

Abstract
Background
Previous studies in heart failure with reduced ejection fraction (HFrEF) suggest that skeletal muscle mitochondrial impairments are associated with exercise intolerance in men. However, the nature of this relationship in female patients remains to be elucidated. This study aimed to determine the relationship between skeletal muscle mitochondrial impairments and exercise intolerance in male and female patients with HFrEF.
Methods
Mitochondrial respiration, enzyme activity, and gene expression were examined in pectoralis major biopsies from age‐matched male (n = 45) and female (n = 11) patients with HFrEF and healthy‐matched male (n = 24) and female (n = 11) controls. Mitochondrial variables were compared between sex and related to peak exercise capacity.
Results
Compared with sex‐matched controls, complex I mitochondrial oxygen flux was 17% (P = 0.030) and 29% (P = 0.013) lower in male and female patients with HFrEF, respectively, which correlated to exercise capacity (r = 0.71; P > 0.0001). Female HFrEF patients had a 32% (P = 0.023) lower mitochondrial content compared with controls. However, after adjusting for mitochondrial content, male patients demonstrated lower complex I function by 15% (P = 0.030). Expression of key mitochondrial genes regulating organelle dynamics and maintenance (i.e. optic atrophy 1, peroxisome proliferator‐activated receptor γ coactivator‐1α, NADH:ubiquinone oxidoreductase core subunit S1/S3, and superoxide dismutase 2) were selectively lower in female HFrEF patients.
Conclusions
These data provide novel evidence that HFrEF induces divergent sex‐specific mitochondrial phenotypes in skeletal muscle that predispose towards exercise intolerance, impacting mitochondrial ‘quantity' in female patients and mitochondrial ‘quality' in male patients. Therapeutic strategies to improve exercise tolerance in HFrEF should consider targeting sex‐specific mitochondrial abnormalities in skeletal muscle.
Keywords: HFrEF, Mitochondrial dysfunction, OPA1, Exercise intolerance, Sex
1. Introduction
Patients with heart failure with reduced ejection fraction (HFrEF) have a complex pathophysiology, which, although underpinned by left ventricular systolic dysfunction, is a multifaceted whole‐body syndrome characterized by exercise intolerance. The prevalence of HFrEF is similar between men and women, yet despite presenting with lower exercise capacity [i.e. peak pulmonary oxygen uptake (V̇O2peak)], women show better clinical outcomes both in the absence and in the presence of an exercise intervention when compared with male counterparts1, 2. The underlying sex‐specific mechanism(s) for this trend remain poorly resolved and seem independent of cardiac function.3, 4 Exercise intolerance in patients with HFrEF is poorly correlated with cardiac dysfunction5, 6, 7 and is accepted that peripheral impairments, including skeletal muscle abnormalities, play a large role in exercise intolerance in HFrEF.8 Mitochondria represent the key site for cellular energy production, and skeletal muscle mitochondrial impairments in HFrEF patients could predispose towards exercise intolerance.
Whether HFrEF induces sex‐specific mitochondrial decrements that contribute to the observed clinical differences between men and women with HFrEF remains poorly explored.9, 10 However, recent advances in the mitochondrial field have given rise to putative mechanisms that could form the basis of any sex‐specific mitochondrial impairments induced by HFrEF. Skeletal muscle oxidative capacity is determined by both mitochondrial ‘quantity' (mitochondrial content) and ‘quality' (intrinsic function normalized to mitochondrial mass). Mitochondrial quality and morphology are dependent on the effective functioning/expression of mitochondrial respiratory chain complexes, which are largely orchestrated by a set of regulatory proteins that act to maintain mitochondrial quality control in a process termed mitochondrial dynamics. Mitochondrial dynamics modulate mitochondrial shape, size, and integrity, which are regulated by fusion proteins [e.g. optic atrophy 1 (OPA1)] and fission proteins [e.g. mitochondrial fission 1 (FIS1)] localized to the inner and outer mitochondrial membranes, respectively.11 One major orchestrator of these mitochondrial processes is the well‐known transcriptional coactivator peroxisome proliferator‐activated receptor γ coactivator‐1α (PGC‐1α), which is considered as the master regulator of mitochondrial biogenesis.12
In the current study, therefore, we assessed skeletal muscle biopsies from a large cohort of both male and female patients with HFrEF compared with sex‐matched controls. We evaluated whether mitochondrial quality, quantity, respiratory chain complexes, and dynamics were impacted by HFrEF in a sex‐specific manner and their relationship to whole‐body exercise intolerance.
2. Methods
2.1. Participants
All participant characteristics are presented in Table 1. We approached consecutive patients with HFrEF of >3 months duration, symptoms corresponding to New York Heart Association (NYHA) functional class ≥ I and a documented left ventricular ejection fraction <50% (following current European Society of Cardiology guidelines13) who were planned for primary electronic cardiac implantable device procedures. All were indicated for device therapy with either an implantable cardioverter defibrillator or cardiac resynchronization therapy according to current indications. Pacemaker requirement was sick sinus syndrome, heart block, or cardiac arrest (implantable cardioverter defibrillator patients). All HFrEF patients undertook a peak symptom‐limited CPX test to volitional exhaustion on a cycle ergometer for determination of pulmonary gas exchange (V̇O2 and V̇CO2) and ventilation (V̇E).14 Control subjects were consecutive patients listed for device implantation of a permanent pacemaker without symptoms of heart failure. Potential participants were excluded if they were unable to provide informed consent due to cognitive dysfunction or had previously been diagnosed with other potentially confounding comorbidities, such as other cardiovascular conditions, chronic obstructive pulmonary disease, or cancer. All patients provided written informed consent, and all procedures were conducted in accordance with the Declaration of Helsinki after receiving local institute ethical approval (11/YH/0291).
Table 1.
