

Abstract
Genetic etiologies of chronic mucocutaneous candidiasis (CMC) disrupt human IL-17A/F-dependent immunity at mucosal surfaces, whereas those of connective tissue disorders (CTD) often impair the TGF-β-dependent homeostasis of connective tissues. The signaling pathways involved are incompletely understood. We report a three-generation family with an autosomal dominant (AD) combination of CMC and a novel CTD that clinically overlaps with Ehlers-Danlos syndrome (EDS). The patients are heterozygous for a private splice-site variant of MAPK8, the gene encoding c-Jun N-terminal kinase 1 (JNK1), a component of the MAPK signaling pathway. This variant is loss-of-expression and loss-of-function in the patients’ fibroblasts, which display AD JNK1 deficiency by haploinsufficiency. These cells have impaired, but not abolished, responses to IL-17A and IL-17F. Moreover, the development of the patients’ TH17 cells was impaired ex vivo and in vitro, probably due to the involvement of JNK1 in the TGF-β-responsive pathway and further accounting for the patients’ CMC. Consistently, the patients’ fibroblasts displayed impaired JNK1- and c-Jun/ATF2-dependent induction of key extracellular matrix (ECM) components and regulators, but not of EDS-causing gene products, in response to TGF-β. Furthermore, they displayed a transcriptional pattern in response to TGF-β different from that of fibroblasts from patients with Loeys-Dietz syndrome and mutations of TGFBR2 or SMAD3, further accounting for the patients’ complex and unusual CTD phenotype. This experiment of Nature indicates that the integrity of the human JNK1-dependent MAPK signaling pathway is essential for IL-17A- and IL-17F-dependent mucocutaneous immunity to Candida, and for the TGF-β-dependent homeostasis of connective tissues.
Introduction
Chronic mucocutaneous candidiasis (CMC) is characterized by recurrent lesions of the skin, nails, oral and genital mucosae caused by Candida albicans (1). Patients with profound and broad inherited T-cell immunodeficiencies present CMC as one of their many infections (2). Most patients heterozygous for dominant-negative STAT3 mutations (3) or gain-of-function STAT1 mutations (4), and most patients with autosomal recessive (AR) RORγT (5) or ZNF341 deficiency (6, 7) present CMC among the infections suffered, the range of which is smaller than for patients with severe T-cell deficiencies. Patients with these various forms of syndromic CMC (SCMC) share a paucity of circulating TH17 cells (5–13). Patients with AR AIRE deficiency display not only autoimmunity but also CMC as their only infection, due to the production of neutralizing autoantibodies against IL-17A and/or IL-17F (14, 15). Finally, isolated forms of CMC (ICMC), in which CMC is the predominant or only clinical manifestation in otherwise healthy individuals, can be due to autosomal dominant (AD) IL-17F deficiency, or inborn errors of the IL-17-responsive pathway, such as AR IL-17RA, IL-17RC, and ACT1 deficiencies (16–20). Fibroblasts and keratinocytes derived from these patients display impaired (AD IL-17F deficiency) (16) or abolished (AR IL-17RA, IL-17RC, or ACT1 deficiency) responses to IL-17A and IL-17F (16–19).
Patients with inherited ICMC do not, however, display any overt signs of connective tissue disorders (CTD), as their skin, joints, bones, and blood vessels are unaffected. Conversely, patients with CTDs, such as Ehlers-Danlos syndrome (EDS), Loeys-Dietz syndrome (LDS), and Marfan syndrome (MS), do not suffer from CMC (21). Whilst the genetic basis of hypermobile EDS (hEDS) is unknown (22), the other 13 subtypes of EDS are caused by various inborn errors of genes, many of which encode collagen or collagen-modifying enzymes (e.g. COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, ADAMTS2, PLOD1) (22, 23). LDS is caused by inborn errors of the TGF-β signaling pathway (TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2, and TGFB3) (24), and MS by inborn errors of FBN1, which encodes fibrillin-1 (25). In these disorders, the homeostasis and integrity of connective tissues are impaired by dysfunctional extracellular matrix (ECM) proteins, the production of which is controlled by TGF-β in fibroblasts (24, 26).
Results
A private heterozygous MAPK8 variant in a kindred with AD CMC and CTD
We studied three patients (P1, P2, and P3) from three generations of a French family with AD CMC and a CTD overlapping with hEDS (Fig. 1A; fig. S1A; table S1; and the “Case reports” section). We performed whole-exome sequencing (WES) and found no rare non-synonymous coding variants in any of the known CMC-, EDS-, LDS-, and MS-causing genes, all of which were well covered by WES (table S2). Under a complete penetrance model, we found 18 heterozygous non-synonymous variants common to the three patients and private to this family, i.e. not previously reported in the 1000 Genomes Project, the Single-Nucleotide Polymorphism Database, the NHLBI GO Exome Sequencing Project, the Exome Aggregation Consortium Genome Aggregation Database, the NHLBI’s TOPMed program (Bravo), or our in-house database of over 6,000 exomes from patients with various infectious diseases (fig. S1B and table S3). The most plausible candidate was a splice-site mutation in the MAPK8 gene, for which the biological distance to six of the eight known SCMC- and ICMC-causing genes other than AIRE (IL17F, IL17RA, ACT1, STAT1, STAT3, and RORC) was shortest in the human gene connectome (HGC), and the distance to the other two (IL17RC and ZNF341) ranked second-shortest (27, 28). The familial segregation of this private mutant MAPK8 allele was consistent with a fully penetrant AD trait (Fig. 1, A and B). This nucleotide substitution (c.311+1G>A), one base pair downstream from exon IV (Fig. 1C), was predicted to affect splicing by altering the donor splice site (29). The c.311+1G>A mutation has a combined annotation-dependent depletion (CADD) score of 26 (30), which is above the mutation significance cutoff (MSC) threshold of 19.034 for MAPK8 (31) (fig. S1C). Moreover, three of the four nonsense or frameshift mutations in MAPK8 present in public databases have a minor allele frequency (MAF) < 10−5, whereas the fourth, with a MAF of 0.0000114, has a CADD score below the MSC threshold (fig. S1C). Consistent with these findings, MAPK8 has a gene damage index (GDI) of 0.32 (32), a neutrality index of 0.06 (33), and a SnIPRE f parameter of 0.329 (within the top 11% of genes within the genome subject to the greatest constraints) (34) (fig. S1D), indicating that this gene is highly conserved in human populations and has evolved under purifying selection. Finally, MAPK8 has a probability of loss-of-function intolerance (pLI) score of 0.98, which is greater than the threshold of 0.9, above which genes are considered to be extremely intolerant to loss-of-function variants (35). The MAPK8 mutation found in this kindred was therefore probably deleterious, with the potential to cause an AD disease.
Fig. 1. Identification of a heterozygous MAPK8 mutation in a kindred with AD CMC and CTD.
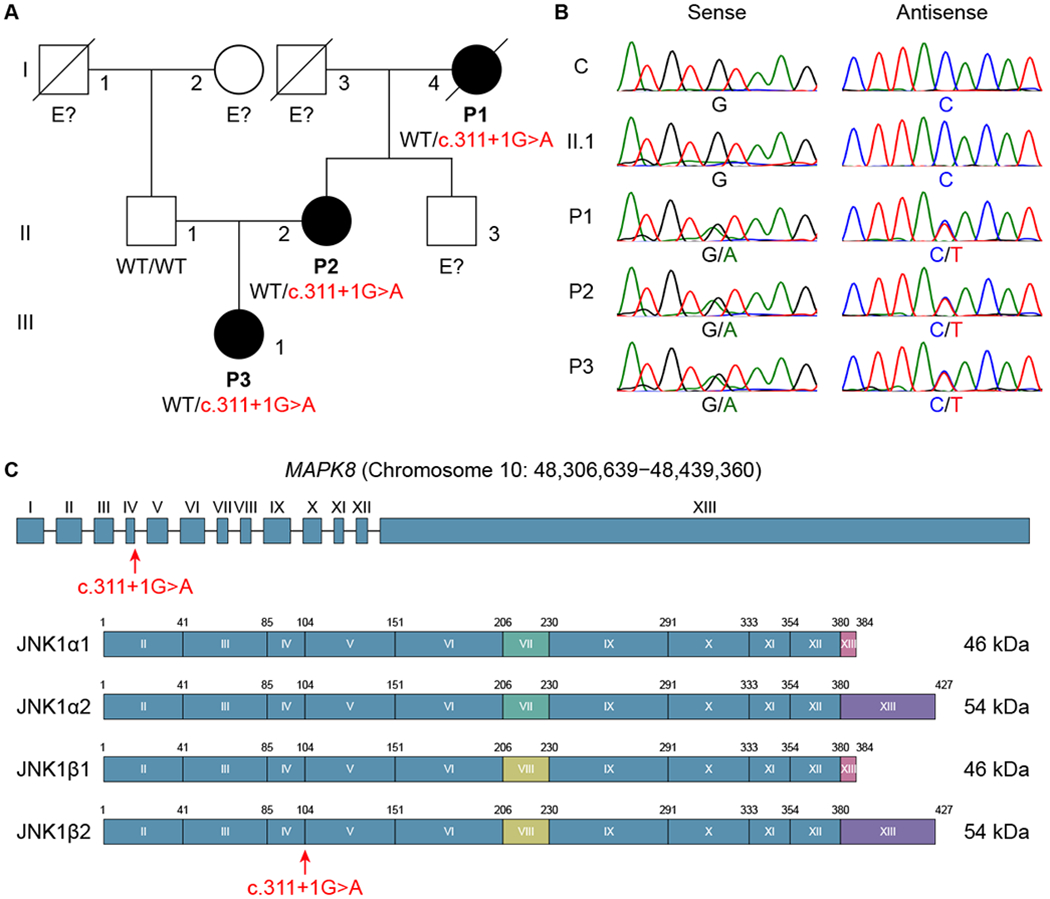
(A) Pedigree and segregation of the MAPK8 mutation. The patients, in black, are heterozygous for the mutation. E? indicates individuals whose genetic status could not be evaluated. (B) Electropherograms of partial sequences of MAPK8 corresponding to the mutation in a healthy control (C) and four members of the kindred (II.1, P1, P2, and P3). (C) Schematic illustration of the genomic locus and of the protein encoded by the MAPK8 gene extracted from the Ensembl database. It has 13 exons (I-XIII), 12 of which are coding exons (II-XIII), encoding four isoforms (JNK1α1, JNK1α2, JNK1β1, and JNK1β2), with alternative usage of exon VII or VIII and alternative splicing of exon XIII. The red arrow indicates the position of the mutation.
A loss-of-expression mutant MAPK8 allele
The MAPK8 gene encodes JNK1, one of the three members of the JNK family. This protein is a component of the mitogen-activated protein kinase (MAPK) pathway that converts extracellular stimuli into cellular responses (36, 37). JNK1 is phosphorylated by upstream MAPK kinases (MAPKK), and in turn phosphorylates downstream activator protein-1 (AP-1) transcription factors, including c-Jun and ATF-2 (37). There are two long (JNK1α2 and JNK1β2, 54 kDa) and two short (JNK1α1 and JNK1β1, 46 kDa) isoforms, generated by alternative usage of exon VII or VIII and alternative splicing of exon XIII (38) (Fig. 1C). We amplified a cDNA fragment extending from exons III to V from Epstein-Barr virus (EBV)-transformed B cells and simian virus 40 (SV40)-transformed fibroblasts from the patients. In addition to the wild-type (WT) transcript (band 4), we detected four aberrant products (bands 1, 2, 3, and 5) (Fig. 2A). TA cloning and subsequent sequencing identified two aberrantly spliced transcripts: one in which intron IV was retained (band 2) and one in which exon IV was skipped (band 5) (Fig. 2A). Bands 1 and 3 were artifacts of heteroduplex formation (39). We then inserted a genomic fragment containing the WT or mutant intron IV together with the surrounding exons (IV and V) into an exon-trapping vector (Fig. 2B). The WT minigene was normally spliced, whereas the mutant minigene generated two aberrant splicing products; one in which exon IV was skipped and another in which intron IV was retained (Fig. 2B). This assay confirmed the direct impact of the c.311+1G>A mutation on MAPK8 mRNA splicing, with no detectable leakiness. Both aberrant mRNAs were predicted to result in the creation of premature stop codons (Fig. 2C). Consistent with this prediction, the levels of WT MAPK8 mRNA and JNK1 protein in the patients’ cells were about half those in control cells (Fig. 2, D and E). Moreover, no truncated proteins were detected in the patients’ cells (Fig. 2E) or in HEK293T cells transfected with the corresponding mutant constructs, with or without the N-terminal Myc tag (Fig. 2F). The three patients were, therefore, heterozygous for a private loss-of-expression MAPK8 allele.
