

Abstract
Activation of the P2X7 receptor (P2X7R) has been found to increase expression of tumor necrosis factor-α (TNF-α) in the joints and synovial lining of patients with rheumatoid arthritis (RA). Increased expression of TNF-α promotes joint destruction through deterioration of type II collagen by matrix metalloproteinases (MMPs), expression of proinflammatory cytokines, oxidative stress, and activation of cellular signaling pathways. In the present study, we exposed fibroblast-like synoviocytes (FLSs) to TNF-α in the presence and absence of the P2X7R antagonist A804598. We then employed real time PCR and western blot analysis to analyze the mRNA and protein expression levels of P2X7R in both control and RA-FLSs. We confirmed that P2X7R is expressed on FLSs and is upregulated in RA-FLSs and FLSs exposed to TNF-α. Importantly, we also demonstrate the ability of P2X7R antagonism using A804598 to suppress oxidative stress, expression of interleukin (IL)-1β, IL-6, MMP-1, MMP-3, MMP-13 as well as activation of the Janus family of tyrosine kinase/signal transducer and activator of transcription (JAK1/STAT3) proinflammatory signaling pathway. These findings implicate a novel role of antagonism of P2X7R as a target for the treatment and prevention of RA.
Keywords: Rheumatoid arthritis, P2X7 receptor, matrix metalloproteinase (MMP), Janus family of tyrosine kinase/signal transducer and activator of transcription (JAK1/STAT3) pathway, fibroblast-like synoviocytes (FLSs)
Introduction
Rheumatoid arthritis (RA) is a systemic inflammatory autoimmune disease that affects approximately 350 million people worldwide. RA causes chronic inflammation and pain in the joints with symptoms including fever, fatigue, stiffness and redness in the joints, asymmetrical presentation of symptoms, nodules on the joints, and in advanced cases, can cause loss of mobility and range of motion [1]. Fibroblast-like synoviocytes (FLSs) are found in the synovial membrane in joints. In normal functioning joints, FLSs work by adhering the tissue of the synovial membrane together to create a thin, two-cell bilayer against the bone. They also secrete a protein called lubricin, which assists in the smooth articulation between joints [2]. RA-FLSs express subdued contact inhibition. The resulting increased proliferation causes hypertrophy of the synovial lining, with the normal bilayer of cells becoming as much as ten to twenty cells thick. The accompanying increased inflammation promotes the differentiation of osteoclasts into an invasive phenotype which then infiltrates the articular cartilage, causing deterioration of the joint and bone erosion near the articulation points [1,3]. FLSs mediated inflammation has been identified as an important hallmark of RA pathogenesis [4]. FLSs play a casual role in regulating the expression and secretion of inflammatory mediators including matrix metalloproteinases (MMPs), which result in chondrocyte dysfunction and matrix degradation [5]. Pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and IL-1β are important for the initiation and progression of RA [6]. Biologic antirheumatic drugs targeting these proinflammatory cytokines have been successfully developed and can significantly improve treatment response [7].
The P2X7 ligand-gated ion channel receptor (P2X7R) is ubiquitously expressed. Upon stimulation, P2X7R triggers the cell membrane to intake calcium and sodium ions and release potassium ions. The passing of ions across the cell membrane causes the permeability to become increasingly non-select, until a large non-select pore is formed in the cell. This pore allows for the free passing of large molecules to cross the cell membrane, most notably in phagocytic immune cells [8]. P2X7R stimulation occurs with abundant amounts of ATP in the extracellular matrix. In certain cell types, this causes the release of pro-inflammation cytokines including tumor necrosis factor-α (TNF-α) [9,10]. TNF-α is considered a master regulator of pro-inflammatory cytokines and has been shown to play a major role in initiating joint destruction in RA [11-13]. TNF-α has also been shown to induce expression of matrix metalloproteinase (MMP) enzymes, which break down proteins in the extracellular matrix including type II collagen and aggrecan [14]. Additionally, TNF-α activates the Janus family of tyrosine kinase (JAK)/signal transducer and activator of transcription (STAT) kinase pathway which phosphorylates intracellular targets leading to inflammation and degradation [15]. TNF-α also contributes to bone emulsion through differentiation of osteoclasts and increased production of FLSs. This excessive proliferation of FLSs causes an increase in protease, thereby assisting in the destruction of cartilage [16,17].
