

Summary
Periodontitis is a common human inflammatory disease. In this condition, microbiota trigger excessive inflammation in oral mucosal tissues surrounding the dentition, resulting in destruction of tooth-supporting structures (connective tissue and bone). While susceptibility factors for common forms of periodontitis are not clearly understood, studies in patients with single genetic defects reveal a critical role for tissue neutrophils in disease susceptibility. Indeed, various genetic defects in the development, egress from the bone marrow, chemotaxis, and extravasation are clearly linked to aggressive/severe periodontitis at an early age. Here, we provide an overview of genetic defects in neutrophil biology that are linked to periodontitis. In particular, we focus on the mechanisms underlying Leukocyte Adhesion Deficiency-I, the prototypic Mendelian defect of impaired neutrophil extravasation and severe periodontitis.
Keywords: inflammation, LAD-I, neutrophils, periodontitis, PID
1 |. PRIMARY IMMUNODEFICIENCY; LESSONS LEARNED IN HUMAN IMMUNIT Y AND DISEASE SUSCEPTIBILIT Y
Studying naturally occurring mutations in humans is a vital step to uncovering the specific role of particular genes/factors and pathways in human biology. In the field of immunology, this includes understanding the inborn genetic errors that trigger primary immunodeficiency diseases (PIDs).1 Indeed, the recent advances in the field of genomics have added a wealth of information with an ever increasing number of identified genetic defects, revealing new genes involved in the function of the immune system and elucidating roles of known genes whose importance was previously unappreciated.2 To date, most described PIDs are monogenic, rare, and recessive traits, although autosomal dominant PIDs have also been reported.2 Over 150 Mendelian conditions associated with ~130 gene defects and an impaired immune response have been described thus far in humans.3 Various PIDs affect different aspects of the innate and adaptive immune system, ranging from microbial immune protection, to immune regulation and tumor surveillance.3
The examination of patients with rare PIDs has evolved from a process of clinical observation to a detailed study of immune mechanisms in humans. To date, the phenotypic characterization and mechanistic study of PIDs has had its greatest impact in the field of infectious disease, where it has played a central role in dissecting pathways involved in microbial surveillance and immunoprotection.3 The study of PIDs has also been exceptionally informative in understanding the role of immune factors that mediate surveillance and tolerance of the commensal microbiota at barrier surfaces, such as the gastrointestinal tract4 and the oral mucosa.2 At the oral mucosal barrier specifically, phenotypic characterization of patients with PIDs has revealed critical elements of oral immunity that mediate immunoprotection from infection as well as genes/pathways that are critical for maintaining the balance between host and the commensal microbiome.2 Immune mechanisms that regulate the balance between microbiome and host at the oral mucosa are of particular relevance in the common human oral disease, periodontitis.
2 |. PERIODONTITIS; MICROBE-TRIGGERED OR AL MUCOSAL IMMUNOPATHOLOGY AND BONE LOSS (FIGURE 1)
FIGURE 1.
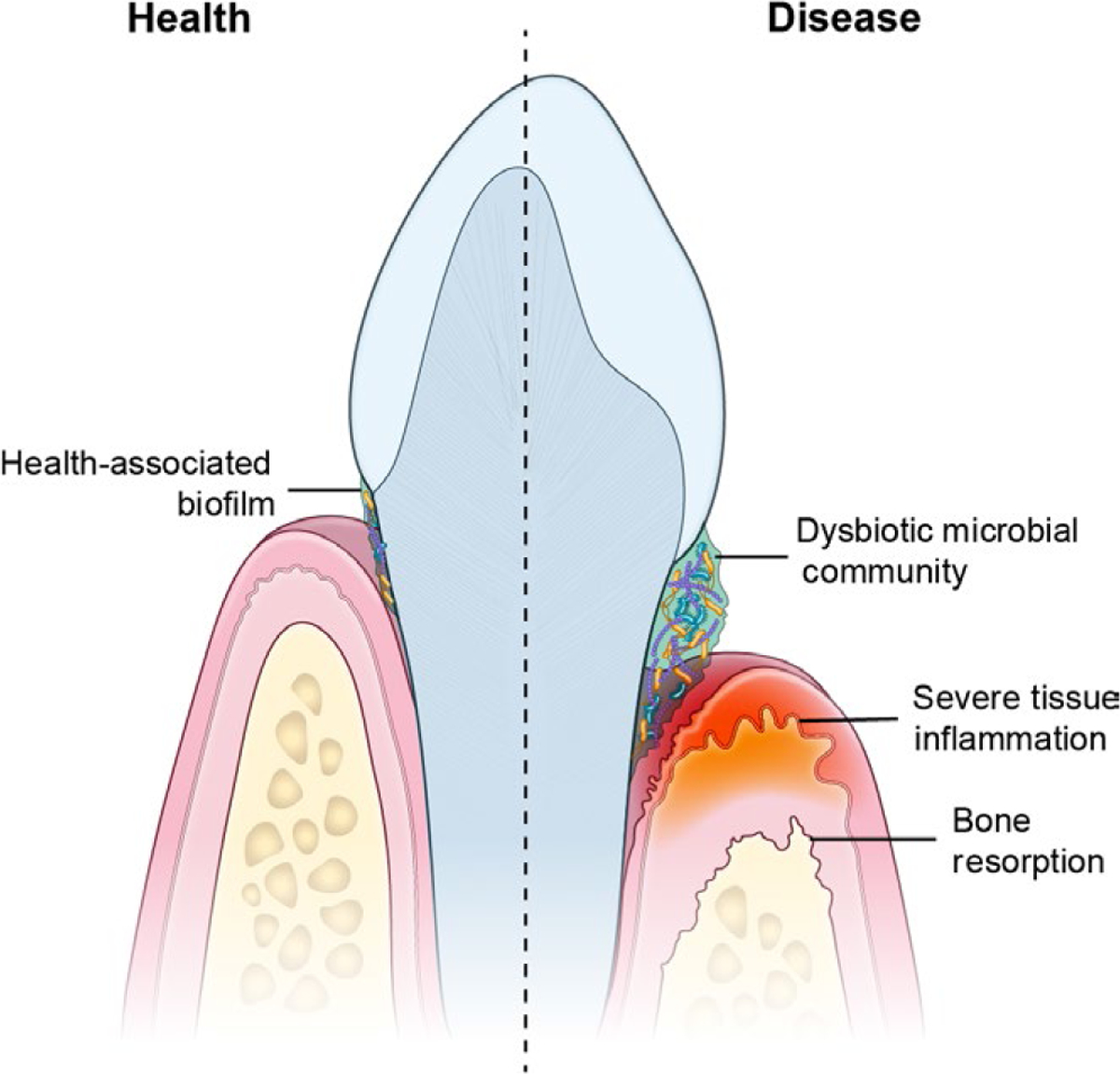
Periodontitis, is a microbiome triggered inflammatory mucosal disease. In periodontitis, a dysbiotic microbiome on the tooth surface triggers inflammation in the mucosal tissues surrounding the tooth. The exaggerated inflammatory response in the mucosa leads to destruction of soft tissues and tooth-supporting bone in susceptible individuals
Periodontitis is one of the most common human inflammatory diseases. In fact, 8%−10% of the general population is documented to present with periodontal disease.5 In this condition, chronic inflammation in mucosal tissues surrounding the dentition (termed gingiva) triggers tissue destruction of the soft tissues and alveolar bone that supports teeth within the maxilla and the mandible (Figure 1). As a consequence of the disease, tooth-supporting structures are destroyed, which in severe cases will lead to loss of the dentition. Interestingly, periodontitis has been proposed as a trigger or aggravating condition in a variety of systemic and distal inflammatory diseases, including diabetes, cardiovascular disease, and Rheumatoid Arthritis.6–8 It is thought that systemic inflammation and/or trans-location of periodontitis-associated microbiota can underlie disease triggering or amplification at distal sites.6
The pathogenesis of periodontitis is not completely understood. However, it is recognized that local tooth-adherent microbial communities are a trigger for disease (Figure 1). Indeed, in periodontitis, microbial communities on the tooth surface become dysbiotic, presenting with an increase in microbial biomass and a shift in abundance of select periodontitis-associated microbial species.6,9,10 Yet, the mechanisms by which local microbial communities trigger exaggerated and destructive inflammatory responses remain unclear.
