

Abstract
Background
Critically ill patients frequently develop muscle atrophy and weakness in the intensive‐care‐unit setting [intensive care unit‐acquired weakness (ICUAW)]. Sepsis, systemic inflammation, and acute‐phase response are major risk factors. We reported earlier that the acute‐phase protein serum amyloid A1 (SAA1) is increased and accumulates in muscle of ICUAW patients, but its relevance was unknown. Our objectives were to identify SAA1 receptors and their downstream signalling pathways in myocytes and skeletal muscle and to investigate the role of SAA1 in inflammation‐induced muscle atrophy.
Methods
We performed cell‐based in vitro and animal in vivo experiments. The atrophic effect of SAA1 on differentiated C2C12 myotubes was investigated by analysing gene expression, protein content, and the atrophy phenotype. We used the cecal ligation and puncture model to induce polymicrobial sepsis in wild type mice, which were treated with the IкB kinase inhibitor Bristol‐Myers Squibb (BMS)‐345541 or vehicle. Morphological and molecular analyses were used to investigate the phenotype of inflammation‐induced muscle atrophy and the effects of BMS‐345541 treatment.
Results
The SAA1 receptors Tlr2, Tlr4, Cd36, P2rx7, Vimp, and Scarb1 were all expressed in myocytes and skeletal muscle. Treatment of differentiated C2C12 myotubes with recombinant SAA1 caused myotube atrophy and increased interleukin 6 (Il6) gene expression. These effects were mediated by Toll‐like receptors (TLR) 2 and 4. SAA1 increased the phosphorylation and activity of the transcription factor nuclear factor ‘kappa‐light‐chain‐enhancer' of activated B‐cells (NF‐κB) p65 via TLR2 and TLR4 leading to an increased binding of NF‐κB to NF‐κB response elements in the promoter region of its target genes resulting in an increased expression of NF‐κB target genes. In polymicrobial sepsis, skeletal muscle mass, tissue morphology, gene expression, and protein content were associated with the atrophy response. Inhibition of NF‐κB signalling by BMS‐345541 increased survival (28.6% vs. 91.7%, P < 0.01). BMS‐345541 diminished inflammation‐induced atrophy as shown by a reduced weight loss of the gastrocnemius/plantaris (vehicle: −21.2% and BMS‐345541: −10.4%; P < 0.05), tibialis anterior (vehicle: −22.7% and BMS‐345541: −17.1%; P < 0.05) and soleus (vehicle: −21.1% and BMS‐345541: −11.3%; P < 0.05) in septic mice. Analysis of the fiber type specific myocyte cross‐sectional area showed that BMS‐345541 reduced inflammation‐induced atrophy of slow/type I and fast/type II myofibers compared with vehicle‐treated septic mice. BMS‐345541 reversed the inflammation‐induced atrophy program as indicated by a reduced expression of the atrogenes Trim63/MuRF1, Fbxo32/Atrogin1, and Fbxo30/MuSA1.
Conclusions
SAA1 activates the TLR2/TLR4//NF‐κB p65 signalling pathway to cause myocyte atrophy. Systemic inhibition of the NF‐κB pathway reduced muscle atrophy and increased survival of septic mice. The SAA1/TLR2/TLR4//NF‐κB p65 atrophy pathway could have utility in combatting ICUAW.
Keywords: Sepsis, NF‐κB, CLP, Muscle atrophy, ICUAW
Introduction
Critically ill intensive care unit (ICU) patients often develop muscle atrophy and weakness [ICU‐acquired weakness (ICUAW)] with increased morbidity and mortality.1, 2 The incidence of ICUAW is up to 90% in severe sepsis patients.3 Sequels persists for up to 5 years after discharge, underscoring substantial morbidity.4 Additionally, ICUAW is associated with high costs for the health care system.4, 5 Inflammation and sepsis clearly increase the risk for ICUAW, and inflammatory cytokines are associated with the incidence of ICUAW.6, 7 Skeletal muscle and skeletal myocytes are targeted by inflammatory cytokines, such as interleukin 1β (IL1β) and IL6,8, 9, 10 causing myocyte atrophy.8, 11, 12 Our attention was drawn to serum amyloid A1 (SAA1) that is associated with inflammation and acute‐phase responses during sepsis.11, 13While the main source of SAA1 is the liver,13 IL6 increases SAA1 expression and secretion in muscle.8, 11, 14 We earlier showed that SAA1 expression is greatly increased in ICUAW and that SAA1 accumulates in the myocyte membrane and in muscular interstitium.11 These findings were also observed in other studies.15, 16, 17, 18, 19 Nonetheless, a role of SAA1 as a perpetrator rather than a mere biomarker in ICUAW is imperfectly defined.20
SAA1 is produced by various tumours21, 22, 23 and is also associated with muscle wasting in cancer cachexia in mice.24 SAA1 cooperates with IL6 to mediate angiotensin II‐induced muscle atrophy,9 which is important for muscle wasting in heart failure patients. SAA1 receptors have been described in immune cells, including the purinergic receptor P2X7 (P2RX7), the CD36 receptor (CD36), the scavenger receptor class b member 1 (SCARB1), the VCP interacting membrane selenoprotein (VIMP), and Toll‐like receptors (TLR) 2 and 4.25, 26, 27, 28, 29, 30, 31 SAA1 activates the NLRP3 inflammasome via P2RX7 and thereby promotes IL1β activation and release from mouse and human macrophages.25 SAA1 also increases IL6 and TNF expression by activating the CD36 receptor in rat macrophages and human embryonic kidney cells.26 SCARB1 mediates internalization of SAA in hepatocytes, and VIMP acts as an SAA1‐receptor in a mouse model of type 2 diabetes.27, 28 In mouse macrophages, SAA1 was shown to increase pro‐inflammatory cytokines, such as IL23α and tumor necrosis factor (TNF) via TLR2, and to increase nitric oxide via TLR4.30, 31, 32 Importantly, SAA1 increases IL6 expression via TLR2 and TLR4 and activates the nuclear factor ‘kappa‐light‐chain‐enhancer' of activated B‐cells (NF‐κB) signalling pathway in dermal fibroblasts.33 However, whether or not the TLR2/TLR4/NF‐κB signalling pathway plays a role in SAA1‐induced myocyte atrophy in vitro and sepsis‐induced muscle atrophy in vivo is not known. We demonstrate that SAA1 acts on myocytes to cause atrophy by the TLR2/TLR4/NF‐κB pathway. We also show that inhibition of NF‐κB by Bristol‐Myers Squibb (BMS)‐345541 increases survival and diminishes skeletal muscle atrophy in septic mice. We suggest that SAA1 via activation of the TLR2/TLR4/NF‐κB atrophy pathway is mechanistically important in ICUAW.
Methods
Patient samples
The institutional ethics committee of the Charité approved the study; it has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Written informed consent was obtained from legal proxy (ICU patients), or the patients themselves (Charité EA2/061/06; http://www.controlled-trials.com, ISRCTN77569430). Clinical data were reported previously.11 Biopsy specimens were obtained at Day 15 after ICU admission; control samples were acquired from patients undergoing elective orthopaedic surgery as described elsewhere.11, 34, 35
Animal model
The Landesamt für Gesundheit und Soziales, Berlin, Germany, approved the animal studies (G207/13, G129/12). Cecal ligation and puncture (CLP) surgery was performed to induce polymicrobial sepsis in 18‐week‐old male C57BL/6J mice as recently described.11, 36, 37 Sham mice were treated identically except for CLP (Supporting Information).
Molecular and cell biology analysis
Routine techniques were performed as previously shown11, 12, 35, 37, 38, 39, 40 and are outlined (Supporting Information).
Cell culture experiments with BMS‐345541
We used BMS‐345541 to investigate if NF‐κB inhibition prevents sepsis‐induced muscle atrophy in the CLP mouse model in vivo.41 BMS‐345541 (4(2'‐aminoethyl)amino‐1,8‐dimethylimidazo(1,2‐a)quinoxaline; CAS‐number: 547757‐23‐3) is a highly selective cell permeable IκB kinase (IKK) 2 and IKK‐1 inhibitor, which blocks NF‐κB‐dependent transcription; it was produced by BMS and purchased from abcam (ab 144822). BMS‐345541 was previously shown to reduce CLP and LPS‐induced lung injury,42, 43 to reduce inflammation in rats with spinal cord injury, and to suppress inflammation in a model of cardiac graft rejection.44, 45 Due to its high bioavailability41 and its anti‐inflammatory characteristics without impairing immune cell activation42 and bacterial clearance, BMS‐345541 was well suited for our studies. A 2.5 mM stock solution of BMS‐345541 dissolved in 10% DMSO was prepared for all cell culture experiments. For short‐term treatment, a final concentration of 25 μM BMS‐345541 was used because Burke et al.41 showed that this concentration abolished lipopolysaccharide (LPS)‐induced production of TNF, IL1β, IL8, and IL6 in THP‐1 cells, a human leukemic monocyte cell line. Likewise, 25 μM BMS‐345541 abolished TNF‐stimulated IκBα‐phosphorylation.41 For long‐term treatment of myocytes (up to 72 h), we used 5 μM BMS‐345541. This concentration was chosen because the NF‐κB pathway is vital for cell function, and its complete inhibition over 72 h provokes toxic side effects. However, 5 μM BMS‐345541 was shown to completely inhibit IKK‐2, reduce IKK‐1 activity by approximately 50%, and substantially decrease LPS‐induced production of TNF, IL 1β, IL‐8, and IL‐6 in THP 1 cells41; 5 μM BMS‐345541 was shown to inhibit IκBα‐phosphorylation in the prostate cancer cell line PC‐3.46
Anti‐MuRF1 antibody
We generated a MuRF1 specific antibody against the C‐terminal Mus musculus MuRF1 cDNA fragment (amino acids 185‐355). The specificity of the antibody was tested by immunoblotting tissue lysates of tibialis anterior from wild type, Trim63/MuRF1 mutant and Trim54/MuRF3//Trim 63/MuRF1 double mutant mice and of lysates from C2C12 cells following siRNA mediated knockdown of MuRF1. For more information about the antibody please refer to Nowak et al.47
Statistical tests
All experiments were performed independently and at least three times using biological triplicates each. The Mann–Whitney or two‐sided Student's t‐test was used to compare quantitative RT‐PCR gene expression data from mouse and cell culture samples. Distribution of myotube diameter of C2C12 myotubes were analysed by the two‐sided Student's t‐test. Survival curves were compared with a Mantel–Cox test. Differences were considered statistically significant at P < 0.05. Data are shown as mean ± standard error of the mean (SEM) in bar plots. GraphPad Prism® program (GraphPad software, version 6.0), Adobe Illustrator, version 16.0.0, and Photoshop, version 13.0, were used for plots and statistical calculations.