Physical, clinical, and treatment characteristics of patients
| CON | HFrEF | |||||
|---|---|---|---|---|---|---|
| Male (n = 24) | Female (n = 11) | P value | Male (n = 45) | Female (n = 13) | P value | |
| Age (years) | 70.1 ± 2.6 | 74.1 ± 3.1 | 0.33 | 71.8 ± 1.8 | 69.9 ± 2.1 | 0.45 |
| BMI (kg·m−2) | 27.4 ± 0.5 | 29.6 ± 2.7 | 0.46 | 28.1 ± 0.9 | 27.2 ± 1.8 | 0.67 |
| V̇O2peak (mL·kg−1·min−1) | — | — | — | 15.1 ± 0.8 | 11.6 ± 0.7 | 0.07 |
| V̇E/V̇CO2 slope | — | — | — | 35.6 ± 1.7 | 37.9 ± 3.0 | 0.56 |
| NYHA functional class [% (n)] | — | — | — | — | — | |
| I | — | — | 13.3 (6) | 7.7 (1) | 0.18 | |
| II | — | — | 60.0 (27) | 38.5 (5) | ||
| III | — | — | 26.7 (12) | 53.8 (7) | ||
| Ischaemic/DCM aetiology [% (n)] | — | — | — | 58/42 (26/19) | 39/61 (5/8) | 0.22 |
| AF [% (n)] | — | — | — | 37.8 (17) | 15.4 (2) | 0.13 |
| CABG [% (n)] | — | — | — | 24.4 (11) | 7.7 (1) | 0.19 |
| Hypertension [% (n)] | — | — | — | 33.3 (15) | 30.8 (4) | 0.86 |
| LVEF (%) | — | — | — | 24.6 ± 1.5 | 25.4 ± 3.4 | 0.83 |
| LVIDd (mm) | — | — | — | 59.3 ± 1.3 | 56.5 ± 1.5 | 0.18 |
| HbA1c (%) | 5.90 ± 0.12 | 5.78 ± 0.13 | 0.53 | 6.01 ± 0.11 | 5.90 ± 0.09 | 0.63 |
| Haemoglobin (g·L−1) | 138.0 ± 3.4 | 124.9 ± 6.2 | 0.08 | 140.5 ± 2.0 | 131.6 ± 3.5 | 0.04 |
| Creatinine (μmol·mL−1) | 83.2 ± 3.1 | 75.9 ± 4.5 | 0.19 | 99.6 ± 6.0 | 82.8 ± 7.8 | 0.10 |
| Pharmacological treatments | — | — | — | — | — | — |
| Beta‐blocker use [% (n)] | — | — | — | 97.8 (44) | 84.6 (11) | 0.06 |
| ACEi or ARB use [% (n)] | — | — | — | 86.7 (39) | 69.2 (9) | 0.14 |
| MRA use [% (n)] | — | — | — | 53.3 (24) | 46.2 (6) | 0.64 |
| ACEi or ARB with beta‐blocker and MRA [% (n)] | — | — | — | 46.7 (21) | 38.5 (5) | 0.52 |
| Loop diuretic use [% (n)] | — | — | — | 87.1 (27) | 30.8 (4) | 0.06 |
| Furosemide equivalent dose (mg) | — | — | — | 45.9 ± 4.2 | 55.0 ± 15.0 | 0.53 |
| Statin use [% (n)] | — | — | — | 60.0 (27) | 69.2 (9) | 0.54 |
| Statin dose (mg) | — | — | — | 48.9 ± 4.8 | 24.4 ± 4.1 | <0.01 |
| Aspirin use [% (n)] | — | — | — | 37.8 (17) | 53.8 (7) | 0.30 |
| Digoxin use [% (n)] | — | — | — | 11.1 (5) | 7.7 (1) | 0.72 |
| Device therapy | — | — | — | — | — | — |
| Pacemaker | 91.7 (22) | 90.9 (10) | 0.94 | 2.2 (1) | 0 (0) | 0.59 |
| ICD [% (n)] | 8.3 (2) | 9.1 (1) | 0.94 | 24.4 (11) | 15.4 (2) | 0.49 |
| CRT [% (n)] | — | — | — | 37.8 (17) | 61.5 (8) | 0.13 |
| CRT‐ICD [% (n)] | — | — | — | 35.6 (16) | 23.1 (3) | 0.40 |
Data are presented as mean ± SEM or % (n). Continuous variables were compared using unpaired Student's t‐tests. Categorical variables were compared using chi‐squared tests (or Fisher's exact test were appropriate). ACEi, angiotensin‐converting enzyme inhibitor; AF, atrial fibrillation; ARB, angiotensin receptor blocker; BMI, body mass index; CABG, coronary artery bypass graft; CRT, cardiac resynchronization therapy; DCM, dilated cardiomyopathy; HbA1c, glycated haemoglobin; ICD, implantable cardioverter‐defibrillator; LVEF, left ventricular ejection fraction; LVIDd, left ventricular internal diameter at diastole; MRA, mineralocorticoid antagonist; NYHA, New York Heart Association; V̇O2peak, peak pulmonary oxygen uptake; V̇E/V̇CO2, ratio of ventilation to carbon dioxide production.
2.2. Skeletal muscle biopsy
Skeletal muscle biopsies of pectoralis major (~100 mg) with a mixed fibre type composition (~35% type I, ~40% type IIA, and ~25% type IIX) were obtained during otherwise routinely performed device implantation procedures. The biopsy was taken within a maximum of 3 months of re‐examination of disease status at the time of decision to implant a device. There were no complications or adverse events with this procedure. One piece of muscle sample was immediately place in 1 mL of ice‐cold specialized preservation solution,15 while another portion was snap frozen in liquid nitrogen and stored at −80°C.
2.3. Mitochondrial function and content
Mitochondrial respiration was assessed in situ from saponin‐permeabilized skeletal muscle fibres using high‐resolution respirometry (Oxygraph‐2K; Oroboros Instruments, Innsbruck, Austria), as described by Wüst et al.16 briefly in the following order: (i) complex I leak respiration was determined by addition of glutamate (10 mM), malate (0.5 mM), and pyruvate (5 mM) (i.e. a measure of proton leak under non‐phosphorylating conditions); (ii) adenosine diphosphate (2.5 mM) added to provide a measure of complex I oxidative phosphorylation (OXPHOS); (iii) outer mitochondrial membrane integrity determined by addition of 10 μM cytochrome c; (iv) succinate (10 mM) as a complex II substrate provided complex I + II OXPHOS; (v) 5 μM FCCP for maximal uncoupled complex I + II respiration; (vi) complex I inhibitor rotenone (0.25 μM) provided uncoupled complex II respiration; and (vii) 2.5 μM antimycin A as a complex III inhibitor for residual oxygen consumption (ROX) to calculate non‐mitochondrial (background) respiration, which was then subtracted from the other data. Mitochondrial content was first determined within the respirometer by the addition of a complex IV activity assay,17 by the addition of 0.5 mM N,N,N′,N′‐tetramethyl‐p‐phenylenediamine dihydrochloride (TMPD) as an artificial electron donor to complex IV in combination with 2 mM ascorbate to maintain TMPD in a reduced state,18 with absolute mitochondrial respiration states thereafter normalized to complex IV activity to provide an index of mitochondrial intrinsic function (i.e. quality). Citrate synthase activity was also measured spectrophotometrically at 30°C in a plate reader at 412 nm in muscle homogenate as a secondary marker of mitochondrial content and specific enzyme activity was calculated.19 Complex IV activity and specific enzyme activity of citrate synthase were expressed as a fold change relative to controls.
