

Abstract
Background and Purpose
Genetics and drug interactions contribute to large interindividual variation in human CYP2D6 activity. Here, we have characterized propranolol inhibition of human and mouse CYP2D using transgenic (TG) mice, which express both mouse CYP2D and human CYP2D6, and wild‐type (WT) mice. Our purpose was to develop a method for in vivo manipulation of CYP2D6 enzyme activity which could be used to investigate the role of CYP2D6 in drug‐induced behaviours.
Experimental Approach
Dextromethorphan metabolism to dextrorphan was used to measure CYP2D activity and to characterize propranolol inhibition in vitro and in vivo. Effects of propranolol pretreatment (24 hr) on serum levels of the CYP2D6 substrate haloperidol and haloperidol‐induced catalepsy were also studied.
Key Results
Dextrorphan formation velocity in vitro was threefold higher in liver microsomes of TG compared to WT mice. Propranolol acted as a mechanism‐based inhibitor (MBI), inactivating CYP2D in liver microsomes from TG and WT mice, and humans. Pretreatment (24 hr) of TG and WT mice with 20 mg·kg−1 intraperitoneal propranolol reduced dextrorphan formation in vivo and by liver microsomes in vitro. Serum haloperidol levels and catalepsy were increased.
Conclusions and Implications
Propranolol was a potent MBI of dextrorphan formation in liver microsomes from TG and WT mice, and humans. The inhibition parameters in TG overlapped with those in WT mice and in humans. Inhibition of CYP2D with propranolol in vivo in TG and WT mice altered drug responses, allowing further investigation of variations in CYP2D6 on drug interactions and drug responses.
Abbreviations
- CLint
intrinsic clearance
- CYP2D
cytochrome P450 2D isoforms from rodent species
- CYP2D6
cytochrome P450 2D isoform from humans
- EM
extensive metabolizers
- MBI
mechanism‐based inhibitor
- PM
poor metabolizers
- TG
transgenic
- UM
ultra‐rapid metabolizers
- WT
wild‐type
What is already known
The humanized CYP2D6‐transgenic mouse can be used to characterize CYP2D6 enzyme activity.
Propranolol is a CYP2D mechanism‐based inhibitor in rats.
What this study adds
Characterization of propranolol as a mechanism‐based inhibitor of mouse and human CYP2D in vitro.
Manipulation of CYP2D in vivo with propranolol alters drug levels and response.
What is the clinical significance
Long‐lasting in vivo inhibition of CYP2D by propranolol suggests potential for drug–drug interactions.
Propranolol pretreatment in transgenic mice can be used to investigate human CYP2D6 variation in vivo.
1. INTRODUCTION
Cytochrome P450 2D6 (CYP2D6) is an important drug metabolizing enzyme estimated to be involved in the oxidation of 20–30% of clinically used drugs (Yu, Idle, & Gonzalez, 2004; Zanger & Schwab, 2013). The CYP2D6 gene is highly genetically polymorphic with numerous CYP2D6 allelic variants; the resulting CYP2D6 activity is used to describe poor metabolizers (PM), intermediate metabolizers, extensive metabolizers (EM), and ultra‐rapid metabolizers (UM; Gaedigk, 2013). CYP2D6 PM have little to no CYP2D6 enzyme activity (Eichelbaum, Spannbrucker, Steincke, & Dengler, 1979; Mahgoub, Dring, Idle, Lancaster, & Smith, 1977), and their CYP2D6 genetic status has been associated with greater side effects when taking antineoplastic drugs (Jung & Lim, 2014; Zeng et al., 2013), antidepressants (de Leon et al., 2005; Llerena, Berecz, Dorado, & de la Rubia, 2004), and antipsychotics (Brockmoller et al., 2002; Schillevoort et al., 2002). PM also experience little analgesia after taking opioids that require metabolic activation by CYP2D6, such as codeine (Zahari & Ismail, 2014).
Phenoconversion is the process by which a genotype‐predicted enzymatic activity is converted to a different phenotype, typically due to enzymatic inhibition or environmental factors such as illness (Shah & Smith, 2015; Zanger & Schwab, 2013). A mechanism‐based inhibitor (MBI), also referred to as an irreversible or suicide inhibitor, is a substrate that is metabolized to a reactive intermediate that then covalently binds to the enzyme, rendering it irreversibly inactivated (Silverman, 1995). Inhibition by MBIs require the synthesis of new enzyme to restore enzymatic activity and thus the inhibition can be long lasting (Liston et al., 2002). Drugs such as methylenedioxymethamphetamine (Heydari et al., 2004), paroxetine (Bertelsen, Venkatakrishnan, Moltke, Obach, & Greenblatt, 2003), or cimetidine (Madeira, Levine, Chang, Mirfazaelian, & Bellward, 2004) are known to be MBIs of CYP2D6 and, when taken, lead to irreversible inhibition of CYP2D6 and the phenoconversion of UM and EM to intermediate metabolizers/PM (Juřica & Žourková, 2013; O'Mathúna et al., 2008).
Propranolol is a β‐adrenoceptor antagonist metabolized by CYP2D6 (Lennard et al., 1984) that requires enzyme‐catalysed metabolism for irreversible inhibition to occur in human liver microsomes (Shaw, Lennard, Tucker, Bax, & Woods, 1987) which suggests that propranolol is an MBI of human CYP2D6. Rowland et al. (1994) found evidence that 4‐hydroxypropranolol, a propranolol metabolite, may be an MBI of CYP2D6 in vitro in human liver microsomes; however, they did not test propranolol itself as an MBI of CYP2D6 in vitro. While propranolol is also metabolized by CYP3A4 and CYP1A2, it does not appear to be an MBI for these enzymes (Herman, Nakamura, Wilkinson, & Wood, 1983; Walle, Walle, Cowart, Conradi, & Gaffney, 1987). Unlike propranolol, we could find no evidence that other β‐adrenoceptor antagonists are suspected to be MBIs of CYP2D6 or other enzymes. Propranolol is an MBI of rat CYP2D (we use CYP2D here to denote the isoform(s) from rodent species) and propranolol pretreatment in rats results in the inhibition of further propranolol metabolism both in vitro and in vivo (Schneck & Pritchard, 1981). In vitro, propranolol pretreatment in rats reduces CYP2D‐specific metabolism of imipramine and debrisoquine by rat liver microsomes (Masubuchi et al., 1991).
A transgenic (TG) CYP2D6 mouse line, which included the complete human CYP2D6 gene and its regulatory sequence, has been created (Corchero et al., 2001). Recently, another TG mouse line was created with a wider CYP2D6 tissue distribution and with tissue‐specific regulation of the human CYP2D6 transgene (Cheng et al., 2013). These TG mice can model overexpression of CYP2D since they have both the mouse Cyp2ds and the human CYP2D6 genes. Propranolol inhibition in TG mice could be a useful tool to investigate CYP2D6 in vitro and in vivo. However, the inhibition and inactivation characteristics of propranolol have not yet been described in mice or humans. These TG mice allow CYP2D6 activity to be manipulated (increased in TG and decreased via propranolol) in order to investigate more effectively the role of CYP2D6 in drug interactions, drug responses, toxicity, therapeutic effect, drug reward, and/or risk of abuse.
The aims of this study were to use TG (Cheng et al., 2013) and wild‐type (WT) mice (a) to determine the relative velocity of hepatic CYP2D metabolism, (b) to assess propranolol as an as an MBI of CYP2D in vitro in liver microsomes from TG and WT mice and humans, and to derive inhibition and inactivation parameters, and (c) to investigate the in vivo metabolism of dextromethorphan and haloperidol (both CYP2D6 substrates; Schmid, Bircher, Preisig, & Küpfer, 1985; Shin, Kane, & Flockhart, 2001). Haloperidol induces catalepsy, an acute behavioural response, which was studied to assess the possible in vivo pharmacodynamic effects of propranolol pretreatment on metabolism catalyzed by CYP2D.
