

Abstract
Background
The majority of patients with advanced cancer develop cachexia, a weight loss syndrome that severely reduces quality of life and limits survival. Our understanding of the underlying mechanisms that cause the condition is limited, and there are currently no treatment options that can completely reverse cachexia. Several tumour‐derived factors and inflammatory mediators have been suggested to contribute to weight loss in cachectic patients. However, inconsistencies between studies are recurrent. Activin A and interleukin 6 (IL‐6) are among the best studied factors that seem to be important, and several studies support their individual role in cachexia development.
Methods
We investigated the interplay between activin A and IL‐6 in the cachexia‐inducing TOV21G cell line, both in culture and in tumours in mice. We previously found that the human TOV21G cells secrete IL‐6 that induces autophagy in reporter cells and cachexia in mice. Using this established cachexia cell model, we targeted autocrine activin A by genetic, chemical, and biological approaches. The secretion of IL‐6 from the cancer cells was determined in both culture and tumour‐bearing mice by a species‐specific ELISA. Autophagy reporter cells were used to monitor the culture medium for autophagy‐inducing activities, and muscle mass changes were evaluated in tumour‐bearing mice.
Results
We show that activin A acts in an autocrine manner to promote the synthesis and secretion of IL‐6 from cancer cells. By inhibiting activin A signalling, the production of IL‐6 from the cancer cells is reduced by 40–50% (up to 42% reduction on protein level, P = 0.0048, and 48% reduction on mRNA level, P = 0.0308). Significantly reduced IL‐6 secretion (P < 0.05) from the cancer cells is consistently observed when using biological, chemical, and genetic approaches to interfere with the autocrine activin A loop. Inhibiting activin signalling also reduces the ability of the cancer cells to accelerate autophagy in non‐cancerous cells (up to 43% reduced autophagy flux, P = 0.0006). Coherent to the in vitro data, the use of an anti‐activin receptor 2 antibody in cachectic tumour‐bearing mice reduces serum levels of cancer cell‐derived IL‐6 by 62% (from 417 to 159 pg/mL, P = 0.03), and, importantly, it reverses cachexia and counteracts loss of all measured muscle groups (P < 0.0005).
Conclusions
Our data support a functional link between activin A and IL‐6 signalling pathways and indicate that interference with activin A‐induced IL‐6 secretion from the tumour has therapeutic potential for cancer‐induced cachexia.
Keywords: Cachexia, Autophagy, IL‐6, Activin, Autocrine loop
Introduction
Cancer cachexia (hereafter referred to as cachexia) severely compromises quality of life and limits survival of up to 80% of patients with advanced cancer.1 Cachexia is characterized by a substantial loss of lean body mass that is often, but not always, accompanied by loss of fat.2 Currently, no therapeutic approach consistently reverses cachexia in humans. The development of therapeutic strategies has been slowed by the incomplete understanding of the underlying mechanisms that lead to the condition.
Several inflammatory and tumour‐derived factors are suggested to drive development of cachexia. One of these is activin A, a member of the transforming growth factor beta (TGFβ) superfamily of proteins. Activin A administration alone is sufficient to cause weight loss (including muscle loss) in mice.3 Also, α‐inhibin‐deficient mice (lacking a central inhibitor of activin) lose weight.4 Furthermore, inhibition of activin receptor 2B (ActRIIB), a receptor for a subset of TGFβ members including activin A, reverses cachexia and increases survival in several mouse models.5 Elevated activin A levels in sera from pancreatic cancer patients are associated with shorter survival.6 This might relate to development of cachexia.
It has been suggested that several members of the TGFβ superfamily, including activin A, can affect skeletal muscle by limiting the differentiation of myoblasts.7, 8 Moreover, the transcription factors SMAD2 and SMAD3, which are activated by, for instance, activin A, can induce atrophy in adult myofibers, in a mechanism involving the AKT/mammalian target of rapamycin pathway.9 Although convincing data underscores a role of activin A in cachexia development, other signalling substances and pathways are likely also involved.
Several studies point to tumour‐derived interleukin 6 (IL‐6) as a central driver of cachexia. We have shown that IL‐6 is secreted from cancer cells, and, via trans‐signalling, it can accelerate the catabolic process autophagy in muscle cells, thereby contributing to cachexia.10 Also, IL‐6 administration is sufficient to drive cachexia,11 and IL‐6‐blocking agents can reverse muscle loss and increase survival in mice.12, 13, 14 Recently, it was shown that the human ovarian cancer cell line ES‐2 induces cachexia in mice in an IL‐6‐dependent manner.15 Moreover, increased IL‐6 levels have been detected in serum from cachectic patients,16 and importantly, case studies in humans show promising results when blocking the IL‐6 receptor, using the neutralizing antibody tocilizumab.17, 18
The existence of convincing data with regard to the cachexia‐causing ability of both activin A and IL‐6 could be due to different mechanisms operating in different contexts and patients but could also be due to a functional relationship between activin A and IL‐6. In order to investigate this, we have used the ovarian carcinoma cell line TOV21G. These cells secrete high amounts of activin A and cause a rapid development of cachexia at a low tumour burden in mice.5, 10 In this model, cachexia can be reversed by injecting activin decoy receptor,5 thereby supporting a role for activin A in TOV21G‐driven cachexia. We recently reported that the TOV21G cells, in addition to activin A, secrete high levels of IL‐6.10 This makes TOV21G a good tool for testing whether there is a functional relationship between activin A and IL‐6 in cachexia development. Here, we present data demonstrating that autocrine activin A signalling in cancer cells is needed for their release and systemic distribution of IL‐6. We also show that activin A‐induced IL‐6 secretion from these cells stimulates autophagy in non‐cancerous cells, a catabolic process suggested by us10 and others19, 20, 21, 22 to be involved in the pathogenesis of cachexia. Furthermore, we find that interference with activin A signalling in cachectic tumour‐bearing mice reduces serum levels of cancer cell‐derived IL‐6 and reverses cachexia. Collectively, our results point to a novel mechanism by which both activin A and IL‐6 can contribute to cachexia and that this interplay may provide targets for treatment of cancer cachexia.
Methods
Reagents
During in vitro cell culture experiments, the following reagents were used as specified: Recombinant (r) human/mouse/rat activin A (R&D Systems, 338‐AC‐010/CF), rHuman IL‐6 (Invitrogen, cat. no. PHC0066), rHuman Activin RIIB Fc Chimera (R&D Systems 339RB/CF), human/mouse/rat activin A (beta A subunit) antibody (R&D Systems, MAB3381), ALK4/5/7 inhibitor (Selleck Chem, cat. no. SB431542), 3‐methyladenine (Sigma, M9281), bafilomycin A1 (Sigma, B1793), Torin 1 (Cayman Chemicals, #10997‐10), and Hanks' Balanced Salt Solution (Sigma, H9269).
CDD866 is a chimeric murinized version of BYM338/bimagrumab, an anti‐ActRII antibody, where the human Fc region of the antibody has been replaced by a mouse Fc (IgG2a Leu234Ala/Leu235Ala), provided by the Novartis Biologic Units.
Cell culture
Cells were maintained in a humidified atmosphere of 5% CO2 and 95% air at 37°C. TOV21G cells from ATCC (CRL‐11730) were cultured in 42.5% medium 199 + GlutamaxTMI (Gibco 41150) and 42.5% medium MCDB 105 [Sigma M6395, dissolved in dH2O to 1 L final volume, added NaHCO3 (1.5 g/L), and adjusted to pH 7.3], supplemented with foetal bovine serum (FBS, 15%) and gentamicin (0.05 mg/mL). HEK293 green fluorescent protein (GFP)‐p62 cells (a kind gift from Prof. T. Johansen and colleagues) were cultured in DMEM (Sigma D5796) supplemented with FBS (10%) and gentamicin (0.05 mg/mL).
