

Graphical abstract

Keywords: Thyroxine, Bisphosphonate, Mutation, Insulin, Skeleton, Growth
Highlights
-
•
The mutations in two different genes should be sought in the patients with complex phenotypes.
-
•
The c.1531G>T in COL1A2 leading to OI and c.364C>T (p.R122W) in LHX4 to CPHD were found in a Thai boy.
-
•
The incomplete penetrance and loss-of-function are the features of p.R122W mutation in LHX4.
-
•
The mutation spectra of COL1A2 and LHX4 and pathomechanism of LHX4 are expanded.
Abstract
Genetic disorders have been shown to co-occur in individual patient. A Thai boy with features of osteogenesis imperfecta (OI) and combined pituitary hormone deficiency (CPHD) was identified. The causative mutations were investigated by whole exome and Sanger sequencing. Pathogenicity and pathomechanism of the variants were studied by luciferase assay. The proband was found to harbor a novel de novo heterozygous missense mutation, c.1531G > T (p.G511C), in COL1A2 leading to OI and a heterozygous missense variant, c.364C > T (p.R122W), in LHX4. The LHX4 p.R122W has never been reported to cause CPHD. The variant was predicted to be deleterious and found in the highly conserved LIM2 domain of LHX4. The luciferase assays revealed that the p.R122W was unable to activate POU1F1, GH1, and TSHB promoters, validating its pathogenic effect in CPHD. Moreover, the variant did not alter the function of wild-type LHX4, indicating its hypomorphic pathomechanism. In conclusion, the novel de novo heterozygous p.G511C mutation in COL1A2 and the heterozygous pathogenic p.R122W mutation in LHX4 were demonstrated in a patient with OI and CPHD. This study proposes that the mutations in two different genes should be sought in the patients with clinical features unable to be explained by a mutation in one gene.
Introduction
Osteogenesis imperfecta (OI) is a rare disease characterized by bone fragility. The prevalence of OI is 6-7/100,000 [1]. The disease is inherited in an autosomal dominant, autosomal recessive, or X-linked recessive manner. Alterations in at least 18 genes have been associated with OI [1], [2]. Mutations in COL1A1 or COL1A2, which encode the pro-alpha1 or pro-alpha2 chain of type I collagen, account for more than 85% of disease-causing variants. Glycine substitutions within the Gly-X-Y repeats of collagen chains are the most common type of mutations leading to abnormal collagen structure [1].
Combined pituitary hormone deficiency (CPHD) is a condition in which the pituitary gland produces insufficient amounts of several hormones, including growth hormone (GH), prolactin production (PRL), luteinizing hormone (LH), follicle-stimulating hormone (FSH), adrenocorticotropic hormone (ACTH), and/or thyroid-stimulating hormone (TSH). Its prevalence is 1/8000 [3]. Up to 2500 variants in 30 genes including PROP1, POU1F1, HESX1, OTX2, GLI2, LHX3, and LHX4 have been associated with CPHD [3]. The LHX4, which is the LIM homeodomain transcription factor, plays an important role in the development of anterior pituitary gland and nervous system. Its expression is found in the Rathke’s pouch. It can regulate POU1F1, GH1, PRL, αGSU, FSHB, and TSHB genes [4]. To date, only thirteen of LHX4 variants have been investigated for their effects in CPHD [3], [4], [5], [6].
A Thai boy manifesting the combined features of OI and CPHD was identified. The study aimed to identify the causative mutations leading to two different Mendelian diseases and to investigate the pathogenicity and pathomechanism of the identified variant causing CPHD.
Materials and methods
Patient characterization and mutation analysis
A Thai boy diagnosed with both OI and CPHD and his parents were recruited. The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (IRB500/61), Faculty of Medicine, Chulalongkorn University. Written informed consents for publication of their clinical details and images were obtained from the participants. Genomic DNA isolated from the peripheral blood was subjected for mutation analyses using whole exome sequencing (WES) according to previous publication [7]. The identified variants were validated using Sanger sequencing.
