

Abstract
Polygenic risk scores (PRSs) have become the standard for quantifying genetic liability in the prediction of disease risks. PRSs are generally constructed as weighted sum scores of risk alleles using effect sizes from genome-wide association studies as their weights. The construction of PRSs is being improved with more appropriate selection of independent single-nucleotide polymorphisms (SNPs) and optimized estimation of their weights but is rarely reflected upon from a theoretical perspective, focusing on the validity of the risk score. Borrowing from psychometrics, this paper discusses the validity of PRSs and introduces the three main types of validity that are considered in the evaluation of tests and measurements: construct, content, and criterion validity. This introduction is followed by a discussion of three topics that challenge the validity of PRS, namely, their claimed independence of clinical risk factors, the consequences of relaxing SNP inclusion thresholds and the selection of SNP weights. This discussion of the validity of PRS reminds us that we need to keep questioning if weighted sums of risk alleles are measuring what we think they are in the various scenarios in which PRSs are used and that we need to keep exploring alternative modeling strategies that might better reflect the underlying biological pathways.
Introduction
Polygenic risk scores (PRSs) aim to quantify the genetic liability of common diseases and traits, the collective of genetic factors that contribute to their development (1). PRSs are typically calculated as a weighted sum of the risk alleles of single-nucleotide polymorphisms (SNPs) and investigated for their potential to improve the prediction of common diseases in clinical care to guide preventive and therapeutic interventions (2).
While the concept of polygenic inheritance is centuries old and long lacked data to prove its merit, the calculation of risk scores developed from an empirical tradition with little attention for its theoretical foundation (3). In the early days of genome-wide association studies (GWASs), researchers considered their few newly identified SNPs as separate variables in the prediction of disease risks (4,5), and PRSs were a practical solution to include larger numbers of variants in the regression analyses (6). Some early studies calculated unweighted risk scores that summed the number of risk alleles, assuming a similar impact on disease risk for all SNPs, but these were rapidly replaced by weighted scores that acknowledge that some SNPs have stronger effect than others (6). In recent years, the construction of PRSs is being improved from a computational perspective, with proposals for a more liberal selection of independent SNPs and a more refined estimation of their weights (7,8).
Even though PRSs typically explain only a small proportion of the genotypic variance (2,9,10), the validity of the PRS as a measurement of polygenic predisposition remains largely undiscussed (3). It may be that researchers are aware and accept that the validity is imperfect as many more genetic associations remain to be identified (2), all models have limitations, and they can be useful even when imperfect. It may also be that researchers feel no reason to question the validity of the weighted sums of risk alleles as (1) the risk distributions are what Fisher predicted for a large number of variants with weak effects (11,12), (2) there seems to be no evidence for strong gene–gene interaction (12) and (3) these PRSs generally hold their discriminative ability in external validation samples (see e.g. (13–15)).
Yet, several methodological concerns about the validity of the PRS have been raised (3,16–19). It is argued that the functional variation underlying SNP associations may not be captured by risk alleles (16) and that the additive model may not adequately capture the polygenic liability (3). Two studies that compared the additive model with alternatives showed that not only the weighted sum of risk alleles was compatible with empirical data but that several other mathematical models fitted as well when effects at single loci are small (17,18). And it is questioned whether the recent trend of including millions of SNPs challenges the validity of the definition of risk alleles as most their effect sizes are too small to produce ‘observable’ changes in the calculation of risks, even by the thousands (19).
In this paper, I discuss the theoretical concepts of validity that are used for evaluating tests and assessments in the social sciences. I introduce the main types of validity and illustrate their relevance for the construction of PRSs by reflecting on several topics in PRS research: the observation of PRSs as independent effects, the consequences of relaxing SNP inclusion thresholds and the accuracy of GWAS weights.
Types of validity
The validity of a test or measurement indicates whether it measures what it intends to measure (20,21). Assessing the validity of a measurement is challenging when what needs to be measured is difficult to describe and cannot be directly observed. When we cannot directly evaluate the validity of a measurement, which is the case for PRSs, then we need to rely on various sorts of indirect evidence to assure that PRSs measure what we want the scores to measure. But what do we want PRSs to assess? And how well are they doing?
In psychometrics, three types of validity are commonly distinguished: construct, content and criterion validity (Fig. 1) (20). Construct validity informs about the intention of the measurement, about what is the construct that needs to be measured. It relates the construction of the PRS, as a weighted sum of risk alleles, to the underlying theoretical views about genetic liability. It questions whether the genetic liability of a disease is quantified by an additive model that combines the risk alleles into a single score. Construct validity is examined by correlating the PRS of a disease with assessments that measure the genetic predisposition of related (convergent validity) or unrelated diseases (discriminant validity).
Figure 1.
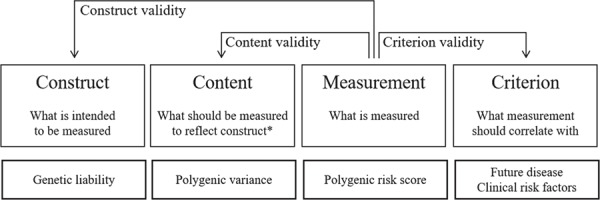
Three types of validity applied to the measurement of polygenic risk scores. Legend: * In the context of the specific application of the measurement.