2. METHODS
2.1. Animals
All animal care and experimental procedures complied with the NIH guidelines for the care and use of laboratory animals and were approved by the University of Toronto Animal Care Committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology. Adult (8–12 weeks old) male TG (Cheng et al., 2013) and WT C57BL/6 mice (Charles River, St‐Constant, Canada) were housed in groups of three to four under a 12 hr light/dark cycle with water and chow supplied ad libitum. All TG mice used for the study were homozygous; all TG pups were genotyped for the presence of the human CYP2D6 transgene prior to use according to the published methods (Cheng et al., 2013). The homozygosity of founders was confirmed via back‐breeding and genotyping for the transgene. All procedures were conducted in the light phase. Equal animal numbers from TG and WT mice were randomly assigned a pretreatment for each experiment; drug levels, enzymatic activity, and drug behavioural responses were assessed blinded to pretreatment group. In the rare case of technical failure, additional animals were assessed for final n = 10 per group, as suggested by Curtis et al. (2018). Based on pilot data for each approach, we determined that n = 10 per mouse line and pretreatment would be sufficient to determine differences.
2.2. In vitro dextrorphan formation
Liver microsomes were prepared as previously described (Siu, Wildenauer, & Tyndale, 2006) using pooled (n = 7) untreated mice from each mouse line for baseline and inhibition assessments. Pooled human liver microsomes (Xenotech, Lenexa, USA) were used to compare in vitro CYP2D metabolism between TG mice and humans. The test substrate dextromethorphan undergoes CYP2D‐specific O‐demethylation to dextrorphan (Schmid et al., 1985). The assay conditions were adapted from Felmlee, Lon, Gonzalez, and Yu (2008), with time and protein concentration optimized for linear dextrorphan formation. Final incubation concentrations contained 100‐mM potassium phosphate buffer (pH 7.4) and 1‐mM NADPH. After preincubation with 50‐μg microsomal protein (final concentration of 0.1 μg·μl−1) from TG, WT, or human liver for 2 min at 37°C, reactions were initiated by adding 50 μl of dextromethorphan (final concentration of 0.3–100 μM) for a total volume of 500 μl. Reactions were stopped with 500 μl of hexane–butanol (95:5 v/v) after 10 min.
2.3. Propranolol inhibition of in vitro dextrorphan formation by liver microsomes
A schematic time course of the preincubation schedule for inhibitory studies can be found in Figure 1. To generate Dixon plots, reaction mixtures containing 50‐μg microsomal protein (final concentration of 0.1 μg·μl−1) from TG or WT liver and propranolol (final concentration of 0–50 nM) in 100‐mM potassium phosphate buffer were prewarmed at 37°C for 2 min before adding NADPH (final concentration of 1 mM). After a 5 min preincubation, reactions were initiated by adding dextromethorphan (final concentration of 2.5, 5, or 10 μM) for a total volume of 500 μl.
Figure 1.
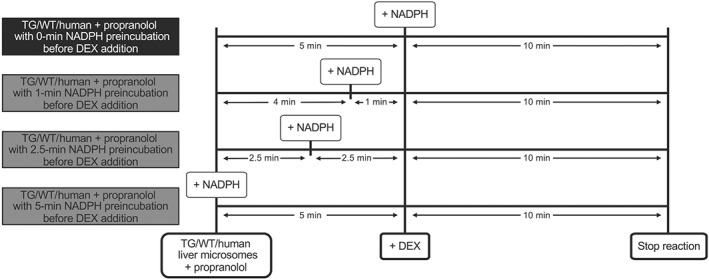
Schematic times courses of the conditions used for propranolol incubations with different preincubation times. Pooled liver microsomes of TG, WT, and humans were co‐incubated with propranolol, and NADPH was added at different times listed above, before dextromethorphan (DEX) was added to each sample, and the reaction was carried out for another 10 min
To generate IC50 plots, reaction mixtures containing 50‐μg microsomal protein (final concentration of 0.1 μg·μl−1) from TG, WT, or human liver and propranolol (final concentration of 0–100 μM) were prewarmed at 37°C for 2 min, before adding NADPH after either 0 or 5 min during a subsequent 5 min preincubation (Figure 1). Reactions were initiated by adding dextromethorphan (final concentration of 5 μM, approximate Km) for a total volume of 500 μl.
To generate inactivation curves, reaction mixtures containing 50‐μg microsomal protein (final concentration of 0.1 μg·μl−1) from TG, WT, or human liver and propranolol (final concentration of 0–10 μM) were prewarmed at 37°C for 2 min, before adding NADPH after 0, 2.5, 4, or 5 min during a subsequent 5 min preincubation (Figure 1). Reactions were initiated by adding dextromethorphan (final concentration of 5 μM) for a total volume of 500 μl.
2.4. In vivo dextrorphan formation
Mice were pretreated i.p. 24 hr before experimental testing with either propranolol hydrochloride (20 mg·kg−1 in saline) or saline alone (n = 10 per group). After 24 hr, dextromethorphan hydrobromide (30 mg·kg−1 in saline i.p.) was injected and saphenous vein blood was collected 30 min later. Blood was collected after dextromethorphan Tmax (estimated as 15 min in mice; Sakai et al., 2014) so that we are at the descending limb of the dextromethorphan curve. Mice were immediately killed, and tissues were collected and stored at −80°C. Dextromethorphan and dextrorphan levels in serum and from in vitro incubates were quantified using HPLC, with standard curves for dextromethorphan and dextrorphan (5–500 ng·ml−1) as previously described (Miksys et al., 2017). The ratio of serum dextrorphan / dextromethorphan was used as an index of in vivo hepatic CYP2D activity. Given that the half‐life of propranolol in mice is ~45 min (Levy, Ngai, Finck, Kawashima, & Spector, 1976), the 24 hr pretreatment represents approximately 32 half‐lives and thus we did not attempt to measure propranolol concentrations.
2.5. Haloperidol‐induced catalepsy
Catalepsy was assessed as the latency to removal of a mouse's forepaws from a surface raised 9 cm above the cage floor, with a 420‐s maximum cut‐off time (Miksys et al., 2017). Twenty‐four hours after i.p. propranolol or saline pretreatment (n = 10 per group), mice received haloperidol (0.1 mg·kg−1 s.c., injectable formulation, 5 mg·ml−1 base in water adjusted to pH 3–3.8 with lactic acid), and catalepsy was measured 120 min later. Timing and dose were selected from dose–response curves (testing from 0 to 180 min against 0.05, 0.1, and 0.2 mg·kg−1 s.c. injections of haloperidol) produced from TG and WT mice to enable detection of increases or decreases in catalepsy. Saphenous vein blood samples were taken at 60 and 180 min after haloperidol injection to assess the effects of pretreatments on serum haloperidol levels. Blood was collected at equal times before and after the time when catalepsy was assessed to minimize the stress from blood collection on catalepsy testing while ensuring that the earliest blood draw was after haloperidol Tmax in mice (Zetler & Baumann, 1985). The effects of vehicle and propranolol pretreatments were tested within animals, with a 2‐week washout period between tests. Serum haloperidol levels were quantified using LC‐MS as previously described (Miksys et al., 2017); no haloperidol metabolites were able to be measured. The internal standard, 0.5‐ng haloperidol‐d4 dissolved in 30:70 of 5‐mM ammonium formate/acetonitrile, was added to each sample and to haloperidol calibration standards (1–500 ng·ml−1).