Pathogenic effect of LHX4 variants
The pTracer-LHX4_WT-HA was a gift from Marie Legendre, France. The LHX4 mutant vectors of p.R122W (the mutation identified in this study) and of two previously reported p.T163P [4] and p.L190R [5] were generated using Q5® Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA). Three luciferase reporter vectors were selected. The proximal promoter regions of human GH1, POU1F1, or TSHB were amplified and cloned into the pGL4.10[luc2] vector (Supplementary Table 1) [4], [5]. Commercial Chinese hamster ovary (CHO)-K1 cells (ATCC®CCL-61TM) were used for transient transfection using X-tremeGENE 9 DNA Transfection Reagent (Roche, Mannheim, Germany).
Pathomechanism of LHX4 variants
The CHO-K1 cells were cotransfected with an equal amount of wild-type and each mutant LHX4. The empty pTracer-CMV expression vector was used as the control. POU1F1 promoter was selected for this experiment. After 48 h, the transcription activity was measured using the Luciferase Assay System (Promega, Madison, WI) and SpectraMax M3 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA). Total amount of protein was measured by the Pierce™ BCA Protein Assay Kit (Thermo Scientific, Rockford, IL). The results of three independent experiments were reported as mean ± SD. The P-value was <0.01.
Western blot analyses
Protein extracted from the cellular lysates of CHO-K1 cells transfected with LHX4 expression vectors were prepared. 20 ug of protein per sample was separated on 12% sodium dodecyl sulfate polyacrylamide gels and then transferred by iBlot™ Transfer Stack, PVDF, regular size (Invitrogen, Carlsbad, CA). Monoclonal anti-HA antibody at 1:250 dilution (cat no. H3663, Sigma-Aldrich, St louis, MO) was used as the primary antibody to detect LHX4-HA proteins, followed by anti-mouse IgG antibody at 1:2000 dilution (cat no. 7076, Cell Signaling Technology, Danvers, MA) as the secondary antibody. Anti-actin antibody (cat no. Mab1501, Sigma-Aldrich, St louis, MO) was used as a positive control to determine gel loading equivalency. Results were visualized using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce Biotechnology, Rockford, IL) and chemiluminescence camera (ImageQuant LAS 4000, Amersham).
Results
Patient characteristics
A 6-year-old Thai boy was born at 37 weeks by caesarian section due to breech presentation, intrauterine growth retardation, and oligohydramnios. His birth weight was 1,840 g (<3rd centile) and length was 41 cm (<3rd centile). His head circumference was 32 cm (25–50th centile). Apgar scores at 1 and 5 min were 5 and 10, respectively. At birth, the proband exhibited deformed limbs, bowed legs, wide anterior fontanelle (4x4 cm), frontal bossing, and blue sclerae. He also developed neonatal sepsis, seizure, and respiratory distress caused by subcostral retraction and chest wall instability. Fractures of his left humerus and angulation of long bones was radiologically observed (Fig. 1A, B). The result of hearing test was normal. Intravenous pamidronate (7.2 mg/kg/year) was started at the age of 2 months and then administered every 2 months. He started to walk unaided at 2 years of age but showed an abnormal gait. Pectus carinatum and flat feet were observed at the age of 3 years (Fig. 1C, D). His primary teeth showed dentinogenesis imperfecta and were severely deteriorated (Fig. 1E, F). He broke his right femur at the age of 5 years 11 months and his left femur at 6 years 2 months caused by falls (Fig. 1G, H). Bone density scans showed that his lumbar spine BMD at the age of 19 months was 0.343 g/cm2 (<-3SD) and at the age of 4 years was 0.491 g/cm2 (-2SD, +42.9%).
Fig. 1.

