

Abstract
Murine models of Mycobacterium tuberculosis (Mtb) infection demonstrate progression of M1-like (proinflammatory) and M2-like (anti-inflammatory) macrophage morphology following primary granuloma formation. The Mtb cell wall cording factor, trehalose 6,6′-dimycolate (TDM), is a physiologically relevant and useful molecule for modeling early macrophage-mediated events during establishment of the tuberculosis-induced granuloma pathogenesis. Here, it is shown that TDM is a major driver of the early M1-like macrophage response as seen during initiation of the granulomas of primary pathology. Proinflammatory cytokines tumor necrosis factor-α, IL-1β, IL-6, and IL-12p40 are produced in lung tissue after administration of TDM to mice. Furthermore, CD11b+CD45+ macrophages with a high surface expression of the M1-like markers CD38 and CD86 were found present in regions of pathology in lungs of mice at 7 days post-TDM introduction. Conversely, only low phenotypic marker expression of M2-like markers CD206 and EGR-2 were present on macrophages. These findings suggest that TDM plays a role in establishment of the M1-like shift in the microenvironment during primary tuberculosis.
Dogma in tuberculosis pathology is the notion that induction of a strong T helper 1 lymphocyte (Th1) phenotype is critical in maintaining protective health during primary mycobacterial infection.1,2 Development of this phenotypic outcome is based on critical aspects of the macrophage and its initial interactions with the infectious organism.3, 4, 5 Multiple organism-induced mechanisms have been identified that function to evade host responses,6,7 essentially limiting effectiveness of this avenue of Th1 lymphocyte response. Many of these evasive properties directly affect macrophage functions.8 The relatively recent realization that macrophage populations can be subdivided into pro- and anti-inflammatory subsets has sparked a revolution in understanding the complexities of innate responding cells to subsequent immune outcomes; this has important consequences on ensuing granuloma pathologies post-infection.9, 10, 11
The primary stages of mycobacterial infection manifest with a robust proinflammatory response to effectively mitigate organism dissemination. Indeed, these host responses induce hemorrhagic inflammation and vascular occlusion,12,13 that coincide with changes to fibrinolytic activity and thrombosis,14 which are in many ways beneficial for limiting spread of microorganisms. Certainly, the establishment of a granuloma is considered protective,15,16 even though it provides a specialized niche within the lung for organisms to replicate.
A major molecule influencing acute reactivity upon entry of Mtb into the host is the mycobacterial cord factor component trehalose-6,6′-dimycolate (TDM). Multiple models of the TDM-induced pulmonary granulomatous response have been studied in mice, all of which have common features to those identified in early acute primary immunopathology of the human host. Initially, Bloch and Noll17 (1955) utilized cord factor to mimic pathologies seen in early primary human disease, including changes to fibrinolytic activity12,13 and thrombosis.14 Perez et al18 successfully repeated these experiments using purified TDM; Donnachie et al19 and Hwang et al20 built on this using molecular tools to further examine early events in extravascular coagulation. Alternative models of the primary TB response were developed using administered TDM to initiate a synchronized transient granulomatous response21, 22, 23; these models are extremely effective to assess monocyte-macrophage factors involved in primary granuloma pathogenesis. Indeed, modifications to this model have allowed further insights into how macrophages utilize required receptors to initiate the cytokine cascades,24 and how these events can push the macrophages to elicit hypersensitive responses.25,26
The common link between the different models is the relatively rapid initiated proinflammatory macrophage responses elicited in vivo by TDM. Here, the authors desired to further examine the nature of the macrophage phenotype response to TDM, and determine the histopathological evidence to support a hypothesis linking TDM to the presence of recruited macrophage polarization to an M1-like phenotype. Evidence presented in this report uses histologic markers and flow cytometry, immunostaining, and enzyme-linked immunoassay methods to link TDM as a driver for the M1-like macrophage phenotype during recruited induction of the primary granulomatous pathology.
Materials and Methods
Mice
Female C57BL/6 mice (Envigo, Houston, TX) were 5 to 6 weeks of age, and weighed approximately 20 g, at study initiation. Animal work was performed at the University of Texas Health Science Center and was approved by the animal welfare committee, according to protocols detailed in approved documents HSC-AWC-16-0140 and HSC-AWC-17-0089.
TDM-Induced Lung Pathology
Mycobacterial-derived TDM (cord factor) (Enzo Life Sciences, Farmingdale, NY) was solubilized in hexane/ethanol at a ratio of 9:1. Material was evaporated by a stream of air. The TDM oil/water emulsion was prepared as previously described.23 Briefly, evaporated TDM (25 μg/mouse) was homogenized in Drakeol (2 μL/mouse) (Penreco, Indianapolis, IN). Then 48 μL/mouse of Dulbecco’s phosphate buffered solution 1× (cellgro; Corning, Corning, NY) with 0.2% Tween-80 (Mallinckrodt, Hazelwood, MO) was added; the mixture was homogenized in a glass tube with a Teflon pestle for 1 minute to produce an oil/water emulsion. The TDM was intravenously given at a volume of 100 μL per animal. Control mice received material formulated with no addition of TDM in oil. Emulsion-only controls did not exhibit inflammation or exhibit cytokines production at the times described here, as was previously reported and detailed.22,27 All mice were sacrificed at times indicated (7 days after injection of formulated material).
Histologic Assessment
Mice were sacrificed, and lungs were immediately perfused with a solution of 1 mmol/L EDTA in Dulbecco’s phosphate buffered solution. Lungs were weighed, sectioned, and evaluated for pathology and histologic results. Tissues were fixed in 10% buffered formalin for histology (Fisher Scientific, Pittsburg, PA). Specimens were processed by the Histology Laboratory at the University of Texas Health Science Center McGovern Medical School (Houston, TX); tissues were embedded in paraffin blocks and then 5-μm-thick sections were subsequently stained with hematoxylin (Surgipath, Richmond, IL) and eosin (Richard-Allen Scientific, Kalamazoo, MI).
