskal–Wallis test corrected for multiple comparisons by controlling the false discovery rate with the Benjamini, Krieger and Yekutieli test. *P < .05 compared to vehicle with Fe, ^P < .05 compared to vehicle with FeII and BCS. (d) Arachidonic acid treated with FeII ± liproxstatin‐1, CuII(atsm), NiII(atsm), or H2(atsm). Data are means ± SEM, n = 5 independent experiments
We hypothesised that CuII(atsm) acts as a RTA to prevent the propagation of lipid peroxyl radicals, as reported for liproxstatin‐1 (Shah, Margison, & Pratt, 2017). Utilising a cell‐free system, we assayed the amount of lipid peroxidation with C11‐BODIPY(581/591) fluorescence after incubation of arachidonic acid with FeII (2 μM). Like liproxstatin‐1, CuII(atsm) and NiII(atsm) inhibited net lipid peroxidation (Figure 4c,d), consistent with RTA activity. H2(atsm) partially prevented lipid peroxidation at the highest doses, indicating that the ligand alone possesses some RTA properties. BCS did not change the inhibition of cell‐free lipid peroxidation (Figure 4c). Malondialdehyde accumulation, another maker of lipid peroxidation, was also prevented by liproxstatin‐1, CuII(atsm), and NiII(atsm) (Figure S7). Whereas liproxstatin‐1, CuII(atsm) and NiII(atsm) prevented ferroptosis at nanomolar concentrations (Figure 4b), micromolar concentrations were required to prevent lipid peroxidation induced by FeII in cell‐free conditions (Figure 4d). CuII(atsm) also prevents FeII‐induced peroxidation of cellular‐derived lipids (Figure S8).
We next analysed which structural components of CuII(atsm) were responsible for its antiferroptotic and radical quenching properties. Liproxstatin‐1 and ferrostatin‐1 each contain an aromatic amine that is involved in rapid H‐atom transfer to autoxidation chain‐carrying lipid peroxy radicals (Figure 1) important for their radical quenching activity (Skouta et al., 2014). Coordination of either CuII or NiII to H2(atsm) involves a double deprotonation of the ligand to form a dianionic delocalised ligand. The ─NH─CH3 functional groups present in both CuII(atsm) and NiII(atsm) are a potential H‐atom donor to give a delocalised stabilised radical analogous to the aromatic amines present in liproxstatin‐1 and ferrostatin‐1 (Figure 1). To investigate this possibility, an analogue where these ─NH─CH3 functional groups were replaced with a ─N (CH3)2 functional group, CuII(atsm2), was evaluated (Figure 1). The ability of CuII(atsm2) to rescue ferroptosis induced by RSL3 or erastin in N27 cells was markedly attenuated when compared to CuII(atsm), although ≈40% survival occurred with doses of CuII(atsm2) ≥ 125 nM (Figure 5a,b). In contrast to CuII(atsm), CuII(atsm2) was unable to prevent lipid peroxidation induced by FeII in cell‐free arachidonic acid (Figure 5c) or in cellular‐derived lipids (Figure S8), consistent with ─NH─CH3 functional groups present in both CuII(atsm) and NiII(atsm) contributing to their radical quenching activity.
Figure 5.
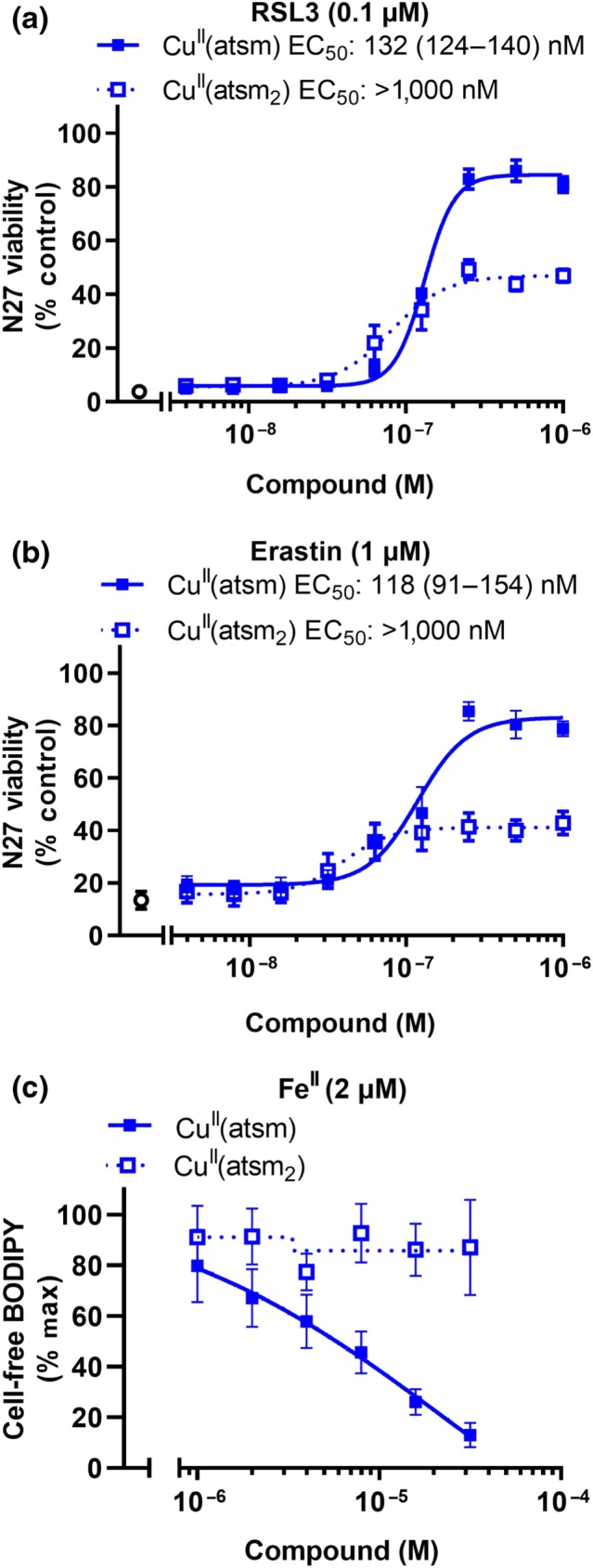
CuII(atsm2) is impaired in preventing ferroptosis in N27 cells or lipid peroxidation induced by FeII. (a, b) Viability in cells treated with RSL3 (a, 0.1 μM) or erastin (b, 1 μM) ± CuII(atsm) or CuII(atsm2) for 24 hr. Data are means ± SEM, n = 5 independent experiments. EC50 was determined by non‐linear regression, with 95% CI shown in parenthesis. CuII(atsm2) EC50 could not be determined as viability did not exceed 50%. (c) Lipid peroxidation measured by C11‐BODIPY(581/591) in Locke's buffer using arachidonic acid (50 μM) treated with FeII (2 μM) ± CuII(atsm) or CuII(atsm2) for 30 min. Data are means ± SEM, n = 5 independent experiments
