

SUMMARY
Follicular dendritic cells (FDCs), a rare and enigmatic stromal cell type in the B cell follicles of secondary lymphoid organs, store and present antigen to B cells. While essential for germinal center (GC) responses, their exact role during GC B cell selection remains unknown. FDCs upregulate the inhibitory IgG Fc receptor FcγRIIB during GC formation. We show that the stromal deficiency of FcγRIIB does not affect GC B cell frequencies compared to wild-type mice. However, in the absence of FcγRIIB on FDCs, GCs show aberrant B cell selection during autoreactive and selective foreign antigen responses. These GCs are more diverse as measured by the AidCreERT2 -confetti system and show the persistence of IgM+ clones with decreased numbers of IgH mutations. Our results show that FDCs can modulate GC B cell diversity by the upregulation of FcγRIIB. Permissive clonal selection and subsequent increased GC diversity may affect epitope spreading during autoimmunity and foreign responses.
Graphical Abstract
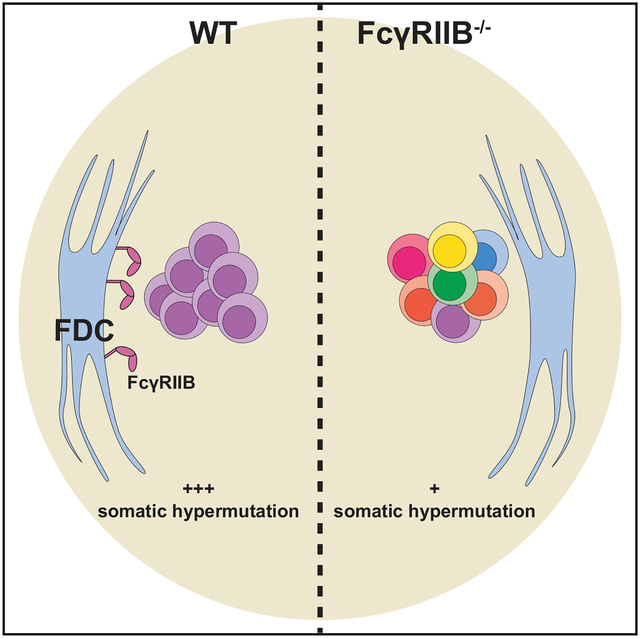
In Brief
van der Poel et al. show that follicular dendritic cells (FDCs) can regulate germinal center diversity through FcγRIIB. In the absence of this receptor, germinal centers appear more diverse. In addition, the loss of FcγRIIB on FDCs leads to the persistence of IgM clones with decreased levels of somatic hypermutation.
INTRODUCTION
Clonal B cell selection in germinal centers (GCs) is central to developing high-affinity antibody responses. In GCs, evolution occurs at a cellular level: high-affinity B cell clones are developed through iterative cycles of stochastic somatic hypermutation (SHM) and selection. These selected cells subsequently differentiate into memory B cells and/or antibody secreting plasma cells. T follicular helper cells (Tfh) in the light zone of the GCs are known to be important in the selection of B cell clones, and T cell-derived signals determine the subsequent proliferation of B cell clones in the dark zone of the GC (McHeyzer-Williams et al., 2015; Mesin et al., 2016; Victora and Nussenzweig, 2012; Vinuesa et al., 2016).
Follicular dendritic cells (FDCs) are a rare type of stromal cell that resides in B cell follicles of secondary lymphoid tissues. FDC, which define the light zone of the GC, are essential for GC formation and maintenance, and are known to bind and store antigen in the form of immune complexes (ICs) for presentation to GC B cells (Suzuki et al., 2009; Wang et al., 2011). In mice, complement receptors (CRs) expressed from the Cr2 gene (CD21 and CD35, CR2 and CR1, respectively) are involved in IC binding by FDCs (Phan et al., 2007), and we have shown previously that periodic internalization of CR1/2 bound IC is important in the storage of these ICs (Heesters et al., 2013). Upon GC formation, FDCs are known to upregulate IC receptors and the integrin ligands intercellular adhesion molecule (ICAM) and vascular cell adhesion molecule (VCAM), which appear to be partly induced by lymphotoxin α1β2 on GC B cells (Myers et al., 2013). The relevance of IC binding and presentation has been an issue for debate as it has been found that in the absence of detectable antigen on FDCs, GCs appear to form normally and affinity maturation is unaffected (Hannum et al., 2000). However, low amounts of ICs below the detection limit may be sufficient to drive most responses. Recent studies have found that GC B cell proliferation depends on both T cell-derived signaling and B cell receptor (BCR) signaling upon binding antigen (Luo et al., 2018).
While FDCs are thought to present antigens to GC B cells in the light zone, a direct role in GC B cell selection has never been demonstrated. For instance, FDCs upregulate adhesion molecules such as ICAM and VCAM upon GC formation, and in silico models have suggested that other than T cell-mediated selection, prolonged FDC-B cell contact through these adhesion molecules could aid in the selection of lower-affinity B cells (Meyer-Hermann et al., 2006). However, experimental studies addressing such interactions did not show any effect on affinity maturation and only modest effects on clonal selection (Wang et al., 2014).
FDCs supporting GCs are known to upregulate the inhibitory Fc receptor for immunoglobulin G (IgG), FcγRIIB (CD32). FcγRIIB on lymphoid and myeloid cells has been well studied, and it is known to inhibit many processes, including BCR signaling and activation of myeloid cells through its immune-receptor tyrosine-based inhibitory motif (ITIM) (Bournazos and Ravetch, 2015; Espéli et al., 2016; Li et al., 2014; Sharp et al., 2013). On FDCs, however, the role of FcγRIIB is less well understood. Bone marrow (BM) chimeras with FcγRIIB-deficient stromal cells are capable of forming GCs, although it has been reported that recall responses may be perturbed in the absence of FcγRIIB on FDCs (Barrington et al., 2002; Qin et al., 2000). However, both studies relied on adoptive transfer to FcγRIIB-deficient mice rather than full BM chimeras and furthermore lacked in-depth studies of GC selection. In addition, reporter mice that allowed confident tracking of memory B cells, such as the AidCreERT2EYFP (enhanced yellow fluorescent protein) line, were not available at the time.
FcγRIIB is known to be involved in the loss of tolerance, and SNPs affecting inhibitory signaling or antibody binding are robustly associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (Baerenwaldt et al., 2011; Chu et al., 2004; Floto et al., 2005; Kyogoku et al., 2002). We have recently described a murine SLE model that enables the study of wild-type (WT) autoreactive GC B cells (Degn et al., 2017). In this model, BM from the ribonuclear protein (RNP) complex reactive 564Igi knockin mice (564Igi) is mixed with WT or reporter BM. Upon transfer to irradiated recipients, spontaneous GCs are formed consisting primarily of WT-derived B cells. We have shown previously that these WT-derived cells are, at least in part, autoreactive. Since this model is based on the generation of BM chimeras, it is ideal to study the role of FDCs during autoreactive responses, as FcγRIIB deficiency will be limited to the stromal compartment in knockout recipients. Using this approach, we find that in the absence of FcγRIIB on FDCs, 564Igi-induced spontaneous GCs are more diverse, suggesting that FDCs can modulate GC selection through FcγRIIB, depending on the type of response.
RESULTS
FcγRIIB Is Upregulated on FDCs and Contributes to IC Binding upon GC Formation
Previous reports have shown that FcγRIIB is upregulated on FDCs supporting GCs. To better understand FcγRIIB expression kinetics on FDCs, we analyzed cryosections of draining popliteal lymph nodes (LNs) at various time points after subcutaneous immunizations. Increased staining of FcγRIIB on FDCs followed GC formation, measured as a GL7+ area, peaked at ~12 days post-immunization (Figures 1A and 1B).
Figure 1. FcγRIIB Is Upregulated on FDCs during GC Responses, but It Does Not Affect GC B Cell Frequencies in 564Igi Mixed BM Chimeras.

(A) Cropped confocal micrograph of a cryosection from a B6 popliteal lymph node 12 days after immunization with NP-CGG/alum. FDC networks are shown in red (CD35), FcγRIIB in green, and GC B cells in blue (Gl7). Scale bar, 100 μm.
(B) FcγRIIB staining on FDC networks and GC size at various days post-immunization. FcγRIIB mean fluorescence intensity (MFI) was measured within FDC masks as defined by CD35 staining. GC size was determined by GL7 staining associated with FDC networks. The absence of GL7 staining near FDCs was scored as an area of zero pixels. Each point contains at least 20 FDC networks from a total of 3–4 mice per time point. ***p < 0.001, ****p < 0.0001, Kruskal-Wallis with Dunn’s post hoc testing, tested versus day 0. Error bars represent SEMs.