It is nevertheless well appreciated that host susceptibility is a critical element for disease initiation and progression. Due to the polygenic nature of the disease—as is the case for the majority of inflammatory diseases—and the contribution of environmental influences (eg, smoking), dissecting host susceptibility in periodontitis has been particularly complex.11 In this regard, studying patients with genetic diseases who present with severe periodontitis at an early age, can reveal specific host factors that lead to periodontitis susceptibility.
Identifying genes and pathways that predispose to severe periodontitis in genetic syndromes is particularly important as it may indeed shed light on pathways involved in polygenic common forms of the disease. Moreover, characterizing host factors involved in periodontitis provides unique insights into human oral mucosal immunity, as well as may reveal genes and factors that are most important in the balance between commensal microbial communities and mucosal immune responses at this barrier surface. In fact, the oral mucosa is home to a diverse and rich microbial community and is the first site of encounter of microbes by the immune system prior to entry in the gastrointestinal tract12; dissecting host factors that mediate homeostatsis at this interface is therefore critical.13 To date, the majority of genetic syndromes identified to predispose to severe periodontitis are associated with defects in the neutrophil immune cell subset, revealing a primary role for this immune cell subtype in periodontal homeostasis.14
3 |. THE ESSENTIAL ROLE OF TISSUE NEUTROPHILS IN PROTECTION FROM PERIODONTITIS (FIGURE 2)
FIGURE 2.
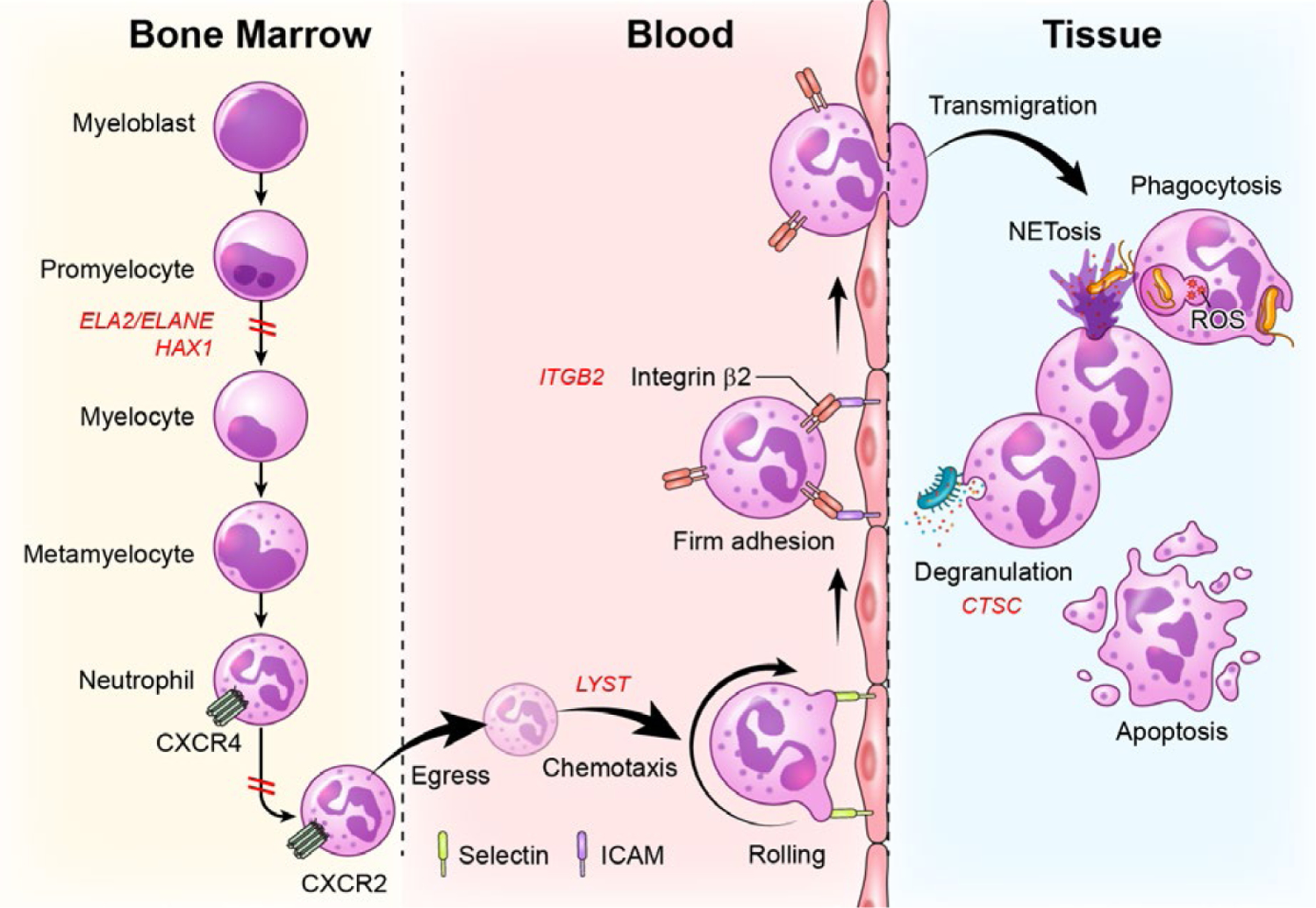
Defects in stages of neutrophil development, trafficking and function are linked to severe periodontitis. Neutrophils develop in the bone marrow in a process termed granulopoiesis. After progressing through discrete steps of maturation (myeloblast, promyelocytes, myelocyte, metamyelocyte, neutrophil), mature neutrophils will downregulate the expression of the chemokine CXCR4 and upregulate CXCR2 to exit the bone marrow via a process termed “egress”. Neutrophils will thereafter follow a chemokine gradient (chemotaxis) and will be led to tissue sites. For entry into tissues, neutrophils will follow discrete steps of tethering and rolling (through interaction with selectins) at the endothelial surface and firm adhesion through β2 integrin-ICAM interactions, which will allow extravasation/transmigration into tissues. In tissues, neutrophils will perform multiple functions, including microbe phagocytosis, degranulation, formation of nuclear extracellular traps (NETs) and will eventually die typically through apoptosis. Genetic defects at all of these stages of development, trafficking and function have been linked to periodontitis. Defects in neutrophil development due to mutations in ELA2 and/or HAX1are linked to severe congenital neutropenia and periodontitis. Defects in neutrophil egress due to gain of function mutations in CXCR4 are linked to WHIM syndrome and periodontitis. Defects in chemotaxis and diapedesis are linked to mutations in LYST in the syndrome Chediak-Higashi. Defects in the process of transmigration into tissues are due to mutations in ITGB2 which disrupts β2 integrin formation leading to Leukocyte Adhesion Deficiency-I and related periodontitis. Finally, defects in the activation and secretion of serine proteases (degranulation) are due to mutations in cysteine protease cathepsin C (CTSC), in the syndrome, Papillon Lefevre, which also predisposes to severe periodontitis at an early age
Multiple genetic defects affecting all stages of neutrophil development, trafficking, transmigration into tissues and in some instances, function have been linked to exceptionally aggressive forms of periodontitis, which present in childhood or adolescence. This current review discusses basic aspects of neutrophil biology and function and presents genetic human defects that affect select aspects of neutrophil development and functionality, predisposing to periodontitis.