Lipid peroxidation associated with ferroptosis is thought to be initiated by the oxidation of cellular FeII (Gaschler et al., 2018). To assess whether CuII(atsm) might stabilise FeII as the mechanism of its antiferroptotic activity, we assayed the change of Fe redox state in the cell‐free lipid peroxidation assay with arachidonic acid. We monitored lipid peroxidation (Figure 6a,b) induced by FeII (Figure 6a,c) and FeIII (Figure 6b,d) solutions over 30 min, and Ferene‐S was used to measure FeII at the beginning and end of this period (Figure 6c,d). FeII induced a large increase in lipid peroxidation, which had a rapid phase that was largely completed by the time the first measurement could be taken (1 min), followed by a slower continual parabolic accumulation (Figure 6a). FeIII induced far less lipid peroxidation, but this slightly accumulated over the 30‐min incubation (Figure 6b), consistent with a small amount of redox cycling in the aerobic buffer. The amount of lipid peroxidation detected by C11‐BODIPY(581/591) over 30 min was not matched by a decrease in FeII detected by Ferene‐S added at the end of the experiment (Figure 6c,d). This indicates that either the C11‐BODIPY(581/591) change is being driven by smaller concentrations of FeII than the Ferene‐S assay can discriminate or that redox cycling of FeII drives the lipid peroxidation with no net oxidation of FeII (Figure 6e). Both liproxstatin‐1 and CuII(atsm) suppressed lipid peroxidation in the presence of either FeII or FeIII (Figure 6a,b). We manipulated the oxidation state of the Fe forms in the experimental system by introducing ceruloplasmin (a ferroxidase that rapidly converts FeII to FeIII) or the strong reducing agent, ascorbate (that rapidly converts FeIII to FeII). Ceruloplasmin decreased FeII levels (Figure 6c), and limited lipid peroxidation induced by either FeII or FeIII (Figure 6a,b). Conversely, ascorbate markedly increased FeII levels when the starting solution contained FeIII (Figure 6d), but not when the starting solution contained FeII (Figure 6c). Ascorbate enhanced FeIII‐induced lipid peroxidation (Figure 6b) but limited FeII‐induced lipid peroxidation (Figure 6a). These findings indicate that hampering the redox cycling of the FeII/FeIII species, by biasing the environment towards one redox state or the other, limits the peroxidation propagated by the Fe species. Liproxstatin‐1 and CuII(atsm) limited lipid peroxidation induced by both FeII and FeIII (Figure 6a,b) without altering FeII levels (Figure 6c,d), indicating that these compounds do not quench lipid peroxidation by modulating Fe oxidation or reduction.
Figure 6.
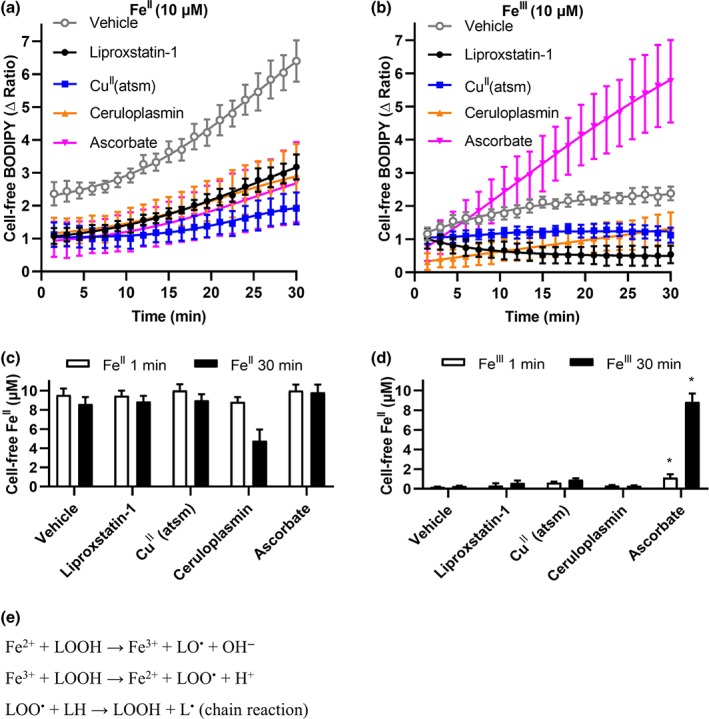
CuII(atsm) does not react with FeII or FeIII to suppress lipid peroxidation. Arachidonic acid (10 μM) was treated with either FeII (10 μM, a, c) or FeIII (10 μM, b, d) ± ceruloplasmin (200 μg·ml−1), ascorbate (1 mM), liproxstatin‐1 (10 μM) or CuII(atsm) (10 μM) in Locke's buffer for 30 min. (a, b) Lipid peroxidation was measured by C11‐BODIPY(581/591) every 90 sec. Data are means ± SEM, n = 5 independent experiments. Non‐linear regression was conducted. (c, d) FeII measured by Ferene‐S after 1 min and 30 min. P values were calculated using the Kruskal–Wallis test corrected for multiple comparisons by controlling the false discovery rate with the Benjamini, Krieger,and Yekutieli test. *P < .05, compared to vehicle treated cells. (e) Redox cycling reactions of Fe species that can lead to the propagation of lipid peroxides and radicals
4. DISCUSSION
Growing evidence implicates ferroptosis in various neurological disorders. Hence, there is interest in developing high potency and selective ferroptosis inhibitors with favourable pharmacokinetic properties and capable of crossing the blood–brain barrier. When ferroptosis was first described in 2012 (Dixon et al., 2012), CuII(atsm) had already commenced development for neurological disorders after showing efficacy in mouse models of ALS (Hilton et al., 2017; McAllum et al., 2013; Roberts et al., 2014; Soon et al., 2011; Vieira et al., 2017), PD (Hung et al., 2012) and stroke (Huuskonen et al., 2017). Phase 1 trial results of CuII(atsm) for ALS (NCT02870634) recently reported promising benefits on ALSFRS‐r score, Edinburgh Cognitive Assessment Scale (ECAS) and forced vital capacity (Rowe et al., 2018). A separate Phase 1 trial of CuII(atsm) for PD (NCT03204929) has more recently reported benefits in disease severity by UPDRS and quality of life by PDQ‐39 (Evans et al., 2019). Further testing will be needed to confirm these results, but our current findings indicate that should CuII(atsm) demonstrate clinical efficacy for these disorders, the drug's mechanism of action could be as a ferroptosis inhibitor with potency approaching that of liproxstatin‐1.