Content validity relates the PRS to what ought to be measured to assess the construct of genetic liability in a certain context. Content considers the measurement of PRS in the practical context of how the score will be applied. For example, when a PRS is used to predict disease in the absence of non-genetic risk factors, then the score needs to capture all polygenic variance. If the score is included in a prediction model with clinical risk factors that mediate part of the polygenic risk, then the score needs to capture the polygenic risk that is not captured by the intermediate phenotypes.
Criterion validity examines to what extent PRSs correlate with other measurements that they are expected to be related to, either at the same time (concurrent validity) or in the future (predictive validity). Most evidence on the validity of PRS is about this predictive validity, and for most common diseases, this predictive validity is modest, except when the PRS includes one or more SNPs that have a strong impact on disease risk. The modest predictive validity may limit the PRS’s potential for clinical utility, but it may be informative enough for establishing criterion validity.
Finally, the widespread adoption and uniform application of PRS suggest that the score certainly does have face validity, the fourth type of validity that is often distinguished: the score seems valid on its appearance (22). Strong face validity in the context of limited construct, criterion and content validity is not enough. Whether the construction of PRSs, as weighted sums of risk alleles, is valid depends on the purpose of assessing the polygenic contribution to disease. In the next sections, I will illustrate why a PRS may be valid for the prediction of risk when used on its own but not when it is combined with clinical risk factors, and why a PRS that has its weights from GWASs or that include millions of SNPs may not be valid in clinical applications to inform people about their genetic risk of disease.
Independent effects
In recent years, researchers often report that PRSs predict the risk of common diseases independently from clinical risk factors. Independent effects have been reported for PRSs in breast cancer (23), coronary heart disease (24,25) and coronary artery disease (26,27) and were based on a formal mediation analysis (25), a statistically significant effect size for PRS after combining with clinical risk factors (26), and the absence of correlations or interactions between PRS and clinical risk scores (23,24). The observation of independent effects suggets that the PRSs were not associated with the clinical risk factors, but none of the studies showed these associations. Associations would however be expected as earlier studies on PRSs that included smaller numbers of selected SNPs did report associations with clinical risk factors (28,29), since PRSs are investigated for these intermediate phenotypes themselves, such as for obesity (30) and hypertension (31).
Independent effects may be expected between PRSs and behavioral and environmental risk factors, such as diet and lifestyle. These independent effects have been observed for lifestyle in stroke (32), coronary heart disease (33,34), adiposity (35,36), diabetes (37) and breast cancer (38), and for stressful life events in depression (39). However, there is less evidence of an independent effect of PRSs when they are combined with clinical risk factors, early symptoms or early-life predictors that represent the outcome. Examples include baseline glucose level to predict type 2 diabetes later in life (40), social impairments to predict psychosis (41), childhood obesity to predict adult obesity (42) and education at younger ages to predict highest educational attainment (43). SNPs that play a role in the pathways that lead to disease through these clinical risk factors or early-life stages may be associated with disease risk through these intermediate variables (44). These clinical risk factors may be measured with various levels of accuracy, which will affect how well they are able to mediate a SNP disease relationship, but to a priori expect that they are independent is incorrect. Developing prediction models that combine genetic and non-genetic risk factors is straightforward, but when the construction of the PRS needs to consider the possible mediating role of various non-genetic risk factors, then more consideration is needed to find out whether and how each of the variables mediates the association between SNPs and disease risk, and how the SNPs are best combined into risk scores.
If we assume that SNPs are biologically related to intermediate clinical risk factors, then we must rule out that the observation of independent effects is an artifact introduced by the method of calculating PRSs. To this end, we must verify whether individual SNPs are associated with the intermediate phenotypes, and find out, if they are, how polygenic predisposition is best quantified in a way that allows the associated SNPs to predispose their intermediate risk factors. Evidently, when none of the SNPs is associated with the clinical risk factors, then a single PRS might suffice.
Figure 2 presents a simplified scenario of how SNPs and PRS relate to coronary artery disease (CAD) with blood pressure and cholesterol as intermediate clinical risk factors. The graph is analogue to the directed acyclic graphs that are used in epidemiological research to express the direct causal relations between study variables (45–47), with the difference that, for prediction, the relationships do not need to be causal. Drawing ‘causal’ graphs helps identifying which variables should be considered in the construction of risk models that combine genetic and clinical risk factors and how they need to be modeled. Figure 2A shows that among the SNPs that predispose to CAD are SNPs for blood pressure and cholesterol. From mediation analyses, we know that the effects of these SNPs on CAD risk decrease when their clinical risk factors are included in the model. We expect that only the SNPs for which no intermediate factors are included will impact CAD risk. ‘Residual’ effects may be observed based on measurement error in the clinical variables and in the genetic data if SNPs do not accurately capture the underlying risk-increasing associations (16).
Figure 2.