4 |. THE NEUTROPHIL
Neutrophils are the most abundant white blood cell in humans.15 Neutrophils were first identified by Paul Elrich, who named them because of their tendency to retain neutral dyes. Metchnikoff, however, was the one that discovered the phagocytic function of neutrophils by demonstrating that injured starfish embryos recruit phagocytic cells, which ingest pathogens and “clear” the injury site.16 This notion that neutrophils are the predominant cell type which rapidly respond to injury and infection is still valid to date. In fact, neutrophils represent the innate immune systems’ first line of defense against the millions of bacteria, fungi, and other microbes that attempt to invade our body daily.
Neutrophils exit the bone marrow as mature cells and their life cycle is primarily orchestrated by granulocyte colony-stimulating factor (G-CSF), a mediator which regulates proliferation, survival, differentiation and trafficking/mobilization.15 Mobilization and recruitment of neutrophils to the inflammatory foci is further mediated through a balance of chemokine signals. Upon activation of diffusing chemical signals from injured or infected sites, neutrophils are recruited to clear microbes and to protect from severe infections. Neutrophils employ four primary mechanisms to kill invading pathogens, namely (a) phagocytosis: uptake of pathogens into a vacuole within the cell; (b) production of reactive oxygen species (ROS) in the pathogen-containing vacuole; (c) fusion and release of neutrophil granules containing various antimicrobial mediators and enzymes to the vacuole; and (d) release of neutrophil extracellular traps (NETs).15,17
Neutrophils are one of the dominant immune cell populations in gingiva in health18,19 and are thought to play important antimicrobial and immune-regulatory roles at this site. In fact, neutrophils continuously extravasate from the circulation into the gingival crevice (pocket), with an estimated 30,000 neutrophils migrating into saliva every minute.20 Interestingly, patients with genetic defects in various stages of neutrophil maturation and function have dominant phenotypes in the oral cavity, particularly in the periodontium (tooth supporting structures), suggesting important site- specific physiological roles for neutrophils in this tissue microenvironemnt. In the remainder of this review, we discuss neutrophil defects in various stages of maturation and function that have been clearly linked to the oral disease, periodontitis.
5 |. DEFECTS IN NEUTROPHIL DEVELOPMENT/GR ANULOPOIESIS: SEVERE COMBINED NEUTROPENIA
5.1 |. Granulopoiesis
Neutrophils develop in the bone marrow from hematopoietic stem cells in a process called “granulopoiesis”.21 Granulopoiesis refers to the formation of granules within the developing neutrophil in the bone marrow between myeloblast and promyelocyte stages of development.22–25 The process of granulopoiesis can be divided into two stages: (a) neutrophil lineage determination and (b) committed granulopoiesis.26 Early neutrophil precursors transition from hematopoietic stem cells (HSCs) to myeloblasts, promyelocytes and immature myelocytes retaining proliferative capabilities. During the transition from myelocytes to metamyelocytes, cells cease to proliferate and commit to the neutrophil lineage.26 Stages of neutrophil development in the bone marrow have been characterized histologically based on cell size, nucleus morphology, and cytosol staining. Granulopoiesis is considered to begin with the appearance of azurophilic (primary) granules in myeloblasts and promyelocytes, which contain large, round nuclei, followed by the production of specific granules in myelocytes and metamyelocytes. During this transition, the round nucleus transforms into a kidney-shaped structure. Next, metamyelocytes transition into band neutrophils, where the nucleus acquires a band-like shape and during this step gelatinase granules are produced. Finally, neutrophils develop Ficollin-1 granules and secretory vesicles and acquire a characteristic segmented nucleus, concluding the process of granulopoiesis.22,24,26–28
5.2 |. Severe Congenital Neutropenia (SCN) and related periodontitis
Defects in the process of granulopoiesis lead to SCN. In SCN, neutrophil differentiation is blocked at the promyelocyte/myelocyte stage (Figure 2),29 with peripheral neutrophil counts below 0.5 × 109/L (1.5–1.8 × 109/L in health) and frequent bacterial/fungal infections. Rolf Kostman first described an autosomal recessive disorder of severe neutropenia in 1956 with absolute neutrophil counts of less than 0.5 × 109/L and severe bacterial infections, later termed “Kostman Syndrome” and associated with mutations in the HAX1 gene.29 However, SCN is mostly associated with autosomal dominant mutations in ELA2/ELANE, which encodes for neutrophil elastase. Although ELA2/ELANE mutations are present in most patients with sporadic severe congenital neutropenia, only some patients with the familial form of the disease carry ELA2/ELANE mutations.30 Neutrophil elastase is a serine protease present in mature myelomonocytes and their immature precursors. It is stored in its active form in azurophilic granules and released upon exposure to inflammatory stimuli.29 ELA2/ELANE mutations are thought to cause neutropenia, as these mutations cause protein misfolding and promote accumulation of neutrophil elastase in the cell endoplasmic reticulum (ER). A feedback signal activates unfolded-protein response (UPR) and ER stress leading to apoptosis.15,29 Mutations in hematopoietic cell-specific Lyn substrate (HCLS) 1-associated gene X1 (HAX1) also lead to SCN. Studies have shown that the interactions among HAX1 and its partner HCLS1 lead to activation of the lymphoid enhancer binding factor (LEF-1) and are essential for G-CSF-triggered granulopoiesis.15,31 Infection susceptibility and prognosis of SCN patients has drastically improved with G-CSF treatment, with more than 90% of patients responding with greater than 1 × 109/L neutrophils in circulation.32
Patients with SCN have been reported to present with severe periodontitis in the primary dentition. Periodontitis in SCN is characterized by gingival inflammation, gingival enlargement and severe bone loss at an early age (in the primary dentition) both in patients with ELA2/ELANE and HAX1 mutations.32 A comprehensive clinical phenotyping study of a cohort of patients with SCN (n = 14) including six patients with ELA2/ELANE mutations and seven with HAX1 mutations,33 revealed severe periodontitis particularly in patients with ELA2/ELANE mutations. Despite G-CSF treatment, all patients with ELA2/ELANE mutations were diagnosed with periodontitis or were rendered edentulous (lacking teeth). Patients with HAX1 mutations treated with G-CSF in this cohort had a lower prevalence of periodontitis compared to patients with ELA2/ELANE mutations.27,33 In general, gingivitis/periodontitis has been reported to persist in SCN even with an increase in neutrophils to 1 × 109/L.32 To date, hematopoietic stem cell transplantation (HSCT) remains the only available cure for SCN, particularly for patients that are refractory to G-CSF treatment.34
6 |. DEFECTS IN NEUTROPHIL EGRESS FROM THE BONE MARROW: WHIM SYNDROME
6.1 |. Egress from the bone marrow
Hematopoietic stem cells (HSC) that develop into neutrophils are located in close proximity to bone-forming osteoblasts in the trabecular regions of long bones near the interface between the bone and bone marrow, called endosteum.35–38 Neutrophils express the chemokine receptor CXCR4, which interacts with its ligand, CXCL12, expressed by perivascular cells and osteoblasts, thus leading to retention of neutrophils in the bone marrow.35,38 As the neutrophil matures, the level of CXCR4 expression decreases, while the chemokine CXCL2 and its receptor, CXCR2, increase (Figure 2).38 This renders them responsive to the chemokine CXCL2 and less responsive to CXCL12 and allows egress from the bone marrow into the circulation.21 Conventional dendritic cells (cDCs) play a role in maintaining neutrophil homeostasis and regulating neutrophil distribution between the bone marrow, peripheral blood and organs via controlled secretion of the chemokines, CXCL1, CXCL2, and CXCL10 and the growth factor, granulocyte colony-stimulating factor (G-CSF), although the exact mechanism remains unexplored.38
6.2 |. WHIM syndrome and periodontitis
WHIM syndrome is considered the prototypic neutrophil egress defect in humans. The term “WHIM” is an acronym for the main clinical manifestations of the syndrome which include warts, hypogammaglobulinemia, recurrent infections, and myelokathexis (impaired egress of mature neutrophils from bone marrow).39,40 WHIM is an autosomal dominant PID caused by mutations in the chemokine receptor CXCR4.41 The signature pathogen in WHIM syndrome is human papillomavirus (HPV), but recurrent bacterial infections are also common and patients are therefore treated with prophylactic antibiotics, intravenous immunoglobulin (IVIg), and G-CSF to prevent infections. Recently, use of the specific CXCR4 antagonist, plerixafor (Mozobil, AMD3100), has shown promise.42 In WHIM syndrome, CXCR4 gain-of-function mutations lead to neutropenia caused by increased retention of neutrophils in the bone marrow.43 In addition to neutropenia, these patients often have lymphopenia and monocytopenia.42
Severe periodontitis, has been reported in multiple cases of WHIM syndrome, with disease presenting as early as puberty.44,45 Periodontitis associated with WHIM syndrome has been reported to progress rapidly, despite standard of care treatment and leads to complete tooth loss at an early age in some cases.45,46 Whether G-CSF treatment and/or use of plerixafor for WHIMS is beneficial to WHIMS-associated periodontitis has not been comprehensively reported to date.