CuII(atsm) prevents lipid peroxidation without altering the oxidation state of Fe, consistent with CuII(atsm) acting like ferrostatin‐1 and liproxstatin‐1 to prevent propagation of the lipid radicals rather than to prevent FeII oxidation. The antiferroptotic activity of ferrostatin‐1 and liproxstatin‐1 is due to their ability to inhibit lipid peroxidation by rapid transfer of a H atom from their aryl amine moieties to lipid radicals, thereby quenching the lipid radical (Shah et al., 2017). Coordination of divalent CuII and NiII to bis(thiosemicarbazone) ligands involves a double deprotonation of the ligand to give a resonance stabilised conjugated conformation of the ligand where the NH‐4‐methyl substituent could be a possible H atom donor that could be considered comparable to the aryl amine present in both ferrostatin‐1 and liproxstatin‐1. The redox activity of bis(thiosemicarbazone)CuII complexes is not confined to the metal ion. Conjugated thiosemicarbazone ligands are redox noninnocent (Haddad, Cronin, Mashuta, Buchanan, & Grapperhaus, 2017; Holland, Green, & Dilworth, 2006; Kowol et al., 2008). The cyclic voltammogram of CuII(atsm) reveals not only a reversible CuII/I process at E0 = −1.20 V versus ferricenium/ferrocene (Fc+/Fc) in dimethylformamide but also a reversible oxidation at higher potentials, E0 = 0.24 V versus Fc+/Fc in DMF (Dearling et al., 2002). This reversible process has been attributed to CuIII/II, but density functional theory calculations suggest that the highest occupied molecular orbital is ligand based with π‐character taking a small contribution from the metal dxy orbital (Holland et al., 2006).
Either of the two NH‐methyl substituents present in CuII(atsm) could serve as H atom donor where the radical generated could be stabilised by delocalisation over the conjugated ligand backbone. This possibility was investigated in two ways. Firstly, the importance of the conjugated ligand backbone was demonstrated by the observation that the analogous NiII(atsm) displayed comparable cytoprotective activity to CuII(atsm). This important observation suggests that the role of the metal ions, in this instance, is to promote the deprotonation of the ligand to provide the delocalised conjugated conformation of the bis(thiosemicarbazone) ligand. Secondly, the importance of the potential H atom donor of the ─N─CH3 substituent was demonstrated by investigating an analogue where this H atom is replaced with a second methyl substituent, ─N (CH3)2, to give CuII(atsm2). The CuII(atsm2) complex, which lacks a potential H donor, was unable to prevent lipid peroxidation induced by FeII in a cell‐free system and was far less efficacious than CuII(atsm) in rescuing ferroptosis in N27 cells and cell‐free lipid peroxidation induced by FeII.
In summary, CuII(atsm) possesses unexpected RTA properties similar to the antiferroptosis agents, liproxstatin‐1 and ferrostatin‐1. This may explain how CuII(atsm) suppressed lipid peroxidation in a myocardial ischaemia/reperfusion model (Wada, Fujibayashi, Tajima, & Yokoyama, 1994), just as liproxstatin‐1 and ferrostatin‐1 rescue models of ischaemia/reperfusion (Friedmann Angeli et al., 2014; Tuo et al., 2017). The potency of CuII(atsm) is within an order of magnitude of the canonical ferroptosis inhibitor, liproxstatin‐1, while its oral bioavailability, preclinical safety, and brain penetrant properties make it an attractive investigational product for clinical trials of ferroptosis‐related diseases. The antiferroptotic properties of CuII(atsm) could be the prospective disease‐modifying mechanism being explored in current ALS and PD clinical trials.
AUTHOR CONTRIBUTIONS
A.S., A.A.B., K.J.B., P.J.C., S.A., P.S.D., and A.I.B. participated in research design; A.S., K.S., K.M.A., K.A.D., and I.V. conducted the research; K.S. and P.D. contributed new reagents; A.S., K.J.B., P.S.D., and A.I.B. performed data analysis; A.S., K.J.B., P.S.D., and A.I.B. contributed to the writing of the manuscript.
CONFLICT OF INTEREST
A.I.B. is a shareholder in Prana Biotechnology Ltd, Cogstate Ltd, Brighton Biotech LLC, Grunbiotics Pty Ltd, Eucalyptus Pty Ltd, and Mesoblast Ltd. He is a paid consultant for, and has a profit share interest in, Collaborative Medicinal Development Pty Ltd. P.S.D. has served as a consultant to Collaborative Medicinal Development LLC. Collaborative Medicinal Development LLC has licensed intellectual property related to this subject from The University of Melbourne, where the inventors include P.S.D. and K.J.B.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Immunoblotting and Immunochemistry, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Primary cortical cultures were predominantly neuronal. Primary cortical cells were grown on poly‐D lysine coated 12 mm glass coverslips until DIV9 and prepared for immunocytochemistry as previously described (Xu et al., 2016). Anti‐neuron‐specific nuclear protein (1:200) and anti‐mouse Alexa 488 (1:400, ThermoFisher Scientific, Scoresby, Australia) were used to identify neurons (red). Anti‐glial fibrillary acidic protein (GFAP, 1:400) and anti‐guinea pig Alexa 647 (1:400, ThermoFisher Scientific) were used to identify astrocytes (green). DAPI (300 nM, ThermoFisher Scientific) was used to detect nuclei (blue). Images were recorded at 20X magnification using an SP8 confocal microscope with Leica Application Suite X software (Leica Microsystems, Macquarie Park, Australia). Images at each wavelength were captured sequentially and multi‐colour XY images were prepared using Image J (Schneider, Rasband et al., 2012). XY images are shown as maximum brightness stacked images. Representative images are shown.