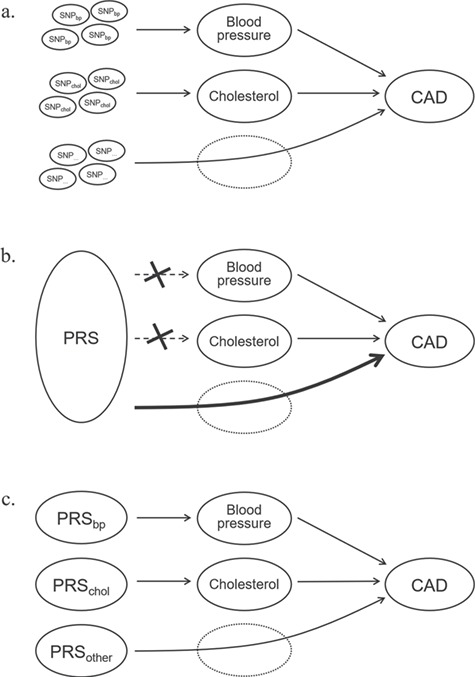
Independent effects between single-nucleotide polymorphisms, polygenic risk scores and clinical risk factors. Legend: PRS, polygenic risk score; SNP, single-nucleotide polymorphism; CAD, coronary artery disease. For illustration purposes, other possible associations between variables are omitted.
When SNPs from various known and unknown pathways are combined into a single PRS, we should expect that the PRS may not be associated with each of the clinical risk factors (Fig. 2B), or the effect may be attenuated (33). The PRS then presents as an independent risk factor, while some of the SNPs in the score may still predispose the clinical risk factors. The effects of the SNPs that predispose blood pressure and cholesterol would be reduced when the SNPs were entered as separate variables in the analysis, but they now remain part of the PRS. Their effects, at least conceptually, are now counted twice: through the clinical risk factor and in the score.
When a PRS is constructed for inclusion in a prediction model with clinical risk factors, the score should not measure the polygenic contribution (as illustrated in Fig. 1) but the part of the polygenic contribution that is not captured by clinical risk factors. PRS needs to quantify the ‘residual’ polygenic contribution. For this, we need alternative methods that capture the effects of SNPs in ways that allow relevant clinical predictors to mediate when their predisposing genes are included in the score. We may need methods that divide the PRS into multiple scores that each are optimized so clinical risk factors can act their predisposing role in pathways (Fig. 2C) (48–50).
Vassy and colleagues investigated pathway specific PRSs in type 2 diabetes (51). They predicted type 2 diabetes using a total PRS consisting of 62 SNPs, as well as using separate, non-overlapping PRSs based on SNPs associated with insulin resistance (10 SNPs) and beta-cell function (20 SNPs). Table 1 shows that the odds ratio of the beta-cell function PRSs were higher than those of the insulin resistance PRSs, which is explained by the fact that these scores included SNPs that had the highest ORs of all SNPs considered in the total PRS. In three different populations, the odds ratios of the overall and beta-cell PRSs remained unchanged after adjustment for clinical variables, but the ORs of all insulin resistance PRSs reduced. This underscores that clinical risk factors may mediate the association between PRS and the risk of disease when the construction or PRS allows for this mediating role.
Table 1.
A comparison of overall and pathway-specific polygenic risk scores in type 2 diabetes
| Model 1 | Model 2 | Model 3 | Model 4 | ||
|---|---|---|---|---|---|
| PRSt | PRSβ | PRSir | PRSβ | PRSir | |
| Framingham offspring study (n = 3471) | |||||
| Demographic model | 1.08 (1.06, 1.10) |
1.11 (1.08, 1.15) |
1.04 (1.00, 1.10) |
1.11 (1.08, 1.15) |
1.05 (1.00, 1.10) |
| Clinical model | 1.06 (1.04, 1.08) |
1.10 (1.06, 1.14) |
0.98 (0.93, 1.04) |
1.10 (1.06, 1.14) |
0.99 (0.93, 1.04) |
| CARDIA study, whites (n = 1650) | |||||
| Demographic model | 1.08 (1.04, 1.12) |
1.09 (1.02, 1.16) |
1.06 (0.96, 1.17) |
1.09 (1.02, 1.16) |
1.06 (0.96, 1.17) |
| Clinical model | 1.06 (1.02, 1.10) |
1.09 (1.02, 1.17) |
1.01 (0.91, 1.12) |
1.09 (1.02, 1.17) |
1.01 (0.91, 1.11) |
| CARDIA study, blacks (n = 820) | |||||
| Demographic model | 1.05 (1.01–1.09) |
1.06 (0.98, 1.14) |
1.09 (1.00, 1.20) |
1.06 (0.98, 1.14) |
1.10 (1.00, 1.20) |
| Clinical model | 1.05 (1.00–1.09) |
1.06 (0.99, 1.15) |
1.05 (0.96, 1.15) |
1.07 (0.99, 1.15) |
1.05 (0.96, 1.16) |
Data are obtained from (51). Values are odds ratios with 95% confidence intervals. Models 1–3 have one PRS in the model; model 4 includes both PRSβ and PRSir. PRS, polygenic risk score; PRSt, PRS total; PRSβ, PRS beta-cell function; PRSir, PRS insulin resistance; CARDIA study, Coronary Artery Risk Development in Young Adults study. Demographic models are adjusted for age and sex, and clinical models are additionally adjusted for parental history of diabetes, body mass index, systolic blood pressure, fasting plasma glucose, high-density lipoprotein and fasting triglycerides. Reprinted with permission from Jason L. Vassy, Marie-France Hivert, Bianca Porneala, Marco Dauriz, Jose C. Florez, Josée Dupuis, David S. Siscovickm Myriam Fornage, Laura J. Rasmussen-Torvik, Claude Bouchard and James B. Meigs: Polygenic Type 2 Diabetes Prediction at the Limit of Common Variant Detection, Diabetes 2014 Jun; 63 (6): 2172–2182: https://doi.org/10.2337/db13-1663. Copyright 2014 by the American Diabetes Association.