Fig. 2. The mutant MAPK8 allele is loss-of-expression.
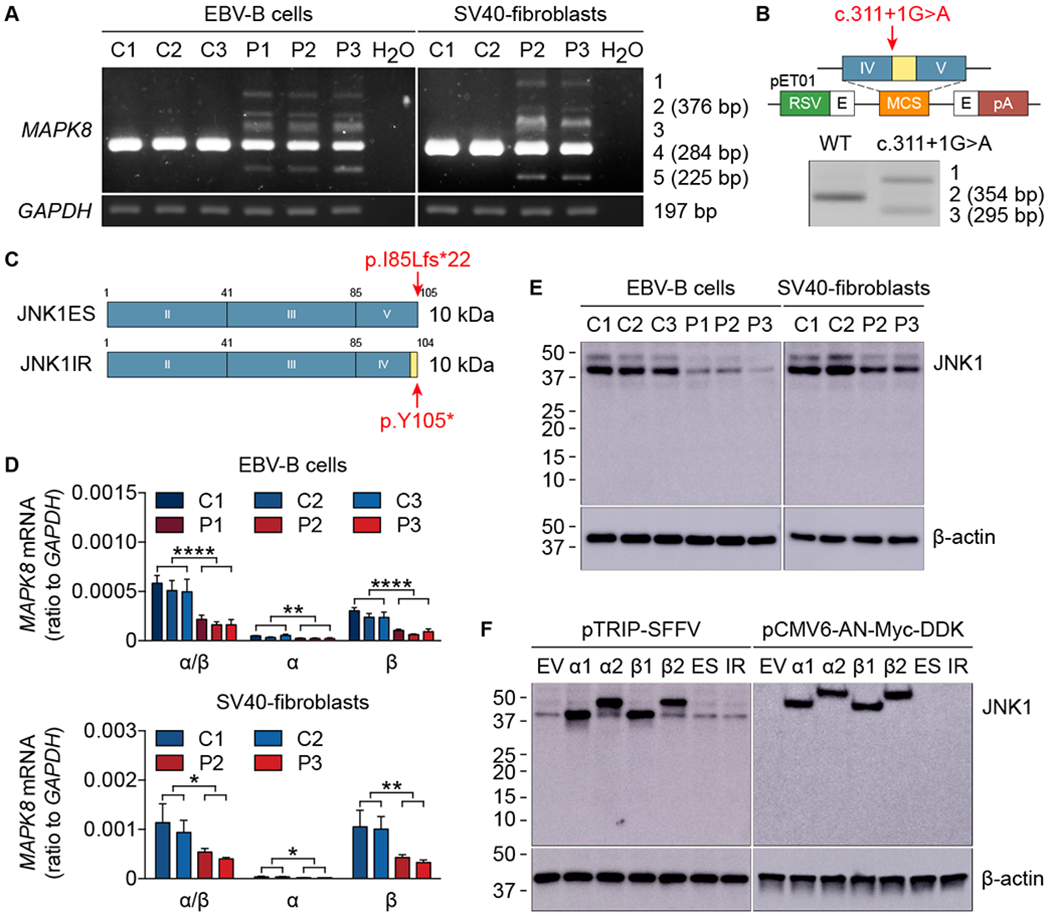
(A) MAPK8 mRNA levels in EBV-B cells and SV40-fibroblasts from healthy controls (C1, C2, and C3) and patients (P1, P2, and P3). TA cloning and subsequent sequencing of the five bands generated by amplification from exon III to exon V identified three spliced transcripts: band 1 corresponding to the WT sequence together with intron IV retention and exon IV skipping; band 2 (376 bp) corresponding to intron IV retention; band 3 corresponding to the WT sequence together with exon IV skipping; band 4 (284 bp) corresponding to the WT sequence; band 5 (225 bp) corresponding to exon IV skipping. (B) Schematic diagram of the constructs used for exon trapping. pET01, exon-trapping vector; RSV, Rous sarcoma virus long terminal repeat promoter; pA, polyadenylation; E in black, exon of the pET01 vector; IV and V in blue, MAPK8 exons IV and V; in yellow, MAPK8 intron IV. The red arrow indicates the position of the mutation. RT-PCR and subsequent sequencing identified three spliced transcripts: band 1 corresponding to intron IV retention and exon IV skipping; band 2 (354 bp) corresponding to the WT sequence; band 3 (295 bp) corresponding to exon IV skipping. (C) Schematic illustration of the mutant proteins. JNK1ES (JNK1 exon skipping) represents exon IV skipping, whereas JNK1IR (JNK1 intron retention) denotes intron IV retention. Both transcripts are predicted to encode proteins of approximately 10 kDa in size. Red arrows indicate the positions of premature stop codons. (D) mRNA levels for MAPK8 isoforms in EBV-B cells (top panel) and SV40-fibroblasts (bottom panel) from healthy controls (C1, C2, and C3) and patients (P1, P2, and P3). Quantitative RT-PCR was performed with primers specific for JNK1α1/JNK1α2 and JNK1β1/JNK1β2 mRNAs. α/β, total mRNA corresponding to JNK1α1, JNK1α2, JNK1β1, and JNK1β2; α, total mRNA corresponding to JNK1α1 and JNK1α2; β, total mRNA corresponding to JNK1β1 and JNK1β2. The values shown are the means ± SEM of three independent experiments. *, P < 0.05, **, P < 0.01, and ****, P < 0.0001; in unpaired t tests. (E and F) Immunoblot of JNK1 in EBV-B cells and SV40-fibroblasts from healthy controls (C1, C2, and C3) and patients (P1, P2, and P3) (E), and in HEK293T cells transfected with plasmids encoding four WT JNK1 isoforms (α1, α2, β1, and β2) and two mutants (ES and IR) inserted into the pTRIP-SFFV vector or the pCMV6-AN-Myc-DDK vector (F). Endogenous JNK1 was detected with an anti-JNK1 antibody recognizing the N-terminus of JNK1. Myc-tagged JNK1 was detected with an anti-Myc antibody. EV, empty vector. The data shown are representative of three independent experiments (A, B, E, and F).
Impaired IL-17A/F signaling in patients’ fibroblasts
Human IL-17A, IL-17F, and IL-17A/F (referred to collectively as IL-17A/F) can activate JNK1 after binding to IL-17RA/IL-17RC, which is mostly expressed in various non-hematopoietic cells, thereby inducing the production of pro-inflammatory cytokines, chemokines, and antimicrobial peptides (40, 41). Upon stimulation with IL-17A/F, SV40-fibroblasts from the patients produced abnormally small amounts of growth-regulated oncogene-α (GRO-α) and IL-6, whereas SV40-fibroblasts from an IL-17RA-deficient patient did not respond at all (Fig. 3A). Similar results were obtained with primary fibroblasts (fig. S2A). The patients’ cells had subnormal-to-normal responses to tumor necrosis factor-α (TNF-α) and IL-1β (Fig. 3B and fig. S2B). Moreover, the activation of AP-1 (c-Jun/ATF-2), unlike that of ERK1/2, p38, and NF-κB, was impaired in the patients’ SV40-fibroblasts following stimulation with IL-17A, as shown by western blotting (fig. S2C). By contrast, AP-1 was normally activated by TNF-α and IL-1β (fig. S2D). Fibroblasts and leukocytes from the patients also responded normally to lymphotoxin α1β2 (LTα1β2) (IL-8 production) and Toll-like receptor (TLR) agonists (IL-6 and IL-8 production), respectively (fig. S2, E to G). Peripheral blood mononuclear cells (PBMCs) responded normally to IL-2 in combination with IL-17E (IL-5 production) (fig. S2H). Lentiviral transduction of the patients’ SV40-fibroblasts with cDNAs encoding WT JNK1 isoforms, JNK1α1 and JNK1β1 in particular, but not with any of the mutant isoforms, restored the response to IL-17A (Fig. 3C and fig. S2I). This finding is consistent with the predominant protein expression of JNK1α1 and JNK1β1 in control SV40-fibroblasts (Fig. 2E). Moreover, the induction of GRO-α and IL-6 in control SV40-fibroblasts was not affected by the overexpression of any mutant JNK1 isoform, suggesting that the mutant allele is not dominant-negative (Fig. 3C and fig. S2I). This is consistent with the purifying selection exerted on the MAPK8 locus (34) (fig. S1D). By contrast, the RNAi-mediated knockdown of MAPK8 impaired the response to IL-17A in control fibroblasts (Fig. 3D and fig. S2, J and K). Finally, we performed RNA-Seq to delineate the range of IL-17A-responsive genes in primary fibroblasts. The number of upregulated or downregulated genes in response to IL-17A was much lower in the patients (fig. S2L). Several IL-17A/F target genes, including CXCL1, CXCL2, IL6, IL8, C3, and ICAM1, were less induced in the patients’ cells (fig. S2M). Approximately 60% of IL-17RA/IL-17RC-dependent genes were JNK1-dependent (fig. S2N). Collectively, these findings indicate that heterozygosity for the private MAPK8 c.311+1G>A loss-of-expression variant underlies a distinctive AD cellular phenotype, with impaired responses to IL-17A/F in fibroblasts, by haploinsufficiency. Moreover, impaired cellular responses to IL-17A/F in fibroblasts, and possibly in other cells, contribute to CMC (42, 43).
Fig. 3. The MAPK8 variant impairs fibroblast responses to IL-17A/F.