Figure S2. CuII(atsm) prevents ferroptosis in SN4741 cells. Cells were derived from E13.5 mouse substantia nigra tissue (Son, Chun et al., 1999) and cultured as per N27 cells. Viability of cells treated with RSL3 (1 μM, A) or erastin (1 μM, B) ± liproxstatin‐1 or CuII(atsm) for 24 hr. Viability was measured with MTT and expressed as a percentage of control cells. Data are means ± SEM, n=5 independent experiments. EC50 was determined by nonlinear regression, with 95% CI shown in parenthesis.
Figure S3. CuII(atsm) prevents ferroptosis in N27 cells. Viability of cells treated with RSL3 (0.1 μM, A‐B) or erastin (1 μM, C‐D) ± liproxstatin‐1 or CuII(atsm) for 24 hr. (A and C) Viability was measured with PrestoBlue according to the manufacturer's instructions (ThermoFisher Scientific) and expressed as a percentage of control cells. (B and D) Viability measured with the Cytotoxicity Detection Kit (LDH) according to the manufacturer's instructions (Merck, Bayswater, Australia) and expressed as a percentage of maximum lactate dehydrogenase (LDH) release. Data are means ± SEM, n=5 independent experiments. EC50 was determined by nonlinear regression, with 95% CI shown in parenthesis.
Figure S4. CuII(atsm) prevents lipid peroxidation in N27 cells. Cells were cultured in 6 well plates and treated in growth media with RSL3 (50 nM) ± liproxstatin‐1 or CuII(atsm) (1 μM) for 4 hr. Cells were washed in PBS and incubated in growth media with C11‐BODIPY(581/591) (125 nM) for 1 hr. Cells were harvested with trypsin and resuspended in PBS containing: EDTA 1 mM, HEPES 25 mM, foetal calf serum 1%, penicillin and streptomycin. Cells were strained through a 35 μm cell strainer (Corning, Mulgrave, Australia) and green fluorescence (excitation 488 nm, emission 525 nm) and red fluorescence (excitation 633 nm, emission 660 nm) were detected using a CytoFLEX S flow cytometer (Beckman Coulter, Mount Waverly, Australia). A minimum of 10000 cells were analysed per condition. Representative data from one of three experiments is shown.
Figure S5. NiII(atsm) does not promote Cu uptake in N27 cells. Cells were treated with CuII(atsm), CuII(atsm2) or NiII(atsm) (1 μM) for 24 hr. Cellular metal levels were measured with inductively coupled plasma mass spectrometry analysis using an Agilent 7700x (Agilent Technologies, Santa Clara, USA) as reported previously (Bica, Liddell et al., 2014). Data are means ± SEM, n=5 independent experiments. P values were calculated using one‐way ANOVA corrected for multiple comparisons with Dunnett's post‐hoc test. A Brown‐Forsythe test confirmed variance was not significantly different between groups and a Kolmogorov‐Smirnov test confirmed residuals were normally distributed. *P<.05 compared to vehicle treated cells.
Figure S6. CuII(atsm) does not elevate GPX4 levels in N27 cells. Cells treated with erastin (1 μM) or RSL3 (0.1 μM) ± liproxstatin‐1 or CuII(atsm) (1 μM) for 24 hr. Samples were lysed in RIPA buffer (Merck) and proteins (15 μg) were resolved on NuPAGE 4‐12% Bis‐Tris gels and transferred to PVDF membranes with an iBlot2 for western immuno‐blotting (ThermoFisher Scientific). Primary antibodies used were rabbit anti‐GPX4 (1:5000, Abcam, Cambridge, USA) and mouse anti‐actin (1:10000, Merck). Horseradish peroxidase coupled secondary antibodies were rabbit anti‐mouse and goat anti‐rabbit (1:7000, Agilent Technologies). Chemiluminescence was detected using ECL (GE Healthcare, Parramatta, Australia) and images were captured with a Fujifilm LAS‐3000 (Fujifilm, Campbellfield, Australia).
Figure S7. CuII(atsm) prevents lipid peroxidation induced by FeII. Locke's buffer with arachidonic acid (50 μM) treated with FeII (2 μM) ± liproxstatin‐1, CuII(atsm), NiII(atsm) or H2(atsm) (10 μM) for 30 min. Thiobarbituric Acid was used to detect malondialdehyde accumulation with the Lipid Peroxidation (MDA) Assay kit (Abcam) according to the manufacturer's instructions. Fluorescence (excitation 532 nm, emission 553 nm) was measured with a FlexStation 3 multi‐mode microplate reader (Molecular Devices). Lipid peroxidation reported by malondialdehyde was normalised to the maximum signal induced by Fe. Data are means ± SEM, n=6 independent experiments. P values were calculated using the Kruskal‐Wallis test corrected for multiple comparisons by controlling the false discovery rate with the Benjamini, Krieger, and Yekutieli test. *P<.05 compared to vehicle treated cells.
Figure S8. CuII(atsm) prevents FeII‐induced peroxidation of cellular‐derived lipids. Cellular‐derived lipids were prepared from 10 million N27 cells snap frozen, resuspended in 10 ml Locke's buffer, and sonicated in a water bath for 30 min. C11‐BODIPY(581/591) (2.5 μM) was added to the homogenate (100 μl) which was treated with FeII (10 μM) ± liproxstatin‐1, CuII(atsm), NiII(atsm), H2(atsm) or CuII(atsm2) for 30 min. C11‐BODIPY(581/591) fluorescence was measured with a FlexStation 3 multi‐mode microplate reader and expressed as the ratio of green to red fluorescence. Lipid peroxidation reported by C11‐BODIPY(581/591) was normalised to the maximum signal induced by Fe. Data are means ± SEM, n=6 independent experiments.
ACKNOWLEDGEMENTS
This project was funded by grants from the National Health and Medical Research Council, Motor Neurone Disease Research Institute of Australia (Betty Laidlaw MND Grant), and Fight MND.