Unfortunately, Vassy et al. did not report the c-statistic as a measure of the discriminative ability for the prediction models that included either one or both pathway-specific PRSs. We do not know if and how considering multiple pathway-specific PRSs changed the c-statistic as in contrast to adding a single PRS to clinical risk factors. The c-statistic may be higher when pathways that are more heritable have PRSs with higher effect sizes that are then not reduced by variants with weaker effects from other pathways. This is illustrated by the data of Vassy et al., which showed that the beta-cell function PRS consistently had higher odds ratio than the total PRS (Table 1). It is also possible that separate PRSs lead to a lower c-statistic when part of the genetic effect is removed after adjustment for clinical risk factors. And it might be that the two approaches yield the same improvement in c-statistic when the contribution of the SNPs was minimal to begin with.
Finally, the extent to which clinical risk factors can mediate the association between SNPs and disease not only depends on how the PRS is constructed but also on the assessment of the clinical risk factors. Adequate assessment is a challenge when biomarkers that fluctuate over time are not measured timely and frequently enough to capture that variation. Such variations may occur based on daily rhythms or be induced by that week’s diet and other relevant lifestyle factors. The development of combined risk models should therefore not only focus on how to optimally assess the genetic contribution but also how to optimally measure the clinical risk factors.
GWAS weights
PRSs are typically constructed using weights from large GWASs. These effect sizes are preferred to using weights obtained from the study in which the PRS is investigated for the robustness of the estimates. Yet, taking weights from GWAS assumes that SNPs have the same impact on disease risk in all populations of the same ethnicity. This is unrealistic. First, GWAS estimates are pooled across multiple studies that differ in study design and study population and that may even differ in the diagnostic criteria and assessment of the disease of interest. The GWAS weight for each SNP may reflect none of the effect sizes of the individual studies. Second, the effect sizes may be overestimated because of winner’s curse and biases (52–54). Even when the identified GWAS hits are true positives, their effect sizes may be attenuated in other populations.
The overestimation of GWAS effect sizes is illustrated in the 23andMe’s white paper about its new PRS for type 2 diabetes (55). 23andMe compared its SNP weights with the weights of the genome-wide significant SNPs in the GWAS of Scott et al. (56). The sample size of the GWAS was about 160 000 individuals and of the 23andMe study about 940 000. Figure 3 shows that all effect sizes were in the same direction, but that most effect sizes of the GWAS were more extreme than the estimates from 23andMe. If the GWAS estimates were used to predict type 2 diabetes risk in the 23andMe population, poorer calibration of the PRS should be expected, especially at the tails of the risk distribution.
Figure 3.
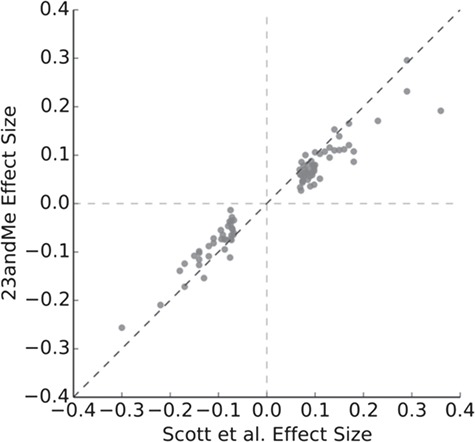
Per allele effect sizes for single-nucleotide polymorphisms in type 2 diabetes. Legend: Picture provided by 23andMe, reproduced from (55). The dots represent the genome-wide significant polymorphisms in the study of Scott et al. (56).
The choice of weights is relevant for the content validity of the PRS. If the PRS is intended as a measure of polygenic variance (Fig. 1), then it can be argued that the PRS needs to be designed such that it reflects the variation in the population that is studied. The weights of the SNPs, the point estimates of their effect sizes, are at the core of the PRS calculation, and their adequate estimation is important. Weights are typically obtained from GWAS, but this may not be the obvious choice when study populations are large enough to be used for estimating their own weights. These cohorts do not need to be used to identify SNPs, but they could be used for re-estimating or adjusting the GWAS effect sizes. We need more insight in the variation of effect sizes within populations of the same ethnicity if we want to understand the generalizability of PRSs (53,54).