7 |. DEFECTS IN NEUTROPHILEXTR AVASATION INTO TISSUES: L AD-I
7.1 |. Neutrophil extravasation
Extravasation of neutrophils involves the following steps: tethering, rolling, adhesion, crawling, and transmigration. In response to inflammatory stimuli, such as proinflammatory cytokines (TNF-α and Il-1β), lipid mediators, damage- or pathogen-associated molecular patterns (DAMPs and PAMPs), endothelial cells express cell surface adhesion molecules, namely selectins (P-selectin/CD62P and E-selectin/CD62E), which bind to glycosylated ligands expressed on the surface of neutrophils.47 This loose binding, called tethering, enables the neutrophils to roll in the direction of the blood flow toward the chemokine-enriched endothelium, where conformational changes in β2 integrins (CD11a/CD18 or LFA-1 and CD11b/CD18 or MAC-1) mediate firm adhesion by binding to ICAM-1 and ICAM-2 on endothelial cells and luminal crawling.48,49 The interaction between ICAM-1 and β2 integrins mediates crawling and transmigration of neutrophils.50 Neutrophils extravasate from blood vessels mostly via junctions between adjacent endothelial cells (paracellular route), whereas a small percentage of neutrophils migrate through endothelial cells (transcellular route).51,52
7.2 |. Leukocyte adhesion deficiency-I (LAD-I) and Periodontitis
Leukocyte adhesion deficiency-I can be considered the prototypic Mendelian defect in neutrophil extravasation into tissues. LAD-I is a rare disorder of leukocyte adhesion and transmigration, which results from mutations in the ITGB2 gene encoding for the β2 integrin component, CD18.53
Deficiencies in CD18 prevent normal integrin dimerization and leukocyte adhesion to endothelial surfaces, a process essential for extravasation. This primarily affects neutrophil transmigration and results in severe tissue neutropenia. In this sense, LAD-I is a model disease toward understanding the role of neutrophils within tissues. LAD-I has a spectrum of severity which largely correlates with the level of CD18 expression on neutrophils.53 Severe LAD-I is generally classified as <2% of CD18-expressing neutrophils and leads to substantial infant mortality. In fact, mortality for severe LAD-I was reported as 75% by the age of 2 years in a multicenter retrospective evaluation.54 On the other hand, most patients with moderate LAD-I (2%−30% CD18-expressing neutrophils) survive childhood, but present with recurrent infections/immunopathology of skin and mucosal surfaces. A recent comprehensive review of all published cases of LAD-I revealed that patients with moderate LAD-I survive childhood without hematopoietic stem cell transplant (HSCT) treatment; the most frequent manifestation is periodontal disease (>50% of cases), followed by otitis media (36%), sepsis (25%), and perianal skin infections and necrotic ulcers (>10%).54 Periodontitis in LAD-I can be exceptionally severe and start at a very young age, as early as the primary or mixed dentition (termed prepubertal periodontitis).55,56 Disease is characterized by enlarged gingiva, with severe inflammation (redness, swelling, spontaneous bleeding on probing) and rapid loss of supporting bone around teeth (alveolar bone) which often leads to tooth mobility, complete bone loss and tooth loss57 (Figure 3). Periodontitis in LAD-I is recalcitrant to standard of care treatment and antibiotics and patients often lose their entire dentition at a very early age (teenage or young adulthood).56 Significant intraoral findings also include recurrent painful oral ulcers that often impede eating.58,59 Until recently, there has not been an effective treatment for LAD-I periodontitis. To date, only hematopoietic stem cell transplant has been reported to be curative for LAD-I-associated periodontitis.60 However, recent work by our group and colleagues in LAD-I has uncovered novel insights into the pathogenesis of LAD-I-associated periodontitis.
FIGURE 3.

Severe periodontitis in leukocyte adhesion deficiency I. (A) Panoramic oral radiograph of a 15-year-old male with LAD-I. Patient has lost the majority of his teeth due to severe periodontitis. The remaining dentition presents with severe bone loss. Dotted white lines indicate the physiologic level of bone and dotted blue lines demonstrate bone levels in this clinical case. (B) Hematoxylin and eosin (H&E) staining of extracted tooth from a LAD-I patient. Soft tissue surrounding the entire tooth is indicative of complete destruction of tooth supporting bone. Diffused inflammatory infiltrate is observed in all surrounding mucosal tissues
7.3 |. Pathogenesis of LAD-I-associated periodontitis
7.3.1 |. LAD-I periodontitis is a microbe-triggered inflammatory disease
For many years, periodontitis in LAD-I was considered and treated as an infection, with systemic broad-spectrum antibiotics and dental cleanings. However, recent work by our group revealed that periodontitis in LAD-I is not a pure infection, but a microbe-triggered inflammatory disease. In fact, lesions of periodontitis in LAD-I do not display a presence of an invasive infection.61,62 However, microbial communities adjacent to inflammatory lesions of LAD-I-associated periodontitis are significantly different from those observed in healthy individuals. We found that the LAD-I-associated tooth adherent biofilm is characterized by an increased microbial biomass and dysbiotic microbial changes.63 Dysbiotic microbial changes in the LAD-I subgingival microbiome include a reduced microbial diversity with loss of health-associated microbial species and an over representation of select periodontitis-associated species such as Parvimonas micra, Porphyromonas endodontalis, Eubacterium brachy, and Treponema species. Pseudomonas aeruginosa, a bacterium not typically found in subgingival plaque was also detected in LAD-I patients.63 Importantly, despite lack of an invasive microbial infection, microbial products, such as LPS were detected within inflammatory lesions of LAD-I periodontitis and in approximation with inflammatory cells,63 suggesting microbial triggering of inflammatory responses. In vitro studies also demonstrated that the subgingival bacterial communities from LAD-I patients displayed increased potential to stimulate inflammatory responses and to particularly trigger the induction of interleukin (IL-) 23-mediated immunity.