RNA interference
For inhibin beta A (INHBA), activin receptor 2A (ACVR2A), activin receptor 2B (ACVR2B), KRAS, and SMAD3 downregulation, TOV21G cells were transfected using 20 nM siRNA (final concentrations), and DharmaFECT transfection reagent 1 (Dharmacon) was diluted in Opti‐MEM 1 Reduced Serum Medium (#31985‐070, Gibco Life Technologies). After 24 h, the transfection medium was exchanged for normal growth medium, and the cells were incubated for 1 or 3 days before conditioned medium (CM) was harvested and protein and RNA were isolated. At this time, cultures were approximately 80% confluent. The following siRNA oligonucleotides were obtained from Dharmacon: L‐011701‐00‐0010 ON‐TARGETplus SMARTpool human INHBA, L‐004926‐00‐0010 ON‐TARGETplus SMARTpool human ACVR2A, L‐004927‐00‐0010 ON‐TARGETplus SMARTpool human ACVR2B, L‐020067‐00‐0005 ON‐TARGETplus SMARTpool human SMAD3, and D‐001210‐01‐20 non‐targeting siRNA. Two siRNA oligonucleotides targeting KRAS were obtained from Ambion: cat. no. 4390824 (ID s7939 and ID s7940).
Immunoblotting
SMAD3 signalling in autophagy reporter cells
HEK293 autophagy reporter cells were exposed to 50 μg/mL recombinant activin A for 5 min at the end of 2 h incubation in normal growth medium containing 0.1% FBS. Cell were lysed by cell scraping on ice in a buffer containing 8 M urea, 0.5% (v/v) Triton X‐100, 100 mM DTT, 1xComplete® protease inhibitor, and 8% phosphatase inhibitor cocktail I and III (Sigma). Protein concentration was determined by BioRad protein assay (BioRad). Equal amounts of proteins were separated using NuPAGE® Novex® 4‐12% Bis‐Tris Gels (Invitrogen) and dry blotted on nitrocellulose membranes. The membrane was blocked, and antibodies were diluted in a 1:1 mixture of Odyssey blocking buffer (Li‐Cor) and TBST (20 mM Tris, pH 7.6, 137 mM NaCl with 0.1 % Tween 20). Bound antibodies were imaged by near infrared fluorescence using appropriate fluorescent dye labelled secondary antibodies and an Odyssey NIR scanner (Li‐Cor Biosciences). Images were processed using the Li‐Cor Odyssey software image studio 2.0. The antibodies used for immunostaining were pSMAD3 (Ser423/Ser425) (Abcam, ab52903), diluted 1:1000, and beta‐tubulin (Abcam, ab6046), diluted 1:5000.
NF‐κB and p38 MAP kinase signalling in TOV21G after interfering with activin signalling
TOV21G cells were transfected with siRNA as described earlier. Three days after removal of transfection reagents, the cells were harvested in urea buffer, and extracts were applied on gels, blotted, and used for western detection as described earlier. Antibodies used for immunostaining were pNF‐κB p65 (Ser536) (Cell Signaling, #3033), phospho‐p38 MAP kinase (Thr180/Tyr182) (Cell Signaling, #9215), and Erk1/2 (Cell Signaling #9107), all diluted 1:1000.
Autophagy reporter system
Autophagy was quantified using flow cytometry measuring GFP fluorescence in live HEK293 cells expressing GFP‐sequestosome 1 (SQSTM1) fusion gene, as previously described.23 HEK293 autophagy reporter cells were seeded in 24‐well plates (25 000 cells per well) and incubated for 1 day. Thereafter, the cells were treated as indicated for 3 days (unless otherwise specified) in the presence of doxycycline (1 ng/mL) before assessment of fusion protein degradation by flow cytometry. When using known autophagy effectors, these were added 17 h before assessment of fusion protein degradation. Protein degradation was determined as the loss of green fluorescent signal compared with control. Triplicate wells were used in all experiments, and signal was measured in 10 000 cells per well.
ELISA
The levels of IL‐6 and activin A in CM from TOV21G cells were determined using human IL‐6 OptEIA ELISA kit (BD Biosciences, cat. no. 555220) and a human/mouse/rat activin A Quantikine ELISA kit (R&D Systems, cat. no. DAC00B), respectively. All analyses were performed according to the manufacturer's protocols.
The levels of human IL‐6 and murine IL‐6 in serum from mice were determined using the U‐PLEX Human IL‐6 Assay (Meso Scale Discovery, cat. no. K151TXK) and the U‐PLEX Mouse IL‐6 Assay (Meso Scale Discovery, cat. no. K152TXK) according to the protocols provided by the manufacturer. The levels of activin A in serum from mice were determined using a self‐made activin A ELISA, which was established according to the protocol of Meso Scale Discovery. The following antibodies were used for this ELISA: anti‐activin A primary antibody (R&D Systems, cat. no. MAB3381; detects human/rat/mouse activin A); biotinylated anti‐activin A secondary antibody (R&D Systems, cat. no. BAM3381; detects human/rat/mouse activin A); for detection, Sulfo‐TAG streptavidin (Meso Scale Discovery, cat. no. R32AD, 0.25 μg/mL); and for the titration of the standard curve, recombinant human activin A (R&D Systems, cat. no. 338 AC/CF). The antibodies in this sandwich ELISA are directed against the same epitope of the INHBA subunit and thus will detect only the homodimer (activin A). They do not recognize the inhibin beta B subunit, and accordingly, there is no cross reactivity towards activin AB. Due to low homology (18%) between INHBA and the alpha subunit found in inhibin A, any cross reactivity towards inhibin A is considered unlikely.
All presented ELISA data are within the range of the respective standard curves (Figure S1). When necessary, the samples were diluted to fall within the range of the standard curve.
RNA isolation and quantitative real‐time PCR
TOV21G cells transfected with NT, INHBA, ACVR2A, ACVR2B, KRAS, or SMAD3 siRNA were left for 1 or 3 days post‐transfection (as explained earlier) before RNA was isolated using RNeasy Mini Kit (Qiagen, cat. no. 74106) according to the protocol from the manufacturer. Purity, quality, and concentration of isolated RNA were confirmed using Nanodrop. cDNA for qPCR analysis was made by using the QuantiTect Reverse Transcription Kit (Qiagen, cat. no. 205311) according to the manufacturer's protocol. mRNA levels were normalized against GAPDH. qPCR was performed in parallel 20 μL reactions containing 10 μL Perfecta qPCR Fast‐Mix ROX (from Quanta, QUNT95077‐012), 1 μL of the respective primer set, and 9 μL template (5 and 25 ng RNA for GAPDH and the remaining primer sets, respectively). The following TaqMan primer sets from Life Technologies were used: human INHBA (Hs01081598_m1, cat. no. 4331182), human AVCR2A (Hs00155658_m1, cat. no. 4331182), human ACVR2B (Hs00609603_m1, cat. no. 4331182), human IL‐6 (Hs00985639_m1, cat. no. 4331182), human KRAS (Hs00364284_g1, cat. no. 4331182), human SMAD3 (Hs00969210_m1, cat. no. 4331182), and human GAPDH (Hs99999905_m1, cat. no. 4331182). The cycling conditions for the StepOne plus system (Applied Biosystems, Foster City, CA, USA) were 45°C for 2 min, 95°C for 30 s, and 40 cycles of 95°C for 1 s and 65°C for 20 s. Relative RNA transcription levels were transformed into linear form by 2−ΔΔCt.
Animal experiments
The animal experiment was performed according to the regulations effective in the Canton of Basel City, Switzerland, under the license number BS‐2186. Eight‐week‐old female mice (Hsd:Athymic Nude‐Foxn1 nu) were purchased from Harlan Laboratories (Horst, Netherlands), acclimated for 7 days housed at 25°C with a 12:12 h light–dark cycle, and provided ad libitum water and a standard laboratory diet containing 18.2% protein and 3.0% fat with an energy content of 15.8 MJ/kg (NAFAG 3890, Kliba, Basel, Switzerland).