Results
Serum amyloid A1, Toll‐like receptors, and NF‐κB
We quantitated the expression of Tlr2, Tlr4, Cd36, P2rx7, Vimp, and Scarb1 in differentiated C2C12 myotubes, and tibialis anterior and gastrocnemius/plantaris by quantitative real‐time polymerase chain reaction (qRT‐PCR). All SAA1 receptors investigated were expressed in myocytes and skeletal muscle (Figure 1A and 1B). We treated differentiated C2C12 myotubes with recombinant SAA1 and vehicle respectively and quantitated the expression of those SAA1 receptors by qRT‐PCR. We found an upregulation of Tlr2 (5.4‐fold, P < 0.05) and a downregulation of Vimp (0.9‐fold, P < 0.05), whereas the expression of Tlr4, Cd36, P2rx7, and Scarb1 remained unchanged (Figure 1A).
Figure 1.
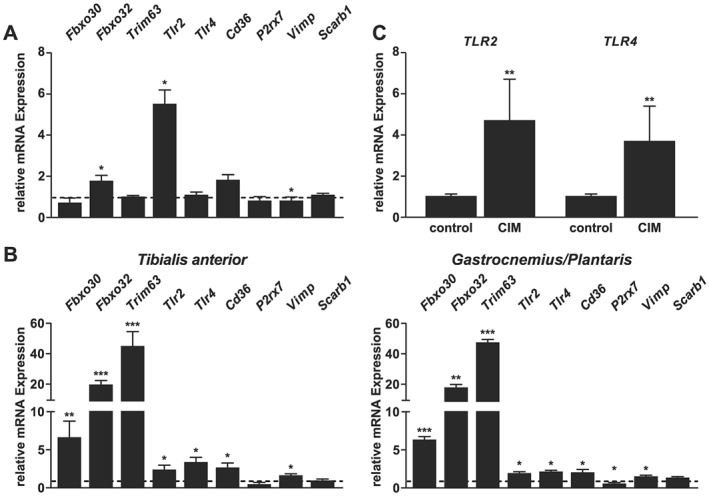
Expression of SAA1 receptors in C2C12 myotubes and skeletal muscle. (A) C2C12 myocytes were differentiated for 5 days and treated for 1 h with human recombinant SAA1 (20 μg/mL). qRT‐PCR analysis of SAA1 receptors Tlr2, Tlr4, Cd36, P2rx7, Vimp, Scarb1, and the atrogenes Fbxo30, Fbxo32, and Trim63. mRNA expression was normalized to Gapdh. Values of vehicle treated cells were set to 1 and are indicated as dotted line. Cd36 indicates cluster of differentiation 36; Fbxo30, F‐box protein 30; Fbxo32, F‐box protein 32; P2rx7, purinergic receptor P2X7; Scarb1, scavenger receptor class b member 1; Tlr2 indicates Toll‐like receptor 2; Trim63, tripartite motif containing 63; Vimp, VCP interacting membrane selenoprotein. (B) Twelve‐week‐old male WT mice were subjected to Sham (n = 3) or CLP (n = 5) surgery for 24 h. Expression of the SAA1 receptors Tlr2, Tlr4, Cd36, P2rx7, Vimp, and Scarb1 and the atrogenes Fbxo30, Fbxo32, and Trim63 was quantitated by qRT‐PCR in tibialis anterior and gastrocnemius/plantaris. mRNA expression was normalized to stably expressed Gapdh. Gene expression of sham treated mice was set to 1 and is indicated as dotted line. (C) qRT‐PCR analyses of TLR2 and TLR4 expression in control (no ICU subjects; n = 5) and CIM (15 days after ICU admission; n = 5) patients. GAPDH expression was used as a reference. Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
To investigate if SAA1 causes atrophy by directly increasing the expression of F‐box only protein 30 (Fbxo30; encoding muscle ubiquitin ligase of the SCF complex in atrophy‐1, MuSA1), Fbxo32 (encoding Atrogin1), and Trim63 (encoding MuRF1), which are bona fide atrogenes48, 49, SAA1 led to an increase in Fbxo32 expression (P < 0.05), whereas the expression of Fbxo30 and Trim63 remained unchanged in differentiated C2C12 myotubes (Figure 1A).
To investigate if polymicrobial sepsis affects the expression of SAA receptors, we studied their expression in tibialis anterior and gastrocnemius/plantaris of mice subjected to CLP surgery. Muscle of sham‐operated mice was used as control. We found an increased expression of Tlr2, Tlr4, Cd36, and Vimp in both muscles of CLP compared with sham‐operated mice. In contrast, P2rx7 was reduced in gastrocnemius/plantaris but remained unchanged in tibialis anterior of septic mice. Sepsis had no effect on Scarb1 expression in both muscles investigated (Figure 1B). In order to investigate if the atrophy program is activated in skeletal muscle during sepsis, we quantitated the expression of Fbxo30, Fbxo32, and Trim63 in both muscles of sham and CLP mice. Fbxo30, Fbxo32, and Trim63 expression was increased in both muscles of septic mice, compared with sham controls, indicating that sepsis leads to an activation of the atrophy program in muscle (Figure 1B). Earlier, we had shown that ICUAW patients showed elevated amounts of SAA1 in skeletal muscle compared with controls.11 Therefore, we investigated whether or not TLR2 and TLR4 are also upregulated in vastus lateralis of ICUAW patients and used biopsy specimens from vastus lateralis of patients that underwent elective orthopaedic surgery as a control.11 QRT‐PCR showed a significant upregulation of TLR2 and TLR4 in muscle of ICUAW patients compared with controls (Figure 1C). In summary, our data show that all SAA receptors investigated are expressed in cultivated myocytes and in mouse skeletal muscle. The expression of Tlr2, Tlr4, Cd36, and Vimp increased in skeletal muscle of septic mice. Because SAA1 induced the expression of Tlr2 in cultivated muscle cells and we found an increased expression of Tlr2 and Tlr4 in muscle of septic mice and critically ill patients, we focused on these SAA1 receptors for further analyses.
To test the hypothesis that SAA1 mediates its atrophic effects through TLR2 and TLR4 on myocytes, we treated C2C12 myotubes with SAA1, performed immunocytochemistry with anti‐myosin antibody, and measured myotube diameters as indicator for atrophy. We found that SAA1‐treatment caused myotube atrophy (Figure 2A) as indicated by a leftward‐shift of the frequency‐distribution histogram (Figure 2B) and a decrease in the mean myotube diameter (Figure 2E). Recently, we reported that inflammation causes a decrease in myosin heavy chain (MyHC) proteins in muscle and that the ubiquitin proteasome system, especially the muscle specific E3‐ligase MuRF1, was involved.12, 34 We therefore next investigated whether SAA1‐induced atrophy coincided with a decrease in MyHC and an increase in MuRF1 protein content. Western blot analysis showed that SAA1‐treatment led to a decrease in Myh7 and an increase in MuRF1 protein contents in C2C12 myotubes (Figure 2F). To test if SAA1‐induced atrophy is mediated by TLR2 and TLR4, we exposed C2C12 myotubes to SAA1 in the presence or absence of anti‐TLR2 and anti‐TLR4 antibody, respectively. Immunocytochemistry and measurements of myotube diameters showed that anti‐TLR2 and anti‐TLR4 antibody attenuated SAA1‐induced atrophy (Figure 2B and 2E). To investigate if both TLR‐receptors individually are capable to cause myocyte atrophy, we exposed C2C12 myotubes to the TLR2‐agonist Pam3Csk4 and the TLR4‐agonist lipopolysaccharide (LPS), respectively, and tested their atrophic response after addition of either anti‐TLR2 or anti‐TLR4 antibody. Both Pam3Csk4 and LPS caused myotube atrophy, which was blocked by their respective inhibitors. These data indicate that TLR2 and TLR4 are equally effective to induce myotube atrophy (Figure 2A and 2C–2E).
Figure 2.
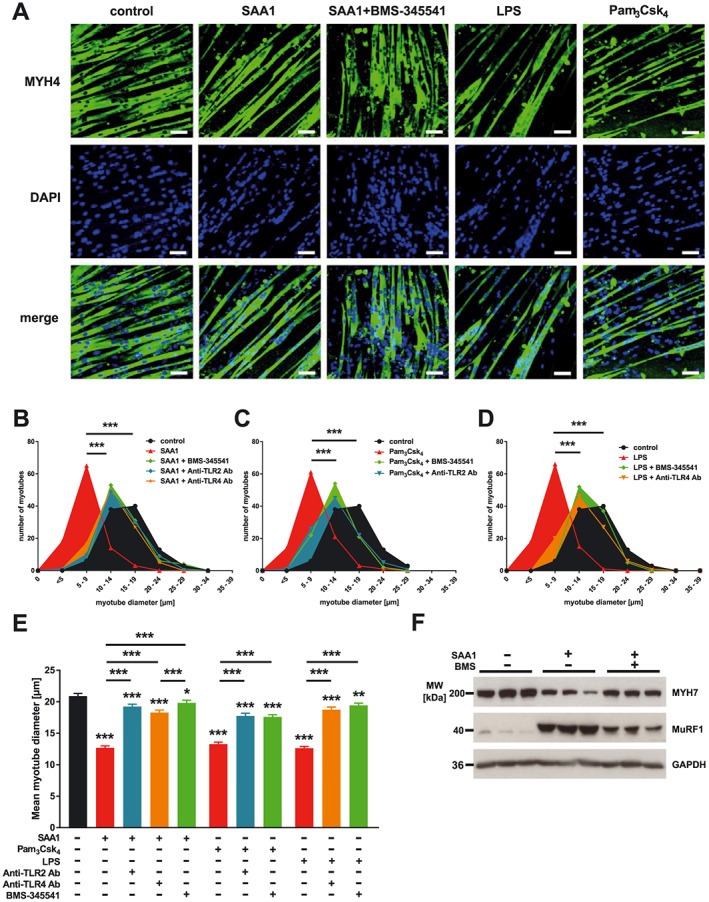
SAA1 causes atrophy of C2C12 myotubes via Toll‐like receptor (TLR) 2 and 4, in an NF‐κB dependent manner. C2C12 cells were differentiated for 5 days and treated with SAA1 (10 μg/mL), Pam3Csk4 (50 ng/mL), lipopolysaccharide (LPS; 1 μg/mL), anti‐TLR2 antibody (10 μg/mL), anti‐TLR4 antibody (20 μg/mL), or BMS‐345541 (5 μM), as indicated, for 72 h. (A) Immunohistochemistry with anti‐myosin heavy chain 4 (MyHC4, clone MF20) antibody. Nuclei were stained with DAPI. Scale bar = 100 μm. (B–D) Frequency distribution histograms of myotube diameters are shown (n = 100 cells per condition). (E) Mean myotube diameter. Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. (F) Western blot analysis with anti‐myosin heavy chain 7 (MyHC7; clone NOQ7.5.4D) and anti‐MuRF1antibody. Specificity of the anti‐MuRF1 antibody was tested by western blot analysis using tissue lysates of tibialis anterior muscle from wild type, Trim63/MuRF1 mutant and Trim54/MuRF3//Trim63/MuRF1 double mutant mice and of lysates from C2C12 cells following siRNA mediated knockdown of MuRF1 (data not shown). For more information, please refer to Nowak et al.47 GAPDH was used as loading control.