The selective P2X7R antagonist A804598 ([3H] 2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) has an equally high affinity for rat, mouse and human P2X7R [18]. In recent years, modulation of P2X7R has been receiving attention as a potential therapeutic target for the treatment of infections, inflammation, cancers, and autoimmune, neurological, and musculoskeletal disorders [19]. In the present study, we determined the effects of P2X7R antagonism by A804598 on various aspects of RA in TNF-α-induced FLSs. Our findings demonstrate an important potential role of P2X7R antagonism by A804598 in the treatment and prevention of RA.
Materials and methods
Fibroblast-like synoviocytes (FLSs) isolation, culture, and treatment
Normal FLSs and RA-FLSs were separated from knee joint synovial tissues from both control (n = 15) and RA patients (n = 8) donors as previously demonstrated [20]. Human tissues were used in accordance with the World Medical Association Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects. This study was approved by the ethics committee of our institute. Written informed consent was signed by all participants in this study. Synovial tissues were cut into small pieces and digested with 0.05% trypsin for 15 min at 37°C. Isolated FLSs were cultured in DMEM supplemented with 5% fetal bovine serum (FBS) and 1% penicillin-streptomycin. To determine the effects of TNF-α on the expression of P2X7R, FLSs were stimulated with 5, 10, and 20 ng/ml TNF-α for 24 h. The P2X7 antagonist A804598 was purchased from Tocris Bioscience, UK and dissolved in dimethyl Sulfoxide (DMSO). To determine the effects of P2X7R on TNF-α-induced insult in FLSs, cells were treated with 10 ng/ml TNF-α in the presence or absence of A804598 at the concentrations of 10 and 20 μM [21] for 24 h.
Semi-quantitative and quantitative polymerase chain reaction (PCR) analysis
Total RNA was isolated from FLSs using a commercial RNA Extraction Kit (#74004, Qiagen). Extracted RNA was examined using a Nanodrop spectrophotometer. Then, 1 µg RNA from each group (test and control groups) was applied to generate cDNA via reverse transcription polymerase chain reaction (RT-PCR) using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, USA). The produced cDNA was then subjected to real-time PCR using the SYBR Green method using a commercial qPCR Master Mix Kit (Thermo Fisher Scientific, USA) to assess the amount of mRNA of human P2X7R, IL-1β, IL-6, MMP-1, MMP-3, and MMP-13. The following primers were used in this study: P2X7R (forward, 5’-GAGGTGACGGAGAATGTC-3’; reverse, 5’-GCGCCTGGGATACTCAG-3’); IL-6 (forward, 5’-GGTACATCCTCGACGGCATCT-3’; reverse, 5’-GTGCCTCTTTGCTGCTTTCAC-3’); IL-1β (forward, 5’-TTCCTGTTGTCTACACCAATGC-3’; reverse, 5’-CGGGCTTTAAGTGAGTAGGAGA-3’); MMP-1 (forward, 5’-CTG AAG GTG ATG AAG CAG CC-3’, reverse, 5’-AGTCCA AGA AATGGCCGAG-3’); MMP-3 (forward: 5’-TTAAAATAAAACTGCTTTT-3’ and reverse: 5’-AACTGGAGCATTTTTT-3’); MMP-13 (forward: 5’-AGG AGC ATG GCG ACT TCT AC-3’; reverse: 5’-TAAAAACAGCTCCGCATCAA-3’); GAPDH (forward, 5’-GGAGCGAGATCCCTCCAAAAT-3’; reverse, 5’-GGCTGTTGTCATACTTCTCATGG-3’).
Western blot analysis
Proteins from FLSs in the different treatment groups were isolated using cell lysis buffer with cocktail inhibitors against protease and phosphatase. The sample concentration was determined. After boiling at 95°C for 5 min, proteins were immobilized by 10% sodium dodecyl sulphate - polyacrylamide gel electrophoresis (SDS-PAGE). The separated samples were then transferred onto polyvinylidene fluoride (PVDF) membranes. Blots were blocked with 10% skim milk for 1 h at room temperature (RT) and incubated with primary antibody at 4°C overnight. Membranes were then washed 3 times with tris-buffered saline (TBS) in tween 20 (TBST) and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at RT. Immunoblotting bands were visualized with a chemiluminescence development kit. The following antibodies were used in this study: P2X7R (1:2000, #ab109054, Abcam, USA); p-JAK1 (1:1000, #3331, Cell signaling technology, USA); JAK1 (1:2000, #29261, Cell signaling technology, USA); p-STAT3 (1:1000, #9145, Cell signaling technology, USA); STAT3 (1:1000, #9139, Cell signaling technology, USA); Anti-rabbit IgG, HRP-linked Antibody (1:2000, #7074, Cell signaling technology, USA); Anti-mouse IgG, HRP-linked Antibody (1:2000, #7072, Cell signaling technology, USA).