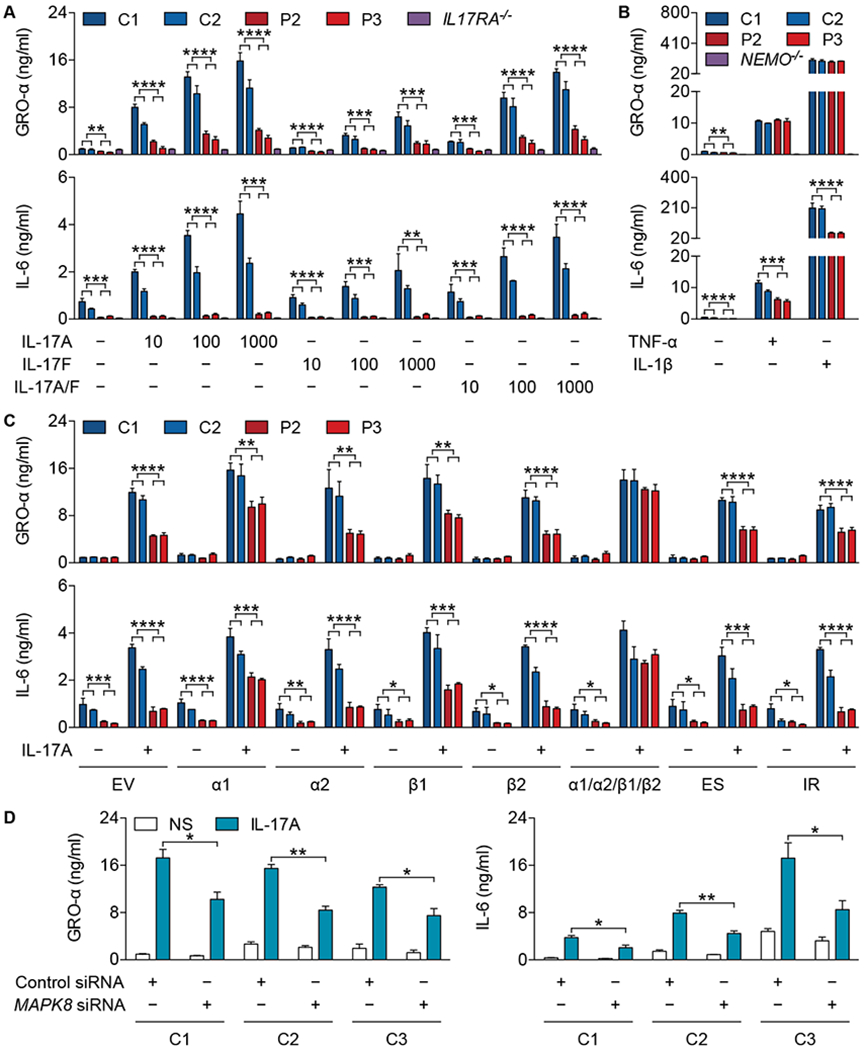
(A) Production of GRO-α (top panel) and IL-6 (bottom panel) by SV40-fibroblasts from healthy controls (C1 and C2), patients (P2 and P3), and an IL-17RA-deficient (IL17RA−/−) patient (16) stimulated with IL-17A, IL-17F, or IL-17A/F (10, 100, or 1000 ng/mL) for 24 h. (B) Production of GRO-α (top panel) and IL-6 (bottom panel) by SV40-fibroblasts from healthy controls (C1 and C2), patients (P2 and P3), and a NEMO-deficient (NEMO−/−) patient (92) stimulated with TNF-α (20 ng/mL) or IL-1β (10 ng/mL) for 24 h. (C) Production of GRO-α (top panel) and IL-6 (bottom panel) by SV40-fibroblasts from healthy controls (C1 and C2) and patients (P2 and P3) transfected with empty vector (EV) or plasmids encoding WT JNK1α1 (α1), JNK1α2 (α2), JNK1β1 (β1), JNK1β2 (β2), all four isoforms (α1/α2/β1/β2), JNK1ES (ES), or JNK1IR (IR), in the presence of IL-17A (100 ng/mL), for 24 h. (D) Production of GRO-α (left panel) and IL-6 (right panel) by primary fibroblasts from healthy controls (C1, C2, and C3) transfected with control siRNA (50 nM) or MAPK8 siRNA (50 nM) for 24 h and then stimulated with IL-17A (100 ng/mL) for an additional 24 h. The values shown are the means ± SEM of three independent experiments (A-D). *, P < 0.05, **, P < 0.01, ***, P < 0.001, and ****, P < 0.0001; in unpaired t tests (A-D).
Low proportions of ex vivo or in vitro differentiated TH17 cells
Given that mouse JNK1 is important for T-cell activation and differentiation (44–46), and that human TGF-β activates JNK1 (47) and is essential for TH17 differentiation in vitro (48–50), we also investigated the development and function of T cells in the patients, testing the hypothesis that impaired TH17 development in the patients might also contribute to their CMC. The frequencies of naïve and CD45RA+ effector memory (EMRA) CD4+ and CD8+ T cells in the patients were slightly higher, whereas those of central (CM) and effector memory (EM) CD4+ and CD8+ T cells were correspondingly slightly lower than those in healthy controls (Fig. 4A). The patients had higher proportions of TH1 cells and lower proportions of TH17 cells than controls, but normal proportions of the TH2, TH1*, TFH, and Treg subsets among circulating CD4+ T cells, as shown by flow cytometry (51) (Fig. 4B). Normal amounts of IL-17A and IL-22 were secreted by whole blood stimulated with PMA plus ionomycin (Fig. 4C). Ex vivo memory CD4+ T cells also expressed IL-17A and IL-17F, albeit in the lower part of the control range, and IFN-γ after stimulation with T-cell activation and expansion beads (TAE; anti-CD2/CD3/CD28 mAbs-conjugated beads) and PMA plus ionomycin (Fig. 4D). The patients’ naïve CD4+ T cells produced less IL-17A and IL-17F than control cells when cultured under TH17-polarizing conditions (Fig. 4, E and F). This difference was more pronounced when memory CD4+ T cells were tested under the same conditions (Fig. 4G). Finally, the percentages of transitional, naïve, and memory B cells, and of class-switched memory B cells were normal in these patients (fig. S3, A and B). The abilities of naïve and memory B cells to differentiate into antibody-secreting cells were also intact (fig. S3, C and D). Overall, the ability of T cells to produce IL-17A and IL-17F was 50% lower (ex vivo) and 75% lower (in vitro) in patients heterozygous for the MAPK8 mutation. The ex vivo development of Treg cells was largely unaffected, consistent with the absence of overt autoimmunity in the patients. The CMC in these patients is, thus, a combined consequence of lower proportions of TH17 cells and impaired cellular responses to IL-17A/F. Both human IL-17A/F- and IL-17RA/IL-17RC-dependent mucocutaneous immunity to C. albicans are, therefore, dependent on JNK1.
Fig. 4. Compromised T-cell differentiation in the patients.
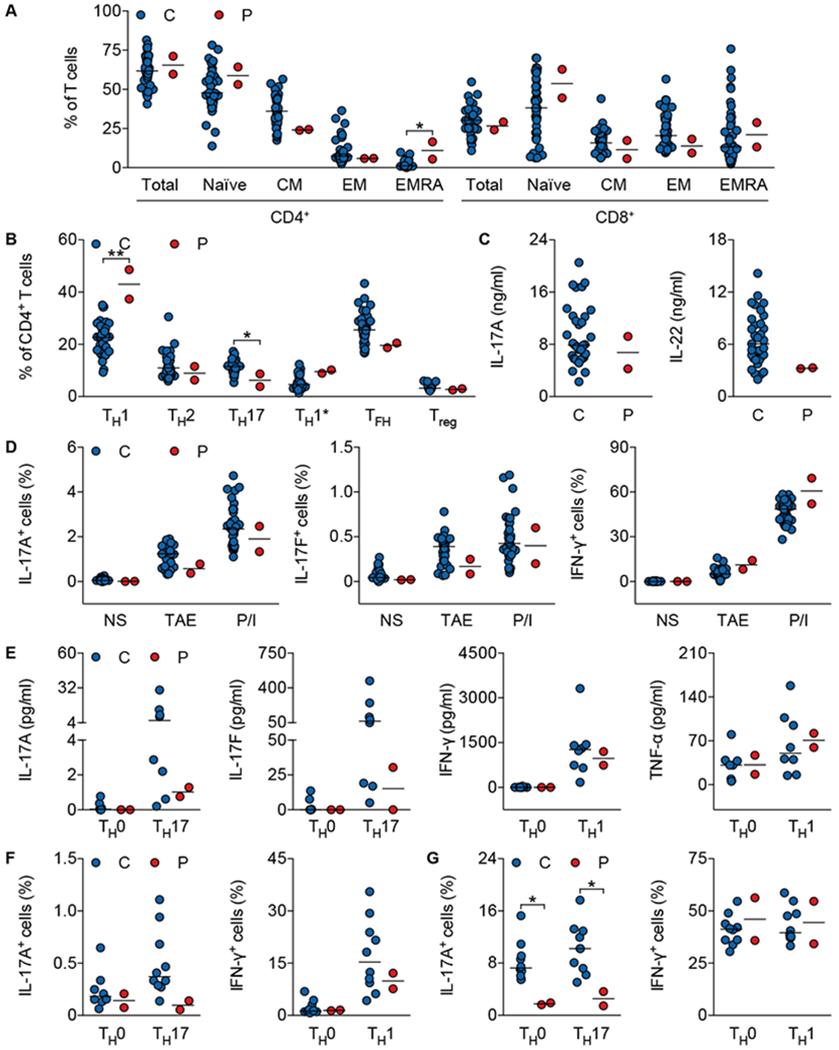
(A) Percentage of total, naïve (CCR7+CD45RA+), central memory (CM; CCR7+CD45RA−), effector memory (EM; CCR7−CD45RA−), or CD45RA+ effector memory (EMRA; CCR7−CD45RA+) CD4+ and CD8+ T cells from healthy controls (n=40) and patients (P2 and P3). (B) Frequency of TH1 (CXCR5−CXCR3+CCR6−), TH2 (CXCR5−CXCR3−CCR6−CCR4+), TH17 (CXCR5−CXCR3−CCR6+CCR4+), TH1* (CXCR5−CXCR3+CCR6+CCR4+), TFH (CXCR5+), and Treg (CD25+FOXP3+) subsets among CD4+ T cells from healthy controls (TH1, TH2, TH17, TH1*, and TFH, n=34; Treg, n=17) and patients (P2 and P3). (C) Production of IL-17A and IL-22 by whole blood from healthy controls (n=33) and patients (P2 and P3) after stimulation with PMA plus ionomycin for 24 h. (D) Percentage of IL-17A+, IL-17F+, and IFN-γ+ cells among memory CD4+ T cells from healthy controls (n=36) and patients (P2 and P3) activated by T-cell activation and expansion (TAE) beads or PMA plus ionomycin (P/I) for 12 h. (E) Cytokine production by naïve CD4+ T cells from healthy controls (n=8) and patients (P2 and P3) cultured under TH0-, TH17- or TH1-polarizing conditions. (F and G) Frequency of IL-17A+ and IFN-γ+ cells among naïve (F) and memory (G) CD4+ T cells from healthy controls (n=10) and patients (P2 and P3) cultured under TH0-, TH17- or TH1-polarizing conditions. C, Healthy controls; P, P2 and P3. Horizontal bars represent median values (A-G). *, P < 0.05 and **, P < 0.01; in two-tailed Mann-Whitney tests (A-G).
Normal extracellular matrix organization but poor migratory capabilities of patients’ fibroblasts
We subsequently investigated the pathogenesis of the novel and complex CTD phenotype of the patients. Previous studies have proposed an in vitro fibroblast phenotype common to most EDS patients, but apparently not observed in other inherited CTDs (52–55). This phenotype is characterized by generalized fibronectin-ECM (FN-ECM) disarray, low levels of expression of the canonical integrin receptor α5β1, and the recruitment of αvβ3 integrin (52–55). EDS fibroblasts also seem to display little or no type III collagen deposition in the ECM (COLLIII-ECM) and a variable disorganization of type V collagen (COLLV-ECM) (52–55). A specific myofibroblast-like phenotype of hEDS has also been proposed, based on the organization of α-smooth muscle actin (α-SMA), cadherin-11 (CAD-11) expression, and enhanced cell migration (56). Unlike cells from EDS patients, the primary fibroblasts of P2 displayed no FN-ECM disarray, and α5β1 integrin was organized as in control fibroblasts (Fig. 5A). Despite the low levels of COLLIII-ECM and a barely detectable organization of COLLV-ECM, P2’s fibroblasts expressed the canonical collagen receptor, α2β1 integrin, normally, unlike EDS cells (Fig. 5A). The myofibroblast-specific markers α-SMA and CAD-11 were absent from the cells of P2, whereas they were present on hEDS fibroblasts (Fig. 5A). Consistent with this finding, the fibroblasts of P2 did not have the enhanced migratory capability reported for some hEDS fibroblasts, as shown by in vitro scratch and Transwell assays (Fig. 5, B and C). Instead, the fibroblasts of P2, like some cEDS cells, migrated poorly (Fig. 5, B and C), probably accounting for the poor wound healing observed in the patients (see the “Case reports” section). Overall, these data suggest that, even though the clinical presentation in these patients overlaps with EDS, and despite the 2017 EDS diagnostic criteria for hEDS being met (22), the in vitro fibroblast phenotype of these patients is apparently different from that proposed for EDS in general, and for hEDS in particular (52–56).
Fig. 5. Impaired response to TGF-β in the patients’ fibroblasts.