(C) Experimental setup of 564Igi mixed BM chimeras. BM cells harvested from CD45.1 and 564Igi homozygous mice were transferred (2:1 ratio) to age-matched, lethally irradiated B6 or FcγRIIB−/− recipients. Eight weeks post-transfer, GC frequency and chimerism were assessed by flow cytometry.
(D) GC B cell frequencies in spleen, inguinal lymph nodes (LNs) and mesenteric lymph nodes (mes) of chimeras, and naive B6 and 564Igi homozygous mice as measured by the frequency of GL7+CD38lo cells within B220 gated lymphocytes. n = 5 mice for B6 recipients and 7 mice for FcγRIIB−/− recipients. Kruskal-Wallis with Dunn’s post hoc testing, tested for each tissue separately. Error bars represent SDs.
(E) Chimerism in peripheral blood in B6 and FcγRIIB−/− recipients showing the percentage of B cells derived from 564Igi BM (anti-idiotype+, in blue) and B cells derived from CD45.1 BM (red). Also shown is the idiotype frequency in a 564Igi homozygous mouse to indicate the idiotype baseline frequency. Error bars represent SDs.
(F) Frequency of CD45.1 BM-derived cells within GC B cells. Error bars represent SDs.
We recently published a model that allows the study of WT B cell repertoires during spontaneous GC responses (Degn et al., 2017). Given the association of FcγRIIB deficiency with the loss of tolerance, we used this model to study FDC function during autoreactive B cell responses. In addition, since this model is based on the generation of mixed BM chimeras, the genotype of the recipient is limited to the radiation-resistant stromal compartment. Using CD45.2 B6 or CD45.2 FcγRIIB-deficient mice as recipients for CD45.2 564Igi and CD45.1 BM, we found spontaneous GC formation in the spleen and to a lesser extent in skin draining lymph nodes (LNs) (Figures 1C and 1D). As documented previously, the WT-derived B cells in this model efficiently outcompete the autoreactive 564Igi cells in the periphery. BM chimeras showed high levels of chimerism in the B cell compartment as indicated by the high level of CD45.1+ circulating B cells 8 weeks post-BM transfer (Figure 1E). Nevertheless, we could detect a small portion of 564Igi-derived B cells as measured by anti-idiotype staining within circulating B cells (Figure 1E). The contribution of this is likely to be a slight underestimation, as in 564Igi homozygous mice the idiotype frequency in peripheral B cells is ~90%. In line with previous results, the majority of GC B cells in this model are derived from the WT (CD45.1) donor (Figure 1F). We did not observe differences in GC B cell frequencies between WT and FcγRIIB-deficient recipients, suggesting that FDC-expressed FcγRIIB does not influence the magnitude of GC formation.
FcγRIIB staining on FDCs in splenic cryosections of 564Igi:CD45.1 mixed BM chimeras was localized to where the GC B cells contacted the FDC network (Figure S1A). The periphery of the FDC network, however, remained mostly negative for FcγRIIB staining. This was also apparent in LNs from 4-hydroxy-3-nitrophenylacetyl-chicken γ-globulin (NP-CGG)-immunized mice (Figure 1A). FDC networks in splenic sections of FcγRIIB-deficient recipients did not stain with the anti-FcγRIII/FcγRIIB antibody (Figures S1A and S1B). This also excludes the potential co-staining of closely associated GC B cells, which, while known to express FcγRIIB, typically do not stain positive with this antibody on cryosections. In addition, since the antibody used also recognizes FcγRIII (CD16), this indicates specific FcγRIIB staining on FDCs under these conditions, as reported previously (Qin et al., 2000). FDCs upregulate the adhesion molecules ICAM and VCAM upon GC formation. In addition to FcγRIIB, these markers are often associated with FDC activation. We did not observe any changes in ICAM or VCAM expression in the absence of FcγRIIB on FDCs compared to WT (Figures S1C and S1D).
Thus, FDC upregulate FcγRIIB during GC formation. However, FcγRIIB on FDCs does not appear to modulate ICAM/VCAM upregulation or the magnitude of spontaneous GC responses.
FcγRIIB on FDC Contributes to IC Binding
FDCs are known to bind and present antigen in the form of ICs. To test whether FcγRIIB contributed to IC binding by FDC in 564Igi mixed BM chimeras, we used a passive anti- phycoerythrin (PE) immunization approach. This approach was chosen to prevent any potential bias that would be associated with the direct immunization of FcγRIIB−/− recipients. Pooled serum from PE-immunized WT mice was injected in 564Igi mixed BM chimeras using B6 or FcγRIIB-deficient mice as recipients. The next day, B-PE as a traceable antigen was injected intravenously (i.v.), and 48 h later, the presence of PE was measured on splenic FDC networks using confocal microscopy. PE was readily observed on FDC networks in B6 recipients (Figure 2A); however, a significant decrease in PE bound to FDCs was observed in FcγRIIB−/− recipients (Figures 2A and 2B). The deposition of PE on FDCs in B6 recipients was dependent on the passive immunization of anti-PE (Figure 2B). Decreased IC binding was not due to decreased FDC network size, as the area of the masks based on CD35 staining was unaltered between B6 and FcγRIIB−/− recipients. We observed a similar decrease in PE IC binding in a direct immunization strategy of full FcγRIIB−/− knockout mice compared to B6 mice (Figure S2).
Figure 2. FcγRIIB Contributes to IC Binding in 564Igi Mixed BM Chimeras.

(A) Confocal micrographs of FDC networks in splenic cryosections from passively immunized 564Igi mixed BM chimeras. The recipients of 564Igi mixed BM chimeras were passively immunized with pooled serum from WT mice immunized with PE in complete Freund’s adjuvant (CFA), followed by i.v. PE injection. The left panels show representative FDC networks from a WT and FcγRIIB−/− recipient with indicated staining. The right panels show the same networks with the mask outline (based on CD35 staining) used for quantification and the PE signal only. Only FDC networks associated with clusters of Ki67+ GC were included in the analysis. The arrowhead in the left panel indicates non-FDC-associated PE, likely derived from a dendritic cell (DC) or macrophage. Scale bar, 100 μm. All of the panels are scaled equally.
(B) Quantification of PE on FDCs in passively immunized 564Igi chimeras. B6, C57BL/6 recipients of 564Igi mixed BM and FcγRIIB−/− (KO, knockout) recipients of 564Igi mixed BM, n = 3 mice per group; each point represents 1 FDC network. No IgG and no PE are negative controls, with B6 564Igi mixed BM chimeras receiving PE only or no IgG and PE, respectively.
(C) Area of each FDC network mask in number of pixels. NS, not significant, ****p < 0.0001, ANOVA with Tukey post hoc test, B6 versus all other groups.
Our data suggest that FcγRIIB on FDCs contributes substantially to IC binding by FDCs.
FcγRIIB on FDCs Enhances Clonal Selection during Spontaneous GC Responses
Since we observed decreased IC binding by FcγRIIB−/− FDC, we investigated whether this decreased IC binding would affect GC B cell selection. AicdaCreERT2Confetti reporter allows the visualization of clonal selection during GC responses. In this reporter, the expression of a tamoxifen-controlled Cre recombinase is limited to GC B cells expressing the Aicda gene product activation-induced cytidine deaminase (AID). Upon tamoxifen treatment, the Cre-ERT2 fusion protein mediates the random selection of 4 fluorescent proteins from 2 separate confetti alleles, which generates 10 different potential color combinations. Tamoxifen treatment in these reporter mice during a B cell response will generate a pool of GC B cells in which each cell will express 1 of 10 color combinations. As daughter cells continue to express the selected combination of fluorescent proteins after tamoxifen treatment, clonal selection can be visualized by measuring the frequencies of each color combination within each GC (Tas et al., 2016). Using this model, we have previously shown a robust clonal selection of spontaneous GC B cells in 564Igi-induced GCs (Degn et al., 2017). To investigate whether FcγRIIB on FDCs is involved in this process, B6 or FcγRIIB−/− mice received a mixture of AicdaCreERT2Confetti and 564Igi BM after lethal irradiation. After 6–7 weeks, GC B cells were induced to express a random fluorescent protein from the confetti cassettes by a single tamoxifen gavage. We assessed clonal selection by analyzing the color distribution of GC B cells at various time points post-tamoxifen induction (Figure 3A). Similar to our previous studies, spontaneous GCs in B6 recipients reached pauci-clonality over a span of 31 days. However, in FcγRIIB−/− recipients, color dominance was consistently depressed at 14 and 31 days post-tamoxifen induction (Figures 3B–3D). A similar trend was observed in the divergence index, which measures the divergence away from the initial color distribution as measured 3 days after tamoxifen induction (Figure 3E). These results suggest that FcγRIIB on FDCs enhances clonal selection in spontaneous GCs.