LWI
The authors’ laboratory utilizes an accepted lung weight index (LWI) that calculates as an approximation of lung inflammation intensity. The following equation was used for calculation of the gross tissue inflammation due to TDM-induced pathology, as used in prior studies22,28,29:
| (1) |
Computerized Analysis
High-resolution scanned images of hematoxylin and eosin–stained slides were scanned for computerized analysis of lung inflammation using Motic Digital Side Assistant software version 1.0.7.44 (Kowloon Bay, Kowloon, Hong Kong). Quantitation of inflammation was performed in two steps using ImageJ software version 1.52o (NIH, Bethesda, MD; https://imagej.nih.gov/ij). Lung area was initially quantified by separation of the image's scale from background. Minimum and maximum values for hue, saturation, and brightness of the image were set as follows: 120, 255; 0, 255; and 0, 255, respectively. Total cell area measurement was calculated using a modified version of the procedure detailed in the online ImageJ stained-sections example directory (NIH, https://imagej.nih.gov/ij/docs/examples/stained-sections/index.html, last accessed September 1, 2019), where the peak threshold was set at 164 for all digitized slides analyzed. Methods were similar to previously published materials.30 Lung inflammation was calculated as a percentage of total area occupied by cell area; values were averaged within treatment groups and normalized to that of group nontreated controls.
Lung Cytokine Production
A weighed section of lung was excised, then homogenized, then incubated at 37°C and 5% CO2 for 4 hours in Dulbecco's Modified Eagle's medium containing 50 μg/mL l-Arginine, 50 μg/mL HEPES, 100 μg/mL penicillin, and 50 μg/mL gentamycin, and 10% fetal bovine serum. Collected supernatants were spun to remove debris, then assessed by enzyme-linked immunoassay. Production of tumor necrosis factor-α (TNF-α), IL-1β, IL-12p40, IL-6, TGF-β, and IL-10 was determined by the manufacturer's instructions (DuoSet kits; R&D Systems, Minneapolis, MN). Supernatants to detect TGF-β were pretreated with a 1:5 supernatant:1N HCl ratio, then neutralized with the same volume of 1.2N NaOH/0.5 mol/L HEPES. The average of duplicate wells was determined using a standard curve produced by reactivity to the manufacturer's supplied recombinant molecules. Detection sensitivity limit was at least 32 pg/mL, according to the manufacturer’s product details.
Immunohistochemical Analysis
The large right lobe of each lung was collected and fixed in 10% buffered formalin. The fixed lung tissue was stained with hematoxylin and eosin using standard procedures. Assessment was performed using immunohistochemistry for integrin family member CD11b (Ab01114-23.0; Absolute Antibody, Wilton, UK),31 diluted at 1:2000, was performed according to modification of the manufacturer's instructions (20 minutes at low pH), and subsequently visualized using standard horseradish peroxidase techniques and 3,3′-diaminobenzidine chromogen using Dako reagents (Dako, Agilent, Santa Clara, CA). In a similar manner, M1-like marker CD38 (14-0381-02; Invitrogen, Thermo Fisher Scientific, Waltham, MA), diluted at 1:1000, was used for visualization on serial slide sections. Hematoxylin-counterstained slides were viewed by a trained pathologist, with descriptive results obtained in an experimentally blinded manner (R.L.H.).
Flow Cytometry Analysis
Lungs were extracted from wild-type and TDM treated mice, homogenized by hand, and underwent a 30-minute incubation with collagenase/hyaluronidase (StemCell Technologies, Vancouver, BC, Canada) and DNAse I (Sigma-Aldrich, St. Louis, MO) in sterile filtered RPMI 1640 medium with 1% 100× pen/strep and 2.5% HEPES (Thermo Fisher Scientific), and 5% heat-inactivated fetal bovine serum (Corning). Enzymatic digestion was quenched with addition of the RPMI mixture before lung tissue was plunged through a 70-μm cell strainer. Cells were pelleted at 4°C for 5 minutes at 450 × g. Cell pellets were resuspended in a 70% Percoll (17-0891-01; GE Healthcare, Chicago, IL) gradient and underlain by 40% Percoll followed by centrifugation at 500 × g for 20 minutes at 22°C with no break and slow acceleration. Cells were collected at the 70–40 Percoll interface before being resuspended in a sterile filtered PBS mixture containing 2% heat-inactivated fetal bovine serum and 1 mL of 0.5 mol/L EDTA (Thermo Fisher Scientific). Surface markers chosen to delineate murine M1-like and M2-like macrophages were based on published data from Jablonski et al,32 later detailed by Orecchioni et al.33 Similar findings were identified in human monocyte-derived macrophages.34 Cells were stained in accordance with the manufacturer’s recommendation, using Live/Dead Aqua stain (L34966; eBioscience, San Diego, CA) for 15 minutes, then FC (antibody receptor) blocked with Anti-Mo CD16/CD32 (14-0161-86; eBioscience) for 15 minutes. Staining used the following antibodies against specific receptors: CD38 (102728; BioLegend, San Diego, CA) APC/Cy7, CD86 (105014; BioLegend) phycoerythrin (PE)/Cy7, CD206 (141704; BioLegend) fluorescein isothiocyanate, EGR2 (17-6691-82; eBioscience) APC, CD11b (101208; BioLegend,) PE, and CD45 (48-0451-82; eBioscience) eFluor 450 for 20 minutes. Cells were fixed with 2% paraformaldehyde and then evaluated on a Beckman Coulter Cytoflex S flow cytometer (B75442; Beckman Coulter, Indianapolis, IN). Data were then analyzed using FlowJo software version 10 (Becton Dickinson, Franklin Lakes, NJ).
Statistical Analysis
Collected data were compared across groups, and against naive mice or mice challenged with vehicle formulated without TDM. Analysis used an unpaired t-test, or used one-way analysis of variance. The differences between means were considered significant at a level of P ≤ 0.05. Generated data points were compiled using GraphPad Prism software version 5.03 (San Diego, CA) and are presented as a representative value obtained from multiple experimental repeats (sets of 2 or 3). Experiments had an N of 4 to 6 mice.