Enzyme-linked immunosorbent assay (ELISA)
FLSs were treated with 10 ng/ml TNF-α in the presence or absence of A804598 at the concentrations of 10 and 20 μM for 24 h. Cell culture medium was collected to determine the protein levels of IL-1β and IL-6. Cell lysate was then prepared to determine the expression of MMP-1, MMP-3, and MMP-13 using commercial kits from R&D Systems. Briefly, 50 μL of assay diluent buffer was added to each well. 50 μl supernatant was then pipetted into each well of the ELISA plate and incubated for 2 h at room temperature (RT). After 4 washes, 100 μL of conjugated antibody was added and incubated for 2 h on the shaker. 200 μL of substrate solution was then added to each well and incubated for 30 min at RT on the benchtop. The reaction was stopped with 50 μL stop solution. Optical density (OD) value was recorded at 450 nm within 30 min to index protein concentration.
Measurement of reactive oxygen species (ROS)
FLSs were treated with 10 ng/ml TNF-α in the presence or absence of A804598 at the concentrations of 10 and 20 μM for 24 h. The dye 2’,7’-dichlorofluorescin diacetate (DCFH-DA) was used to examine intracellular ROS. Briefly, after the indicated treatment, cells were washed 3 times with PBS and loaded with 5 μM DCFH-DA for 30 min in darkness. Cells were then washed 3 times, and fluorescent signals were recorded using a DM500 fluorescence microscope (Leica Microsystems, Germany). Fluorescent intensity of ROS staining was quantified using the Image J software. Briefly, regions of interest (ROI) in the fluorescent figures were defined and the average number of cells present in the defined ROI was counted. The integrated density value (IDV) in ROI was calculated. The IDV was divided by the average number of cells and was used to index the average level of intracellular ROS.
Measurement of intracellular reduced glutathione (GSH)
Reduced GSH in FLSs was assessed using the fluorimetric method as previously described [22]. FLSs were seeded onto a 6-well plate and maintained in normal medium for 12 h. Cells were treated with 10 ng/ml TNF-α in the presence or absence of A804598 at the concentrations of 10 and 20 μM for 24 h. Cell lysate was prepared and briefly centrifuged at 15,000 × g at 4°C for 15 min. Equal volumes (100 µL) of the supernatant and ortho-phthaldehyde (0.1% w/v in methanol) were mixed together in 1.8 ml of 100 mM Na2HPO4 and incubated for 20 min at RT. Fluorescent signals were recorded using a spectrofluorometer (emission: 420 nm; excitation: 350 nm).
Statistical analysis
Experimental results are presented as means ± standard error of measurement (S.E.M.). The software SPSS 16.0 was used to perform statistical analysis. Statistical significance was determined using analysis of variance (ANOVA), followed by the Bonferroni post-hoc test. A difference of P < 0.05 was considered statistically significant.
Results
First, we confirmed that P2X7R is expressed in human FLSs at the protein level using β-actin as a control. Human Caco-2 cells were used as a positive control as previously described [23] (Figure 1). Next, we determined whether expression of P2X7R is upregulated in RA-FLSs as both the mRNA and protein levels using PCR and western blot analyses, respectively. As shown in Figure 2, RA-FLSs had roughly 4-fold higher expression of P2X7R at the mRNA level as compared to normal FLSs (Figure 2A). Additionally, RA-FLSs had approximately 3.75-fold higher expression of P2X7R at the protein level (P < 0.01) (Figure 2B). As demonstrated by the results of real-time PCR and western blot analyses in Figure 3 and using β-actin as a control, stimulation of FLSs with 5, 10 and 20 ng/mL TNF-α resulted in approximately 2-, 3-, and 4-fold higher expression of P2X7R at both the mRNA and protein levels as compared to the control. These findings demonstrate that P2X7R is expressed on both normal and RA-FLSs, with RA-FLSs exhibiting significantly higher P2X7R expression. Additionally, insult from TNF-α greatly increased expression of P2X7R in a dose-dependent manner.
Figure 1.