2.4. Gene and protein expression analyses
RNA was extracted and purified from ~30 to 40 mg snap‐frozen muscle tissue using the RNeasy® Fibrous Tissue Mini Kit (Qiagen, Hilden, Germany). RNA concentrations (ng·μL−1) were quantified and reverse transcribed to cDNA and mRNA expression was determined using real‐time quantitative PCR with SYBR® Green ROX™ quantitative PCR Mastermix (QIAGEN, Hilden, Germany) and a quantitative PCR system (Applied Biosystems Prism 7900HT, Foster City, CA). Primers were purchased from Qiagen including PGC‐1α, superoxide dismutase 2 (SOD2), FIS1, OPA1, NADH:ubiquinone oxidoreductase core subunit S1 (NDUFS1), and NADH:ubiquinone oxidoreductase core subunit S3 (NDUFS3). Expression levels were normalized to an endogenous control, beta‐actin, using the Δ‐Δ‐CT method20 and then expressed relative to controls. All real‐time quantitative PCR primers used in this study are commercially available (Qiagen Ltd) and are as follows: PPARGC1A, detected transcript NM_013261 (6318 bp) (PPH00461F); OPA1, detected transcript NM_015560 (6345 bp) (PPH12084A‐200); FIS1, detected transcript NM_016068 (785 bp) PPH19947A‐200; SOD2, detected transcript NM_000636 (1593 bp) PPH01716B‐200; NDUFS1, detected transcript NM_001199981 (3365 bp) (PPH19871A‐200); and NDUFS3, detected transcript NM_004551 (971 bp) PPH05975A‐200. For protein expression, homogenized muscle samples underwent sodium dodecyl sulfate (SDS)‐polyacrylamide gel electrophoresis, membrane transfer, and were incubated overnight at 4°C with a primary antibody against PGC‐1α (1/200; GeneTex) and subsequently with a horseradish peroxidase‐conjugated secondary antibody before visualization by enzymatic chemiluminescence and quantification using densitometry, as previously described.19 Blots were normalized to the loading control GAPDH (1/5000, Abcam).
2.5. Statistical analyses
Homogeneity of variance and normal distribution was first confirmed, then one‐way analysis of variance was used to compare all four patient cohorts. Differences in V̇O2peak, NYHA functional class, echocardiography, and clinical and treatment variables between HFrEF men and HFrEF women were determined using unpaired Student's t‐tests. Categorical variables were compared using chi‐squared tests (or Fisher's exact test were appropriate). For all mitochondrial function and content data as well as gene expression analyses, Student's t‐tests were employed for within‐sex comparisons. Pearson correlations were included to examine relationships between measures of mitochondrial content and function with V̇O2peak. Statistical significance was accepted as P < 0.05, and all data were analysed using commercial software (SPSS for Windows Version 25.0; IBM Corporation, Armonk, NY).
3. Results
3.1. Patient characteristics
Characteristics for all patients are presented in Table 1. The four cohorts showed no differences between age and body mass index. Patients with HFrEF had symptoms of exercise intolerance and left ventricular systolic dysfunction, with no significant differences between sexes in terms of NYHA functional class, aetiology, glycated haemoglobin, additional comorbidities, V̇O2peak, and cardiac function. Pharmacological treatments and device therapies were also not different between men and women in the HFrEF group.
3.2. Mitochondrial respiration and content
Compared with sex‐matched control subjects, mitochondrial respiration with substrates supporting complex I OXPHOS was lower by 17.2% (P = 0.030; Figure 1A) in men with HFrEF and 28.9% (P = 0.013; Figure 1B) in women with HFrEF. Oxygen flux with complex I leak substrates was also 35.2% (P = 0.010; Figure 1B) lower in women with HFrEF compared with women in the control group. Mitochondrial content, as assessed by complex IV activity, was 31.5% (P = 0.023; Figure 2A) lower in women with HFrEF compared with female controls, whereas complex IV activity was not different in the male patients with HFrEF compared with controls (P > 0.05; Figure 2A). We also confirmed this by measuring citrate synthase enzyme activity as a secondary, independent measure of mitochondrial content (Figure 2B). This measure demonstrated that mitochondrial content was 40.9% lower in female patients with HFrEF (P = 0.011; Figure 2B) while there were no differences between the two male cohorts (P > 0.05; Figure 2B). There was a positive and significant correlation between complex IV activity and citrate synthase activity (r = 0.420; P = 0.012), thereby validating these two independent measures of mitochondrial content. When the mitochondrial respiration was normalized to complex IV activity (and therefore mitochondrial content), there were no differences between women with and without HFrEF (all P > 0.05; Figure 3); however, complex I OXPHOS was still 15.4% (P = 0.030; Figure 3) lower in men with HFrEF than male controls. Overall, these data show women with HFrEF have lower mitochondrial content whereas men with HFrEF have complex I‐specific impaired mitochondrial quality. The respiratory control ratio (ratio of complex I OXPHOS to leak respiration) was not different between control and HFrEF groups in both men (5.30 ± 0.67 vs. 5.25 ± 0.62; P > 0.05) and women (4.57 ± 0.46 vs. 5.06 ± 0.56; P > 0.05). While flux control ratio (ratio of individual complex respiration to maximal uncoupled respiration) was not different between male groups for complex I (0.51 ± 0.04 vs. 0.45 ± 0.02, P > 0.05), it was lower in the women with CHF relative to the controls (0.55 ± 0.05 vs. 0.44 ± 0.03; P = 0.045). No other differences in the flux control ratio across the respiratory states was otherwise noted.
Figure 1.
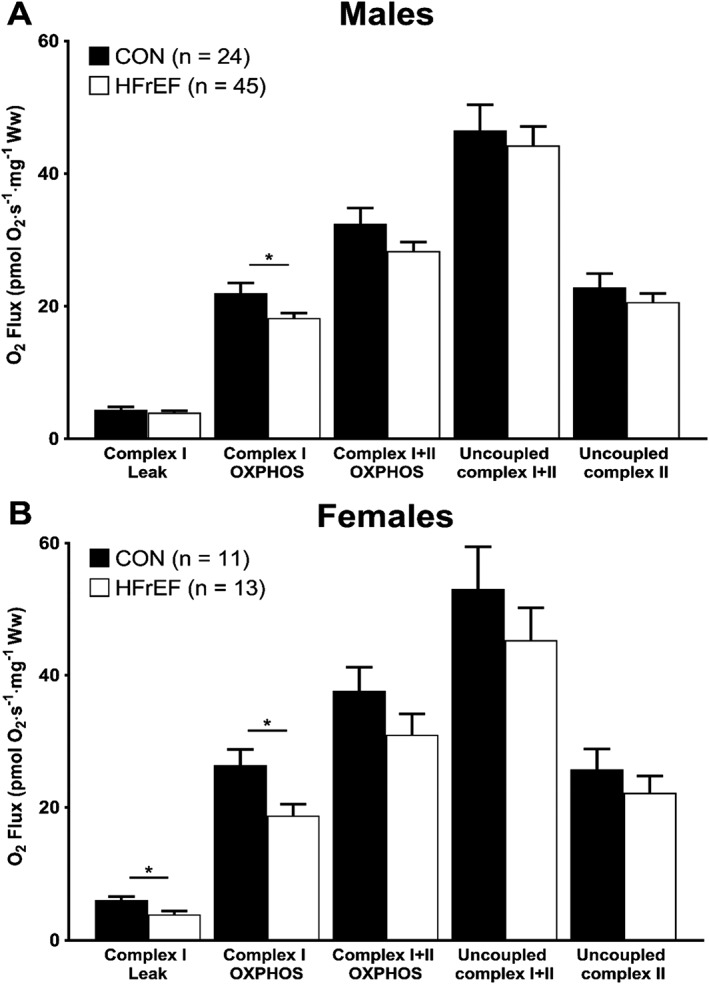
Skeletal muscle mitochondrial O2 flux across all respiratory states in male and female patients. Mitochondrial oxygen flux with complex I substrates is lower in both male (A) and female (B) patients with HFrEF compared with age‐matched and sex‐matched controls. Data are mean ± SEM. *P < 0.05 using unpaired Student's t‐tests. HFrEF, heart failure with reduced ejection fraction; OXPHOS, oxidative phosphorylation.