TOV21G cells were harvested by trypsin treatment and suspended in a solution containing 50% PBS and 50% BD Matrigel™ matrix without phenol red (cat. no. 356237, BD Biosciences, Bedford, MA) at a density of 3 × 107/mL. A 0.1 mL of cell suspension containing 3 × 106 cells was inoculated subcutaneously into the left flank of mice anaesthetized with Forane® (isoflurane; Abbott AG, Baar, Switzerland) using a Station anaesthesia minihub (Tem Sega, Pessac, France). When tumours were palpable, length and width of tumours were measured through skin, and tumour volumes were calculated according to the formula (length × width2)/2. Seven days after cell inoculation, mice bearing tumours with acceptable morphology and size (mean volume of approximately 130 mm3) were randomized to groups containing 10 mice per group, and the treatments were initiated on the day of randomization (Day 0). CDD866 was administered at 20 mg/kg s.c., once weekly in a volume of 5 mL/kg. Body weight and tumour volume were measured two to three times per week. At the end of the experiment (Day 35), the mice were euthanized with CO2. In the group of vehicle‐treated mice, three mice had to be sacrificed at Days 28, 30, and 32, respectively, for animal welfare reasons. For all animals, tumour, tibialis anterior, gastrocnemius–soleus–plantaris complex, quadriceps, heart, liver, kidney, perigonadal white adipose tissue, spleen, and brain were collected and weighed. The brain weight was measured for normalization of organ weights.24
Before performing our analyses, we omitted three mice from each of the two groups inoculated with TOV21G cells due to very large or small tumours (larger or smaller than average ±SD).
cBioPortal
cBioPortal (http://www.cbioportal.org/) is an open‐access database that allows visualization and analysis of large‐scale cancer genomics data sets.25, 26 Our analyses utilize the co‐expression visualization to identify co‐expression of IL‐6 and INHBA mRNA. For the analysis, we used the TCGA Provisional data set for ovarian serous cystadenocarcinoma.
Statistics
Statistical work was performed using SPSS or Microsoft Excel. The statistical tests performed are specified when individual results are presented.
Data presentation
All figures were mounted using Canvas 14 (ACD Systems).
Results
Activin A promotes secretion of autophagy‐accelerating factors from tumour cells
We have previously found that weight loss in cancer patients is associated with autophagy‐inducing bioactivity in the blood stream.10 These findings implicate a causal role in cachexia development for tumour‐derived signalling substances that increase autophagy in normal cells. We have shown that IL‐6 is abundantly secreted from certain cancer cells and potently accelerates autophagy in muscle cells when complexed to soluble IL‐6 receptor.10 This way of IL‐6 signalling may contribute to weight loss. Because tumour‐derived activin A is proposed to be a mediator of cancer cachexia,3, 5 we hypothesized that also activin A contributes to cachexia by accelerating autophagy in host cells.
To quantify autophagic flux, we utilized an autophagy reporter cell system,23 HEK293 cells with an inducible expression of GFP fused to SQSTM1 (also known as p62). SQSTM1 is specifically targeted by autophagy, and its time‐dependent decline (and consequently GFP decline/loss of green fluorescence) is a measure of autophagic activity and can be detected by flow cytometry. The autophagy reporter cells responded to recombinant activin A, evident as increased SMAD3 phosphorylation (Figure 1A, upper panel). However, activin A did not accelerate autophagy at any of the tested concentrations (Figure 1A, lower panel). This could mean that activin A contributes to cachexia in a manner that is independent of autophagy in host cells. Alternatively, activin A can still accelerate autophagy in vivo but in an indirect manner by influencing the abundancy of other autophagy‐inducing factors.
Figure 1.
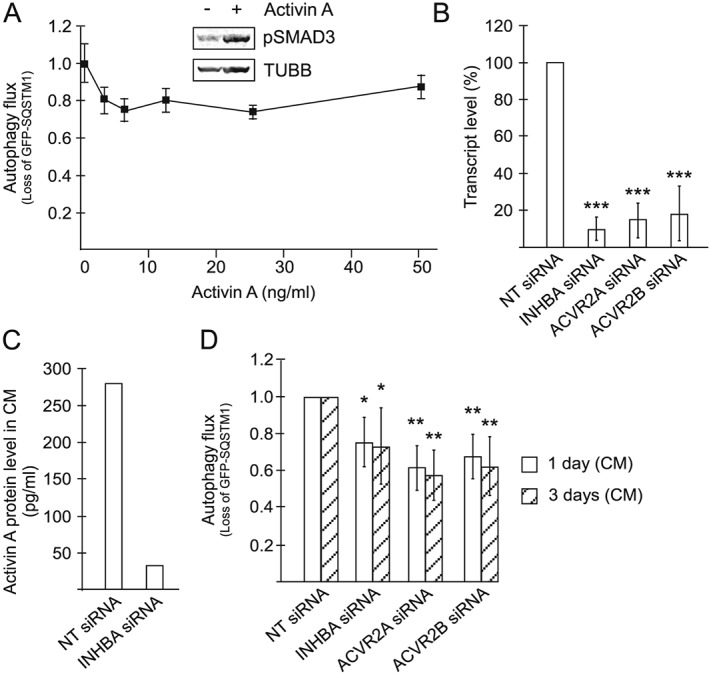
Activin A acts in an autocrine or paracrine loop to promote the secretion of autophagy‐accelerating compounds from TOV21G cells. (A) Lower panel: Autophagy flux in autophagy reporter cells treated with recombinant activin A for 3 days at indicated concentrations. Mean from three independent experiments, each using triplicate wells ±SD. Upper panel: Level of phosphorylated SMAD3 (Ser423/Ser425) in autophagy reporter cells following 5 min exposure to activin A (50 ng/mL). β‐Tubulin is used as a loading control. (B) INHBA, ACVR2A, and ACVR2B transcript levels in TOV21G cells treated with siRNA targeting INHBA, ACVR2A, and ACVR2B, respectively, 1 day post‐transfection, relative to non‐targeting (NT) siRNA‐treated TOV21G cells. Measured using qPCR. Mean from six independent experiments ±SD. *** P < 0.0005 vs. NT siRNA (Student's t‐test). (C) Level of activin A protein in CM from TOV21G cells treated with INHBA siRNA, 3 days post‐transfection. Mean from two experiments. (D) Autophagy flux in autophagy reporter cells treated with CM from TOV21G cells. CM had been collected 1 or 3 days post‐transfection using NT siRNA or siRNA targeting INHBA, ACVR2A, or ACVR2B. Mean from six independent experiments, each using triplicate wells. * P < 0.05, ** P < 0.005 vs. respective NT siRNA (Student's t‐test).
In mice, cachexia is reversed by activin receptor antagonists.5, 27 This may be due to the ability of these agents to prevent activin A‐induced signalling in muscle cells (and other host cells). Alternatively, the receptor antagonist could inhibit activin A signalling in the tumour and affect the secretion of other cachexia‐inducing factor(s). Because we observed no direct effect of activin A on autophagy, we hypothesized that activin A may control the secretion of other factors that accelerate autophagy and cause cachexia.
TOV21G ovarian cancer cells have been shown to secrete high amounts of activin A.5 Moreover, we have shown that CM from these cells potently accelerates autophagy in other cells,10 and, as shown by us10 and others,5 TOV21G tumours potently cause weight loss in mice. Importantly, cachexia in these mice can be reversed by interfering with activin A signalling.5 To test if autocrine signalling could affect the autophagy‐inducing activity of the CM, activin A signalling was targeted by siRNA directed towards either INHBA or ACVR2A/ACVR2B (also called ACTRIIA/ACTRIIB, respectively). INHBA encodes the INHBA subunit that is needed to form bioactive activin A homodimers. Activin A initiates signalling via type 1 and 2 activin receptors and of the type 2 receptors activin A can bind to both ActRIIA and B.28 All siRNAs efficiently reduced the level of their target transcripts (Figure 1B), and the reduction in mRNA levels was maintained for at least 3 days post‐transfection (data not shown). No apparent effects of the different siRNAs were noted on cell proliferation or survival (data not shown). Consistent with the depletion of the target transcripts, INHBA siRNA reduced the activin A protein secreted by TOV21G by about 90% (from an average of 278 to 32 pg/mL), measured by ELISA (Figure 1C). Having successfully depleted components of autocrine activin A signalling, we asked whether this reduced the autophagy‐accelerating ability of CM from the cells. When using the autophagy reporter cell system, we found that the CM from TOV21G cells treated with INHBA, ACVR2A, or ACVR2B siRNA was clearly less potent in accelerating autophagy as compared with CM from cells treated with non‐targeting siRNA. Approximately 40% reduced autophagy flux was observed already 1 day post‐transfection, and this effect was sustained for at least 3 days (Figure 1D). By comparison, the autophagy inhibitors 3‐methyladenine or bafilomycin A1 (17 h) reduce the autophagy flux in the reporter cells by 30% and 40%, respectively (Figure S2). Our results indicate that although activin A may not activate autophagy directly, it can act in an autocrine or paracrine manner to promote secretion of other autophagy‐accelerating factors from the cancer cells.