Phenotype and genotype of the proband. (A, B) Radiographs at birth showed the fractures of the left humerus and deformities of lower extremities (C, D) The proband at 6 years of age exhibited pectus carinatum and flat feet. (E, F) Oral photographs and radiographs at the age of 4 years showed dentinogenesis imperfecta. (G, H) Radiographs showed right femur fracture at the age of 5 years 11 months and left femur fracture at the age of 6 years 2 months. (I) Growth curve from birth to 3 years of age. (J, K) Brain MRI showed hypoplasia of the pituitary gland. (L) Growth curve from 2.5 to 6.5 years of age. Red arrow indicated the initiation of growth hormone and thyroid hormone therapy. (M) Sanger sequencing showed a novel de novo heterozygous missense p.G511C (c.1531G > T) mutation in COL1A2 and a heterozygous missense p.R122W (c.364C > T) variant in LHX4 which was inherited from his healthy father. (N) Alignment of the amino acid sequence of LHX4 among several species. (O) Schematic diagram of LHX4. The top panel showed the p.R122W variant identified in this study. The bottom panel showed the variants previously reported with functional studies. (P) Family pedigree of the proband. Symbol filled with black represents a subject with osteogenesis imperfecta; symbol filled with gray represents a subject with combined pituitary hormone deficiency; and empty symbols represent healthy subject. An arrow indicates the proband. Underlined letters are genotypes of the COL1A2 while those which are not underlined are genotypes of LHX4. W, wild-type allele.
Apart from short stature and bone fragility which are the main features of OI, slow growth velocity was noticed (Fig. 1I). Physical examination of the proband at the age of 3 years 5 months showed that his upper lower body ratio was 1.19:1 and arm span was 72 cm. Further endocrine evaluation revealed that the proband had low level of free thyroxine (FT4) (0.51 ng/dl; normal 1.0–1.8). The levels of insulin-like growth factor-1 (IGF-1) and insulin-like growth factor-binding protein 3 (IGFBP3) were in the low end of normal range. Growth hormone (GH) stimulation test showed the peak value of GH at 1.01 ng/ml (normal > 10). The levels of cortisol and adrenocorticotropic hormone (ACTH) were within normal limit (Table 1). Renal function test and blood biochemistry were normal. Brain MRI identified the hypoplastic pituitary gland and stalk and mildly diffused parenchymal volume loss (Fig. 1J, K). Based on the clinical and laboratory investigations, the proband was diagnosed with the deficiencies of growth hormone and thyroid hormone, indicating CPHD. The administration of growth hormone (Norditropin® 0.3 mg/day subcutaneously) and thyroid hormone (Eltroxin, 25 ug/day) was started when he was 3 years 9 months. Since hormonal replacement was initiated, his growth had been improved (Fig. 1L).
Table 1.
Hormonal tests of the proband.
| Treatment | Age | TSH mIU/l | FT4 (ng/dl) | FT3 (pg/dl) | IGF1 (ng/ml) | IGFBP3 (ug/ml) | GH (ng/ml) | Cortisol (ug/dl) | ACTH (pg/ml) |
|---|---|---|---|---|---|---|---|---|---|
| No treatment | 3 days | 7.52 (0.7–15.2) | 1.02 (0.9–2.5) | – | – | – | – | – | – |
| 3 years 5 months | 4.3 (0.7–6.0) | 0.51* (1.0–1.8) | – | <25 (22–229) | 0.9 (0.9–4.3) | – | 4.2* (5–25) | – | |
| 3 years 6 months | 5.09 (0.7–6.0) | 0.62* (1.0–1.8) | – | – | – | 0.50* [1] (>10) | 6.2 [1] (5–25) | 29.9 (7.2–63) | |
| – | – | – | – | – | 0.66* [2] (>10) | 16.6 [2] (5–25) | – | ||
| – | – | – | – | – | 0.88* [3] (>10) | – | – | ||
| – | – | – | – | – | 0.85* [4] (>10) | – | – | ||
| – | – | – | – | – | 1.01* [5] (>10) | – | – | ||
| Norditropin® and Eltroxin | 3 years 9 months | <0.005 (0.7–6.0) | 1.31 (1.0–1.8) | 4.68 (2.8–4.4) | – | – | – | – | – |
| 3 years 11 months | – | 1.11 (1.0–1.8) | – | <25 (22–229) | 1.2 (0.9–4.3) | – | – | – | |
| 5 years 10 months | – | – | – | 45 (39–250) | 2.4 (1.1–5.2) | – | – | – | |
| 6 years 6 months | <0.0025 (0.6–4.8) | 1.38 (1.0–1.7) | – | 107 (47–275) | – | – | – | – | |
| 6 years 9 months | – | 1.41 (1.0–1.7) | – | – | – | – | – | – | |
| 6 years 10 months | – | 1.77 (1.0–1.7) | – | 79.6 (47–275) | – | – | – | – | |
*not in normal range
Numbers in parentheses are normal ranges.
