

Abstract
Background
Ctns−/− mice, a mouse model of infantile nephropathic cystinosis, exhibit hypermetabolism with adipose tissue browning and profound muscle wasting. Ctns−/− mice are 25(OH)D3 and 1,25(OH)2D3 insufficient. We investigated whether vitamin D repletion could ameliorate adipose tissue browning and muscle wasting in Ctns−/− mice.
Methods
Twelve‐month‐old Ctns−/− mice and wild‐type controls were treated with 25(OH)D3 and 1,25(OH)2D3 (75 μg/kg/day and 60 ng/kg/day, respectively) or an ethylene glycol vehicle for 6 weeks. Serum chemistry and parameters of energy homeostasis were measured. We quantitated total fat mass and studied expression of molecules regulating adipose tissue browning, energy metabolism, and inflammation. We measured lean mass content, skeletal muscle fibre size, in vivo muscle function (grip strength and rotarod activity), and expression of molecules regulating muscle metabolism. We also analysed the transcriptome of skeletal muscle in Ctns−/− mice using RNAseq.
Results
Supplementation of 25(OH)D3 and 1,25(OH)2D3 normalized serum concentration of 25(OH)D3 and 1,25(OH)2D3 in Ctns−/− mice, respectively. Repletion of vitamin D partially or fully normalized food intake, weight gain, gain of fat, and lean mass, improved energy homeostasis, and attenuated perturbations of uncoupling proteins and adenosine triphosphate content in adipose tissue and muscle in Ctns−/− mice. Vitamin D repletion attenuated elevated expression of beige adipose cell biomarkers (UCP‐1, CD137, Tmem26, and Tbx1) as well as aberrant expression of molecules implicated in adipose tissue browning (Cox2, Pgf2α, and NF‐κB pathway) in inguinal white adipose tissue in Ctns−/− mice. Vitamin D repletion normalized skeletal muscle fibre size and improved in vivo muscle function in Ctns−/− mice. This was accompanied by correcting the increased muscle catabolic signalling (increased protein contents of IL‐1β, IL‐6, and TNF‐α as well as an increased gene expression of Murf‐2, atrogin‐1, and myostatin) and promoting the decreased muscle regeneration and myogenesis process (decreased gene expression of Igf1, Pax7, and MyoD) in skeletal muscles of Ctns−/− mice. Muscle RNAseq analysis revealed aberrant gene expression profiles associated with reduced muscle and neuron regeneration, increased energy metabolism, and fibrosis in Ctns−/− mice. Importantly, repletion of 25(OH)D3 and 1,25(OH)2D3 normalized the top 20 differentially expressed genes in Ctns−/− mice.
Conclusions
We report the novel findings that correction of 25(OH)D3 and 1,25(OH)2D3 insufficiency reverses cachexia and may improve quality of life by restoring muscle function in an animal model of infantile nephropathic cystinosis. Mechanistically, vitamin D repletion attenuates adipose tissue browning and muscle wasting in Ctns−/− mice via multiple cellular and molecular mechanisms.
Keywords: Infantile nephropathic cystinosis, Vitamin D insufficiency, Cachexia, Adipose tissue browning, Muscle wasting
Introduction
Cystinosis is caused by mutations of the CTNS gene (17p13) and is inherited as an autosomal recessive disease.1, 2 It is a multisystem genetic disorder characterized by the accumulation of cystine in different tissues and organs. Infantile nephropathic cystinosis (INC) is the most common and severe form of cystinosis.3 Patients suffering from INC exhibit signs and symptoms of renal Fanconi syndrome and chronic kidney disease in early childhood.4 Metabolic abnormalities are common complications in patients with INC, which are associated with poor quality of life and mortality and for which there is no current therapy.3, 4 In order to elucidate disease mechanisms, we previously characterized the metabolic phenotype in Ctns−/− mice, a mouse model of INC. We showed that Ctns−/− mice exhibited cachexia characterized by hypermetabolism, profound adipose tissue browning, and muscle wasting.5 Serum concentrations of 25(OH)D3 and 1,25(OH)2D3 were significantly lower in patients with INC relative to normal subjects.6, 7, 8 In addition to its classic function of maintaining calcium and phosphate homeostasis, vitamin D plays a very extensive role as a cell differentiating and anti‐proliferative factor with actions in a variety of tissues, including renal, cardiovascular, immune systems, adipose tissue, and muscles.9, 10 Vitamin D insufficiency has been implicated in a wide range of metabolic disorders.11, 12 Vitamin D plays a significant role in adipogenesis and inflammation.13 Furthermore, vitamin D has been proposed to modulate muscle growth and muscle function.14, 15 Vitamin D deficiency is associated with decreased muscle size and strength, reduced physical function, and increased falls in the elderly.14, 15, 16 These deficits in muscle function can be improved by vitamin D supplementation.16 Serum 25(OH)D3 may exert both paracrine and autocrine effects via 1α hydroxylase and vitamin D receptor (VDR).14 Low serum 25(OH)D3 values are associated with muscle weakness and increased risk of falls in patients on dialysis, independent of 1,25(OH)2D3 values.16 Concentration of serum 25(OH)D3 exhibited significant seasonal variation, and deficiency of 25(OH)D3 is common in patients with chronic kidney disease and is associated with poor outcomes such as progression to end‐stage kidney disease, infections, fracture rates, hospitalizations, and all‐cause mortality.17, 18 Apart from the classical VDR signalling pathway, other cytosol receptors such as annexin II and membrane‐associated rapid response steroid‐binding proteins could induce rapid effects of vitamin D in muscle.19, 20 In this study, we investigated the effects of vitamin D repletion in a mouse model of INC‐associated cachexia, with particular focus on adipose tissue browning and muscle wasting.
Materials and methods
Study design
C57BL/6 Ctns−/− mice were kindly provided by Professor Corinne Antignac.21 Wild‐type (WT) C57BL/6 control mice were acquired from Jackson Lab. Only male mice were used for this study. Mice were treated with 25(OH)D3 (Sigma, Catalogue 739650‐1ML), 1,25(OH)2D3 (Sigma, Catalogue 740578‐1ML), or ethylene glycol as a vehicle control using subcutaneous osmotic Alzet 2006 pump. The study period was 6 weeks. Mice were fed with LabDiet 5015 Diet (3.83 kcal/g, 3.3 IU/g vitamin D3). Ctns−/− mice and WT mice were treated with 25(OH)D3 and 1,25(OH)2D3 (75 μg/kg/day and 60 ng/kg/day, respectively) or vehicle. Two sets of experiments were performed. In the first set, all animals were fed ad libitum. In the second set, the food intake was all kept the same (pair feeding) (details were provided in the Results section). Whole‐body fat and lean mass of mice were determined by quantitative magnetic resonance analysis (EchoMRI‐100TM, Echo Medical System). A grip strength meter (Model 47106, UGO Basile) and AccuRotor Rota Rod (model RRF/SP, AccuScan Instrument) were used to quantify forelimb grip strength and motor coordination in mice, respectively. Oxygen consumption (VO2) and energy expenditure were measured in mice using Oxymax calorimetry (Columbus Instrument).5 The study protocol was in compliance with institutional guidelines for the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee at the University of California, San Diego.
Serum and blood chemistry
Serum concentration of bicarbonate, Ca, Pi, and blood urea nitrogen was assessed using VetScan2 VS2 Comprehensive Diagnostic Concentration of serum 25(OH)D3, 1,25(OH)2D3 and parathyroid hormone were also analysed (Supporting Information, Table S1 ).
Protein assay for adipose and muscle tissue
Adipose and muscle tissue was prepared in a homogenizer tube (USA Scientific, Catalogue 1420‐9600) containing ceramic beads (Omni International, Catalogue 19‐646) using a Bead Mill Homogenizer (Omni International). Protein concentration of tissue homogenate was assayed using Pierce BAC Protein Assay Kit (Thermo Scientific, Catalogue 23227). Uncoupling protein (UCP) protein content as well as adenosine triphosphate (ATP) concentration in adipose tissue and muscle homogenates were assayed. Protein concentration of CD137, Tmem26, Tbx‐1, Cox2, Pgf2α, NF‐κB phosphorylated Ser337 p50 and total NF‐κB p50, NF‐κB phosphorylated Ser536 p65 and total NF‐κB p65, and Iκκα phosphorylated Thr23, toll‐like receptor 2 (Tlr2), myeloid differentiation primary response 88 (MyD88), and TNF receptor‐associated factor 6 (Traf6) in adipose tissue homogenates and protein concentration of IL‐1β, IL‐6, TNF‐α, and collagen content in muscle homogenates were assayed (Supporting Information, Table S1 ).
