

Graphical abstract
Keywords: Microbiome, Endophytic bacteria, Solanacea, Horticulture, Microbial ecology
Highlights
-
•
A common core microbiome was found in seeds of diverging N. tabacum cultivars.
-
•
Enterobacteriaceae accounted for the predominant fraction of this core microbiome.
-
•
Cultivars from the same breeding line shared the highest number of bacterial taxa.
-
•
Seed-endophytic communities were extended by distinct taxa in each cultivar.
Abstract
Seed endophytes of crop plants have recently received increased attention due to their implications in plant health and the potential to be included in agro-biotechnological applications. While previous studies indicated that plants from the Solanaceae family harbor a highly diverse seed microbiome, genotype-specific effects on the community composition and structure remained largely unexplored. The present study revealed Enterobacteriaceae-dominated seed-endophytic communities in four Nicotiana tabacum L. cultivars originating from Brazil, China, and the USA. When the dissimilarity of bacterial communities was assessed, none of the cultivars showed significant differences in microbial community composition. Various unusual endophyte signatures were represented by Spirochaetaceae family members and the genera Mycobacterium, Clostridium, and Staphylococcus. The bacterial fraction shared by all cultivars was dominated by members of the phyla Proteobacteria and Firmicutes. In total, 29 OTUs were present in all investigated cultivars and accounted for 65.5% of the combined core microbiome reads. Cultivars from the same breeding line were shown to share a higher number of common OTUs than more distant lines. Moreover, the Chinese cultivar Yunyan 87 contained the highest number (33 taxa) of unique signatures. Our results indicate that a distinct proportion of the seed microbiome of N. tabacum remained unaffected by breeding approaches of the last century, while a substantial proportion co-diverged with the plant genotype. Moreover, they provide the basis to identify plant-specific endophytes that could be addressed for upcoming biotechnological approaches in agriculture.
1. Introduction
In human history the cultivation of Nicotiana tabacum L. represents a long tradition across different cultures and continents. Its seeds were recovered from historic settlements (1500–1000 BC), providing evidence for tobacco cultivation during the early Chiripa phase in Southern America [1]. Centuries of genetic selection and breeding resulted in new cultivars which have larger leaves and higher nicotine content than earlier wild varieties [2]. Nowadays, its cultivation mainly relies on hybrid plants that originate from Southern as well as Northern America [3]. In addition to the important role as a model plant, N. tabacum is still an important agricultural crop in different countries that provide suitable environmental conditions for its cultivation [4]. The four most important cultivation areas are currently found in Asia and the Americas; China, Brazil, India, and the United Sates are currently the countries with the largest cultivation areas [5].
Similar to other plants from the Solanaceae family, various fungi, oomycetes, bacteria, viruses, as well as nematodes, can cause high yield losses during tobacco cultivation [6]. Due to the limitations of plant breeding towards more resistant cultivars and conventional disease management, the plant microbiota has emerged as a promising basis to increase plant resilience against prevailing pathogens. Recently, the tobacco microbiome was explored in fields with different incidences of bacteria wilt caused by Ralstonia solanacearum [6], [7], [8]. The authors concluded that the soil microbiota plays an important role in the bacteria wilt incidence as well as its management [9], [10]. Recent studies with different crop plants have shown that early colonizers of germinating seedlings can substantially contribute to plant development and that they originate from different sources [11]. Here, the indigenous seed microbiota constitutes a core element, because it is intrinsically connected to its host and it can be vertically transmitted between plant generations [12]. For a long time, it was assumed that a plant seedling’s microbiome is exclusively recruited from microbes located in the surrounding environment; especially from soils. This hypothesis was based on the concept of sterile seeds and omitted potential legacy effects of parental lines, because modern seed production includes surface sterilization as an integral process step [12]. However, distinct microorganisms colonize the integument, endosperm or embryo of seeds and thus are not affected by the robust sterilization procedures [13]. These seed endophytes are largely uncultivable and plant metabolites that are released when seeds are disrupted further aggravate the detection of microbes based on cultivation-dependent methods [14]. Therefore, seed endophyte studies have substantially profited from the recent development of cultivation-independent methods that allow community-level assessments [15].
So far, seed endophytes were explored in several plants, including maize, pumpkins, beans, and radish [e.g. [16], [17], [18]]. Each of the plant species harbored distinct bacterial communities that showed substantial differences in their composition at phylum level. However, relatively high proportions of Gammaproteobacteria and Enterobacteriaceae therein were identified as a common feature irrespective of species and cultivar. Studies that focused on plant breeding have shown that it not only affects root-associated microbial communities and the biocontrol potential of beneficial microbes against pathogens [19], [20], but also has an impact on the seed microbiome [16]. Tomato plants were used as model systems to verify vertical transmission of beneficial endophytes from one generation to the next [21]. However, little is known about the structure of seed-associated microbial assemblages in other plants of the Solanaceae family, which includes tobacco as a prominent member. The tobacco plant offers a highly specific microenvironment for endophytes due to the presence of high nicotine concentration in its tissues, which can substantially vary among different cultivars [22]. Diverging breeding approaches within the same plant species provide an ideal basis to explore the impact of the plant genotype on seed-endophytic communities. In the present study, we used four different tobacco cultivars, originating from Asia and the Americas, to characterize their seed-endophytic bacterial communities and to provide new insights into genotype-specific differences related to their diversity and structure. The cultivars originate from different breeding approaches, which is reflected by their phenotypes. We aimed to investigate the implication of different N. tabacum genotypes on the composition and structure of their seed-endophytic bacterial communities.