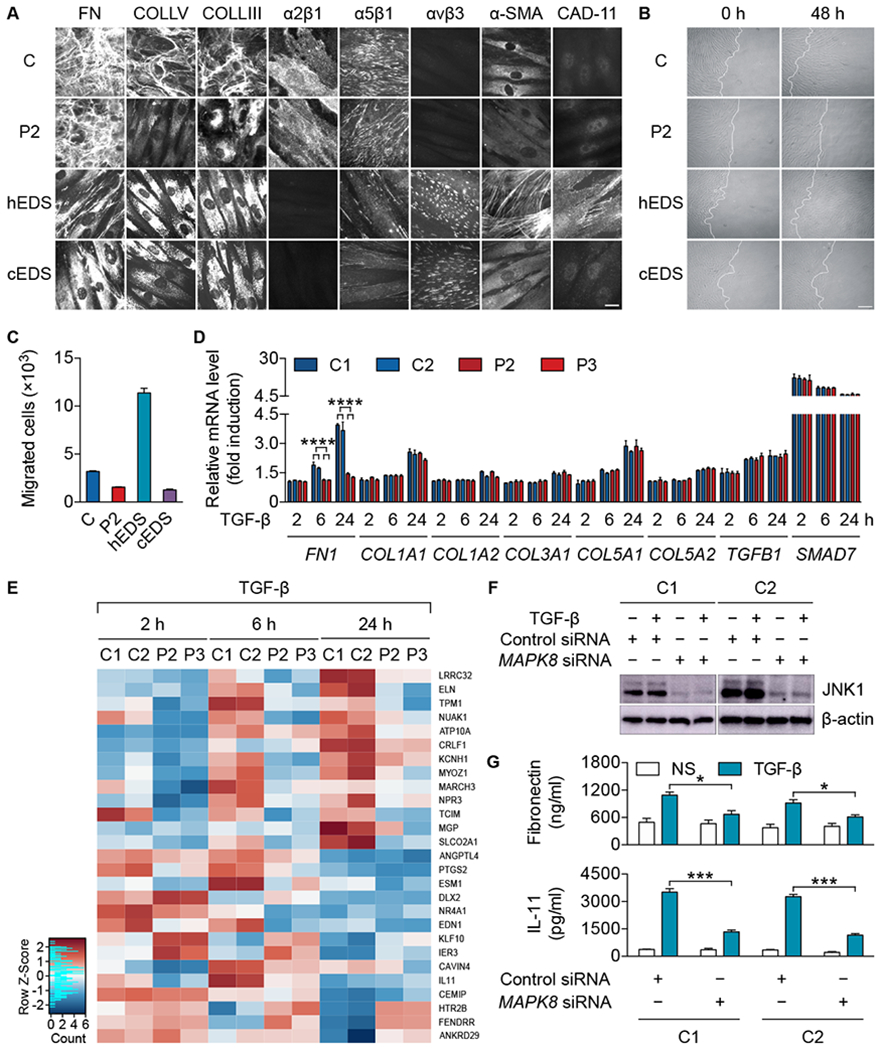
(A) Immunofluorescence of fibronectin (FN), type V collagen (COLLV), type III collagen (COLLIII), α2β1, α5β1, and αvβ3 integrins, α-smooth muscle actin (α-SMA), and cadherin-11 (CAD-11) in primary fibroblasts from a healthy control (C), P2, a patient with hEDS (hEDS) (56), and a patient with cEDS (cEDS) (93). Scale bar: 10 μm. (B) In vitro scratch assay with primary fibroblasts from a healthy control (C), P2, a patient with hEDS (hEDS) (56), and a patient with cEDS (cEDS) (93). Images were captured at 0 and 48 h after scratching. Scale bar: 100 μm. (C) Transwell assay with primary fibroblasts from a healthy control (C), P2, a patient with hEDS (hEDS) (56), and a patient with cEDS (cEDS) (93). (D) mRNA induction in primary fibroblasts from healthy controls (C1 and C2) and patients (P2 and P3) stimulated with TGF-β (10 ng/mL) for the indicated times. (E) The top 10 upregulated or downregulated genes in terms of absolute fold change, in primary fibroblasts from healthy controls (C1 and C2) stimulated with TGF-β (10 ng/mL) for 2, 6, and 24 h, with a greater than 1.5-fold change relative to patients (P2 and P3) at each time point. (F and G) Expression of JNK1 protein (F) and production of fibronectin (top panel) and IL-11 (bottom panel) (G) by primary fibroblasts from healthy controls (C1 and C2) transfected with control siRNA (50 nM) or MAPK8 siRNA (50 nM) for 48 h and then stimulated with TGF-β (10 ng/mL) for an additional 24 h. NS, non-stimulated conditions. The values shown are the means ± SEM of two (C) or three (D and G) independent experiments. *, P < 0.05, ***, P < 0.001, and ****, P < 0.0001; in unpaired t tests (D and G).
Impaired TGF-β signaling in patients’ fibroblasts
We tested the hypothesis that the patients’ CTD resulted from dysfunctional TGF-β signaling, as this pathway controls the expression of key genes involved in the development and maintenance of the ECM (24). Upon TGF-β stimulation, the patients’ SV40-fibroblasts displayed impaired AP-1 (c-Jun/ATF-2) activation, whereas ERK1/2, p38, and SMAD2/3, were normally activated, as shown by western blotting (fig. S4A). Previous reports have suggested that TGF-β induces the expression of FN in a JNK1-dependent manner (57, 58). Consistent with these findings, the induction of FN production by TGF-β was impaired at both the mRNA and protein levels in the patients’ fibroblasts (Fig. 5D and fig. S4, B and C). The patients did not display spondylometaphyseal dysplasia (SMD), which can be caused by heterozygous FN1 mutations (59), probably because their baseline FN-ECM organization levels were normal (Fig. 5A). By contrast, various SMAD2/3-dependent TGF-β target genes (58, 60), such as COL1A1, COL1A2, COL3A1, COL5A1, and COL5A2, encoding key components of the ECM and mutated in patients with cEDS and other forms of EDS (22), were normally induced by TGF-β in the patients’ cells (Fig. 5D and fig. S4, B and C). Finally, we performed a transcriptomic analysis of the cellular response to TGF-β in primary fibroblasts. The genome-wide transcriptional response to TGF-β was impaired in the patients’ cells (fig. S4D). A number of TGF-β-responsive genes, including ELN, EDN1, IL11, and COMP were not induced in the patients’ cells (Fig. 5E and fig. S4E). Consistently, their induction in control fibroblasts stimulated with TGF-β was impaired by the RNAi-mediated knockdown of MAPK8 (Fig. 5, F and G and fig. S4F). These findings are consistent with previous reports of the presence of AP-1-binding motifs in the regulatory regions of COMP and ELN (61, 62), or of the AP-1-dependent induction of EDN1 and IL11 by TGF-β (63, 64). Mutations in these genes (59, 65–68) or in those encoding the corresponding receptors (69, 70) have already been reported in patients with various CTDs other than EDS, LDS, and MS (table S4). The study of the patients’ fibroblasts thus delineated the transcriptomic impact of impaired JNK1-dependent, SMAD2/3-independent TGF-β signaling. Moreover, fibroblasts from patients with LDS, heterozygous for mutations in TGFBR2 or SMAD3, also showed impaired responses to TGF-β (fig. S4D), consistent with previous studies showing these mutations to be loss-of-function in vitro (71–73). However, their impact differed from that of JNK1 haploinsufficiency, as about 40% of JNK1-dependent genes were TGFBR2/SMAD3-independent (fig. S4G). This is consistent with the clinical differences observed between our patient’s particular CTD (displaying some overlap with hEDS) and LDS. In addition, about 30% of TGFBR2-dependent genes were SMAD3-independent (fig. S4H), potentially accounting for some of the phenotypic differences between LDS patients with TGFBR2 and SMAD3 mutations. Our findings provide a molecular and cellular basis for the complex new form of CTD displayed by the patients, with an impairment of the TGF-β-dependent induction of key ECM components and regulators different from that of patients with another CTD, LDS, who are heterozygous for TGFBR2 or SMAD3 mutations.
Discussion
We have discovered a heterozygous loss-of-expression and loss-of-function mutation of MAPK8 in a three-generation multiplex kindred with a rare combination of classic CMC and novel CTD (Fig. 6). Human JNK1 haploinsufficiency impairs IL-17A/F immunity in two ways, by reducing the responses of fibroblasts to IL-17RA/IL-17RC ligation and by compromising the TGF-β-dependent development of TH17 cells, accounting for the impaired mucocutaneous immunity to C. albicans and subsequent development of CMC in these patients. These findings indicate that IL-17RA/IL-17RC-dependent protective mucocutaneous immunity to C. albicans is JNK1-dependent. We previously described CMC patients with biallelic mutations of ACT1 (19). The findings reported here identify JNK1 as a key component of this antifungal pathway acting downstream from ACT1. They also indicate that haploinsufficiency at the JNK1 locus has an impact on the development of TH17 cells, probably due to the involvement of JNK1 in the TGF-β pathway.
Fig. 6. JNK1-dependent IL-17 and TGF-β signaling.
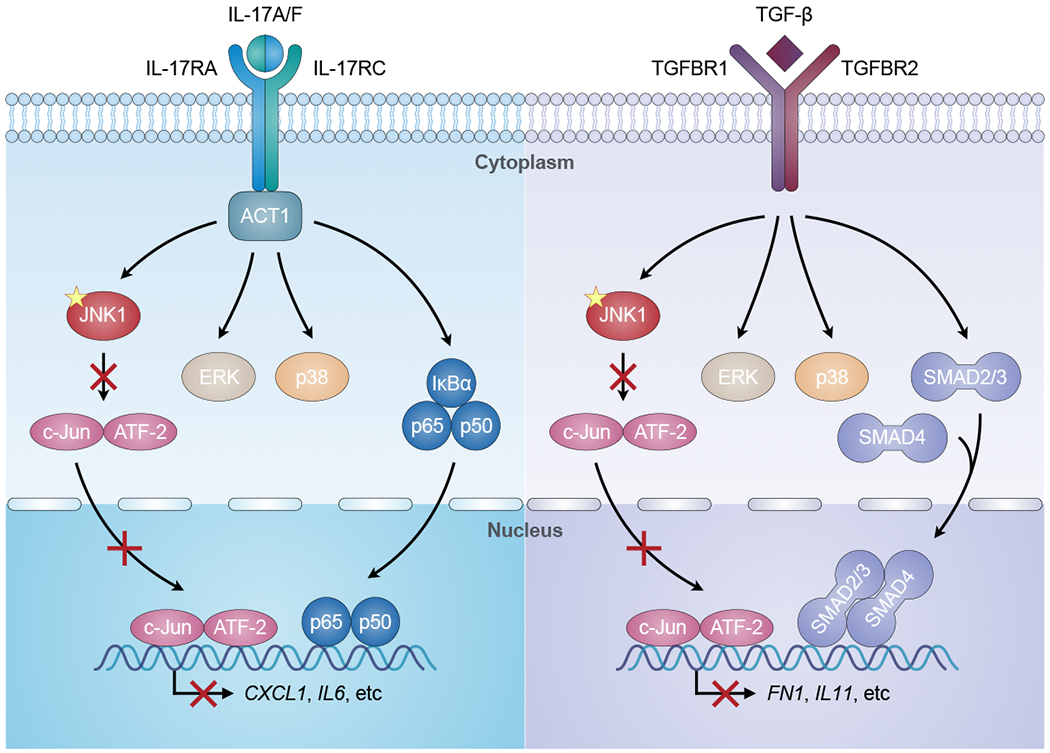
The binding of IL-17A/F to the IL-17RA/IL-17RC receptor facilitates the recruitment of ACT1 to the receptor, which mediates the activation of JNK1, ERK, p38, and NF-κB (p65/p50) signaling, leading to the production of pro-inflammatory cytokines and chemokines (e.g. CXCL1, IL6). Similarly, TGF-β binds to its receptor (TGFBR1/TGFBR2), leading to the activation of JNK1, ERK, p38, and SMAD (SMAD2/3/4) signaling. This pathway ultimately results in the production of extracellular matrix proteins and regulators (e.g. FN1, IL11). The mutation (yellow star) in JNK1 impairs the JNK1-dependent activation of downstream AP-1 (c-Jun/ATF-2), thereby reducing the JNK1-dependent cellular responses to IL-17 and TGF-β.