Muscle fibre size and muscle collagen content
Fibre cross‐sectional area of soleus muscles was measured. Excised soleus muscles were snap frozen in isopentane cooled by liquid nitrogen and stored at −80 °C for subsequent analysis. Muscle cross sections (10 μm thick) were taken from muscle midbelly. Sections were first treated with 1% bovine serum albumin and normal goat and mouse serum as blocking agents. Sections were incubated overnight with a polyclonal anti‐laminin antibody (Sigma, dilution 1:1000) and then with the secondary antibody, Alexa Fluor 594 goat anti‐rabbit immunoglobulin G (Invitrogen, dilution 1:200). The laminin antibody was used to label the fibre perimeter and facilitate semiautomated fibre area quantification.23 Sections were imaged with a microscope (Leica CTR 6500, Buffalo Grove) fit with a fluorescent camera (Leica DFC365 FX) set for 594 emission fluorescence using a 10× objective. Fibre cross‐sectional areas were measured using a custom‐written macro in ImageJ (National Institutes of Health). Filtering criteria were applied to ensure measurement of actual muscle fibres. These criteria rejected regions with areas below 50 μm2 and above 5000 μm2 to eliminate neurovascular structures and ‘optically fused' fibres, respectively. Excised soleus muscles were hydrolyzed, and hydroxyproline content was calculated using a colorimetric assay.24
Quantitative real‐time PCR
Total adipose and muscle RNA was isolated using TRIzol (Life Technology) and further purified with Direct‐zol RNA MiniPre Kit (Zymo Research). cDNA was synthesized using SuperScript III Reverse Transcriptase and oligo (dT)12‐18 primer (Invitrogen). The PCR reaction based on SYBR (KAPA Biosystems, SYBR FAST Universal qPCR kit, Catalogue KK4602) was performed with a 7300 Real‐Time PCR System (ABI Applied Biosystems) using the reaction procedure as follows: 95 °C for 1 min followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Appropriate primers for target genes are listed (Supporting Information, Table S2 ). The comparative 2−ΔΔCt method was used to determine the relative quantity of each target gene. Final results were expressed in arbitrary units, with one unit being the mean mRNA level in WT control mice.
Muscle RNAseq analysis
Total gastrocnemius muscle RNA was isolated in the experimental mice (three mice in each group) using TRIzol (Life Technology) followed by RNeasy mini kit (Qiagen) for further purification. The extracted muscle RNA samples were analysed using Agilent 2100 Bioanalyzer (Agilent RNA 6000 Nano Kit) with RNA integrity number >8.5 and rRNA 28S/18S >1.0. Samples were used to construct cDNA libraries (Illumina) and sequenced through an Illumina HiSeq2000 platform at BGI Hong Kong (http://www.bgi.com). Approximately 5.88 GB of raw reads was generated for each of the samples. The raw RNAseq data were filtered into clean reads, followed by mapping to the mouse reference genome using HISAT. On average, 93.45% reads were mapped, and the uniformity of the mapping results for each sample suggested that the samples were comparable. The gene expression level for each sample was analysed using RSEM quantification tool. Based on the gene expression level, differentially expressed genes (DEG) between experimental groups were identified using DESeq2 algorithms. Biological function analysis of the DGE was enriched by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes pathway. To identify pathways related to phenotypic differences of the muscle between Ctns−/− and WT control mice, DEG between these two groups of mice were analysed using the Ingenuity Pathway Analysis software (Ingenuity Systems, http://www.ingenuity.com).
Results
Vitamin D supplementation normalizes serum vitamin D concentrations in Ctns−/− mice
Ctns−/− mice, at the age of 12 months, were both 25(OH)D3 and 1,25(OH)2D3 insufficient (Table 1). We show that 25(OH)D3 (75 μg/kg/day for 6 weeks) or 1,25(OH)2D3 (60 ng/kg/day for 6 weeks) alone did not normalize serum 25(OH)D3 and1,25(OH)2D3 concentrations in Ctns−/− mice (Supporting Information, Table S3 ). However, supplementation of 25(OH)D3 and 1,25(OH)2D3 (75 μg/kg/day and 60 ng/kg/day, respectively, for 6 weeks) normalized serum vitamin D concentrations in Ctns−/− mice (Table 1). Vitamin D repletion normalized serum concentration of calcium and phosphorus in Ctns−/− mice. Vitamin D repletion in Ctns−/− mice significantly decrease but did not completely normalize serum parathyroid hormone levels.
Table 1.
Serum and blood chemistry of mice
| WT + vehicle | WT + 25(OH)D3 + 1,25(OH)2D3 | Ctns−/− + vehicle | Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 | |
|---|---|---|---|---|
| n = 8 | n = 8 | n = 8 | n = 8 | |
| BUN (mg/dL) | 32.4 ± 5.5 | 32.5 ± 3.8 | 79.6 ± 14.6a | 80.2 ± 9.8a |
| Ca (mg/dL) | 11.2 ± 0.6 | 11.3 ± 0.8 | 9.1 ± 0.5b | 10.6 ± 0.4 |
| Creatinine (mg/dL) | <0.2 ± 1.1 | 0.3 ± 0.2 | 0.7 ± 0.2a | 0.8 ± 0.2a |
| Bicarbonate (mmol/L) | 27.8 ± 0.5 | 27.6 ± 1.7 | 26.8 ± 1.1 | 26.7 ± 2.4 |
| Pi (mg/dL) | 7.5 ± 0.5 | 7.6 ± 0.3 | 9.8 ± 0.4a | 8.2 ± 0.5c |
| PTH (pg/mL) | 108.4 ± 9.4 | 118.6 ± 18.4 | 364.1 ± 21.4a | 227.3 ± 17.5a,c |
| 25(OH)D3 (ng/mL) | 113.2 ± 12.3 | 103.7 ± 23.4 | 43.5 ± 15.4b | 117.8 ± 15.3c |
| 1,25(OH)2D3 (pg/mL) | 298.4 ± 23.4 | 278.4 ± 26.9 | 105.6 ± 24.8b | 302.4 ± 37.8c |
BUN, blood urea nitrogen; PTH, parathyroid hormone; WT, wild‐type.
Twelve‐month‐old, male, WT, and Ctns−/− mice were treated with 25(OH)D3 and 1,25(OH)2D3 (75 μg/kg/day and 60 ng/kg/day, respectively) or ethylene glycol as vehicle for 6 weeks. Four groups of mice were included: WT + vehicle, WT + 25(OH)D3 + 1,25(OH)2D3, Ctns−/− + vehicle, and Ctns−/− + 25(OH)D3 + 1,25(OH)2D3. Ctns−/− + vehicle mice were fed ad libitum, while other groups of mice were fed the same food intake as that of Ctns−/− + vehicle mice. Data are expressed as mean ± standard error of the mean.
P < 0.05, significantly higher in Ctns−/− + vehicle and Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 mice vs. WT + vehicle and WT + 25(OH)D3 + 1,25(OH)2D3 mice, respectively.
P < 0.05, significantly lower in Ctns−/− + vehicle and Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 mice vs. WT + vehicle and WT + 25(OH)D3 + 1,25(OH)2D3 mice, respectively.
P < 0.05, significantly different between Ctns−/− + vehicle vs. Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 mice.
Vitamin D repletion improves the cachexia phenotype in Ctns−/− mice
Cachexia is defined by weight loss in adults.25 However, it is characterized by lack of adequate weight gain in growing children and mice.26, 27 Similar to our previous published studies, Ctns−/− mice exhibited the cachexia phenotype consisting of anorexia, reduced weight gain, abnormal body mass (manifested as reduced gain of lean and fat mass), and hypermetabolism (manifested as elevated energy expenditure).5 We studied the impact of vitamin D repletion on the cachexia phenotype. First, we fed all four groups of mice ad libitum. The food intake of Ctns−/− mice supplemented with 25(OH)D3 and 1,25(OH)2D3 was significantly increased compared with Ctns−/− mice and was not different from WT controls with or without 25(OH)D3 and 1,25(OH)2D3. The weight gain in the Ctns−/− mice with 25(OH)D3 and 1,25(OH)2D3 was also increased compared with Ctns−/− mice control but still less than the WT mice with or without 25(OH)D3 and 1,25(OH)2D3 (Supporting Information, Figure S1 ). We then repeated the experiments using a pair‐feeding strategy. Ctns−/− mice treated with vehicle were given an ad libitum amount of food, and detailed daily intake was recorded. Subsequently, the other three groups of mice were given an equivalent amount of food as vehicle treated Ctns−/− mice (Figure 1). With equal food intake in all groups, supplementation of 25(OH)D3 and 1,25(OH)2D3 normalized weight gain as well as lean and fat mass gains in the Ctns−/− mice. Muscle function (grip strength and rotarod activity), muscle fibre size (cross‐sectional area), and collagen content were decreased in Ctns−/− mice and were normalized following 25(OH)D3 and 1,25(OH)2D3 supplementation. Light‐phase and dark‐phase volume of oxygen consumption (VO2) as well as respective energy expenditure were significantly elevated in Ctns−/− mice relative to WT mice and were normalized with 25(OH)D3 and 1,25(OH)2D3. Taken together, these results show that vitamin D supplementation corrects the cachexia phenotype in Ctns−/− mice.
Figure 1.