2.6. Data and statistical analyses
The data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). The data were analysed using GraphPad Prism (RRID:SCR_002798; version 6.0c; La Jolla, California, USA). Michaelis–Menten parameters, Km and Vmax, as well as the corresponding standard error for both parameters were estimated using non‐linear regression. Michaelis–Menten parameters were also confirmed using the Eadie–Hofstee method. Intrinsic clearance (CLint) was calculated as the ratio of Vmax to Km. The inhibition constant Ki was determined according to Dixon (1953). IC50 was estimated using non‐linear regression and calculated as the concentration of propranolol that was halfway between the top and bottom plateaus of the curve for a one‐site enzyme (Neubig, Spedding, Kenakin, & Christopoulos, 2003). For two‐enzyme systems, IC50,High and IC50,Low are defined as IC50 for the high affinity sites and the low affinity sites, respectively. To estimate inactivation constants, the initial rate constant of inactivation of dextrorphan formation by each propranolol concentration (Kobs) was assessed by linear regression analysis of the natural logarithm of the percentage of activity remaining against preincubation time data (Kitz & Wilson, 1962). Thereafter, the Kobs values were used to determine the inhibitor concentration needed to cause a half‐maximal rate of enzyme inactivation (KI) and the maximal rate of inactivation (kinactivation). The KI and kinactivation were estimated by non‐linear regression using the following equation (Jones et al., 1999): . Serum dextrorphan / dextromethorphan ratios and hepatic dextrorphan formation velocities were analysed by two‐tailed, unpaired sample t tests, while catalepsy scores and serum haloperidol levels were analysed by two‐tailed, paired sample t tests. Statistical analyses were only performed with measures that had at least five animals per group; n = 10 individual animals per group (treatment and mouse line) were the basis of the independent values, using within or between statistical analyses as indicated. All values are expressed as mean ± SEM unless otherwise stated. A P value of <.05 was considered statistically significant. Data were normalized to mean saline pretreatment controls (fold mean of the controls) within each mouse line to illustrate the effect of propranolol pretreatment; however, all statistical analyses were run with raw, non‐normalized data. Outliers were included in data analysis and figures.
2.7. Materials
We obtained propranolol hydrochloride and dextromethorphan hydrobromide from Sigma‐Aldrich Canada (Oakville, Canada); haloperidol from Omega (Montreal, Canada) and haloperidol‐d4 from Toronto Research Chemicals (Toronto, Canada).
2.8. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
3. RESULTS
3.1. TG mice exhibit faster in vitro dextrorphan formation compared to WT mice
Using liver microsomes, Vmax values of 2.8 nmol·mg−1·min−1 for TG and of 0.9 nmol·mg−1·min−1 for WT mice and a Km of 4.5 μM for TG and of 3.6 μM for WT mice were derived from Michaelis–Menten and Eadie–Hofstee plots (Figure 2a,b). The CLint was estimated to be threefold faster in TG compared to WT mice. The kinetic parameters for each mouse line are summarized in Figure 2c.
Figure 2.
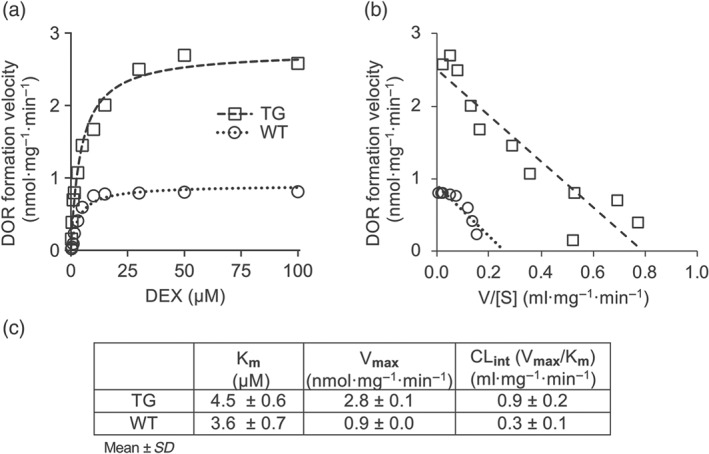
TG mice show faster dextrorphan (DOR) formation by liver microsomes compared to WT mice. Michaelis–Menten (a) and Eadie–Hofstee (b) plots of liver microsomal formation of dextrorphan from dextromethorphan (DEX). Enzyme kinetic values were obtained from these plots (c)
3.2. Propranolol irreversibly inhibits in vitro dextrorphan formation by liver microsomes
A Ki of 35 nM for TG and of 10 nM for WT mouse liver microsomes was observed for propranolol, with dextromethorphan as substrate (Figure 3a,b). The IC50 plots demonstrated that the inhibitory effect of propranolol on dextrorphan formation velocity in TG and WT mouse liver microsomes was increased by preincubation with NADPH. The data suggested propranolol inhibition of a one‐site system for WT and of a two‐site system for TG mouse liver microsomes. Propranolol preincubation with NADPH decreased the IC50,High (3.8‐fold) and IC50,Low (1.4‐fold) in TG mouse liver microsomes and decreased the IC50 (8.3‐fold) in WT mouse liver microsomes (Figure 3c,d). Propranolol inhibited in vitro dextrorphan formation velocity in a concentration‐ and time‐dependent manner for TG and WT mouse liver microsomes (Figure 4a,b). The kinactivation and KI for CYP2D were 0.11 min−1 and 84 nM for TG and 0.24 min−1 and 11 nM for WT mouse liver microsomes, respectively (Figure 4c,d).
Figure 3.
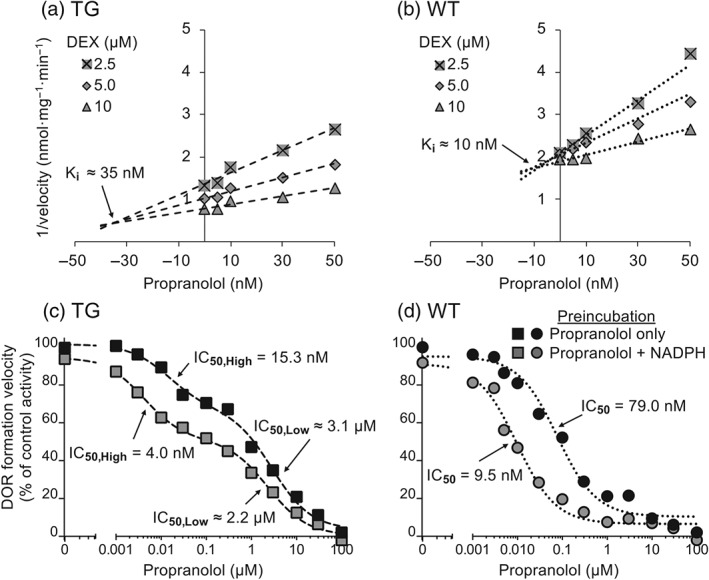
Propranolol acts as an MBI of dextrorphan (DOR) formation in TG and WT mouse liver microsomes in vitro. Dixon (a and b) and IC50 plots (c and d) of dextrorphan formation by pooled TG (a and c) and WT (b and d) mouse liver microsomes. In (a) and (b), pooled TG or WT mouse liver microsomes (0.1 mg·ml−1) were incubated with propranolol (0–50 nM) for 5 min before 2.5, 5, or 10 μM of dextromethorphan (DEX) was added. In (c) and (d), pooled TG or WT mouse liver microsomes (0.1 mg·ml−1) were incubated with propranolol (0–100 μM), with or without NADPH, for 5 min before dextromethorphan (5 µM) was added. The difference in dextrorphan formation between propranolol preincubation, with and without NADPH, before the dextromethorphan was added, suggests that propranolol acts as an MBI of CYP2D in liver microsomes from TG (c) and WT mice (d)
Figure 4.