Figure 3. Increased GC B Cell Diversity in the Absence of FcγRIIB on FDCs.

(A) Experimental setup to assess the role of FcγRIIB on FDCs during B cell selection. B6 or FcγRIIB−/− mice received mixed BM cells from 564Igi and AicdaCreERT2Confetti mice (1:2 ratio) after lethal irradiation. Six to seven weeks after BM transfer, mice were treated with a single gavage of tamoxifen to induce the confetti cassettes in GC B cells. Splenic GC in ~2-mm-thick splenic sections were imaged ex vivo at the indicated time points post-tamoxifen treatment.
(B) Representative images cropped from multiphoton optical sectioning of spleens from B6 or FcγRIIB−/− recipients 3, 14, or 31 days post-tamoxifen treatment. Scale bar, 100 μm. All of the images are scaled equally.
(C–E) Quantification of color dominance in splenic GCs from B6 (red circles) or FcγRIIB−/− (blue squares) mixed BM recipients. Images were blinded for recipient genotype before counting. The graphs depict the frequency of the most dominant color (C), the sum of frequencies of 1st and 2nd most dominant clone (D), and the divergence index (E). The color distribution for B6 recipients at day 3 was used to calculate the divergence index for all of the points. Each point represents one GC; the line indicates the median. Pooled data from 4 independent experiments, n = 3–6 mice for each genotype at each time point. Kruskal-Wallis with Dunn’s post hoc testing for statistical analysis. Asterisks above the graphs indicate significance relative to day 3 post-tamoxifen within genotypes; asterisks below the graphs indicate the significance between genotypes within time points. The tests comparing between time points and between genotypes were included in adjusted p value calculations, but they are not indicated.
FcγRIIB Enhances Clonal Selection during Specific Foreign Responses
We next tested whether the observed effect of FDC-expressed FcγRIIB on clonal selection extended beyond autoreactive responses. B6 and FcγRIIB−/− mice were irradiated and received AicdaCreERT2Confetti BM. GC diversity after sheep red blood cell (SRBC) (Figure 4A) or influenza virus hemagglutinin (HA/alum; Figure S3A) immunizations was subsequently assessed. GCs induced upon SRBC immunization showed increased color diversity at day 14 post-tamoxifen, reaching pauci-clonality at day 31 in B6 recipients. In FcγRIIB-deficient recipients, however, we observed decreased frequencies of the most dominant color at day 14, indicating less stringent clonal selection (Figure 3B). This trend continued up to 31 days post-tamoxifen treatment. At this time point, the difference between B6 and FcγRIIB-deficient recipients did not reach statistical significance, possibly due to some GCs in FcγRIIB-deficient recipients reaching pauci-clonality, while many others remained diverse (Figure 4C). This bimodal distribution at day 31 persisted when summing the frequencies of both the 1st and 2nd most dominant clones (Figure 4D). Divergence indices trended toward decreased clonality at days 14 and 31 in FcγRIIB-deficient recipients, although this was not statistically significant.
Figure 4. Increased GC Diversity in FcγRIIB−/− Recipients during Foreign Responses.

(A) Experimental setup to measure the role of FcγRIIB on FDCs during B cell selection in GCs directed against foreign antigens. Indicated AicdaCreERT2Confetti BM chimeras were immunized intraperitoneally (i.p.) with 109 SRBCs. The mice received a single gavage of tamoxifen to induce the random expression of fluorescent proteins from the confetti cassettes 4 days post-immunization. An additional boost of 107 SRBC was given to maintain robust GC 21 days post-tamoxifen.
(B) Representative cropped images of SRBC-induced splenic GCs at day 14 post-tamoxifen induction in B6 (left) or FcγRIIB recipients (right). Scale bar, 100 μm. All of the images are scaled equally.
(C–E) Quantification of color dominance in splenic GCs shown as the frequency of the most dominant color (C), the summed frequency of 1st and 2nd most dominant color (D), and the divergence index (E). The divergence index was calculated based on the color distribution of GCs in B6 recipients at day 3 post-tamoxifen. Pooled data from 2 independent experiments; n = 3–4 mice per group per genotype. Kruskal-Wallis with Dunn’s post hoc was used for statistical testing. Asterisks above the graphs indicate the significance relative to day 3 post-tamoxifen within genotypes; asterisks below the graphs indicate the significance between the genotypes within time points. The tests comparing between time points and between genotypes were included in adjusted p value calculations, but they are not indicated.
Upon immunizations with HA/alum in AicdaCreERT2Confetti chimeras, we observed a large spread in clonalities in both B6 and FcγRIIB-deficient recipients at day 14 post-tamoxifen (Figure S3B), with many GCs remaining as diverse as 3 days post-tamoxifen. However, at day 31, most GCs reached pauci-clonality. The HA/alum immunization schedule did not result in altered GC diversity in FcγRIIB-deficient recipients (Figures S3C–S3E).
Appearance of Low SHM Clones in the Absence of FcγRIIB on FDCs in 564Igi-Induced GCs
To investigate in greater detail the impact that the loss of FcγRIIB on FDCs has on spontaneous GCs, we sequenced BCRs from single B cells from individual GCs. This was accomplished by photoactivating GCs in spleen sections from B6 or FcγRIIB−/− recipients of a 1:2 mix of 564Igi and photoactivatable GFP (paGFP) BM. The subsequent magnetic enrichment of GC B cells followed by fluorescence-activated cell sorting (FACS) of photoactivated GCB cells allowed us to obtain sequences from clones from individual GCs (Figure 5A and 5B). Overall, this approach yielded IgH-derived sequences from 9 GCs from B6 recipients and 6 GCs from FcγRIIB−/− recipients containing ≥10 sequences. Several photoactivated GCs that yielded ≤9 sequences were excluded from analysis. Most GCs showed selection toward select heavy chain V regions (VH) per GC, as we have observed previously (Figure S4; Degn et al., 2017). While the overall response appeared diverse in terms of VH usage, we observed VH regions that were found in our previous studies, such as J558.22.112, and to a lesser extent, VHQ52.a27.79. In addition, some GCs contained clones bearing VH regions J558.53.146 and J558.75.117. These particular VH regions were found previously in autoreactive antibodies cloned from 564Igi-induced WT-derived GC B cells (Figure S4; Degn et al., 2017).
Figure 5. Decreased SHM in IgM+ B Cell Clones in FcγRIIB−/− 564Igi Mixed BM Chimeras.

(A) Experimental setup to analyze B cell clones from individual GCs. B6 or FcγRIIB−/− mice received a mix of 564Igi- and paGFP-derived BM cells (1:2 ratio) after lethal irradiation.
(B) Sorting strategy for sorting photoactivated GC B cells from single GCs in the spleen. Nine weeks post-BM transfer, mice were euthanized and ~2-mm-thick spleen sections were imaged by 2-photon microscopy. Mice had received phycoerythrin immune complexes (PE:IC) 1 day before the endpoint and CD169-PE 30 min before the endpoint to visualize FDC networks and marginal zone macrophages bordering the follicles. GCs were identified by the presence of auto-fluorescent tingible body macrophages. One GC was photoactivated per spleen section. After generating single-cell suspensions and staining for GC markers, photoactivated Gl7hiCD38lo cells were sorted. Scale bar, 100 μm.
(C) Global distribution of VH usage between GC B cells from B6 and FcγRIIB−/− recipients. VH segments found to be involved in 564Igi-induced GCs in previous studies are indicated (Degn et al., 2017). The numbers in the center of the charts reflect the total analyzed sequences (n = 3 mice per group). (D) Somatic hypermutation within Fr1-CDR3 as measured by the number of mismatched nucleotides compared to best matched germline cassettes. Follicular B cells (FoBs) from a naive paGFP mouse were used as a negative control. Each point indicates one IgH sequence; the line indicates the median. ****p < 0.0001, Kruskal-Wallis with Dunn’s post hoc testing, compared to all of the groups within the graph. Not indicated is the statistical significance between the IgM FoB group and the FcγRIIB−/− IgM group; p = 0.03.