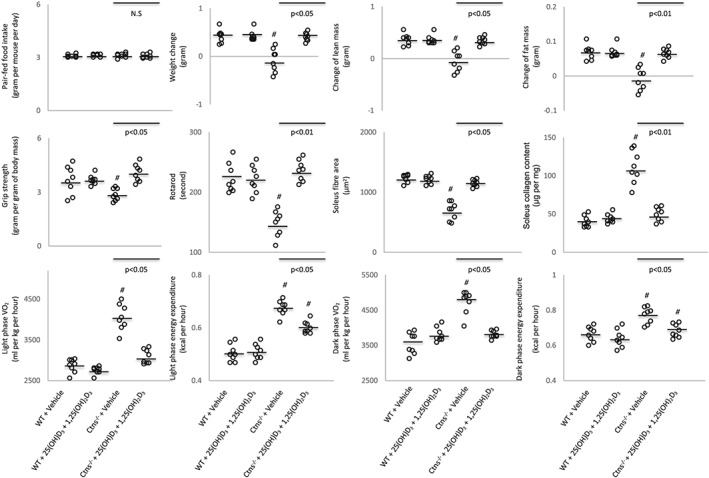
Repletion of vitamin D attenuates cachexia in Ctns−/− mice. Twelve‐month‐old wild‐type (WT) and Ctns−/− mice were treated with 25(OH)D3 and 1,25(OH)2D3 (75 μg/kg/day and 60 ng/kg/day, respectively) or ethylene glycol as vehicle for 6 weeks. Four groups of mice were included: WT + vehicle (n = 8), WT + 25(OH)D3 + 1,25(OH)2D3 (n = 8), Ctns−/− + vehicle (n = 8), and Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 (n = 8). Ctns−/− + vehicle mice were fed ad libitum, whereas other groups of mice were fed the same amount of food intake as that of Ctns−/− + vehicle mice. Data are expressed as mean ± standard error of the mean. # P < 0.05, significantly different in Ctns−/− + vehicle and Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 mice vs. WT + vehicle and WT + 25(OH)D3 + 1,25(OH)2D3 mice, respectively. Results of Ctns−/− + vehicle mice were also compared with Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 mice.
Vitamin D repletion normalized uncoupling protein content and adenosine triphosphate in Ctns−/− mice
We measured adipose and muscle protein contents of UCPs and ATP in Ctns−/− mice. Protein content of UCPs in adipose tissue [inguinal white adipose tissue (WAT) and intercapsular brown adipose tissue] and gastrocnemius muscle was elevated in Ctns−/− mice relative to control mice (Figure 2). In contrast, ATP content in adipose tissue and muscle was significantly decreased in Ctns−/− mice relative to control mice. Repletion of 25(OH)D3 and 1,25(OH)2D3 normalized UCPs and ATP content in adipose tissue and muscle in Ctns−/− mice relative to control mice.
Figure 2.
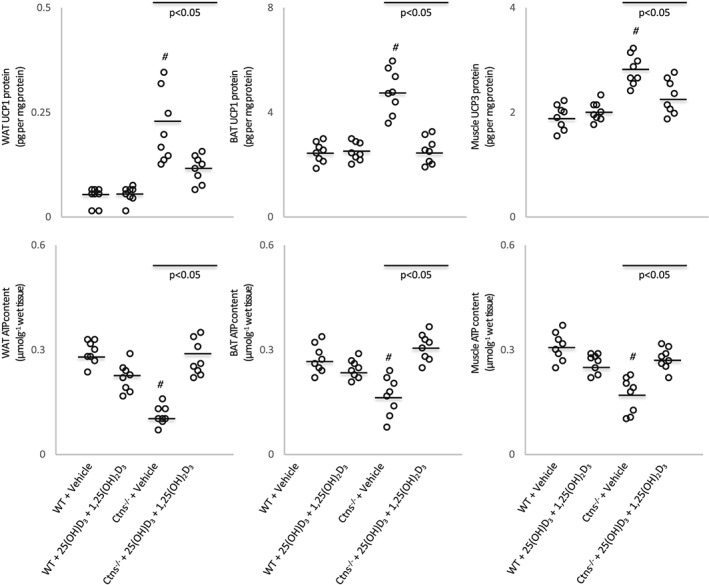
Vitamin D repletion ameliorates uncoupling protein content and adenosine triphosphate in Ctns−/− mice. UCP and ATP content in inguinal white adipose tissue, brown adipose tissue, and gastrocnemius muscle were measured. Results are analysed and expressed as in Figure 1. BAT, brown adipose tissue; WAT, white adipose tissue; WT, wild‐type.
Vitamin D repletion attenuates adipose tissue browning in Ctns−/− mice
The mRNA and protein content of beige adipose cell surface markers (CD137, Tmem26, and Tbx1) were elevated in inguinal WAT in Ctns−/− mice compared with control mice (Figure 3), and this is consistent with increased UCP‐1 in WAT shown in Figure 2, which is a marker for beige adipocyte, and usually undetected in WAT. Collectively, our results demonstrate the presence of beige adipocytes in Ctns−/− mice. Repletion of 25(OH)D3 and 1,25(OH)2D3 attenuated browning of beige adipocytes in Ctns−/− mice. Protein content of UCP‐1 and mRNA and protein content of CD137, Tmem26, and Tbx1 were normalized in Ctns−/− mice treated with 25(OH)D3 and 1,25(OH)2D3 relative to control mice (Figures 2 and 3). In addition, repletion of 25(OH)D3 and 1,25(OH)2D3 ameliorated aberrant levels of key molecules implicated in adipose tissue browning in Ctns−/− mice. Inguinal WAT of Ctns−/− mice displayed higher inguinal WAT mRNA expression and protein content of Cox2 and Pgf2α than that of controls (Figure 4). Inflammation has been implicated in the pathogenesis of adipose tissue browning. One of the most important inflammatory pathways in mammalian cells is that of the NF‐κB pathway, and therefore, we studied expression of transcription factors involved in pro‐inflammatory pathways in Ctns−/− mice. Inguinal WAT NF‐κB p50 (phosphorylated Ser337)/total NF‐κκB p50 ratio, NF‐κB p65 (phosphorylated S536)/total NF‐κB p65 ratio, protein content of Iκκ‐α (phosphorylated Thr23), mRNA expression, and protein content of Tlr2, MyD88, and Traf6 were up‐regulated in Ctns−/− mice relative to control mice. Repletion of 25(OH)D3 and 1,25(OH)2D3 partially or fully normalized mRNA expression and protein content of the entire Tlr/NF‐κB pathway in Ctns−/− mice relative to control mice (Figure 4).
Figure 3.
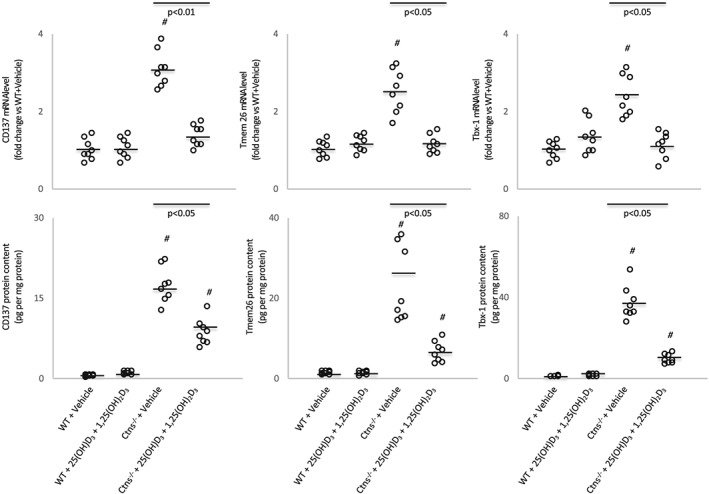
Vitamin D repletion attenuates adipose tissue browning in Ctns−/− mice. Gene expression and protein content of beige adipocyte markers (CD137, Tmen 26, and Tbx‐1) in inguinal white adipose tissue were measured. Results are analysed and expressed as in Figure 1. WT, wild‐type.
Figure 4.
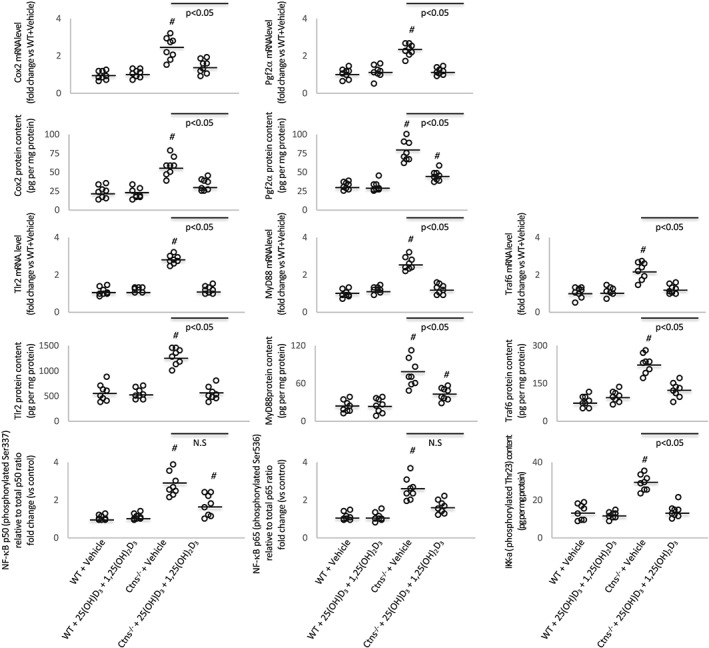
Vitamin D repletion modulates expression of key molecules implicated in adipose tissue browning in Ctns−/− mice. Gene expression and protein content of Cox2 signalling pathway (Cox2 and Pgf2α) and Tlr/NF‐κB pathway (Tlr2, MyD88, and Traf6 as well as relative NF‐κB phosphorylated Ser337 p50/total p50 ratio, relative NF‐κB phosphorylated Ser536 p65/total p65 ratio, and Iκκ‐α phosphorylated Thr23) in inguinal white adipose tissue were measured. For relative NF‐κB phosphorylated Ser337 p50/total p50 ratio and relative NF‐κB phosphorylated Ser536 p65/total p65 ratio in mice, value was normalized with respect to wild‐type (WT) control mice. Final results were expressed in arbitrary units, with one unit being the mean level in WT control mice. Results are analysed and expressed as in Figure 1.