Because TLR2 and TLR4 activate NF‐κB signalling in immune cells,50, 51 we investigated if SAA1 activates NF‐κB via TLR2 and TLR4 in myocytes. Western blot analysis showed that SAA1‐treatment caused an increased phosphorylation of the NF‐κB transcription factor p65 in C2C12 myotubes (Figure 3A). Electrophoretic mobility shift assay (EMSA) revealed that SAA1‐treatment led to an increased binding of activated NF‐κB p65 to NF‐ĸB response elements (NRE) (Figure 3B). Addition of an anti‐p65 antibody to the EMSA‐assay resulted in a super‐shift of the NF‐κB p65/NRE complex underscoring the specificity of this assay (Figure 3B). To further investigate the NF‐κB p65 pathway, we used the IκB kinase (IKK) inhibitor BMS‐345541 that abolishes IKK‐dependent phosphorylation of p65 and of IκBα and subsequent degradation of IκBα and activation of NF‐κB.41 We found that SAA1‐induced phosphorylation of NF‐κB p65 was attenuated by BMS‐345541 (Figure 3A) indicating that IKK is involved in this process. Likewise, BMS‐345541 abolished SAA1‐induced NF‐κB p65/NRE complex formation and the super‐shift in EMSA (Figure 3B).
Figure 3.
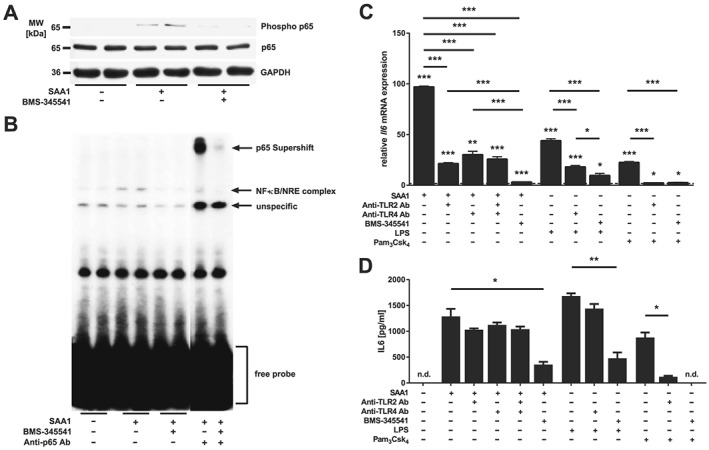
SAA1 increases Il6 expression and secretion via Toll‐like receptor (TLR) 2, TLR4 and NF‐κB p65. (A) C2C12 cells were treated with SAA1 (10 μg/mL) for 25 min in the presence or absence of BMS‐345541 (25 μM). Western blot analysis with anti‐phospho p65 antibody and anti‐p65 antibody are shown. GAPDH was used as loading control. (B) C2C12 cells were treated with SAA1 (10 μg/mL) for 25 min in the presence or absence of BMS‐345541 (25 μM). NF‐кB p65/NF‐κB‐response element complex formation was analysed by EMSA. (C) Five‐day‐differentiated C2C12 myotubes were preincubated with anti‐TLR2 antibody (10 μg/mL), anti‐TLR4 antibody (20 μg/mL), or BMS‐345541 (25 μM) as indicated. Afterwards cells were treated with SAA1 (10 μg/mL), LPS (1 μg/mL), or Pam3Csk4 (50 ng/mL) as indicated. qRT‐PCR analysis of Il6. mRNA expression was normalized to Gapdh. Control values were set to 1 and are indicated as dotted line. Data are presented as mean fold change ± SEM. (D) Quantification of IL6 in cell culture supernatants by ELISA. Cells were preincubated with anti‐TLR2 antibody (10 μg/mL), anti‐TLR4 antibody (20 μg/mL), or BMS‐345541 (25 μM) as indicated prior to SAA1 treatment (10 μg/mL). Data are presented as the mean ± SEM. n.d., not detectable; *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Although NF‐κB p65 mediates denervation and tumour‐induced atrophy52, 53 and its inhibition prevents muscle atrophy in mice with acute lung injury,54 whether or not NF‐κB p65 mediates SAA1‐induced atrophy is unknown. We therefore treated C2C12 myotubes with SAA1 in the presence or absence of BMS‐345541 and quantitated the atrophic response. Indeed, BMS‐345541 abolished SAA1‐induced atrophy (Figure 2A, 2B, and 2E), which was accompanied by an increase in Myh7 and a decrease in MuRF1 proteins as shown by western blot (Figure 2F). These data indicate that SAA1‐induced atrophy is mediated by TLR2 and TLR4 and that the NF‐κB p65 pathway is involved.
Recently, we reported that ICUAW patients and septic mice show increased muscular IL6 and SAA1 levels.11 Interestingly, IL6‐treatment causes an increase in SAA1 expression in myocytes,11 and conversely SAA1 induces IL6 in fibroblasts.33 These data suggested a positive feedback loop between SAA1 and IL6. To test if this feedback loop is present in myocytes and depends on TLR2‐mediated and TLR4‐mediated NF‐κB activation, we investigated if SAA1 induces IL6 in these cells. Using qRT‐PCR, we found that SAA1 increased Il6 expression in C2C12 myotubes. Anti‐TLR2 and TLR4 antibodies, as well as BMS‐345541 treatment, attenuated SAA1‐induced Il6 expression (Figure 3C). ELISA‐based quantification of the IL6 content in C2C12 cell culture supernatant revealed that SAA1‐induced IL6‐secretion was diminished by BMS‐345541 but remained unaffected by anti‐TLR2 and TLR4 antibody (Figure 3D). To further test if TLR2 and TLR4 activation regulate both IL6 expression and secretion, we treated C2C12 myotubes with the TLR2‐agonist Pam3Csk4 and the TLR4‐agonist lipopolysaccharide (LPS), respectively, in the presence or absence of anti‐TLR2 or TLR4 antibody. The anti‐TLR2 antibody reduced Pam3Csk4‐induced IL6 expression and secretion. In contrast, the anti‐TLR4 antibody did not inhibit LPS‐induced IL6‐secretion (Figure 3C and 3D). BMS‐345541 attenuated Pam3Csk4‐induced and LPS‐induced Il6 expression and secretion (Figure 3C and 3D). These data suggest that SAA1‐induced Il6 expression is mediated by TLR2 and TLR4 and that the NF‐κB p65 pathway is involved in this process.
Sepsis‐induced muscle atrophy is reduced by BMS‐345541 treatment in vivo
Because our data indicated that SAA1 causes myocyte atrophy via the TLR2/TLR4/NF‐κB pathway, we hypothesized that inhibition of the NF‐κB pathway could prevent muscle atrophy in septic mice. To test this hypothesis, we subjected male WT mice to sham or CLP surgery and treated them with either vehicle or BMS‐345541, which were administered by oral gavage.41 BMS‐345541 treatment resulted in highly significant improvement of survival of CLP‐operated compared with vehicle‐treated septic mice (Figure 4A). Sepsis‐induced reduction in tibialis anterior (−25%, P < 0.05), gastrocnemius/plantaris (−49%, P < 0.05), and soleus (−45%, P < 0.05) weights was inhibited by BMS‐345541. Sepsis caused a reduction in heart weight as well (sham + vehicle vs. sham + BMS‐345541: −20.4%, P < 0.05; sham + BMS‐345541 vs. CLP + BMS‐345541: −14.4%, P < 0.05). BMS‐345541 treatment was accompanied by a reduced cardiac weight loss which was not significant (P = 0.3) (Figures 4B and S1). As expected, western blot analysis showed an increased NF‐κB p65 phosphorylation in tibialis anterior of septic compared with sham operated mice, which was attenuated by BMS‐34551 (Figure 4C). Our data show that inhibition of sepsis‐induced NF‐κB activation by BMS‐345541 improves survival and attenuates muscular weight loss during sepsis.
Figure 4.
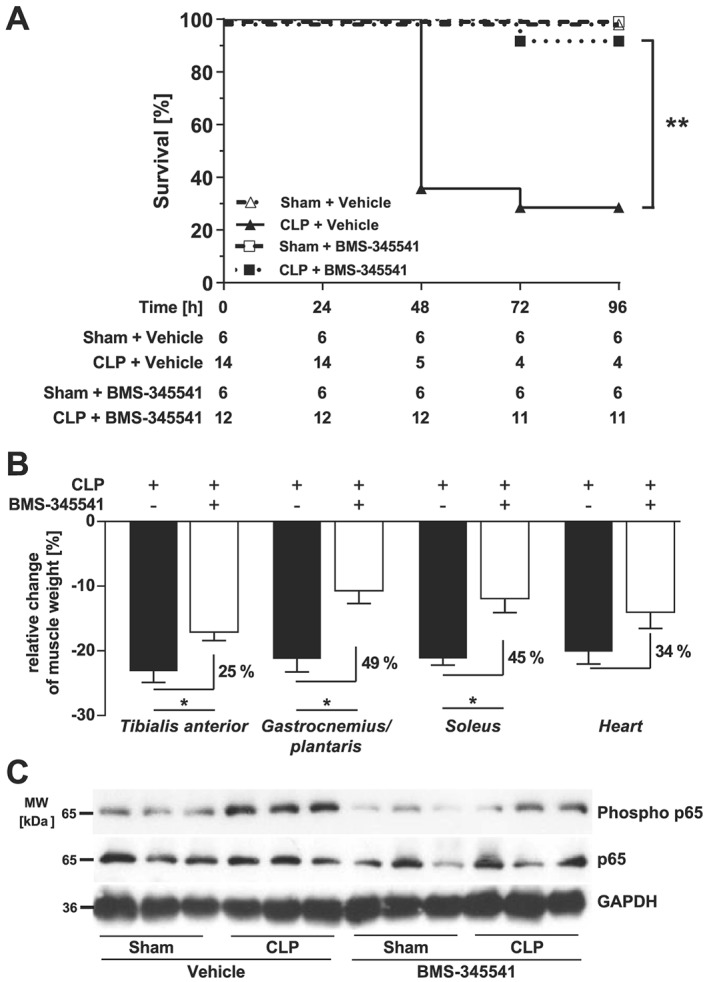
BMS‐345541 treatment increases survival and attenuates muscle atrophy in septic mice in vivo. (A–C) Eighteen‐week‐old male WT mice were either sham or CLP operated as indicated. Sham‐operated and CLP‐operated mice were divided into two groups each which then received either vehicle (5% TWEEN 20 in distilled H2O, pH 7.4) or BMS‐345541 30 min prior to (30 mg/kg body weight) and 6 h after (15 mg/kg body weight) surgery p.o.; experimental groups are as follows: sham + vehicle (n = 6), CLP + vehicle (n = 14), sham + BMS‐345541 (n = 6), and CLP + BMS‐345531 (n = 12). After 96 h, all mice were sacrificed. (A) Survival of all animals. Statistical analysis was performed using the log‐rank test. **P ≤ 0.01. (B) Weights of tibialis anterior, gastrocnemius/plantaris, soleus, and the heart normalized to tibia length. Data are presented as mean ± SEM. *P ≤ 0.05. (C) Western blot analysis of tibialis anterior with anti‐phospho‐p65 and anti‐p65 antibody. GAPDH was used as loading control.