Activin A is important for secretion of the autophagy‐accelerating factor IL‐6 from cancer cells
We have previously found that the CM from TOV21G cells accelerates autophagy in other cells and have largely attributed this effect to their secretion of IL‐6.10 TOV21G cells are known to have an activating KRAS mutation, and this has been shown to be important for IL‐6 production, especially in murine lung cancer models.29, 30 We therefore wanted to establish whether activated KRAS was a major contributor to the IL‐6 production in our model system. Two different siRNAs for KRAS were shown to very efficiently take down the level of KRAS transcripts (Figure 2A), and this was accompanied by clearly reduced levels of IL‐6 transcripts in the cells (Figure 2B). Also, IL‐6 protein in CM 1 day post‐transfection was reduced about 43% from an average of 23.5 ng/mL in CM from non‐targeting siRNA transfected cells (Figures 2C and S3). To investigate if activin‐mediated signalling could also influence IL‐6 production, we performed siRNA‐mediated knockdown of either INHBA or ACVR2A/ACVR2B in TOV21G cells. Indeed, all siRNAs caused reduced levels of IL‐6 in the CM (about 20% reduction from an average of 23.5 ng/mL in CM from non‐targeting siRNA transfected cells), as measured by an IL‐6 ELISA 1 day post‐transfection (Figures 3A and S3) and confirmed by a bioplex assay (data not shown). The effect was sustained for at least 3 days post‐transfection (24–47% reduction, depending on the siRNA, from an average of 49.1 ng/mL in CM from non‐targeting siRNA transfected cells) (Figures 3A and S3).
Figure 2.
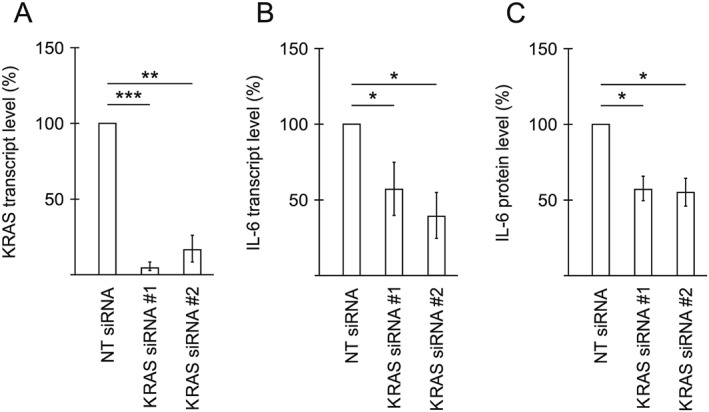
KRAS affects the secretion of the autophagy‐inducing cytokine IL‐6 from TOV21G cells. (A) KRAS transcript level in TOV21G cells treated with two different siRNAs targeting KRAS, 1 day post‐transfection, relative to non‐targeting (NT) siRNA‐treated TOV21G cells. Measured using qPCR. Mean from three independent experiments ±SD. *** P < 0.0005 and ** P < 0.005 vs. NT siRNA (Student's t‐test). (B) IL‐6 transcript level in TOV21G cells treated with siRNAs targeting KRAS, 1 day post‐transfection, relative to NT siRNA‐treated TOV21G cells. Measured using qPCR. Mean from three independent experiments ±SD. * P < 0.05 vs. NT siRNA (Student's t‐test). (C) Relative level of IL‐6, measured using an ELISA assay, in CM from TOV21G cells treated with NT siRNA or siRNAs targeting KRAS. CM was harvested 1 day post‐transfection. Mean from three experiments ±SD. * P < 0.05 vs. NT siRNA (Student's t‐test).
Figure 3.
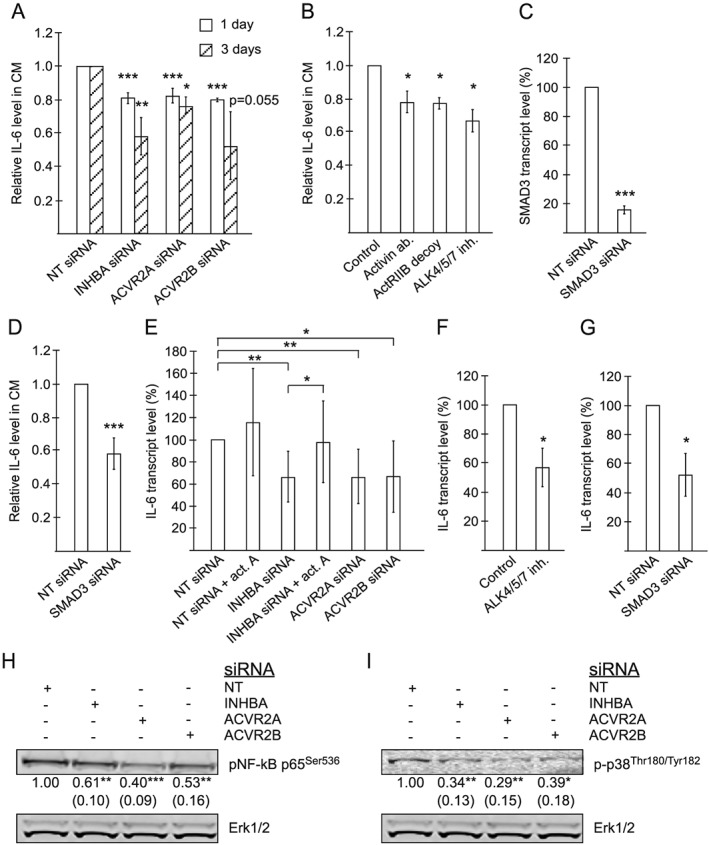
Activin A affects the secretion of the autophagy‐inducing cytokine IL‐6 from TOV21G cells. (A) Relative level of IL‐6, measured using an ELISA assay, in CM from TOV21G cells treated with non‐targeting (NT) siRNA or siRNA targeting INHBA, ACVR2A, or ACVR2B. CM was harvested 1 or 3 days post‐transfection. Mean from three experiments [ACVR2A (3 days) and ACVR2B (1 day and 3 days)], four experiments [ACVR2A (1 day) and INHBA (3 days)], or six experiments [INHBA (1 day)] ±SD. * P < 0.05, ** P < 0.005, *** P < 0.0005 vs. respective NT siRNA (Student's t‐test). (B) Relative level of IL‐6, measured using an ELISA assay, in CM from TOV21G cells treated with 1.6 μg/mL activin neutralizing antibody, 1.8 μg/mL ActRIIB decoy receptor, or 10 μM ALK4/5/7 inhibitor (SB431542). CM was harvested after 1 day of treatment. Mean from three experiments ±SD. * P < 0.05 vs. control (Student's t‐test). (C) SMAD3 transcript level in TOV21G cells treated with siRNA targeting SMAD3, 1 day post‐transfection, relative to NT siRNA‐treated TOV21G cells. Measured using qPCR. Mean from three independent experiments ±SD. *** P < 0.0005 vs. NT siRNA (Student's t‐test). (D) Relative level of IL‐6, measured using an ELISA assay, in CM from TOV21G cells treated with NT siRNA or siRNA targeting SMAD3. CM was harvested 1 day post‐transfection. Mean from six experiments ±SD. *** P < 0.0005 vs. NT siRNA (Student's t‐test). (E) IL‐6 transcript level in TOV21G cells treated with NT siRNA (±50 ng/mL recombinant activin A) or siRNA targeting INHBA (±50 ng/mL recombinant activin A), ACVR2A, or ACVR2B. Measured using qPCR. RNA was isolated 1 or 3 days post‐transfection. Mean from 10 independent experiments (seven for recombinant activin A) ±SD. * P < 0.05, ** P < 0.005 (Student's t‐test). (F) IL‐6 transcript level in TOV21G cells treated with 10 μM ALK4/5/7 inhibitor (SB431542) for 1 day, relative to vehicle‐treated TOV21G cells. Measured using qPCR. Mean from three independent experiments ±SD. * P < 0.05 vs. vehicle (Student's t‐test). (G) IL‐6 transcript level in TOV21G cells treated with siRNA targeting SMAD3, 1 day post‐transfection, relative to NT siRNA‐treated TOV21G cells. Measured using qPCR. Mean from three independent experiments ±SD. * P < 0.05 vs. NT siRNA (Student's t‐test). (H, I) Levels of pNF‐κB p65 (Ser536) and Erk1/2, and p‐p38 (Thr180/Tyr182) and Erk1/2, respectively, in TOV21G cells treated with NT siRNA or siRNA targeting INHBA, ACVR2A, or ACVR2B. Extracts were made 3 days post‐transfection. Representative blots are shown with numbers representing mean values from five [panel (H)] or four [panel (I)] independent experiments (and SD) after normalization against signals from Erk1/2. * P < 0.05, ** P < 0.005, *** P < 0.0005 (Student's t‐test).