Results
TDM Induces an Acute Granulomatous Pathology
C57BL/6 mice injected with Mtb-derived TDM (25 μg/mouse) in oil/water given intravenously developed inflammation in lung tissue (Figure 1). Broad lung inflammation was assessed as LWI at 7 days after i.v. TDM challenge; this time point reflects peak granulomatous response induced by TDM.22 Acute treatment increased LWI significantly. TDM-treated mice had an average LWI of 1.60 ± 0.49 units, compared with naive controls (0.97 ± 0.03 units; P ≤ 0.05). The LWI was similar to computerized assessments of lung inflammation. Analysis of digitized lung histograms post-TDM administration confirmed significant parenchymal inflammation and presence of granulomas throughout pulmonary tissue.
Figure 1.

Comparative gross pulmonary inflammation during TDM-induced pathology. Lungs from mice given i.v. TDM (25 μg) were assessed after 7 days and compared with control naive mice. A: The lung weight index was calculated to quantify gross pulmonary inflammation. B: Digital analysis of area occupied due to cellularity confirms induction of inflammatory response. Similar data were obtained in repeated experiments. C and D: Histologic examination of lungs at day 7 post-TDM administration reveals acute granulomatous response culminating in high levels of monocytic infiltration, with increased presence of focal macrophages (D), versus non–TDM-treated control mouse lungs (C). Higher magnification reveals foamy vesiculated macrophages aggregating between regions of relatively normal parenchyma. Formalin-fixed lung sections were stained with hematoxylin and eosin. Sections represent data obtained from repeated experiments. Data are expressed as means ± SEM. n = 3 experiments; n = 4 to 6 mice per group, per experiment. *P < 0.05 versus control. Scale bars = 300 μm. Original magnification: ×10 (main image); ×40 (insets).
Histologic Assessment of TDM-Induced Pathology
Mice administered i.v. TDM showed a focal accumulation of macrophages at day 7 post-injection (Figure 1). The pathologic reactivity demonstrated widespread inflammation and severely reduced open alveolar space. Small focal hemorrhagic petechiae were present as part of the inflammatory response. Lymphocytic infiltration to lung tissue occurred around regions where granulomas coincided with vasculature. Slight hemorrhage was present throughout the tissue. There was visual evidence of occlusion of intermediate or small blood vessels. Activated macrophages with intracellular vesicles were predominant in regions of reactivity; limited-to-negligible accumulation of lymphocytes occurred within the focal response. General edema was not a major component of the response. Naive (no TDM) mice did not demonstrate changes to lung architecture. Lungs from control mice exhibited normal pulmonary parenchyma; there were no noticeable cellular infiltrates, and limited presence of monocytic or leukocytic foci.
Pro-Inflammatory Response in Pulmonary Tissue of TDM-Treated Mice
Administration of TDM results in a strong proinflammatory response.23 Cytokine assessment by enzyme-linked immunoassay confirmed these findings. Lungs were examined at 7 days post-administration of TDM. Significant production of proinflammatory mediators TNF-α, IL-1β, and IL-12p40 was observed relative to control mice; IL-6 was also elevated in the TDM-treated group (Figure 2). Anti-inflammatory mediating cytokines were also evaluated; whereas there was a minor increase in production of TGF-β, it was not a significant change. There was no change, relative to controls, in IL-10 production (Figure 2).
Figure 2.

Pro- and Anti-Inflammatory Cytokine Mediators in Lung Tissue during TDM-Induced Response. Enzyme-linked immunoassay assessment of inflammatory cytokines reveals increase in production of tumor necrosis factor (TNF)-α, IL-1β, IL-12p40, and IL-6 at day 7 post-administration of TDM, compared with non–TDM-treated control mice. Lungs of mice receiving TDM do not reveal significant changes in production of transforming growth factor (TGF)-β and IL-10. Data are expressed as means ± SEM. n = 3 (experiments); n = 4 to 6 mice per group, per experiment. *P < 0.05 versus control.
Identification of the M1-Like Macrophage Phenotype within Regions of TDM Granulomas
The extent of macrophage phenotypic polarization was investigated using flow cytometry. Lungs treated with TDM were dissociated, and individual cells were further examined for the presence of M1-like and M2-like surface markers. Populations were initially gated on infiltrating monocytic macrophages (CD11bhiCD45hi).35,36 Figure 3 depicts data collected for the M1-like markers CD38 and CD86, along with assessment for expression of the M2-like marker CD206 and the early growth response gene-2 (EGR-2). Treatment with TDM resulted in an overall accumulation of CD11bhiCD45hi cells that expressed higher M1-like markers, but not the M2-like surface proteins. Specifically, CD38 was present on 39.50 ± 3.33% on infiltrating macrophages, which was significantly elevated when compared with the nontreated wild-type group controls (7.76 ± 2.24%; P ≤ 0.001). Additionally, CD86 was present on 19.34 ± 1.48% macrophages, compared with nontreated controls (5.31 ± 0.61%; P ≤ 0.001). Figure 3 also lists the values for the markers examined. CD14 was also highly elevated in this population (57.74 ± 4.06%; data not shown).
Figure 3.

Flow Cytometry Assessment of M1/M2-Like Markers on Infiltrating Monocyte-Macrophages. Isolated cells obtained from lungs at 7 days post-administration of TDM were examined by flow cytometry for expression of M1-like markers CD38 and CD86 (top), and for M2-like markers CD206 and EGR-2 (bottom). Isolated cells were first gated to identify CD11bhiCD45hi macrophages, then further analyzed for surface expression; Representative histographic plots of data accompany dot plots, side scatter (SSC) is shown versus signal intensity, for TDM-treated (blue) or wild type (WT) controls (orange). Analysis on the right indicates average (Avg) percent values with SD for each marker indicated. n = 4 to 6 mice.***P < 0.001 versus control and SD.