The proband was the only son of non-consanguineous and healthy Thai parents. They were 31 years of age when the proband was born. Paternal height was 164.8 cm and maternal height was 159 cm. Physical and dental manifestations of the parents were unremarkable. The proband’s father had normal growth and no infertility problems. His sexual maturity was at Tanner stage V. Endocrine evaluation of the father at age 37 years showed normal levels of IGF1, FT4, thyroid stimulating hormone (TSH), follicle stimulating hormone (FSH), and testosterone.
Mutation analyses
Exome sequencing revealed that the proband possessed a novel de novo heterozygous missense mutation, c.1531G > T (p.G511C), in COL1A2. The variant was not found in ExAC and our in-house database of 1,876 Thai exomes. Several lines of evidences have supported the pathogenicity of the COL1A2 p.G511C. The variant was 1) de novo which is a strong evidence of its etiologic role, 2) absent from controls in multiple variant databases and in-house database, 3) highly conserved among several species, 4) predicted to be deleterious based on multiple lines of computational evidences, 5) corresponding to the patient’s phenotype for OI, and 6) located in the triple helical domain of alpha 2 chains of type I collagen. According to American College of Medical Genetics and Genomics (ACMG) guidelines, the p.G511C variant is considered pathogenic [8]. Changes of glycine, the smallest amino acid, in Gly-X-Y triplets of collagen chain have been shown to disturb triple helical assembly and collagen chain stability [9], [10]. These therefore provide compelling evidence that p.G511C in COL1A2 is the causative variant of OI in the proband. In addition, a missense variant, c.364C > T (p.R122W), in LHX4 was detected in the proband and his healthy father (Fig. 1M). The p.R122W variant was found in 1 out of 121,208 alleles in ExAC. However, it was not observed in our in-house exome database and had never been reported to cause any diseases. The p.R122W was highly conserved among several species and located in the LIM2 domain of LHX4 (Fig. 1N, O). Both COL1A2 and LHX4 variants were predicted to be deleterious (Sorting Intolerant From Tolerant/SIFT), probably damaging (Polymorphism Phenotyping/PolyPhen), and possibly pathogenic (Mendelian Clinically Applicable Pathogenicity/M-CAP). The phenotypic and genotypic findings of the proband lead to the diagnosis of combined OI and CPHD (Fig. 1P). Mutation analyses were demonstrated in Table 2, Table 3.
Table 2.
Filtering criteria for the exome sequencing of the proband.
| Total number of variants after exclusion of variants with quality score <20 | 87,959 | ||||
| After exclusion of variants with read depth <10 | 72,310 | ||||
| After exclusion of variants not in or close to the coding regions | 12,478 | ||||
| After exclusion of variants with allele frequency >1% in the database (Exome Variant Server, Exome Aggregation Consortium, 1000 Genomes Project Consortium, dbSNPs) | 1,969 | ||||
| After exclusion of variants found in an in-house database of 1,876 Thai exomes | 726 | ||||
| After selection of variants associated with phenotypes (Supplementary Table 4) | osteogenesis imperfecta | combined pituitary hormone deficiency | |||
| 1 | 4 | ||||
| Gene | COL1A2 | LHX4 | WNT4 | ALK | IGF1R |
| Variant | G > G/T | C > C/T | G > G/A | T > T/C | G > G/A |
| Coordinate | 7:94042422 | 1:180235642 | 1:22447789 | 2:29449866 | 15:99473472 |
| Genotype | heterozygous | heterozygous | heterozygous | heterozygous | heterozygous |
| Transcript | NM_000089.3 | NM_033343.3 | NM_030761.4 | NM_004304.4 | NM_000875.3 |
| Consequence | missense variant | missense variant | missense variant | missense variant | missense variant |
| cDNA | c.1531G > T | c.364C > T | c.503C > T | c.2989A > G | c.2894G > A |
| Protein | p.G511C | p.R122W | p.S168L | p.M997V | p.S965N |
| Sift | deleterious (0) | deleterious (0) | deleterious (0.042) | tolerated (0.184) | tolerated (0.438) |
| PolyPhen-2 | probably damaging (1) | probably damaging (1) | possibly damaging (0.867) | benign (0.001) | benign (0) |
| M-CAP | possibly pathogenic (0.956) | possibly pathogenic (0.196) | possibly pathogenic (0.096) | likely benign (0.021) | likely benign (0.011) |
| Associated diseases | OI | CPHD | Mullerian aplasia and hyperandrogenism | Neuroblastoma, susceptibility to, 3 | Insulin-like growth factor I, resistance to |
| SERKAL syndrome | |||||
Table 3.