The P2X7 receptor (P2X7R) is expressed in human fibroblast-like synoviocytes (FLSs). Human Caco-2 cells were used as a positive control. A. RT-PCR analysis of P2X7R; B. Western blot analysis of P2X7R.
Figure 2.

The expression of P2X7R is increased in rheumatoid arthritis (RA)-FLSs compared to normal FLSs. A. Real-time PCR analysis of P2X7R at the mRNA level; B. Western blot analysis of P2X7R at the protein level (*, P < 0.01 vs. normal group).
Figure 3.

The expression of P2X7R in normal FLSs is increased in response to TNF-α treatment. FLSs were stimulated with various concentrations (5, 10, 20 ng/ml) TNF-α for 24 h. A. mRNA of P2X7R was determined by real-time PCR analysis; B. Protein of P2X7R was determined by western blot analysis (#, &, *, P < 0.01 vs. previous column group).
Next, we investigated the effects of the addition of the P2X7R antagonist A804598 on two major markers of oxidative stress in FLSs-production of ROS and reduced GSH antioxidant. The molecular structure of A804598 is shown in Figure 4. FLSs were exposed to insult from 10 ng/mL TNF-α in the presence or absence of 10 and 20 µM A804598 for 24 h. As demonstrated by the results of DCFH-DA in Figure 5A, TNF-α gave rise to an increase in intracellular ROS of more than 4-fold basal levels, which was ameliorated by treatment with 10 and 20 µM A804598 in a dose-dependent manner. Additionally, the results in Figure 5B show that TNF-α reduced the level of GSH by roughly half, which was also rescued by treatment with 10 and 20 µM A804598 in a dose-dependent manner. These findings demonstrate a powerful antioxidant property of A804598 against TNF-α-induced oxidative stress.
Figure 4.

Molecular structure of the P2X7 receptor (P2X7R) antagonist A804598.
Figure 5.

The P2X7 receptor (P2X7R) antagonist A804598 ameliorated TNF-α-induced oxidative stress in human FLSs. FLSs were treated with 10 ng/ml TNF-α in the presence or absence of A804598 at the concentrations of 10 and 20 μM for 24 h. A. Intracellular reactive oxygen species (ROS) was measured by DCFH-DA; Scale bar, 100 μm; B. Intracellular GSH levels (#, &, *, P < 0.01 vs. previous column group).
Increased expression of IL-1β and IL-6 plays a major role in the pathogenesis of RA. To determine the effects of A804598 on TNF-α-induced upregulation of IL-1β and IL-6, we exposed FLSs to insult from 10 ng/mL TNF-α for 24 h. As shown in Figure 6A, TNF-α caused an increase in the expression of IL-1β and IL-6 at the mRNA level by approximately 4.5- and 4.75-fold, respectively, which was ameliorated by treatment with 10 and 20 µM A804598 in a dose-dependent manner. Concordantly, the results of ELISA analysis in Figure 6B show that protein expression of IL-1β and IL-6 was increased by approximately 4.5- and 4-fold upon exposure to TNF-α, which was ameliorated by 10 and 20 µM A804598 in a dose-dependent manner. These findings demonstrate the involvement of P2X7R expression in TNF-α-mediated upregulation of IL-1β and IL-6. Therefore, blockade of P2X7R by its antagonist A804598 may be a novel treatment against expression of proinflammatory cytokines in RA.
Figure 6.

The P2X7 receptor (P2X7R) antagonist A804598 inhibited TNF-α-induced production of IL-1β and IL-6 in human FLSs. FLSs were treated with 10 ng/ml TNF-α in the presence or absence of A804598 at the concentrations of 10 and 20 μM for 24 h. A. Real-time PCR analysis of IL-1β and IL-6; B. Secretions of IL-1β and IL-6 determined by ELISA (#, &, *, P < 0.01 vs. previous column group).
Degradation of the cartilage extracellular matrix is largely driven by overexpression of degradative enzymes such as MMP-1, MMP-3 and MMP-13 and plays a vital role in the pathogenesis of RA. To determine the effects of P2X7R on TNF-α-induced upregulation of MMPs, FLSs were exposed to 10 ng/mL TNF-α in the presence or absence of 10 and 20 µM A804598 for 24 h. As demonstrated by the results of real-time PCR analysis in Figure 7A, TNF-α insult led to increases in the expressions of MMP-1, MMP-3 and MMP-13 of roughly 4.8-, 4.7, and 4.5-fold, respectively, at the mRNA level. However, treatment with 10 and 20 µM A804598 rescued the expression of these three MMPs to roughly 2-fold basal levels. Additionally, the results in Figure 7B show that this effect was also present at the protein level, as demonstrated by ELISA analysis. These findings implicate a potential role of P2X7R antagonism using A804598 in preventing cartilage degradation by downregulating TNF-α-induced expression of MMP-1, MMP-3 and MMP-13 in FLSs.