2. Materials and methods
2.1. Sample description
N. tabacum seeds were acquired during the growing season in 2018 from the seed producing companies Yuxi Zhong Yan Seed Co., Ltd (Yuxi, Yunnan; N24°19′54.32″, E102°31′44.95″) and Variety Production Base owned by Guizhou Tobacco Company (Guiyang, Guizhou; N26°52′30.48″, E107°05′50.79″). All obtained seeds were produced for conventional cultivation in January 2018 and treated with a specific seed coating as required by China’s State Tobacco Monopoly Administration. The coatings include colored talcum powder as structural element and following nutrients and trace elements: NO3, NH4, P2O5, K2O, Mg, S, Fe, Mn, B, Cu, Zn, and Mo. In addition, the coating includes carbendazim as fungicide to protect the seedlings during germination. Exact proportions are not provided for any of the components (Chinese patent: CN1561740A). The hybrid cultivars K326 and PVH1452 (obtained from Yuxi Zhong Yan Seed Co., Ltd) as well as Yunyan87 and Bina1 (obtained from Variety Production Base of Guizhou Tobacco Company) are currently the prevalent N. tabacum genotypes in China’s main cultivation areas and were therefore selected as models for the present study. They originate from different breeding lines and show different levels of resistance towards nematodes, oomycetes, and viruses (Table 1). For the Brazilian cultivar PVH1452, the hybrid’s pedigree was not publicly available, while the other relevant information was directly obtained from the seed producer.
Table 1.
Cultivar details for the utilized N. tabacum seeds. All seeds were produced in the same season at the indicated seed production facilities in China. Information related to disease resistance was obtained from the State Tobacco Monopoly Administration (http://www.tobacco.gov.cn). The nicotine content refers to the proportion in dry tobacco leaves that were cultivated in Guizhou/China (Xia et al., 2017).
| Genotype name | Breeding type | Pedigree | Geographic origin of seeds | Location of seed production/harvest year | Disease resistance of cultivar | Nicotine content [%] |
|---|---|---|---|---|---|---|
| Bina1 | Hybrid | Yunyan2 × K326 (mutant line) | Guizhou, China | 26°52′56″ N, 107°05′6″ E/2017 | Black shank: moderately resistant; Nematodes: moderately resistant; TMV: moderately susceptible |
1.69 |
| K326 | Hybrid | McNair30 × NC95 | Kentucky, USA | 24°20′2″ N, 102°31′50″ E/2017 | Black shank: highly resistant; Nematodes: moderately resistant; TMV: highly resistant |
2.24 |
| PVH1452 | Hybrid | Information not available |
Rio Grande do Sul, Brazil | 24°20′2″ N, 102°31′50″ E/2017 | Black shank: moderately resistant; Nematodes: moderately susceptible; TMV: moderately susceptible |
2.39 |
| Yunyan87 | Hybrid | Yunyan2 × K326 | Yunnan, China | 26°52′56″ N, 107°05′6″ E/2017 | Black shank: moderately resistant; Nematodes: moderately resistant; TMV: highly susceptible |
2.02 |
2.2. Extraction of total community DNA from seeds and construction of the amplicon library
In the first processing step, the seed coating was removed from all seeds by washing seeds in sterile ddH2O. The washing step was conducted in 50-ml tubes with manual shaking until no residues were visible by visual inspection. Subsequently, a surface sterilization was conducted to remove all non-endophytic microorganisms from the seed surface. The seeds were collected with a sterile sieve and transferred into 20 ml of an aqueous sodium hypochlorite solution (4% NaOCl) in sterile 50-ml reaction tubes. After placing the tubes horizontally on a shaker (120 rpm) for 3 min, the seeds were again collected with a sterile sieve and washed three times in 20 ml sterile ddH2O for 3 min at 120 rpm. For each composite sample a total of 20 seeds were pooled and treated for 5 min with an automatic homogenizer (Sangon Biotech, China) and sterile, disposable pestles. The total community DNA was extracted with the FastDNA SPIN Kit for Soil (MP Biomedicals, USA) from five composite samples from each tobacco cultivar following the manufacturer’s protocol. Subsequently samples were analyzed with NanoDrop (Thermo Fisher Scientific, USA) to monitor the DNA recovery. The samples were amplified with the primers 515f (5′ GTGYCAGCMGCCGCGGTAA) and 806r (5′ GGACTACHVGGGTWTCTAAT) according to the Earth Microbiome Project protocol (www.earthmicrobiome.org) [23] with sample-specific barcodes and Illumina sequencing adaptors. In addition, specific peptide nucleic acid (PNA) oligomers were added to the PCR mix to prevent the amplification of mitochondrial (mPNA) or plastidial (pPNA) RNA from eukaryotes [24]. The sequencing was conducted on an Illumina PE250 platform (2 × 250 bp paired-end reads) by Novogene (Beijing, China) with a minimum output of 100,000 quality-filtered (Q30 ≥ 75%) reads per sample.