Southon A, Szostak K, Acevedo KM, et al. CuII(atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br J Pharmacol. 2020;177:656–667. 10.1111/bph.14881
REFERENCES
- Adlard, P. A. , Cherny, R. A. , Finkelstein, D. I. , Gautier, E. , Robb, E. , Cortes, M. , … Bush, A. I. (2008). Rapid restoration of cognition in Alzheimer's transgenic mice with 8‐hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron, 59, 43–55. 10.1016/j.neuron.2008.06.018 [DOI] [PubMed] [Google Scholar]
- Ayton, S. , Fazlollahi, A. , Bourgeat, P. , Raniga, P. , Ng, A. , Lim, Y. Y. , … Bush, A. I. (2017). Cerebral quantitative susceptibility mapping predicts amyloid‐β‐related cognitive decline. Brain, 140, 2112–2119. 10.1093/brain/awx137 [DOI] [PubMed] [Google Scholar]
- Ayton, S. , Lei, P. , Hare, D. J. , Duce, J. A. , George, J. L. , Adlard, P. A. , … Bush, A. I. (2015). Parkinson's disease iron deposition caused by nitric oxide‐induced loss of β‐amyloid precursor protein. The Journal of Neuroscience, 35, 3591–3597. 10.1523/JNEUROSCI.3439-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraldo, H. , Boyd, L. P. , & West, D. X. (1998). Copper(II) and nickel(II) complexes of glyoxaldehyde bis(N(3)‐substituted thiosemicarbazones). Transition Metal Chemistry, 23, 67–71. [Google Scholar]
- Cherubini, A. , Ruggiero, C. , Polidori, M. C. , & Mecocci, P. (2005). Potential markers of oxidative stress in stroke. Free Radical Biology & Medicine, 39, 841–852. [DOI] [PubMed] [Google Scholar]
- Choi, I. Y. , Lee, P. , Statland, J. , McVey, A. , Dimachkie, M. , Brooks, W. , & Barohn, R. (2015). Reduction in cerebral antioxidant, glutathione (GSH), in patients with ALS: A preliminary study. Neurology, 84(14 Supplement), P6.105. [Google Scholar]
- Crouch, P. J. , Hung, L. W. , Adlard, P. A. , Cortes, M. , Lal, V. , Filiz, G. , … Barnham, K. J. (2009). Increasing Cu bioavailability inhibits Aβ oligomers and τ phosphorylation. Proceedings of the National Academy of Sciences of the United States of America, 106, 381–386. 10.1073/pnas.0809057106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearling, J. L. , Lewis, J. S. , Mullen, G. E. , Welch, M. J. , & Blower, P. J. (2002). Copper bis(thiosemicarbazone) complexes as hypoxia imaging agents: Structure–activity relationships. Journal of Biological Inorganic Chemistry, 7, 249–259. [DOI] [PubMed] [Google Scholar]
- Dexter, D. T. , Carter, C. J. , Wells, F. R. , Javoy‐Agid, F. , Agid, Y. , Lees, A. , … Marsden, C. D. (1989). Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. Journal of Neurochemistry, 52, 381–389. 10.1111/j.1471-4159.1989.tb09133.x [DOI] [PubMed] [Google Scholar]
- Ding, H. , Yan, C. Z. , Shi, H. , Zhao, Y. S. , Chang, S. Y. , Yu, P. , … Duan, X. L. (2011). Hepcidin is involved in iron regulation in the ischemic brain. PLoS ONE, 6, e25324‐25333 10.1371/journal.pone.0025324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, S. J. , Lemberg, K. M. , Lamprecht, M. R. , Skouta, R. , Zaitsev, E. M. , Gleason, C. E. , … Stockwell, B. R. (2012). Ferroptosis: An iron‐dependent form of nonapoptotic cell death. Cell, 149, 1060–1072. 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Van, B. , Gouel, F. , Jonneaux, A. , Timmerman, K. , Gele, P. , Petrault, M. , … Devedjian, J. C. (2016). Ferroptosis, a newly characterized form of cell death in Parkinson's disease that is regulated by PKC. Neurobiology of Disease, 94, 169–178. 10.1016/j.nbd.2016.05.011 [DOI] [PubMed] [Google Scholar]
- Donnelly, P. S. , Caragounis, A. , Du, T. , Laughton, K. M. , Volitakis, I. , Cherny, R. A. , … White, A. R. (2008). Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid‐β peptide. The Journal of Biological Chemistry, 283, 4568–4577. 10.1074/jbc.M705957200 [DOI] [PubMed] [Google Scholar]
- Evans, .A , Rowe, D. , Lee, W. , Noel, K. , & Rosenfeld, C. (2019). Preliminary evidence of CuATSM treatment benefit in Parkinson's disease. In XXIV World Congress on Parkinson‘s Disease and Related Disorders. Montreal, Canada, pp 119–120.