Figure 7.
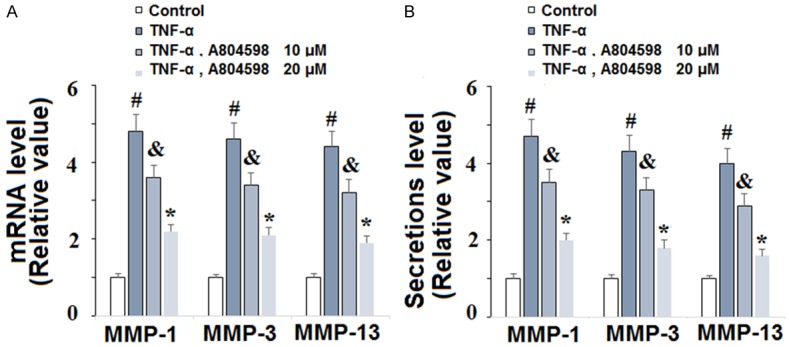
The P2X7 receptor (P2X7R) antagonist A804598 reduced TNF-α-induced production of MMP-1, MMP-3, and MMP-13 in human FLSs. FLSs were treated with 10 ng/ml TNF-α in the presence or absence of A804598 at the concentrations of 10 and 20 μM for 24 h. A. Real-time PCR analysis of MMP-1, MMP-3, and MMP-13; B. Secretions of MMP-1, MMP-3, and MMP-13 determined by ELISA (#, &, *, P < 0.01 vs. previous column group).
Finally, we explored the role of P2X7R antagonism in TNF-α-induced activation of the JAK1/STAT3 proinflammatory pathway in FLSs. Activation of JAK1/STAT3 in RA-FLSs by IL-6 has been shown to promote differentiation into the inflammatory Th17 phenotype [24]. To determine the involvement of P2X7R in activation of JAK1/STAT3 signaling, we exposed FLSs to 10 ng/mL TNF-α in the presence or absence of 10 and 20 µM A804598 for 2 h. Using β-actin as a control, the results of western blot analysis in Figure 8 show that TNF-α induced significant phosphorylation of both JAK1 and STAT3 protein by approximately 5-fold basal levels, while total JAK1 and STAT3 remained constant. However, treatment with A804598 could ameliorate TNF-α-induced phosphorylation of JAK1/STAT3 in a dose-dependent manner, with the higher dose reducing p-JAK1/STAT3 to roughly 2-fold basal levels. These findings suggest a novel role of A804598 in ameliorating inflammation via modulation of the JAK1/STAT3 signaling pathway.
Figure 8.

The P2X7 receptor (P2X7R) antagonist A804598 reduced TNF-α-induced activation of JAK1/STAT3 in human FLSs. FLSs were treated with 10 ng/ml TNF-α in the presence or absence of A804598 at the concentrations of 10 and 20 μM for 2 h. Phosphorylated JAK1 and STAT3 was determined by western blot analysis (#, &, *, P < 0.01 vs. previous column group).
Discussion
The inflammatory pathways involved in the pathogenesis of RA are numerous and intricate. Current medication treatments focus on symptom reduction through the use of NSAIDs and corticosteroids, and on slowing disease progression through the use of disease-modifying antirheumatic drugs (DMARs) and a newer class of DMARs called biologic agents [24]. Current biologic agents target TNF-α cytokine inhibition utilizing monoclonal antibodies [25]. However, the majority of patients eventually develop resistance to these therapies, and thus new and novel treatment strategies are desperately needed [26]. In the present study, we identified a novel role of antagonism of P2X7R using its specific antagonist A804598. Our findings demonstrate an important role of P2X7R in driving TNF-α-induced inflammation in RA.