2.3. Demultiplexing of the 16S rRNA gene fragment amplicons and OTU table construction
The data was subjected to a standardized workflow for further dereplication and quality filtering. In order to demultiplex the 16S rRNA gene fragment library, paired-end reads were assigned to samples based on their unique barcode and truncated by removing their barcode and primer sequence from the raw reads. Corresponding paired-end reads were merged using FLASH v1.2.7 [25] before further bioinformatic processing. Subsequently, quality filtering of the raw tags was performed under strict filtering conditions to obtain high-quality tags [26]. The resulting output was used to identify and remove all chimeric sequences using the UCHIME algorithm [27], [28]. The taxonomic analysis was performed after the sequences were clustered at 97% similarity with the Uparse software [29]. The assignments were based on a naïve-Bayes RDP classifier [30] clustered at 97% similarities with the Greengenes Database 13.8 [31].
2.4. Detailed bioinformatic processing of the amplicon libraries and network generation
Highly abundant bacterial operational taxonomic units (OTUs), which remained without taxonomic assignments, were manually blasted against the NCBI nucleotide collection database (www.ncbi.nlm.nih.gov/nucleotide). Results indicated that these OTUs had a high similarity to plant mitochondrial sequences (95% identity to accession number XM_027917124.1) and were therefore excluded of further analyses. Moreover, OTUs assigned to the bacterial phylum Oxyphotobacteria and the order Rickettsiales were also manually removed from the dataset due to their sequence similarity to mitochondrial and chloroplast sequences of N. tabacum (NCBI id: txid4097). After removal of the host plant-derived sequences, the dataset was normalized to 2,795 reads in order to account for read number variations in the samples reaching from 23,700 to 2,795 reads. The normalized OTU table served as input for alpha and beta diversity analyses and statistical tests (t-test, ANOSIM). Principal coordinate analyses (PCoA) plots and non-metric multidimensional scaling (nMDS) plots were constructed by calculating the unweighted UniFrac distance matrix within QIIME (1.9.0). The alpha diversity was assessed by the observed OTUs and Shannon diversity indices. Significant differences in relative abundance of OTUs were calculated using DESeq2 [32]. As input for calculating differently abundant OTUs, a non-normalized OTU table excluding mitochondrial sequences was used to fulfill the input requirements for DESeq2. For generating the bar charts, OTUs were grouped on genus level (level 6). The resulting bar charts represent the highly abundant fraction of the amplicon dataset (>0.5% mean rel. abundance). For OTU network construction, the OTU table was further reduced by retaining only OTUs occurring in at least 80% of respective replicates (further referred to as core OTUs). The network was generated using the QIIME (1.9.0) pipeline with the ‘make_OTU_network.py’ script and visualized using Cytoscape version 3.7.0 [33].
3. Results
3.1. Diversity of bacterial seed endophytes in N. Tabacum seeds
After removing mitochondrial sequences, the dataset comprised a total of 196,226 reads, which were clustered into 3344 OTUs. Following normalization to 2795 reads per sample, 2423 OTUs were retained. The OTUs were then grouped on genus level resulting in 1030 taxa. Alpha rarefaction analyses revealed the highest Shannon index for cultivar Yunyan 87 (H′ = 6.3 ± 0.8) and the lowest for cultivar K326 (H′ = 4.5 ± 1.1). The same results were obtained when assessing bacterial diversity based on observed OTUs. The lowest number of observed OTUs was shown for K326 (306 ± 65) while the highest number of OTUs could be observed for Yunyan 87 (375 ± 140). Differences in alpha-diversity were not significant (t-test; p < 0.05) when Shannon diversity indices and observed OTU counts were compared for different cultivars (Fig. S1). The beta-diversity was assessed based on an unweighted UniFrac distance matrix. PCoA and NMDS plots were generated based on this matrix in order to visualize potential clustering of sample groups (Fig. 1). Pairwise comparison of unweighted UniFrac distance matrices using ANOSIM (999 permutations) showed no statistically significant differences in beta diversity among the different cultivars.
Fig. 1.

Principal coordinates analysis (A, B) and non-metric multidimensional scaling plot (C) of the bacterial community in seeds of different tobacco cultivars. The community clustering is based on Bray-Curtis dissimilarities (unweighted UniFrac). Each of the four N. tabacum cultivars is labelled with a different color.
3.2. Structure of the bacterial community in different tobacco cultivars
Bacterial taxa with a mean relative abundance of at least 0.5% in the whole dataset were assessed in more detail (Fig. 2) and additionally visualized for each sample individually (Fig. S2). The predominant bacterial fraction included six phyla, with Proteobacteria (39.53%–56.30%) being the most dominant in all investigated cultivars. The second most abundant phylum was the Firmicutes (15.26%–34.02%) followed by Spirochaetes that were unevenly distributed among the different cultivars. The highest abundance of Spirochaetes, with a not further identified Spirochaetaceae (family level) as only representative, was detected in cultivar K326 (34.56%), compared to a low relative abundance in cultivars PVH1452 (0.96%), Bina1 (1.39%), and Yunyan 87 (0.09%). When assessed in more detail, it became evident that the occurrence of this taxonomic group was not evenly distributed in K326 seeds with an occurrence ranging from 0 to 56.7%.