Figure 2.
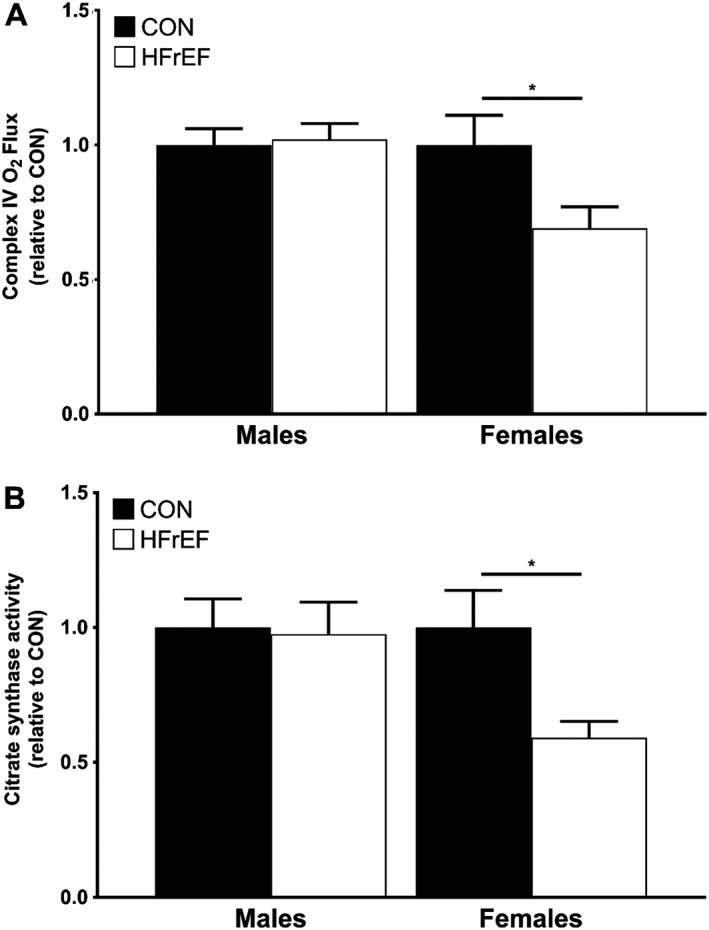
Relative measures of complex IV and citrate synthase activities as markers of mitochondrial content. Mitochondrial content determined from complex IV activity (A) was lower in women with HFrEF (n = 13) compared with female controls (n = 11) but did not differ between men with (n = 45) or without (n = 24) HFrEF. Similarly, citrate synthase activity (B) was also lower in women with HFrEF (n = 10) compared with women (n = 8) but men with HFrEF (n = 9) did not differ to sex‐matched controls (n = 8). Data are mean ± SEM. *P < 0.05 using unpaired Student's t‐tests. HFrEF, heart failure with reduced ejection fraction.
Figure 3.
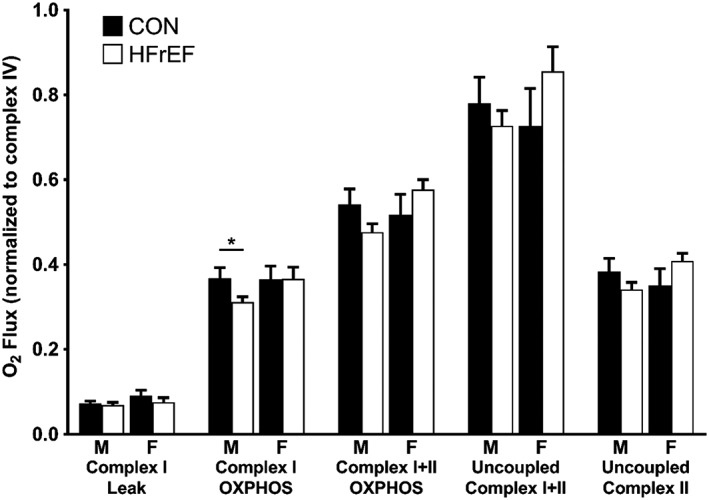
Skeletal muscle mitochondrial O2 flux across all respiratory states after adjusting the data to complex IV activity. After adjusting for mitochondrial content, complex I OXPHOS O2 flux was only lower in male (M) patients with HFrEF (n = 45) compared with male controls (n = 24). There were no differences between female (F) patients with HFrEF (n = 13) and female controls (n = 11). Data are mean ± SEM. *P < 0.05 using unpaired Student's t‐tests. HFrEF, heart failure with reduced ejection fraction; OXPHOS, oxidative phosphorylation.
Additional between‐group comparisons for mitochondrial measures between all cohorts revealed further differences, including female controls having higher values than CHF men for mitochondrial respiration during leak respiration (P = 0.02), complex I OXPHOS (P = 0.001), and complex I + II OXPHOS (P = 0.03) (Figure 1). No further differences between all cohorts were detected in terms of mitochondrial density (Figure 2), normalized mitochondrial function (Figure 3), or in the respiratory or flux control ratios (all P > 0.05).