Immunohistochemical staining allowed localization of the CD11b+ macrophage population to regions of granulomatous response (Figure 4A), and specifically to areas of inflammation. Furthermore, staining for the CD38 surface glycoprotein marker on serial sections demonstrated the presence of the M1-like population in a pattern overlapping that for CD11b, coinciding with regions of high macrophage activation within the granulomas architecture (Figure 4B). The CD11b marker is present throughout the inflammatory foci, with diffuse staining in most of the monocytic cells. There is also a number of CD11b+ cells that are heavily stained; the presence of the CD38 marker appears to coincide with cells also expressing the higher levels of CD11b. Of note, not all macrophages within the granuloma exhibit CD38. Cells were also stained with anti-CD206 and anti–EGR-2; however, the presence of these M2-markers was not detected (data not shown). Taken together with the flow analysis, this suggests that the macrophages recruited to the focal inflammation during the pulmonary granulomatous response induced by the TDM are of the M1-like phenotype.
Figure 4.
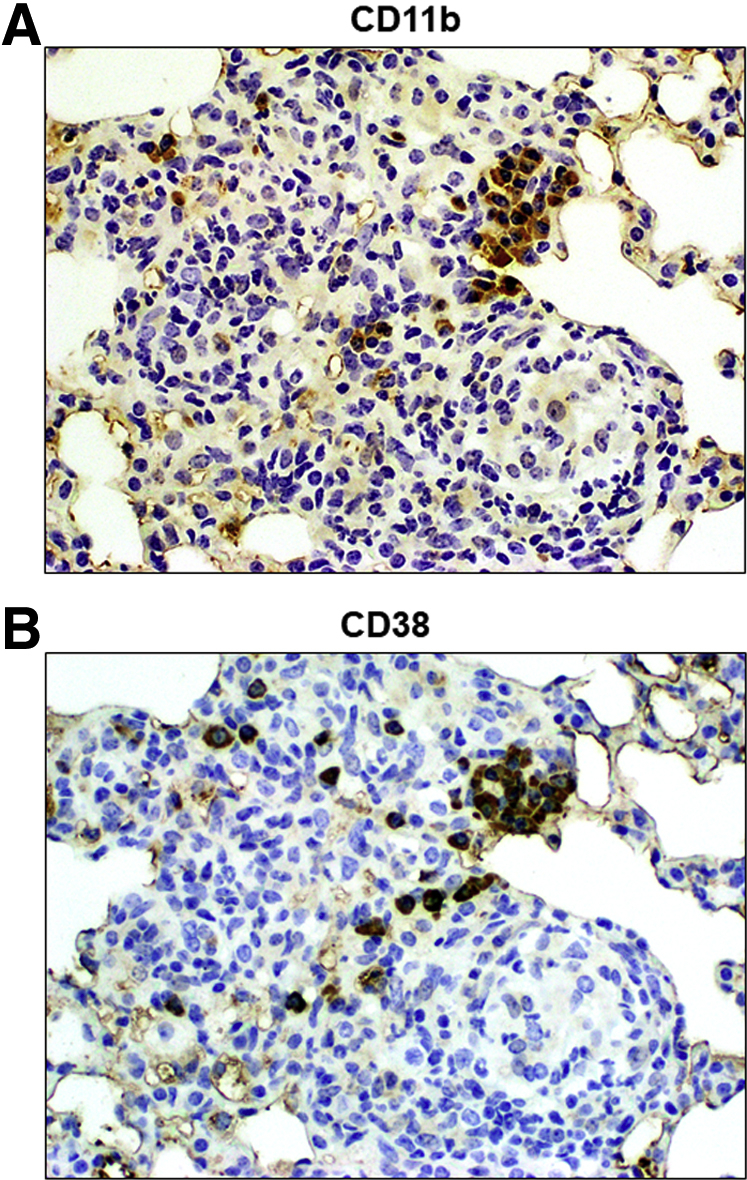
Localization of CD11b+ and CD38+ Cells to Pulmonary Regions of Focal TDM-Induced Inflammation. Serial sections of formalin-fixed lung tissue were reacted with antibody to CD11b (A) or CD38 (B), and subsequently visualized by way of standard horseradish peroxidase staining techniques. The acute regions of inflammation demonstrate the overlapping presence of both populations of cells within focal granulomatous regions. Original magnification, ×100.
Discussion
The establishment of the Mtb-induced granuloma is a complex interaction of multiple cell phenotypes responding to a myriad of antigenic stimuli.37 A multitude of studies have focused on mechanisms underlying development of the induced inflammatory response leading to development of this pathology. Clearly, the influence of any one specific molecule may not form an essential pathological bias in vivo during infection. However, a deficiency in production of purified cord factor, trehalose 6,6′-dimycolate,38 or manipulation of its structure,39 has certainly been shown to affect development and morphology of the early granulomatous pathology.
TDM has been identified as an immune mediator of both innate and adaptive inflammatory responses, and has been detailed as a virulence factor in reports focused on animal models.40 Others have examined its link and relevance to human disease by descriptive comparisons to human pathological manifestations.41 The nature of the induced pathology is clearly defined by physical parameters.42,43 Experiments using deficient mice allowed delineation of events that lead to establishment of the TDM-induced inflammatory granulomatous response.23 Furthermore, investigators identified innate receptors that trigger distinct immune events.44 This is the first report to our knowledge that examines the influence of TDM on accumulated M1-like or M2-like phenotype macrophages to the induced pulmonary foci. The results indicate that the presence of the M1-like surface marker expression was significantly up-regulated in TDM-treated animals, as opposed to the wild-type control animals, following the TDM-induced granulomatous response.