Fig. 2.

Taxonomic classification of the highly abundant (>0.5%) members of the microbiome inhabiting seeds of different tobacco cultivars. Bar charts represent the bacterial composition on genus level where assignments were possible, otherwise taxa were labelled at the lowest assignable taxonomic level.
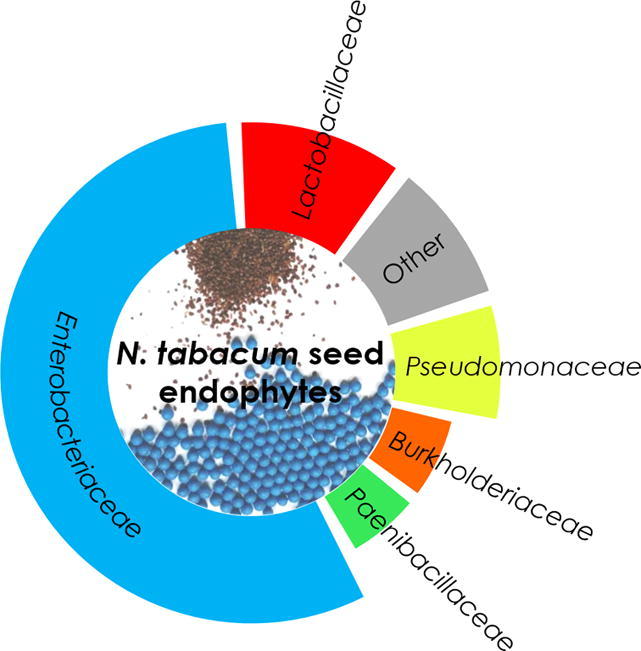
The cultivar Yunyan 87 harbored the highest relative amount of Actinobacteria (12.53%) compared to all other cultivars investigated (0.26%–2.43%). Bacteroidetes, including only members of the Bacteroidia class, were most dominant in Bina1 (22.0%) compared to the other cultivars (2.27%–3.61%). The phylum Tenericutes (0.47%–6.87%), which was not part of the microbiota of the cultivar Yunyan 87 and which included Mycoplasma as the only genus in this group turned out as second most abundant phylum in cultivar PVH1452 while being less represented in all other cultivars (<1%). The prevalent fraction of the microbiome comprised nine classes, while high proportions of Gammaproteobacteria (38.60%–51.61%) were observed in all cultivars. Relative abundance of Betaproteobacteria reached from 2.23% in cultivar Bina1 to 4.42% in cultivar PVH1452. Bacilli were least abundant in cultivar PVH1452 (1.11%) compared to all other cultivars (8.51%–30.29%). The remaining fraction of the microbiome consisted of, Clostridia (1.93%–17.13%), Actinobacteria (0.18%–12.53%), and Alphaproteobacteria (0.22%–5.21%). The predominant order was identified as Enterobacteriales (23.81%–49.41%) with highest relative abundance in cultivar PHV1452 and least in K326. Lactobacillales (0.84%–26.52%) were most frequently observed in cultivar Bina1, while being least abundant in cultivar PVH1452. Pseudomonadales (1.90%–13.15%) was identified as fourth most abundant order followed by Bacteroidales (22.0% rel. abundance in Bina1, 2.55%–3.61% in all other cultivars), Clostridiales (1.94%–17.13%), Mycoplasmatales (0%–21.76%), Bacillales (0.27%–12.24%), Burkholderiales (2.23%–5.67%), Micrococcales (0.19%–12.52%), Xanthomonadales (0.36%–9.76%), Sphingomonadales (0.18%–2.99%) and Rhizobiales (0.03%–3.41%). The two most prevalent families were assigned to Enterobacteriaceae (23.81%–49.41%) comprising two not further classified genera within Enterobacteriaceae and two Lactobacillus spp. within Lactobacillaceae (0.67%–23.58%).
3.3. Detection of cultivar-specific entophyte signatures
A grouped OTU table (level 6) served as input file to generate a phylogenetic tree graph (Fig. 3) that comprised taxa with a mean relative abundance of 0.01% over the whole dataset. By applying this threshold, 384 taxa that were found in at least one N. tabacum cultivar were included in the tree. The highest number of unique signatures was identified for cultivar Yunyan 87, it included 33 taxa that were not present in the other cultivars. Most of the unique signatures were found for Proteobacteria (13 taxa), Actinobacteria (13 taxa), Bacteroidetes (3 taxa) and Firmicutes (3 taxa). Cultivar K236 included the second highest number of unique taxa (31) with the most signatures found within Proteobacteria (13 taxa), followed by Actinobacteria (5 taxa) and Firmicutes (4 taxa). Cultivars Bina1 and PVH 1452 harbored less unique signatures with the most being assigned to Proteobacteria with 6 and 2 taxa respectively and Firmicutes (2 and 5 taxa respectively). Among all cultivars, Proteobacteria and the class of Alphaproteobacteria therein harbored the highest number of unique signatures (11 taxa).