Vitamin D repletion attenuates aberrant muscle mass signalling pathway in Ctns−/− mice
Muscle catabolic signalling (muscle lysate protein levels of inflammatory cytokines IL‐1β, IL‐6, and TNF‐α and expression of muscle proteolytic genes, Murf‐1, atrogin‐1, and myostatin) was significantly increased in Ctns−/− mice than that in controls (Figure 5). In contrast, gene expression for myogenesis and skeletal muscle regeneration (Igf1, Pax7, and MyoD) was decreased in gastrocnemius muscle of Ctns−/− mice relative to controls. Repletion of 25(OH)D3 and 1,25(OH)2D3 normalized aberrant levels of key molecules implicated in muscle wasting pathways in Ctns−/− mice.
Figure 5.
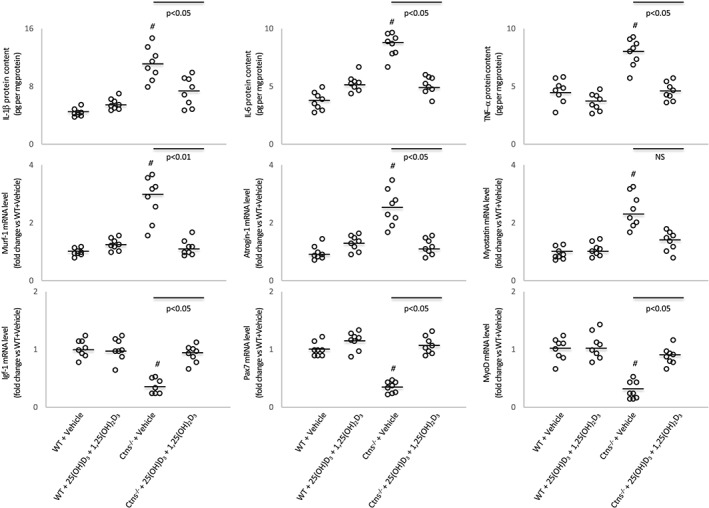
Vitamin D repletion corrects aberrant muscle mass signalling pathways in Ctns−/− mice. Protein concentrations of inflammatory cytokine (IL‐1β, IL‐6, and TNF‐α) and mRNA levels of key molecules associated with myogenesis (Murf‐1, atrogin‐1, and myostatin) and skeletal muscle regeneration (IGF‐1, Pax7, and MyoD) in gastrocnemius muscle were shown. Results are analysed and expressed as in Figure 1. WT, wild‐type.
Differentially expressed genes in muscle from Ctns−/− mice
To characterize muscle transcriptome changes associated with vitamin D repletion in Ctns−/− mice, we performed RNAseq analysis on the gastrocnemius muscle. We identified ~17 000 genes in all experimental groups of mice. We compared DEG in muscle between Ctns−/− mice and WT mice. We identified 214 genes up‐regulated and 76 genes down‐regulated in Ctns−/− mice relative to WT mice (Figure 6). Completed information for those DEG were listed (Supporting Information, Table S4 ). These DEG are displayed in the heatmap by hierarchical clustering to illustrate the high degree of reproducibility in individual samples of the same experimental groups of mice. The DEG data were further analysed using the GO pathway through Kyoto Encyclopedia of Genes and Genomes. Detailed GO of biological process and molecular function in Ctns−/− mice vs. WT mice revealed significant differential expression of genes implicated in cellular processes, biological regulation and processes, and metabolic processes. Similarly, we also identified gastrocnemius muscle DEG in Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 mice vs. WT + 25(OH)D3 + 1,25(OH)2D3 mice as well as Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 mice vs. Ctns−/− + vehicle mice. Results are shown (Figure 6). Completed information for those DEG were listed (Supporting Information, Tables S5 and S6 ).
Figure 6.
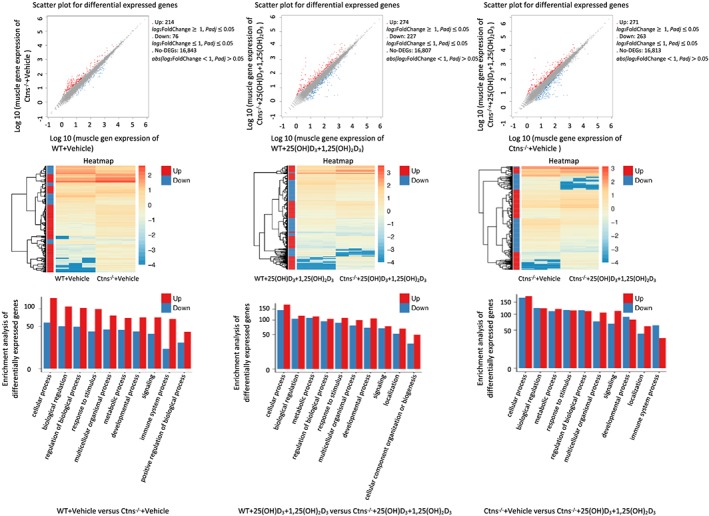
RNAseq analysis of gastrocnemius muscle. Summary of the number of up‐regulated and down‐regulated genes in individual‐group comparison. Wild‐type (WT) + vehicle mice vs. Ctns−/− + vehicle mice, WT + 25(OH)D3 + 1,25(OH)2D3 mice vs. Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 mice, and Ctns−/− + vehicle mice vs. Ctns−/− + 25(OH)D3 + 1,25(OH)2D3 mice. Hierarchical clustering heatmap of differentially expressed genes in individual‐group comparisons. Gene Ontology enrichment analysis in individual‐group comparisons based on the fold changes of differentially expression of genes. All significantly up‐regulated and down‐regulated genes are used for gene ontology enrichment analysis.
Canonical signalling pathways analysis in muscle from Ctns−/− mice
We performed Ingenuity Pathway Analysis enrichment tests for DEG in muscle from Ctns−/− mice vs. WT mice. We particularly focused on pathways related to energy metabolism, skeletal and muscular system development and function, and organismal injury and abnormalities. Results are shown in Figure 7. Up‐regulated genes include ANKRD2, CSRP3, CYFIP2, FHL1, LY6A, MUP1, MYL2, MYL3, PDK4, SELL, SLN, SPP1, TNNC1, TNNI1, and TPM3, whereas down‐regulated genes are ATF3, CIDEA, FOS, SNCG,s and TBC1D1 in Ctns−/− mice relative to WT control mice. Functional significance of those DEG is listed (Table 2). Taken together, gene expression associated with regeneration of muscle and neurons is compromised in Ctns−/− mice. Furthermore, expression of genes related to enhanced muscle thermogenesis and fibrosis is increased in Ctns−/− mice. Importantly, repletion of 25(OH)D3 and 1,25(OH)2D3 normalized the top 20 DEG as listed in Table 2 in Ctns−/− mice.
Figure 7.
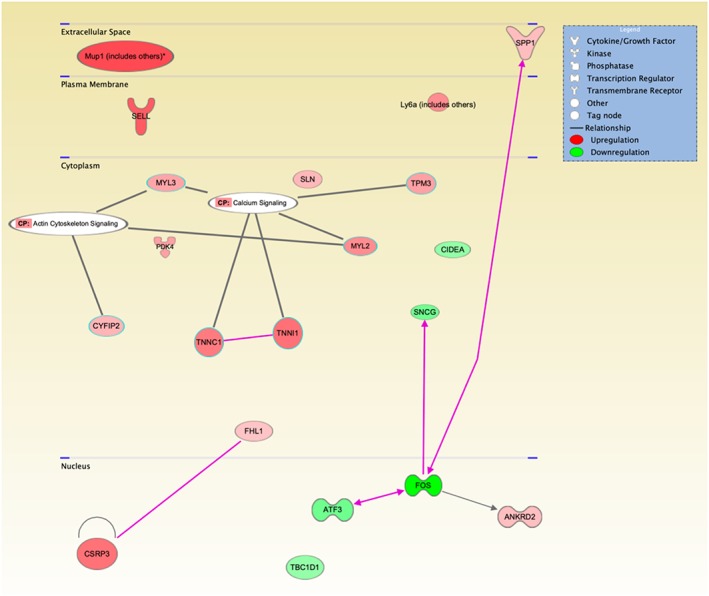
Functional annotation network. Ingenuity Pathway Analysis of alternations in energy metabolism, skeletal and muscular system development and function, and organismal injury and abnormalities in Ctns−/− mice relative to wild‐type mice. The coloured genes in the networks are differentially expressed between Ctns−/− mice relative to wild‐type mice. Node colour represents the expression status. Red: up‐regulated in Ctns−/− mice relative to wild‐type mice; green: down‐regulated in Ctns−/− mice relative to wild‐type mice. Increased expression of genes are ANKRD2, CSRP3, CYFIP2, FHL1, LY6A, MUP1, MYL2, MYL3, PDK4, SELL, SLN, SPP1, TNNC1, TNNI1, and TPM3, and decreased expression of genes are ATF3, CIDEA, FOS, SNCG, and TBC1D1.