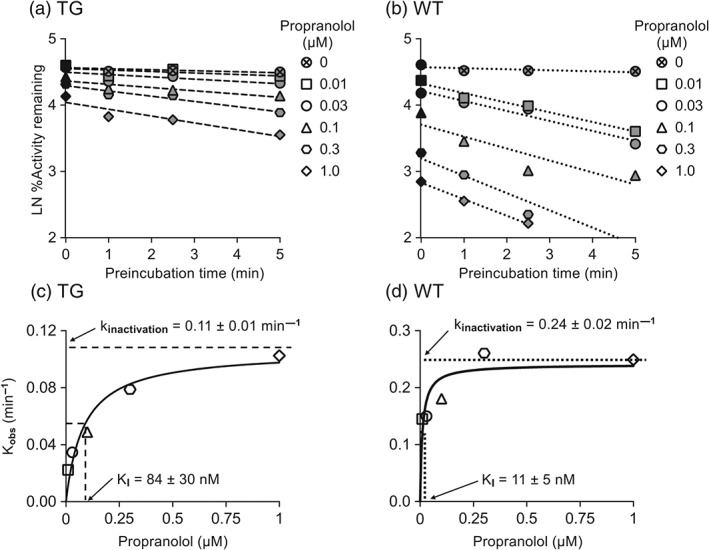
Propranolol inhibits dextrorphan formation velocity by liver microsomes from TG and WT mice in a time‐ and concentration‐dependent manner. In (a) and (b), propranolol (0‐1 µM) inhibition of dextrorphan formation velocity by pooled TG and WT mouse liver microsomes (0.1 mg·ml−1) was preincubation time and dose‐dependent. In (c) and (d), the rate of inactivation of CYP2D (Kobs) by each propranolol concentration was plotted to determine kinactivation and KI
Propranolol preincubation with NADPH decreased the IC50 in human liver microsomes (1.8‐fold) compared to preincubation with propranolol alone (Figure 5a). Propranolol also inhibited in vitro dextrorphan formation velocity in a concentration‐ and time‐dependent manner for human liver microsomes (Figure 5b), and the kinactivation and KI for CYP2D6 were 0.05 min−1 and 420 nM, respectively (Figure 5c).
Figure 5.
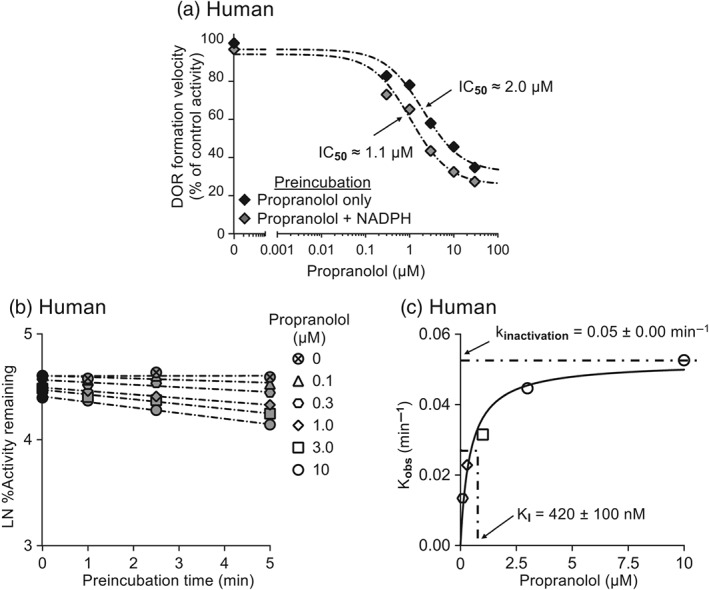
Propranolol acts as an MBI of CYP2D6 in human liver microsomes in vitro. In (a), pooled human liver microsomes (0.1 mg·ml−1) were preincubated with propranolol (0–30 μM) for 5 min before dextromethorphan (5 μM) was added. The difference in dextrorphan (DOR) formation between propranolol preincubation with and without NADPH, before dextromethorphan was added, suggests that propranolol acts as an MBI of CYP2D6 in liver microsomes from humans. In (b), propranolol (0–10 μM) inhibition of dextrorphan formation velocity by pooled human liver microsomes (0.1 mg*ml̂ ‐1) was preincubation time and dose‐dependent. In (c), the rate of inactivation of CYP2D6 (Kobs) by each propranolol concentration was plotted to determine kinactivation and KI
3.3. Propranolol pretreatment irreversibly inhibits dextrorphan formation in TG and WT mice in vivo and in vitro
The serum dextrorphan / dextromethorphan ratio was significantly reduced in both TG (by 26%, Figure 6a) and WT mice (by 33%, Figure 6b) by 24 hr i.p. pretreatment with propranolol, compared to saline. Using the liver microsomes from these pretreated mice, dextrorphan formation velocity in vitro was significantly reduced in both TG (by 22%, Figure 6c) and WT mice (by 25%, Figure 6d). Our results ‐ the long‐lasting (greater than 24 hr) inhibition by propranolol of subsequent in vivo and in vitro dextrorphan formation ‐ suggest that propranolol pretreatment irreversibly inhibits CYP2D in TG and WT mice in vivo, consistent with the inhibition previously observed in vitro (Figure 4a,b).
Figure 6.
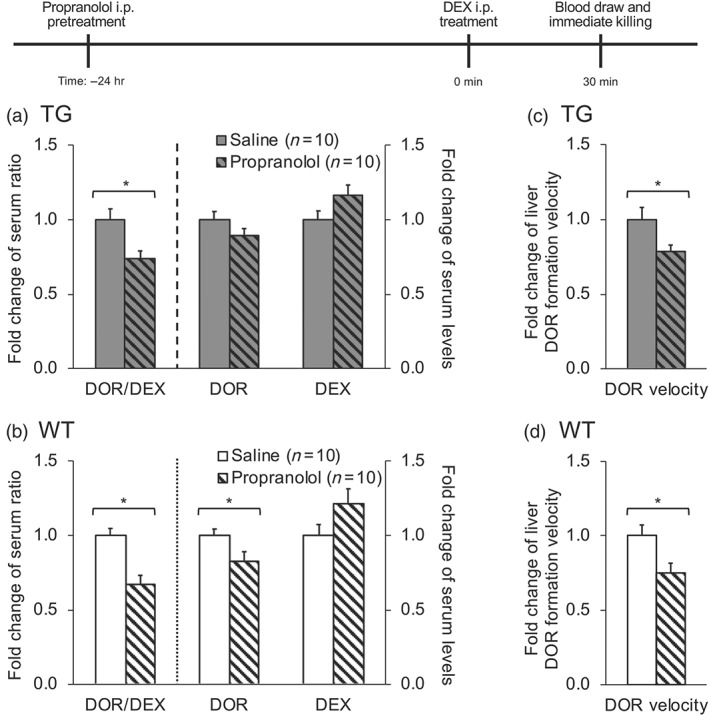
Pretreatment (24 hr) with 20 mg·kg−1 i.p. propranolol inhibited dextrorphan (DOR) formation in vivo and in vitro in liver microsomes from TG and WT mice. After 24 hr i.p. pretreatment with either saline or propranolol, all mice were given an i.p. injection of 30 mg·kg−1 of dextromethorphan (DEX) and blood was collected via saphenous vein 30 min after dextromethorphan injection. In (a) and (b), the in vivo serum dextrorphan/dextromethorphan ratio, serum dextrorphan and serum dextromethorphan levels in TG and WT mice were assessed. In (c) and (d), the dextrorphan formation by liver microsomes (0.1 mg·ml−1) prepared from pretreated mice was assessed after incubation with dextromethorphan (5 µM) for 10 min. Data presented are expressed as mean (with SEM) values of individual animals normalized to the saline pretreated group, within the same mouse line. *P < .05, significantly different from saline pretreated group; two‐tailed, unpaired t test on the raw data
3.4. Propranolol pretreatment increases serum haloperidol levels and haloperidol‐induced catalepsy
Serum haloperidol levels were significantly increased by 24 hr i.p. pretreatment with propranolol, compared to saline pretreatment within animals, in both TG (by 19% and 28%, at 60 and 180 min, respectively, Figure 7a; AUC60–180 by 22%, Figure 7c) and WT mice (by 30% and 37%, at 60 and 180 min, respectively, Figure 7b; AUC60–180 by 32%, Figure 7d). Similarly, the mean catalepsy scores at 120 min after the haloperidol injection were significantly increased by 24 hr i.p. pretreatment with propranolol, compared to saline pretreatment within animals, in both TG (by 31%, Figure 7e) and WT mice (by 35%, Figure 7f).