- Friedmann Angeli, J. P. , Schneider, M. , Proneth, B. , Tyurina, Y. Y. , Tyurin, V. A. , Hammond, V. J. , … Conrad, M. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature Cell Biology, 16, 1180–1191. 10.1038/ncb3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler, M. M. , Andia, A. A. , Liu, H. , Csuka, J. M. , Hurlocker, B. , Vaiana, C. A. , … Stockwell, B. R. (2018). FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nature Chemical Biology, 14, 507–515. 10.1038/s41589-018-0031-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras, B. A. , Suprunchuk, T. , & Bayley, C. H. (1962). The preparation of some thiosemicarbazones and their copper complexes. Canadian Journal of Chemistry, 40, 1053–1059. [Google Scholar]
- Grolez, G. , Moreau, C. , Danel‐Brunaud, V. , Delmaire, C. , Lopes, R. , Pradat, P. F. , … Devos, D. (2016). The value of magnetic resonance imaging as a biomarker for amyotrophic lateral sclerosis: A systematic review. BMC Neurology, 16, 155–171. 10.1186/s12883-016-0672-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiney, S. J. , Adlard, P. A. , Bush, A. I. , Finkelstein, D. I. , & Ayton, S. (2017). Ferroptosis and cell death mechanisms in Parkinson's disease. Neurochemistry International, 104, 34–48. [DOI] [PubMed] [Google Scholar]
- Haddad, A. Z. , Cronin, S. P. , Mashuta, M. S. , Buchanan, R. M. , & Grapperhaus, C. A. (2017). Metal‐assisted ligand‐centered electrocatalytic hydrogen evolution upon reduction of a bis(thiosemicarbazonato)Cu(II) complex. Inorganic Chemistry, 56, 11254–11265. [DOI] [PubMed] [Google Scholar]
- Hilton, J. B. , Mercer, S. W. , Lim, N. K. , Faux, N. G. , Buncic, G. , Beckman, J. S. , … Crouch, P. J. (2017). CuII(atsm) improves the neurological phenotype and survival of SOD1G93A mice and selectively increases enzymatically active SOD1 in the spinal cord. Scientific Reports, 7, 42292‐42302 10.1038/srep42292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, J. P. , Green, J. C. , & Dilworth, J. R. (2006). Probing the mechanism of hypoxia selectivity of copper bis(thiosemicarbazonato) complexes: DFT calculation of redox potentials and absolute acidities in solution. Dalton Transactions, 783–794. [DOI] [PubMed] [Google Scholar]
- Homma, T. , Kobayashi, S. , Sato, H. , & Fujii, J. (2019). Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Experimental Cell Research, 384, 111592‐111600 10.1016/j.yexcr.2019.111592 [DOI] [PubMed] [Google Scholar]
- Hung, L. W. , Villemagne, V. L. , Cheng, L. , Sherratt, N. A. , Ayton, S. , White, A. R. , … Barnham, K. J. (2012). The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson's disease. The Journal of Experimental Medicine, 209, 837–854. 10.1084/jem.20112285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huuskonen, M. T. , Tuo, Q. Z. , Loppi, S. , Dhungana, H. , Korhonen, P. , McInnes, L. E. , … Kanninen, K. M. (2017). The copper bis(thiosemicarbazone) complex CuII(atsm) is protective against cerebral ischemia through modulation of the inflammatory milieu. Neurotherapeutics, 14, 519–532. 10.1007/s13311-016-0504-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa, M. , Okazawa, H. , Kudo, T. , Kuriyama, M. , Fujibayashi, Y. , & Yoneda, M. (2011). Evaluation of striatal oxidative stress in patients with Parkinson's disease using [62Cu]ATSM PET. Nuclear Medicine and Biology, 38, 945–951. [DOI] [PubMed] [Google Scholar]
- Ikawa, M. , Okazawa, H. , Tsujikawa, T. , Matsunaga, A. , Yamamura, O. , Mori, T. , … Yoneda, M. (2015). Increased oxidative stress is related to disease severity in the ALS motor cortex: A PET study. Neurology, 84, 2033–2039. 10.1212/WNL.0000000000001588 [DOI] [PubMed] [Google Scholar]
- Jenner, P. , Dexter, D. T. , Sian, J. , Schapira, A. H. , & Marsden, C. D. (1992). Oxidative stress as a cause of nigral cell death in Parkinson's disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson's Disease Research Group. Annals of Neurology, 32(Suppl), S82–S87. 10.1002/ana.410320714 [DOI] [PubMed] [Google Scholar]
- Kowol, C. R. , Reisner, E. , Chiorescu, I. , Arion, V. B. , Galanski, M. , Deubel, D. V. , & Keppler, B. K. (2008). An electrochemical study of antineoplastic gallium, iron and ruthenium complexes with redox noninnocent α‐N‐heterocyclic chalcogensemicarbazones. Inorganic Chemistry, 47, 11032–11047. [DOI] [PubMed] [Google Scholar]
- Mandal, P. K. , Saharan, S. , Tripathi, M. , & Murari, G. (2015). Brain glutathione levels—A novel biomarker for mild cognitive impairment and Alzheimer's disease. Biological Psychiatry, 78, 702–710. [DOI] [PubMed] [Google Scholar]
- Masaldan, S. , Bush, A. I. , Devos, D. , Rolland, A. S. , & Moreau, C. (2018). Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radical Biology & Medicine. 133, 221–233. [DOI] [PubMed] [Google Scholar]
- McAllum, E. J. , Lim, N. K. , Hickey, J. L. , Paterson, B. M. , Donnelly, P. S. , Li, Q. X. , … Crouch, P. J. (2013). Therapeutic effects of CuII(atsm) in the SOD1‐G37R mouse model of amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration, 14, 586–590. 10.3109/21678421.2013.824000 [DOI] [PubMed] [Google Scholar]
- McCleverty, J. A. , & Jones, C. J. (1970). Complexes of transition metals with Schiff bases and the factors influencing their redox properties. I. Nickel and copper complexes of some diketone bis‐thiosemicarbazones. Journal of the Chemical Society [Section] a: Inorganic, Physical, Theoretical, 2829–2836. [Google Scholar]
- Pap, E. H. , Drummen, G. P. , Winter, V. J. , Kooij, T. W. , Rijken, P. , Wirtz, K. W. , … Post, J. A. (1999). Ratio‐fluorescence microscopy of lipid oxidation in living cells using C11‐BODIPY581/591 . FEBS Letters, 453, 278–282. 10.1016/S0014-5793(99)00696-1 [DOI] [PubMed] [Google Scholar]
- Park, U. J. , Lee, Y. A. , Won, S. M. , Lee, J. H. , Kang, S. H. , Springer, J. E. , … Gwag, B. J. (2011). Blood‐derived iron mediates free radical production and neuronal death in the hippocampal CA1 area following transient forebrain ischemia in rat. Acta Neuropathologica, 121, 459–473. 10.1007/s00401-010-0785-8 [DOI] [PubMed] [Google Scholar]
- Pyatigorskaya, N. , Sharman, M. , Corvol, J. C. , Valabregue, R. , Yahia‐Cherif, L. , Poupon, F. , … Lehericy, S. (2015). High nigral iron deposition in LRRK2 and Parkin mutation carriers using R2* relaxometry. Movement Disorders, 30, 1077–1084. 10.1002/mds.26218 [DOI] [PubMed] [Google Scholar]
- Raven, E. P. , Lu, P. H. , Tishler, T. A. , Heydari, P. , & Bartzokis, G. (2013). Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer's disease detected in vivo with magnetic resonance imaging. Journal of Alzheimer's Disease, 37, 127–136. [DOI] [PubMed] [Google Scholar]