Consistent with an autocrine function of activin A in the regulation of IL‐6 secretion from the cancer cells, an activin A neutralizing antibody reduced the level of IL‐6 in CMs by 22% from 6.1 ng/mL in untreated cells (Figures 3B and S3). It should be noted that this antibody is directed towards the INHBA subunit that is also found in activin AB,31 potentially also recognizing activin AB. However, based on transcriptome characterization (data not shown), the expression level of the beta B subunit in these cells is very low (below detection limit), indicating that activin AB is not of any significance in this system. Further, an ActRIIB decoy receptor and an ALK4/5/7 inhibitor reduced IL‐6 levels in CM by 23% and 33%, respectively, from 6.1 ng/mL in untreated cells (Figures 3B and S3). Moreover, SMAD3 siRNA efficiently reduced the level of SMAD3 transcripts and significantly reduced the secretion of IL‐6 from TOV21G cells (42% reduction from an average of 23.5 ng/mL in CM from non‐targeting siRNA transfected cells) (Figures 3C and 3D and S3), showing that canonical activin A signalling, involving ALK4/5/7‐induced SMAD3, is important for the induced secretion of IL‐6.
Because autocrine activin A signalling clearly stimulates IL‐6 secretion from the cancer cells, we asked if this involved increased transcription of the IL‐6 gene. qPCR analysis of NT, INHBA, ACVR2A, and ACVR2B siRNA‐treated TOV21G cells demonstrated that all siRNAs targeting activin A signalling caused reduced mRNA level of IL‐6, supporting that autocrine activin A signalling promotes IL‐6 transcription (Figure 3E). In accordance, ALK4/5/7 inhibition or siRNA‐mediated SMAD3 knockdown reduced the level of IL‐6 transcripts (Figure 3F and 3G). Importantly, treatment with recombinant activin A could significantly counteract the effect of INHBA siRNA on IL‐6 transcript level (Figure 3E), demonstrating that the effects on IL‐6 transcription are regulated by autocrine activin A signalling. The IL‐6 gene is among the typical NF‐κB controlled genes. Thus, we asked if the autocrine activin A‐induced IL‐6 transcription could be controlled by NF‐κB. Interference with activin signalling using siRNAs causes a clear reduction in the levels of pNF‐κB (p65), which is in accordance with the effects observed on IL‐6 transcript levels (Figure 3H). NF‐κB has been shown to collaborate with p38 MAPK to induce IL‐6 expression.32 Also, p38 MAPK is one of the signalling mediators downstream of activin A.33 We found that coinciding with reduced pNF‐κB and IL‐6 levels, interference with activin A signalling reduced the level of phospho‐p38 in the cancer cells (Figure 3I). These data suggest that p38 MAPK and NF‐κB play a role in activin A‐induced production of IL‐6.
Interference with activin A signalling reduces tumour‐derived IL‐6 in sera and reverses cachexia in mice
Our results clearly demonstrate that autocrine activin A signalling in the cancer cell culture promotes the release of IL‐6. Thus, we tested whether activin A antagonism is able to reduce the systemic (serum) level of IL‐6 in tumour‐bearing animals and whether a putative reduction in IL‐6 level is associated with reversal of cachexia. We treated TOV21G tumour‐bearing mice with an ActRII neutralizing antibody (CDD866). CDD866 efficiently inhibits the binding of ligands, including activin A and myostatin, to both ActRIIA and ActRIIB.27, 34
As expected, the TOV21G tumour‐bearing mice rapidly developed cachexia, evident as loss of body weight, muscle (tibialis, gastrocnemius complex, quadriceps, and heart), and white adipose tissue (Figure 4A–4I). Importantly, CDD866 treatment clearly reversed cachexia; the mice gained body weight, heart, and muscle mass, even to an extent that surpassed weight and muscle mass of non‐tumour‐bearing mice (Figure 4A–4F). Similar observations were also made for kidney and liver, while there was no significant change in the weight of spleen after the CDD866 treatment (Figure S4). Of note, CDD866 treatment also led to reduced tumour size (Figure 4G and 4H). Similar to the observed reversal of loss of muscle, the CDD866‐treated mice also seemed to have reduced loss of white adipose tissue; however, this effect was not significant (P = 0.06) (Figure 4I).
Figure 4.
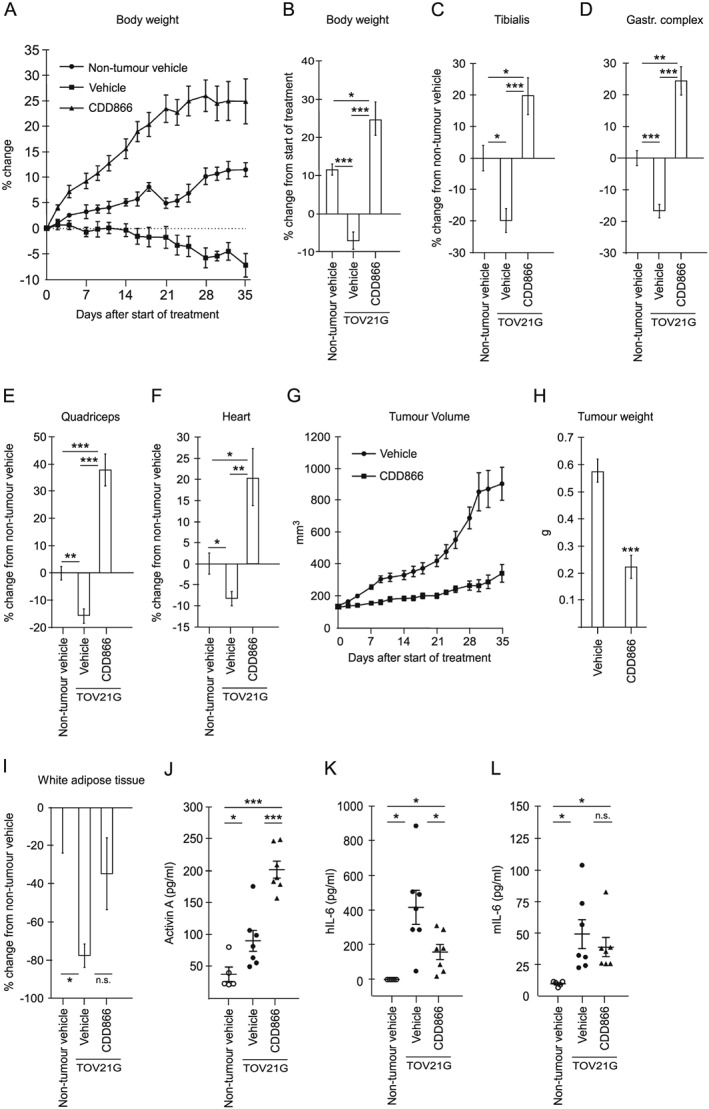
Interference with activin A signalling reduces tumour‐derived IL‐6 in sera and reverses cachexia in mice. (A, B) Mean relative body weight (at indicated time intervals) and mean relative change in body weight ±SEM, respectively, of non‐tumour control mice (n = 5) and vehicle‐treated (n = 7) and CDD866‐treated (n = 7) TOV21G tumour‐bearing mice. For the vehicle group, the data on Days 30, 32, and 35 are based on six, five, and four mice, respectively. (C–F) Mean relative change in tibialis, gastrocnemius complex, quadriceps, and heart weight, respectively, ±SEM of vehicle‐treated (n = 7) and CDD866‐treated (n = 7) TOV21G tumour‐bearing mice relative to non‐tumour control mice (n = 5). (G, H) Mean tumour volume and tumour weight ±SEM, respectively, of vehicle‐treated (n = 7) and CDD866‐treated (n = 7) TOV21G tumour‐bearing mice. (I) Mean relative change in white adipose tissue weight ±SEM of vehicle‐treated (n = 7) and CDD866‐treated (n = 7) TOV21G tumour‐bearing mice relative to non‐tumour control mice (n = 5). (J–L) Activin A, human IL‐6 (hIL‐6), and murine IL‐6 (mIL‐6) protein level, respectively, in sera from non‐tumour control mice (n = 5) and vehicle‐treated (n = 7) and CDD866‐treated (n = 7) TOV21G tumour‐bearing mice. Mean ± SEM are indicated. * P < 0.05, ** P < 0.005, *** P < 0.0005 (Student's t‐test), n.s. = non‐significant.