Figure 7.
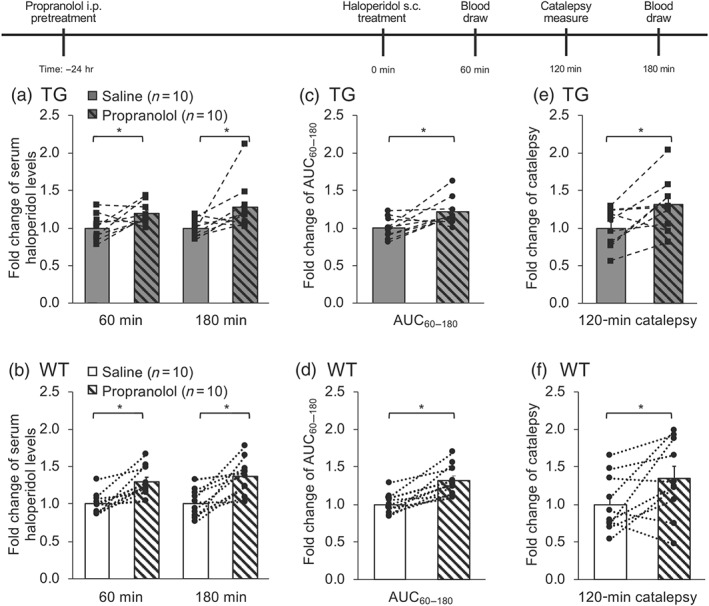
Serum haloperidol, serum haloperidol AUC60–180, and acute haloperidol‐induced catalepsy are increased (within animals) in TG and WT mice after 24 hr i.p. propranolol pretreatment. After 24 hr i.p. pretreatment with either saline or propranolol, all mice were given a s.c. injection of 0.1 mg·kg−1 of haloperidol, and serum haloperidol levels were assessed at 60 and 180 min after the haloperidol injection (a and b), and serum haloperidol AUC from 60 to 180 min was derived (c and d). Catalepsy was tested at 120 min after haloperidol injection (e and f). Data are expressed as mean (with SEM) values of each individual animal normalized to the saline pretreated group, within the same mouse line. Pretreatments were given 2 weeks apart. *P < .05, significantly different from saline pretreated group; two‐tailed, paired t test on the raw data
4. DISCUSSION
We have shown that TG have faster hepatic dextrorphan formation in vitro compared to WT mice, and that propranolol irreversibly inhibits dextrorphan formation in liver microsomes of TG and WT mice and humans in vitro. Pretreatment of the mice with propranolol (24 hr, i.p.) resulted in a significant reduction of dextrorphan formation in vivo in both TG and WT mice, and a significant reduction of dextrorphan formation velocity was observed in liver microsomes prepared from these pretreated animals. After showing that propranolol acts as an MBI of CYP2D‐mediated dextromethorphan metabolism to dextrorphan, haloperidol was used to confirm the effects of propranolol mechanism‐based inhibition of CYP2D by measuring serum haloperidol levels and response to halperidol. Propranolol pretreatment resulted in a significant increase in serum haloperidol levels and haloperidol‐induced catalepsy. These data indicate that propranolol exerts mechanism‐based inhibition of hepatic CYP2D in vitro and in vivo in TG and WT mice, and that this inhibition is sufficient to alter drug‐induced behaviour (catalepsy).
The CYP2D6 MBI paroxetine is known to have long‐lasting effects in vivo in humans, as a short‐term (6 weeks) paroxetine treatment required a 4‐week washout period before CYP2D6 disinhibition (Juřica & Žourková, 2013). Longer term (~18 weeks) paroxetine use was reported to need an even longer 6‐week washout period, and CYP2D6 inhibition was still present in three of eight patients after 6‐weeks (Juřica & Žourková, 2013). Co‐medication with propranolol increased drug concentrations of other CYP2D6 substrates (Greendyke & Kanter, 1987; Jones et al., 1999; Kiss et al., 2019) and increased adverse events (Drake & Gordon, 1994; Zhou, Anthony, Roden, & Wood, 1990). These results taken together suggest that patients taking propranolol with other CYP2D6 substrates, especially when those substrates are CYP2D6 MBIs themselves (e.g., methylenedioxymethamphetamine, paroxetine, and cimetidine), are at greater risk for adverse events. Here, we have derived inactivation parameters of CYP2D6 mechanism‐based inhibition by propranolol in human liver microsomes. These data can be useful in improving our understanding of the clinical effects of CYP2D6 inhibition by propranolol. Careful consideration of treatment with CYP2D6 substrates may be required in patients taking propranolol, even if propranolol has been discontinued prior to the administration of the CYP2D6 substrate.
Propranolol acts as an MBI against mouse CYP2D in TG and WT, both in vitro and in vivo. We did not investigate whether other CYPs were also inhibited by propranolol pretreatment as our conditions for in vitro and in vivo testing were optimized to specifically investigate CYP2D activity (as measured by dextrorphan formation and the dextrorphan / dextromethorphan ratio). The propranolol IC50 plots with WT mouse liver microsomes suggest one‐site inhibition, while the propranolol IC50 plots with TG mouse liver microsomes suggest two‐site inhibition. These two sites in TG mouse liver microsomes may correspond to mouse CYP2D and human CYP2D6, but it is difficult to distinguish between the two as the dextrorphan formation Km of mouse CYP2D (Felmlee et al., 2008) and human CYP2D6 (Kerry, Somogyi, Bochner, & Mikus, 1994) overlap. The higher affinity site in TG has a similar IC50 value to WT mice, suggesting that this higher affinity site represents the mouse CYP2D. The lower affinity site in TG mouse liver microsomes has a similar IC50 value to human liver microsomes, and similar to the IC50 value previously observed (6.6 μM; Obach et al., 2006), suggesting that the lower affinity site represents human CYP2D6. The mouse Cyp2d cluster is composed of nine genes (Cyp2d9, d10, 2d11, 2d12, 2d13, 2d22, 2d26, 2d34, and 2d40; Nelson et al., 2004) and the mRNA of six are transcribed to different extents in the liver (Cyp2d9, 2d10, 2d11, 2d22, 2d26, and 2d40; Renaud, Cui, Khan, & Klaassen, 2011). Other organs in WT mice also express CYP2D isozymes (Miksys, Cheung, Gonzalez, & Tyndale, 2005), and this variation in organ‐specific expression is even greater in TG mice with the addition of the differential expression of human CYP2D6 (Cheng et al., 2013). Inhibition of CYP2D by propranolol may be useful in studying the role of CYP2D and CYP2D6 metabolism within other organs, such as intestine, heart, and brain.