Relaxing SNP inclusion thresholds
A recent trend in the construction of PRSs is to use millions of SNPs in the prediction of risks (27,57,58). These PRSs go beyond only including genome-wide significant SNPs from GWASs and beyond setting lower thresholds (higher P-values) than genome-wide significance for the selection of SNPs (7). These PRSs are constructed using methods like LDpred that optimize the weights for all SNPs using their GWAS weights, linkage disequilibrium and an estimate of the proportion of SNPs that are expected to have non-zero weights (8). The often millions of SNPs that are assumed to have zero weights are generally kept in the PRS calculation even when their weights are small and would have been zero if the weights would be quantified using, say, only three or four digits after the decimal point.
Most of these millions of SNPs have such negligible impact on risk that their inclusion in the score does not affect the predictions: excluding all SNPs except, say, the genome-wide significant SNPS or the top 1000 SNPs with the largest weights will unlikely change individual predictions (27,59–61). When Khera and colleagues constructed 30 PRSs using up to 7 million SNPs for each of five common diseases (27), most PRSs had lower c-statistics than the PRSs based on genome-wide significant variants only (19). Considering millions of SNPs that have negligible impact on disease risk might be a non-issue computationally, but it is not a non-issue if the scores need to reflect the underlying theoretical views on the genetic predisposition of the disease of interest.
Construct and content validity are about semantics and labeling. When the weights of ‘risk’ alleles are as low as 0.0000001 (and lower), do we consider these SNPs to be associated with disease risk, and should they be included in the PRS? When a PRS is constructed based on 2 million of SNPs of which, say, 100 are able to change predicted risks before the decimal, will we tell a patient that their risk is calculated using 2 million or 100 SNPs?
When researchers relax the SNP inclusion thresholds to include millions of SNPs, they often aim for and select the PRS with the highest proportion of explained variance or the highest c-statistic, even when the differences in these metrics are minimal and adding millions of SNPs is unlikely to change predicted risks for individuals (24,27,58,62). These minimally higher proportions of explained variance or minimal improvements of the c-statistic do not evidently translate into better health or more healthcare benefits. There may be no benefit for relaxing threshold beyond GWA significance (63), and there may be no good reason to go beyond the genome-wide significant SNPs for PRSs that are to be used in healthcare. If it is deemed valid to include millions of SNPs in PRSs, then we need to challenge ourselves to specify what is basis for this validity judgment.
Conclusion
PRSs do not ‘exist’ in the same way blood pressure and cholesterol level exist. The latter may be measured inaccurately, but blood flow has a pressure, and blood may contain more or less cholesterol. PRS is constructed, a pragmatic solution introduced when the number of SNPs became too large to be considered as separate variables in a regression analysis. PRS might be valid as an algorithm for predicting risk when used alone or in combination with variables we expect to be independent, such as age, sex and behavioral risk factors. Yet, when modeled together with clinical risk factors that are associated with its SNPs as intermediate phenotypes, the construction of PRS should be such that these risk factors can act as intermediate phenotypes, capturing the effects of the SNPs that predispose them.
PRSs do not ‘exist’ in the same way clinical risk models do not exist either. The validity of clinical risk models needs to be demonstrated, and the choices in the model development need to be justified (64). Clinical risk models need to be developed and externally validated in relevant settings so that they predict what they intend to predict in the population where the risk model is intended to be used. They need to be compared with other risk models that calculate the same risks using different algorithms. The demonstration of validity should be no less rigorous for PRS.
This paper aimed to reflect on the validity of PRSs by introducing the types of validity that are deemed important in the design of measurements, tests or questionnaires (20). PRSs have strong face validity; they intuitively seem to make sense, but this apparent face validity is not enough. More comparative research is needed to investigate the construct, content and criterion validity of PRS; to explore alternative ways of quantifying polygenic risk; and to rigorously compare new and current methods (3). A critical reflection of what needs to be measured by PRSs, from a theoretical perspective to assure their construct validity and from a practical perspective to assure their content validity, will help evaluating whether the PRSs that are constructed are the ones that were intended.
Models are simplifications of reality. They can be useful even when they are wrong. The same holds for PRSs, but we need to keep questioning if what we assess is what we think we do and to seek for alternative modeling strategies that might better reflect the underlying biological pathways. The construction of PRSs needs to acknowledge the biological reality, not create a new one.