Table 2.
Muscle ingenuity analysis of canonical signalling pathways in Ctns−/− mice
| Up‐regulated gene | Functional significance and reference |
| ANKRD2 | Induced by skeletal muscle denervation and muscle injury60 |
| CSRP3 | Inhibition of myotube differentiation61 |
| CYFIP2 | Promotes apoptosis73 |
| FHL1 | Activates myostatin signalling and promotes atrophy in skeletal muscle78 |
| LY6A | Associated with increased fibrosis in aged skeletal muscle72 |
| MUP1 | Increases energy expenditure in skeletal muscle68 |
| MYL2 | Associated with cardiac cycling kinetics62 |
| MYL3 | Biomarker for skeletal muscle toxicity77 |
| PDK4 | Associated with skeletal muscle energy deprivation via a FOXO1‐dependent pathway76 |
| SELL | Promotes progression of cancer cachexia75 |
| SLN | Interacts with ATPase and promotes muscle non‐shivering thermogenesis69 |
| SPP1 | Shares molecular network with myostatin and inhibits muscle regeneration63 |
| TNNC1 | Biomarker for muscle depolarization64 |
| TNNI1 | Controls striated muscle contraction and relaxation and disease marker for aged skeletal muscle65 |
| TPM3 | Promotes slow myofiber hypotrophy and associated with generalized muscle weakness79 |
| Down‐regulated gene | Functional significance and reference |
| ATF3 | Marker of neural injury and reduces the regeneration of neurons67 |
| CIDEA | Increases metabolic rates, lipolysis in brown adipose tissue, and higher core temperature70 |
| FOS | Associated with decreased skeletal muscle regeneration66 |
| SNCG | Increases energy expenditure, particularly in BAT and WAT71 |
| TBC1D1 | Impaired glucose transport in skeletal muscle74 |
BAT, brown adipose tissue; WAT, white adipose tissue.
We focus on pathways related to energy metabolism, skeletal and muscular system development and function, nervous system development and function, and organismal injury and abnormalities. Muscle differentially expressed genes in Ctns−/− mice relative to WT mice as well as functional significance and relevant references for those differentially expressed genes are listed.
Discussion
We previously described cachexia characterized by adipose tissue browning and muscle wasting in Ctns−/− mice, an established mouse model of INC, but the aetiology was not clear. This study showed that Ctns−/− mice, at 12 months of age, were both 25(OH)D3 and 1,25(OH)2D3 insufficient. We show that vitamin D supplementation corrects the cachexia phenotype in Ctns−/− mice. Importantly, vitamin D repletion attenuates adipose tissue browning and skeletal muscle wasting in Ctns−/− mice. These findings are novel.
Muscle wasting is a life‐threatening complication in patients with cystinosis. Muscle wasting is prevalent in patients with INC and may be associated with significant morbidity and mortality. In post‐transplant cystinotic patients who did not receive long‐term cystine‐depleting therapy, cystinotic myopathy have been associated with swallowing myopathy and, as a result, aspiration pneumonia, which constitutes a severe and potentially lethal complication.28, 29 Vitamin D insufficiency has been associated with muscle wasting and impaired muscle strength,30, 31 and low vitamin D suppresses skeletal muscle tropism and contraction.14 Therefore, we studied the effect of vitamin D repletion on muscle fibre histomorphometry in Ctns−/− mice. We showed that 25(OH)D3 and 1,25(OH)2D3 repletion normalized mean soleus muscle cross‐sectional area in Ctns−/− mice (Figure 1).
Skeletal muscle fibrosis, a major pathological hallmark of myopathies, is evident in Ctns−/− mice. Repletion of vitamin D normalized soleus collagen content in Ctns−/− mice. Our results were consistent with a recent report in which vitamin D treatment significantly increased cross‐sectional size of cultured mouse myoblast C2C12 cells.32 Vitamin D reduced the expression of collagen (collagen I, III, and other collagen informs) and key profibrotic factors (TGF‐β1 and plasminogen activator inhibitor) in mesenchymal multipotent cells.33
Adipose tissues and muscle are important in energy metabolism. UCP1 contributes to adaptive thermogenesis, while UCP2 and UCP3 are involved in the resting metabolic rate.34 We, and others, have previously described the increased thermogenesis and up‐regulation of UCPs in adipose tissues and muscle in rodent models of cachexia.35, 36, 37, 38, 39 Up‐regulation of UCP expression promotes protein leak and reduces ATP production in exchange for the generation of heat.34, 38 Protein contents of UCPs were increased in adipose tissues and muscle of Ctns−/− mice compared with controls (Figure 2). In contrast, ATP contents in adipose tissues and muscle were decreased in Ctns−/− mice relative to controls. Vitamin D insufficiency exacerbates adipose tissue and muscle metabolism. 25(OH)D3 and 1,25(OH)2D3 repletion attenuated perturbations of UCPs and ATP contents in adipose tissues and muscle in Ctns−/− mice. 1,25(OH)2D3 suppresses UCPs expression in primary brown adipocyte and suppresses differentiation and mitochondrial respiration of brown adipocyte39, 40 1,25(OH)2D3/VDR by binding to the promoter region of the UCP3 gene and modulating UCP3 gene transcription and subsequent energy metabolism in muscle.41
Adipose tissue browning is an important cause of hypermetabolism in patients with chronic cachectic disorders.42, 43 Recent studies revealed the detrimental effects of adipose tissue browning in the context of cachexia. Adipose tissue browning and its associated increases in energy expenditure precedes skeletal muscle atrophy and is important for the development and progression of cachexia.44, 45 We demonstrated the presence of brown adipose tissue marker UCP1 protein as well as an increased mRNA expression and protein content of unique beige adipose cell markers (CD137, Tmem26, and Tbx1) in inguinal WAT of Ctns−/− mice (Figures 2 and 3). Importantly, repletion of 25(OH)D3 and 1,25(OH)2D3 attenuated expression of beige adipose cell markers in inguinal WAT of Ctns−/− mice. Several mechanisms are important for biogenesis of WAT browning, including Cox2 signalling pathway and chronic inflammation. Activation of Cox2, a downstream effector of β‐adrenergic signalling, is crucial for the induction of beige cells in WAT depots.46 Cox2 produces prostaglandins that enhance mitochondrial biogenesis and increase the uncoupling capacity when activated with adrenergics.47 We showed that 25(OH)D3 and 1,25(OH)2D3 repletion normalized elevated inguinal WAT gene expression and protein content of Cox2 and Pgf2α in Ctns−/− mice (Figure 4). In addition, increased expression of key molecules implicated in the pro‐inflammatory pathways in inguinal WAT (NF‐κB p50 and p65, Iκκ‐α, Tlr2, MyD88, and Traf6) was normalized in Ctns−/− mice treated with 25(OH)D3 and 1,25(OH)2D3 relative to control mice.