Recently, others and we showed that primarily fast‐twitch fibers undergo atrophy in ICUAW patients and septic mice.12, 34 Therefore, we tested if BMS‐345541 protects fast/type‐II myofibers from atrophy in sepsis. Analyses of tibialis anterior and gastrocnemius/plantaris cross sections and quantification of myocyte cross‐sectional area (MCSA) showed that CLP treated animals had a pronounced atrophy of fast/type‐II myofibers (Figures 5A, 5B, 6A, and 6B). CLP operated mice that were treated with BMS‐345541 showed less myofiber atrophy compared with vehicle‐treated CLP mice as indicated by a significantly lower reduction in MCSA in tibialis anterior and gastrocnemius/plantaris (Figures 5A, 5B, 6A, and 6B). Because the soleus contains both slow/type‐I and fast/type‐II myofibers, we performed histological cross sections of the soleus and quantitated the MCSA for both fiber types following metachromatic ATPase staining. We found a reduction in the MCSA of slow/type‐I (sham + vehicle vs. CLP + vehicle: −17%, P < 0.001; sham + BMS‐345541 vs. CLP + BMS‐345541: −15%, P < 0.001) and fast/type‐II myofibers (sham + vehicle vs. CLP + vehicle: −28%, P < 0.001; sham + BMS‐345541 vs. CLP + BMS‐345541: −20%, P < 0.001). We found a significantly lower MCSA reduction of slow/type‐I (P < 0.001) and fast/type‐II myofibers (P < 0.001) in soleus of BMS‐345541‐treated septic mice (Figure 7A and 7B). Our results show that fast/type‐II myofibers are more susceptible than slow/type‐I myofibers to sepsis‐induced atrophy and that BMS‐345541 treatment protects from sepsis‐induced myofiber atrophy in mice in vivo.
Figure 5.
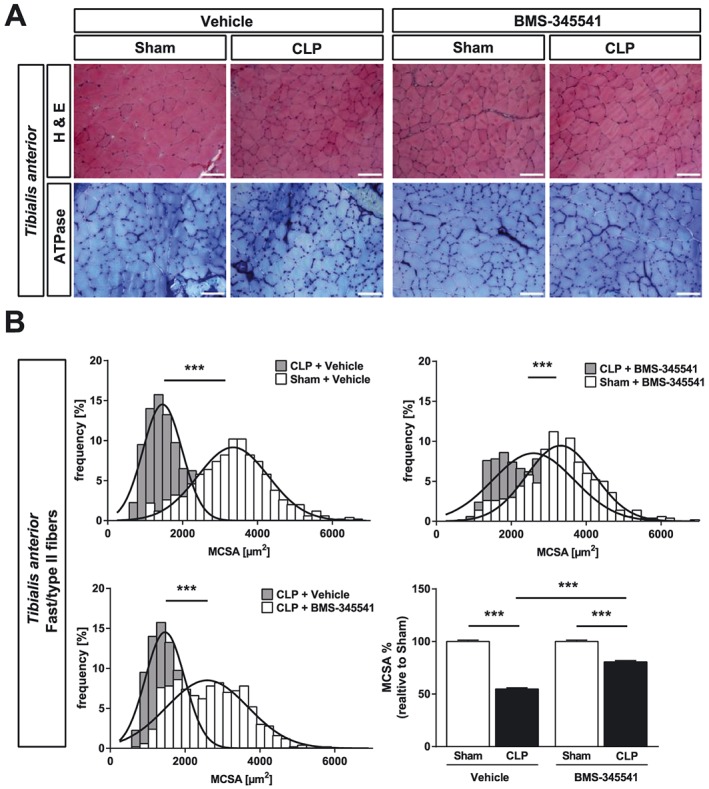
BMS‐345541 treated mice are protected from sepsis‐induced atrophy of the tibialis anterior muscle. Eighteen‐week‐old male WT mice were either sham or CLP operated as indicated. Per surgical group mice were divided into two groups each which than received either vehicle (5% TWEEN 20 in distilled H2O, pH 7.4) or BMS‐345541 30 min prior to (30 mg/kg body weight) and 6 h after (15 mg/kg body weight) surgery p.o.; experimental groups are as follows: sham + vehicle (n = 6), CLP + vehicle (n = 4), sham + BMS‐345541 (n = 6), and CLP + BMS‐345531 (n = 11). After 96 h, the mice were sacrificed. (A) Haematoxylin and eosin (H&E) and ATPase staining of histological cross sections from tibialis anterior (TA). Scale bar = 100 μm. (B) Quantification of fast/type II myofiber cross‐sectional area (MCSA) from metachromatic ATPase staining (as depicted in panel A). One hundred myofibers per animal were measured. ***P ≤ 0.001.
Figure 6.
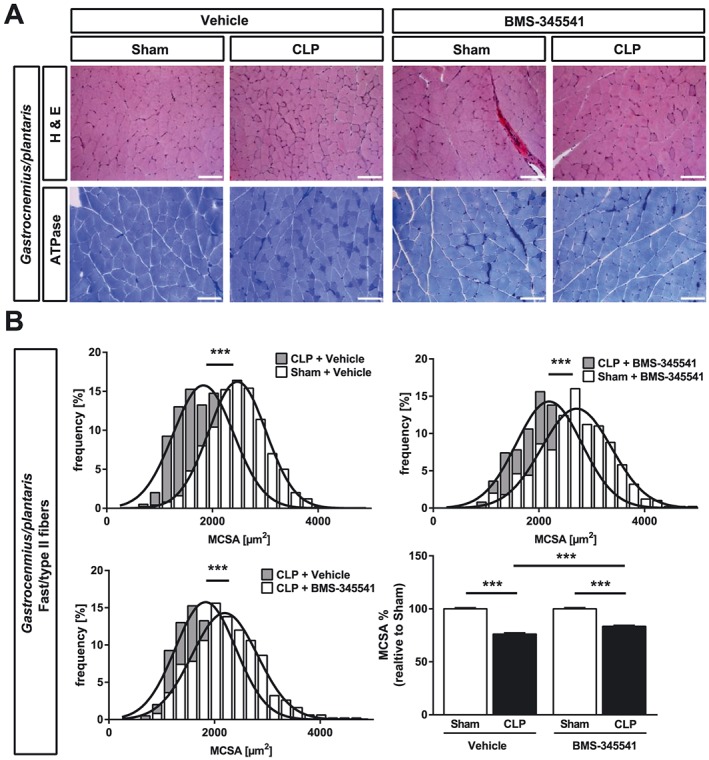
BMS‐345541 treated mice are protected from sepsis‐induced muscle atrophy of the gastrocnemius/plantaris. Eighteen‐week‐old male WT mice were either sham or CLP operated as indicated. Per surgical group, mice were divided into two groups each which then received either vehicle (5% TWEEN 20 in distilled H2O, pH 7.4) or BMS‐345541 30 min prior to (30 mg/kg body weight) and 6 h after (15 mg/kg body weight) surgery p.o.; experimental groups are as follows: sham + vehicle (n = 6), CLP + vehicle (n = 4), sham + BMS‐345541 (n = 6), and CLP + BMS‐345531 (n = 11). After 96 h, the mice were sacrificed. (A) Haematoxylin and eosin (H&E) and ATPase stain of histological cross sections from gastrocnemius/plantaris (GP). Scale bar = 100 μm. (B) Quantification of fast/type II myofiber cross‐sectional area (MCSA) from metachromatic ATPase staining (as depicted in panel A). One hundred myofibers per animal were measured. ***P ≤ 0.001.
Figure 7.
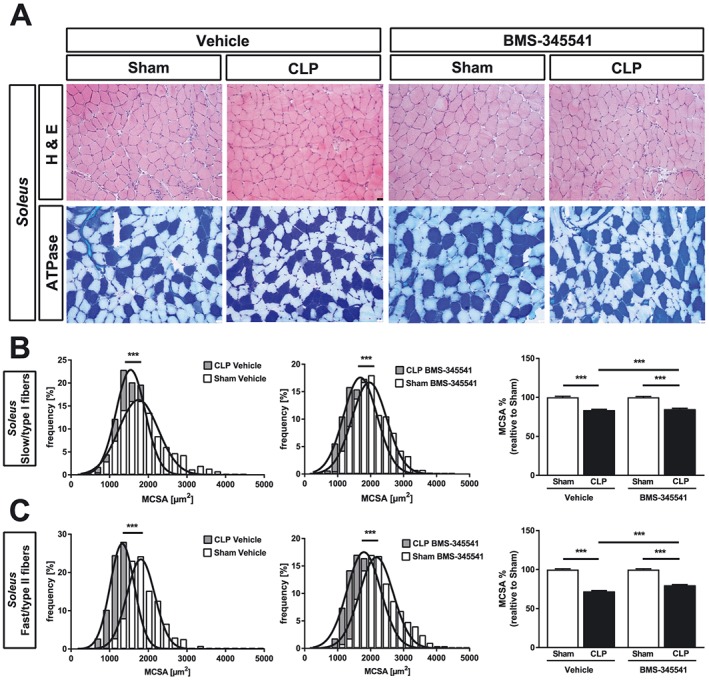
BMS‐345541 treated mice are protected from sepsis‐induced muscle atrophy of the soleus. Eighteen‐week‐old male WT mice were either sham or CLP operated as indicated. Per surgical group, mice were divided into two groups each which then received either vehicle (5% TWEEN 20 in distilled H2O, pH 7.4) or BMS‐345541 30 min prior to (30 mg/kg body weight) and 6 h after (15 mg/kg body weight) surgery p.o.; experimental groups are as follows: sham + vehicle (n = 6), CLP + vehicle (n = 4), sham + BMS‐345541 (n = 6), and CLP + BMS‐345531 (n = 11). After 96 h, the mice were sacrificed. (A) Haematoxylin and eosin (H&E) and ATPase stain of histological cross sections from soleus. Scale bar = 100 μm. (B) Quantification of slow/type I and fast/type II myofiber cross‐sectional area (MCSA) from metachromatic ATPase staining (as depicted in panel A). One hundred myofibers per animal were measured. ***P ≤ 0.001.