Reference
- 1. De La Vega F.M. and Bustamante C.D. (2018) Polygenic risk scores: a biased prediction? Genome Med., 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Torkamani A., Wineinger N.E. and Topol E.J. (2018) The personal and clinical utility of polygenic risk scores. Nat Rev Genet., 19, 581–590. [DOI] [PubMed] [Google Scholar]
- 3. Nelson R.M., Pettersson M.E. and Carlborg O. (2013) A century after Fisher: time for a new paradigm in quantitative genetics. Trend Genet., 29, 669–676. [DOI] [PubMed] [Google Scholar]
- 4. Lyssenko V., Almgren P., Anevski D., Orho-Melander M., Sjogren M., Saloranta C., Tuomi T., Groop L. and Botnia Study G. (2005) Genetic prediction of future type 2 diabetes. PLoS Med., 2, e345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seddon J.M., George S., Rosner B. and Klein M.L. (2006) CFH gene variant, Y402H, and smoking, body mass index, environmental associations with advanced age-related macular degeneration. Hum Hered., 61, 157–165. [DOI] [PubMed] [Google Scholar]
- 6. Janssens A.C. and van Duijn C.M. (2009) Genome-based prediction of common diseases: methodological considerations for future research. Genome Med., 1, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. International Schizophrenia, C, Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O'Donovan M.C., Sullivan P.F. and Sklar P. (2009) Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature, 460, 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vilhjalmsson B.J., Yang J., Finucane H.K., Gusev A., Lindstrom S., Ripke S., Genovese G., Loh P.R., Bhatia G., Do R. et al. (2015) Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am J Hum Genet., 97, 576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martin A.R., Daly M.J., Robinson E.B., Hyman S.E. and Neale B.M. (2019) Predicting polygenic risk of psychiatric disorders. Biol Psychiat., 86, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roberts M.R., Asgari M.M. and Toland A.E. (2019) Genome-wide association studies and polygenic risk scores for skin cancer: clinically useful yet? Br J Dermatol., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fisher R.A. (2012) XV.—the correlation between relatives on the supposition of Mendelian inheritance. Trans R Soc Edinb., 52, 399–433. [Google Scholar]
- 12. Visscher P.M. and Wray N.R. (2015) Concepts and misconceptions about the polygenic additive model applied to disease. Hum Hered., 80, 165–170. [DOI] [PubMed] [Google Scholar]
- 13. Mavaddat N., Michailidou K., Dennis J., Lush M., Fachal L., Lee A., Tyrer J.P., Chen T.H., Wang Q., Bolla M.K. et al. (2019) Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am J Hum Genet., 104, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen G.B., Lee S.H., Montgomery G.W., Wray N.R., Visscher P.M., Gearry R.B., Lawrance I.C., Andrews J.M., Bampton P., Mahy G. et al. (2017) Performance of risk prediction for inflammatory bowel disease based on genotyping platform and genomic risk score method. BMC Med Genet., 18, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malik R., Bevan S., Nalls M.A., Holliday E.G., Devan W.J., Cheng Y.C., Ibrahim-Verbaas C.A., Verhaaren B.F., Bis J.C., Joon A.Y. et al. (2014) Multilocus genetic risk score associates with ischemic stroke in case-control and prospective cohort studies. Stroke, 45, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Genin E. and Clerget-Darpoux F. (2015) Revisiting the polygenic additive liability model through the example of diabetes mellitus. Hum Hered., 80, 171–177. [DOI] [PubMed] [Google Scholar]
- 17. Wray N.R. and Goddard M. (2010) Multi-locus models of genetic risk of disease. Genome Med., 2, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moonesinghe R., Khoury M.J., Liu T. and Janssens A.C. (2011) Discriminative accuracy of genomic profiling comparing multiplicative and additive risk models. Eur J Hum Genet., 19, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janssens A.C. and Joyner M.J. (2019) Polygenic risk scores that predict common diseases using millions of single nucleotide polymorphisms: is more, better? Clin Chem., 65, 609–611. [DOI] [PubMed] [Google Scholar]
- 20. Cronbach L.J. and Meehl P.E. (1955) Construct validity in psychological tests. Psychol Bull., 52, 281–302. [DOI] [PubMed] [Google Scholar]
- 21. Terwee C.B., Bot S.D.M., De Boer M.R., Van Der Windt D.A.W.M., Knol D.L., Dekker J., Bouter L.M. and De Vet H.C.W. (2007) Quality criteria were proposed for measurement properties of health status questionnaires. J Clin Epidemiol., 60, 34–42. [DOI] [PubMed] [Google Scholar]
- 22. Mosier C.I. (1947) A critical examination of the concepts of face validity. Educ Psychol Meas., 7, 191–205. [DOI] [PubMed] [Google Scholar]