Gene lists associated with osteogenesis imperfecta and combined pituitary hormone deficiency.
| Phenotype | Genes |
|---|---|
| Osteogenesis imperfecta | FKBP10, LEPRE1, PPIB, BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, IFITM5, MBTPS2, PLOD2, SERPINF1, SERPINH1, SP7, TMEM38B, WNT1, SEC24D, SPARC |
| Combined pituitary hormone deficiency | AAAS, AANAT, AES, ALK, ANXA1, AR, ARNT2, BGLAP, BLVRB, CDA, CRH, CSF2, CSHL1, CUL4B, CYP19A1, DRD2, ELANE, EPO, ERBB2, ESR1, F2, FOXA2, GH1, GH2, GHR, GHRH, GHRHR, GHSR, GLI2, GLIS3, HESX1, IGF1, IGF1R, IGFBP3, IGHD, IGSF1, INSM1, INSR, KLK3, LHX3, LHX4, MC2R, MCHR1, MRAP, NBEA, NF1, NKX2-1, NKX2-5, NOG, OTP, OTX2, PAX8, PLAT, POMC, POU1F1, POU3F2, PRL, PROP1, PTTG1IP, REN, SERPINA1, SHOX, SHOXY, SIM1, SIX6, SLC6A3, SOX2, SOX3, SST, STAT5B, SYTL4, TBG, THRA, TNFSF11, TRH, TSHB, TSHR, VWF, WNT4, XRCC4, ZIC2 (HP:0000871, HP:0000824, HP:0000851) |
Pathogenicity of the LHX4 missense variant, p.R122W
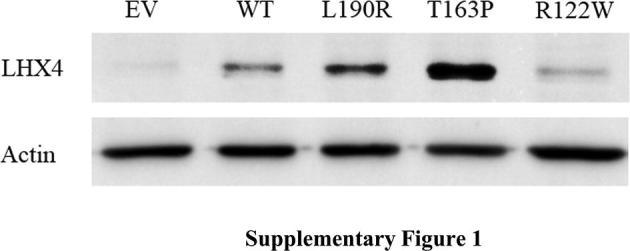
Western blotting showed that the wild-type, p.L190R, p.T163P, and p.R122W LHX4 proteins were detected at similar sizes (50 kDa) (Supplementary Fig. 1). The luciferase assays showed that the wild-type LHX4 was able to activate the POU1F1, GH1, and TSHB promoters. In contrast, the luciferase activities of the p.R122W variant and two previously reported mutations, p.T163P and p.L190R, were significantly lower than the wild-type (Fig. 2A–C).
Fig. 2.

Luciferase reporter assays demonstrating pathogenicity and pathomechanism of the p. R122W variant in LHX4. (A–C) The LHX4 mutations reduced the transcriptional activities of POU1F1, GH1, and TSHB promoters, compared to the wild-type LHX4. (D) The mutations did not show a dominant negative effect on the wild-type LHX4. EV, empty vector; WT, wild-type LHX4; p.L190R and p.T163P, the variants previously reported; p.R122W, the variant identified in this study; (A-C) P < 0.01 compared with WT (*); (D) P < 0.01 compared with WT + EV (*).
Pathomechanism of the LHX4 missense variant, p.R122W
Cotransfection of wild-type and each mutant LHX4 (p.R112W, p.T163P, and p.L190R) showed comparable luciferase activities to the cotransfection of wild-type LHX4 and empty pTracer-CMV expression vector. These suggest that these LHX4 mutations do not have a dominant negative effect on the wild-type function and they are likely to cause the disease by loss-of-function mechanism (Fig. 2D).