We had also observed that an increased protein degradation and a decreased protein synthesis contributes to muscle atrophy in ICUAW patients and septic mice.12 To test if the inhibition of septic muscle atrophy by BMS‐345541 was accompanied by an improved protein homeostasis, we quantitated Myh4 and Myh7 expression in tibialis anterior by qRT‐PCR. Sepsis led to a decreased Myh4 expression (sham + vehicle vs. CLP + vehicle: −56%, P < 0.01; sham + BMS‐345541 vs. CLP + BMS‐345541: −15%, n.s.), which was attenuated by 34% by BMS‐345541 (P < 0.05). Myh7 expression followed the same trend that did not reach significance (Figure 8A). To investigate the effect of BMS‐345541 treatment on protein degradation, we quantified the expression of Fbxo30, Fbxo32, and Trim63 in tibialis anterior and gastrocnemius/plantaris. CLP significantly increased Fbxo32 and Trim63 expression, and BMS‐345541 treatment attenuated this response in both muscles. Fbxo30 was also significantly increased in tibialis anterior and gastrocnemius/plantaris of vehicle‐treated septic mice but remained unchanged in BMS‐345541 treated animals (Figure 8B). The Atrogin1/Fbxo32 and MuRF1/Trim63 protein content increased during sepsis in tibialis anterior and BMS‐345541 attenuated this response (Figure 8C). Our data indicate that the attenuation of sepsis‐induced atrophy by BMS‐345541 involved an improved protein homeostasis.
Figure 8.
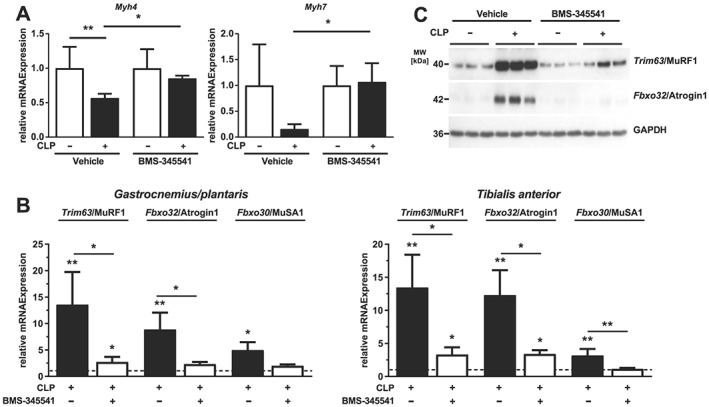
BMS‐345541 treatment attenuates sepsis‐induced protein degradation. Eighteen‐week‐old male WT mice were Sham or CLP operated. Mice received vehicle or BMS‐345541 (30 mg/kg) 30 min prior surgery and 6 h later vehicle or BMS‐345541 (15 mg/kg) by oral gavage. After 96 h, the mice were sacrificed. (A and B) Sham + vehicle (n = 6), CLP + vehicle (n = 4), sham + BMS‐345541 (n = 6), and CLP + BMS‐345531 (n = 11). (A) qRT‐PCR analysis of myosin heavy chain (Myh) 4 and Myh7 expression in tibialis anterior muscle. mRNA expression was normalized to Gapdh. (B) qRT‐PCR analysis of Trim63/MuRF1, Fbxo32/Atrogin1, and Fbxo30/MuSA1 expression in tibialis anterior and gastrocnemius/plantaris muscle. mRNA expression was normalized to Gapdh. Data are presented as fold change ± SEM. *P ≤ 0.05 and **P ≤ 0.01. (C) Western blot analysis of proteins isolated from tibialis anterior with anti‐MuRF1 and anti‐Atrogin1 antibody. GAPDH was used as loading control.
Increased NF‐κB activity plays a pivotal role for the regulation of immune cell function during a bacterial insult.55, 56 Indeed, immune cell activation is important for bacterial clearance in sepsis.42, 57 To investigate if BMS‐345541 treatment affected immune cells, we performed flow cytometric analyses of splenocytes. Sepsis caused an increase in spleen weight, which was unaffected by BMS‐345541 (Figure S2). The total numbers and proportions of dendritic cells (CD11chiMHCII+), B‐cells (CD19+B220+), and natural killer cells (NK, NK1.1+) decreased (Figure S3A–S3C), while neutrophil numbers (Ly6G+) increased in septic mice (Figure S3D). We also observed a significant reduction in the proportions of CD3+ T‐cells, which was due to decreased percentages of CD4+ helper T‐cells in the spleens of septic mice, whereas CD8+ cytotoxic T‐cells remained unchanged (Figure S4A–S4C). Finally, percentages of Foxp3+CD25+CD4+ regulatory T‐cells were increased, and Foxp3‐/CD4+ effective T‐cells were decreased in the spleen of septic mice, and both T‐cell subpopulations were activated, as shown by the increased CD69 expression. In the case of vehicle‐treated animals, this increase did not reach significance (Figure S5A–S5C). Notably, BMS‐345541 treatment had no significant effect on sepsis‐induced alterations of numbers, proportions, and activation of splenic immune cells, indicating that its strong impact on survival and muscle atrophy were independent of the immune cell responses to sepsis.
Discussion
Serum amyloid A1 (SAA1) accumulation in muscle is associated with ICUAW.11 However, neither the putative receptors for SAA1 nor the involved pathways have been described in myocytes or muscle. We identified Toll‐like receptors 2 and 4 as SAA1 receptors on myocytes in vitro. We show that SAA1 via TLR2 and TLR4 activates the NF‐κB p65 pathway that mediates the SAA1‐response of myocytes. Specifically, we demonstrate that SAA1‐induced myocyte atrophy is attenuated by blocking either TLR2 or TLR4 or by inhibition of the NF‐κB p65 pathway with BMS‐345541 in vitro. We show that inhibition of NF‐κB activity by BMS‐345541 increased survival and reduced muscle atrophy in a mouse model of polymicrobial sepsis in vivo. Our findings suggest that inhibition of NF‐κB during sepsis may increase survival and attenuate SAA1‐induced muscle atrophy.
Although the function of SAA1 is well described for liver and immune cells,58 any role in muscle is less well understood. For example, SAA1 can bind to several receptors, such as TLR2, TLR4, CD36, P2RX7, VIMP, and SCARB1, which are expressed on hepatocytes, keratinocytes, fibroblasts, and macrophages.25, 26, 27, 28, 30, 31, 33, 59 Whether or not these receptors are involved in SAA1 signalling in myocytes was uncertain. Here, we show that TLR2, TLR4, CD36, P2RX7, VIMP, and SCARB1 are also expressed in myocytes and muscle and that muscular expression of TLR2, TLR4, CD36, and VIMP increases during sepsis. We found that SAA1 causes myocyte atrophy in vitro, which was mediated by TLR2 and TLR4 as well as NF‐κB. Together with our recent report showing that SAA1 expression, synthesis, and secretion increase in muscle of ICUAW patients and septic mice, our data indicate that muscle is targeted by SAA1 in a paracrine manner and that TLR2, TLR4, and NF‐κB are involved in SAA1‐induced atrophy.
TLR2 was shown to mediate SAA1‐induced Il6 expression in fibroblasts33 and to mediate Il6 expression and atrophy in myocytes.8 Activation of TLR2 and TLR4 by specific agonists were found to mediate IL6 secretion from C2C12 myotubes.60 Recently, we reported that SAA1 increases Il6 expression in murine and human myocytes in vitro.11 In addition, IL6 expression is increased in muscle of ICUAW patients and septic mice.11, 12 We found that muscular TLR2 and TLR4 expression is increased during sepsis in patients and mice, that SAA1 increases Tlr2 and Tlr4 expression in myotubes, and that SAA1 via TLR2 and TLR4 increases Il6 expression in myocytes. Based on these data, we hypothesize that during sepsis, SAA1 participates in a positive feedback loop of self‐perpetuating inflammation in muscle not only by increasing IL6 expression but also by increasing the expression of its own receptors, which further augments IL6 expression. Moreover, we showed that SAA1 induces IL6 expression in myocytes and vice versa, which further supports a self‐augmenting positive feedback loop in muscle during inflammation. Considering that SAA1 accumulation correlates with muscle atrophy in ICUAW patients, these feedback mechanisms could worsen IL6‐mediated atrophy.
Because LPS elevates NF‐κB‐dependent gene expression via TLR4,61, 62 we investigated whether SAA1 activates this pathway in myocytes. We show that SAA1 activates the canonical NF‐κB p65 signalling pathway via TLR2 and TLR4 resulting in an increased expression of NF‐κB p65 target genes and myocyte atrophy. For our analyses, we used the highly selective IKK inhibitor BMS‐345541. IKK phosphorylates IκBα and NF‐κB p65, leading to the proteasomal degradation of IκBα and release of active NF‐κB.63 Indeed, BMS‐345541 decreased SAA1‐induced NF‐κB p65 phosphorylation, inhibited formation of NF‐κB‐DNA complexes, and attenuated SAA1‐induced Il6 expression in myotubes as well as NF‐κB p65 phosphorylation in muscle of septic mice. These data indicate that BMS‐345541 effectively inhibited NF‐κB activity in myocytes. In addition, BMS‐345541 attenuated myotube atrophy as well as the SAA1‐induced molecular changes consistently described as atrophic response with an increase in MuRF1 and a decrease in MyHC.34, 48, 64, 65, 66 Our results indicate that SAA1‐induced atrophy was mediated by NF‐κB. Of note, NF‐κB p65 was shown to mediate denervation and tumour‐induced muscle atrophy,52, 53 and its inhibition prevented atrophy in a mouse model of acute lung injury.54 Our data corroborate and extend these findings (i) by demonstrating that NF‐κB has an important role in sepsis‐induced atrophy as well and (ii) by identifying SAA1 as a facilitator of the NF‐κB p65 pathway, which mediates most of its effects on myocytes in vitro.