(E) Mutation analysis as in (D) at the amino acid level, ****p < 0.0001, Kruskal-Wallis with Dunn’s post hoc testing, compared to all of the groups within the graph.
(F) Number of mutations per GC per isotype. The linked points represent the mutation rates in IgM+ and IgG+ clones in one GC. Error bars represent SDs.
(G and H) Distribution of SHM at the nucleotide level for IgM (G) and IgG (H). Sequences were obtained from 3 mice per group for chimeras and 1 naive paGFP mouse as a WT sequencing control. Number of sequences analyzed per group: n = 86 for IgM FoB sequences, n = 64 for IgM B6 recipient sequences, n = 127 for IgG B6 recipient sequences, n = 39 for IgM FcγRIIB−/− recipient sequences, and n = 141 for IgG FcγRIIB−/− recipient sequences.
Globally, we did not observe large differences in diversity at the VH level between B6 and FcγRIIB−/− recipients, indicating that differences observed in the confetti system are likely the result of a more complex selection process (Figure 5C). However, when comparing SHM levels, we noted fewer mutations in IgM+ clones in FcγRIIB−/− recipients at both the nucleotide and the amino acid level (Figures 5D and 5E). Grouping clones by their GCs and plotting SHM for either the IgM or IgG isotype showed that all but one GC in the FcγRIIB−/− recipients contained IgM+ clones of lower SHM (Figure 5F). By examining the distribution of SHM in each group, a population of low SHM IgG+ clones in FcγRIIB−/− recipients was also observed (Figures 5G and 5H).
Persistence of Low SHM Clones in Spontaneous GCs of FcγRIIB−/− Recipient Mice
The presence of predominantly low SHM, IgM+ GC B cells in the absence of FcγRIIB on FDCs could result from either an increased influx of naive clones or a persistence of low SHM clones within the GC. We used the AicdaCreERT2-flox-stop-flox EYFP (AicdaCreERT2EYFP) model to measure SHM in clones that persist or reenter in GCs. In this model, tamoxifen induces the irreversible expression of EYFP in a portion of AID expressing GC B cells. By sorting and subsequently sequencing EYFP+ GC B cells, we measured SHM in a population that persisted (or reentered) in the GC since the time of the tamoxifen induction. Chimeras were generated with B6 or FcγRIIB−/− recipients of mixed 564Igi and AicdaCreERT2EYFP BM (Figure 6A). Single EYFP+ GC B cells from chimeras treated with tamoxifen 1 or 2 weeks earlier were sorted (Figure 6B). Similar to our previous results (Figure 1D), we did not observe a difference in the frequency of total GC B or EYFP+ GC B cells between B6 or FcγRIIB−/− recipients (Figures 6C, S5A, and S5C). The presence of EYFP GC B cells was largely dependent on 564Igi BM since BM chimeras receiving a mix of B6 and AicdaCreERT2EYFP BM showed an ~3-fold lower frequency of EYFP+ GC B cells, although some background GC formation was observed compared to tamoxifen-treated naive AicdaCreERT2EYFP mice (Figure 6C). A similar trend was observed when gating on all GC B cells, irrespective of EYFP expression (Figure S5A). We used the irreversible labeling of GC B cells by EYFP to measure the amount of memory B cells that arose from GCs, defined as EYFP+ cells negative for GC markers (Gl7loCD38hi). Although the overall frequency of these cells was low, we found no clear differences in the frequencies of these cells between B6 and FcγRIIB−/− recipients (Figure S5B). Similarly, at 2 weeks post-tamoxifen, there was no difference between recipients in the frequency of YFP+ GC B cells, YFP+ memory cells, or YFP+ CD138+ plasma cells/blasts relative to the total B cell population (Figures S5C–S5F).
Figure 6. Low SHM IgM+ GC B Cell Clones Persist in the Absence of FcγRIIB on FDCs.

(A) Experimental setup to label and follow 564Igi-induced, autoreactive GC B cells. B6 or FcγRIIB−/− mice were lethally irradiated and subsequently received a 1:2 mixture of 564Igi and AicdaCreERT2EYFP BM or a 1:2 mixture of B6 and AicdaCreERT2EYFP BM. After 9 weeks, mice were tamoxifen induced (tamox) and splenic EYFP+ GC B cells were sorted at days 7 or 14 post-tamoxifen.
(B) Gating strategy used to sort EYFP+ GC B cells. Cells were gated on forward versus side scatter (FSC/SSC) for lymphocytes, singlets, and live-dead stain negative cells before the indicated gates.
(C) Frequency of EYFP+ GC B cells relative to measured B220+ B cell population in indicated chimeras and naive AicdaCreERT2-EYFP mice (naiv) and 564Igi mice at the 1 week post-tamoxifen time point. Recip, recipient; KO, FcγRIIB−/−. All of the chimeras received 2 parts AicdaCreERT2-EYFP (not shown) and 1 part 564Igi (+564) or 1 part B6 (+B6). Pooled data from 3 independent experiments. n = 6 mice for 564Igi+AicdaCreERT2 EYFP chimeras, 4 mice for B6+AicdaCreERT2EYFP recipients, naive AicdaCreERT2EYFP, and 564Igi mice. Error bars represent SDs.
(D and E) The number of nucleotide mutations (D) or amino acid mutations (E) in the VDJ region from EYFP+ GC B cell heavy chains obtained from 564Igi+AicdaCreERT2EYFP mixed BM chimeras 1 week post-tamoxifen. Sequences from three mice per group.
(F and G) Nucleotide mutations (F) or amino acid mutations (G) 2 weeks post-tamoxifen treatment. Sequences from two mice per group. Kruskal-Wallis with Dunn’s post hoc testing. The number of sequences analyzed for this figure is listed in Table S1.
Upon the sequencing of EYFP+ GC B cells, suppressed SHM in IgM+ clones in FcγRIIB−/− recipients at 1 and 2 weeks post-tamoxifen induction was observed (Figures 6D–6G). One week after tamoxifen treatment, roughly 30% of the recovered sequences were IgM derived irrespective of recipient genotype. At 2 weeks post-tamoxifen, the frequency of IgM sequences decreased to ~12% and 18% for B6 and FcγRIIB-deficient recipients, respectively, although the variability within the groups was high (Table S1). This pulse-chase experiment reveals that in the absence of FcγRIIB on FDCs, low SHM IgM+ clones reenter and/or persist in spontaneous GCs with timescales similar to the increased GC diversity observed in FcγRIIB-deficient recipients (Figure 2). In addition, since we observed comparable total GC B cell frequencies as well as comparable YFP+ GC B cell frequencies between B6 and FcγRIIB−/− recipients, there is no difference in the GC entry of naive clones between these groups.
DISCUSSION
FDCs upregulate FcγRIIB during GC responses, yet the role of this inhibitory IgG receptor on FDCs is poorly understood. Using the AicdaCreERT2Confetti reporter system, we found that FDCs modulate GC B cell selection through this receptor. This appears to be specific to certain responses, as we observed this phenomenon in splenic 564Igi and SRBC-induced GCs. Despite the effects of FDC-expressed FcγRIIB on clonal selection, GCs appear remarkably similar in terms of overall GC B cell frequencies and differentiation toward memory B cells and plasma cells. Upon the sequencing of B cells in spontaneous GCs, we found low SHM IgM+ clones in FcγRIIB-deficient chimeras. BCR sequencing from GC B cells sorted from the recipients of mixed 564Igi and AicdaCreERT2-EYFP BM suggests that these low SHM clones can persist up to at least 2 weeks, a time frame in which B6 recipients showed efficient GC-driven affinity maturation.
The increased diversity observed in the FcγRIIB-deficient recipients of AicdaCreERT2Confetti BM was not mirrored by a more diverse VH usage (Figures 5C and S4). Given the chronic nature of the 564Igi-induced GC, it is likely that extensive clonal selection occurred before tamoxifen induction. As a result, the observed diversity in FcγRIIB-deficient chimeras may reflect not only altered competition between but also within subclones. To address this at the sequence level would require a highly efficient sampling of B cells from single GCs, something that is technically challenging.