7.3.2 |. LAD-I periodontitis pathogenesis is mediated by exaggerated IL23-IL17 inflammation
Consistent with microbial induction of IL-23-mediated responses in LAD-I, our studies documented an exaggerated IL-23 and IL-17 signature within the inflammatory periodontitis lesions. We found high levels of the cytokines IL-23 and IL-17 and induction of downstream IL-17-dependent neutrophil granulopoiesis factors, such as G-CSF and chemoattractants, such as CXCL1, CXCL2 and CXCL5.64 Importantly, this IL-17 exaggerated response was localized to the mucosal tissues and was not evident in the systemic circulation, revealing the tissue-specific nature of these responses.
From a mechanistic standpoint, IL-23/IL-17 immunity is a shared response at mucosal barrier tissues and typically induced by microbial triggers.65 IL-17 mediates barrier immunity through induction of antimicrobial defenses, maintenance of barrier integrity and largely through recruitment of neutrophils.65,66 It is therefore to be expected that at mucosal sites, where the IL-17 axis is physiologically activated by microbes, the response becomes amplified in a continuous (but unsatisfied) effort to bring neutrophils into the tissue. In fact, there is a well-established mechanism by which neutrophil recruitment is regulated and lack of neutrophil presence is sensed in tissues, termed the “neutrostat”. According to the neutrostat, IL-23/IL-17 triggers neutrophil recruitment by induction of G-CSF and neutrophil chemoattractants.67 After neutrophils enter the tissue and perform their function, apoptosis of tissue neutrophils and subsequent engulfment (efferocytosis) by tissue macrophages becomes the signal for downregulation of IL-23 and downstream responses.67 In LAD-I, in the absence of tissue neutrophils, the IL-23 response fails to downregulate and continuously induces IL-17 and related inflammation. Indeed, preclinical studies by our laboratory and colleagues, have demonstrated that aberrant IL-23/IL-17 responses are the drivers of periodontal immunopathology in LAD-I. Importantly, IL-23 and IL-17 antibody blockade was successful in inhibiting periodontal disease progression in experimental models of LAD-I-associated periodontal disease.64
7.3.3 |. IL23/IL-17 as a plausible therapeutic target for LAD-I-associated immunopathology
These human observations and preclinical studies became the basis of IL-23 blockade in LAD-I-associated periodontitis. Recently, our group and collaborators treated a single LAD-I patient with ustekinumab, an antibody that binds the p40 subunit of interleukin-23 and interleukin-12 and thereby blocks the activity of these cytokines, inhibiting interleukin-23-dependent production of interleukin-17.59 In this case, our patient was a 19-year-old male with moderate LAD-I disease, but severe periodontitis and a deep non-healing cutaneous sacral wound, which had progressed over 2 years despite numerous courses of antibiotics and surgical debridements. Treatment with IL-12/IL-23 blockade (ustekinumab), resulted in significant reduction in oral inflammation and complete resolution of the deep cutaneous ulcer without significant adverse reactions.59 This work revealed IL-23-mediated inflammation as the driving force of mucosal and cutaneous disease in LAD-I and suggested further exploration of IL-23 blockade for the treatment of LAD-I-associated disease. Based on these studies, an interventional protocol for the treatment of LAD-I immunopathology has been recently initiated ().
8 |. COMBINED NEUTROPHIL DEFECTS IN DEVELOPMENT, RECRUITMENT AND EXTR AVASATION: CHEDIAK-HIGASHI SYNDROME (CHS)
Chediak-Higashi syndrome is a rare, autosomal recessive disorder caused by mutations in the lysosomal trafficking regulator gene (CHS1/LYST), which is part of the Beige and Chediak-Higashi (BEACH) family of vesicle trafficking regulatory proteins.68,69 This defect leads to impaired intracellular lysosomal trafficking and results in the formation of characteristic giant granules within cells. Disease diagnosis is made by examination of a peripheral blood smear for detection of pathognomonic giant cytoplasmic granules in leukocytes and platelets and molecular analysis of the CHS1/LYST gene. Defective lysosomal trafficking predisposes to intramedullary destruction of neutrophils early in myelopoiesis and results in moderate neutropenia. Beyond neutropenia, neutrophils in Chediak Higashi also display decreased chemotactic responses, impaired diapedesis and reduced ability for intracellular killing of bacteria.70,71 Consistent with neutrophil defects, patients show not only susceptibility to bacterial infections and immunodeficiency, but also display a variety of non-hematopoietic manifestations, including oculocutaneous albinism and neurological dysfunction.72 The vast majority of patients develop severe disease (85%−90%), while a smaller proportion develop mild “atypical” disease.73 Severe-aggressive forms of periodontitis have been documented in several reports of Chediak-Higashi Syndrome. In the reported cases, periodontal disease presented at an early age with severe generalized destruction of tooth supporting bone by teenage years.74–76 Severe bone destruction led to tooth mobility and was the cause of either premature tooth exfoliation or indicated full mouth extractions. In most cases, periodontal disease progressed despite intense efforts for treatment including antimicrobial rinses, intense non-surgical periodontal therapy such as scaling and root planning and systemic antibiotic administration.74–76 In one case, periodontal bone loss progressed despite professional dental cleanings every 2 weeks and local administration of minocycline into periodontal pockets.77 However, a recent study has shown that periodontitis can be mild in “atypical” forms of Chediak-Higashi, indicating that periodontitis severity may correlate with overall severity of disease and levels of neutrophil dysfunction.73 Furthermore, this study revealed absence of periodontitis in Chediak-Higashi patients treated early in life with bone marrow hematopoietic cell transplantation, further confirming the primary role of the hematopoietic (neutrophil) compartment in Chediak-Higashi-associated periodontitis.73
9 |. DEFECTS IN NEUTROPHILDEGR ANUL ATION: PAPILLON LEFÈVRE SYNDROME (PLS)
9.1 |. Degranulation
Neutrophils are enriched with granules containing proteinases and antimicrobial peptides that fuse with the phagosome during pathogen uptake. In fact, fusion and extracellular release of secretory vesicles and tertiary vesicles occurs even during neutrophil activation, whereas extracellular release of secondary and primary granules occurs through unintended release from the phagosome, most commonly during “frustrated phagocytosis”.78 These granules also play a role in removing physical barriers to neutrophil egress during transmigration by facilitating degradation of basement membrane collagen. There are three main types of neutrophil granules (azyrophilic, specific, and tertiary granules). Azurophilic granules are named for their ability to take up the basic dye azure A and contain myeloperoxidase (MPO), an enzyme critical in the oxidative burst.16 Myeloperoxidase catalyzes the reaction of hydrogen peroxide with chloride generating hypochlorous acid (HOCl). This oxygen derivative plays a critical role in killing of the pathogenic bacteria and fungi.79,80 These granules also contain defensins, lysozyme, bactericidal/permeability-increasing protein (BPI), and a number of serine proteases: neutrophil elastase (NE), proteinase 3 (PR3), cathepsin G (CG), and neutrophil serine protease 4 (NSP4). The second class of granules, known as specific granules, are characterized by the presence of the glycoprotein lactoferrin. The third class, the gelatinase (tertiary) granules, serve as a storage location for a number of metalloproteases, such as gelatinase, matrix metalloprotease 9 (MMP9) and leukolysin, MMP25.16,81 Aside from their largely antimicrobial effects, some contents of neutrophil granules, such as the antibacterial peptide LL-37, show functions in immune regulation, including stimulation of neutrophil chemotaxis, induction of chemokine receptor expression, induction of cytokine production and suppression of neutrophil apoptosis.