As anticipated, TOV21G‐bearing mice had increased serum level of activin A, and an additional rise in serum activin A was detected following CDD866 treatment (Figure 4J). By the use of species‐specific ELISAs, we also found that tumour‐bearing mice were subjected to a massive rise in the level of serum IL‐6 derived from the cancer cells (human IL‐6) (Figure 4K) and a significant, but clearly less substantial, increase in host‐derived murine IL‐6 (Figure 4L). Because activin A was important for IL‐6 secretion from the cancer cells in vitro, we tested whether the ActRII neutralizing antibody (CDD866) could reduce the level of IL‐6 in sera from TOV21G‐bearing mice. In accordance with our in vitro findings, inhibition of ActRII caused a significant reduction of cancer‐derived human IL‐6 in serum from the tumour‐bearing mice (Figure 4K). Interestingly, despite the increase in mouse IL‐6 levels in serum, treatment with the ActRII neutralizing antibody did not significantly reduce this pool of IL‐6 (Figure 4L). Together, this shows that disrupting activin A signalling in the tumour decreases systemic abundance of tumour‐derived IL‐6 and coincides with reversal of cachexia in the mice.
Association between INHBA and IL‐6 gene expression in human ovarian tumours
Finding that activin signalling is needed for release of IL‐6 in vitro and in the TOV21G murine cachexia model, we investigated the possible association between activin A and IL‐6 in human ovarian cancer patients. By evaluating the levels of transcripts encoding IL‐6 and INHBA in tumour biopsies from ovarian cancer patients (n = 307) using the cBioPortal website, we found a significant association between the expression of these genes (Figure 5). This indicates that activin A may also modulate the expression of IL‐6 in human tumours.
Figure 5.
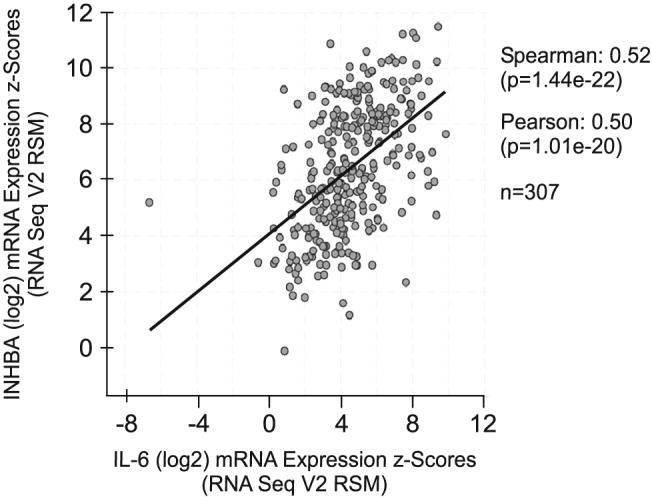
Association between INHBA and IL‐6 gene expression in human ovarian tumours. IL‐6 and INHBA mRNA expression in human ovarian serous cystadenocarcinoma (n = 307). From the cBioPortal database (http://www.cbioportal.org/).
Discussion
Activin A and IL‐6 are both suggested to contribute to cachexia, and accordingly, their individual role in development of this condition has been a central topic of research. In mouse models of cancer‐induced cachexia, it has been demonstrated that both activin A and IL‐6 are contributing, and neutralizing each axis reverses the muscle wasting.5, 11, 12, 13, 14, 27, 35, 36, 37, 38, 39 Here, we show for the first time that intratumoural activin A signalling promotes IL‐6 secretion from the cancer cells, thus demonstrating a direct link between the two factors. We show that interference with activin A signalling reduces IL‐6 transcription, secretion, and systemic distribution. Reduced IL‐6 levels following obstruction of activin A activities associate with reduced ability of the cancer cells to accelerate autophagy in non‐cancerous cells in vitro and the ability to cause cachexia in mice. This study presents a so far unexplored mechanism to drive cachexia that may be targeted in treatment.
The importance of intratumoural signalling activities in cachexia development has been highlighted also by others. Johnston et al.40 demonstrated that TWEAK/Fn14 signalling in the tumour, rather than the host, is important to cause cachexia in mice and that interference with these factors increases survival. Similar to our findings, the authors suggest that Fn14 stimulates the secretion of cachexia‐inducing factors from the tumour. However, the secreted cachexia‐inducing factor(s) were not identified. In hepatic stellate cells, the Fn14 ligand TWEAK has been shown to upregulate both transcription and secretion of inflammatory cytokines, including IL‐6.41 This suggests that TWEAK and activin A may both work in a paracrine or autocrine manner in the tumours to stimulate IL‐6 secretion from cancer cells and facilitate the development of cachexia.
TWEAK‐induced IL‐6 involves activation of NF‐κB.42 Similarly, we find that interfering with the autocrine activin signalling causes a clear reduction in the active form of NF‐κB. Furthermore, SMAD3 seems to be involved in this pathway, and preliminary data from our lab indicate that SMAD3 is upstream of activation of NF‐κB. Such a sequential order of events is in line with data from mouse keratinocytes where TGFβ‐induced Alk5 and SMAD3 are required for NF‐κB‐dependent gene expression.43 Interestingly, Qin et al. showed binding of SMAD4 to the IL‐6 promoter after TGFβ stimulation of ovarian surface epithelial cells.44 This suggests that SMAD signalling may also directly control IL‐6 gene expression.
The involvement of activin A in IL‐6 secretion has also been described in non‐cancerous cells, like mouse neutrophils, where the cells in response to LPS secrete IL‐6 in an activin A/SMAD3‐dependent manner.45 IL‐6 may act systemically and has been suggested to be one of the major contributors to cachexia.12, 13, 14, 17, 18, 46, 47, 48 We have shown that IL‐6 accelerates autophagy10 and this may in part explain the potent cachexia‐inducing effect of this cytokine. Consistently, we could demonstrate that interference with autocrine activin A signalling in the cancer cells is sufficient to reduce the ability of the cells to accelerate autophagy in non‐cancerous cells.
Our in vitro data show that activin A acts in an autocrine or paracrine manner to promote secretion of IL‐6 from the cancer cells. Due to the heterogeneous nature of a tumour in vivo, with a variety of infiltrating cell types, it is likely that activin A signalling also occurs between cancer cells and stromal cells. Such crosstalk could potentially contribute to a higher IL‐6 secretion than what can be caused by the transformed cancer cells themselves. Activin A may also act systemically to induce secretion of IL‐6 from other cell types located distantly from the tumour. For instance, muscle cells may secrete IL‐6,49 and an effect of activin A on IL‐6 levels may occur as a result of myocyte signalling. In line with this, preliminary data from our lab show that activin A can induce release of IL‐6 from cardiomyocytes in vitro (data not shown). Surprisingly, we found that although host IL‐6 levels increased in sera of cachectic tumour‐bearing mice, the levels were not significantly affected by interference with activin A signalling. It should, however, be noted that we used immune‐compromised nude mice with subcutaneous flank implantation of TOV21G, similarly to Zhou et al. who reported the ability of TOV21G to cause cachexia in an activin A‐driven mechanism.5 We acknowledge, however, that activin A may affect both cancer cell‐derived and host‐derived IL‐6 differently in the presence of a complete immune system and when the cancer cells are growing in the pelvic/abdominal cavity.
In this study, we used TOV21G cells to induce cancer cachexia. In a recent paper, Pin et al. used another ovarian clear cell carcinoma cell line, ES‐2, and argued that this is more similar to human ovarian cancer.15 The ES‐2 cell line also gives cachexia, and bioactive IL‐6 seems to be causal. A mechanism for IL‐6 regulation in the ES‐2 model has not yet been proposed, but activation of the RAS/RAF/MEK/ERK signalling pathway is a common feature of the two cell models. While TOV21G cells harbour activated KRAS, ES‐2 have activated BRAF.15 Consistently, we find that IL‐6 secretion from the TOV21G cells is driven by activin A and oncogenic RAS. Whether activin A and/or BRAF is involved in the IL‐6 secretion from ES‐2 is not known.