Our data also suggest that JNK1 haploinsufficiency impairs the c-Jun/ATF-2-dependent, and SMAD2/3-independent, TGF-β-responsive pathway in fibroblasts, a novel cellular phenotype that probably accounts for the patients’ complex and unusual CTD phenotype. Interestingly, the induction of collagen genes mutated in cEDS and other forms of EDS, such as COL1A1 and COL5A1, was intact, whereas that of other ECM proteins, such as COMP and ELN, mutated in patients with other types of CTD (65, 66), was impaired. The impaired induction of genes encoding ECM regulators, such as EDN1 and IL11, may also contribute to the patients’ CTD phenotype. It is also relevant that the impact of heterozygous mutations of MAPK8 differed from that of the TGFBR2 or SMAD3 of patients with LDS, in terms of the transcriptional response to TGF-β. Haploinsufficiency for JNK1 probably defines a novel CTD entity encompassing various clinical manifestations, some of which overlap with EDS, but not LDS. Cellular responses to cytokines other than IL-17A/F and TGF-β were apparently intact in cells from the patients. JNK1-deficient mice have defects of innate and adaptive immunity to various infections (74–76), but their connective tissues have not been studied. MAPK8-heterozygous mice have rarely been studied and seem to be normal (77). In conclusion, the integrity of the human JNK1 pathway is essential for IL-17A/F-dependent mucocutaneous immunity to Candida and for the TGF-β-dependent homeostasis of connective tissues.
Materials and Methods
Study design
We studied three patients from a kindred suffering from CMC and CTD. We analyzed this kindred by WES and found that the patients were heterozygous for a private splice-site mutation in MAPK8, the gene encoding JNK1. We evaluated the impact of this mutation in an overexpression system and in the patients’ cells. We assessed the cellular responses to IL-17A/F and TGF-β of the patients’ fibroblasts, the development and the differentiation properties of the patients’ T and B cells.
Human subjects
The patients (P1, P2, and P3) were followed in their country of residence, France. Another family member (II.1) also participated to the genetic study. Informed consent was obtained from each patient, in accordance with local regulations and a protocol for research on human subjects approved by the institutional review board (IRB) of INSERM. Experiments were performed on samples from human subjects in the United States, France, Italy, and Australia, in accordance with local regulations and with the approval of the IRB of The Rockefeller University, the IRB of INSERM, the local ethical committee of Brescia, and the Sydney South West Area Health Service, respectively.
Whole-exome sequencing
Genomic DNA was extracted from whole blood and sheared with an S2 focused ultrasonicator (Covaris). An adaptor-ligated library was prepared with the TruSeq DNA Sample Prep Kit (Illumina). Exome capture was performed with the SureSelect Human All Exon V5 kit (Agilent Technologies). Paired-end sequencing was performed on a HiSeq 2500 System (Illumina) generating 100-base reads. The sequences were aligned with the GRCh37 build of the human genome reference sequence, with the Burrows-Wheeler Aligner (78). Downstream processing and variant calling were performed with the Genome Analysis Toolkit (79), SAMtools (80), and Picard tools (http://broadinstitute.github.io/picard/). All variants were annotated with in-house annotation software.
Cell culture and transfection
Primary fibroblasts were obtained from skin biopsy specimens and cultured in Dulbecco’s modified Eagle medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco). Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by density gradient centrifugation on Ficoll-Paque PLUS (GE Healthcare Life Sciences). Immortalized simian virus 40 (SV40)-transformed fibroblasts (SV40-fibroblasts) and Epstein-Barr virus (EBV)-transformed B (EBV-B) cells were generated as previously described (81). Human embryonic kidney 293T (HEK293T) (ATCC) and GP2-293 retroviral packaging cells (Clontech) were maintained in DMEM containing 10% FBS. HEK293T and GP2-293 cells were transiently transfected with the aid of X-tremeGENE 9 DNA Transfection Reagent (Roche). Primary fibroblasts were transfected with siRNA in the presence of Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific), in accordance with the manufacturer’s instructions.
Molecular genetics
Genomic DNA was isolated from primary fibroblasts or EBV-B cells with the QIAamp DNA Mini Kit (QIAGEN). A fragment encompassing exon IV and intron IV of MAPK8 was amplified by PCR with specific primers (table S5). The PCR products were analyzed by electrophoresis in 1% agarose gels and sequenced with the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems). Sequencing products were purified by gel filtration on Sephadex G-50 Superfine columns (GE Healthcare Life Sciences) and sequences were analyzed in an ABI 3730 DNA Analyzer (Applied Biosystems).
Plasmids and siRNAs
JNK1α1 and JNK1α2 were amplified from pCDNA3 FLAG JNK1α1 (Addgene) and pCDNA3 FLAG JNK1α2 (Addgene), respectively. JNK1β1 and JNK1β2 were amplified from the cDNA derived from SV40-fibroblasts. The full-length WT isoforms and truncated mutants were inserted into pTRIP-SFFV (82) and the pCMV6-AN-Myc-DDK tagged vector (OriGene), respectively. TA cloning and exon trapping were performed with the pCR4-TOPO vector (Thermo Fisher Scientific) and the pET01 vector (MoBiTec GmbH), respectively, according to the manufacturer’s instructions. Control siRNA (D-001810-10) and MAPK8 siRNA (L-003514-00) were obtained from Dharmacon.
Cell stimulation and cytokine production
SV40- and primary fibroblasts were plated on 24-well plates at a density of 6×104 cells per well, in 0.5 mL DMEM supplemented with 10% FBS. After 24 h, cells were left unstimulated or were stimulated with recombinant human (rh) IL-17A (317-ILB; R&D Systems), rh IL-17F (1335-IL; R&D Systems), rh IL-17A/F (5194-IL; R&D Systems), rh TNF-α (210-TA; R&D Systems), rh IL-1β (201-LB; R&D Systems), rh lymphotoxin α1/β2 (8884-LY; R&D Systems), LTA-SA (tlrl-slta; InvivoGen), Pam3CSK4 (tlrl-pms; InvivoGen), FSL-1 (tlrl-fsl; InvivoGen), Pam2CSK4 (tlrl-pm2s-1; InvivoGen), and lipopolysaccharide (LPS) (L9764; Sigma-Aldrich) for a further 24 h. ELISA kits were used to determine the levels of GRO-α (DY275; R&D Systems), IL-6 (88-7066; Invitrogen), and IL-8 (M9318; Sanquin) in the supernatants. SV40- and primary fibroblasts were cultured in DMEM supplemented with 1% FBS for 24 h and then stimulated with recombinant human TGF-β1 (240-B-002; R&D Systems) for various time periods. Protein levels were determined by ELISA for fibronectin (DY1918-05; R&D Systems), procollagen I (α1) (DY6220-05; R&D Systems), and IL-11 (DY218; R&D Systems). Whole blood was stimulated with IL-1β, Pam3CSK4, heat-killed Staphylococcus aureus (HKSA) (tlrl-hksa; InvivoGen), FSL-1, Pam2CSK4, LPS, R848 (tlrl-r848; InvivoGen), and PMA (P1585; Sigma-Aldrich) plus ionomycin (I3909; Sigma-Aldrich) for 24 h and IL-6 production was measured by ELISA. PBMCs were cultured in X-VIVO 15 (Lonza) containing 5% human serum AB (Lonza) and 100 ng/mL recombinant human thymic stromal lymphopoietin (TSLP) (1398-TS/CF; R&D Systems) for 24 h. Cells were washed and plated on 48-well plates, at a density of 4×106 cells per well, in 0.5 mL of X-VIVO 15 supplemented with 5% human serum AB in the presence of 10 ng/mL recombinant human IL-2 (202-IL; R&D Systems) and 10 ng/mL recombinant human IL-17E (1258-IL; R&D Systems). After 72 h, the amount of IL-5 present in each well was determined with an ELISA kit (DY205; R&D Systems).
Reverse transcription and PCR (RT-PCR)
Total RNA was extracted with the RNeasy Mini Kit (QIAGEN), according to the manufacturer’s instructions. Reverse transcription was carried out with the SuperScript III First-Strand Synthesis System (Invitrogen). Conventional PCR was performed with the ChoiceTaq Blue DNA Polymerase (Denville Scientific) and the amplicons were analyzed by electrophoresis in 2% agarose gels. Quantitative PCR was performed with Fast SYBR Green Master Mix (Applied Biosystems) in the 7500 Fast Real-Time PCR System (Applied Biosystems). The primer pairs used for conventional and quantitative PCR are listed in table S5.
Western blotting
Whole-cell lysates were prepared in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with cOmplete Protease Inhibitor Cocktail (Roche). Proteins were separated by electrophoresis in either 10% Criterion XT Bis-Tris Protein Gels (Bio-Rad) or 4-20% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad) and the resulting bands were transferred onto Immobilon-P PVDF Membrane (Millipore). All blots were incubated overnight with primary antibodies and developed with the Pierce ECL Western Blotting Substrate (Thermo Scientific). The antibodies used in this study included antibodies (from Cell Signaling Technology) against JNK1 (3708), pc-Jun (2361), c-Jun (9165), pATF-2 (9221), ATF-2 (9226); pIκBα (9246), pp65 (3033), pp38 (9211), p38 (9212), pERK1/2 (4370), ERK1/2 (4695), pSMAD2 (3101), SMAD2 (5339), pSMAD3 (9520), SMAD3 (9523), SMAD4 (38454), Myc (2040); as well as IκBα (610690; BD Biosciences), p65 (sc-372; Santa Cruz Biotechnology), and β-actin (AM1829B; Abgent), and the following secondary antibodies: Amersham ECL Mouse IgG, HRP-linked whole Ab (from sheep) (NA931; GE Healthcare Life Sciences) and Amersham ECL Rabbit IgG, HRP-linked whole Ab (from donkey) (NA934; GE Healthcare Life Sciences).
Ex vivo T-cell activation
PBMCs were cultured in 48-well plates, at a density of 3×106 cells per mL, in RPMI 1640 medium (Gibco) containing 10% FBS with T-cell activation and expansion beads (TAE) (130-091-441; Miltenyi Biotec) or PMA plus ionomycin, in the presence of a protein transport inhibitor (GolgiPlug; BD Biosciences). After 12 h, the cells were collected and their expression of the indicated cytokines was assessed by flow cytometry, as previously described (17).
In vitro T-cell differentiation
Naïve and memory CD4+ T cells were isolated and cultured under polarizing conditions, as previously described (6, 83). Briefly, cells were cultured with TAE beads alone (TH0) or under TH1 (IL-12 [20 ng/mL; R&D Systems]) or TH17 (TGF-β1 [2.5 ng/mL; Peprotech], IL-1β [20 ng/mL; Peprotech], IL-6 [50 ng/mL; PeproTech], IL-21 [50 ng/mL; PeproTech], IL-23 [20 ng/mL; eBioscience]) polarizing conditions. After 5 d, the supernatants were harvested and the cells were restimulated with PMA/ionomycin for 6 h. The levels of specific cytokines were determined by intracellular staining and flow cytometry. The secretion of the indicated cytokines was determined with a cytometric bead array (BD Biosciences).
In vitro B-cell differentiation
Naïve and memory B cells were sorted and cultured in the presence of CD40L (200 ng/mL; R&D systems), with or without IL-21 (50 ng/mL; Peprotech) for 7 d, as previously described (83). The production of IgA, IgG, and IgM was assessed by Ig heavy chain-specific ELISA (83).