The in vivo studies show a significant increase of CD86 expression on CD11b+CD45+ cells within pulmonary foci. This population may have been recruited from alveolar spaces, as shown to occur by Cohen et al45 during Mtb infection. At this time, it is unclear whether the identified macrophages arrive as a committed M1-like population, or the environment induces relative M1/M2-like pathology progression once cells arrive at the focus of inflammation. Other groups have identified cellular plasticity in immune cell phenotypes post-arrival to pulmonary tissue post-infection.46 This may explain conflicting data from Kan-Sutton et al,47 where they found that TDM produced nearly identical strong proinflammatory effects in purified bone marrow–derived macrophages without inducing changes in the surface expression of CD86. Although it is difficult to compare in vivo and in vitro effects, it may be that the discrepancy between these studies was due to the nature of the presented TDM. Kan-Sutton et al47 used TDM adhered to the surface of beads; this format of exposure to TDM likely measured internalized effects on intracellular mechanisms associated with mycobacterial mycolic acid function, rather than those triggered via extracellular receptors. Also, TDM impacts intracellular trafficking events.48,49 Indeed, the nature of the engagement of macrophages can influence phenotypic and functional outcome.50
A brief discussion on the evidence for specific cellular receptors for TDM engagement is warranted. These data, along with previous findings on host macrophage receptors macrophage receptor with collagenous structure (MARCO)24 and Mincle,51, 52, 53 and Mincle-related Clec4d,54 suggest a potential link between the existence of the M1-like phenotype and engagement of those putative TDM receptors. An intriguing finding by Bowdish et al24 indicated that TDM engagement with MARCO activates the TLR2 signaling pathway, with the end result of production of similar proinflammatory cytokines to those identified in this study; macrophages from mice lacking MARCO also produced a significantly reduced amount of TNF-α, IL-6, and IL-1β in response to virulent Mtb. In our model of TDM infection, a similarly strong proinflammatory response was observed in lung tissue. However, engagement of the Mincle receptor could just as likely predict an M2-like outcome; Schoenen et al55 elegantly linked Cebpβ and Hif1α nuclear signaling pathways to TDM responsiveness in macrophages, with the end result of Egr (M2-like marker) synthesis. It is likely that phenotypic outcome is regulated through multiple receptors. A related Mincle ligand, the shorter chain TDM analog trehalose dibehenate, was also shown to differentially modulate M1-like (and M2-like) macrophage phenotypes through Syk signaling processes.56 Both Syk and CARD9 pathway interactions have shown to then further engage development of directed hypersensitive responses,57,58 as well as inflammatory signaling pathways.59 Perhaps additional experiments comparing trehalose 6-monomycolate (TMM) or galactose-galactose 6,6′ dimycolate (GDM) may shed light on the inflammatory response leading to the presence of M1-like phenotype found in this study. However, these molecules have previously shown limits, as they do not induce Mycobacterium tuberculosis–like primary granulomas in oil-in-water emulsions. They do not engage macrophages in the same manner as TDM.48,60 Therefore, although little change in M1 markers might be expected on recruited cells due to TMM or GDM because of the lack of inflammation, those molecules may provide a handle on pathways related to development of the M1-like phenotype following TDM administration.
The rationale for induction of a strong macrophage proinflammatory response has been suggested to promote signals resulting in induction of differentiated hypersensitive populations.61 Mtb has been shown to regulate internal pathways that may dictate polarity of macrophages.62 Multiple antigens from tuberculosis spp, in addition to the mycolic acids, have been shown to trigger strong proinflammatory responses, which, again, are likely dependent upon receptor engagement. For example, specific engagement of CD38 affects polarization of Th1 immune responses to M. avium63 Other molecules, such as ESAT-6, demonstrated a mixed reported transitional function. Refai et al64 demonstrated that ESAT-6 drives macrophages to the M1-like phenotype, although in the presence of TLR-2 engagement, it may exert anti-inflammatory M2-like polarization activity. Huang et al65 reported that ESAT-6 contributes to primary innate granuloma formation by inducing an M1-type differentiation, occurring in the presence of strong interferon-γ activity. However, they also indicated that this was likely dependent on the stage of infection in which this molecule was examined (as well as the presence of external mediators); ESAT-6 was shown to drive macrophages toward an M2-like phenotype at the later stages of the infection. Regarding Mtb antigens in total, they have been shown to induce a robust M2-like phenotype of human macrophages over time, with a shift away from CD86 toward CD206 surface expression in in vitro models. Those findings were consistent with the presence of M2-like antigens seen on cells in various stages of post-primary granulomas from infected patients. However, it should be noted that these represent stages of infection occurring well after granuloma formation and Th1 infiltration, which is not seen in the authors’ results. Indeed, reactivity to M2-like markers CD206 or EGR-2 was not seen in this study’s histological sections. Therefore, the findings discussed do not preclude the results here.
Overall, the presented results support the hypothesis that purified TDM by itself can be a strong inducer of a M1-like polarization of macrophages. As such, it continues to be an effective tool to experimentally define the cross-regulation of inflammatory molecules and cellular recruitment cascades which culminate in pathological changes in models of lung granulomatous disease caused by Mycobacterium tuberculosis infection.
Acknowledgment
We thank Dr. Louise D. McCullough, for assistance with flow cytometry and for the use of her facilities and reagents for cellular analysis.
Footnotes
Supported in part by NIH grant 1R41-AI117990-02 (J.K.A.).
Disclosures: None declared.
This research was performed in part to fulfill requirements for the MS degree from The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences (T.K.T.N.; University of Texas Medical Center, Houston, TX).