In the absence of FcγRIIB on FDCs, low SHM frequency clones were mainly observed within IgM+ GC B cells. Class switching by GC B cells is thought to be dependent on CD40-mediated stimulation from Tfh (Kawabe et al., 1994). It is possible that these IgM+ clones did not receive proper T cell help, yet despite this, they persisted in the GCs of FcγRIIB-deficient recipient mice. A decrease in the frequencies of IgM+ in the YFP+ cells over time suggests that eventually some of these clones exit this population through differentiation, class switching to IgG, cell death, or a combination of these. Chronic GCs can in principle be replenished by naive clones. These new arrivals would not contribute to the diversity observed in the confetti system because these cells would not have expressed CreERT2 recombinase at the time of tamoxifen treatment. Since we did not observe decreased YFP+ GCB cells in the FcγRIIB-deficient recipients of AicdaCreERTEYFP BM (Figure 6C), we do not expect an increased entry of naive B cells in the absence of FcγRIIB on FDCs. However, we cannot exclude an increased exchange of antigen-experienced B cells between GCs, perhaps through a memory B cell intermediate, in FcγRIIB-deficient recipients. This “GC hopping” (denoted by Mesin et al., 2016) may be a feature of chronic or long-lived GCs such as the SRBC- or 564Igi-induced GCs we have described in this study. While we were not able to fully elucidate the nature of the increased diversity at the sequence level, it is likely that persistence or continued reentry of low SHM clones could at least contribute to this phenomenon, as we found that these cells persist at similar timescales as the readout for the confetti system (Figures 6F and 6G).
While the absence of FcγRIIB on FDCs resulted in an overall increased diversity in GCs, we did observe GCs in these mice that reached near or complete color homogeneity. This could either reflect under-sampling of diversity due to limited color space of the confetti system or represent the presence of actual pauci-clonal GCs. The coexistence of both diverse and homogeneous GCs appears not to be limited to spontaneous GCs, as we observed similar heterogeneity within responses against SRBCs. This phenomenon has been described before and it has been suggested that switching between birth-limited and death-limited selection during GC responses may in part explain these differences (Amitai et al., 2017; Tas et al., 2016). It should be noted that while some of the observed differences in the confetti system in FcγRIIB-deficient mice reported here seem minor, in addition to the apparently inherent noise in GC clonality, the dynamic range is limited due to the preferential usage of YFP cassettes (Tas et al., 2016).
There has been discussion on the relation between the level of antigen presented by FDCs and the selection stringency of GC B cells. The idea that limited antigen availability leads to increased selection stringency has been contested (Victora et al., 2010). In our experiments, we noted a significant decrease in IC binding by FcγRIIB-deficient FDCs when using murine IgG to generate ICs. Despite this lowered potential of antigen presentation, GCs appeared more diverse, suggesting lower selection thresholds rather than more stringent selection. This implies that other mechanisms rather than antigen availability are driving the observed phenotype in GC diversity. Interpretation of the consequence of lowered antigen presentation on GC selection is further complicated by the autoreactive model: unlike immunization approaches with foreign antigens, there is a practically endless supply of autoantigen in the autoreactive 564Igi mixed BM model. Thus, other mechanisms such as antigen turnover or perhaps a direct negative regulatory effect of FcγRIIB on FDCs should be considered in future studies.
Although further study is needed to fully clarify the molecular mechanism involved in FDC-mediated clonal selection, RNA sensing through TLR7 or other mechanisms may be involved either directly or indirectly. Autoreactive B cell responses are known to be dependent on TLR signaling, and we and others have previously confirmed this in the 564Igi mixed BM model (Berland et al., 2006; Degn et al., 2017; Lau et al., 2005; Leadbetter et al., 2002). In addition, we recently described that FDCs can sense RNA during autoreactive responses and produce type I interferon (IFN) in response (Das et al., 2017). Further study is needed to elucidate the molecular pathways that dictate the regulation of GC diversity.
It is not known whether the observed GC phenotype is dependent on inhibitory signaling by the Fc receptor. Therefore, it is difficult to predict whether lowered FcγRIIB inhibitory function due to I232T mutations would result in increased GC diversity. The absence of FcγRIIB on FDCs does not perturb clonal selection altogether. Even if the observed phenotype was dependent on inhibitory signaling, chronic GC could still produce high SHM and high-affinity antibodies. We did not observe large shifts in VH usage in the 564Igi-induced GC B cells supported by FcγRIIB-deficient FDCs. Nevertheless, the persistence of low SHM clones could in theory alter the autoreactive antibody landscape and broaden epitope spreading.
Previous studies investigating the role of FcγRIIB on FDCs reported perturbed recall responses and modest changes in global serum IgG affinities upon the transfer of splenocytes to FcγRIIB-deficient mice (Barrington et al., 2002; Qin et al., 2000). These studies relied mainly on adoptive transfers of splenocytes to sublethally irradiated mice rather than the full BM chimeras we describe here. Thus, the interpretation of previous studies is complicated by the presence of FcγRIIB-deficient B cells and antigen-presenting cells. In particular, the presence of FcγRIIB-deficient competitor B cells, which would not receive normal inhibitory signaling, confound the readout. This, in addition to the differing immunization approaches, may in part explain why we did not find evidence for changes in memory B cell output or maintenance in the absence of FcγRIIB on FDCs.
FcγRIIB is upregulated concomitantly with the formation of the GC and the emergence of antigen-specific IgG. FcγRIIB on FDCs could function as a way to increase selection stringency after the initial seeding of the GC. The absence of the receptor during the onset of the response may help in the GC growth phase, allowing a broader pool of clones to seed the GC. As antigen-specific IgG titers increase during the ensuing response and FcγRIIB surface expression on FDCs is upregulated, clonal selection becomes stricter, preventing the escape of lower-affinity clones back to the dark zone of the GC.
Our data demonstrate that FDCs can modulate GC diversity and somatic hypermutation. This finding may furnish a deeper understanding of clonal selection during GC responses, both in foreign and autoimmune settings. Future investigations should further clarify the underlying mechanism and may indicate whether the modulation of GC B cell selection by FDCs extends to other molecular pathways.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael C. Carroll (michael.carroll@childrens.harvard.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C57BL/6J FcγRIIBfl/fl were provided by JS Verbeek (Toin University of Yokohama, Japan) and maintained in house (Boross et al., 2011). FcγRIIB−/− mice were obtained due to germline Cre “leakiness” after crossing with CD21Cre mice (Schmidt-Supprian and Rajewsky, 2007). FcγRIIBfl/- mice were bred with C57BL/6J to remove the CD21Cre locus before cross breeding. C57BL/6J mice, B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice and Photo-activatable paGFP mice (Victora et al., 2010) were from Jackson Laboratories and maintained in house. The AicdaCreERT2 flox-stop-flox- EYFP (AicdaCreERT2 EYFP) mouse line was provided by Claude-Agne’s Reynaud and Jean-Claude Weill (Institut Necker)(Dogan et al., 2009). AicdaCreERT2 EYFP mice were maintained as homozygous mice and F1 from crosses with C57BL/6 or CD45.1 mice were used for experiments. 564Igi mice (Berland et al., 2006) were provided by Theresa Imanishi-Kari (Tufts University) and maintained in-house. AicdaCreERT2-Confetti (Tas et al., 2016) donors were from Gabriel D. Victora (Rockefeller University). Mice were bred and maintained in an AAALAC accredited facility at Harvard Medical School under specific pathogen-free (SPF) conditions. Both female and male mice were used in the study. Mice were on a standard 12hr light/dark cycle and were kept on a low-fat diet. Mice were 6–10 weeks old at the start of experiments. Mice were age matched when comparing different strains (max 1 week between experimental and control groups). The local IACUC of Harvard Medical School approved all procedures as described in protocol number IS00000111.
METHOD DETAILS
Bone Marrow Chimeras
Bone marrow chimeras were generated by lethal irradiation of 6–10 week recipients (1030 rads). Mice were kept on antibiotics (sulfamethoxazole/trimethoprim) through drinking water for 10 days after irradiation. Femurs, tibia and humerus from donor mice were cleaned from muscle tissue and subsequently rinsed with BM buffer (HBSS with 10mM HEPES, 1mM EDTA and 2% heat inactivated FBS). All bones were crushed using mortar and pestle and the cells were passed through a 70 mM sterile filter. Erythrocytes were lysed in a small sample to allow accurate counting using a hemacytometer. Where applicable, BM were mixed prior to spinning down and resuspension. Unless stated differently, all 564Igi mixed BM chimeras were generated with a 1:2 ratio of 564Igi and WT/reporter BM respectively. BM recipients received 15–20E6 cells i.v. in 100 μL BM buffer through retro-orbital injection approximately 8 hours post irradiation.