Flow cytometry
Cells were surface-labeled with CD4-APC-Vio770 anti-human CD4 (clone M-T321; Miltenyi Biotec), Brilliant Violet 421 anti-human CD197 (CCR7) (clone G043H7; BioLegend), PE-CF594 anti-human CD45RA (clone HI100; BD Biosciences), and LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (L34957; Thermo Fisher Scientific). Intracellular staining was performed with the Fixation/Permeabilization Solution Kit (BD Biosciences) and antibodies including Alexa Fluor 488 anti-IL-17A (clone eBio64DEC17; eBioscience), PE anti-IL-17F (clone SHLR17; eBioscience), and Alexa Fluor 700 anti-IFN-γ (clone 4S.B3; eBioscience). Samples were analyzed with a Gallios Flow Cytometer (Beckman Coulter) and FlowJo software.
Immunofluorescence microscopy
Primary fibroblasts were fixed with ice-cold methanol and incubated with antibodies against fibronectin (Sigma-Aldrich), type III collagen (Chemicon), and type V collagen (LifeSpan BioSciences) at a dilution of 1:100, and with anti-α-smooth muscle actin antibody (A2547; Sigma-Aldrich) at a concentration of 2 μg/mL, as previously described (52, 56, 84). For analysis of the α2β1, α5β1, and αvβ3 integrins, cells were fixed with 3% paraformaldehyde (PFA)/60 mM sucrose and permeabilized with 0.5% Triton X-100, as previously reported (84). In particular, cells were incubated with anti-α5β1 (MAB1969; Chemicon), anti-αvβ3 (MAB1976; Chemicon), and anti-α2β1 (MAB1998; Chemicon) integrin antibodies at a concentration of 4 μg/mL for 1 h. Cadherin-11 levels were investigated by fixing cells by incubation with 4% PFA/10 mM sucrose for 10 min, permeabilizing them by incubation with 0.1% Triton X-100 for 10 min, blocking them with by incubation with 2% BSA in PBS for 1 h, and then incubating them with anti-CDH11/cadherin OB antibody (Thermo Fisher Scientific) at a concentration of 2 μg/mL for 3 h, as previously described (56). The cells were washed and then stained with Alexa Fluor 488 anti-rabbit and Alexa Fluor 594 anti-mouse antibodies (Thermo Fisher Scientific), or with rhodamine-conjugated anti-goat IgG antibody (Chemicon) for 1 h. Immunofluorescence signals were acquired with a black-and-white CCD TV camera (SensiCam; PCO Computer Optics GmbH) mounted on a Zeiss Axiovert fluorescence microscope, and digitized with Image-Pro Plus software (Media Cybernetics).
In vitro scratch assay
Primary fibroblasts were plated on 35-mm Petri dishes at a density of 3×104 cells per dish and grown to confluence. The cell monolayers were wounded with a rubber policeman to generate an acellular area and dishes were marked to ensure the recording of the correct area. The monolayers were washed with PBS, rinsed in DMEM plus 10% FBS, and photographed with an inverted microscope at 0 and 48 h after scratching.
Transwell assay
Cell migration was evaluated in a Transwell assay with an 8 μm-pore filter (Corning Costar). Primary fibroblasts (5×104 cells) were resuspended in DMEM without FBS, placed in the upper chamber, and allowed to migrate for 6 h through the polycarbonate membrane into the bottom well, which was filled with DMEM containing 10% FBS. The cells that did not migrate were removed from the upper surface with a cotton swab. The cells that had migrated were collected in the bottom chamber. They were fixed in methanol, stained with the Diff-Quik staining kit (Medion Diagnostic GmbH), and quantified in 10 non-overlapping fields of 1 mm2 with a light microscope.
Microarray and RNA-Seq analyses
Total RNA was extracted with the RNeasy Plus Micro Kit (QIAGEN), according to the manufacturer’s instructions. Microarray analysis was performed with the GeneChip Human Gene 2.0 ST Array (Thermo Fisher Scientific). The raw expression data were normalized in R with the robust multi-array average (RMA) method (85) and the affy R package (86), and processed as previously described (87). RNA-Seq analysis was performed with TruSeq Stranded mRNA (Illumina) and standard polyA-based methods for library preparation. Paired-end sequencing with a read length of 150 bp and ~19 million reads per sample was carried out with a HiSeq 4000 system (Illumina). Raw reads were aligned to the human genome assembly (hg38) with STAR aligner (88). The number of reads mapping to each gene feature was determined with HTSeq (89). Differential expression was analyzed with an in-house script in R with DESeq2 (90) and ComplexHeatmap (91). In brief, fold changes in expression between non-stimulated and stimulated conditions were calculated for each individual and time point separately, and genes were further filtered based on a minimal 1.5-fold change in expression (upregulation or downregulation). The residual responses of the patients were calculated based on the number of responsive genes passing the above filter in both healthy controls (number of responsive genes in a subject / total number of responsive genes in healthy controls) × 100).
Statistical analysis
Unpaired t tests and two-tailed Mann-Whitney tests were used for comparisons of two groups. P < 0.05 was considered statistically significant in all tests performed with Prism software (GraphPad).
Supplementary Material
Case reports
Fig. S1. Identification of a private MAPK8 variant in the patients.
Fig. S2. Impaired IL-17A/F signaling in the patients’ fibroblasts.
Fig. S3. Normal B-cell differentiation in the patients.
Fig. S4. Impaired TGF-β signaling in the patients’ fibroblasts.
Table S1. Immunological parameters of P1, P2, and P3.
Table S2. Careful WES analysis of rare (MAF < 1%) non-synonymous coding variants in the known CMC-, EDS-, LDS-, and MS-causing genes.
Table S3. Heterozygous non-synonymous variants common to P1, P2, and P3.
Table S4. Clinical presentations of disorders caused by mutations in JNK1-dependent TGF-β target genes or in genes encoding the corresponding receptors.
Table S5. Primers used for Sanger sequencing, RT-PCR, and exon trapping.
Data file S1. Raw data used to generate all graphs.
Acknowledgments:
We warmly thank the patients and their family for participating in the study. We thank R. Döffinger, J. Reichenbach, S. Dupuis-Girod, M. Beaudoin, A.-E. Fargeton and I. Meyts for their help searching for additional JNK1-deficient patients. We also thank all the members of the Laboratory of Human Genetics of Infectious Diseases for fruitful discussions and the members of the genomics core facility at Sidra Medicine for their contributions to Illumina library preparation and RNA sequencing.
Funding: This work was funded by the French National Research Agency (ANR) under the “Investments for the future” program (ANR-10-IAHU-01), the HGDIFD project (ANR-14-CE15-0006-01), the EURO-CMC project (ANR-14-RARE-0005-02), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), the Institut National de la Santé et de la Recherche Médicale (INSERM), Paris Descartes University, The Rockefeller University, Sidra Medicine, the Jeffrey Modell Foundation Translational Research Program, the Jeffrey Modell Centers Network, the St. Giles Foundation, the National Center for Research Resources of the National Institutes of Health (NIH), the National Center for Advancing Translational Sciences (NCATS) of the NIH (UL1TR001866), and the National Institute of Allergy and Infectious Diseases (NIAID) of the NIH (R01AI127564). C.S.M. was supported by an Early-Mid Career Research Fellowship from the Office of Health and Medical Research of the New South Wales State Government. T.F. and L.G. were supported by the Ministry of Health of the Czech Republic (16-34414A). D.S. and F.M. were supported by the Research Foundation Flanders (FWO) of Belgium. S.G.T. was supported by the National Health and Medical Research Council of Australia. A.P. was supported by an AP-HP transversal research contract.
Footnotes
Final version of paper: https://immunology.sciencemag.org/content/4/41/eaax7965.long
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The WES data are available from the Sequence Read Archive via accession number PRJNA563623. The microarray and RNA-Seq data have been deposited to the Gene Expression Omnibus and are accessible under accession number GSE137110.
References and Notes
- 1.Li J, Vinh DC, Casanova JL, Puel A, Inborn errors of immunity underlying fungal diseases in otherwise healthy individuals. Curr. Opin. Microbiol 40, 46–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puel A, Cypowyj S, Marodi L, Abel L, Picard C, Casanova JL, Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr. Opin. Allergy Clin. Immunol 12, 616–622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeman AF, Holland SM, Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes. Pediatr. Res 65, 32R–37R (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, Ouachee-Chardin M, Fouyssac F, Girisha KM, Etzioni A, Van Montfrans J, Camcioglu Y, Kerns LA, Belohradsky B, Blanche S, Bousfiha A, Rodriguez-Gallego C, Meyts I, Kisand K, Reichenbach J, Renner ED, Rosenzweig S, Grimbacher B, van de Veerdonk FL, Traidl-Hoffmann C, Picard C, Marodi L, Morio T, Kobayashi M, Lilic D, Milner JD, Holland S, Casanova JL, Puel A, S. G.-o.-F. S. G. International, Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 127, 3154–3164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, Alzahrani M, Al-Muhsen S, Halwani R, Ma CS, Wong N, Soudais C, Henderson LA, Marzouqa H, Shamma J, Gonzalez M, Martinez-Barricarte R, Okada C, Avery DT, Latorre D, Deswarte C, Jabot-Hanin F, Torrado E, Fountain J, Belkadi A, Itan Y, Boisson B, Migaud M, Arlehamn CS, Sette A, Breton S, McCluskey J, Rossjohn J, de Villartay JP, Moshous D, Hambleton S, Latour S, Arkwright PD, Picard C, Lantz O, Engelhard D, Kobayashi M, Abel L, Cooper AM, Notarangelo LD, Boisson-Dupuis S, Puel A, Sallusto F, Bustamante J, Tangye SG, Casanova JL, IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 349, 606–613 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, Pellier I, Zoghi S, Baris S, Keles S, Gray P, Du N, Wang Y, Zerbib Y, Levy R, Leclercq T, About F, Lim AI, Rao G, Payne K, Pelham SJ, Avery DT, Deenick EK, Pillay B, Chou J, Guery R, Belkadi A, Guerin A, Migaud M, Rattina V, Ailal F, Benhsaien I, Bouaziz M, Habib T, Chaussabel D, Marr N, El-Benna J, Grimbacher B, Wargon O, Bustamante J, Boisson B, Muller-Fleckenstein I, Fleckenstein B, Chandesris MO, Titeux M, Fraitag S, Alyanakian MA, Leruez-Ville M, Picard C, Meyts I, Di Santo JP, Hovnanian A, Somer A, Ozen A, Rezaei N, Chatila TA, Abel L, Leonard WJ, Tangye SG, Puel A, Casanova JL, A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci. Immunol 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frey-Jakobs S, Hartberger JM, Fliegauf M, Bossen C, Wehmeyer ML, Neubauer JC, Bulashevska A, Proietti M, Frobel P, Noltner C, Yang L, Rojas-Restrepo J, Langer N, Winzer S, Engelhardt KR, Glocker C, Pfeifer D, Klein A, Schaffer AA, Lagovsky I, Lachover-Roth I, Beziat V, Puel A, Casanova JL, Fleckenstein B, Weidinger S, Kilic SS, Garty BZ, Etzioni A, Grimbacher B, ZNF341 controls STAT3 expression and thereby immunocompetence. Sci. Immunol 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H, Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448, 1058–1062 (2007). [DOI] [PubMed] [Google Scholar]