3.3. Gene and protein expression
Compared with female controls, women with HFrEF had a significantly lower relative mRNA expression in several key mitochondrial and respiratory complex genes involved in mitochondrial fusion, biogenesis, and oxidative stress (Figure 4). OPA1, a key fusion protein required for normal mitochondrial network integrity and dynamics, was 50.4% lower (P = 0.022; Figure 4A) in women with HFrEF. In contrast, no differences were observed in transcript levels of the key protein regulating mitochondrial fragmentation, FIS1 (P > 0.05; Figure 4B). The master regulator of mitochondrial biogenesis, PGC‐1α, was also lower in women with HFrEF by 71.7 % (P = 0.035; Figure 4C), while a similar trend was also found for the key mitochondrial‐anti‐oxidant SOD2, which was lower by 49.9% (P = 0.009; Figure 4D) in women with HFrEF. Finally, the expression of NDUFS1 and NDUFS3, both of which encode for core subunit proteins in complex I, were also 39.4% (P = 0.048; Figure 4E) and 32.2% (P = 0.034; Figure 4F) lower in women with HFrEF, respectively. In contrast, male patients with HFrEF did not differ in the gene expression measurements compared with controls (Figure 4A–4F) suggesting a systematic reduction in key mitochondrial gene expression specific to the skeletal muscle of female patients with HFrEF. We followed up these findings by confirming that protein expression of PGC‐1α was also lower in female patients (Figure 5; P = 0.022) but not in male patients (Figure 5; P = 0.398), when compared with controls.
Figure 4.
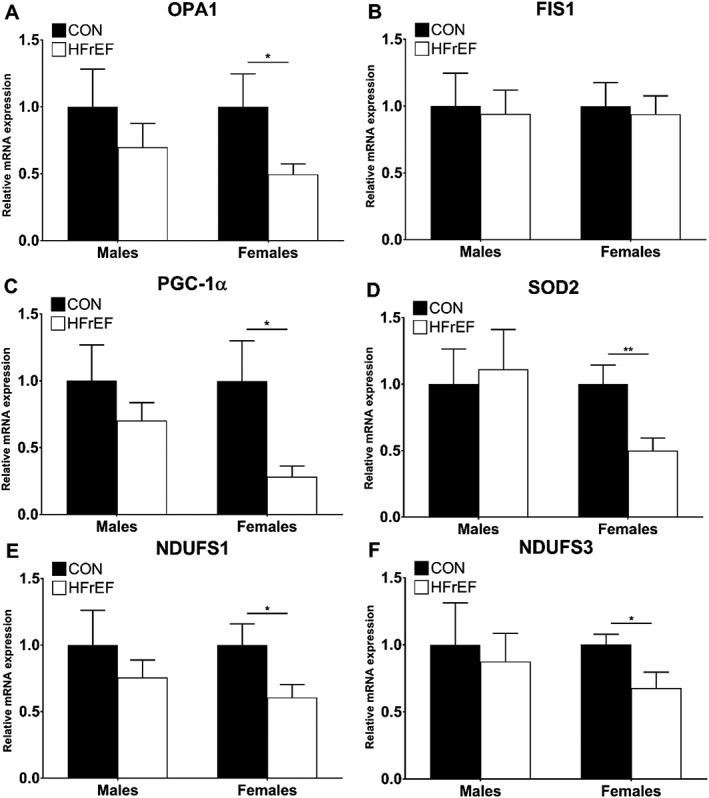
Relative mRNA expression levels of several key mitochondrial gene transcripts. Compared with controls, female patients with HFrEF have lower relative mRNA expression for OPA1 (A), PGC‐1α (C), SOD2 (D), NDUFS1 (E), and NDUFS3 (F) (n = 10 per group for each sex). *P < 0.05 using unpaired Student's t‐tests. ** P < 0.01 using unpaired Student's t‐tests. FIS1, mitochondrial fission 1; OPA1, optic atrophy 1; NDUFS1, NADH:ubiquinone oxidoreductase core subunit S1; NDUFS3, NADH:ubiquinone oxidoreductase core subunit S3; PGC‐1α, peroxisome proliferator‐activated receptor γ coactivator‐1α; SOD2, superoxide dismutase 2.
Figure 5.
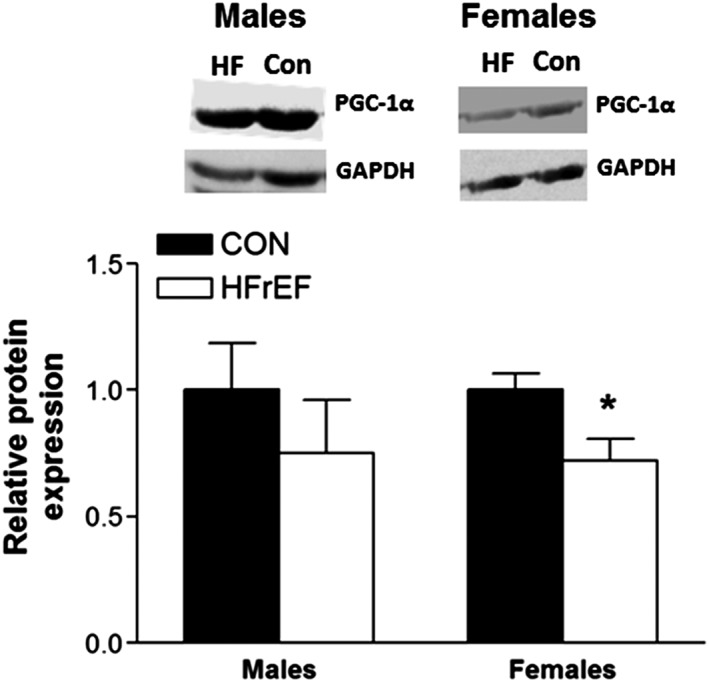
Relative protein expression levels of PGC‐1α. Compared with controls, female patients with HFrEF have lower relative protein expression while no difference was found in male patients (n = 8 per group for each sex). *P < 0.05 using unpaired Student's t‐tests. HFrEF, heart failure with reduced ejection fraction; PGC‐1α, peroxisome proliferator‐activated receptor γ coactivator‐1α.
3.4. Relationships between mitochondrial function, content, and gene transcripts
Measures of mitochondrial respiration and mitochondrial content were compared against the V̇O2peak measurements obtained from HFrEF patients during the exercise test (Figure 6). When comparing all HFrEF patients, there was a significant positive correlation between mitochondrial complex I respiration and V̇O2peak (Figure 6A). Similarly, there was a significant positive correlation between mitochondrial complex IV respiration, as a marker of mitochondrial content, and V̇O2peak (Figure 6B) across all patients with HFrEF. We also explored the relationships between the gene transcript expression levels and V̇O2peak as well as mitochondrial function measurements in all HFrEF patients (Table 2). Of all the gene expression measurements, the only marker to show a trend for a correlation with V̇O2peak was OPA1 (Table 2). OPA1, FIS1, NDUFS1, and NDUFS3 were all significantly correlated with complex I O2 flux (Table 2). Similarly, OPA1, PGC‐1α, SOD2, and NDUFS1 were all correlated with complex IV O2 flux (Table 2).