In this study, we used a two‐step method without dilution at lower microsome and inhibitor concentrations to determine if propranolol was an MBI. This two‐step method has the advantage of using a lower microsome concentration to reduce inhibitor binding to microsomes, as well as to reduce reactive‐intermediate formation in the primary incubation that can affect the secondary incubation (Parkinson et al., 2011). The disadvantage of this method is that without a dilution step, there is an increased chance of inactivation and reversible inhibition occurring in the secondary incubation with the test substrate, thereby decreasing sensitivity (Grimm et al., 2009). Another approach to determine mechanism‐based inhibition is to start with high microsome and inhibitor concentrations and then to dilute them 10‐fold after a primary preincubation step, prior to the addition of the test substrate to assay the CYP activity in a secondary incubation (Grimm et al., 2009). As described earlier, the dilution step reduces the degree of inactivation and reversible inhibition, and should thus result in increased sensitivity. However, concerns have been raised about this dilution step, given that higher initial microsome concentrations can lead to higher amounts of reactive intermediates formed in the primary incubation which can potentially inflate the extent of protein inactivation that can occur in the secondary incubation (Parkinson et al., 2011). Thus, in this study, we used the former approach.
Our data showing that TG have threefold higher Vmax and turnover than WT mice suggest that the TG mouse could be used to represent CYP2D6 UM, while the WT mouse could model CYP2D6 EM, as previously suggested (Cheng et al., 2013; Corchero et al., 2001). A similar fold difference in activity was observed by Kiss et al. (2018) in human liver microsomes where the rate of dextrorphan formation from dextromethorphan in UM was ~2.5‐fold higher compared to EM. For a number of drugs, high drug levels in vivo in CYP2D6 PM, as a result of genetic status (Brockmoller et al., 2002; Llerena et al., 2004), or from CYP2D6 MBIs (Juřica & Žourková, 2013; O'Mathúna et al., 2008), could increase the occurrence of adverse events. Using the long‐lasting, mechanism‐based inhibition by propranolol of mouse CYP2D and human CYP2D6 in TG, and of mouse CYP2D in WT, we can model the reduction of CYP2D6 UM and EM metabolism from an acute exposure to a CYP2D6 MBI. Propranolol pretreatment was sufficient to alter in vivo dextromethorphan metabolism; the decrease in serum dextrorphan / dextromethorphan ratio of saline pretreated TG mice (0.43), compared to propranolol pretreated TG mice (0.31) resulted in ratios that were similar to saline pretreated WT mice (0.33). Propranolol pretreatment was also sufficient to affect in vivo haloperidol metabolism (as seen in the modest increase in serum haloperidol AUC) and behaviour (as seen with the significant increase in haloperidol‐induced catalepsy), and was sufficient to show clear proof of MBI inactivation (as seen within the in vitro inhibition of liver microsomes from these mice). This was observed despite this propranolol pretreatment being given 24 hr prior to testing. This 24 hr propranolol pretreatment did not affect baseline catalepsy response, suggesting that haloperidol‐induced catalepsy was increased due to irreversible inhibition of CYP2D‐mediated haloperidol metabolism and not through the effects of propranolol itself. If even under these circumstances we see a modest shift in in vivo metabolism and response in propranolol pretreated mice, then chronic or concurrent propranolol treatments in patients may have a substantial effect on their overall CYP2D6 metabolism. For example, propranolol inhibited the metabolism of another CYP2D6 substrate (debrisoquine) in vivo in humans taking propranolo for a week (Rowland et al., 1994). This approach could also be useful for investigating novel CYP2D substrates—using both the relative rate of metabolism between the WT and TG mice, and the effects of propranolol's CYP2D mechanism‐based inhibition of in vivo drug metabolism (e.g., changes in serum drug levels) and drug response, including side effects, as we have done here using haloperidol‐induced catalepsy. This approach of giving an MBI such as propranolol 24 hr prior to testing is also useful for avoiding the possible confounding pharmacodynamic effects that competitive inhibitors have in vivo which can hinder their usefulness in modelling decreases in activity. The combined use of propranolol and TG mice could also be used to investigate drug interactions, therapeutic effects, and risk for drug dependence and abuse.
In conclusion, we provide evidence for mouse lines that can be used to model extensive and ultra‐rapid CYP2D6 metabolism, and the effect of acute propranolol (mechanism‐based inhibition) in vitro and in vivo on resulting drug levels and response. Our data also provides evidence for long‐lasting inhibition of CYP2D by propranolol, suggesting caution when prescribing propranolol with other CYP2D6 substrates.
AUTHOR CONTRIBUTIONS
E.C.T. performed the experiments and analysed the data. E.C.T., S.M., and R.F.T. contributed to the study design. E.C.T., S.M., F.G., and R.F.T. contributed to the manuscript writing. All authors approved the final version of this paper.
CONFLICT OF INTEREST
R.F.T. has consulted in the past for Quinn Emanuel and Ethismos on unrelated topics. E.C.T., S.M., and F.G. declare no conflict of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
ACKNOWLEDGEMENTS
We thank Dr. Bin Zhao for his technical assistance with the LC‐MS assay, Dr. Qian Zhou for genotyping the transgenic animals, and Fariba Baghai Wadji for her expert support with animal husbandry and procedures. This work was supported by a Canada Research Chair in Pharmacogenomics, the Canadian Institutes of Health Research (FDN 154294 and FRN 132557), the Campbell Family Mental Health Research Institute of the Centre for Addiction and Mental Health (CAMH)and the CAMH Foundation.