Recombinant HA Expression and Purification
Recombinant A/Hong Kong/1/1968 X31 (H3N2) hemagglutinin (HA) construct was expressed by infection of insect cells with recombinant baculovirus as previously described (Bajic et al., 2019; Raymond et al., 2018; Schmidt et al., 2013). In brief, synthetic DNA corresponding to the full-length ectodomain HA was subcloned into a pFastBac vector modified to encode an N-terminal gp67 secretion signal, and C-terminal thrombin cleavage site, a T4 fibritin (foldon) trimerization tag, and a 6xHis tag. Supernatant from recombinant baculovirus infected High Five (Trichoplusia ni) cells was harvested 72 h post infection and clarified by centrifugation. Protein was purified by affinity chromatography on cobalt-nitrilotriacetic acid (Co2+-NTA) agarose resin (Clontech), followed by a wash in buffer A (20 mM HEPES pH 7.5, 150 mM NaCl), a second wash with buffer A supplemented with 5mM imidazole, elution in buffer A plus 500mM imidazole and gel filtration chromatography on a Superdex 200 column (GE Healthcare) in 10 mM Tris pH 7.5, 150 mM NaCl. The eluted sample was incubated overnight with thrombin protease resin (Sigma) at a 100 μL of slurry per 1 mg of protein to remove the foldon and 6xHis purification tag. HA was further purified by orthogonal Co2+−NTA agarose chromatography, followed by gel filtration chromatography in PBS pH 7.4 and the elution fractions were concentrated to > 2 mg/ml and stored at 4°C. HA was not activated and the resulting protein was uncleaved HA0.
Immunizations
For studies with HA, mice were immunized i.p. with 50 μg HA or 100μg NP-CGG in alum (Imject, Thermo Fisher Sci). Antigen in PBS was added 1:1 (V/V) to alum and shaken vigorously for 30 min prior to injection.
For SRBC immunizations, 10–15 mL of Sheep blood in Alsevers (Colorado Serum Company) was washed two times in HBSS prior to counting SRBC using a Neubauer hemacytometer. Mice received a single injection of 1E9 SRBC in HBSS i.p followed by a boost of 1E7 SRBC 25 days later.
PE:IC Studies
To generate anti-PE sera, three C57/Bl6 mice were immunized SQ with 50mg B-PE (Anaspec) in CFA (Sigma-Aldrich). 3 weeks later, mice were boosted with 50μg B-PE in PBS. 6 days later, serum from these mice was pooled and injected IP into WT and FcγRIIB deficient recipients of 564Igi: AidCreERT2EYFP CD45.1/2 BM (10 weeks post BM transfer). The next day, these passively immunized mice received 5μg of B-PE in PBS IV. After 48 hours, spleens were freshly frozen in OCT. 12μm Sections were fixed with 4% PFA for 15min, permeabilized with 0.1% Triton, 1% BSA in PBS and subsequently stained O/N with ki67 (8c12)-biotin (BD sciences) and Ki67-Fitc (Biolegend) in PBS containing 1% BSA. After washing, sections were incubated for an hour with streptavidin pacific blue for 30 min. Sections were washed again and coverslips were mounted with fluorogel (EM sciences). At least 2 individual sections were imaged per mouse by confocal microscopy (Olympus) each at least 100 μm apart. On average 7 FDC networks were imaged per mouse per section. Cell profiler was used to quantify PE MFI within the CD35 staining defined mask. Only FDC networks which had GC (Ki67 positive groups) associated were included in the analysis.
Photoactivation for Single GC Sorting
B cells from single splenic GCs were sorted as reported previously (Degn et al., 2017). In brief, mice received PE:IC i.v. the day before sorting to label FDC networks. The next day, 30min prior to euthanasia, mice received 800pg CD169-PE IV (Biolegend) to label marginal zone macrophages. Approximately 2mm thick splenic sections were cut using surgical scissors and placed in PBS in chambers made with vacuum grease on a slide. A coverslip was placed over the chambers and the slide was kept on ice prior to photo activation by multiphoton microscopy using an Olympus FV1200 MPE multiphoton system microscope fitted with a 20X 0.95NA Plan water-immersion objective, a MaiTai HP DeepSee Ti-Sapphire laser (Spectraphysics), and 4 non-descanned detectors (2 GaAsP and 2 regular PMTs). Single follicles were identified by the presence of FDC networks surrounded by a ring of CD169 positive macrophages indicating the marginal zone. A single follicle per slice was photo-activated if large auto-fluorescent tingible body macrophages were present, indicating a local GC. After photoactivation of 5–6 slices for each mouse, a single cell suspension was prepared by mashing through a 70 μm filter. Erythrocytes were shocked using RBC lysis buffer (155 mM NH4Cl, 12 mM NaHCO3, 0.1 mM EDTA), and cells were washed in FACS buffer (2% FCS, 1mM EDTA in PBS). Cells were stained with GC markers in addition to CD3ε-bio and IgD-bio. GC B cells were subsequently enriched using streptavidin magnetic beads (Promega) and an easysep magnet (Stemcell) before sorting as described in the single cell sorting paragraph.
Single Cell Sorting
Regular or photoactivated GC B cells (B220+, Gl7hi, CD38lo and paGFP+) were sorted to semiskirted 96 well plates using a single cell sorting mask on a 5-laser FACSARIA II Special Order System (355, 405, 488, 640, 594 nm laser lines; FCS-PMT module and enhanced optics and digital focusing), using a single-cell sort mask. Each well contained 5 mL TCL buffer (QIAGEN) supplemented with 1% Beta-mercaptoethanol (Sigma). At least 4 wells dispersed over the plate were kept empty to serve as negative controls for subsequent PCRs. After sorting, plates were immediately sealed, spun down briefly and stored at −80°C.
Single Cell BCR Sequence PCR and Analysis
Amplification and sequencing of single cell BCR was performed as described previously (Degn et al., 2017). In brief, 96 well plates with single cell sorted B cells were thawed on ice and spun down briefly. RNA was isolated using 11 μL of SPRI beads (Agencourt RNAclean XP) and a homemade 96 well plate magnet. After 3 washes with 80% EtOH, beads were air-dried and RNA was eluted in a mix of anchored oligo-dT, dNTP and RNaseout. Maxima H- Reverse transcriptase (Thermo Fisher), Betaine and MgCl2 was added and reverse transcription was performed for 1hr at 50°C. BCRs were amplified from cDNA using a semi nested approach with specific primers for IgM, IgG and Kappa light chain (Busse et al., 2014; Tiller et al., 2009). Sanger sequencing was performed on precipitated PCR products from the second amplification round by the DF/HCC DNA resource core.
Sequences were manually curated based on chromatograms to screen out double or ambiguous reads. Alternatively, a custom script in Python was used to screen for sequences with > 200 consecutive nucleotides with a PHRED score ≥ 20. Sequences were then run through a custom Python script that used FASTX to generate reverse complement sequences and clip of the 3′end of the msVHE primer. Sequences were then passed to Igblastn using the NCBI VDJ dataset as reference. Igblast’s output including V(D)J usage and mutation frequency (FR1-CDR3) was parsed automatically to a tabular form allowing analysis by excel. In addition, the python script used the biopython library for translation of the query and reference sequence to calculate mutation rates at the peptide level.
Confetti Imaging and Analysis
For induction of the Aicda driven CreERT2 recombinase, mice received a single gavage of 500 μl tamoxifen (Sigma) in sunflower oil at 30mg/ml. At various endpoints, splenic sections were made using surgical scissors and placed in PBS in vacuum grease chambers on a slide. A coverslip was placed over the chambers and the sections were imaged using the multiphoton microscope described in the photo-activation paragraph. Z stacks were acquired at 3 mm interval with a laser wavelength of 940nm. Only complete GC (no or few cells at the section surface) were imaged and depth permitting, the entire volume of the GC was acquired. Typically, 12–15 GC were acquired per spleen. GC with low density of confetti positive cells (high amount of dark clones) were not acquired or omitted from analysis. Scoring of confetti positive cells was done blinded by renaming files to a random, 10 digit alphanumerical filename using a bash script and random strings generated by https://www.random.org. Images were analyzed using Fiji (ImageJ) and the cell counter plugin. Calculation of divergence index was performed as described by Tas et al. (2016) with 3 days post tamoxifen data used as baseline color distribution.
Flow Cytometry
For analysis by flow-cytometry, roughly one quarter of the spleen or lymph nodes were mashed in FACS buffer and filtered through a 70 μm filter. After shocking of erythrocytes (spleens only), cells were stained in FACS buffer for at least 30 min on ice. All stainings included a live/dead control in the APC/Cy7 channel. Anti-idiotype (clone 9D11) was purified in house and labeled with AF647 for facs stainings (Chatterjee et al., 2013). After washing, cells were analyzed on a FACS ARIAII (see single cell sorting) or a FACS Canto II equipped with 405, 488 and 640nm lasers.