Fig. 3.

Tree graph comprising taxa with at least a mean relative abundance of 0.01% over the whole dataset. OTUs were collapsed at genus level to generate the phylogenetic tree. Coloration in the outer rings indicates the occurrence of distinct taxa in the core seed microbiome (≥80% of the samples) of the respective cultivar. Node sizes correspond to the mean relative abundance of distinct taxa over the whole dataset.
3.4. Overlaps in the bacterial endophyte community of N. tabacum seeds
Following further filtering for OTUs that were present in at least 80% of the respective cultivar replicates, a total of 172 OTUs remained. The reduced OTU table was used for network generation (Fig. 4). OTUs shared by all cultivars were primarily identified as members of the phyla Proteobacteria and Firmicutes. In total, 29 OTUs were present in all cultivars, including Lactobacillus, Ralstonia, Romboutsia, Saccharibacillus, Pseudomonas, Sphingomonas, Staphylococcus, Microbacterium, Acinetobacter, Morganiella, Stenotrophomonas, Delftia, Massilia, not further classified Enterobacteriaceae, Enterococcaceae, Sanguibacteriaceae and Burkholderiales. Moreover, 16 of those core OTUs occurred with a mean relative abundance of at least 1%. The cultivars Bina1 and Yunyan 87 had the highest number of OTUs in common, they shared 24 OTUs that were not present in the other two cultivars. These OTUs included seven members of Actinobacteria and three members of Bacteroidetes; both bacterial phyla were not present in the core microbiome shared by all N. tabacum cultivars. The combinations Bina1 with PVH1452 and Bina1 with K326 shared 11 and six unique OTUs respectively. Bina1, K326, and Yunyan 87 had 11 distinct OTUs in common that were not present in PVH1452. Furthermore, a total of 72 OTUs that were mostly shared by at least two cultivars, differed significantly (p ≤ 0.05) in their relative abundance among the cultivars (details are provided in Table S1). Significantly different abundant OTUs with a mean relative abundance of at least 0.5% were mainly identified as members of the phyla Proteobacteria, Firmicutes, and Actinobacteria (Fig. 5). A total of six predominant OTUs that were part of the core microbiome and assigned to Enterobacteriaceae occurred in different abundances among the cultivars.
Fig. 4.

OTU network with core OTUs that occurred in at least 80% of the respective replicates of each N. tabacum cultivar. OTUs with a mean relative abundance of at least 5% were labeled with their taxonomic assignment at the lowest assignable level (family or genus level). The node size corresponds to the relative abundance of the respective OTU in the whole dataset.
Fig. 5.

Highly abundant (>0.5% abundance in the whole dataset) OTUs which differ significantly in their relative abundance among different cultivars were identified according to DESeq2 analyses (p < 0.05). Node size represents the relative abundance of each taxon.
4. Discussion
Although we are gradually beginning to understand the implications of seed endophytes as vertically transmitted, natural inocula of different agricultural plants, we are still lacking possibilities to exploit them biotechnologically. Previous studies have shown that plant seeds provide a viable basis for microbiota engineering in terms of introducing beneficial microorganisms to crop plants through targeted approaches [34]. Cultivation of agricultural crops would benefit from microbiota-engineered seeds with improved resilience against biotic and abiotic stress. However, the development of novel biotechnological applications that make use of naturally co-evolved transmission mechanisms requires a detailed understanding of the composition and functioning of microbial seed colonizers. The present study provides novel insights into bacterial endophytes of N. tabacum and diverging breeding lines thereof. When compared to related studies with different plant species that have found different degrees of genotype specificity [16], [35], [36], N. tabacum seeds had a relatively low proportion of 17% shared OTUs. However, these OTUs included all predominant taxa accounting for 65.5% of the combined core microbiome reads, while cultivar-specific seed endophytes only occurred in lower abundances. Interestingly, the cultivars Bina1 and Yunyan 87, which have the same ancestors in their breeding lines, shared the highest proportion (37.2%) of common OTUs. This provides a strong indication for co-divergence of the seed microbiome with the host during conventional plant breeding. The overall results are in agreement with previous observations, which showed that plants with an individual-specific seed microbiome can harbor a low number of common OTUs that still proportionally account for a substantial share of the microbial population [35].
When the core microbiome was assessed, N. tabacum seeds were shown to harbor a high proportion of Enterobacteriaceae. Interestingly, a similar taxonomic structure was also found for pumpkin seed endophytes [16]. The seed community of different Cucurbita pepo cultivars was shown to harbor a high proportion of Enterobacteriaceae, whereas more than 60% of the seed-specific OTUs were not found in other plant tissues. C. pepo was also the first plant model used to study the effects of breeding on seed endophytes. It was shown that the seed microbiome of the plant had a higher genotype specificity than its rhizosphere microbiome. The present study provides further evidence that breeding affects the seed microbiome across plant species. In addition to being the predominant fraction in the core microbiome, the Enterobacteriaceae family also accounted for the most OTUs with a differing abundance among the analyzed tobacco cultivars. Further studies will be required in order to elucidate if these bacteria are specifically selected and maintained between plant generations or if the plant recruits available members from the soil community. When compared to the tomato seed microbiome [21], which is another important representative in the Solanaceae family, it becomes evident that both plant species harbor a high proportion of Proteobacteria in their seeds. However, tomato seeds were shown to be primarily colonized by members of Betaproteobacteria [21], while Gammaproteobacteria and more specifically Enterobacteriaceae constituted the core microbiome in tobacco plants.