- 23. Vachon C.M., Pankratz V.S., Scott C.G., Haeberle L., Ziv E., Jensen M.R., Brandt K.R., Whaley D.H., Olson J.E., Heusinger K. et al. (2015) The contributions of breast density and common genetic variation to breast cancer risk. J Nat Cancer Inst., 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abraham G., Havulinna A.S., Bhalala O.G., Byars S.G., De Livera A.M., Yetukuri L., Tikkanen E., Perola M., Schunkert H., Sijbrands E.J. et al. (2016) Genomic prediction of coronary heart disease. Eur Heart J., 37, 3267–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fritz J., Shiffman D., Melander O., Tada H. and Ulmer H. (2017) Metabolic mediators of the effects of family history and genetic risk score on coronary heart disease-findings from the malmo diet and cancer study. J Am Heart Assoc., 6, e005254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pereira A., Mendonca M.I., Borges S., Sousa A.C., Freitas S., Henriques E., Rodrigues M., Freitas A.I., Guerra G., Freitas C. et al. (2018) Additional value of a combined genetic risk score to standard cardiovascular stratification. Genet Mol Biol., 41, 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khera A.V., Chaffin M., Aragam K.G., Haas M.E., Roselli C., Choi S.H., Natarajan P., Lander E.S., Lubitz S.A., Ellinor P.T. et al. (2018) Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet., 50, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kathiresan S., Melander O., Anevski D., Guiducci C., Burtt N.P., Roos C., Hirschhorn J.N., Berglund G., Hedblad B., Groop L. et al. (2008) Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med., 358, 1240–1249. [DOI] [PubMed] [Google Scholar]
- 29. Paynter N.P., Chasman D.I., Pare G., Buring J.E., Cook N.R., Miletich J.P. and Ridker P.M. (2010) Association between a literature-based genetic risk score and cardiovascular events in women. JAMA, 303, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loos R.J.F. and Janssens A. (2017) Predicting polygenic obesity using genetic information. Cell Metab, 25, 535–543. [DOI] [PubMed] [Google Scholar]
- 31. Lukács Krogager M., Skals R.K., Appel E.V.R., Schnurr T.M., Engelbrechtsen L., Have C.T., Pedersen O., Engstrøm T., Roden D.M., Gislason G. et al. (2018) Hypertension genetic risk score is associated with burden of coronary heart disease among patients referred for coronary angiography. PLOS One, 13, e0208645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rutten-Jacobs L.C., Larsson S.C., Malik R., Rannikmae K., Consortium M., International Stroke Genetics, C, Sudlow C.L., Dichgans M., Markus H.S. and Traylor M. (2018) Genetic risk, incident stroke, and the benefits of adhering to a healthy lifestyle: cohort study of 306 473 UK Biobank participants. BMJ, 363, k4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morieri M.L., Gao H., Pigeyre M., Shah H.S., Sjaarda J., Mendonca C., Hastings T., Buranasupkajorn P., Motsinger-Reif A.A., Rotroff D.M. et al. (2018) Genetic tools for coronary risk assessment in type 2 diabetes: a cohort study from the ACCORD Clinical Trial. Diabetes Care, 41, 2404–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khera A.V., Emdin C.A., Drake I., Natarajan P., Bick A.G., Cook N.R., Chasman D.I., Baber U., Mehran R., Rader D.J. et al. (2016) Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med., 375, 2349–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Konttinen H., Llewellyn C., Silventoinen K., Joensuu A., Mannisto S., Salomaa V., Jousilahti P., Kaprio J., Perola M. and Haukkala A. (2018) Genetic predisposition to obesity, restrained eating and changes in body weight: a population-based prospective study. Int J Obes. (Lond), 42, 858–865. [DOI] [PubMed] [Google Scholar]
- 36. Calvin C.M., Hagenaars S.P., Gallacher J., Harris S.E., Davies G., Liewald D.C., Gale C.R. and Deary I.J. (2019) Sex-specific moderation by lifestyle and psychosocial factors on the genetic contributions to adiposity in 112,151 individuals from UK Biobank. Sci Rep., 9, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ericson U., Hindy G., Drake I., Schulz C.-A., Brunkwall L., Hellstrand S., Almgren P. and Orho-Melander M. (2018) Dietary and genetic risk scores and incidence of type 2 diabetes. Genes Nutr., 13, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee A., Mavaddat N., Wilcox A.N., Cunningham A.P., Carver T., Hartley S., Babb de Villiers C., Izquierdo A., Simard J., Schmidt M.K. et al. (2019) BOADICEA: a comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet Med, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Musliner K.L., Seifuddin F., Judy J.A., Pirooznia M., Goes F.S. and Zandi P.P. (2015) Polygenic risk, stressful life events and depressive symptoms in older adults: a polygenic score analysis. Psychol Med., 45, 1709–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meigs J.B., Shrader P., Sullivan L.M., McAteer J.B., Fox C.S., Dupuis J., Manning A.K., Florez J.C., Wilson P.W., D'Agostino R.B. Sr. et al. (2008) Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med., 359, 2208–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Velthorst E., Froudist-Walsh S., Stahl E., Ruderfer D., Ivanov I., Buxbaum J., iPsych-Broad Asd Group, t.I.c, Banaschewski T., Bokde A.L.W. and Dipl-Psych, U.B. et al. (2018) Genetic risk for schizophrenia and autism, social impairment and developmental pathways to psychosis. Transl Psychiatry, 8, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Belsky D.W., Moffitt T.E., Houts R., Bennett G.G., Biddle A.K., Blumenthal J.A., Evans J.P., Harrington H., Sugden K., Williams B. et al. (2012) Polygenic risk, rapid childhood growth, and the development of obesity: evidence from a 4-decade longitudinal study. Arch Pediatr Adolesc Med., 166, 515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morris T.T., Davies N.M. and Davey Smith G. (2019) Can education be personalised using pupils’ genetic data? bioRxiv, 645218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Janssens A.C. and Duijn C.M. (2008) Genome-based prediction of common diseases: advances and prospects. Hum Mol Genet., 17, R166–R173. [DOI] [PubMed] [Google Scholar]