We investigated the effect of vitamin D repletion on muscle mass regulatory signalling pathways in Ctns−/− mice. Repletion of 25(OH)D3 and 1,25(OH)2D3 normalized molecular signatures of processes associated with muscle wasting, including increased expression of pro‐inflammatory muscle cytokines (IL‐1β, IL‐6, and TNF‐α) and muscle proteolysis (Murf‐1, atrogin‐1, and myostatin) as well as decreased gene expression associated with myogenesis and skeletal muscle regeneration (Igf1, Pax7, and MyoD) in skeletal muscle of Ctns−/− mice relative to controls (Figure 5). Elevation of pro‐inflammatory cytokines has been implicated in many forms of muscle wasting including chronic kidney disease and age‐related sarcopenia. We acknowledge that increased pro‐inflammatory cytokines may not be casually related to muscle wasting in Ctns−/− mice. Vitamin D administration has been associated with reduced concentration of serum TNF‐α in renal patients.48 Epidemiological studies suggest an inverse association between serum 25(OH)D3 and inflammatory markers, including C‐reactive protein and IL‐6.49 Supplemental vitamin D decreased levels of inflammatory biomarkers.48, 50, 51 Vitamin D inhibited lipopolysaccharide‐induced IL‐6 and TNF‐α production in a dose‐dependent manner in monocytes by inhibiting the activation of NF‐κB.52
Vitamin D also inhibits T‐lymphocyte proliferation and the production of pro‐inflammatory cytokines.53 Interestingly, T‐cell cytokines modulate vitamin D metabolism in macrophages. IFN‐γ, a Th1 cytokine, activates the macrophage CYP27B1, leading to the enhanced conversion of 25(OH)D3 to 1,25(OH)2D3. In contrast, the Th2 cytokine IL‐4 induces catabolism of 25(OH)D3 to the inactive metabolite 24,25(OH)2D3, suggesting a potential mechanism by which vitamin D metabolism links the cell‐mediated immune responses to the immune responses.54
Vitamin D deficiency decreased muscle Pax7 expression while increased the expression of MuRF‐1 and atrogin‐1 in the gastrocnemius muscle of diabetic mice.55 Combined treatment of vitamin D and low‐intensity aerobic exercise attenuated osteopenia and muscle atrophy by enhancing muscle anabolic markers (Pax7, MyoD, and myogenin) and decreasing expression of catabolic markers (atrogin‐1, MuRF‐1, and REDD1) in diabetic rats.56 1,25(OH)2D3 promotes cultured skeletal muscle differentiation by promoting the expression of myogenic markers and myotube formation. Myostatin is a TGF‐β family member that acts as a negative regulator of skeletal muscle mass. Follistatin, a myostatin‐binding protein, inhibits myostatin activity and promotes muscle growth.57 1,25(OH)2D3 promotes myogenic differentiation by increasing follistatin expression and decreasing the expression of myostatin in C2C12 skeletal muscle cells.58 Results of a recent investigation showed that skeletal muscle‐derived satellite cells from C57BL/6J mice expressed VDR and its expression, and nuclear translocation was induced upon incubating the cells with 1,25(OH)2D3. Furthermore, incubation of 1,25(OH)2D3 enhanced myogenic differentiation in satellite cells by promoting the expression of myogenic markers (MYH1, TNNI1, TNNI3, and MMP9), marker of myotube formation (BMP4), and marker of myogenic cell migration and engraftment (MMP9) and by modulating myogenic growth factors (IGF‐1, IGF‐2, FGF2, and FGF2). Additional pro‐myogenic effects of vitamin D on satellite cells were further demonstrated by the increased expression of myogenic markers including MyoD, myogenin, and follistatin while inhibiting the expression of myostatin.59 Vitamin D deficiency has been associated with muscle wasting in diabetic mice.
We performed RNAseq analysis on muscle in Ctns−/− mice. Our data demonstrate clear differentiation in muscle transcriptomics in Ctns−/− mice relative to control WT mice (Figures 6 and 7 and Supporting Information, Table S3 ). We identified muscle DEG associated with reduced regeneration of muscle and neurons as well as elevated energy metabolism and fibrosis in Ctns−/− mice (Table 2). Elevated muscle ANKRD2, CSRP3, MYL2, SPP1, TNNC1, and TNNI1 and decreased FOS expression are associated with reduced regeneration of muscle, while decreased expression of ATF3 has been implicated in reduced regeneration of neurons.60, 61, 62, 63, 64, 65, 66, 67 Increased muscle MUP1 and SLN and decreased CIDEA and SNCG expression has been associated with elevation of muscle energy metabolism.68, 69, 70, 71 Increased muscle LY6A and CYFIP expression promotes fibrosis and apoptosis, respectively.72, 73 Down‐regulation of TBC1D1 reduces glucose transport in skeletal muscle.74 Increased expression of SELL has been shown to promote cancer cachexia.75 Up‐regulation of PDK4 expression is a biomarker for muscle energy deprivation.76 Increased expression of MYL3 has been used as a biomarker for muscle injury.77 In addition, increased expression of muscle FHL1 and TPM3 promotes muscle atrophy and muscle weakness.78, 79 Importantly, repletion of 25(OH)D3 and 1,25(OH)2D3 normalized the top 20 DEG as listed in Table 2 in Ctns−/− mice.
Conclusion
We report the novel findings that correction of 25(OH)D3 and 1,25(OH)2D3 insufficiency reverses cachexia and may improve quality of life by restoring muscle function in an animal model of INC. Mechanistically, vitamin D repletion attenuates adipose tissue browning and muscle wasting in Ctns−/− mice via multiple cellular and molecular mechanisms.
Funding
This work is supported in part by funding from the Cystinosis Research Foundation. P.Z. was supported by Department of Education, Heilongjiang Province (no. 12541324), Harbin Science and Technology Bureau (no. 2014RFXYJ077), and China Scholarship Council (no. 201308230141). S.L. was supported by Yangzhou University Overseas Study Program. R.L.L. received funding from the National Institutes of Health (NIH) grant R24‐HD050837. This work was supported by NIH grant R24‐HD050837 to create the National Skeletal Muscle Research Center (NSMRC) at University of California, San Diego. R.H.M. received funding from NIH U01DK03012.
Conflicts of interest
None declared.
Supporting information
Table S1. Immunoassay information for serum chemistry, adipose and muscle tissue homogenate assay analysis.
Table S2. PCR primer information.
Table S3. Serum and blood chemistry of mice. Twelve‐month‐old, male, wild type (WT) and Ctns‐/‐ mice were treated with 25(OH)D3 (75 μg/kg per day) or 1,25(OH)2D3 (60 ng/kg per day) versus ethylene glycol as vehicle for 6 weeks. Six groups of mice were included: WT + Vehicle, WT + 25(OH)D3, WT++1,25(OH)2D3, Ctns‐/‐ + Vehicle, Ctns‐/‐ + 25(OH)D3, Ctns‐/‐ + 1,25(OH)2D3. All mice were fed ad libitum. Data are expressed as mean SEM. Ap < 0.05, significantly higher in Ctns‐/‐ + Vehicle, Ctns‐/‐ + 25(OH)D3 or Ctns‐/‐ +1,25(OH)2D3 versus WT + Vehicle, WT + 25(OH)D3 or WT + 1,25(OH)2D3, respectively. Bp < 0.05, significantly lower in Ctns‐/‐ + Vehicle, Ctns‐/‐ + 25(OH)D3 or Ctns‐/‐ + 1,25(OH)2D3 versus WT + Vehicle, WT + 25(OH)D3 or WT + 1,25(OH)2D3, respectively. cp < 0.05, significantly difference between Ctns‐/‐ + 25(OH)D3 versus Ctns‐/‐ + Vehicle or Ctns‐/‐ + 1,25(OH)2D3 versus Ctns‐/‐ + Vehicle.
Table S4. List of differential expressed genes in gastrocnemius muscle from Ctns‐/‐ + Vehicle versus WT + Vehicle mice
Table S5. List of differential expressed genes in gastrocnemius muscle from Ctns‐/‐ + 25(OH)D3 + 1,25(OH)2D3 versus WT + 25(OH)D3 + 1,25(OH)2D3 mice
Table S6. List of differential expressed genes in gastrocnemius muscle from Ctns‐/‐ + 25(OH)D3+ 1,25(OH)2D3 versus Ctns‐/‐ + Vehicle mice
Figure S1. Ad libitum food intake and weight gain in Ctns‐/‐ mice.
Acknowledgement
All authors of this manuscript certify that they complied with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle update 2017.80
Cheung W. W., Hao S., Wang Z., Ding W., Zheng R., Gonzalez A., Zhan J.‐Y., Zhou P., Li S., Esparza M. C., Hoffman H. M., Lieber R. L., and Mak R. H. (2020) Vitamin D repletion ameliorates adipose tissue browning and muscle wasting in infantile nephropathic cystinosis‐associated cachexia, Journal of Cachexia, Sarcopenia and Muscle, 11: 120–134. 10.1002/jcsm.12497.