- Reed, T. T. , Pierce, W. M. , Markesbery, W. R. , & Butterfield, D. A. (2009). Proteomic identification of HNE‐bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Research, 1274, 66–76. [DOI] [PubMed] [Google Scholar]
- Roberts, B. R. , Lim, N. K. , McAllum, E. J. , Donnelly, P. S. , Hare, D. J. , Doble, P. A. , … Crouch, P. J. (2014). Oral treatment with CuII(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. The Journal of Neuroscience, 34, 8021–8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe, D. , Mathers, S. , Smith, G. , Windebank, E. , Rogers, M. , Noel, K. , & Rosenfeld, C. (2018). Modification of ALS disease progression in a phase 1 trial of CuATSM. Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration, 19(Suppl. S1), 280–281. [Google Scholar]
- Shah, R. , Margison, K. , & Pratt, D. A. (2017). The potency of diarylamine radical‐trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chemical Biology, 12, 2538–2545. [DOI] [PubMed] [Google Scholar]
- Sheng, X. , Shan, C. , Liu, J. , Yang, J. , Sun, B. , & Chen, D. (2017). Theoretical insights into the mechanism of ferroptosis suppression via inactivation of a lipid peroxide radical by liproxstatin‐1. Physical Chemistry Chemical Physics, 19, 13153–13159. [DOI] [PubMed] [Google Scholar]
- Sian, J. , Dexter, D. T. , Lees, A. J. , Daniel, S. , Agid, Y. , Javoy‐Agid, F. , … Marsden, C. D. (1994). Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Annals of Neurology, 36, 348–355. [DOI] [PubMed] [Google Scholar]
- Simpson, E. P. , Henry, Y. K. , Henkel, J. S. , Smith, R. G. , & Appel, S. H. (2004). Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology, 62, 1758–1765. [DOI] [PubMed] [Google Scholar]
- Skouta, R. , Dixon, S. J. , Wang, J. , Dunn, D. E. , Orman, M. , Shimada, K. , … Stockwell, B. R. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. Journal of the American Chemical Society, 136, 4551–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soon, C. P. , Donnelly, P. S. , Turner, B. J. , Hung, L. W. , Crouch, P. J. , Sherratt, N. A. , … Li, Q. X. (2011). Diacetylbis(N(4)‐methylthiosemicarbazonato) copper (II) (CuII(atsm)) protects against peroxynitrite‐induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. The Journal of Biological Chemistry, 286, 44035–44044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell, B. R. , Friedmann Angeli, J. P. , Bayir, H. , Bush, A. I. , Conrad, M. , Dixon, S. J. , … Zhang, D. D. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell, 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, Y. , Wang, Y. , Rogers, J. T. , & Wang, F. (2014). Perturbed iron distribution in Alzheimer's disease serum, cerebrospinal fluid, and selected brain regions: A systematic review and meta‐analysis. Journal of Alzheimer's Disease, 42, 679–690. [DOI] [PubMed] [Google Scholar]
- Tohgi, H. , Abe, T. , Yamazaki, K. , Murata, T. , Ishizaki, E. , & Isobe, C. (1999). Increase in oxidized NO products and reduction in oxidized glutathione in cerebrospinal fluid from patients with sporadic form of amyotrophic lateral sclerosis. Neuroscience Letters, 260, 204–206. [DOI] [PubMed] [Google Scholar]
- Tuo, Q. Z. , Lei, P. , Jackman, K. A. , Li, X. L. , Xiong, H. , Li, X. L. , … Bush, A. I. (2017). τ‐mediated iron export prevents ferroptotic damage after ischemic stroke. Molecular Psychiatry, 22, 1520–1530. [DOI] [PubMed] [Google Scholar]
- Vieira, F. G. , Hatzipetros, T. , Thompson, K. , Moreno, A. J. , Kidd, J. D. , Tassinari, V. R. , … Gill, A. (2017). CuATSM efficacy is independently replicated in a SOD1 mouse model of ALS while unmetallated ATSM therapy fails to reveal benefits. IBRO Rep, 2, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada, K. , Fujibayashi, Y. , Tajima, N. , & Yokoyama, A. (1994). Cu‐ATSM, an intracellular‐accessible superoxide dismutase (SOD)‐like copper complex: Evaluation in an ischemia‐reperfusion injury model. Biological & Pharmaceutical Bulletin, 17, 701–704. [DOI] [PubMed] [Google Scholar]
- West, D. X. , Ives, J. S. , Bain, G. A. , Liberta, A. E. , Valdes‐Martinez, J. , Ebert, K. H. , & Hernandez‐Ortega, S. (1997). Copper(II) and nickel(II) complexes of 2,3‐butanedione bis(N(3)‐substituted thiosemicarbazones). Polyhedron, 16, 1895–1905. [Google Scholar]
- Wong, B. X. , Ayton, S. , Lam, L. Q. , Lei, P. , Adlard, P. A. , Bush, A. I. , & Duce, J. A. (2014). A comparison of ceruloplasmin to biological polyanions in promoting the oxidation of Fe2+ under physiologically relevant conditions. Biochimica et Biophysica Acta, 1840, 3299–3310. [DOI] [PubMed] [Google Scholar]
- Xiao, Z. , Brose, J. , Schimo, S. , Ackland, S. M. , La Fontaine, S. , & Wedd, A. G. (2011). Unification of the copper(I) binding affinities of the metallo‐chaperones Atx1, Atox1, and related proteins: Detection probes and affinity standards. The Journal of Biological Chemistry, 286, 11047–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H. , Perreau, V. M. , Dent, K. A. , Bush, A. I. , Finkelstein, D. I. , & Adlard, P. A. (2016). Iron regulates apolipoprotein E expression and secretion in neurons and astrocytes. Journal of Alzheimer's Disease, 51, 471–487. [DOI] [PubMed] [Google Scholar]
- Yu, G. , Liang, Y. , Huang, Z. , Jones, D. W. , Pritchard, K. A. Jr. , & Zhang, H. (2016). Inhibition of myeloperoxidase oxidant production by N‐acetyl lysyltyrosylcysteine amide reduces brain damage in a murine model of stroke. Journal of Neuroinflammation, 13, 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilka, O. , Shah, R. , Li, B. , Friedmann Angeli, J. P. , Griesser, M. , Conrad, M. , & Pratt, D. A. (2017). On the mechanism of cytoprotection by ferrostatin‐1 and liproxstatin‐1 and the role of lipid peroxidation in ferroptotic cell death. ACS Central Science, 3, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Primary cortical cultures were predominantly neuronal. Primary cortical cells were grown on poly‐D lysine coated 12 mm glass coverslips until DIV9 and prepared for immunocytochemistry as previously described (Xu et al., 2016). Anti‐neuron‐specific nuclear protein (1:200) and anti‐mouse Alexa 488 (1:400, ThermoFisher Scientific, Scoresby, Australia) were used to identify neurons (red). Anti‐glial fibrillary acidic protein (GFAP, 1:400) and anti‐guinea pig Alexa 647 (1:400, ThermoFisher Scientific) were used to identify astrocytes (green). DAPI (300 nM, ThermoFisher Scientific) was used to detect nuclei (blue). Images were recorded at 20X magnification using an SP8 confocal microscope with Leica Application Suite X software (Leica Microsystems, Macquarie Park, Australia). Images at each wavelength were captured sequentially and multi‐colour XY images were prepared using Image J (Schneider, Rasband et al., 2012). XY images are shown as maximum brightness stacked images. Representative images are shown.