Microbiome studies have supported the establishment of different enterobacterial lineages as indigenous members of the plant microbiome. This bacterial group is widespread and commonly occurs in the intestines of humans and animals, but also colonizes the endosphere of different plant tissues, which indicates a co-adaption [37]. While it is not completely understood if enterobacterial populations associated with plants are distinguishable from those found in other environments, different isolates from this group were shown to be efficient plant growth promoters or biocontrol agents [38], [39]. However, the implications of enterobacterial colonization of crop plants for human health are not fully understood. It was recently found that they can serve as natural reservoirs of antibiotic resistances [40]. It remains to be elucidated if these plant colonizers play an indirect role, i.e. transfer of resistance genes to human pathogens, in disease outbreaks when enterobacteria are the causing agents. Lactobacilli constituted another prevalent group in the core microbiome of tobacco seeds. These Gram-positive bacteria occur in rather low abundances in plant microbiomes, but can have positive effects on seed germination and plant growth [41]. Such characteristics might have facilitated positive selection and potentially enrichment of these bacteria in the seed endosphere of the analyzed cultivars.
Our data showed a substantial overlap in the presence of several predominant taxa as common seed endophytes in all four cultivars. This indicates a conserved acquisition mechanisms of a distinct fraction of the seed microbiota in N. tabacum that was only partially affected by plant breeding. Similar observations were made when the rhizosphere microbiomes of lettuce, common bean, and sugar beet plants were analyzed [42], [43], [44]. Different cultivars of the same plant species were shown to harbor a common core microbiome with different degrees of genotype-specific extensions. The present study provides further evidence, in addition to the findings for pumpkin seeds [16], that genotype specificity of the plant microbiome is also reflected by seed endophytes. The tobacco seed microbiome harbored several unexpected bacterial lineages. For example, cultivar K326 showed an exceptionally high abundance of Spirochaetaceae in some of the analyzed samples. This bacterial family is primarily known for its human-pathogenic representatives, e.g. Borrelia and Treponema, but has also free-living members [45]. Although not commonly associated with plants, Spirochaetaceae were found to be part of the sugarcane microbiome [46]. Further assessments are required in order to clarify the role of this bacterial group in tobacco seeds and to explore if they have any implication for health and disease in the plant.
Three of the cultivars harbored OTUs assigned to the bacterial genus Mycoplasma in their seeds. This genus is known for its parasitic lifestyle and small genome size, but was also found to be a seaweed endophyte [47]. Due to the lack of a cell wall, this bacterial group is mainly found in favorable habitats, which includes eukaryotic hosts; however, plants were so far not reported to harbor larger proportions of these often parasitic microorganisms. Interestingly, also the order Clostridiales was found to be present in the seed communities. The bacterial family Lachnospiraceae, which constituted the main fraction of Clostridiales in the seeds, was also found in association with a native plant in China [48]. It remains to be elucidated if this rather unusual fraction of the seed microbiome is favored by the specific phytochemical environment in N. tabacum tissues, i.e. the high concentration of nicotine.
The Chinese cultivar Yunyan 87 harbored the highest number of unique OTUs in its seed microbiome, wherein members of Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria prevailed. Various members from these taxonomic groups commonly constitute the plant phyllosphere [49], therefore it is likely that they are acquired from above ground parts of N. tabacum plants. In general, our data suggests that this cultivar is more permissive for different seed endophytes than the other cultivars; however, this must be confirmed with deepening studies.
5. Conclusions
Our assessment of seed endophyte diversity and composition has provided novel insights related to the impact of breeding on the plant microbiota. While a common core of predominant bacterial taxa was maintained in tobacco seeds of the analyzed cultivars, they also harbored a genotype-specific extension of this microbiome. This is in analogy to previous findings targeting other plant tissues and the rhizosphere of different genotypes from the same species. N. tabacum seeds were shown to be predominately colonized by Enterobacteriaceae. This was so far not observed within the Solanaceae plant family and might be a specific feature of the tobacco plant. Moreover, different bacterial lineages were found in the seeds that primarily include members that are known for a parasitic lifestyle. Their implications for plant health remains to be elucidated.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 31701836, 31960555), Guizhou Provincial Science and Technology Program (Grant No. 2019-1410, 2018-1050, 2017-5788), “The Scientific Research Foundation for Talent Introduced in Guizhou University” (Grant No. 2016-68), and Guizhou Tobacco Company Research Program (Grant No. 201826).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.01.004.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
The amplicon dataset that was used for this study was deposited at ENA (https://www.ebi.ac.uk/ena) under the accession number PRJEB31125.