9.2 |. Papillon Lefèvre Syndrome and periodontitis
Papillon-Lefèvre Syndrome is a rare autosomal recessive genetic disorder, caused by mutations in the gene encoding lysosomal cysteine protease cathepsin C (CTSC).82 CTSC is necessary for posttranslational modification and activation of serine proteases stored primarily in azurophilic granules, such as neutrophil elastase (NE), cathepsin G (CTSG), proteinase 3 (PR3), and neutrophil serine protease 4 (NSP4).83 CTSC is also essential for activation of granzymes A and B in cytotoxic lymphocytes.84 Mature PLS neutrophils lack all serine protease activity.85 However, despite lack of active serine proteases in neutrophils and cytotoxic T lymphocytes in patients with PLS, the associated immunodeficiency is remarkably mild.86,87 The dominating clinical phenotype of PLS is a severe periodontal disease that results in loss of both primary and permanent teeth at an early age and in most cases also in a skin condition known as keratosis palmoplantaris.88 In PLS, biosynthesis of CTSC and neutrophil serine proteases appeared to primarily affect mature neutrophils, but not the neutrophil progenitors, indicating physiological granulopoiesis.85 These recent data support the notion that PLS phenotypes arise from functional defects of neutrophils within tissues. Indeed, neutrophils in PLS have been shown to be incapable of producing NETs in response to ROS and processing endogenous cathelicidin hCAP-18 into the antibacterial peptide LL-37 in response to ionomycin. Although periodontitis in PLS has been associated with functional defects in neutrophils, the mechanisms by which CTSC deficiency leads to periodontitis have not been mechanistically dissected to date.
Periodontitis in Papillon-Lefevre syndrome patients has been reported to be exceptionally severe. Disease presents in the primary dentition with severe inflammation and bone destruction as early as 3–4 years of age. Multiple cases of children with severe periodontitis, generalized bone loss and tooth mobility have been reported from 3 to 11 years of age. In the majority of cases reported, patients progress to tooth loss in the primary and mixed dentition even in the presence of continuous dental care and antibiotic regimens.89–91 Microbiological evaluation of periodontal biofilms in several cases have identified the presence of known periodontitis-associated bacteria, such as Fusobacterium nucleatum, Peptostreptococcus micros, Prevotella nigrescens and also Aggregatibacter actinomycetemcomitans, a microbe classically associated with aggressive periodontitis at an early age.92 Herpesviruses has also been detected in the periodontal pockets of PLS patients.89,93 Despite many cases of severe periodontitis, which were proven recalcitrant to treatment, in one case, stringent dental care in conjunction with systemic amoxicillin-metronidazole therapy (250 mg of each/3 times daily/10 days) was shown to arrest or delay disease progression over a period of 16 months.93 Another encouraging case has documented successful treatment with osteointegrated implants in a patient with Papillon-Lefevre syndrome. In this case, dental implants were placed in a fully edentulous PLS patient and were reported to be stable, without signs of infection over a period of 4–5 years.94
Given that periodontitis is a dominant feature of PLS disease and is particularly severe, further understanding of disease pathogenesis is of great importance to identify opportunities for therapeutic targeting.
10 |. CONCLUDING: THE PAR ADOX OF IMMUNE DEFICIENCY AND INFL AMMATION IN PERIODONTITIS
While the mechanisms by which various defects in neutrophils predispose to periodontitis have not been dissected, extensive work in Leukocyte Adhesion Deficiency-I revealed a mechanism by which absence of tissue neutrophils leads to periodontal inflammation and bone loss.59,64 Therefore, it is conceivable that periodontitis in patients with neutrophil defects, ultimately leading to tissue neutropenia, because of either severe reduction in neutrophil numbers, defective neutrophil recruitment or impaired neutrophil transmigration, may be mediated by a similar mechanism as LAD-I. In this paradigm, immune deficiency ultimately predisposes to exaggerated inflammatory responses and indeed, is increasingly recognized today in the field of primary immunodeficiencies (PIDs).95 A recent analysis of French National Primary Immunodeficiencies Registry (CEDEDIH) showed that one or more autoimmune/inflammatory symptoms are observed in 26.2% of patients with primary immunodeficiency.96 This paradox of immune deficiency leading to compensatory inflammation is only recently appreciated, as traditionally immune deficiency was only associated with infection susceptibility.
In fact, in the majority of cases where chronic inflammation is linked to primary immunodeficiency, it is speculated that the lack of appropriate immune responses to infection/insult (due to immunodeficiency) will trigger the excessive induction of compensatory immune reponses. These compensatory response are often unable to appropriately regulate/contain the incoming insult and therefore, become excessive, misplaced and drive immunopathology. In the case of neutrophil defects, the lack of innate surveillance of commensal microbes at barrier sites induces excessive IL-17-mediated inflammation, which ultimately evolves into a tissue destructive process. In this scenario, lack of tissue neutrophils fails to regulate not only microbial triggering but also IL-23/IL-17 responses, which then perpetuate and become pathologic.
Appreciation of this concept, that immunodeficiency can lead to excessive inflammation and immunopathology, raises the question of whether a similar mechanism could be a plausible underlying cause in polygenic mucosal (barrier) inflammatory diseases. In the case of chronic (polygenic forms) periodontitis, it is therefore possible that immune deficiency in mechanisms related to surveillance of the commensal microbiota may be the underlying cause for excessive microbial triggering and exacerbated destructive inflammatory responses. However, in common forms of periodontitis, given that the disease is diagnosed after the occurrence of immunopathology, it is likely that studies of disease lesions will define compensatory inflammatory reactions rather than underlying initiating mechanisms. Ongoing studies in genomics of chronic forms of periodontitis are more likely to define immune-genetic variants linked to periodontitis susceptibility in the near future. In the interim, the discovery of additional single gene defects linked to periodontitis susceptibility and the mechanistic understanding of how these genetic defects lead to disease, will give clear insights into human periodontal immunity and disease susceptibility.
ACKNOWLEDGEMENTS
This work was funded by the Intramural Program of the National Institute of Dental and Craniofacial Research. The authors would like to acknowledge Erina He at the NIH Medical Arts Department and Ms. Teresa Wild and Dr Thomas H Bugge for critically reviewing the manuscript.
Funding information
National Institute of Dental and Craniofacial Research, Grant/Award Number: 1ZIADE000732
Footnotes
This article is part of a series of reviews covering Lessons primary immunodeficiencies teach about the healthy and diseased immune system appearing in Volume 287 of Immunological Reviews.
CONFLIC T OF INTEREST
Authors declare no competing financial interests.