Tolledo EC, Miksys S, Gonzalez FJ, Tyndale RF. Propranolol is a mechanism‐based inhibitor of CYP2D and CYP2D6 in humanized CYP2D6‐transgenic mice: Effects on activity and drug responses. Br J Pharmacol. 2020;177:701–712. 10.1111/bph.14884
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen, K. M. , Venkatakrishnan, K. , Moltke, L. L. V. , Obach, R. S. , & Greenblatt, D. J. (2003). Apparent mechanism‐based inhibition of human CYP2D6 in vitro by paroxetine: Comparison with fluoxetine and quinidine. Drug Metabolism and Disposition, 31(3), 289–293. [DOI] [PubMed] [Google Scholar]
- Brockmoller, J. , Kirchheiner, J. , Schmider, J. , Walter, S. , Sachse, C. , Muller‐Oerlinghausen, B. , & Roots, I. (2002). The impact of the CYP2D6 polymorphism on haloperidol pharmacokinetics and on the outcome of haloperidol treatment. Clinical Pharmacology & Therapeutics, 72(4), 438–452. [DOI] [PubMed] [Google Scholar]
- Cheng, J. , Zhen, Y. , Miksys, S. , Beyoğlu, D. , Krausz, K. W. , Tyndale, R. F. , … Gonzalez, F. J. (2013). Potential role of CYP2D6 in the central nervous system. Xenobiotica, 43(11), 973–984. 10.3109/00498254.2013.791410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corchero, J. , Granvil, C. P. , Akiyama, T. E. , Hayhurst, G. P. , Pimprale, S. , Feigenbaum, L. , … Gonzalez, F. J. (2001). The CYP2D6 humanized mouse: Effect of the human CYP2D6 transgene and HNF4 α on the disposition of debrisoquine in the mouse. Molecular Pharmacology, 60(6), 1260–1267. 10.1124/mol.60.6.1260 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon, J. , Susce, M. T. , Pan, R.‐M. , Fairchild, M. , Koch, W. H. , & Wedlund, P. J. (2005). The CYP2D6 poor metabolizer phenotype may be associated with risperidone adverse drug reactions and discontinuation. The Journal of Clinical Psychiatry, 66(1), 15–27. [DOI] [PubMed] [Google Scholar]
- Dixon, M. (1953). The determination of enzyme inhibitor constants. Biochemical Journal, 55(1), 170–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake, W. M. , & Gordon, G. D. (1994). Heart block in a patient on propranolol and fluoxetine. The Lancet, 343(8894), 425–426. [DOI] [PubMed] [Google Scholar]
- Eichelbaum, M. , Spannbrucker, N. , Steincke, B. , & Dengler, H. J. (1979). Defective N‐oxidation of sparteine in man: A new pharmacogenetic. European Journal of Clinical Pharmacology, 16(3), 183–187. [DOI] [PubMed] [Google Scholar]
- Felmlee, M. A. , Lon, H. K. , Gonzalez, F. J. , & Yu, A. (2008). Cytochrome P450 expression and regulation in CYP3A4/CYP2D6 double transgenic humanized mice. Drug Metabolism and Disposition, 36(2), 435–441. [DOI] [PubMed] [Google Scholar]
- Gaedigk, A. (2013). Complexities of CYP2D6 gene analysis and interpretation. International Review of Psychiatry, 25(5), 534–553. [DOI] [PubMed] [Google Scholar]
- Greendyke, R. M. , & Kanter, D. R. (1987). Plasma propranolol levels and their effect on plasma thioridazine and haloperidol concentrations. Journal of Clinical Psychopharmacology, 7(3), 178–182. [PubMed] [Google Scholar]
- Grimm, S. W. , Einolf, H. J. , Hall, S. D. , He, K. , Lim, H. K. , Ling, K. H. J. , … Obach, R. S. (2009). The conduct of in vitro studies to address time‐dependent inhibition of drug‐metabolizing enzymes: A perspective of the pharmaceutical research and manufacturers of America. Drug Metabolism and Disposition, 37(7), 1355–1370. 10.1124/dmd.109.026716 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman, R. , Nakamura, K. , Wilkinson, G. , & Wood, A. (1983). Induction of propranolol metabolism by rifampicin. British Journal of Clinical Pharmacology, 16(5), 565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydari, A. , Yeo, K. R. , Lennard, M. S. , Ellis, S. W. , Tucker, G. T. , & Rostami‐Hodjegan, A. (2004). Mechanism‐based inactivation of CYP2D6 by methylenedioxymethamphetamine. Drug Metabolism and Disposition, 32(11), 1213–1217. [DOI] [PubMed] [Google Scholar]
- Jones, D. R. , Gorski, J. C. , Hamman, M. A. , Mayhew, B. S. , Rider, S. , & Hall, S. D. (1999). Diltiazem inhibition of cytochrome P‐450 3A activity is due to metabolite intermediate complex formation. Journal of Pharmacology and Experimental Therapeutics, 290(3), 1116–1125. [PubMed] [Google Scholar]
- Jung, J.‐A. , & Lim, H.‐S. (2014). Association between CYP2D6 genotypes and the clinical outcomes of adjuvant tamoxifen for breast cancer: A meta‐analysis. Pharmacogenomics, 15(1), 49–60. [DOI] [PubMed] [Google Scholar]
- Juřica, J. , & Žourková, A. (2013). Dynamics and persistence of CYP2D6 inhibition by paroxetine. Journal of Clinical Pharmacy and Therapeutics, 38(4), 294–300. [DOI] [PubMed] [Google Scholar]
- Kerry, N. , Somogyi, A. , Bochner, F. , & Mikus, G. (1994). The role of CYP2D6 in primary and secondary oxidative metabolism of dextromethorphan: in vitro studies using human liver microsomes. British Journal of Clinical Pharmacology, 38(3), 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss, Á. , Menus, Á. , Tóth, K. , Déri, M. , Sirok, D. , Gabri, E. , … Monostory, K. (2019). Phenoconversion of CYP2D6 by inhibitors modifies aripiprazole exposure. European Archives of Psychiatry and Clinical Neuroscience, 1–12. 10.1007/s00406-018-0975-2 [DOI] [PubMed] [Google Scholar]
- Kiss, Á. F. , Tóth, K. , Juhász, C. , Temesvári, M. , Paulik, J. , Hirka, G. , & Monostory, K. (2018). Is CYP2D6 phenotype predictable from CYP2D6 genotype? Microchemical Journal, 136, 209–214. [Google Scholar]
- Kitz, R. , & Wilson, I. B. (1962). Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. The Journal of Biological Chemistry, 237, 3245–3249. [PubMed] [Google Scholar]
- Lennard, M. S. , Jackson, P. R. , Freestone, S. , Tucker, G. T. , Ramsay, L. E. , & Woods, H. F. (1984). The relationship between debrisoquine oxidation phenotype and the pharmacokinetics and pharmacodynamics of propranolol. British Journal of Clinical Pharmacology, 17(6), 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, A. , Ngai, S. , Finck, A. , Kawashima, K. , & Spector, S. (1976). Disposition of propranolol isomers in mice. European Journal of Pharmacology, 40(1), 93–100. [DOI] [PubMed] [Google Scholar]
- Liston, H. L. , DeVane, C. L. , Boulton, D. W. , Risch, S. C. , Markowitz, J. S. , & Goldman, J. (2002). Differential time course of cytochrome P450 2D6 enzyme inhibition by fluoxetine, sertraline, and paroxetine in healthy volunteers. Journal of Clinical Psychopharmacology, 22(2), 169–173. [DOI] [PubMed] [Google Scholar]
- Llerena, A. , Berecz, R. , Dorado, P. , & de la Rubia, A. (2004). QTc interval, CYP2D6 and CYP2C9 genotypes and risperidone plasma concentrations. Journal of Psychopharmacology, 18(2), 189–193. [DOI] [PubMed] [Google Scholar]
- Madeira, M. , Levine, M. , Chang, T. K. H. , Mirfazaelian, A. , & Bellward, G. D. (2004). The effect of cimetidine on dextromethorphan o‐demethylase activity of human liver microsomes and recombinant CYP2D6. Drug Metabolism and Disposition, 32(4), 460–467. [DOI] [PubMed] [Google Scholar]
- Mahgoub, A. , Dring, L. G. , Idle, J. R. , Lancaster, R. , & Smith, R. L. (1977). Polymorphic hydroxylation of debrisoquine in man. The Lancet, 310(8038), 584–586. [DOI] [PubMed] [Google Scholar]