Among the many factors driving the development and progression of RA, recent studies have highlighted the importance of oxidative stress resulting from increased generation of ROS and decreased intracellular levels of the antioxidant GSH in RA [27,28]. Our findings demonstrate that antagonism of P2X7R can modulate overproduction of ROS and suppression of GSH induced by TNF-α in FLSs, thereby suggesting P2X7R as a potential target for regulating oxidative stress in RA. While there are numerous factors contributing to chronic inflammation as seen in RA, a contemporary study suggested inhibition of IL-1β and IL-6 as a potential therapeutic target in RA [29]. IL-1β has been shown to play a pivotal role in bone resorption via stimulation of osteoclastogenesis by receptor activator of nuclear factor kappa-B ligand (RANKL), while IL-6 is considered a preferred target for preventing inflammation of RA patients who have only partial response to TNF-α inhibitors [30,31]. Here, we demonstrate that antagonism of P2X7R exerts powerful suppression of IL-1β and IL-6 at both the mRNA and protein levels in FLSs stimulated with TNF-α, thus implying the potential of P2X7R antagonism using A804598 as a novel therapy for RA, especially in patients with partial or no response to TNF-α inhibitors. TNF-α is well-recognized as an activator of MMP enzyme expression, which plays a major role in joint destruction in RA [32-34]. In the present study, we show that antagonism of P2X7R significantly inhibits expression of MMP-1, MMP-3 and MMP-13. While MPP-1 and MMP-3 are the major MMPs produced in fibroblasts and macrophage-like synoviocytes, these two enzymes not only drive degradation of cartilage and subchondral bone, but also induce expression of other MMPs including MMP-13 [35]. MMP-13 plays a pivotal role in cartilage degradation in RA, with one experiment reporting a 75% reduction in cartilage destruction after inhibition of MMP-13 in an RA mouse model [36]. Our findings demonstrate a powerful ability of P2X7R antagonism to reduce expression of MMP-1, MMP-3 and MMP-13 induced by TNF-α, with the higher dose of A804598 reducing expression of these enzymes by nearly 75%. Finally, we show that A804598 also significantly reduced TNF-α-induced phosphorylation of JAK1/STAT3. Activation of the JAK1/STAT3 cellular signaling pathway plays a critical role in IL-6 signaling and blockade of JAK1/STAT3 signaling has recently been suggested as a promising target for the treatment of RA [37,38]. The findings of the present study demonstrate a strong capacity of P2X7R antagonism by A804598 to prevent phosphorylation of JAK1/STAT3, with the higher dose reducing the level of p-JAK1/STAT3 by roughly 75%. Thus, selective inhibition of P2X7R activation by A804598 may serve as a novel therapeutic strategy for preventing oxidative stress, expression of key proinflammatory cytokines, cartilage degradation, and activation of the proinflammatory JAK1/STAT3 signaling pathway induced by TNF-α in RA.
The major limitation of the present study is that our findings are based on an in vitro primary FLSs culture model. However, it should be realized that the pathological mechanisms of RA are complex. In this study, we treated FLSs with TNF-α to induce inflammatory conditions. However, there are a variety of risk factors involved in the pathological development of RA, including aging, genetics, and joint injuries. Additionally, other pro-inflammatory cytokines such as interleukin-1β (IL-1β) and IL-6 are associated with the pathogenesis of RA. Animal models have been widely used to explore the pathological mechanisms and therapy of RA. Therefore, future in vivo investigations will further verify the biological function of P2X7R in RA.
Acknowledgements
This research was supported by the National Nature Science Foundation of China (81873990, 81873991 and 81672238), the Jiangsu Provincial Medical Youth Talent (QNRC2016751), and the Natural Science Foundation of Jiangsu province (BK20180001).
Disclosure of conflict of interest
None.