Figure 6.
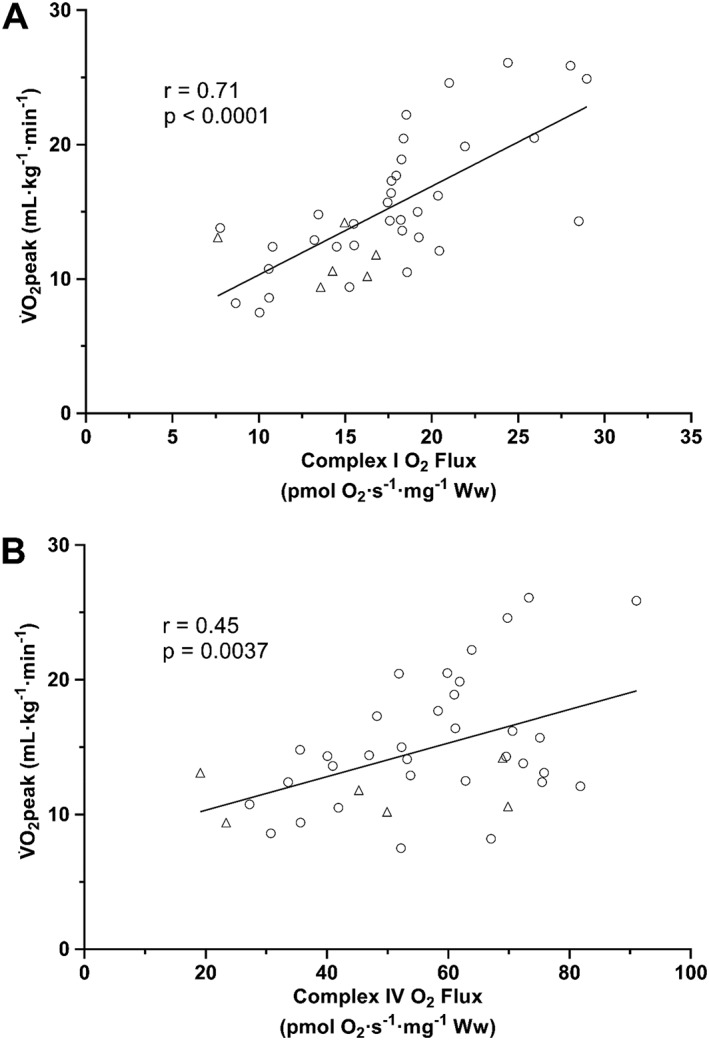
Measures of skeletal muscle mitochondrial function and content correlate with peak pulmonary oxygen uptake (V̇O2peak). Complex I O2 flux (A) and complex IV O2 flux (B) correlate with V̇O2peak in HFrEF men (circles; n = 34) and women (triangles; n = 6).
Table 2.
Correlations between gene transcript expression levels with peak pulmonary oxygen uptake (V̇O2peak) and measures of mitochondrial function and content in heart failure with reduced ejection fraction patients
| OPA1 | FIS1 | PGC‐1α | SOD2 | NDUFS1 | NDUFS3 | ||
|---|---|---|---|---|---|---|---|
| V̇O2peak | r | 0.550 | −0.118 | 0.043 | −0.253 | 0.111 | 0.242 |
| p | 0.080 | 0.729 | 0.900 | 0.453 | 0.746 | 0.501 | |
| Complex I O2 flux | r | 0.642 | 0.616 | 0.448 | 0.293 | 0.646 | 0.584 |
| p | 0.007* | 0.011* | 0.071 | 0.254 | 0.005* | 0.017* | |
| Complex IV O2 flux | r | 0.498 | 0.436 | 0.572 | 0.534 | 0.490 | 0.384 |
| p | 0.049* | 0.092 | 0.016* | 0.027* | 0.046 | 0.142 |
FIS1, mitochondrial fission 1; OPA1, optic atrophy 1; NDUFS1, NADH:ubiquinone oxidoreductase core subunit S1; NDUFS3, NADH:ubiquinone oxidoreductase core subunit S3; PGC‐1α, peroxisome proliferator‐activated receptor γ coactivator‐1α; SOD2, superoxide dismutase 2.
P < 0.05 using Pearson correlation.
4. Discussion
These data provide novel evidence that HFrEF induces divergent sex‐specific pathophysiological mitochondrial phenotypes in skeletal muscle that would be expected to predispose towards exercise intolerance, impacting mitochondrial ‘quantity' in women and mitochondrial ‘quality' in men. Major findings of the present study revealed that female patients with HFrEF demonstrated a lower mitochondrial content when compared with matched‐controls, which corresponded to a downregulation in the gene/protein expression of the key regulator of mitochondrial biogenesis PGC‐1α. In contrast, male patients with HFrEF presented with clear intrinsic mitochondrial dysfunction when compared with matched controls, with no alterations in mitochondrial content or aberrant gene/protein expression. Mitochondrial deficits closely correlated to V̇O2peak across HFrEF patients, suggesting their clinical relevance in driving symptoms of exercise intolerance.
4.1. Impact of sex on skeletal muscle mitochondrial alterations in heart failure with reduced ejection fraction
A novel finding of the present study showed that, when compared with age‐matched controls, female HFrEF patients had lower mitochondrial complex I respiration (i.e. flux per wet muscle mass), but these differences were negated after adjusting for the significantly lower mitochondrial content (i.e. flux per mitochondrion). This finding therefore suggests that any differences in mitochondrial oxidative capacity in female HFrEF patients may be attributed to lower mitochondrial content rather than intrinsic mitochondrial dysfunction as observed in men. As such, female patients that have substantial muscle wasting (and by inference a reduction in mitochondrial content) would likely show the most severe impairments to exercise intolerance.12 Compared with age‐matched controls, previous studies measuring mitochondrial enzymes in patients with HFrEF have shown either no major changes9 or more pronounced deficits in men, the latter indicative of reduced content.10 However, these studies were concluded based on just a couple of oxidative enzyme measures/ limited mitochondrial functional data, inclusion of young patients (<60 years), and poorly matched cardiac function. In contrast to the female patients, we found that male patients with HFrEF exhibited a mitochondrial complex I‐specific impairment that was independent of mitochondrial content (i.e. an intrinsic qualitative deficit). This finding suggests that male and female patients with HFrEF have divergent responses to HFrEF in terms of skeletal muscle mitochondrial impairments. Thus, the current study significantly expands our understanding of the impact of sex on mitochondrial impairments in HFrEF. Furthermore, our data showed a significant association between complex I function and V̇O2peak in patients, which firmly supports the evidence that mitochondrial dysfunction as an important mechanism of exercise intolerance in HFrEF.21, 22, 23 Future studies are therefore warranted on validating the current findings via non‐invasive or more accessible clinical approaches (e.g. near‐infrared spectroscopy14; Technetium‐99m sestamibi, 99mTc‐MIBI and computed tomography,24, 25 or magnetic resonance spectroscopy26) in order to accommodate the wider CHF population (i.e. non‐pacemaker) and enable more targeted treatment of peripheral and/or central mechanisms limiting exercise intolerance in patients with CHF.