References
- 1.Orme I.M., Robinson R.T., Cooper A.M. The balance between protective and pathogenic immune responses in the TB-infected lung. Nat Immunol. 2015;16:57–63. doi: 10.1038/ni.3048. [DOI] [PubMed] [Google Scholar]
- 2.Jasenosky L.D., Scriba T.J., Hanekom W.A., Goldfeld A.E. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunol Rev. 2015;264:74–87. doi: 10.1111/imr.12274. [DOI] [PubMed] [Google Scholar]
- 3.McClean C.M., Tobin D.M. Macrophage form, function, and phenotype in mycobacterial infection: lessons from tuberculosis and other diseases. Pathog Dis. 2016;74:ftw068. doi: 10.1093/femspd/ftw068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.BoseDasgupta S., Pieters J. Macrophage-microbe interaction: lessons learned from the pathogen Mycobacterium tuberculosis. Semin Immunopathol. 2018;40:577–591. doi: 10.1007/s00281-018-0710-0. [DOI] [PubMed] [Google Scholar]
- 5.Marakalala M.J., Martinez F.O., Pluddemann A., Gordon S. Macrophage heterogeneity in the immunopathogenesis of tuberculosis. Front Microbiol. 2018;9:1028. doi: 10.3389/fmicb.2018.01028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldberg M.F., Saini N.K., Porcelli S.A. Evasion of innate and adaptive immunity by Mycobacterium tuberculosis. Microbiol Spectr. 2014;2 doi: 10.1128/microbiolspec.MGM2-0005-2013. MGM2-0005-2013. [DOI] [PubMed] [Google Scholar]
- 7.Hmama Z., Pena-Diaz S., Joseph S., Av-Gay Y. Immunoevasion and immunosuppression of the macrophage by Mycobacterium tuberculosis. Immunol Rev. 2015;264:220–232. doi: 10.1111/imr.12268. [DOI] [PubMed] [Google Scholar]
- 8.Upadhyay S., Mittal E., Philips J.A. Tuberculosis and the art of macrophage manipulation. Pathog Dis. 2018;76:fty037. doi: 10.1093/femspd/fty037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hao W., Schlesinger L.S., Friedman A. Modeling granulomas in response to infection in the lung. PLoS One. 2016;11:e0148738. doi: 10.1371/journal.pone.0148738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marino S., Cilfone N.A., Mattila J.T., Linderman J.J., Flynn J.L., Kirschner D.E. Macrophage polarization drives granuloma outcome during Mycobacterium tuberculosis infection. Infect Immun. 2015;83:324–338. doi: 10.1128/IAI.02494-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan A., Singh V.K., Hunter R.L., Jagannath C. Macrophage heterogeneity and plasticity in tuberculosis. J Leukoc Biol. 2019;106:275–282. doi: 10.1002/JLB.MR0318-095RR. [DOI] [PubMed] [Google Scholar]
- 12.Turken O., Kunter E., Sezer M., Solmazgul E., Cerrahoglu K., Bozkanat E., Ozturk A., Ilvan A. Hemostatic changes in active pulmonary tuberculosis. Int J Tuberc Lung Dis. 2002;6:927–932. [PubMed] [Google Scholar]
- 13.Sezer M., Ozturk A., Ilvan A., Ozkan M., Uskent N. The hemostatic changes in active pulmonary tuberculosis. Turk J Haematol. 2001;18:95–100. [PubMed] [Google Scholar]
- 14.Kager L.M., Blok D.C., Lede I.O., Rahman W., Afroz R., Bresser P., van der Zee J.S., Ghose A., Visser C.E., de Jong M.D., Tanck M.W., Zahed A.S., Alam K.M., Hassan M., Hossain A., Lutter R., Veer C.V., Dondorp A.M., Meijers J.C., van der Poll T. Pulmonary tuberculosis induces a systemic hypercoagulable state. J Infect. 2015;70:324–334. doi: 10.1016/j.jinf.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Russell D.G. Who puts the tubercle in tuberculosis? Nat Rev Microbiol. 2007;5:39–47. doi: 10.1038/nrmicro1538. [DOI] [PubMed] [Google Scholar]
- 16.Dorhoi A., Reece S.T., Kaufmann S.H. For better or for worse: the immune response against Mycobacterium tuberculosis balances pathology and protection. Immunol Rev. 2011;240:235–251. doi: 10.1111/j.1600-065X.2010.00994.x. [DOI] [PubMed] [Google Scholar]
- 17.Bloch H., Noll H. Studies on the virulence of Tubercle bacilli; the effect of cord factor on murine tuberculosis. Br J Exp Pathol. 1955;36:8–17. [PMC free article] [PubMed] [Google Scholar]
- 18.Perez R.L., Roman J., Staton G.W., Jr., Hunter R.L. Extravascular coagulation and fibrinolysis in murine lung inflammation induced by the mycobacterial cord factor trehalose-6,6'-dimycolate. Am J Respir Crit Care Med. 1994;149:510–518. doi: 10.1164/ajrccm.149.2.8306054. [DOI] [PubMed] [Google Scholar]
- 19.Donnachie E., Fedotova E.P., Hwang S.A. Trehalose 6,6-dimycolate from mycobacterium tuberculosis induces hypercoagulation. Am J Pathol. 2016;186:1221–1233. doi: 10.1016/j.ajpath.2015.12.019. [DOI] [PubMed] [Google Scholar]
- 20.Hwang S.A., Byerly C.D., Actor J.K. Mycobacterial trehalose 6,6'-dimycolate induced vascular occlusion is accompanied by subendothelial inflammation. Tuberculosis (Edinb) 2019;116S:S118–S122. doi: 10.1016/j.tube.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geisel R.E., Sakamoto K., Russell D.G., Rhoades E.R. In vivo activity of released cell wall lipids of Mycobacterium bovis bacillus Calmette-Guerin is due principally to trehalose mycolates. J Immunol. 2005;174:5007–5015. doi: 10.4049/jimmunol.174.8.5007. [DOI] [PubMed] [Google Scholar]
- 22.Perez R.L., Roman J., Roser S., Little C., Olsen M., Indrigo J., Hunter R.L., Actor J.K. Cytokine message and protein expression during lung granuloma formation and resolution induced by the mycobacterial cord factor trehalose-6,6'-dimycolate. J Interferon Cytokine Res. 2000;20:795–804. doi: 10.1089/10799900050151067. [DOI] [PubMed] [Google Scholar]