- Masubuchi, Y. , Fujita, S. , Chiba, M. , Kagimoto, N. , Umeda, S. , & Suzuki, T. (1991). Impairment of debrisoquine 4‐hydroxylase and related monooxygenase activities in the rat following treatment with propranolol. Biochemical Pharmacology, 41(6), 861–865. [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172(13), 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miksys, S. , Cheung, C. , Gonzalez, F. J. , & Tyndale, R. F. (2005). Human CYP2D6 and mouse CYP2Ds: Organ distribution in a humanized mouse model. Drug Metabolism and Disposition, 33(10), 1495–1502. [DOI] [PubMed] [Google Scholar]
- Miksys, S. , Wadji, F. B. , Tolledo, E. C. , Remington, G. , Nobrega, J. N. , & Tyndale, R. F. (2017). Rat brain CYP2D enzymatic metabolism alters acute and chronic haloperidol side‐effects by different mechanisms. Progress in Neuropsychopharmacology & Biological Psychiatry, 78, 140–148. [DOI] [PubMed] [Google Scholar]
- Nelson, D. R. , Zeldin, D. C. , Hoffman, S. M. G. , Maltais, L. J. , Wain, H. M. , & Nebert, D. W. (2004). Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative‐splice variants. Pharmacogenetics, 14(1), 1–18. [DOI] [PubMed] [Google Scholar]
- Neubig, R. R. , Spedding, M. , Kenakin, T. , & Christopoulos, A. (2003). International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacological Reviews, 55(4), 597–606. [DOI] [PubMed] [Google Scholar]
- Obach, R. S. , Walsky, R. L. , Venkatakrishnan, K. , Gaman, E. A. , Houston, J. B. , & Tremaine, L. M. (2006). The utility of in vitro cytochrome P450 inhibition data in the prediction of drug‐drug interactions. Journal of Pharmacology and Experimental Therapeutics, 316(1), 336–348. [DOI] [PubMed] [Google Scholar]
- O'Mathúna, B. , Farré, M. , Rostami‐Hodjegan, A. , Yang, J. , Cuyàs, E. , Torrens, M. , … de la Torre, R. (2008). The consequences of 3,4‐methylenedioxymethamphetamine induced CYP2D6 inhibition in humans. Journal of Clinical Psychopharmacology, 28(5), 523–529. 10.1097/JCP.0b013e318184ff6e [DOI] [PubMed] [Google Scholar]
- Parkinson, A. , Kazmi, F. , Buckley, D. B. , Yerino, P. , Paris, B. L. , Holsapple, J. , … Ogilvie, B. W. (2011). An evaluation of the dilution method for identifying metabolism‐dependent inhibitors of cytochrome P450 enzymes. Drug Metabolism and Disposition, 39(8), 1370–1387. 10.1124/dmd.111.038596 [DOI] [PubMed] [Google Scholar]
- Renaud, H. J. , Cui, J. Y. , Khan, M. , & Klaassen, C. D. (2011). Tissue distribution and gender‐divergent expression of 78 cytochrome P450 mRNAs in mice. Toxicological Sciences, 124, 261–262, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland, K. , Yeo, W. W. , Ellis, S. W. , Chadwick, I. G. , Haq, I. , Lennard, M. S. , … Tucker, G. T. (1994). Inhibition of CYP2D6 activity by treatment with propranolol and the role of 4‐hydroxy propranolol. British Journal of Clinical Pharmacology, 38(1), 9–14. 10.1111/j.1365-2125.1994.tb04315.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, C. , Iwano, S. , Yamazaki, Y. , Ando, A. , Nakane, F. , Kouno, M. , … Miyamoto, Y. (2014). Species differences in the pharmacokinetic parameters of cytochrome P450 probe substrates between experimental animals, such as mice, rats, dogs, monkeys, and microminipigs, and humans. Journal of Drug Metabolism and Toxiciology, 5(6), 1–12. [Google Scholar]
- Schillevoort, I. , de Boer, A. , van der Weide, J. , Steijns, L. , Roos, R. , Jansen, P. , & Leufkens, H. (2002). Antipsychotic‐induced extrapyramidal syndromes and cytochrome P450 2D6 genotype: A case‐control study. Pharmacogenetics, 12(3), 235–240. [DOI] [PubMed] [Google Scholar]
- Schmid, B. , Bircher, J. , Preisig, R. , & Küpfer, A. (1985). Polymorphic dextromethorphan metabolism: Co‐segregation of oxidative O‐demethylation with debrisoquin hydroxylation. Clinical Pharmacology and Therapeutics, 38(6), 618–624. 10.1038/clpt.1985.235 [DOI] [PubMed] [Google Scholar]
- Schneck, D. W. , & Pritchard, J. F. (1981). The inhibitory effect of propranolol pretreatment on its own metabolism in the rat. Journal of Pharmacology and Experimental Therapeutics, 218(3), 575–581. [PubMed] [Google Scholar]
- Shah, R. R. , & Smith, R. L. (2015). Addressing phenoconversion: The Achilles heel of personalized medicine: Impact of phenoconversion. British Journal of Clinical Pharmacology, 79(2), 222–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, L. , Lennard, M. S. , Tucker, G. T. , Bax, N. D. , & Woods, H. F. (1987). Irreversible binding and metabolism of propranolol by human liver microsomes—Relationship to polymorphic oxidation. Biochemical Pharmacology, 36(14), 2283–2288. [DOI] [PubMed] [Google Scholar]
- Shin, J.‐G. , Kane, K. , & Flockhart, D. A. (2001). Potent inhibition of CYP2D6 by haloperidol metabolites: Stereoselective inhibition by reduced haloperidol. British Journal of Clinical Pharmacology, 51(1), 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman, R. B. (1995). Mechanism‐based enzyme inactivators In Purich D. L. (Ed.), Enzyme kinetics and mechanism part D: Developments in enzyme dynamics (Vol. 249) (pp. 240–283). United States: Academic Press. [DOI] [PubMed] [Google Scholar]
- Siu, E. C. K. , Wildenauer, D. B. , & Tyndale, R. F. (2006). Nicotine self‐administration in mice is associated with rates of nicotine inactivation by CYP2A5. Psychopharmacology, 184(3), 401–408. [DOI] [PubMed] [Google Scholar]
- Walle, T. , Walle, U. K. , Cowart, T. D. , Conradi, E. C. , & Gaffney, T. E. (1987). Selective induction of propranolol metabolism by smoking: Additional effects on renal clearance of metabolites. Journal of Pharmacology and Experimental Therapeutics, 241(3), 928–933. [PubMed] [Google Scholar]
- Yu, A.‐M. , Idle, J. R. , & Gonzalez, F. J. (2004). Polymorphic cytochrome P450 2D6: Humanized mouse model and endogenous substrates. Drug Metabolism Reviews, 36(2), 243–277. [DOI] [PubMed] [Google Scholar]
- Zahari, Z. , & Ismail, R. (2014). Influence of Cytochrome P450, Family 2, Subfamily D, Polypeptide 6 (CYP2D6) polymorphisms on pain sensitivity and clinical response to weak opioid analgesics. Drug Metabolism and Pharmacokinetics, 29(1), 29–43. [DOI] [PubMed] [Google Scholar]
- Zanger, U. M. , & Schwab, M. (2013). Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacology & Therapeutics, 138(2013), 103–141. [DOI] [PubMed] [Google Scholar]
- Zeng, Z. , Liu, Y. , Liu, Z. , You, J. , Chen, Z. , Wang, J. , … Qin, X. (2013). CYP2D6 polymorphisms influence tamoxifen treatment outcomes in breast cancer patients: A meta‐analysis. Cancer Chemotherapy and Pharmacology, 72(2), 287–303. 10.1007/s00280-013-2195-9 [DOI] [PubMed] [Google Scholar]
- Zetler, G. , & Baumann, G. H. (1985). Pharmacokinetics and effects of haloperidol in the isolated mouse. Pharmacology, 31(6), 318–327. [DOI] [PubMed] [Google Scholar]
- Zhou, H. H. , Anthony, L. , Roden, D. M. , & Wood, A. J. J. (1990). Quinidine reduces clearance of (+)‐propranolol more than (−)‐propranolol through marked reduction in 4‐hydroxylation. Clinical Pharmacology & Therapeutics, 47(6), 686–693. [DOI] [PubMed] [Google Scholar]