References
- 1.Groark K.P. The angel in the gourd: ritual, therapeutic, and protective uses of tobacco (Nicotiana tabacum) among the Tzeltal and Tzotzil Maya of Chiapas, Mexico. J Ethnobiol. 2010;30:5–30. [Google Scholar]
- 2.Winter J.C. University of Oklahoma Press; Norman, OK: 2000. Tobacco use by Native North Americans: Sacred smoke and silent killer; pp. 305–330. [Google Scholar]
- 3.Tushingham Shannon, Snyder Charles M., Brownstein Korey J., Damitio William J., Gang David R. Biomolecular archaeology reveals ancient origins of indigenous tobacco smoking in North American Plateau. Proc Natl Acad Sci USA. 2018;115(46):11742–11747. doi: 10.1073/pnas.1813796115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Food and Agriculture Organization of the United Nations. FAOSTAT Statistics Database. http://www.fao.org/faostat/en/#data/QC/visualize (Retrieved 21 May 2019).
- 5.Ma L., Zhang H.Y., Zhou X.K., Yang C.G., Zheng S.C. Biological control tobacco bacterial wilt and black shank and root colonization by bio-organic fertilizer containing bacterium Pseudomonas aeruginosa NXHG29. Appl Soil Ecol. 2018;129:136–144. [Google Scholar]
- 6.Wu K., Yuan S., Wang L., Shi J., Zhao J. Effects of bio-organic fertilizer plus soil amendment on the control of tobacco bacterial wilt and composition of soil bacterial communities. Biol Fertil Soils. 2014;50:961–971. [Google Scholar]
- 7.Liu X., Zhang S., Jiang Q., Bai Y., Shen G. Using community analysis to explore bacterial indicators for disease suppression of tobacco bacterial wilt. Sci Rep. 2016;6:36773. doi: 10.1038/srep36773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang H., Li J., Xiao Y., Gu Y., Liu H. An integrated insight into the relationship between soil microbial community and tobacco bacterial wilt disease. Front Microbiol. 2017;8:2179. doi: 10.3389/fmicb.2017.02179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang G., Wei Z., Xu J., Chen H., Zhang Y. Bacterial wilt in China: history, current status, and future perspectives. Front Plant Sci. 2017;8:1549. doi: 10.3389/fpls.2017.01549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen G., Zhang S., Liu X., Jiang Q., Ding W. Soil acidification amendments change the rhizosphere bacterial community of tobacco in a bacterial wilt affected field. Appl Microbiol Biotechnol. 2018;102:9781–9791. doi: 10.1007/s00253-018-9347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson E.B. The seed microbiome: Origins, interactions, and impacts. Plant Soil. 2018;422:7–34. [Google Scholar]
- 12.Berg G., Raaijmakers J.M. Saving seed microbiomes. The ISME Journal. 2018;12:1167. doi: 10.1038/s41396-017-0028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson R.J., Fraaije B.A., Clark I.M., Jackson R.W., Hirsch P.R. Wheat seed embryo excision enables the creation of axenic seedlings and Koch’s postulates testing of putative bacterial endophytes. Sci Rep. 2016;6:25581. doi: 10.1038/srep25581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shade A., Jacques M.A., Barret M. Ecological patterns of seed microbiome diversity, transmission, and assembly. Curr Opin Microbiol. 2017;37:15–22. doi: 10.1016/j.mib.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Bergna A., Cernava T., Zachow C., Berg G. Analysing seed endophytes for biotechnology. In: Schouten A., editor. Endophyte biotechnology: promise for agriculture and pharmacology. CABI; Wallingford: 2019. pp. 42–58. [Google Scholar]
- 16.Adam E., Bernhart M., Müller H., Winkler J., Berg G. The Cucurbita pepo seed microbiome: genotype-specific composition and implications for breeding. Plant Soil. 2018;422:35–49. [Google Scholar]
- 17.Johnston-Monje D., Raizada M.N. Conservation and diversity of seed associated endophytes in Zea across boundaries of evolution, ethnography and ecology. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0020396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torres-Cortes G., Bonneau S., Bouchez O., Genthon C., Briand M. Functional microbial features driving community assembly during seed germination and emergence. Front Plant Sci. 2018;9:902. doi: 10.3389/fpls.2018.00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aleklett K., Hart M. The root microbiota- a fingerprint in the soil? Plant Soil. 2013;370:671–686. [Google Scholar]
- 20.Barret M., Briand M., Bonneau S., Preveaux A., Valiere S. Emergence shapes the structure of the seed microbiota. Appl Environ Microbiol. 2015;81:1257–1266. doi: 10.1128/AEM.03722-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergna A., Cernava T., Rändler M., Grosch R., Zachow C. Tomato seeds preferably transmit plant beneficial endophytes. Phytobiomes Journal. 2018;2:183–193. [Google Scholar]
- 22.Xia Z., Wang J., Liu R., Liu Y., Nie Q. Adaptability of different tobacco cultivars grown in Zunyi Area. Guizhou Agricultural Sciences. 2017;45:75–78. [Google Scholar]