To investigate if NF‐κB inhibition prevents sepsis‐induced muscle atrophy in vivo, we tested BMS‐34554141 in the CLP mouse model. The expression of many pro‐inflammatory genes is regulated by NF‐κB. NF‐κB normally resides in the cytoplasm of unstimulated cells as an inactive complex with a member of the IκB inhibitory protein family. This class of protein includes IκBα, IκBβ, and IκBε, which all form a complex with NF‐κB (for a review, see Whiteside and Israel67). Stimulation of cells with compounds that activate NF‐κB‐dependent gene expression results in the phosphorylation of IκBα at Ser‐32 and Ser‐36.68 This is critical for subsequent ubiquitination and proteolysis of IκBα, which then leaves NF‐κB free to translocate to the nucleus and promote gene expression.69 Analogous serines have been identified in both IκBβ and IκBε, and phosphorylation at these residues appears to regulate the proteolytic degradation of these proteins by a mechanism similar to that of IκBα.70 Phosphorylation of IκBα at Ser‐32 and Ser‐36 is mediated by a high molecular mass (500–900 kDa) multisubunit IκB kinase (termed IKK).71 Two catalytic subunits (termed IKK‐1 and IKK‐2) of IKK have recently been identified.72 Several working groups have shown that all known pro‐inflammatory stimuli, including cytokines, viruses, and LPS, require IKK for NF‐κB activation (for a review, see Ghosh and Karin73). Previously, BMS‐345541, a highly selective cell permeable IKK‐2 and IKK‐1 inhibitor that inhibits NF‐κB‐dependent transcription of pro‐inflammatory cytokines both in vitro and in vivo was identified. Subsequently, BMS‐345541 was shown to attenuate LPS‐induced production of TNF, IL1β, IL8, and IL6 in THP‐1 cells.41 BMS‐345541 was also shown to abolish TNF‐stimulated IκBα‐phosphorylation in THP‐1 cells41 and the prostate cancer cell line PC‐3.46 BMS‐345541 was previously shown to reduce CLP and LPS‐induced lung injury,42, 43 to reduce inflammation in rats with spinal cord injury, and to suppress inflammation in a model of cardiac graft rejection.44, 45 Due to its high bioavailability41 and its anti‐inflammatory characteristics without impairing immune cell activation42 and bacterial clearance, BMS‐345541 was well suited for our studies. Indeed, we show that NF‐κB p65 was activated in muscle of septic mice, which is in accordance with previously published work demonstrating NF‐κB activation in muscle of CLP operated rats.74 Next, we confirmed that BMS‐345541 prevented activation of NF‐κB p65 in muscle of septic mice, which is in line with a recent report where BMS‐345541 was shown to inhibit LPS‐induced NF‐κB‐dependent gene expression in mice41 and proves its biological activity. Interestingly, BMS‐345541 treatment resulted in a 63% percent higher survival rate of septic mice. Because increased NF‐κB activity in peripheral blood mononuclear cells predicted fatal outcome in septic patients and inhibition of the IKK‐NF‐κB axis was shown to increase survival in an LPS‐induced toxic shock model in mice,75 this result was somewhat expected. Importantly, BMS‐345541 reduced the degree of sepsis‐induced muscle atrophy. Accordingly, sepsis‐induced reduction in Myh4 expression and induction of Trim63/MuRF1 and Fbxo32/Atrogin1 expression was reversed by BMS‐345541. MuRF1 mediates proteasomal degradation of muscular proteins, like MyHC2 and 4,64 and as an atrophy marker is increased in muscle of septic patients and mice.34, 37 A previous report showed that mice overexpressing activated IKK2 in muscle, which increases NF‐κB activity, displayed increased MuRF1 expression and aggravated muscle atrophy. When these mice were crossed to MuRF1 knockout mice, atrophy was greatly reduced.52 Together with our report, these data suggest that NF‐κB signalling contributes to sepsis‐induced muscle atrophy, which involves MuRF1‐dependent protein degradation.
Our data implicate that inhibition of NF‐κB by BMS‐345541 could be effective to decrease muscle wasting in sepsis. Because NF‐κB is a key regulator of inflammatory responses and involved in other forms of muscle wasting,52, 53, 76 we hypothesize that BMS‐345541 could be useful to prevent or treat other forms of muscle wasting as well. For example, end‐stage cancer patients often suffer from cachexia, which is accompanied with a poor prognosis. Among the best studied inflammatory cytokines promoting cancer cachexia are TNF, IL6,77 IL1, and interferon gamma,78 which are all elevated79 and may together trigger muscle wasting by increasing NF‐κB activity.80, 81, 82 For instance, TNF promotes anorexia83 and skeletal muscle wasting mainly through activation of the NF‐κB pathway.84 However, the precise mechanism how NF‐κB mediates muscle wasting in cancer is less well understood. One explanation is that NF‐κB signalling and UPS‐mediated protein degradation are closely connected to each other. On one side, NF‐κB increases the expression of atrogenes and proteasome‐mediated protein degradation in muscle. On the other side, proteasome inhibitors interfere with the NF‐κB pathway because they inhibit IκB degradation, which in turn prevents NF‐κB activation.85 Accordingly, an increased Fbxo32/Atrogin1 gene expression86 and protein ubiquitination86 was associated with an activated UPS in a rat model of cachexia, induced by the Yoshida ascites hepatoma cells. In tumour‐bearing mice, this UPS activation was mediated by an increased NF‐κB activity, which stimulated Trim63/MuRF1 expression and muscle wasting.52 These data implicate that NF‐κB inhibition could be useful to prevent cancer cachexia. Indeed, synthetic double‐stranded oligodeoxynucleotides, which block NF‐κB binding to promoter regions, inhibited cachexia in a mouse tumour model.87 However, if BMS‐3445541 is useful to prevent muscle wasting in cancer needs to be shown in the future. Furthermore, because NF‐κB activation is important for denervation‐induced muscle wasting, many approaches were undertaken to reduce its activity to attenuate this phenotype. For example, misexpression of IκB that was used to inhibit NF‐κB activity reduced denervation‐induced muscle wasting in tumour‐bearing mice.52 Denervation‐induced atrophy was also attenuated in mice lacking IKKβ.53 Inhibition of NF‐κB by BMS‐3445541 could therefore be useful to prevent denervation‐induced muscle wasting as well.
Finally, and in agreement with previous reports,42 BMS‐345541 did not affect the immune cell responses to sepsis.42 In summary, our data indicate that inhibition of the IKK‐NF‐κB pathway holds promise to prevent both mortality and muscle atrophy in sepsis.
Conclusions
During sepsis, SAA1 is increased in muscle and directly acts on myocytes via the TLR2/TLR4/NF‐κB p65 axis. Blockade of SAA1‐mediated NF‐κB activation by BMS‐345541 attenuated atrophy and inhibited the SAA1‐IL6 feedback loop in myocytes in vitro. Likewise, BMS‐345541 increased survival and attenuated skeletal muscle atrophy in septic mice (Figure 9). We suggest that monitoring SAA1 and inhibition of the SAA1/TLR2/TLR4/NF‐κB atrophy pathway could ameliorate ICUAW. Further studies are needed to determine how the SAA1 pathway is mechanistically connected to other atrophy pathways such as MuRF1 and Atrogin1 that we found to be affected.
Figure 9.
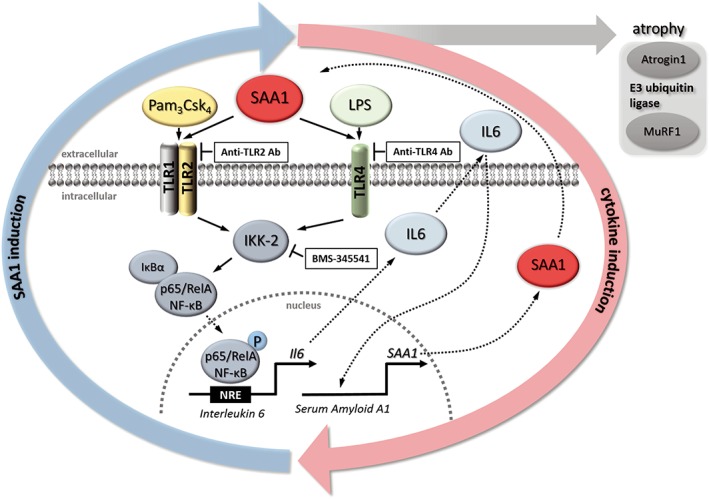
Proposed mechanism of serum amyloid A 1 (SAA1) induced muscle atrophy. SAA1 binds to muscular Toll‐like receptors (TLR) 2 and 4, which results in an activation of the canonical NF‐κB p65 pathway leading to NF‐κB p65/RelA phosphorylation and its translocation into the nucleus. In the nucleus, NF‐κB p65/RelA binds to NF‐κB response elements (NRE), which leads to increased expression and secretion of the pro‐inflammatory cytokine Il6. IL6 in turn induces expression and secretion of SAA1 resulting in a positive feedback loop. In addition, SAA1 causes an increase of MuRF1/Trim63 and Atrogin1/Fbxo32, which mediate muscle atrophy in sepsis. Anti‐TLR2 or anti‐TLR4 antibodies inhibit SAA1‐induced Il6 expression. The specific IκB kinase subunit IKK‐2 inhibitor BMS‐345541 inhibits SAA1‐induced and NF‐κB dependent gene expression, thereby reducing SAA1‐mediated atrophy and inhibiting the self‐augmenting SAA1‐IL6 feedback loop.
Conflict of interest
A.H., M.K., C.P.‐T., M.T., M.W., S.S., K.S., I.J., M.N., E.J., T.S., B.M.B., C.S., S.W.‐C., C.B., and F.C.L. declare that they have no conflict of interest. J.F. has received research grants from the Deutsche Forschungsgemeinschaft. J.F. and S.B.F. have received research grants from the German Center for Cardiovascular Research.
Authors' contributions
A.H., M.K., C.P.T., M.T., M.W., S.S., K.S., I.J., M.N., E.J. and T.S. designed, performed, and analysed experiments and discussed data. A.H., M.K., and J.F. prepared and edited the manuscript. B.B., S.B.F., C.S., S.W.‐C., C.B., F.C.L., and J.F. designed experiments, discussed data, and edited the manuscript. All authors declare that the submitted work has not been published before (neither in English nor in any other language) and that the work is not under consideration for publication elsewhere.