In our mouse model, we find that treating tumour‐bearing mice with an ACVR2 neutralizing antibody not only reverts muscle mass to normal level but also causes a significant increase in muscle mass. Possibly, this relates to the ability of this antibody to block the binding of both activin A and myostatin to the ACVR2.27, 34, 50 Myostatin deficiency has previously been shown to be highly anabolic.51, 52 Whether myostatin also has a role in regulating IL‐6 secretion, and thereby is influencing cachexia development, cannot be excluded but is beyond the scope of this study.
A complicating factor when interpreting the reduced hIL‐6 levels in circulation after interfering with the ACVR2 is that the tumour size is significantly influenced. The reduced levels of tumour‐derived IL‐6 in circulation could thus, to an unknown extent, be a result of reduced number of IL‐6‐producing TOV21G cells. Regardless, in vitro, we show that there is a direct effect of activin A signalling on IL‐6 gene transcription and IL‐6 protein secretion from the cancer cells. It is likely that this mechanism also is of relevance in the in vivo model.
Based on results from murine models, recent clinical trials have aimed to unravel the efficiency of blocking activin signalling in patients with cachexia or muscle dystrophy. Clinical trials involving ACVR2B decoy receptors, however, were terminated due to severe adverse effects, including bleeding.53 More specific antibodies directed towards the ACVR2B are being tested in clinical trials53, 54, 55; however, because ACVR2 are central receptors for multiple TGFβ family members, and our study points to the central role of activin A, specific inhibition of activin A itself may prove less invasive and equally efficient.
The present study provides novel insights into the interplay between activin A and IL‐6 secretion from cancer cells that may be important for the development of future therapeutic strategies to reverse cancer cachexia.
Conflict of interest
None declared.
Funding
This study was financially supported by grants from The Norwegian Cancer Society, The Research Council of Norway through its Centres of Excellence funding scheme (223255/F50), and The Research Council of Norway (139610/300).
Supporting information
Figure S1. Standard curves from activin A and IL‐6 ELISA. Standard curve for activin A (A) and IL‐6 (B) ELISA used when analyzing TOV21G conditioned medium, and for activin A ELISA used for analyzing serum samples (C).
Figure S2. Autophagy flux in autophagy reporter cells treated with autophagy effectors. Autophagy flux in autophagy reporter cells treated for 17 hours with 3‐methyladenine (3‐MA, 3 mM), bafilomycin A1 (BafA1, 100 nM), Hanks' Balanced Salt Solution (HBSS) or Torin 1 (150 nM).
Figure S3. IL‐6 protein level in conditioned medium (CM) from TOV21G cells. Level of IL‐6 in CM from untreated TOV21G cells and TOV21G cells treated with non‐targeting (NT) siRNA. CM from 1 or 3 days post‐seeding (untreated) or post‐transfection (NT siRNA treated).
Figure S4. In TOV21G tumor‐bearing mice, injection of the ActRII neutralizing antibody does not affect the weight of the spleen but display an anabolic effect on kidney and liver. Mean relative weight change of spleen, kidney and liver, respectively ±SEM of vehicle‐treated (n = 7) and CDD866‐treated (n = 7) TOV21G tumor‐bearing mice relative to non‐tumor, vehicle‐treated control mice (n = 5). *p < 0.05, ***p < 0.0005 (Student t‐test), n.s. = non‐significant.
Acknowledgements
We thank Prof. Terje Johansen and the Molecular Cancer Research Group (The Arctic University of Norway, Norway) for HEK293 GFP‐SQSTM1 cells and for helpful discussions and Dr. Toril Holien (Norwegian University of Science and Technology, Norway) for valuable advice and experimental support. The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017.56
Pettersen K., Andersen S., van der Veen A., Nonstad U., Hatakeyama S., Lambert C., Lach‐Trifilieff E., Moestue S., Kim J., Grønberg B. H., Schilb A., Jacobi C., and Bjørkøy G. (2020) Autocrine activin A signalling in ovarian cancer cells regulates secretion of interleukin 6, autophagy, and cachexia, Journal of Cachexia, Sarcopenia and Muscle, 11: 195–207. 10.1002/jcsm.12489.
References
- 1. von Haehling S, Anker SD. Prevalence, incidence and clinical impact of cachexia: facts and numbers—update 2014. J Cachexia Sarcopenia Muscle 2014;5:261–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 3. Chen JL, Walton KL, Winbanks CE, Murphy KT, Thomson RE, Makanji Y, et al. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J: official publication of the Federation of American Societies for Experimental Biology 2014;28:1711–1723. [DOI] [PubMed] [Google Scholar]
- 4. Matzuk MM, Finegold MJ, Mather JP, Krummen L, Lu H, Bradley A. Development of cancer cachexia‐like syndrome and adrenal tumors in inhibin‐deficient mice. Proc Natl Acad Sci U S A 1994;91:8817–8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010;142:531–543. [DOI] [PubMed] [Google Scholar]
- 6. Togashi Y, Kogita A, Sakamoto H, Hayashi H, Terashima M, de Velasco MA, et al. Activin signal promotes cancer progression and is involved in cachexia in a subset of pancreatic cancer. Cancer Lett 2015;356( Pt B:819–827. [DOI] [PubMed] [Google Scholar]
- 7. Trendelenburg AU, Meyer A, Jacobi C, Feige JN, Glass DJ. TAK‐1/p38/nNFκB signaling inhibits myoblast differentiation by increasing levels of Activin A. Skelet muscle 2012;2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol 2009;296:C1258–C1270. [DOI] [PubMed] [Google Scholar]
- 9. Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, et al. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol 2009;296:C1248–C1257. [DOI] [PubMed] [Google Scholar]
- 10. Pettersen K, Andersen S, Degen S, Tadini V, Grosjean J, Hatakeyama S, et al. Cancer cachexia associates with a systemic autophagy‐inducing activity mimicked by cancer cell‐derived IL‐6 trans‐signaling. Sci Rep 2017;7:2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL‐6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 2012;303:E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Strassmann G, Fong M, Freter CE, Windsor S, D'Alessandro F, Nordan RP. Suramin interferes with interleukin‐6 receptor binding in vitro and inhibits colon‐26‐mediated experimental cancer cachexia in vivo. J Clin Invest 1993;92:2152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Strassmann G, Fong M, Kenney JS, Jacob CO. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest 1992;89:1681–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tamura S, Ouchi KF, Mori K, Endo M, Matsumoto T, Eda H, et al. Involvement of human interleukin 6 in experimental cachexia induced by a human uterine cervical carcinoma xenograft. Clin Cancer Res 1995;1:1353–1358. [PubMed] [Google Scholar]
- 15. Pin F, Barreto R, Kitase Y, Mitra S, Erne CE, Novinger LJ, et al. Growth of ovarian cancer xenografts causes loss of muscle and bone mass: a new model for the study of cancer cachexia. J Cachexia Sarcopenia Muscle 2018;9:685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martignoni ME, Kunze P, Hildebrandt W, Kunzli B, Berberat P, Giese T, et al. Role of mononuclear cells and inflammatory cytokines in pancreatic cancer‐related cachexia. Clin Cancer Res 2005;11:5802–5808. [DOI] [PubMed] [Google Scholar]
- 17. Hirata H, Tetsumoto S, Kijima T, Kida H, Kumagai T, Takahashi R, et al. Favorable responses to tocilizumab in two patients with cancer‐related cachexia. J Pain Symptom Manage 2013;46:e9–e13. [DOI] [PubMed] [Google Scholar]
- 18. Ando K, Takahashi F, Motojima S, Nakashima K, Kaneko N, Hoshi K, et al. Possible role for tocilizumab, an anti‐interleukin‐6 receptor antibody, in treating cancer cachexia. J Clin Oncol Off J Am Soc Clin Oncol 2013;31:e69–e72. [DOI] [PubMed] [Google Scholar]
- 19. Aversa Z, Pin F, Lucia S, Penna F, Verzaro R, Fazi M, et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci Rep 2016;6:30340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Penna F, Baccino FM, Costelli P. Coming back: autophagy in cachexia. Curr Opin Clin Nutr Metab Care 2014;17:241–246. [DOI] [PubMed] [Google Scholar]