- 23.Thompson L.R., Sanders J.G., McDonald D., Amir A., Ladau J. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551:457. doi: 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lundberg D.S., Yourstone S., Mieczkowski P., Jones C.D., Dangl J.L. Practical innovations for high-throughput amplicon sequencing. Nat Methods. 2013;10:999. doi: 10.1038/nmeth.2634. [DOI] [PubMed] [Google Scholar]
- 25.Magoč T., Salzberg S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bokulich N.A., Subramanian S., Faith J.J., Gevers D., Gordon J.I. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10:57. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgar R.C., Haas B.J., Clemente J.C., Quince C., Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haas B.J., Gevers D., Earl A.M., Feldgarden M., Ward D.V. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edgar R.C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q., Garrity G.M., Tiedje J.M., Cole J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeSantis T.Z., Hugenholtz P., Larsen N., Rojas M., Brodie E.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitter B., Pfaffenbichler N., Flavell R., Compant S., Antonielli L. A new approach to modify plant microbiomes and traits by introducing beneficial bacteria at flowering into progeny seeds. Front Microbiol. 2017;8:11. doi: 10.3389/fmicb.2017.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rezki S., Campion C., Simoneau P., Jacques M.A., Shade A. Assembly of seed associated microbial communities within and across successive plant generations. Plant Soil. 2018;422:67–79. [Google Scholar]
- 36.Wassermann B., Cernava T., Müller H., Berg C., Berg G. Seeds of native alpine plants host unique microbial communities embedded in cross-kingdom networks. Microbiome. 2019;7:108. doi: 10.1186/s40168-019-0723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lodewyckx C., Vangronsveld J., Porteous F., Moore E.R., Taghavi S. Endophytic bacteria and their potential applications. Crit Rev Plant Sci. 2002;21:583–606. [Google Scholar]
- 38.Kalbe C., Marten P., Berg G. Strains of the genus Serratia as beneficial rhizobacteria of oilseed rape with antifungal properties. Microbiol Res. 1996;151:433–439. doi: 10.1016/S0944-5013(96)80014-0. [DOI] [PubMed] [Google Scholar]
- 39.Shoebitz M., Ribaudo C.M., Pardo M.A., Cantore M.L., Ciampi L. Plant growth promoting properties of a strain of Enterobacter ludwigii isolated from Lolium perenne rhizosphere. Soil Biol Biochem. 2009;41:1768–1774. [Google Scholar]
- 40.Cernava T., Erlacher A., Soh J., Sensen C.W., Grube M. Enterobacteriaceae dominate the core microbiome and contribute to the resistome of arugula (Eruca sativa Mill.) Microbiome. 2019;7:13. doi: 10.1186/s40168-019-0624-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lamont J.R., Wilkins O., Bywater-Ekegärd M., Smith D.L. From yogurt to yield: Potential applications of lactic acid bacteria in plant production. Soil Biol Biochem. 2017;111:1–9. [Google Scholar]
- 42.Cardinale M., Grube M., Erlacher A., Quehenberger J., Berg G. Bacterial networks and co-occurrence relationships in the lettuce root microbiota. Environ Microbiol. 2015;17:239–252. doi: 10.1111/1462-2920.12686. [DOI] [PubMed] [Google Scholar]
- 43.Mendes L.W., Raaijmakers J.M., De Hollander M., Mendes R., Tsai S.M. Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. The ISME Journal. 2018;12:212. doi: 10.1038/ismej.2017.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zachow C., Müller H., Tilcher R., Berg G. Differences between the rhizosphere microbiome of Beta vulgaris ssp. Maritima-ancestor of all beet crops-and modern sugar beets. Front Microbiol. 2014;5:415. doi: 10.3389/fmicb.2014.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leschine S., Paster B.J., Canale-Parola E. Free-living saccharolytic spirochetes: the genus Spirochaeta. In: Dworkin M., Falkow S., Rosenberg E., Schleifer K.-.H., Stackebrandt E., editors. The Prokaryotes: Volume 7: Proteobacteria: Delta, Epsilon Subclass. Springer; New York, NY: 2006. pp. 195–210. [Google Scholar]
- 46.Yeoh Y.K., Paungfoo-Lonhienne C., Dennis P.G., Robinson N., Ragan M.A. The core root microbiome of sugarcanes cultivated under varying nitrogen fertilizer application. Environ Microbiol. 2016;18:1338–1351. doi: 10.1111/1462-2920.12925. [DOI] [PubMed] [Google Scholar]
- 47.Hollants J., Leroux O., Leliaert F., Decleyre H., De Clerck O. Who is in there? Exploration of endophytic bacteria within the siphonous green seaweed Bryopsis (Bryopsidales, Chlorophyta) PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0026458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen P., Zhao M., Tang F., Hu Y., Peng X. The effect of environment on the microbiome associated with the roots of a native woody plant under different climate types in China. Appl Microbiol Biotechnol. 2019;103:1–15. doi: 10.1007/s00253-019-09747-6. [DOI] [PubMed] [Google Scholar]
- 49.Vorholt J.A. Microbial life in the phyllosphere. Nat Rev Microbiol. 2012;10:828. doi: 10.1038/nrmicro2910. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The amplicon dataset that was used for this study was deposited at ENA (https://www.ebi.ac.uk/ena) under the accession number PRJEB31125.