Declarations
Ethics approval and consent to participate
The institutional review board of the Charité approved the study, and written informed consent was obtained from legal proxy (ICU patients), or the patients themselves (Charité EA2/061/06; http://www.controlled-trials.com, ISRCTN77569430).
The Landesamt für Gesundheit und Soziales, Berlin, Germany (G207/13, G129/12), approved the animal studies (NIH publication No. 86‐23, revised 1985).
Supporting information
Data S1. Supporting Information
Figure S1. Supporting Information
Figure S2. Supporting Information
Figure S3. Supporting Information
Figure S4. Supporting Information
Acknowledgements
We thank Sebastian Wundersitz, Lukas Zanders, Elisa Martin, and Benjamin Sünkel for insightful discussion and excellent technical assistance. This study was supported by the Deutsche Forschungsgemeinschaft [FI 965/4‐1, FI 965/5‐1, and FI 965/5‐2 (to J.F.)] and the German Center for Cardiovascular Research, Partner Site Greifswald [DZHK 81Z5400153 (to S.B.F. and J.F.)]. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle.88
Hahn A., Kny M., Pablo‐Tortola C., Todiras M., Willenbrock M., Schmidt S., Schmoeckel K., Jorde I., Nowak M., Jarosch E., Sommer T., Bröker B. M., Felix S. B., Scheidereit C., Weber‐Carstens S., Butter C., Luft F. C., and Fielitz J. (2020) Serum amyloid A1 mediates myotube atrophy via Toll‐like receptors, Journal of Cachexia, Sarcopenia and Muscle, 11: 103–119. 10.1002/jcsm.12491.
References
- 1. Latronico N, Bolton CF. Critical illness polyneuropathy and myopathy: a major cause of muscle weakness and paralysis. Lancet Neurol 2011;10:931–941. [DOI] [PubMed] [Google Scholar]
- 2. Ali NA, O'Brien JM Jr, Hoffmann SP, Phillips G, Garland A, Finley JC, et al. Acquired weakness, handgrip strength, and mortality in critically ill patients. Am J Respir Crit Care Med 2008;178:261–268. [DOI] [PubMed] [Google Scholar]
- 3. Tennila A, Salmi T, Pettila V, Roine RO, Varpula T, Takkunen O. Early signs of critical illness polyneuropathy in ICU patients with systemic inflammatory response syndrome or sepsis. Intensive Care Med 2000;26:1360–1363. [DOI] [PubMed] [Google Scholar]
- 4. Herridge MS, Tansey CM, Matté A, Tomlinson G, Diaz‐Granados N, Cooper A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 2011;364:1293–1304. [DOI] [PubMed] [Google Scholar]
- 5. De Jonghe B, Sharshar T, Lefaucheur J, Authier FJ, Durand‐Zaleski I, Boussarsar M, et al. Paresis acquired in the intensive care unit: a prospective multicenter study. JAMA 2002;288:2859–2867. [DOI] [PubMed] [Google Scholar]
- 6. Weber‐Carstens S, Deja M, Koch S, Spranger J, Bubser F, Wernecke KD, et al. Risk factors in critical illness myopathy during the early course of critical illness: a prospective observational study. Crit Care 2010;14:R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winkelman C. The role of inflammation in ICU‐acquired weakness. Crit Care 2010;14:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Passey SL, Bozinovski S, Vlahos R, Anderson GP, Hansen MJ. Serum amyloid A induces toll‐like receptor 2‐dependent inflammatory cytokine expression and atrophy in C2C12 skeletal muscle myotubes. PLoS ONE 2016;11:e0146882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, et al. IL‐6 and serum amyloid A synergy mediates angiotensin II‐induced muscle wasting. J Am Soc Nephrol 2009;20:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haddad F, Zaldivar F, Cooper DM, Adams GR. IL‐6‐induced skeletal muscle atrophy. J Appl Physiol (1985) 2005;98:911–917. [DOI] [PubMed] [Google Scholar]
- 11. Langhans C, Weber‐Carstens S, Schmidt F, Hamati J, Kny M, Zhu X, et al. Inflammation‐induced acute phase response in skeletal muscle and critical illness myopathy. PLoS ONE 2014;9:e92048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang N, Kny M, Riediger F, Busch K, Schmidt S, Luft FC, et al. Deletion of Nlrp3 protects from inflammation‐induced skeletal muscle atrophy. Intensive Care Med Exp 2017;5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sack GH Jr. Serum amyloid A—a review. Mol Med (Cambridge, Mass) 2018;24:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute‐phase reactant. Eur J Biochem 1999;265:501–523. [DOI] [PubMed] [Google Scholar]
- 15. Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, et al. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J Clin Invest 1996;97:244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adams V, Mangner N, Gasch A, Krohne C, Gielen S, Hirner S, et al. Induction of MuRF1 is essential for TNF‐alpha‐induced loss of muscle function in mice. J Mol Biol 2008;384:48–59. [DOI] [PubMed] [Google Scholar]
- 17. Zamir O, Hasselgren PO, Kunkel SL, Frederick J, Higashiguchi T, Fischer JE. Evidence that tumor necrosis factor participates in the regulation of muscle proteolysis during sepsis. Arch Surg (Chicago, Ill: 1960) 1992;127:170–174. [DOI] [PubMed] [Google Scholar]
- 18. Goodman MN. Interleukin‐6 induces skeletal muscle protein breakdown in rats. Proc Soc Exp Biol Med Soc Exp Biol Med (New York, NY) 1994;205:182–185. [DOI] [PubMed] [Google Scholar]
- 19. Goodman MN. Tumor necrosis factor induces skeletal muscle protein breakdown in rats. Am J Physiol 1991;260:E727–E730. [DOI] [PubMed] [Google Scholar]
- 20. Garcia‐Obregon S, Azkargorta M, Seijas I, Pilar‐Orive J, Borrego F, Elortza F, et al. Identification of a panel of serum protein markers in early stage of sepsis and its validation in a cohort of patients. J Microbiol Immunol Infect 2018;51:465–472. [DOI] [PubMed] [Google Scholar]
- 21. Gutfeld O, Prus D, Ackerman Z, Dishon S, Linke RP, Levin M, et al. Expression of serum amyloid A, in normal, dysplastic, and neoplastic human colonic mucosa: implication for a role in colonic tumorigenesis. J Histochem Cytochem 2006;54:63–73. [DOI] [PubMed] [Google Scholar]
- 22. Urieli‐Shoval S, Finci‐Yeheskel Z, Dishon S, Galinsky D, Linke RP, Ariel I, et al. Expression of serum amyloid a in human ovarian epithelial tumors: implication for a role in ovarian tumorigenesis. J Histochem Cytochem 2010;58:1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cocco E, Bellone S, El‐Sahwi K, Cargnelutti M, Casagrande F, Buza N, et al. Serum amyloid A (SAA): a novel biomarker for uterine serous papillary cancer. Br J Cancer 2009;101:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 2011;6:e22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Niemi K, Teirila L, Lappalainen J, Rajamaki K, Baumann MH, Oorni K, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B‐sensitive pathway. Journal of immunology (Baltimore, Md: 1950). 2011;186(11):6119‐28. [DOI] [PubMed]
- 26. Baranova IN, Bocharov AV, Vishnyakova TG, Kurlander R, Chen Z, Fu D, et al. CD36 is a novel serum amyloid A (SAA) receptor mediating SAA binding and SAA‐induced signaling in human and rodent cells. J Biol Chem 2010;285:8492–8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cai L, de Beer MC, de Beer FC, van der Westhuyzen DR. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J Biol Chem 2005;280:2954–2961. [DOI] [PubMed] [Google Scholar]
- 28. Walder K, Kantham L, McMillan JS, Trevaskis J, Kerr L, De Silva A, et al. Tanis: a link between type 2 diabetes and inflammation? Diabetes 2002;51:1859–1866. [DOI] [PubMed] [Google Scholar]
- 29. Chen M, Zhou H, Cheng N, Qian F, Ye RD. Serum amyloid A1 isoforms display different efficacy at Toll‐like receptor 2 and formyl peptide receptor 2. Immunobiology 2014;219:916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute‐phase serum amyloid A. J Immunol (Baltimore, Md: 1950) 2008;181:22–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol 2008;83:1174–1180. [DOI] [PubMed] [Google Scholar]
- 32. Chen ES, Song Z, Willett MH, Heine S, Yung RC, Liu MC, et al. Serum amyloid A regulates granulomatous inflammation in sarcoidosis through Toll‐like receptor‐2. Am J Respir Crit Care Med 2010;181:360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. O'Reilly S, Cant R, Ciechomska M, Finnigan J, Oakley F, Hambleton S, et al. Serum amyloid A induces interleukin‐6 in dermal fibroblasts via Toll‐like receptor 2, interleukin‐1 receptor‐associated kinase 4 and nuclear factor‐kappaB. Immunology 2014;143:331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wollersheim T, Woehlecke J, Krebs M, Hamati J, Lodka D, Luther‐Schroeder A, et al. Dynamics of myosin degradation in intensive care unit‐acquired weakness during severe critical illness. Intensive Care Med 2014;40:528–538. [DOI] [PubMed] [Google Scholar]
- 35. Wollersheim T, Grunow JJ, Carbon NM, Haas K, Malleike J, Ramme SF, et al. Muscle wasting and function after muscle activation and early protocol‐based physiotherapy: an explorative trial. J Cachexia Sarcopenia Muscle 2019; 10.1002/jcsm.12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rittirsch D, Huber‐Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 2009;4:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schmidt F, Kny M, Zhu X, Wollersheim T, Persicke K, Langhans C, et al. The E3 ubiquitin ligase TRIM62 and inflammation‐induced skeletal muscle atrophy. Crit Care 2014;18:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Du Bois P, Pablo Tortola C, Lodka D, Kny M, Schmidt F, Song K, et al. Angiotensin II induces skeletal muscle atrophy by activating TFEB‐mediated MuRF1 expression. Circ Res 2015;117:424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lodka D, Pahuja A, Geers‐Knorr C, Scheibe RJ, Nowak M, Hamati J, et al. Muscle RING‐finger 2 and 3 maintain striated‐muscle structure and function. J Cachexia Sarcopenia Muscle 2016;7:165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu X, Kny M, Schmidt F, Hahn A, Wollersheim T, Kleber C, et al. Secreted frizzled‐related protein 2 and inflammation‐induced skeletal muscle atrophy. Crit Care Med 2017;45:e169–e183. [DOI] [PubMed] [Google Scholar]