- 23.Welsh K.J., Abbott A.N., Hwang S.A., Indrigo J., Armitige L.Y., Blackburn M.R., Hunter R.L., Jr., Actor J.K. A role for tumour necrosis factor-alpha, complement C5 and interleukin-6 in the initiation and development of the mycobacterial cord factor trehalose 6,6'-dimycolate induced granulomatous response. Microbiology. 2008;154:1813–1824. doi: 10.1099/mic.0.2008/016923-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowdish D.M., Sakamoto K., Kim M.J., Kroos M., Mukhopadhyay S., Leifer C.A., Tryggvason K., Gordon S., Russell D.G. MARCO, TLR2, and CD14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and Mycobacterium tuberculosis. PLoS Pathog. 2009;5:e1000474. doi: 10.1371/journal.ppat.1000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guidry T.V., Hunter R.L., Jr., Actor J.K. Mycobacterial glycolipid trehalose 6,6'-dimycolate-induced hypersensitive granulomas: contribution of CD4+ lymphocytes. Microbiology. 2007;153:3360–3369. doi: 10.1099/mic.0.2007/010850-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamagami H., Matsumoto T., Fujiwara N., Arakawa T., Kaneda K., Yano I., Kobayashi K. Trehalose 6,6'-dimycolate (cord factor) of Mycobacterium tuberculosis induces foreign-body- and hypersensitivity-type granulomas in mice. Infect Immun. 2001;69:810–815. doi: 10.1128/IAI.69.2.810-815.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guidry T.V., Olsen M., Kil K.S., Hunter R.L., Jr., Geng Y.J., Actor J.K. Failure of CD1D-/- mice to elicit hypersensitive granulomas to mycobacterial cord factor trehalose 6,6'-dimycolate. J Interferon Cytokine Res. 2004;24:362–371. doi: 10.1089/107999004323142222. [DOI] [PubMed] [Google Scholar]
- 28.Pelletier M., Forget A., Bourassa D., Gros P., Skamene E. Immunopathology of BCG infection in genetically resistant and susceptible mouse strains. J Immunol. 1982;129:2179–2185. [PubMed] [Google Scholar]
- 29.Collins F.M., Congdon C.C., Morrison N.E. Growth of mycobacterium bovis (BCG) in T lymphocyte-depleted mice. Infect Immun. 1975;11:57–64. doi: 10.1128/iai.11.1.57-64.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yue Y., Huang W., Liang J., Guo J., Ji J., Yao Y., Zheng M., Cai Z., Lu L., Wang J. IL4I1 is a novel regulator of M2 macrophage polarization that can inhibit T cell activation via L-tryptophan and arginine depletion and IL-10 production. PLoS One. 2015;10:e0142979. doi: 10.1371/journal.pone.0142979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jablonski K.A., Amici S.A., Webb L.M., Ruiz-Rosado Jde D., Popovich P.G., Partida-Sanchez S., Guerau-de-Arellano M. Novel markers to delineate murine M1 and M2 macrophages. PLoS One. 2015;10:e0145342. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orecchioni M., Ghosheh Y., Pramod A.B., Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol. 2019;10:1084. doi: 10.3389/fimmu.2019.01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amici S.A., Young N.A., Narvaez-Miranda J., Jablonski K.A., Arcos J., Rosas L., Papenfuss T.L., Torrelles J.B., Jarjour W.N., Guerau-de-Arellano M. CD38 is robustly induced in human macrophages and monocytes in inflammatory conditions. Front Immunol. 2018;9:1593. doi: 10.3389/fimmu.2018.01593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koeniger T., Kuerten S. Splitting the “unsplittable”: dissecting resident and infiltrating macrophages in experimental autoimmune encephalomyelitis. Int J Mol Sci. 2017;18:E2072. doi: 10.3390/ijms18102072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheng J., Ruedl C., Karjalainen K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity. 2015;43:382–393. doi: 10.1016/j.immuni.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 37.Dorhoi A., Kaufmann S.H. Versatile myeloid cell subsets contribute to tuberculosis-associated inflammation. Eur J Immunol. 2015;45:2191–2202. doi: 10.1002/eji.201545493. [DOI] [PubMed] [Google Scholar]
- 38.Copenhaver R.H., Sepulveda E., Armitige L.Y., Actor J.K., Wanger A., Norris S.J., Hunter R.L., Jagannath C. A mutant of Mycobacterium tuberculosis H37Rv that lacks expression of antigen 85A is attenuated in mice but retains vaccinogenic potential. Infect Immun. 2004;72:7084–7095. doi: 10.1128/IAI.72.12.7084-7095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glickman M.S., Cox J.S., Jacobs W.R., Jr. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5:717–727. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- 40.Ryll R., Kumazawa Y., Yano I. Immunological properties of trehalose dimycolate (cord factor) and other mycolic acid-containing glycolipids–a review. Microbiol Immunol. 2001;45:801–811. doi: 10.1111/j.1348-0421.2001.tb01319.x. [DOI] [PubMed] [Google Scholar]
- 41.Hunter R.L., Olsen M.R., Jagannath C., Actor J.K. Multiple roles of cord factor in the pathogenesis of primary, secondary, and cavitary tuberculosis, including a revised description of the pathology of secondary disease. Ann Clin Lab Sci. 2006;36:371–386. [PubMed] [Google Scholar]
- 42.Schabbing R.W., Garcia A., Hunter R.L. Characterization of the trehalose 6,6'-dimycolate surface monolayer by scanning tunneling microscopy. Infect Immun. 1994;62:754–756. doi: 10.1128/iai.62.2.754-756.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Syed S.S., Hunter R.L., Jr. Studies on the toxic effects of quartz and a mycobacterial glycolipid, trehalose 6,6'-dimycolate. Ann Clin Lab Sci. 1997;27:375–383. [PubMed] [Google Scholar]