References
- 1. Town M, Jean G, Cherqui S, Attard M, Forestier L, Whitmore SA, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet 1998;18:319–324. [DOI] [PubMed] [Google Scholar]
- 2. Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med 2002;347:111–121. [DOI] [PubMed] [Google Scholar]
- 3. Nesterova G, Gahl W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol 2008;23:863–878. [DOI] [PubMed] [Google Scholar]
- 4. Theodoropoulos DS, Krasnewich D, Kaiser‐Kupfer MI, Gahl WA. Classic nephropathic cystinosis as an adult disease. JAMA 1993;270:2200–2204. [PubMed] [Google Scholar]
- 5. Cheung W, Cherqui S, Ding W, Esparza W, Zhou P, Shao J, et al. Muscle wasting and adipose tissue browning in infantile nephropathic cystinosis. J Cachexia Sarcopenia Muscle 2016;7:152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katzir Z, Shivil Y, Landau H, Kidrony G, Popovtzer MM. Nephrogenic diabetes insipidus, cystinosis, and vitamin D. Arch Dis Child 1988;63:548–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Steinherz R, Chesney RW, Schulman JD, DeLuca HF, Phelps M. Circulating vitamin D metabolites in nephropathic cystinosis. J Pediatr 1983;102:592–594. [PubMed] [Google Scholar]
- 8. Chesney RW, Hamstra J, Mazess RB, Rose P, DeLuca HF. Circulating vitamin D metabolites concentrations in childhood renal disease. Kidney Int 1982;21:65–69. [DOI] [PubMed] [Google Scholar]
- 9. Pavlovic D, Katicic D, Gulin T, Josipović J. Vitamin D in the patients with chronic kidney disease: when, to whom and in which form. Mater Sociomed 2015;27:122–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Williams S, Malatesta K, Norris K. Vitamin D and chronic kidney disease. Ethn Dis 2009;19:S5, ‐8‐11. [PMC free article] [PubMed] [Google Scholar]
- 11. Autier P, Boniol M, Pizot C, Mullie P. Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol 2014;2:76–89. [DOI] [PubMed] [Google Scholar]
- 12. Prasad P, Kochhar A. Interplay of vitamin D and metabolic syndrome: a review. Diabetes Metab Syndr 2016;10:105–112. [DOI] [PubMed] [Google Scholar]
- 13. Mutt SJ, Hyppönen E, Saarnio J, Järvelin MR, Herzig KH. Vitamin D and adipose tissue—more than storage. Front Physiol 2014;5:228, eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ceglia L. Vitamin D and its role in skeletal muscle. Curr Opin Clin Nutr Metab Care 2009;12:628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rejnmark L. Effects of vitamin D on muscle function and performance: a review of evidence from randomized controlled trials. Ther Adv Chronic Dis 2011;2:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gordon OL, Sakkas PK, Doyle JW, Shubert T, Johansen KL. The Relationship between vitamin D and muscle size and strength in patients on hemodialysis. J Ren Nutr 2007;17:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kleine CE, Obi Y, Streja E, Hsiung JT, Park C, Holick MF, et al. Seasonal variation of serum 25‐hydroxyvitamin D and parameters of bone and mineral disorder in dialysis patients. Bone 2019;124:158–165. [DOI] [PubMed] [Google Scholar]
- 18. Melamed ML, Chonchol M, Gutiérrez OM, Kalantar‐Zadeh K, Kendrick J, Norris K, et al. The role of vitamin D in CKD stages 3 to 4: report of a scientific workshop sponsored by the National Kidney Foundation. Am J Kidney Dis 2018;72:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baran DT, Quail JM, Ray R, Honeyman T. Annexin II is the membrane receptor that mediates the rapid actions of 1α,25‐dihydroxyvitamin D3 . J Cell Biochem 2000;78:34–46. [PubMed] [Google Scholar]
- 20. Khanal R, Nemere I. Membrane receptors for vitamin D metabolites. Crit Rev Eukaryot Gene Expr 2007;17:31–47. [DOI] [PubMed] [Google Scholar]
- 21. Cherqui S, Sevin C, Hamard G, Kalatzis V, Sich M, Pequignot MO, et al. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol 2002;22:7622–7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Young S, Struys E, Wood T. Quantification of creatinine and guanidinoacetate using GC‐MS and LC‐MS/MS for the detection of cerebral creatinine deficiency syndromes. Curr Protoc Hum Genet 2007;2007:17.3.1–17.3.18. [DOI] [PubMed] [Google Scholar]
- 23. Minamoto VB, Hulst JB, Lim M, Peace WJ, Bremner SN, Ward SR, et al. Increased efficacy and decreased systemic‐effects of botulinum toxin A injection after active or passive muscle manipulation. Dev Med Child Neurol 2007;49:907–914. [DOI] [PubMed] [Google Scholar]
- 24. Reddy GK, Enwemeka CS. A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem 1996;29:225–229. [DOI] [PubMed] [Google Scholar]
- 25. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 26. Cheung W, Yu PX, Little BM, Cone RD, Marks DL, Mak RH. Role of leptin and melanocortin signaling in uremia‐associated cachexia. J Clin Invest 2005;115:1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mak RH. Cachexia in children with chronic kidney disease: challenges in diagnosis and treatment. Curr Opin Support Palliat Care 2016;10:293–297. [DOI] [PubMed] [Google Scholar]
- 28. Anikster Y, Lacbawan F, Brantly M, Gochuico BL, Avila NA, Travis W, et al. Pulmonary dysfunction in adults with nephropathic cystinosis. Chest 2001;119:394–401. [DOI] [PubMed] [Google Scholar]
- 29. Sonies BC, Almajid P, Kleta R, Bernardini I, Gahl WA. Swallowing dysfunction in 101 patients with nephropathic cystinosis: benefit of long‐term cysteamine therapy. Medicine (Baltimore) 2005;84:137–146. [DOI] [PubMed] [Google Scholar]
- 30. Murad MH, Elamin KB, Abu Elnour NO, Elamin MDB, Alkatib AA, Fatourechi MM, et al. Clinical review: the effect of vitamin D on falls: a systematic review and meta‐analysis. J Clin Endocrinol Metab 2011;96:2997–3006. [DOI] [PubMed] [Google Scholar]
- 31. Gerdhem P, Ringsberg KA, Obrant KJ, Akesson K. Association between 25‐hydroxy vitamin D levels, physical activity, muscle strength and fractures in the prospective population based OPRA Study of elderly women. Osteoporos Int 2005;16:1425–1431. [DOI] [PubMed] [Google Scholar]
- 32. Girgis CM, Clifton‐Bligh RJ, Mokbel N, Cheng K, Gunton JE. Vitamin D signaling regulates proliferation, differentiation, and myotube size in C2C12 skeletal muscle cells. Endocrinology 2014;155:347–357. [DOI] [PubMed] [Google Scholar]
- 33. Artaza JN, Norris KC. Vitamin D reduces the expression of collagen and key profibrotic factors by inducing an antifibrotic phenotype in mesenchymal multipotent cells. J Endocrinol 2009;200:207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rousset S, Alves‐Guerra MC, Mozo J, Miroux B, Cassard‐Doulcier AM, Bouillaud F, et al. The biology of mitochondrial uncoupling proteins. Diabetes 2004;53:S130–S135. [DOI] [PubMed] [Google Scholar]
- 35. Cheung WW, Ding W, Gunta SS, Gu Y, Tabakman R, Klapper LN, et al. A pegylated leptin antagonist ameliorates CKD‐associated cachexia in mice. J Am Soc Nephrol 2014;25:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cheung WW, Kuo HJ, Markison S, Chen C, Foster AC, Marks DL, et al. Peripheral Administration of the Melanocortin‐4 Receptor Antagonist NBI‐12i Ameliorates Uremia‐Associated Cachexia in Mice. J Am Soc Nephrol 2007;18:2517–2524. [DOI] [PubMed] [Google Scholar]
- 37. Bing C, Brown M, King P, Collins P, Tisdale MJ, Williams G. Increased gene expression of brown fat uncoupling protein (UCP)1 and skeletal muscle UCP2 and UCP3 in MAC16‐induced cancer cachexia. Cancer Res 2000;60:2405–2410. [PubMed] [Google Scholar]
- 38. Sluse FE. Uncoupling proteins: molecular, functional, regulatory, physiological and pathological aspects. Adv Exp Med Biol 2012;942:137–156. [DOI] [PubMed] [Google Scholar]
- 39. Wong KE, Szeto FL, Zhang W, Ye H, Kong J, Zhang Z, et al. Involvement of the vitamin D receptor in energy metabolism: regulation of uncoupling proteins. Am J Physiol Endocrinol Metab 2009;296:E820–E828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ricciardi CJ, Bae J, Esposito D, Komamytsky S, Hu P, Chen J, et al. 1,25‐Dihydroxyvitamin D3/vitamin D receptor suppresses brown adipocyte differentiation and mitochondrial respiration. Eur J Nutr 2015;54:1001–1012. [DOI] [PubMed] [Google Scholar]
- 41. Fan Y, Futawaka K, Koyama R, Hayashi M, Imanmoto M, Miyawaki T, et al. Vitamin D3/VDR resists diet‐induced obesity by modulating UCP3 expression in muscle. J Biomed Sci 2016;23:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012;150:366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mulya A, Kirwan JP. Brown and beige adipose tissue, therapy for obesity and its comorbidities? Endocrinol Metab Clin North Am 2016;45:605–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Petruzzelli M, Wagner EF. Mechanisms of metabolic dysfunction in cancer cachexia. Genes Dev 2016;30:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, et al. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab 2016;23:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vegiopoulos A, Müller‐Decker K, Strzoda D, Schmitt I, Chicheknitskiy E, Ostertag A, et al. Cyclooxygenase‐2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 2010;328:1158–1161. [DOI] [PubMed] [Google Scholar]