- 45. Pearl J. (1995) Causal diagrams for empirical research. Biometrika, 82, 669–688. [Google Scholar]
- 46. Hernan M.A., Hernandez-Diaz S., Werler M.M. and Mitchell A.A. (2002) Causal knowledge as a prerequisite for confounding evaluation: an application to birth defects epidemiology. Am J Epidemiol., 155, 176–184. [DOI] [PubMed] [Google Scholar]
- 47. Konigorski S., Wang Y., Cigsar C. and Yilmaz Y.E. (2017) Estimating and testing direct genetic effects in directed acyclic graphs using estimating equations. Genet Epidemiol., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang K., Li M. and Hakonarson H. (2010) Analysing biological pathways in genome-wide association studies. Nat Rev Genet., 11, 843–854. [DOI] [PubMed] [Google Scholar]
- 49. Cantor R.M., Lange K. and Sinsheimer J.S. (2010) Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am J Hum Genet., 86, 6–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang L., Jia P., Wolfinger R.D., Chen X. and Zhao Z. (2011) Gene set analysis of genome-wide association studies: methodological issues and perspectives. Genomics, 98, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vassy J.L., Hivert M.F., Porneala B., Dauriz M., Florez J.C., Dupuis J., Siscovick D.S., Fornage M., Rasmussen-Torvik L.J., Bouchard C. et al. (2014) Polygenic type 2 diabetes prediction at the limit of common variant detection. Diabetes, 63, 2172–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shi J., Park J.-H., Duan J., Berndt S.T., Moy W., Yu K., Song L., Wheeler W., Hua X., Silverman D. et al. (2016) Winner’s curse correction and variable thresholding improve performance of polygenic risk modeling based on genome-wide association study summary-level data. PLOS Genet., 12, e1006493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Berg J.J., Harpak A., Sinnott-Armstrong N., Joergensen A.M., Mostafavi H., Field Y., Boyle E.A., Zhang X., Racimo F., Pritchard J.K. et al. (2019) Reduced signal for polygenic adaptation of height in UK Biobank. eLife, 8, e39725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sohail M., Maier R.M., Ganna A., Bloemendal A., Martin A.R., Turchin M.C., Chiang C.W., Hirschhorn J., Daly M.J., Patterson N. et al. (2019) Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. eLife, 8, e39702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Multhaup M.L., Kita R., Krock B., Eriksson N., Fontanillas P., Aslibekyan S., Del Gobbo L., Shelton J.F., Tennen R.I., Lehman A. et al. (2019) Estimating the likelihood of developing type 2 diabetes with polygenic models In White Paper: the Science Behind 23andMe’s Type 2 Diabetes Report.
- 56. Scott R.A., Scott L.J., Magi R., Marullo L., Gaulton K.J., Kaakinen M., Pervjakova N., Pers T.H., Johnson A.D., Eicher J.D. et al. (2017) An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes, 66, 2888–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Inouye M., Abraham G., Nelson C.P., Wood A.M., Sweeting M.J., Dudbridge F., Lai F.Y., Kaptoge S., Brozynska M., Wang T. et al. (2018) Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J Am Coll Cardiol., 72, 1883–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Khera A.V., Chaffin M., Wade K.H., Zahid S., Brancale J., Xia R., Distefano M., Senol-Cosar O., Haas M.E., Bick A. et al. (2019) Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell, 177, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fritsche L.G., Beesley L.J., Vandehaar P., Peng R.B., Salvatore M., Zawistowski M., Gagliano Taliun S.A., Das S., Lefaive J., Kaleba E.O. et al. (2019) Exploring various polygenic risk scores for skin cancer in the phenomes of the Michigan genomics initiative and the UK Biobank with a visual catalog: PRSWeb. PLOS Genet., 15, e1008202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Allegrini A.G., Selzam S., Rimfeld K., Von Stumm S., Pingault J.B. and Plomin R. (2019) Genomic prediction of cognitive traits in childhood and adolescence. Mol Psychiatr., 24, 819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chung W., Chen J., Turman C., Lindstrom S., Zhu Z., Loh P.-R., Kraft P. and Liang L. (2019) Efficient cross-trait penalized regression increases prediction accuracy in large cohorts using secondary phenotypes. Nat Comm., 10, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee J.J., Wedow R., Okbay A., Kong E., Maghzian O., Zacher M., Nguyen-Viet T.A., Bowers P., Sidorenko J., Karlsson Linnér R. et al. (2018) Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet., 50, 1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rosenberg M.A., Lubitz S.A., Lin H., Kosova G., Castro V.M., Huang P., Ellinor P.T., Perlis R.H. and Newton-Cheh C. (2017) Validation of polygenic scores for qt interval in clinical populations. Circ Cardiovasc Genet., 10, e001724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Moons K.G., Altman D.G., Reitsma J.B., Ioannidis J.P., Macaskill P., Steyerberg E.W., Vickers A.J., Ransohoff D.F. and Collins G.S. (2015) Transparent reporting of a multivariable prediction model for Individual Prognosis or Diagnosis (TRIPOD): explanation and elaboration. Ann Intern Med., 162, W1–W73. [DOI] [PubMed] [Google Scholar]