- 44.Ishikawa E., Mori D., Yamasaki S. Recognition of mycobacterial lipids by immune receptors. Trends Immunol. 2017;38:66–76. doi: 10.1016/j.it.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 45.Cohen S.B., Gern B.H., Delahaye J.L., Adams K.N., Plumlee C.R., Winkler J.K., Sherman D.R., Gerner M.Y., Urdahl K.B. Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. 2018;24:439–446.e4. doi: 10.1016/j.chom.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis M.J., Tsang T.M., Qiu Y., Dayrit J.K., Freij J.B., Huffnagle G.B., Olszewski M.A. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio. 2013;4 doi: 10.1128/mBio.00264-13. e00264–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kan-Sutton C., Jagannath C., Hunter R.L., Jr. Trehalose 6,6'-dimycolate on the surface of Mycobacterium tuberculosis modulates surface marker expression for antigen presentation and costimulation in murine macrophages. Microbes Infect. 2009;11:40–48. doi: 10.1016/j.micinf.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Indrigo J., Hunter R.L., Jr., Actor J.K. Cord factor trehalose 6,6'-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology. 2003;149:2049–2059. doi: 10.1099/mic.0.26226-0. [DOI] [PubMed] [Google Scholar]
- 49.Indrigo J., Hunter R.L., Jr., Actor J.K. Influence of trehalose 6,6'-dimycolate (TDM) during mycobacterial infection of bone marrow macrophages. Microbiology. 2002;148:1991–1998. doi: 10.1099/00221287-148-7-1991. [DOI] [PubMed] [Google Scholar]
- 50.Rajaram M.V., Ni B., Dodd C.E., Schlesinger L.S. Macrophage immunoregulatory pathways in tuberculosis. Semin Immunol. 2014;26:471–485. doi: 10.1016/j.smim.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schoenen H., Bodendorfer B., Hitchens K., Manzanero S., Werninghaus K., Nimmerjahn F., Agger E.M., Stenger S., Andersen P., Ruland J., Brown G.D., Wells C., Lang R. Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol. 2010;184:2756–2760. doi: 10.4049/jimmunol.0904013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsunaga I., Moody D.B. Mincle is a long sought receptor for mycobacterial cord factor. J Exp Med. 2009;206:2865–2868. doi: 10.1084/jem.20092533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishikawa E., Ishikawa T., Morita Y.S., Toyonaga K., Yamada H., Takeuchi O., Kinoshita T., Akira S., Yoshikai Y., Yamasaki S. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med. 2009;206:2879–2888. doi: 10.1084/jem.20091750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyake Y., Toyonaga K., Mori D., Kakuta S., Hoshino Y., Oyamada A., Yamada H., Ono K., Suyama M., Iwakura Y., Yoshikai Y., Yamasaki S. C-type lectin MCL is an FcRgamma-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity. 2013;38:1050–1062. doi: 10.1016/j.immuni.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 55.Schoenen H., Huber A., Sonda N., Zimmermann S., Jantsch J., Lepenies B., Bronte V., Lang R. Differential control of Mincle-dependent cord factor recognition and macrophage responses by the transcription factors C/EBPbeta and HIF1alpha. J Immunol. 2014;193:3664–3675. doi: 10.4049/jimmunol.1301593. [DOI] [PubMed] [Google Scholar]
- 56.Kodar K., Harper J.L., McConnell M.J., Timmer M.S.M., Stocker B.L. The Mincle ligand trehalose dibehenate differentially modulates M1-like and M2-like macrophage phenotype and function via Syk signaling. Immun Inflamm Dis. 2017;5:503–514. doi: 10.1002/iid3.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao X.Q., Zhu L.L., Chang Q., Jiang C., You Y., Luo T., Jia X.M., Lin X. C-type lectin receptor dectin-3 mediates trehalose 6,6'-dimycolate (TDM)-induced Mincle expression through CARD9/Bcl10/MALT1-dependent nuclear factor (NF)-kappaB activation. J Biol Chem. 2014;289:30052–30062. doi: 10.1074/jbc.M114.588574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.LeibundGut-Landmann S., Gross O., Robinson M.J., Osorio F., Slack E.C., Tsoni S.V., Schweighoffer E., Tybulewicz V., Brown G.D., Ruland J., Reis e Sousa C. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 59.Schweneker K., Gorka O., Schweneker M., Poeck H., Tschopp J., Peschel C., Ruland J., Gross O. The mycobacterial cord factor adjuvant analogue trehalose-6,6'-dibehenate (TDB) activates the Nlrp3 inflammasome. Immunobiology. 2013;218:664–673. doi: 10.1016/j.imbio.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 60.Behling C.A., Perez R.L., Kidd M.R., Staton G.W., Jr., Hunter R.L. Induction of pulmonary granulomas, macrophage procoagulant activity, and tumor necrosis factor-alpha by trehalose glycolipids. Ann Clin Lab Sci. 1993;23:256–266. [PubMed] [Google Scholar]
- 61.Tomioka H., Tatano Y., Maw W.W., Sano C., Kanehiro Y., Shimizu T. Characteristics of suppressor macrophages induced by mycobacterial and protozoal infections in relation to alternatively activated M2 macrophages. Clin Dev Immunol. 2012;2012:635451. doi: 10.1155/2012/635451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen P., Li Q., Ma J., Tian M., Hong F., Zhai X., Li J., Huang H., Shi C. IRAK-M alters the polarity of macrophages to facilitate the survival of Mycobacterium tuberculosis. BMC Microbiol. 2017;17:185. doi: 10.1186/s12866-017-1095-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Viegas M.S., do Carmo A., Silva T., Seco F., Serra V., Lacerda M., Martins T.C. CD38 plays a role in effective containment of mycobacteria within granulomata and polarization of Th1 immune responses against Mycobacterium avium. Microbes Infect. 2007;9:847–854. doi: 10.1016/j.micinf.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 64.Refai A., Gritli S., Barbouche M.R., Essafi M. Mycobacterium tuberculosis virulent factor ESAT-6 drives macrophage differentiation toward the pro-inflammatory M1 phenotype and subsequently switches it to the anti-inflammatory M2 phenotype. Front Cell Infect Microbiol. 2018;8:327. doi: 10.3389/fcimb.2018.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang Z., Luo Q., Guo Y., Chen J., Xiong G., Peng Y., Ye J., Li J. Mycobacterium tuberculosis-induced polarization of human macrophage orchestrates the formation and development of tuberculous granulomas in vitro. PLoS One. 2015;10:e0129744. doi: 10.1371/journal.pone.0129744. [DOI] [PMC free article] [PubMed] [Google Scholar]