- 47. Barbatelli G, Murano I, Madsen L, Hao Q, Jimenez M, Kristansen K, et al. The emergence of cold‐induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am J Physiol Ebdocrinol Metab 2010;298:E1244–E1253. [DOI] [PubMed] [Google Scholar]
- 48. Dusso A, González EA, Martin KJ. Vitamin D in chronic kidney disease. Best Pract Res Clin Endocrinol Metab 2011;25:647–655. [DOI] [PubMed] [Google Scholar]
- 49. Yin K, Agrawal DK. Vitamin D and inflammatory diseases. J Inflamm Res 2014;7:69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu LC, Voors AA, van Veldhuisen DJ, van der Veer E, Belonje AM, Szymanski MK, et al. Vitamin D status and outcomes in heart failure patients. Eur J Heart Fail 2011;13:619–625. [DOI] [PubMed] [Google Scholar]
- 51. Björkhem‐Bergman L, Nylén H, Norlin AC, Lindh JD, Ekström L, Eliasson E, et al. Serum Levels of 25‐Hydroxyvitamin D and the CYP3A Biomarker 4β‐Hydroxycholesterol in a High‐Dose Vitamin D Supplementation Study. Drug Metab Dispos 2013;41:704–708. [DOI] [PubMed] [Google Scholar]
- 52. Zhang Y, Leung DY, Richers BN, Liu Y, Regigio LK, Riches DW, et al. Vitamin D Inhibits Monocyte/Macrophage Proinflammatory Cytokine Production by Targeting MAPK Phosphatase‐1. J Immunol 2012;188:2127–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mayne CG, Spanier JA, Relland LM, Williams CB, Hayes CE. 1,25‐dihydroxyvitamin D3 acts directly on the T lymphocyte vitamin D receptor to inhibit experimental autoimmune encephalomyelitis. Eur J Immunol 2011;41:822–832. [DOI] [PubMed] [Google Scholar]
- 54. Edfeldt K, Liu PT, Chun R, Fabri M, Schenk M, Wheelwright M, et al. T‐cell cytokines differentially control human monocyte antimicrobial responses by regulating vitamin D metabolism. Proc Natl Acad Sci U S A 2010;107:22593–22598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tamura Y, Fujito H, Kawao N, Kaji H. Vitamin D deficiency aggravates diabetes‐induced muscle wasting in female mice. Diabetol Int 2017;8:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Akagawa M, Miyakoshi N, Kasukawa Y, Ono Y, Yuasa Y, Nagahata I, et al. Effects of activated vitamin D, alfacalcidol, and low‐intensity aerobic exercise on osteopenia and muscle atrophy in type 2 diabetes mellitus model rats. PLoS ONE 2018;13:e0204857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad USA 2001;98:9306–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Garcia LA, King KK, Ferrini MG, Norris KC, Artaza JN. 1,25(OH)2 vitamin D3 stimulates myogenic differentiation by inhibiting cell proliferation and modulating the expression of promyogenic growth factors and myostatin in C2C12 skeletal muscle cells. Endocrinology 2011;152:2976–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Braga M, Simmons Z, Norris KC, Ferrini MG, Artaza JN. Vitamin D induces myogenic differentiation in skeletal muscle derived stem cells. Endocr Connect 2017;6:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tsukamoto Y, Senda T, Nakano T, Nakada C, Hida T, Ishiguro N, et al. Arpp, a new homolog of carp, is preferentially expressed in type 1 skeletal muscle fibers and is markedly induced by denervation. Lab Invest 2002;82:645–655. [DOI] [PubMed] [Google Scholar]
- 61. Vafiadaki E, Arvanitis DA, Papalouka V, Terzis G, Roumeliotis TI, Spengos K, et al. Muscle lim protein isoform negatively regulates striated muscle actin dynamics and differentiation. FEBS J 2014;281:3261–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sheikh F, Lyon RC, Chen J. Functions of myosin light chain‐2 in cardiac muscle and disease. Gene 2015;569:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nghiem PP, Kornegay JN, Uaesoontrachoon K, Bello L, Yin Y, Kesari A, et al. Osteopontin is linked with AKT, Foxo1, and myostatin in skeletal muscle cell. Muscle Nerve 2017;56:1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Juretić N, Urzúa U, Munroe DJ, Jaimovich E, Riveros N. Differential gene expression in skeletal muscle cells after membrane depolarization. J Cell Physiol 2007;210:819–830. [DOI] [PubMed] [Google Scholar]
- 65. Lin IH, Chang JL, Hua K, Huang WC, Hsu MT, Chen YF. Skeletal muscle in aged mice reveals extensive transformation of muscle gene expression. BMC Genet 2018;19:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kami K, Noguchi K, Senba E. Localization of myogenin, c‐fos, c‐jun, and muscle‐specific gene mRNAs in regenerating rat skeletal muscle. Cell Tissue Res 1995;280:11–19. [DOI] [PubMed] [Google Scholar]
- 67. Lindå H, Sköld MK, Ochsmann T. Activating transcription factor 3, a useful marker for regenerative response after nerve root injury. Front Neurol 2011;2:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hui X, Zhu W, Wang Y, Lam KS, Zhang J, Wu D, et al. Major Urinary Protein‐1 Increases Energy Expenditure and Improves Glucose Intolerance through Enhancing Mitochondrial Function in Skeletal Muscle of Diabetic Mice. J Biol Chem 2009;284:14050–14057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bal NC, Sahoo SK, Maurya SK, Periasamy M. The Role of Sarcolipin in Muscle Non‐shivering Thermogenesis. Front Physiol 2018;9:1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhou Z, Yon Toh S, Chen Z, Guo K, Ng CP, Ponniah S, et al. Cidea‐deficient mice have lean phenotype and are resistant to obesity. Nat Genet 2003;35:49–56. [DOI] [PubMed] [Google Scholar]
- 71. Millership S, Ninkina N, Rochford JJ, Buchman VL. γ‐synuclein is a novel player in the control of body lipid metabolism. Adipocyte 2013;2:276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hidestrand M, Richards‐Malcolm S, Gurley CM, Nolen G, Grimes B, Waterstrat A, et al. Sca‐1‐expressing nonmyogenic cells contribute to fibrosis in aged skeletal muscle. J Gerontol A Biol Sci Med Sci 2008;63:566–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Saller E, Tom E, Brunori M, Otter M, Estreicher A, Mack DH, et al. Increased apoptosis induction by 121F mutant p53. EMBO J 1999;18:4424–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. An D, Toyoda T, Taylor EB, Yu H, Fujii N, Hirshman MF, et al. TBC1D1 regulates insulin‐ and contraction‐induced glucose transport in mouse skeletal muscle. Diabetes 2010;59:1358–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tan BH, Fladvad T, Braun TP, Vigano A, Strasser F, Deans DA, et al. P‐selectin genotype is associated with the development of cancer cachexia. EMBO Mol Med 2012;4:462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)‐dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J 2003;375:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bodié K, Buck WR, Pieh J, Liguori MJ, Popp A. Biomarker evaluation of skeletal muscle toxicity following clofibrate administration in rats. Exp Toxicol Pathol 2016;68:289–299. [DOI] [PubMed] [Google Scholar]
- 78. Lee JY, Lori D, Wells DJ, Kemp PR. FHL1 activates myostatin signaling in skeletal muscle and promotes atrophy. FEBS Open Bio 2015;5:753–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yuen M, Cooper ST, Marston SB, Nowak KJ, McNamara E, Mokbel N, et al. Muscle weakness in TPM3‐myopathy is due to reduced Ca2+ sensitivity and impaired acto‐myosin cross‐bridge cycling in slow fibres. Hum Mol Genet 2015;24:6278–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Immunoassay information for serum chemistry, adipose and muscle tissue homogenate assay analysis.
Table S2. PCR primer information.
Table S3. Serum and blood chemistry of mice. Twelve‐month‐old, male, wild type (WT) and Ctns‐/‐ mice were treated with 25(OH)D3 (75 μg/kg per day) or 1,25(OH)2D3 (60 ng/kg per day) versus ethylene glycol as vehicle for 6 weeks. Six groups of mice were included: WT + Vehicle, WT + 25(OH)D3, WT++1,25(OH)2D3, Ctns‐/‐ + Vehicle, Ctns‐/‐ + 25(OH)D3, Ctns‐/‐ + 1,25(OH)2D3. All mice were fed ad libitum. Data are expressed as mean SEM. Ap < 0.05, significantly higher in Ctns‐/‐ + Vehicle, Ctns‐/‐ + 25(OH)D3 or Ctns‐/‐ +1,25(OH)2D3 versus WT + Vehicle, WT + 25(OH)D3 or WT + 1,25(OH)2D3, respectively. Bp < 0.05, significantly lower in Ctns‐/‐ + Vehicle, Ctns‐/‐ + 25(OH)D3 or Ctns‐/‐ + 1,25(OH)2D3 versus WT + Vehicle, WT + 25(OH)D3 or WT + 1,25(OH)2D3, respectively. cp < 0.05, significantly difference between Ctns‐/‐ + 25(OH)D3 versus Ctns‐/‐ + Vehicle or Ctns‐/‐ + 1,25(OH)2D3 versus Ctns‐/‐ + Vehicle.
Table S4. List of differential expressed genes in gastrocnemius muscle from Ctns‐/‐ + Vehicle versus WT + Vehicle mice
Table S5. List of differential expressed genes in gastrocnemius muscle from Ctns‐/‐ + 25(OH)D3 + 1,25(OH)2D3 versus WT + 25(OH)D3 + 1,25(OH)2D3 mice
Table S6. List of differential expressed genes in gastrocnemius muscle from Ctns‐/‐ + 25(OH)D3+ 1,25(OH)2D3 versus Ctns‐/‐ + Vehicle mice
Figure S1. Ad libitum food intake and weight gain in Ctns‐/‐ mice.