4.2. Mechanisms of sex‐specific mitochondrial alterations in heart failure with reduced ejection fraction
The expression levels of several key mitochondrial gene transcripts involved in mitochondrial shaping and maintenance (i.e. OPA1, PGC‐1α, SOD2, NDUFS1, and NDUFS3) were uniquely lower in female HFrEF patients, which corresponded to lower levels of mitochondrial content but not intrinsic dysfunction. These findings were not replicated in male patients with HFrEF, which clearly indicates sex‐specific divergences in potential molecular control mechanisms. In particular, we detected a significantly lower level of both gene and protein expression in the master regulator of mitochondrial biogenesis PGC‐1α. Given the key role PGC‐1α plays in maintaining mitochondrial content and exercise tolerance in disease,12 this finding could provide one mechanism for the sex‐specific differences we observed in terms of mitochondrial content between male and female CHF patients, and further why women present with worse exercise intolerance but respond better to exercise training compared with their male counter parts.1, 2 In addition, female CHF patients showed lower gene expression of OPA1 (a regulator of mitochondrial fusion) and SOD2 (a mitochondrial‐specific antioxidant enzyme), while FIS1 expression (a regulator of mitochondrial fission) was unchanged that suggests an equilibrium shift towards mitochondrial fragmentation and disruption of the mitochondrial network. Impaired OPA1 expression in skeletal muscle is known to impair mitochondrial morphology, induce apoptosis, and decrease mitochondrial content,27 while OPA1 is closely correlated to mitochondrial impairments and exercise intolerance in ageing.11 Further transcripts of specific respiratory complex I subunits (i.e. NDUFS1 and NDUFS3) were also downregulated in women with HFrEF, suggesting a widespread disruption of the mitochondrial signalling pathway is not induced within male patients. The finding that several genes were comparably lower in female patients with HFrEF would appear to suggest a coordinated downregulation in mitochondrial content, which we confirmed in further independent assays (i.e. complex IV and citrate synthase). That most of these mitochondrial gene transcripts were well correlated to mitochondrial respiration and content (Table 2) provide strong evidence these underpin, at least in part, the lower mitochondrial quantity and quality observed in patients with HFrEF. Interestingly, OPA1 also tended (P = 0.08) to be closely correlated to whole‐body V̇O2peak, supporting evidence that impairments to mitochondrial fusion may impair not only mitochondrial capacity but also overall skeletal muscle morphology, to impact exercise tolerance.11
The underlying mechanisms of why women respond differently in their mitochondrial morphology and function compared with male patients with HFrEF remains unclear but may be related to the sex hormone oestrogen or the preferential oxidation of fatty acids in women that may increase intrinsic function.28 The mechanism behind the lower intrinsic function of complex I in male patients with HFrEF remains unclear, but it may be related to a downregulation in complex activity/expression as transcriptional markers were not impacted.29 As such, future studies are warranted to help provide further mechanistic insight (e.g. signalling cascades) into the sex‐specific mitochondrial derangements induced by CHF.
4.3. Study limitations
This study was limited by the observational cross‐sectional design, which allowed characterization of variables and their relationships rather than prove causation and direct mechanistic insight. Therefore, an interventional study is warranted to address how skeletal muscle mitochondrial content and function change longitudinally in both male and female patients with HFrEF. When applying our findings to the wider CHF population, it should be considered that the data were collected from a moderate sample size where other confounding factors may play a role (e.g. cohort matching and physical activity levels). For example, we did not match participants for physical activity levels, which may have influenced our results.30, 31 However, detraining cannot fully explain mitochondrial deficits in HFrEF,32, 33 and we obtained samples from pectoralis major, which is less susceptible to the effects of detraining compared with locomotor muscles. Furthermore, we also did not have a measure of exercise intolerance (i.e. V̇O2peak) in controls to evaluate their current levels of aerobic fitness, while the assessment of muscle mass would have proved valuable at providing insight into the involvement of muscle atrophy into sex‐specific mitochondrial derangements.
5. Conclusions
This study presents novel evidence to indicate that male and female patients with HFrEF exhibit divergent mitochondrial responses in terms of content, intrinsic function, and gene/protein dynamics, which closely correlated with V̇O2peak. As such, these findings support the rationale for sex‐specific treatment therapies in HFrEF patients, with interventions targeting mitochondrial content in women and mitochondrial intrinsic function in men as an alternative approach to ameliorate symptoms of exercise intolerance.
Conflict of Interest
None declared.
Acknowledgements
L.D.R. acknowledges the support of the Diabetes UK RD Lawrence Fellowship (16/0005382). J.G. holds an NIHR‐HCS Fellowship Award. M.P. holds an NIHR‐ICA fellowship Award. R.M.C. is a British Heart Foundation Intermediate Research Fellow (FS/12/80/29821). M.T.K. holds a British Heart Foundation Chair of Cardiology (CH/13/1/30086). T.S.B. acknowledges support from the Fondation Leducq (TNE 13CVD04) and Medical Research Council, UK (MR/S025472/1). During the period of data collection, K.K.W. held an NIHR Clinician Scientist Award (NIHR‐CS‐012‐032). The authors certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia, and Muscle.34
Garnham J. O., Roberts L. D., Caspi T., Al‐Owais M. M., Bullock M., Swoboda P. P., Koshy A., Gierula J., Paton M. F., Cubbon R. M., Kearney M. T., Bowen T. S., and Witte K. K. (2020) Divergent skeletal muscle mitochondrial phenotype between male and female patients with chronic heart failure, Journal of Cachexia, Sarcopenia and Muscle, 11: 79–88. 10.1002/jcsm.12488.