Immunofluorescence and Confocal Microscopy
Spleens were fresh frozen in OCT (Fisher Healthcare) and stored at −80°C. Lymph nodes were fixed for 2 hours in 4% PFA on ice followed by 2 hr incubation in 30% sucrose on ice. LN were embedded in OCT and oriented using a stereo microscope prior to storage at −80°C. For all tissue 10–15 μm sections were cut and for spleen either fixed with ice cold acetone or 4% PFA on slide for 20 min. Sections were stained in blocked and stained in block buffer (0.1% TX100, 2% BSA in PBS). Typically, primary antibodies were allowed to stain overnight at 4°C. The next day, streptavidin conjugates or secondary antibodies were incubated for 2 hr at RT in blockbuffer after washing the slides in blockbuffer. Slides were washed 3 times in blockbuffer and coverslips were mounted with fluorogel (Electron microscopy services). Slides were allowed to dry and imaged the same day or the following day using an Olympus F1000 confocal system with an Olympus IX81 inverted microscope and 405, 457, 488, 515, 559, 635nm laser lines. Cellprofiler (Lamprecht et al., 2007) and Fiji (Schindelin et al., 2012) were used to analyze and view images post acquisition.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of confocal images was performed using CellProfiler. Quantification of confetti GC was done after assigning the file names a randomized letter-digit combination as described in the “Confetti Imaging and Analysis” section. All Statistical analysis were performed in Prism Graphpad. Figure legends indicate the test used for each dataset. A p value ≤ 0.05 was scored as statistically significant. For graphs showing GC diversity based on the confetti system, each point denotes one GC, lines indicate the median of the GC diversity per group. N indicates the number of mice used to obtain the GC data. In graphs describing sequencing data (somatic hypermutation), each point represents a curated sequence. Lines indicate the median of the group.
DATA AND CODE AVAILABILITY
Source IgH VDJ sequence data for Figures 5 and 6 in the paper is available in supplemental Tables S2 and S3, respectively. Scripts generated during this study to aid with IgBlast analysis of sequences are available at Github, https://doi.org/10.5281/zenodo.3483838
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-mouse/human GL7 antigen-PacBlue (clone GL7) | Biolegend | Cat# 144613; RRID: AB_2563291 |
| Rat monoclonal anti-mouse/human CD45R/B220-PerCP/Cy5.5 | Biolegend | Cat# 103235; RRID: AB_893356 |
| Mouse monoclonal anti-mouse CD45.1-FITC (clone A20) | Biolegend | Cat# 110705; RRID: AB_313494 |
| Mouse monoclonal anti-mouse CD45.2-APC (clone 104) | Biolegend | Cat# 109813; RRID: AB_389210 |
| Rat monoclonal anti-mouse CD16/32 PE | Biolegend | Cat# 101307; RRID: AB_312806 |
| Rat monoclonal anti-mouse CD16/32 Alexafluor 647 | Biolegend | Cat# 101314; RRID: AB_2278396 |
| Rat monoclonal anti-mouse CD35 biotin | BD Biosciences | Cat# 553816; RRID: AB_395068 |
| Rat monoclonal anti-mouse Ki-67 FITC | Biolegend | Cat# 652409; RRID: AB_2562140 |
| Streptavidin- Pacific Blue | ThermoFisher Scientific | Cat# S11222 |
| Rat monoclonal anti-mouse CD169 PE | Biolegend | Cat# 3142403; RRID: AB_10915470 |
| Rat monoclonal anti-mouse CD38 PE/Cy7 | Biolegend | Cat# 102717; RRID: AB_2072892 |
| Rat monoclonal anti-mouse CD54 (ICAM) PE | Biolegend | Cat# 116107; RRID: AB_313698 |
| Rat monoclonal anti-mouse CD106(VCAM) FITC | Biolegend | Cat# 105705; RRID: AB_313206 |
| Rat monoclonal anti-mouse CD138 PE | Biolegend | Cat# 142503; RRID: AB_10915989 |
| Mouse monoclonal anti-idiotype (clone 9D11) | (Chatterjee et al., 2013) | N/A |
| Rat monoclonal anti-mouse IgD biotin | Biolegend | Cat# 405733; RRID: AB_2563343 |
| Armenian Hamster monoclonal anti-mouse CD3e biotin | Biolegend | Cat# 100304; RRID: AB_312669 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| B - Phycoerythrin | Anaspec | Cat# AS-82001 |
| Tamoxifen | SigmaAldrich | Cat# T5648–5G |
| Sunflower Seed oil | Spectrum Chemical | Cat# S1929–500MLGL |
| Fixable Viability Dye eFluor® 780 | Thermofisher Scientific | Cat# 65–0865-18 |
| Imject Alum Adjuvant | Thermofisher Scientific | Cat# 77161 |
| Np-Osu | Bioresearch technologies | Cat# N-1010–100 |
| Chicken gamma globulin | Rockland Immunochemicals | Cat# D602–0100 |
| Experimental Models: Cell Lines | ||
| T. ni: High Five | ThermoFisher | Cat# B85502 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Aid-CreERT2 EYFP: AicdaCreERT2 flox-stop-flox-EYFP | (Dogan et al., 2009) | N/A |
| Mouse: FcgRIIB flox/flox | (Boross et al., 2011) | N/A |
| Mouse: PA-GFP: B6.Cg-Ptprca Tg(UBC-PA-GFP)1Mnz/J | The Jackson Laboratory | JAX:022486 |
| Mouse: 564Igi: 564 HiKi | (Berland et al., 2006) | N/A |
| Mouse: Aid-CreERT2 Confetti: AicdaCreERT2-Rosa26Confetti | (Tas et al., 2016) | N/A |
| Mouse: C57BL/6J | The Jackson laboratory | JAX: 000664 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson laboratory | JAX: 002014 |
| Oligonucleotides | ||
| Primers for Ig PCR and sequencing | (Busse et al., 2014; Degn et al., 2017; Tiller et al., 2009) | N/A |
| Recombinant DNA | ||
| pFB_H3_Aichi_2_1968_(X31)_FLsE | (Bajic et al., 2019) | N/A |
| Software and Algorithms | ||
| Igblast tools | This paper | https://doi.org/10.5281/zenodo.3483838 |
| FIJI (ImageJ) | (Schindelin et al., 2012) | https://fiji.sc |
| CellProfiler | (Lamprecht et al., 2007) | https://cellprofiler.org/ |
| Python | Python Software Foundation | https://www.python.org |
| Other | ||
| Agencourt RNAClean XP | Beckman Coulter | Cat# A63987 |
| Maxima H Minus Reverse Transcriptase | Thermo Fisher Scientific | Cat# EP0752 |
| TCL buffer | QIAGEN | Cat# 1031576 |
Highlights.
Stromal FcγRIIB deficiency causes increased germinal center diversity
Absence of FcγRIIB on FDCs supports persistence of low SHM B cell clones
ACKNOWLEDGMENTS
We thank J.S. Verbeek for providing the FcγRIIB fl/fl mouse, Elisabeth Alicot for administrative and technical support, Khristina Holscher for technical support, and all of the members of the Carroll lab for helpful discussions. Imaging was performed at the Program in Cellular and Molecular Medicine (PCMM) microscopy core, with technical support from Harry Leung. Flow cytometry and cell sorting were performed at the PCMM flow cytometry core, with technical support from Natasha Barteneva, Kenneth Ketman, and Jodene K. Moore. This work was funded by NIH grants R01AI119006 (G.D.V.), R01AI130307 (M.C.C.), and R01AR074105 (M.C.C.). C.E.v.d.P. was supported by a GlaxoSmithKline (GSK) Early Talent Fellowship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.086.
DECLARATION OF INTERESTS
C.D.E. and G. Bouma are employees and shareholders of GlaxoSmithKline. All of the other authors declare no competing interests.