- 9.de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Janniere L, Fieschi C, Stephan JL, Boileau C, Lyonnet S, Jondeau G, Cormier-Daire V, Le Merrer M, Hoarau C, Lebranchu Y, Lortholary O, Chandesris MO, Tron F, Gambineri E, Bianchi L, Rodriguez-Gallego C, Zitnik SE, Vasconcelos J, Guedes M, Vitor AB, Marodi L, Chapel H, Reid B, Roifman C, Nadal D, Reichenbach J, Caragol I, Garty BZ, Dogu F, Camcioglu Y, Gulle S, Sanal O, Fischer A, Abel L, Stockinger B, Picard C, Casanova JL, Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J. Exp. Med 205, 1543–1550 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC, Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med 205, 1551–1557 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’Shea J, Holland SM, Paul WE, Douek DC, Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452, 773–776 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, Flatot J, Migaud M, Chrabieh M, Kochetkov T, Bolze A, Borghesi A, Toulon A, Hiller J, Eyerich S, Eyerich K, Gulacsy V, Chernyshova L, Chernyshov V, Bondarenko A, Grimaldo RM, Blancas-Galicia L, Beas IM, Roesler J, Magdorf K, Engelhard D, Thumerelle C, Burgel PR, Hoernes M, Drexel B, Seger R, Kusuma T, Jansson AF, Sawalle-Belohradsky J, Belohradsky B, Jouanguy E, Bustamante J, Bue M, Karin N, Wildbaum G, Bodemer C, Lortholary O, Fischer A, Blanche S, Al-Muhsen S, Reichenbach J, Kobayashi M, Rosales FE, Lozano CT, Kilic SS, Oleastro M, Etzioni A, Traidl-Hoffmann C, Renner ED, Abel L, Picard C, Marodi L, Boisson-Dupuis S, Puel A, Casanova JL, Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med 208, 1635–1648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-van der Graaf CA, Kullberg BJ, van der Meer JW, Lilic D, Veltman JA, Netea MG, STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med 365, 54–61 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, Ersvaer E, Perheentupa J, Erichsen MM, Bratanic N, Meloni A, Cetani F, Perniola R, Ergun-Longmire B, Maclaren N, Krohn KJ, Pura M, Schalke B, Strobel P, Leite MI, Battelino T, Husebye ES, Peterson P, Willcox N, Meager A, Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med 207, 299–308 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachee-Chardin M, Toulon A, Bustamante J, Al-Muhsen S, Al-Owain M, Arkwright PD, Costigan C, McConnell V, Cant AJ, Abinun M, Polak M, Bougneres PF, Kumararatne D, Marodi L, Nahum A, Roifman C, Blanche S, Fischer A, Bodemer C, Abel L, Lilic D, Casanova JL, Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med 207, 291–297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova JL, Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332, 65–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levy R, Okada S, Beziat V, Moriya K, Liu C, Chai LY, Migaud M, Hauck F, Al Ali A, Cyrus C, Vatte C, Patiroglu T, Unal E, Ferneiny M, Hyakuna N, Nepesov S, Oleastro M, Ikinciogullari A, Dogu F, Asano T, Ohara O, Yun L, Della Mina E, Bronnimann D, Itan Y, Gothe F, Bustamante J, Boisson-Dupuis S, Tahuil N, Aytekin C, Salhi A, Al Muhsen S, Kobayashi M, Toubiana J, Abel L, Li X, Camcioglu Y, Celmeli F, Klein C, AlKhater SA, Casanova JL, Puel A, Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc. Natl. Acad. Sci. U.S.A 113, E8277–E8285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, Ikinciogullari A, Dogu F, Belkadi A, Levy R, Migaud M, Boisson B, Bolze A, Itan Y, Goudin N, Cottineau J, Picard C, Abel L, Bustamante J, Casanova JL, Puel A, Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med 212, 619–631 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, Belkadi A, Picard C, Abel L, Fieschi C, Puel A, Li X, Casanova JL, An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 39, 676–686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhattad S, Dinakar C, Pinnamaraju H, Ganapathy A, Mannan A, Chronic Mucocutaneous Candidiasis in an Adolescent Boy Due to a Novel Mutation in TRAF3IP2. J. Clin. Immunol 39, 596–599 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Meester JAN, Verstraeten A, Schepers D, Alaerts M, Van Laer L, Loeys BL, Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann. Cardiothorac. Surg 6, 582–594 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B, The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet 175, 8–26 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Blackburn PR, Xu Z, Tumelty KE, Zhao RW, Monis WJ, Harris KG, Gass JM, Cousin MA, Boczek NJ, Mitkov MV, Cappel MA, Francomano CA, Parisi JE, Klee EW, Faqeih E, Alkuraya FS, Layne MD, McDonnell NB, Atwal PS, Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome. Am. J. Hum. Genet 102, 696–705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacFarlane EG, Haupt J, Dietz HC, Shore EM, TGF-beta Family Signaling in Connective Tissue and Skeletal Diseases. Cold Spring Harb. Perspect. Biol 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, et al. , Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352, 337–339 (1991). [DOI] [PubMed] [Google Scholar]
- 26.Verrecchia F, Mauviel A, Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J. Invest. Dermatol 118, 211–215 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Itan Y, Zhang SY, Vogt G, Abhyankar A, Herman M, Nitschke P, Fried D, Quintana-Murci L, Abel L, Casanova JL, The human gene connectome as a map of short cuts for morbid allele discovery. Proc. Natl. Acad. Sci. U.S.A 110, 5558–5563 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itan Y, Mazel M, Mazel B, Abhyankar A, Nitschke P, Quintana-Murci L, Boisson-Dupuis S, Boisson B, Abel L, Zhang SY, Casanova JL, HGCS: an online tool for prioritizing disease-causing gene variants by biological distance. BMC Genomics 15, 256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C, Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37, e67 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J, A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, Scott E, Shah I, Stenson PD, Gleeson J, Cooper DN, Quintana-Murci L, Zhang SY, Abel L, Casanova JL, The mutation significance cutoff: gene-level thresholds for variant predictions. Nat. Methods 13, 109–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itan Y, Shang L, Boisson B, Patin E, Bolze A, Moncada-Velez M, Scott E, Ciancanelli MJ, Lafaille FG, Markle JG, Martinez-Barricarte R, de Jong SJ, Kong XF, Nitschke P, Belkadi A, Bustamante J, Puel A, Boisson-Dupuis S, Stenson PD, Gleeson JG, Cooper DN, Quintana-Murci L, Claverie JM, Zhang SY, Abel L, Casanova JL, The human gene damage index as a gene-level approach to prioritizing exome variants. Proc. Natl. Acad. Sci. U.S.A 112, 13615–13620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald JH, Kreitman M, Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654 (1991). [DOI] [PubMed] [Google Scholar]
- 34.Eilertson KE, Booth JG, Bustamante CD, SnIPRE: selection inference using a Poisson random effects model. PLoS Comput. Biol 8, e1002806 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation C, Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson GL, Nakamura K, The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim. Biophys. Acta 1773, 1341–1348 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hotamisligil GS, Davis RJ, Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol 8, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manning AM, Davis RJ, Targeting JNK for therapeutic benefit: from junk to gold? Nat. Rev. Drug Discov 2, 554–565 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Eckhart L, Ban J, Ballaun C, Weninger W, Tschachler E, Reverse transcription-polymerase chain reaction products of alternatively spliced mRNAs form DNA heteroduplexes and heteroduplex complexes. J. Biol. Chem 274, 2613–2615 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Gaffen SL, Jain R, Garg AV, Cua DJ, The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol 14, 585–600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Casanova JL, Puel A, Mucocutaneous IL-17 immunity in mice and humans: host defense vs. excessive inflammation. Mucosal Immunol 11, 581–589 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puel A, Picard C, Cypowyj S, Lilic D, Abel L, Casanova JL, Inborn errors of mucocutaneous immunity to Candida albicans in humans: a role for IL-17 cytokines? Curr. Opin. Immunol 22, 467–474 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hernandez-Santos N, Gaffen SL, Th17 cells in immunity to Candida albicans. Cell Host Microbe 11, 425–435 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA, Defective T cell differentiation in the absence of Jnk1. Science 282, 2092–2095 (1998). [DOI] [PubMed] [Google Scholar]
- 45.Conze D, Krahl T, Kennedy N, Weiss L, Lumsden J, Hess P, Flavell RA, Le Gros G, Davis RJ, Rincon M, c-Jun NH(2)-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8(+) T cell activation. J. Exp. Med 195, 811–823 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ichiyama K, Sekiya T, Inoue N, Tamiya T, Kashiwagi I, Kimura A, Morita R, Muto G, Shichita T, Takahashi R, Yoshimura A, Transcription factor Smad-independent T helper 17 cell induction by transforming-growth factor-beta is mediated by suppression of eomesodermin. Immunity 34, 741–754 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Zhang YE, Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manel N, Unutmaz D, Littman DR, The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol 9, 641–649 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V, A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol 9, 650–657 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA, IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature 454, 350–352 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, Bustamante J, Okada S, Stoddard JL, Deenick EK, Pelham SJ, Payne K, Boisson-Dupuis S, Puel A, Kobayashi M, Arkwright PD, Kilic SS, El Baghdadi J, Nonoyama S, Minegishi Y, Mahdaviani SA, Mansouri D, Bousfiha A, Blincoe AK, French MA, Hsu P, Campbell DE, Stormon MO, Wong M, Adelstein S, Smart JM, Fulcher DA, Cook MC, Phan TG, Stepensky P, Boztug K, Kansu A, Ikinciogullari A, Baumann U, Beier R, Roscioli T, Ziegler JB, Gray P, Picard C, Grimbacher B, Warnatz K, Holland SM, Casanova JL, Uzel G, Tangye SG, Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J. Allergy Clin. Immunol 136, 993–1006 e1001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiarelli N, Carini G, Zoppi N, Dordoni C, Ritelli M, Venturini M, Castori M, Colombi M, Transcriptome-Wide Expression Profiling in Skin Fibroblasts of Patients with Joint Hypermobility Syndrome/Ehlers-Danlos Syndrome Hypermobility Type. PLoS One 11, e0161347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zoppi N, Chiarelli N, Ritelli M, Colombi M, Multifaced Roles of the alphavbeta3 Integrin in Ehlers-Danlos and Arterial Tortuosity Syndromes’ Dermal Fibroblasts. Int. J. Mol. Sci. 19, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mendoza-Londono R, Chitayat D, Kahr WH, Hinek A, Blaser S, Dupuis L, Goh E, Badilla-Porras R, Howard A, Mittaz L, Superti-Furga A, Unger S, Nishimura G, Bonafe L, Extracellular matrix and platelet function in patients with musculocontractural Ehlers-Danlos syndrome caused by mutations in the CHST14 gene. Am. J. Med. Genet. A 158A, 1344–1354 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Syx D, Van Damme T, Symoens S, Maiburg MC, van de Laar I, Morton J, Suri M, Del Campo M, Hausser I, Hermanns-Le T, De Paepe A, Malfait F, Genetic heterogeneity and clinical variability in musculocontractural Ehlers-Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat 36, 535–547 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Zoppi N, Chiarelli N, Binetti S, Ritelli M, Colombi M, Dermal fibroblast-to-myofibroblast transition sustained by alphavss3 integrin-ILK-Snail1/Slug signaling is a common feature for hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. Biochim. Biophys. Acta 1864, 1010–1023 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Hocevar BA, Brown TL, Howe PH, TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 18, 1345–1356 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M, Deng C, Kucherlapati R, Bottinger EP, Roberts AB, Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J. Biol. Chem 