Discussion
A Thai boy with the complex phenotypes was identified. His main features including short stature, bone deformity, skeletal fracture, low BMD, blue sclerae, and dentinogenesis imperfecta led to the primary diagnosis of OI. After intravenous pamidronate was administered every 2 months, his BMD had increased. However, low height velocity was observed. Further laboratory investigations showing pituitary hypoplasia and deficiencies of GH and TSH indicated the secondary diagnosis of CPHD. These suggest that attentive monitoring a child’s growth benefits an early diagnosis of combined rare diseases. In addition, the replacement therapy with GH apart from IV bisphosphonate results in an increased height velocity for this patient.
More than forty OI patients have been identified in our Genetics Clinic of King Chulalongkorn Memorial Hospital. Those were associated with the mutations in various genes including COL1A1, BMP1, P4HB, WNT1, and MBTPS2 [2], [11], [12], [13]. In the proband, WES analysis revealed the novel de novo heterozygous missense mutation, c.1531G > T (p.G511C), in COL1A2, corresponding to OI. This COL1A2 variant was not found in his healthy parents. Glycine is considered to be the smallest amino acid allowing it to be incorporated in the triple helix structure of collagen. Therefore, the change of glycine to cysteine is expected to impede the folding and formation of collagen resulting in abnormal collagen structure.
The complex phenotype of the proband was unexplainable by the single mutation in COL1A2. He was the only one out of our 40 Thai OI patients that exhibited CPHD features. Further genetic investigation revealed that the proband also possessed the heterozygous missense mutation, c.364C > T (p.R122W), in LHX4, which was inherited from his healthy father. Interestingly, the mutations in LHX4 causing CPHD have been reported with a high rate of incomplete penetrance [4], [14]. The pathogenicity of LHX4 variant, p.R122W, was then investigated. The p.R122W was predicted to be deleterious and located in the highly conserved LIM2 domain playing important role in protein-protein interactions [15]. Using the luciferase reporter assays, the LHX4 p.R122W was shown to lose its ability to activate POU1F1, GH1, and TSHB promoters, validating its pathogenicity. These explain the deficiencies of GH and TSH found in the proband. Western blot analyses showed that the wild-type and mutant LHX4 proteins of expected size (50 kDa) were detected. The level of expression of p.R122W was lower than that of the wild-type. However, this might not be the cause for its low level of luciferase activity as the control mutants that were expressed at higher levels also showed low activities. Next, the pathomechanism study of p.R122W showed that it did not interfere with the function of wild-type LHX4, similar to the other two LHX4 mutations, L190R and T163P. These indicate that it possesses loss-of-function or haploinsufficiency mechanism rather than dominant negative effect in CPHD. Notably, LHX4 has been proposed to have random monoallelic expression [16]. The single expression of normal LHX4 allele could therefore explain the absence of disease in the proband’s father who also carries the heterozygous p.R122W variant.
Patients with mutations in two genes leading to clinical manifestations of two or more Mendelian disorders are not uncommon. Recently, it was reported that these patients accounted for five percent of individuals with informative exome [17].
In conclusion, this study demonstrated the first patient with combined OI and CPHD. While OI was caused by the de novo heterozygous COL1A2 mutation, CPHD was by the heterozygous LHX4 mutation inherited from the healthy father. The incomplete penetrance and loss-of-function are the characteristics of p.R122W mutation in LHX4. This study expands the mutation spectra of COL1A2 and LHX4 and demonstrates the pathogenicity of the LHX4 p.R122W mutation. We propose here that exome sequencing could be a promising tool to discover pathogenic variants for complex phenotypes leading to precise diagnosis of combined disorders.
Compliance with ethics requirements
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008 (5). Informed consent was obtained from all patients for being included in the study.