References
- 1.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 2.Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233:233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arend WP. The pathophysiology and treatment of rheumatoid arthritis. Arthritis Rheum. 1997;40:595–597. doi: 10.1002/art.1780400402. [DOI] [PubMed] [Google Scholar]
- 4.Doody KM, Bottini N, Firestein GS. Epigenetic alterations in rheumatoid arthritis fibroblast-like synoviocytes. Epigenomics. 2017;9:479–492. doi: 10.2217/epi-2016-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korb-Pap A, Bertrand J, Sherwood J, Pap T. Stable activation of fibroblasts in rheumatic arthritis-causes and consequences. Rheumatology. 2016;55(Suppl 2):ii64–ii67. doi: 10.1093/rheumatology/kew347. [DOI] [PubMed] [Google Scholar]
- 6.Chen Z, Bozec A, Ramming A, Schett G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat Rev Rheumatol. 2019;15:9–17. doi: 10.1038/s41584-018-0109-2. [DOI] [PubMed] [Google Scholar]
- 7.Furst DE, Emery P. Rheumatoid arthritis pathophysiology: update on emerging cytokine and cytokine-associated cell targets. Rheumatology (Oxford) 2014;53:1560–1569. doi: 10.1093/rheumatology/ket414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 9.Sekar P, Huang DY, Chang SF, Lin WW. Coordinate effects of P2X7 and extracellular acidification in microglial cells. Oncotarget. 2018;9:12718. doi: 10.18632/oncotarget.24331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karasawa A, Kawate T. Structural basis for subtype-specific inhibition of the P2X7 receptor. Elife. 2016;5 doi: 10.7554/eLife.22153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barberà-Cremades M, Gómez AI, Baroja-Mazo A, Martínez-Alarcón L, Martínez CM, de Torre-Minguela C, Pelegrín P. P2X7 receptor induces tumor necrosis factor-α converting enzyme activation and release to boost TNF-α production. Front Immunol. 2017;8:862. doi: 10.3389/fimmu.2017.00862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parameswaran N, Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20:87–103. doi: 10.1615/critreveukargeneexpr.v20.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu H, He Y, Yang X, Liang L, Zhan Z, Ye Y, Yang X, Lian F, Sun L. Anti-malarial agent artesunate inhibits TNF-α-induced production of proinflammatory cytokines via inhibition of NF-κB and PI3 kinase/Akt signal pathway in human rheumatoid arthritis fibroblast-like synoviocytes. Rheumatology (Oxford) 2007;46:920–926. doi: 10.1093/rheumatology/kem014. [DOI] [PubMed] [Google Scholar]
- 14.Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol. 2007;82:1375–1381. doi: 10.1189/jlb.0607338. [DOI] [PubMed] [Google Scholar]
- 15.Miscia S, Marchisio M, Grilli A, Valerio VD, Centurione L, Sabatino G, Garaci F, Zauli G, Bonvini E, Baldassarre AD. Tumor necrosis factor a (TNF-a) activates Jak1/Stat3-Stat5B signaling through TNFR-1 in human B cells. Cell Growth Differ. 2002;13:13–18. [PubMed] [Google Scholar]
- 16.Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106:1481–1488. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM. Mechanisms of TNF-α- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest. 2003;111:821–831. doi: 10.1172/JCI16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donnelly-Roberts DL, Namovic MT, Surber B, Vaidyanathan SX, Perez-Medrano A, Wang Y, Carroll WA, Jarvis MF. [3H] A-804598 ([3H] 2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) is a novel, potent, and selective antagonist radioligand for P2X7 receptors. Neuropharmacology. 2009;56:223–229. doi: 10.1016/j.neuropharm.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 19.De Marchi E, Orioli E, Dal Ben D, Adinolfi E. P2X7 receptor as a therapeutic target. Adv Protein Chem Struct Biol. 2016;104:39–79. doi: 10.1016/bs.apcsb.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Andreassen SM, Berg LC, Nielsen SS, Kristensen AT, Jacobsen S. mRNA expression of genes involved in inflammation and haemostasis in equine fibroblast-like synoviocytes following exposure to lipopolysaccharide, fibrinogen and thrombin. BMC Vet Res. 2015;11:141. doi: 10.1186/s12917-015-0448-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fabbrizio P, Amadio S, Apolloni S, Volonté C. P2X7 receptor activation modulates autophagy in SOD1-G93A mouse microglia. Front Cell Neurosci. 2017;11:249. doi: 10.3389/fncel.2017.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hissin PJ, Hilf R. A fl uorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem. 1976;74:214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- 23.Diezmos EF, Markus I, Perera DS, Gan S, Zhang L, Sandow SL, Bertrand PP, Liu L. Blockade of Pannexin-1 channels and purinergic P2X7 receptors shows protective effects against cytokines-induced colitis of human colonic mucosa. Front Pharmacol. 2018;9:865. doi: 10.3389/fphar.2018.00865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin J, Zhou Z, Huo R, Xiao L, Ouyang G, Wang L, Sun Y, Shen B, Li D, Li N. Cyr61 induces IL-6 production by fibroblast-like synoviocytes promoting Th17 differentiation in rheumatoid arthritis. J Immunol. 2012;188:5776–5784. doi: 10.4049/jimmunol.1103201. [DOI] [PubMed] [Google Scholar]