- 21. Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013;182:1367–1378. [DOI] [PubMed] [Google Scholar]
- 22. Tardif N, Klaude M, Lundell L, Thorell A, Rooyackers O. Autophagic‐lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients. Am J Clin Nutr 2013;98:1485–1492. [DOI] [PubMed] [Google Scholar]
- 23. Larsen KB, Lamark T, Overvatn A, Harneshaug I, Johansen T, Bjorkoy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 2010;6:784–793. [DOI] [PubMed] [Google Scholar]
- 24. Sellers RS, Morton D, Michael B, Roome N, Johnson JK, Yano BL, et al. Society of Toxicologic Pathology position paper: organ weight recommendations for toxicology studies. Toxicol Pathol 2007;35:751–755. [DOI] [PubMed] [Google Scholar]
- 25. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hatakeyama S, Summermatter S, Jourdain M, Melly S, Minetti GC, Lach‐Trifilieff E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti‐cancer treatments. Skeletal muscle 2016;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wakefield LM, Hill CS. Beyond TGFβ: roles of other TGFβ superfamily members in cancer. Nat Rev Cancer 2013;13:328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller A, McLeod L, Alhayyani S, Szczepny A, Watkins DN, Chen W, et al. Blockade of the IL‐6 trans‐signalling/STAT3 axis suppresses cachexia in Kras‐induced lung adenocarcinoma. Oncogene 2016;36:3059. [DOI] [PubMed] [Google Scholar]
- 30. Caetano MS, Zhang H, Cumpian AM, Gong L, Unver N, Ostrin EJ, et al. IL6 blockade reprograms the lung tumor microenvironment to limit the development and progression of K‐ras‐mutant lung cancer. Cancer Res 2016;76:3189–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morikawa M, Derynck R, Miyazono K. TGF‐β and the TGF‐β Family: Context‐Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb Perspect Biol 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Craig R, Larkin A, Mingo AM, Thuerauf DJ, Andrews C, McDonough PM, et al. p38 MAPK and NF‐κB Collaborate to Induce Interleukin‐6 Gene Expression and Release. J Biol Chem 2000;275:23814–23824. [DOI] [PubMed] [Google Scholar]
- 33. Ding H, Zhang G, Sin KW, Liu Z, Lin RK, Li M, et al. Activin A induces skeletal muscle catabolism via p38β mitogen‐activated protein kinase. J Cachexia Sarcopenia Muscle 2017;8:202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morvan F, Rondeau JM, Zou C, Minetti G, Scheufler C, Scharenberg M, et al. Blockade of activin type II receptors with a dual anti‐ActRIIA/IIB antibody is critical to promote maximal skeletal muscle hypertrophy. Proc Natl Acad Sci U S A 2017;114:12448–12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marino FE, Risbridger G, Gold E. The therapeutic potential of blocking the activin signalling pathway. Cytokine Growth Factor Rev 2013;24:477–484. [DOI] [PubMed] [Google Scholar]
- 36. Han HQ, Zhou X, Mitch WE, Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol 2013;45:2333–2347. [DOI] [PubMed] [Google Scholar]
- 37. Busquets S, Toledo M, Orpi M, Massa D, Porta M, Capdevila E, et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J Cachexia Sarcopenia Muscle 2012;3:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Q, Kumar R, Underwood K, O'Connor AE, Loveland KL, Seehra JS, et al. Prevention of cachexia‐like syndrome development and reduction of tumor progression in inhibin‐deficient mice following administration of a chimeric activin receptor type II‐murine Fc protein. Mol Hum Reprod 2007;13:675–683. [DOI] [PubMed] [Google Scholar]
- 39. White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, et al. The Regulation of Skeletal Muscle Protein Turnover during the Progression of Cancer Cachexia in the ApcMin/+ Mouse. PLoS ONE 2011;6:e24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnston AJ, Murphy KT, Jenkinson L, Laine D, Emmrich K, Faou P, et al. Targeting of Fn14 prevents cancer‐induced cachexia and prolongs survival. Cell. 2015;162(6):1365–78. [DOI] [PubMed] [Google Scholar]
- 41. Wang A, Zhang F, Xu H, Xu M, Cao Y, Wang C, et al. TWEAK/Fn14 promotes pro‐inflammatory cytokine secretion in hepatic stellate cells via NF‐κB/STAT3 pathways. Mol Immunol 2017;87:67–75. [DOI] [PubMed] [Google Scholar]
- 42. Hu G, Zeng W, Xia Y. TWEAK/Fn14 signaling in tumors. Tumour Biol 2017;39: 1010428317714624, 101042831771462. [DOI] [PubMed] [Google Scholar]
- 43. Hogan KA, Ravindran A, Podolsky MA, Glick AB. The TGFβ1 pathway is required for NFκB dependent gene expression in mouse keratinocytes. Cytokine 2013;64:652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qin H, Chan MW, Liyanarachchi S, Balch C, Potter D, Souriraj IJ, et al. An integrative ChIP‐chip and gene expression profiling to model SMAD regulatory modules. BMC Syst Biol 2009;3:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Qi Y, Ge J, Ma C, Wu N, Cui X, Liu Z. Activin A regulates activation of mouse neutrophils by Smad3 signalling. Open Biol 2017;7:160342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 47. Scott HR, McMillan DC, Crilly A, McArdle CS, Milroy R. The relationship between weight loss and interleukin 6 in non‐small‐cell lung cancer. Br J Cancer 1996;73:1560–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Narsale AA, Carson JA. Role of interleukin‐6 in cachexia: therapeutic implications. Curr Opin Support Palliat Care 2014;8:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pal M, Febbraio MA, Whitham M. From cytokine to myokine: the emerging role of interleukin‐6 in metabolic regulation. Immunol Cell Biol 2014;92:331–339. [DOI] [PubMed] [Google Scholar]
- 50. Lach‐Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, et al. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol 2014;34:606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A 1997;94:12457–12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 2004;350:2682–2688. [DOI] [PubMed] [Google Scholar]
- 53. Aversa Z, Costelli P, Muscaritoli M. Cancer‐induced muscle wasting: latest findings in prevention and treatment. Ther Adv Medical Oncol 2017;9:369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rooks DS, Laurent D, Praestgaard J, Rasmussen S, Bartlett M, Tanko LB. Effect of bimagrumab on thigh muscle volume and composition in men with casting‐induced atrophy. J Cachexia Sarcopenia Muscle 2017;8:727–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rooks D, Praestgaard J, Hariry S, Laurent D, Petricoul O, Perry RG, et al. Treatment of sarcopenia with bimagrumab: results from a phase II, randomized, controlled, proof‐of‐concept study. J Am Geriatr Soc 2017;65:1988–1995. [DOI] [PubMed] [Google Scholar]
- 56. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Standard curves from activin A and IL‐6 ELISA. Standard curve for activin A (A) and IL‐6 (B) ELISA used when analyzing TOV21G conditioned medium, and for activin A ELISA used for analyzing serum samples (C).
Figure S2. Autophagy flux in autophagy reporter cells treated with autophagy effectors. Autophagy flux in autophagy reporter cells treated for 17 hours with 3‐methyladenine (3‐MA, 3 mM), bafilomycin A1 (BafA1, 100 nM), Hanks' Balanced Salt Solution (HBSS) or Torin 1 (150 nM).
Figure S3. IL‐6 protein level in conditioned medium (CM) from TOV21G cells. Level of IL‐6 in CM from untreated TOV21G cells and TOV21G cells treated with non‐targeting (NT) siRNA. CM from 1 or 3 days post‐seeding (untreated) or post‐transfection (NT siRNA treated).
Figure S4. In TOV21G tumor‐bearing mice, injection of the ActRII neutralizing antibody does not affect the weight of the spleen but display an anabolic effect on kidney and liver. Mean relative weight change of spleen, kidney and liver, respectively ±SEM of vehicle‐treated (n = 7) and CDD866‐treated (n = 7) TOV21G tumor‐bearing mice relative to non‐tumor, vehicle‐treated control mice (n = 5). *p < 0.05, ***p < 0.0005 (Student t‐test), n.s. = non‐significant.