REFERENCES
- Amitai A, Mesin L, Victora GD, Kardar M, and Chakraborty AK (2017). A Population Dynamics Model for Clonal Diversity in a Germinal Center. Front. Microbiol 8, 1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenwaldt A, Lux A, Danzer H, Spriewald BM, Ullrich E, Heidkamp G, Dudziak D, and Nimmerjahn F (2011). Fcg receptor IIB (FcγRIIB) maintains humoral tolerance in the human immune system in vivo. Proc. Natl. Acad. Sci. USA 108, 18772–18777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic G, Maron MJ, Adachi Y, Onodera T, McCarthy KR, McGee CE, Sempowski GD, Takahashi Y, Kelsoe G, Kuraoka M, and Schmidt AG (2019). Influenza Antigen Engineering Focuses Immune Responses to a Sub-dominant but Broadly Protective Viral Epitope. Cell Host Microbe 25, 827–835.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrington RA, Pozdnyakova O, Zafari MR, Benjamin CD, and Carroll MC (2002). B lymphocyte memory: role of stromal cell complement and FcgammaRIIB receptors. J. Exp. Med 196, 1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, Wortis HH, Kearney JF, Ucci AA, and Imanishi-Kari T (2006). Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity 25, 429–440. [DOI] [PubMed] [Google Scholar]
- Boross P, Arandhara VL, Martin-Ramirez J, Santiago-Raber ML, Carlucci F, Flierman R, van der Kaa J, Breukel C, Claassens JW, Camps M, et al. (2011). The inhibiting Fc receptor for IgG, FcγRIIB, is a modifier of autoimmune susceptibility. J. Immunol 187, 1304–1313. [DOI] [PubMed] [Google Scholar]
- Bournazos S, and Ravetch JV (2015). Fcg receptor pathways during active and passive immunization. Immunol. Rev 268, 88–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse CE, Czogiel I, Braun P, Arndt PF, and Wardemann H (2014). Single-cell based high-throughput sequencing of full-length immunoglobulin heavy and light chain genes. Eur. J. Immunol 44, 597–603. [DOI] [PubMed] [Google Scholar]
- Chatterjee P, Agyemang AF, Alimzhanov MB, Degn S, Tsiftsoglou SA, Alicot E, Jones SA, Ma M, and Carroll MC (2013). Complement C4 maintains peripheral B-cell tolerance in a myeloid cell dependent manner. Eur. J. Immunol 43, 2441–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu ZT, Tsuchiya N, Kyogoku C, Ohashi J, Qian YP, Xu SB, Mao CZ, Chu JY, and Tokunaga K (2004). Association of Fcgamma receptor IIb polymorphism with susceptibility to systemic lupus erythematosus in Chinese: a common susceptibility gene in the Asian populations. Tissue Antigens 63, 21–27. [DOI] [PubMed] [Google Scholar]
- Das A, Heesters BA, Bialas A, O’Flynn J, Rifkin IR, Ochando J, Mittereder N, Carlesso G, Herbst R, and Carroll MC (2017). Follicular Dendritic Cell Activation by TLR Ligands Promotes Autoreactive B Cell Responses. Immunity 46, 106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degn SE, van der Poel CE, Firl DJ, Ayoglu B, Al Qureshah FA, Bajic G, Mesin L, Reynaud CA, Weill JC, Utz PJ, et al. (2017). Clonal Evolution of Autoreactive Germinal Centers. Cell 170, 913–926 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, Reynaud CA, and Weill JC (2009). Multiple layers of B cell memory with different effector functions. Nat. Immunol 10, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Espéli M, Smith KG, and Clatworthy MR (2016). FcγRIIB and autoimmunity. Immunol. Rev 269, 194–211. [DOI] [PubMed] [Google Scholar]
- Floto RA, Clatworthy MR, Heilbronn KR, Rosner DR, MacAry PA, Rankin A, Lehner PJ, Ouwehand WH, Allen JM, Watkins NA, and Smith KG (2005). Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat. Med 11, 1056–1058. [DOI] [PubMed] [Google Scholar]
- Hannum LG, Haberman AM, Anderson SM, and Shlomchik MJ (2000). Germinal center initiation, variable gene region hypermutation, and mutant B cell selection without detectable immune complexes on follicular dendritic cells. J. Exp. Med 192, 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, and Carroll MC (2013). Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity 38, 1164–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, and Kikutani H (1994). The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1, 167–178. [DOI] [PubMed] [Google Scholar]
- Kyogoku C, Dijstelbloem HM, Tsuchiya N, Hatta Y, Kato H, Yamaguchi A, Fukazawa T, Jansen MD, Hashimoto H, van de Winkel JG, et al. (2002). Fcgamma receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis Rheum. 46, 1242–1254. [DOI] [PubMed] [Google Scholar]
- Lamprecht MR, Sabatini DM, and Carpenter AE (2007). CellProfiler: free, versatile software for automated biological image analysis. Biotechniques 42, 71–75. [DOI] [PubMed] [Google Scholar]
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, and Marshak-Rothstein A (2005). RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J. Exp. Med 202, 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, and Marshak-Rothstein A (2002). Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416, 603–607. [DOI] [PubMed] [Google Scholar]
- Li F, Smith P, and Ravetch JV (2014). Inhibitory Fcg receptor is required for the maintenance of tolerance through distinct mechanisms. J. Immunol 192, 3021–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Weisel F, and Shlomchik MJ (2018). B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the c-Myc Transcription Factor in Germinal Center B Cells. Immunity 48, 313–326 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHeyzer-Williams LJ, Milpied PJ, Okitsu SL, and McHeyzer-Williams MG (2015). Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat. Immunol 16, 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesin L, Ersching J, and Victora GD (2016). Germinal Center B Cell Dynamics. Immunity 45, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Hermann ME, Maini PK, and Iber D (2006). An analysis of B cell selection mechanisms in germinal centers. Math. Med. Biol 23, 255–277. [DOI] [PubMed] [Google Scholar]
- Myers RC, King RG, Carter RH, and Justement LB (2013). Lymphotoxin a1b2 expression on B cells is required for follicular dendritic cell activation during the germinal center response. Eur. J. Immunol 43, 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan TG, Grigorova I, Okada T, and Cyster JG (2007). Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat. Immunol 8, 992–1000. [DOI] [PubMed] [Google Scholar]
- Qin D, Wu J, Vora KA, Ravetch JV, Szakal AK, Manser T, and Tew JG (2000). Fc gamma receptor IIB on follicular dendritic cells regulates the B cell recall response. J. Immunol 164, 6268–6275. [DOI] [PubMed] [Google Scholar]
- Raymond DD, Bajic G, Ferdman J, Suphaphiphat P, Settembre EC, Moody MA, Schmidt AG, and Harrison SC (2018). Conserved epitope on influenza-virus hemagglutinin head defined by a vaccine-induced antibody. Proc. Natl. Acad. Sci. USA 115, 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AG, Xu H, Khan AR, O’Donnell T, Khurana S, King LR, Manischewitz J, Golding H, Suphaphiphat P, Carfi A, et al. (2013). Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc. Natl. Acad. Sci. USA 110, 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Supprian M, and Rajewsky K (2007). Vagaries of conditional gene targeting. Nat. Immunol 8, 665–668. [DOI] [PubMed] [Google Scholar]
- Sharp PEH, Martin-Ramirez J, Mangsbo SM, Boross P, Pusey CD, Touw IP, Cook HT, Verbeek JS, and Tarzi RM (2013). FcγRIIB on myeloid cells and intrinsic renal cells rather than B cells protects from nephrotoxic nephritis. J. Immunol 190, 340–348. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Grigorova I, Phan TG, Kelly LM, and Cyster JG (2009). Visualizing B cell capture of cognate antigen from follicular dendritic cells. J. Exp. Med 206, 1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tas JM, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM, Chen CS, Weill JC, Reynaud CA, Browne EP, et al. (2016). Visualizing antibody affinity maturation in germinal centers. Science 351, 1048–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiller T, Busse CE, and Wardemann H (2009). Cloning and expression of murine Ig genes from single B cells. J. Immunol. Methods 350, 183–193. [DOI] [PubMed] [Google Scholar]
- Victora GD, and Nussenzweig MC (2012). Germinal centers. Annu. Rev. Immunol 30, 429–457. [DOI] [PubMed] [Google Scholar]
- Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, and Nussenzweig MC (2010). Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143, 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinuesa CG, Linterman MA, Yu D, and MacLennan ICM (2016). Follicular Helper T Cells. Annu. Rev. Immunol 34, 335–368. [DOI] [PubMed] [Google Scholar]
- Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, and Cyster JG (2011). Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J. Exp. Med 208, 2497–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Rodda LB, Bannard O, and Cyster JG (2014). Integrin-mediated interactions between B cells and follicular dendritic cells influence germinal center B cell fitness. J. Immunol 192, 4601–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source IgH VDJ sequence data for Figures 5 and 6 in the paper is available in supplemental Tables S2 and S3, respectively. Scripts generated during this study to aid with IgBlast analysis of sequences are available at Github, https://doi.org/10.5281/zenodo.3483838