- 41. Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, et al. BMS‐345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF‐kappa B‐dependent transcription in mice. J Biol Chem 2003;278:1450–1456. [DOI] [PubMed] [Google Scholar]
- 42. Li H, Han W, Polosukhin V, Yull FE, Segal BH, Xie CM, et al. NF‐kappaB inhibition after cecal ligation and puncture reduces sepsis‐associated lung injury without altering bacterial host defense. Mediators Inflamm 2013;2013:503213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Everhart MB, Han W, Sherrill TP, Arutiunov M, Polosukhin VV, Burke JR, et al. Duration and intensity of NF‐kappaB activity determine the severity of endotoxin‐induced acute lung injury. J Immunol (Baltimore, Md: 1950) 2006;176:4995–5005. [DOI] [PubMed] [Google Scholar]
- 44. Han X, Lu M, Wang S, Lv D, Liu H. Targeting IKK/NF‐kappaB pathway reduces infiltration of inflammatory cells and apoptosis after spinal cord injury in rats. Neurosci Lett 2012;511:28–32. [DOI] [PubMed] [Google Scholar]
- 45. Townsend RM, Postelnek J, Susulic V, McIntyre KW, Shuster DJ, Qiu Y, et al. A highly selective inhibitor of IkappaB kinase, BMS‐345541, augments graft survival mediated by suboptimal immunosuppression in a murine model of cardiac graft rejection. Transplantation 2004;77:1090–1094. [DOI] [PubMed] [Google Scholar]
- 46. Ping H, Yang F, Wang M, Niu Y, Xing N. IKK inhibitor suppresses epithelial‐mesenchymal transition and induces cell death in prostate cancer. Oncol Rep 2016;36:1658–1664. [DOI] [PubMed] [Google Scholar]
- 47. Nowak M, Suenkel B, Porras P, Migotti R, Schmidt F, Kny M, et al. DCAF8, a novel MuRF1 interaction partner, promotes muscle atrophy. J Cell Sci 2019; jcs.233395doi: 10.1242/jcs.233395 Published 7 August 2019. [DOI] [PubMed] [Google Scholar]
- 48. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science (New York, NY) 2001;294:1704–1708. [DOI] [PubMed] [Google Scholar]
- 49. Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, et al. BMP signaling controls muscle mass. Nat Genet 2013;45:1309–1318. [DOI] [PubMed] [Google Scholar]
- 50. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88‐deficient mice to endotoxin. Immunity 1999;11:115–122. [DOI] [PubMed] [Google Scholar]
- 51. Shimizu T, Kida Y, Kuwano K. A dipalmitoylated lipoprotein from Mycoplasma pneumoniae activates NF‐kappa B through TLR1, TLR2, and TLR6. J Immunol (Baltimore, Md: 1950) 2005;175:4641–4646. [DOI] [PubMed] [Google Scholar]
- 52. Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, et al. IKKbeta/NF‐kappaB activation causes severe muscle wasting in mice. Cell 2004;119:285–298. [DOI] [PubMed] [Google Scholar]
- 53. Mourkioti F, Kratsios P, Luedde T, Song YH, Delafontaine P, Adami R, et al. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest 2006;116:2945–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Langen RC, Haegens A, Vernooy JH, Wouters EF, de Winther MP, Carlsen H, et al. NF‐kappaB activation is required for the transition of pulmonary inflammation to muscle atrophy. Am J Respir Cell Mol Biol 2012;47:288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu SF, Malik AB. NF‐kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol 2006;290:L622–l45. [DOI] [PubMed] [Google Scholar]
- 56. Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF‐kappa B leads to multifocal defects in immune responses. Cell 1995;80:321–330. [DOI] [PubMed] [Google Scholar]
- 57. Craciun FL, Schuller ER, Remick DG. Early enhanced local neutrophil recruitment in peritonitis‐induced sepsis improves bacterial clearance and survival. J Immunol (Baltimore, Md: 1950) 2010;185:6930–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Eklund KK, Niemi K, Kovanen PT. Immune functions of serum amyloid A. Crit Rev Immunol 2012;32:335–348. [DOI] [PubMed] [Google Scholar]
- 59. Yu N, Liu S, Yi X, Zhang S, Ding Y. Serum amyloid A induces interleukin‐1beta secretion from keratinocytes via the NACHT, LRR and PYD domains‐containing protein 3 inflammasome. Clin Exp Immunol 2015;179:344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frost RA, Nystrom GJ, Lang CH. Multiple Toll‐like receptor ligands induce an IL‐6 transcriptional response in skeletal myocytes. Am J Physiol Regul Integr Comp Physiol 2006;290:R773–R784. [DOI] [PubMed] [Google Scholar]
- 61. Verzola D, Bonanni A, Sofia A, Montecucco F, D'Amato E, Cademartori V, et al. Toll‐like receptor 4 signalling mediates inflammation in skeletal muscle of patients with chronic kidney disease. J Cachexia Sarcopenia Muscle 2017;8:131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ono Y, Sakamoto K. Lipopolysaccharide inhibits myogenic differentiation of C2C12 myoblasts through the Toll‐like receptor 4‐nuclear factor‐kappaB signaling pathway and myoblast‐derived tumor necrosis factor‐alpha. PLoS ONE 2017;12:e0182040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin‐conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000;103:351–361. [DOI] [PubMed] [Google Scholar]
- 64. Fielitz J, Kim M‐S, Shelton JM, Latif S, Spencer JA, Glass DJ, et al. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J Clin Invest 2007;117:2486–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, et al. The E3 ligase MuRF1 degrades myosin heavy chain protein in dexamethasone‐treated skeletal muscle. Cell Metab 2007;6:376–385. [DOI] [PubMed] [Google Scholar]
- 66. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- 67. Whiteside ST, Israel A. I kappa B proteins: structure, function and regulation. Semin Cancer Biol 1997;8:75–82. [DOI] [PubMed] [Google Scholar]
- 68. Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B‐alpha proteolysis by site‐specific, signal‐induced phosphorylation. Science (New York, NY) 1995;267:1485–1488. [DOI] [PubMed] [Google Scholar]
- 69. Finco TS, Beg AA, Baldwin AS Jr. Inducible phosphorylation of I kappa B alpha is not sufficient for its dissociation from NF‐kappa B and is inhibited by protease inhibitors. Proc Natl Acad Sci U S A 1994;91:11884–11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Whiteside ST, Epinat JC, Rice NR, Israel A. I kappa B epsilon, a novel member of the I kappa B family, controls RelA and cRel NF‐kappa B activity. EMBO J 1997;16:1413–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen ZJ, Parent L, Maniatis T. Site‐specific phosphorylation of IkappaBalpha by a novel ubiquitination‐dependent protein kinase activity. Cell 1996;84:853–862. [DOI] [PubMed] [Google Scholar]
- 72. Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF‐kappaB activation. Cell 1997;91:243–252. [DOI] [PubMed] [Google Scholar]
- 73. Ghosh S, Karin M. Missing pieces in the NF‐kappaB puzzle. Cell 2002;109:S81–S96. [DOI] [PubMed] [Google Scholar]
- 74. Penner CG, Gang G, Wray C, Fischer JE, Hasselgren PO. The transcription factors NF‐kappab and AP‐1 are differentially regulated in skeletal muscle during sepsis. Biochem Biophys Res Commun 2001;281:1331–1336. [DOI] [PubMed] [Google Scholar]
- 75. Bohrer H, Qiu F, Zimmermann T, Zhang Y, Jllmer T, Mannel D, et al. Role of NFkappaB in the mortality of sepsis. J Clin Invest 1997;100:972–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hunter RB, Kandarian SC. Disruption of either the Nfkb1 or the Bcl3 gene inhibits skeletal muscle atrophy. J Clin Invest 2004;114:1504–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Baltgalvis KA, Berger FG, Pena MM, Davis JM, Muga SJ, Carson JA. Interleukin‐6 and cachexia in ApcMin/+ mice. Am J Physiol Regul Integr Comp Physiol 2008;294:R393–R401. [DOI] [PubMed] [Google Scholar]
- 78. Mantovani G, Maccio A, Mura L, Massa E, Mudu MC, Mulas C, et al. Serum levels of leptin and proinflammatory cytokines in patients with advanced‐stage cancer at different sites. J Mol Med (Berl) 2000;78:554–561. [DOI] [PubMed] [Google Scholar]
- 79. Reid MB, Li YP. Tumor necrosis factor‐alpha and muscle wasting: a cellular perspective. Respir Res 2001;2:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yamaki T, Wu CL, Gustin M, Lim J, Jackman RW, Kandarian SC. Rel A/p65 is required for cytokine‐induced myotube atrophy. Am J Physiol Cell Physiol 2012;303:C135–C142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang L, Pan J, Dong Y, Tweardy DJ, Dong Y, Garibotto G, et al. Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass. Cell Metab 2013;18:368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fielitz J. Cancer cachexia‐when proteasomal inhibition is not enough. J Cachexia Sarcopenia Muscle 2016;7:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jakubowski AA, Casper ES, Gabrilove JL, Templeton MA, Sherwin SA, Oettgen HF. Phase I trial of intramuscularly administered tumor necrosis factor in patients with advanced cancer. J Clin Oncol Off J Am Soc Clin Oncol 1989;7:298–303. [DOI] [PubMed] [Google Scholar]
- 84. Han Y, Weinman S, Boldogh I, Walker RK, Brasier AR. Tumor necrosis factor‐alpha‐inducible IkappaBalpha proteolysis mediated by cytosolic m‐calpain. A mechanism parallel to the ubiquitin‐proteasome pathway for nuclear factor‐kappab activation. J Biol Chem 1999;274:787–794. [DOI] [PubMed] [Google Scholar]
- 85. Traenckner EB, Wilk S, Baeuerle PA. A proteasome inhibitor prevents activation of NF‐kappa B and stabilizes a newly phosphorylated form of I kappa B‐alpha that is still bound to NF‐kappa B. EMBO J 1994;13:5433–5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Baracos VE, DeVivo C, Hoyle DH, Goldberg AL. Activation of the ATP‐ubiquitin‐proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol 1995;268:E996–E1006. [DOI] [PubMed] [Google Scholar]
- 87. Kawamura I, Morishita R, Tomita N, Lacey E, Aketa M, Tsujimoto S, et al. Intratumoral injection of oligonucleotides to the NF kappa B binding site inhibits cachexia in a mouse tumor model. Gene Ther 1999;6:91–97. [DOI] [PubMed] [Google Scholar]
- 88. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information
Figure S1. Supporting Information
Figure S2. Supporting Information
Figure S3. Supporting Information
Figure S4. Supporting Information