- 25.Tabeling HJ, Dolwick MF. Rheumatoid arthritis: diagnosis and treatment. Fla Dent J. 1985;56:16–18. [PubMed] [Google Scholar]
- 26.Lamanna WC, Mayer RE, Rupprechter A, Fuchs M, Higel F, Fritsch C, Vogelsang C, Seidl A, Toll H, Schiestl M, Holzmann J. The structure-function relationship of disulfide bonds in etanercept. Sci Rep. 2017;7:3951. doi: 10.1038/s41598-017-04320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mateen S, Moin S, Khan AQ, Zafar A, Fatima N. Increased reactive oxygen species formation and oxidative stress in rheumatoid arthritis. PLoS One. 2016;11:e0152925. doi: 10.1371/journal.pone.0152925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quiñonez-Flores CM, González-Chávez SA, Del Río Nájera D, Pacheco-Tena C. Oxidative stress relevance in the pathogenesis of the rheumatoid arthritis: a systematic review. Biomed Res Int. 2016;2016:6097417. doi: 10.1155/2016/6097417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong H, Zeng Y, Jian W, Li L, Lin L, Mo Y, Liu M, Fang S, Xia Y. CDK 7 inhibition suppresses rheumatoid arthritis inflammation via blockage of NF-κB activation and IL-1β/IL-6 secretion. J Cell Mol Med. 2018;22:1292–1301. doi: 10.1111/jcmm.13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruscitti P, Cipriani P, Carubbi F, Liakouli V, Zazzeroni F, Di Benedetto P, Berardicurti O, Alesse E, Giacomelli R. The role of IL-1β in the bone loss during rheumatic diseases. Mediators Inflamm. 2015;2015:782382. doi: 10.1155/2015/782382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim GW, Lee NR, Pi RH, Lim YS, Lee YM, Lee JM, Jeong HS, Chung SH. IL-6 inhibitors for treatment of rheumatoid arthritis: past, present, and future. Arch Pharm Res. 2015;38:575–584. doi: 10.1007/s12272-015-0569-8. [DOI] [PubMed] [Google Scholar]
- 32.Singh JA, Saag KG, Bridges SL Jr, Akl EA, Bannuru RR, Sullivan MC, Vaysbrot E, McNaughton C, Osani M, Shmerling RH, Curtis JR. 2015 American college of rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016;68:1–26. doi: 10.1002/art.39480. [DOI] [PubMed] [Google Scholar]
- 33.Han YP, Tuan TL, Wu H, Hughes M, Garner WL. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-(kappa) B mediated induction of MT1-MMP. J Cell Sci. 2001;114:131–139. doi: 10.1242/jcs.114.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Catrina AI, Lampa J, Ernestam S, Af Klint E, Bratt J, Klareskog L, Ulfgren AK. Anti-tumour necrosis factor (TNF)-α therapy (etanercept) down-regulates serum matrix metalloproteinase (MMP)-3 and MMP-1 in rheumatoid arthritis. Rheumatology. 2002;41:484–489. doi: 10.1093/rheumatology/41.5.484. [DOI] [PubMed] [Google Scholar]
- 35.Hanabayashi M, Takahashi N, Sobue Y, Hirabara S, Ishiguro N, Kojima T. Hyaluronan oligosaccharides induce MMP-1 and-3 via transcriptional activation of NF-κB and p38 MAPK in rheumatoid synovial fibroblasts. PLoS One. 2016;11:e0161875. doi: 10.1371/journal.pone.0161875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jüngel A, Ospelt C, Lesch M, Thiel M, Sunyer T, Schorr O, Michel BA, Gay RE, Kolling C, Flory C, Gay S. Effect of the oral application of a highly selective MMP-13 inhibitor in three different animal models of rheumatoid arthritis. Ann Rheum Dis. 2010;69:898–902. doi: 10.1136/ard.2008.106021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim NH, Lee MY, Park SJ, Choi JS, Oh MK, Kim IS. Auranofin blocks interleukin-6 signalling by inhibiting phosphorylation of JAK1 and STAT3. Immunology. 2007;122:607–614. doi: 10.1111/j.1365-2567.2007.02679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H, Yan S, Chen J, Luo X, Li P, Jia X, Dai X, Wang C, Huang Q, Liu L, Zhang Y. JAK1-STAT3 blockade by JAK inhibitor SHR0302 attenuates inflammatory responses of adjuvant-induced arthritis rats and decreases Th17 and total B cells. Joint Bone Spine. 2016;83:525–532. doi: 10.1016/j.jbspin.2015.09.002. [DOI] [PubMed] [Google Scholar]