REFERENCES
- 1.Casanova JL, Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317:617–619. [DOI] [PubMed] [Google Scholar]
- 2.Moutsopoulos NM, Lionakis MS, Hajishengallis G. Inborn errors in immunity: unique natural models to dissect oral immunity. J Dent Res 2015;94:753–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischer A Human primary immunodeficiency diseases. Immunity. 2007;27:835–845. [DOI] [PubMed] [Google Scholar]
- 4.Uhlig HH. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut. 2013;62:1795–1805. [DOI] [PubMed] [Google Scholar]
- 5.Eke PI, Dye BA, Wei L, et al. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res 2012;91:914–920. [DOI] [PubMed] [Google Scholar]
- 6.Hajishengallis G Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. 2015;15:30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Offenbacher S, Beck JD. Commentary: changing paradigms in the oral disease-systemic disease relationship. J Periodontol. 2014;85:761–764. [DOI] [PubMed] [Google Scholar]
- 8.Konig MF, Abusleme L, Reinholdt J, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. 2016;8:369ra176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abusleme L, Dupuy AK, Dutzan N, et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013;7:1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–490. [DOI] [PubMed] [Google Scholar]
- 11.Divaris K, Monda KL, North KE, et al. Exploring the genetic basis of chronic periodontitis: a genome-wide association study. Hum Mol Genet. 2013;22:2312–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moutsopoulos NM, Konkel JE. Tissue-specific immunity at the oral mucosal barrier. Trends Immunol. 2017;39:276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abusleme L, Diaz PI, Freeman AF, et al. Human defects in STAT3 promote oral mucosal fungal and bacterial dysbiosis. JCI Insight. 2018;3:e122061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jepsen S, Caton JG, Albandar JM, et al. Periodontal manifestations of systemic diseases and developmental and acquired conditions: consensus report of workgroup 3 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Periodontol. 2018;89(Suppl 1):S237–S248. [DOI] [PubMed] [Google Scholar]
- 15.Kruger P, Saffarzadeh M, Weber AN, et al. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015;11:e1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459–489. [DOI] [PubMed] [Google Scholar]
- 17.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. [DOI] [PubMed] [Google Scholar]
- 18.Delima AJ, Van Dyke TE. Origin and function of the cellular components in gingival crevice fluid. Periodontol 2000. 2003;31:55–76. [DOI] [PubMed] [Google Scholar]
- 19.Dutzan N, Konkel JE, Greenwell-Wild T, Moutsopoulos NM. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol. 2016;9:1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schiott CR, Loe H. The origin and variation in number of leukocytes in the human saliva. J Periodontal Res. 1970;5:36–41. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence SM, Corriden R, Nizet V. The ontogeny of a neutrophil: mechanisms of granulopoiesis and homeostasis. Microbiol Mol Biol Rev. 2018;82:e00057–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res. 2009;43:25–61. [DOI] [PubMed] [Google Scholar]
- 23.Summers C, Rankin SM, Condliffe AM, et al. Neutrophil kinetics in health and disease. Trends Immunol. 2010;31:318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5:1317–1327. [DOI] [PubMed] [Google Scholar]
- 25.Bainton DF, Ullyot JL, Farquhar MG. The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J Exp Med. 1971;134:907–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. 2016;273:11–28. [DOI] [PubMed] [Google Scholar]
- 27.Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. [DOI] [PubMed] [Google Scholar]
- 28.Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. 1997;89:3503–3521. [PubMed] [Google Scholar]
- 29.Klein C Genetic defects in severe congenital neutropenia: emerging insights into life and death of human neutrophil granulocytes. Annu Rev Immunol. 2011;29:399–413. [DOI] [PubMed] [Google Scholar]
- 30.Ancliff PJ, Gale RE, Liesner R, Hann IM, Linch DC. Mutations in the ELA2 gene encoding neutrophil elastase are present in most patients with sporadic severe congenital neutropenia but only in some patients with the familial form of the disease. Blood. 2001;98:2645–2650. [DOI] [PubMed] [Google Scholar]
- 31.Skokowa J, Klimiankou M, Klimenkova O, et al. Interactions among HCLS1, HAX1 and LEF-1 proteins are essential for G-CSF-triggered granulopoiesis. Nat Med. 2012;18:1550–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeidler C, Germeshausen M, Klein C, Welte K. Clinical implications of ELA2-, HAX1-, and G-CSF-receptor (CSF3R) mutations in severe congenital neutropenia. Br J Haematol. 2009;144:459–467. [DOI] [PubMed] [Google Scholar]
- 33.Ye Y, Carlsson G, Wondimu B, et al. Mutations in the ELANE gene are associated with development of periodontitis in patients with severe congenital neutropenia. J Clin Immunol. 2011;31:936–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Putsep K, Carlsson G, Boman HG, Andersson M. Deficiency of antibacterial peptides in patients with morbus Kostmann: an observation study. Lancet. 2002;360:1144–1149. [DOI] [PubMed] [Google Scholar]
- 35.Luis TC, Killmann NM, Staal FJ. Signal transduction pathways regulating hematopoietic stem cell biology: introduction to a series of Spotlight Reviews. Leukemia. 2012;26:86–90. [DOI] [PubMed] [Google Scholar]
- 36.Gong JK. Endosteal marrow: a rich source of hematopoietic stem cells. Science. 1978;199:1443–1445. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. [DOI] [PubMed] [Google Scholar]
- 38.Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. 2014;15:602–611. [DOI] [PubMed] [Google Scholar]
- 39.Wetzler M, Talpaz M, Kleinerman ES, et al. A new familial immunodeficiency disorder characterized by severe neutropenia, a defective marrow release mechanism, and hypogammaglobulinemia. Am J Med. 1990;89:663–672. [DOI] [PubMed] [Google Scholar]
- 40.Zuelzer WW. “Myelokathexis” – a new form of chronic granulocytopenia. Report of a case. N Engl J Med. 1964;270:699–704. [DOI] [PubMed] [Google Scholar]
- 41.Hernandez PA, Gorlin RJ, Lukens JN, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34:70–74. [DOI] [PubMed] [Google Scholar]
- 42.McDermott DH, Liu Q, Ulrick J, et al. The CXCR4 antagonist plerixafor corrects panleukopenia in patients with WHIM syndrome. Blood. 2011;118:4957–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gulino AV, Moratto D, Sozzani S, et al. Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood. 2004;104:444–452. [DOI] [PubMed] [Google Scholar]
- 44.Mc Guire PJ, Cunningham-Rundles C, Ochs H, Diaz GA. Oligoclonality, impaired class switch and B-cell memory responses in WHIM syndrome. Clin Immunol. 2010;135:412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gorlin RJ, Gelb B, Diaz GA, et al. WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet. 2000;91:368–376. [PubMed] [Google Scholar]
- 46.Hajishengallis E, Hajishengallis G. Neutrophil homeostasis and periodontal health in children and adults. J Dent Res. 2014;93:231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17:1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Voisin MB, Nourshargh S. Neutrophil transmigration: emergence of an adhesive cascade within venular walls. J Innate Immun. 2013;5:336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Constantin G, Majeed M, Giagulli C, et al. Chemokines trigger immediate beta2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 2000;13:759–769. [DOI] [PubMed] [Google Scholar]
- 50.Phillipson M, Heit B, Colarusso P, et al. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203:2569–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woodfin A, Voisin MB, Beyrau M, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. 2011;12:761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nourshargh S, Renshaw SA, Imhof BA. Reverse migration of neutrophils: where, when, how, and why? Trends Immunol. 2016;37:273–286. [DOI] [PubMed] [Google Scholar]