References
- 1. Corra U, Mezzani A, Giordano A, Pistono M, Gnemmi M, Caruso R, et al. Peak oxygen consumption and prognosis in heart failure: 14 mL/kg/min is not a ‘gender‐neutral' reference. Int J Cardiol 2013;167:157–161. [DOI] [PubMed] [Google Scholar]
- 2. Piña IL, Bittner V, Clare RM, Swank A, Kao A, Safford R, et al. Effects of exercise training on outcomes in women with heart failure: analysis of HF‐ACTION (Heart Failure‐A Controlled Trial Investigating Outcomes of Exercise TraiNing) by sex. J Am Coll Cardiol HF 2014;2:180–186. [DOI] [PubMed] [Google Scholar]
- 3. O'Meara E, Clayton T, McEntegart MB, McMurray JJ, Pina IL, Granger CB, et al. Sex differences in clinical characteristics and prognosis in a broad spectrum of patients with heart failure: results of the Candesartan in heart failure: assessment of reduction in mortality and morbidity (CHARM) program. Circulation 2007;115:3111–3120. [DOI] [PubMed] [Google Scholar]
- 4. Roger VL, Weston SA, Redfield MM, Hellermann‐Homan JP, Killian J, Yawn BP, et al. Trends in heart failure incidence and survival in a community‐based population. JAMA 2004;292:344–350. [DOI] [PubMed] [Google Scholar]
- 5. Clark AL, Davies LC, Francis DP, Coats AJS. Ventilatory capacity and exercise tolerance in patients with chronic stable heart failure. Eur J Heart Fail 2000;2:47–51. [DOI] [PubMed] [Google Scholar]
- 6. Franciosa JA, Park M, Levine TB. Lack of correlation between exercise capacity and indexes of resting left ventricular performance in heart failure. Am J Cardiol 1981;47:33–39. [DOI] [PubMed] [Google Scholar]
- 7. Witte KK, Nikitin NP, De Silva R, Cleland JG, Clark AL. Exercise capacity and cardiac function assessed by tissue Doppler imaging in chronic heart failure. Heart 2004;90:1144–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haykowsky MJ, Tomczak CR, Scott JM, Paterson DI, Kitzman DW. Determinants of exercise intolerance in patients with heart failure and reduced or preserved ejection fraction. J Appl Physiol (1985) 2015;119:739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duscha BD, Annex BH, Keteyian SJ, Green HJ, Sullivan MJ, Samsa GP, et al. Differences in skeletal muscle between men and women with chronic heart failure. J Appl Physiol (1985) 2001;90:280–286. [DOI] [PubMed] [Google Scholar]
- 10. Duscha BD, Annex BH, Green HJ, Pippen AM, Kraus WE. Deconditioning fails to explain peripheral skeletal muscle alterations in men with chronic heart failure. J Am Coll Cardiol 2002;39:1170–1174. [DOI] [PubMed] [Google Scholar]
- 11. Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, et al. Age‐associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab 2017;25:1374–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bowen TS, Schuler G, Adams V. Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J Cachexia Sarcopenia Muscle 2015;6:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
- 14. Bowen TS, Cannon DT, Murgatroyd SR, Birch KM, Witte KK, Rossiter HB. The intramuscular contribution to the slow oxygen uptake kinetics during exercise in chronic heart failure is related to the severity of the condition. J Appl Physiol (1985) 2012;112:378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fontana‐Ayoub M, Fasching M, Gnaiger E. Selected media and chemicals for respirometry with mitochondrial preparations. Mitochondr Physiol Network 2014;3:1–9. [Google Scholar]
- 16. Wüst RC, Myers DS, Stones R, Benoist D, Robinson PA, Boyle JP, et al. Regional skeletal muscle remodeling and mitochondrial dysfunction in right ventricular heart failure. Am J Physiol Heart Circ Physiol 2012;302:H402–H411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 2012;590:3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuznetsov AV, Gnaiger E. Oxygraph assay of cytochrome c oxidase activity: chemical background correction. Mitochondr Physiol Netw 2010;6:1–4. [Google Scholar]
- 19. Bowen TS, Rolim NP, Fischer T, Bækkerud FH, Medeiros A, Werner S, et al. Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur J Heart Fail 2015;17:263–272. [DOI] [PubMed] [Google Scholar]
- 20. Roberts LD, Bostrom P, O'Sullivan JF, Schinzel RT, Lewis GD, Dejam A, et al. β‐Aminoisobutyric acid induces browning of white fat and hepatic β‐oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab 2014;19:96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sullivan MJ, Green HJ, Cobb FR. Skeletal muscle biochemistry and histology in ambulatory patients with long‐term heart failure. Circulation 1990;81:518–527. [DOI] [PubMed] [Google Scholar]
- 22. Drexler H, Riede U, Münzel T, König H, Funke E, Just H. Alterations of skeletal muscle in chronic heart failure. Circulation 1992;85:1751–1759. [DOI] [PubMed] [Google Scholar]
- 23. Hambrecht R, Niebauer J, Fiehn E, Kälberer B, Offner B, Hauer K, et al. Physical training in patients with stable chronic heart failure: effects on cardiorespiratory fitness and ultrastructural abnormalities of leg muscles. J Am Coll Cardiol 1995;25:1239–1249. [DOI] [PubMed] [Google Scholar]
- 24. Kato T, Nakane E, Funasako M, Miyamoto S, Izumi T, Haruna T, et al. A potential linkage between mitochondrial function of the heart and leg muscles in patients with heart failure. Int J Cardiol 2015;188:67–69. [DOI] [PubMed] [Google Scholar]
- 25. Matsuo S, Nakae I, Tsutamoto T, Okamoto N, Horie M. A novel clinical indicator using Tc‐99m sestamibi for evaluating cardiac mitochondrial function in patients with cardiomyopathies. J Nucl Cardiol 2007;14:215–220. [DOI] [PubMed] [Google Scholar]
- 26. Weiss K, Schar M, Panjrath GS, Zhang Y, Sharma K, Bottomley PA, et al. Fatigability, exercise intolerance, and abnormal skeletal muscle energetics in heart failure. Circ Heart Fail 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 2003;278:7743–7746. [DOI] [PubMed] [Google Scholar]
- 28. Cardinale DA, Larsen FJ, Schiffer TA, Morales‐Alamo D, Ekblom B, Calbet JAL, et al. Superior intrinsic mitochondrial respiration in women than in men. Front Physiol 2018;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Galkin A, Meyer B, Wittig I, Karas M, Schagger H, Vinogradov A, et al. Identification of the mitochondrial ND3 subunit as a structural component involved in the active/deactive enzyme transition of respiratory complex I. J Biol Chem 2008;283:20907–20913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toth MJ, Miller MS, Ward KA, Ades PA. Skeletal muscle mitochondrial density, gene expression, and enzyme activities in human heart failure: minimal effects of the disease and resistance training. J Appl Physiol (1985) 2012;112:1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mettauer B, Zoll J, Sanchez H, Lampert E, Ribera F, Veksler V, et al. Oxidative capacity of skeletal muscle in heart failure patients versus sedentary or active control subjects. J Am Coll Cardiol 2001;38:947–954. [DOI] [PubMed] [Google Scholar]
- 32. Simonini A, Long CS, Dudley GA, Yue P, McElhinny J, Massie BM. Heart failure in rats causes changes in skeletal muscle morphology and gene expression that are not explained by reduced activity. Circ Res 1996;79:128–136. [DOI] [PubMed] [Google Scholar]
- 33. Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura‐Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 2003;551:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]