276, 19945–19953 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Lee CS, Fu H, Baratang N, Rousseau J, Kumra H, Sutton VR, Niceta M, Ciolfi A, Yamamoto G, Bertola D, Marcelis CL, Lugtenberg D, Bartuli A, Kim C, Hoover-Fong J, Sobreira N, Pauli R, Bacino C, Krakow D, Parboosingh J, Yap P, Kariminejad A, McDonald MT, Aracena MI, Lausch E, Unger S, Superti-Furga A, Lu JT, Baylor-Hopkins G Center for Mendelian, Cohn DH, Tartaglia M, Lee BH, Reinhardt DP, Campeau PM, Mutations in Fibronectin Cause a Subtype of Spondylometaphyseal Dysplasia with “Corner Fractures”. Am. J. Hum. Genet 101, 815–823 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verrecchia F, Chu ML, Mauviel A, Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem 276, 17058–17062 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Deere M, Rhoades Hall C, Gunning KB, LeFebvre V, Ridall AL, Hecht JT, Analysis of the promoter region of human cartilage oligomeric matrix protein (COMP). Matrix Biol. 19, 783–792 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Oleggini R, Gastaldo N, Di Donato A, Regulation of elastin promoter by lysyl oxidase and growth factors: cross control of lysyl oxidase on TGF-beta1 effects. Matrix Biol. 26, 494–505 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Shi-Wen X, Rodriguez-Pascual F, Lamas S, Holmes A, Howat S, Pearson JD, Dashwood MR, du Bois RM, Denton CP, Black CM, Abraham DJ, Leask A, Constitutive ALK5-independent c-Jun N-terminal kinase activation contributes to endothelin-1 overexpression in pulmonary fibrosis: evidence of an autocrine endothelin loop operating through the endothelin A and B receptors. Mol. Cell. Biol 26, 5518–5527 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang W, Yang L, Yang YC, Leng SX, Elias JA, Transforming growth factor-beta stimulates interleukin-11 transcription via complex activating protein-1-dependent pathways. J. Biol. Chem 273, 5506–5513 (1998). [DOI] [PubMed] [Google Scholar]
- 65.Briggs MD, Hoffman SM, King LM, Olsen AS, Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines ES, et al. , Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat. Genet 10, 330–336 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Zhang MC, He L, Giro M, Yong SL, Tiller GE, Davidson JM, Cutis laxa arising from frameshift mutations in exon 30 of the elastin gene (ELN). J. Biol. Chem 274, 981–986 (1999). [DOI] [PubMed] [Google Scholar]
- 67.Gordon CT, Petit F, Kroisel PM, Jakobsen L, Zechi-Ceide RM, Oufadem M, Bole-Feysot C, Pruvost S, Masson C, Tores F, Hieu T, Nitschke P, Lindholm P, Pellerin P, Guion-Almeida ML, Kokitsu-Nakata NM, Vendramini-Pittoli S, Munnich A, Lyonnet S, Holder-Espinasse M, Amiel J, Mutations in endothelin 1 cause recessive auriculocondylar syndrome and dominant isolated question-mark ears. Am. J. Hum. Genet 93, 1118–1125 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munroe PB, Olgunturk RO, Fryns JP, Van Maldergem L, Ziereisen F, Yuksel B, Gardiner RM, Chung E, Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat. Genet 21, 142–144 (1999). [DOI] [PubMed] [Google Scholar]
- 69.Gordon CT, Weaver KN, Zechi-Ceide RM, Madsen EC, Tavares AL, Oufadem M, Kurihara Y, Adameyko I, Picard A, Breton S, Pierrot S, Biosse-Duplan M, Voisin N, Masson C, Bole-Feysot C, Nitschke P, Delrue MA, Lacombe D, Guion-Almeida ML, Moura PP, Garib DG, Munnich A, Ernfors P, Hufnagel RB, Hopkin RJ, Kurihara H, Saal HM, Weaver DD, Katsanis N, Lyonnet S, Golzio C, Clouthier DE, Amiel J, Mutations in the endothelin receptor type A cause mandibulofacial dysostosis with alopecia. Am. J. Hum. Genet 96, 519–531 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nieminen P, Morgan NV, Fenwick AL, Parmanen S, Veistinen L, Mikkola ML, van der Spek PJ, Giraud A, Judd L, Arte S, Brueton LA, Wall SA, Mathijssen IM, Maher ER, Wilkie AO, Kreiborg S, Thesleff I, Inactivation of IL11 signaling causes craniosynostosis, delayed tooth eruption, and supernumerary teeth. Am. J. Hum. Genet 89, 67–81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, Hoedemaekers YM, Willemsen R, Severijnen LA, Venselaar H, Vriend G, Pattynama PM, Collee M, Majoor-Krakauer D, Poldermans D, Frohn-Mulder IM, Micha D, Timmermans J, Hilhorst-Hofstee Y, Bierma-Zeinstra SM, Willems PJ, Kros JM, Oei EH, Oostra BA, Wessels MW, Bertoli-Avella AM, Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet 43, 121–126 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, Kempers MJ, Fishman EK, Chen Y, Myers L, Bjeda D, Oswald G, Elias AF, Levy HP, Anderlid BM, Yang MH, Bongers EM, Timmermans J, Braverman AC, Canham N, Mortier GR, Brunner HG, Byers PH, Van Eyk J, Van Laer L, Dietz HC, Loeys BL, Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet 44, 922–927 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meester JA, Vandeweyer G, Pintelon I, Lammens M, Van Hoorick L, De Belder S, Waitzman K, Young L, Markham LW, Vogt J, Richer J, Beauchesne LM, Unger S, Superti-Furga A, Prsa M, Dhillon R, Reyniers E, Dietz HC, Wuyts W, Mortier G, Verstraeten A, Van Laer L, Loeys BL, Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med 19, 386–395 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rincon M, Davis RJ, Regulation of the immune response by stress-activated protein kinases. Immunol. Rev 228, 212–224 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Van der Velden J, Janssen-Heininger YM, Mandalapu S, Scheller EV, Kolls JK, Alcorn JF, Differential requirement for c-Jun N-terminal kinase 1 in lung inflammation and host defense. PLoS One 7, e34638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang YQ, Ma X, Lu L, Zhao L, Zhang X, Xu Q, Wang Y, Defective antiviral CD8 T-cell response and viral clearance in the absence of c-Jun N-terminal kinases. Immunology 142, 603–613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sabapathy K, Kallunki T, David JP, Graef I, Karin M, Wagner EF, c-Jun NH2-terminal kinase (JNK)1 and JNK2 have similar and stage-dependent roles in regulating T cell apoptosis and proliferation. J. Exp. Med 193, 317–328 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA, The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome S Project Data Processing, The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang SY, Clark NE, Freije CA, Pauwels E, Taggart AJ, Okada S, Mandel H, Garcia P, Ciancanelli MJ, Biran A, Lafaille FG, Tsumura M, Cobat A, Luo J, Volpi S, Zimmer B, Sakata S, Dinis A, Ohara O, Garcia Reino EJ, Dobbs K, Hasek M, Holloway SP, McCammon K, Hussong SA, DeRosa N, Van Skike CE, Katolik A, Lorenzo L, Hyodo M, Faria E, Halwani R, Fukuhara R, Smith GA, Galvan V, Damha MJ, Al-Muhsen S, Itan Y, Boeke JD, Notarangelo LD, Studer L, Kobayashi M, Diogo L, Fairbrother WG, Abel L, Rosenberg BR, Hart PJ, Etzioni A, Casanova JL, Inborn Errors of RNA Lariat Metabolism in Humans with Brainstem Viral Infection. Cell 172, 952–965 e918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lahaye X, Satoh T, Gentili M, Cerboni S, Silvin A, Conrad C, Ahmed-Belkacem A, Rodriguez EC, Guichou JF, Bosquet N, Piel M, Le Grand R, King MC, Pawlotsky JM, Manel N, Nuclear Envelope Protein SUN2 Promotes Cyclophilin-A-Dependent Steps of HIV Replication. Cell Rep. 15, 879–892 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma CS, Wong N, Rao G, Nguyen A, Avery DT, Payne K, Torpy J, O’Young P, Deenick E, Bustamante J, Puel A, Okada S, Kobayashi M, Martinez-Barricarte R, Elliott M, Sebnem Kilic S, El Baghdadi J, Minegishi Y, Bousfiha A, Robertson N, Hambleton S, Arkwright PD, French M, Blincoe AK, Hsu P, Campbell DE, Stormon MO, Wong M, Adelstein S, Fulcher DA, Cook MC, Stepensky P, Boztug K, Beier R, Ikinciogullari A, Ziegler JB, Gray P, Picard C, Boisson-Dupuis S, Phan TG, Grimbacher B, Warnatz K, Holland SM, Uzel G, Casanova JL, Tangye SG, Unique and shared signaling pathways cooperate to regulate the differentiation of human CD4+ T cells into distinct effector subsets. J. Exp. Med 213, 1589–1608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zoppi N, Gardella R, De Paepe A, Barlati S, Colombi M, Human fibroblasts with mutations in COL5A1 and COL3A1 genes do not organize collagens and fibronectin in the extracellular matrix, down-regulate alpha2beta1 integrin, and recruit alphavbeta3 Instead of alpha5beta1 integrin. J. Biol. Chem 279, 18157–18168 (2004). [DOI] [PubMed] [Google Scholar]
- 85.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP, Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003). [DOI] [PubMed] [Google Scholar]
- 86.Gautier L, Cope L, Bolstad BM, Irizarry RA, affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Alsina L, Israelsson E, Altman MC, Dang KK, Ghandil P, Israel L, von Bernuth H, Baldwin N, Qin H, Jin Z, Banchereau R, Anguiano E, Ionan A, Abel L, Puel A, Picard C, Pascual V, Casanova JL, Chaussabel D, A narrow repertoire of transcriptional modules responsive to pyogenic bacteria is impaired in patients carrying loss-of-function mutations in MYD88 or IRAK4. Nat. Immunol 15, 1134–1142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anders S, Pyl PT, Huber W, HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gu Z, Eils R, Schlesner M, Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 92.Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, Israel A, Heiss NS, Klauck SM, Kioschis P, Wiemann S, Poustka A, Esposito T, Bardaro T, Gianfrancesco F, Ciccodicola A, D’Urso M, Woffendin H, Jakins T, Donnai D, Stewart H, Kenwrick SJ, Aradhya S, Yamagata T, Levy M, Lewis RA, Nelson DL, Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 405, 466–472 (2000). [DOI] [PubMed] [Google Scholar]
- 93.Ritelli M, Dordoni C, Venturini M, Chiarelli N, Quinzani S, Traversa M, Zoppi N, Vascellaro A, Wischmeijer A, Manfredini E, Garavelli L, Calzavara-Pinton P, Colombi M, Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet J. Rare Dis 8, 58 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, Chrabieh M, Mustapha IB, Ghandil P, Camcioglu Y, Vasconcelos J, Sirvent N, Guedes M, Vitor AB, Herrero-Mata MJ, Arostegui JI, Rodrigo C, Alsina L, Ruiz-Ortiz E, Juan M, Fortuny C, Yague J, Anton J, Pascal M, Chang HH, Janniere L, Rose Y, Garty BZ, Chapel H, Issekutz A, Marodi L, Rodriguez-Gallego C, Banchereau J, Abel L, Li X, Chaussabel D, Puel A, Casanova JL, Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321, 691–696 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van de Laar IM, van der Linde D, Oei EH, Bos PK, Bessems JH, Bierma-Zeinstra SM, van Meer BL, Pals G, Oldenburg RA, Bekkers JA, Moelker A, de Graaf BM, Matyas G, Frohn-Mulder IM, Timmermans J, Hilhorst-Hofstee Y, Cobben JM, Bruggenwirth HT, van Laer L, Loeys B, De Backer J, Coucke PJ, Dietz HC, Willems PJ, Oostra BA, De Paepe A, Roos-Hesselink JW, Bertoli-Avella AM, Wessels MW, Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J. Med. Genet 49, 47–57 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Case reports
Fig. S1. Identification of a private MAPK8 variant in the patients.
Fig. S2. Impaired IL-17A/F signaling in the patients’ fibroblasts.
Fig. S3. Normal B-cell differentiation in the patients.
Fig. S4. Impaired TGF-β signaling in the patients’ fibroblasts.
Table S1. Immunological parameters of P1, P2, and P3.
Table S2. Careful WES analysis of rare (MAF < 1%) non-synonymous coding variants in the known CMC-, EDS-, LDS-, and MS-causing genes.
Table S3. Heterozygous non-synonymous variants common to P1, P2, and P3.
Table S4. Clinical presentations of disorders caused by mutations in JNK1-dependent TGF-β target genes or in genes encoding the corresponding receptors.
Table S5. Primers used for Sanger sequencing, RT-PCR, and exon trapping.
Data file S1. Raw data used to generate all graphs.