Figure S2. CuII(atsm) prevents ferroptosis in SN4741 cells. Cells were derived from E13.5 mouse substantia nigra tissue (Son, Chun et al., 1999) and cultured as per N27 cells. Viability of cells treated with RSL3 (1 μM, A) or erastin (1 μM, B) ± liproxstatin‐1 or CuII(atsm) for 24 hr. Viability was measured with MTT and expressed as a percentage of control cells. Data are means ± SEM, n=5 independent experiments. EC50 was determined by nonlinear regression, with 95% CI shown in parenthesis.
Figure S3. CuII(atsm) prevents ferroptosis in N27 cells. Viability of cells treated with RSL3 (0.1 μM, A‐B) or erastin (1 μM, C‐D) ± liproxstatin‐1 or CuII(atsm) for 24 hr. (A and C) Viability was measured with PrestoBlue according to the manufacturer's instructions (ThermoFisher Scientific) and expressed as a percentage of control cells. (B and D) Viability measured with the Cytotoxicity Detection Kit (LDH) according to the manufacturer's instructions (Merck, Bayswater, Australia) and expressed as a percentage of maximum lactate dehydrogenase (LDH) release. Data are means ± SEM, n=5 independent experiments. EC50 was determined by nonlinear regression, with 95% CI shown in parenthesis.
Figure S4. CuII(atsm) prevents lipid peroxidation in N27 cells. Cells were cultured in 6 well plates and treated in growth media with RSL3 (50 nM) ± liproxstatin‐1 or CuII(atsm) (1 μM) for 4 hr. Cells were washed in PBS and incubated in growth media with C11‐BODIPY(581/591) (125 nM) for 1 hr. Cells were harvested with trypsin and resuspended in PBS containing: EDTA 1 mM, HEPES 25 mM, foetal calf serum 1%, penicillin and streptomycin. Cells were strained through a 35 μm cell strainer (Corning, Mulgrave, Australia) and green fluorescence (excitation 488 nm, emission 525 nm) and red fluorescence (excitation 633 nm, emission 660 nm) were detected using a CytoFLEX S flow cytometer (Beckman Coulter, Mount Waverly, Australia). A minimum of 10000 cells were analysed per condition. Representative data from one of three experiments is shown.
Figure S5. NiII(atsm) does not promote Cu uptake in N27 cells. Cells were treated with CuII(atsm), CuII(atsm2) or NiII(atsm) (1 μM) for 24 hr. Cellular metal levels were measured with inductively coupled plasma mass spectrometry analysis using an Agilent 7700x (Agilent Technologies, Santa Clara, USA) as reported previously (Bica, Liddell et al., 2014). Data are means ± SEM, n=5 independent experiments. P values were calculated using one‐way ANOVA corrected for multiple comparisons with Dunnett's post‐hoc test. A Brown‐Forsythe test confirmed variance was not significantly different between groups and a Kolmogorov‐Smirnov test confirmed residuals were normally distributed. *P<.05 compared to vehicle treated cells.
Figure S6. CuII(atsm) does not elevate GPX4 levels in N27 cells. Cells treated with erastin (1 μM) or RSL3 (0.1 μM) ± liproxstatin‐1 or CuII(atsm) (1 μM) for 24 hr. Samples were lysed in RIPA buffer (Merck) and proteins (15 μg) were resolved on NuPAGE 4‐12% Bis‐Tris gels and transferred to PVDF membranes with an iBlot2 for western immuno‐blotting (ThermoFisher Scientific). Primary antibodies used were rabbit anti‐GPX4 (1:5000, Abcam, Cambridge, USA) and mouse anti‐actin (1:10000, Merck). Horseradish peroxidase coupled secondary antibodies were rabbit anti‐mouse and goat anti‐rabbit (1:7000, Agilent Technologies). Chemiluminescence was detected using ECL (GE Healthcare, Parramatta, Australia) and images were captured with a Fujifilm LAS‐3000 (Fujifilm, Campbellfield, Australia).
Figure S7. CuII(atsm) prevents lipid peroxidation induced by FeII. Locke's buffer with arachidonic acid (50 μM) treated with FeII (2 μM) ± liproxstatin‐1, CuII(atsm), NiII(atsm) or H2(atsm) (10 μM) for 30 min. Thiobarbituric Acid was used to detect malondialdehyde accumulation with the Lipid Peroxidation (MDA) Assay kit (Abcam) according to the manufacturer's instructions. Fluorescence (excitation 532 nm, emission 553 nm) was measured with a FlexStation 3 multi‐mode microplate reader (Molecular Devices). Lipid peroxidation reported by malondialdehyde was normalised to the maximum signal induced by Fe. Data are means ± SEM, n=6 independent experiments. P values were calculated using the Kruskal‐Wallis test corrected for multiple comparisons by controlling the false discovery rate with the Benjamini, Krieger, and Yekutieli test. *P<.05 compared to vehicle treated cells.
Figure S8. CuII(atsm) prevents FeII‐induced peroxidation of cellular‐derived lipids. Cellular‐derived lipids were prepared from 10 million N27 cells snap frozen, resuspended in 10 ml Locke's buffer, and sonicated in a water bath for 30 min. C11‐BODIPY(581/591) (2.5 μM) was added to the homogenate (100 μl) which was treated with FeII (10 μM) ± liproxstatin‐1, CuII(atsm), NiII(atsm), H2(atsm) or CuII(atsm2) for 30 min. C11‐BODIPY(581/591) fluorescence was measured with a FlexStation 3 multi‐mode microplate reader and expressed as the ratio of green to red fluorescence. Lipid peroxidation reported by C11‐BODIPY(581/591) was normalised to the maximum signal induced by Fe. Data are means ± SEM, n=6 independent experiments.