- 53.Hanna S, Etzioni A. Leukocyte adhesion deficiencies. Ann N Y Acad Sci. 2012;1250:50–55. [DOI] [PubMed] [Google Scholar]
- 54.Almarza Novoa E, Kasbekar S, Thrasher AJ, et al. Leukocyte adhesion deficiency-I: a comprehensive review of all published cases. J Allergy Clin Immunol Pract. 2018;6:1418–1420 e1410. [DOI] [PubMed] [Google Scholar]
- 55.Meyle J Leukocyte adhesion deficiency and prepubertal periodontitis. Periodontol 2000. 1994;6:26–36. [DOI] [PubMed] [Google Scholar]
- 56.Dababneh R, Al-Wahadneh AM, Hamadneh S, Khouri A, Bissada NF. Periodontal manifestation of leukocyte adhesion deficiency type I. J Periodontol. 2008;79:764–768. [DOI] [PubMed] [Google Scholar]
- 57.Roberts MW, Atkinson JC. Oral manifestations associated with leukocyte adhesion deficiency: a five-year case study. Pediatr Dent. 1990;12:107–111. [PubMed] [Google Scholar]
- 58.Yashoda-Devi BK, Rakesh N, Devaraju D, Santana N. Leukocyte adhesion deficiency type I – a focus on oral disease in a young child. Med Oral Patol Oral Cir Bucal. 2011;16:e153–e157. [DOI] [PubMed] [Google Scholar]
- 59.Moutsopoulos NM, Zerbe CS, Wild T, et al. Interleukin-12 and inter-leukin-23 blockade in leukocyte adhesion deficiency type 1. N Engl J Med. 2017;376:1141–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thomas C, Le Deist F, Cavazzana-Calvo M, et al. Results of allogeneic bone marrow transplantation in patients with leukocyte adhesion deficiency. Blood. 1995;86:1629–1635. [PubMed] [Google Scholar]
- 61.Hajishengallis G, Moutsopoulos NM. Etiology of leukocyte adhesion deficiency-associated periodontitis revisited: not a raging infection but a raging inflammatory response. Expert Rev Clin Immunol. 2014;10:973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hajishengallis G, Moutsopoulos NM. Role of bacteria in leukocyte adhesion deficiency-associated periodontitis. Microb Pathog. 2016;94:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moutsopoulos NM, Chalmers NI, Barb JJ, et al. Subgingival microbial communities in Leukocyte Adhesion Deficiency and their relationship with local immunopathology. PLoS Pathog. 2015;11:e1004698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moutsopoulos NM, Konkel J, Sarmadi M, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. 2014;6:229ra240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Interleukin Veldhoen M. 17 is a chief orchestrator of immunity. Nat Immunol. 2017;18:612–621. [DOI] [PubMed] [Google Scholar]
- 66.Abusleme L, Moutsopoulos NM. IL-17: overview and role in oral immunity and microbiome. Oral Dis. 2016;23:854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stark MA, Huo Y, Burcin TL, et al. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22:285–294. [DOI] [PubMed] [Google Scholar]
- 68.Barbosa MD, Nguyen QA, Tchernev VT, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996;382:262–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nagle DL, Karim MA, Woolf EA, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet. 1996;14:307–311. [DOI] [PubMed] [Google Scholar]
- 70.Blume RS, Bennett JM, Yankee RA, Wolff SM. Defective granulocyte regulation in the Chediak-Higashi syndrome. N Engl J Med. 1968;279:1009–1015. [DOI] [PubMed] [Google Scholar]
- 71.Root RK, Rosenthal AS, Balestra DJ. Abnormal bactericidal, metabolic, and lysosomal functions of Chediak-Higashi Syndrome leukocytes. J Clin Invest. 1972;51:649–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab. 1999;68:283–303. [DOI] [PubMed] [Google Scholar]
- 73.Thumbigere Math V, Reboucas P, Giovani PA, et al. Periodontitis in Chediak-Higashi Syndrome: an altered immunoinflammatory response. JDR Clin Trans Res. 2018;3:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Delcourt-Debruyne EM, Boutigny HR, Hildebrand HF. Features of severe periodontal disease in a teenager with Chediak-Higashi syndrome. J Periodontol. 2000;71:816–824. [DOI] [PubMed] [Google Scholar]
- 75.Bailleul-Forestier I, Monod-Broca J, Benkerrou M, Mora F, Picard B. Generalized periodontitis associated with Chediak-Higashi syndrome. J Periodontol. 2008;79:1263–1270. [DOI] [PubMed] [Google Scholar]
- 76.Khocht A, Viera-Negron YE, Ameri A, Abdelsayed R. Periodontitis associated with Chediak-Higashi syndrome in a young African American male. J Int Acad Periodontol. 2010;12:49–55. [PubMed] [Google Scholar]
- 77.Shibutani T, Gen K, Shibata M, et al. Long-term follow-up of periodontitis in a patient with Chediak-Higashi syndrome. A case report. J Periodontol. 2000;71:1024–1028. [DOI] [PubMed] [Google Scholar]
- 78.Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. 2014;9:181–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. [DOI] [PubMed] [Google Scholar]
- 80.Williams R Killing controversy. J Exp Med. 2006;203:2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Choi KY, Chow LN, Mookherjee N. Cationic host defence peptides: multifaceted role in immune modulation and inflammation. J Innate Immun. 2012;4:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Toomes C, James J, Wood AJ, et al. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat Genet. 1999;23:421–424. [DOI] [PubMed] [Google Scholar]
- 83.McGuire MJ, Lipsky PE, Thiele DL. Generation of active myeloid and lymphoid granule serine proteases requires processing by the granule thiol protease dipeptidyl peptidase I. J Biol Chem. 1993;268:2458–2467. [PubMed] [Google Scholar]
- 84.Pham CT, Ley TJ. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc Natl Acad Sci USA. 1999;96:8627–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sorensen OE, Clemmensen SN, Dahl SL, et al. Papillon-Lefevre syndrome patient reveals species-dependent requirements for neutrophil defenses. J Clin Invest. 2014;124:4539–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dalgic B, Bukulmez A, Sari S. Eponym: Papillon-Lefevre syndrome. Eur J Pediatr. 2011;170:689–691. [DOI] [PubMed] [Google Scholar]
- 87.Pham CT, Ivanovich JL, Raptis SZ, Zehnbauer B, Ley TJ. Papillon-Lefevre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. J Immunol. 2004;173:7277–7281. [DOI] [PubMed] [Google Scholar]
- 88.Haneke E The Papillon-Lefevre syndrome: keratosis palmoplantaris with periodontopathy. Report of a case and review of the cases in the literature. Hum Genet. 1979;51:1–35. [DOI] [PubMed] [Google Scholar]
- 89.Ishikawa I, Umeda M, Laosrisin N. Clinical, bacteriological, and immunological examinations and the treatment process of two Papillon-Lefevre syndrome patients. J Periodontol. 1994;65:364–371. [DOI] [PubMed] [Google Scholar]
- 90.Bashiardes S, Thaiss CA, Elinav E. It’s in the milk: feeding the microbiome to promote infant growth. Cell Metab. 2016;23:393–394. [DOI] [PubMed] [Google Scholar]
- 91.Hart TC, Shapira L. Papillon-Lefevre syndrome. Periodontol 2000. 1994;6:88–100. [DOI] [PubMed] [Google Scholar]
- 92.Fine DH, Markowitz K, Furgang D, et al. Aggregatibacter actionmycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J Clin Microbiol. 2007;45:3859–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pacheco JJ, Coelho C, Salazar F, et al. Treatment of Papillon-Lefevre syndrome periodontitis. J Clin Periodontol. 2002;29:370–374. [DOI] [PubMed] [Google Scholar]
- 94.Ullbro C, Crossner CG, Lundgren T, Stalblad PA, Renvert S. Osseointegrated implants in a patient with Papillon-Lefevre syndrome. A 4 1/2-year follow up. J Clin Periodontol. 2000;27:951–954. [DOI] [PubMed] [Google Scholar]
- 95.Schmidt RE, Grimbacher B, Witte T. Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat Rev Rheumatol. 2017;14:7–18. [DOI] [PubMed] [Google Scholar]
- 96.Fischer A, Provot J, Jais JP, et al. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol. 2017;140:1388–1393 e1388. [DOI] [PubMed] [Google Scholar]