Author’s contributions
Hemwong N, Shotelersuk V, Porntaveetus T contributed to study design, data analysis, and drafting the manuscript; Phokaew C, Srichomthong C, Tongkobpetch S, Suphapeetiporn K contributed to analysis and interpretation of data; Srilanchakon K, Supornsilchai V contributed to patient’s examination and data analysis. All authors revised the manuscript critically, gave final approval, and agreed to be accountable for all aspects of the work.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This study was supported by the Thailand Research Fund (RSA6280001, DPG6180001), Ratchadapisek Sompoch Endowment Fund (2019) under Medical Genomics Cluster, Chulalongkorn Academic Advancement Into Its 2nd Century Project, Faculty of Dentistry, Chulalongkorn University (DRF62003), and Newton Fund. We are grateful to Dr Marie Legendre, U.F. de Génétique Moléculaire, Hôpital Trousseau, France for giving us the pTracer-LHX4_WT-HA vector to use in the experiments.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2019.10.006.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
Supplementary Fig. 1.
References
- 1.Marini J.C., Forlino A., Bächinger H.P., Bishop N.J., Byers P.H., Paepe A.D. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. doi: 10.1038/nrdp.2017.52. [DOI] [PubMed] [Google Scholar]
- 2.Lindert U., Cabral W.A., Ausavarat S., Tongkobpetch S., Ludin K., Barnes A.M. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun. 2016;7:11920. doi: 10.1038/ncomms11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fang Q., George A.S., Brinkmeier M.L., Mortensen A.H., Gergics P., Cheung L.Y. Genetics of combined pituitary hormone deficiency: roadmap into the genome era. Endocr Rev. 2016;37(6):636–675. doi: 10.1210/er.2016-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen E., Maghnie M., Collot N., Leger J., Dastot F., Polak M. Contribution of LHX4 mutations to pituitary deficits in a cohort of 417 unrelated patients. J Clin Endocrinol Metab. 2017;102(1):290–301. doi: 10.1210/jc.2016-3158. [DOI] [PubMed] [Google Scholar]
- 5.Pfaeffle R.W., Hunter C.S., Savage J.J., Duran-Prado M., Mullen R.D., Neeb Z.P. Three novel missense mutations within the LHX4 gene are associated with variable pituitary hormone deficiencies. J Clin Endocrinol Metab. 2008;93(3):1062–1071. doi: 10.1210/jc.2007-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gregory L.C., Humayun K.N., Turton J.P., McCabe M.J., Rhodes S.J., Dattani M.T. Novel lethal form of congenital hypopituitarism associated with the first recessive LHX4 mutation. J Clin Endocrinol Metab. 2015;100(6):2158–2164. doi: 10.1210/jc.2014-4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porntaveetus T., Srichomthong C., Suphapeetiporn K., Shotelersuk V. Monoallelic FGFR3 and Biallelic ALPL mutations in a Thai girl with hypochondroplasia and hypophosphatasia. Am J Med Genet A. 2017;173(10):2747–2752. doi: 10.1002/ajmg.a.38370. [DOI] [PubMed] [Google Scholar]
- 8.Clements K.A., Acevedo-Jake A.M., Walker D.R., Hartgerink J.D. Glycine substitutions in collagen heterotrimers alter triple helical assembly. Biomacromolecules. 2017;18(2):617–624. doi: 10.1021/acs.biomac.6b01808. [DOI] [PubMed] [Google Scholar]
- 9.Wenstrup R.J., Shrago-Howe A.W., Lever L.W., Phillips C.L., Byers P.H., Cohn D.H. The effects of different cysteine for glycine substitutions within alpha 2(I) chains. Evidence of distinct structural domains within the type I collagen triple helix. J Biol Chem. 1991;266(4):2590–2594. [PubMed] [Google Scholar]
- 10.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porntaveetus T., Theerapanon T., Srichomthong C., Shotelersuk V. Cole-Carpenter syndrome in a patient from Thailand. Am J Med Genet A. 2018;176(8):1706–1710. doi: 10.1002/ajmg.a.40358. [DOI] [PubMed] [Google Scholar]
- 12.Tongkobpetch S., Limpaphayom N., Sangsin A., Porntaveetus T., Suphapeetiporn K., Shotelersuk V. A novel de novo COL1A1 mutation in a Thai boy with osteogenesis imperfecta born to consanguineous parents. Genet Mol Biol. 2017;40(4):763–767. doi: 10.1590/1678-4685-GMB-2016-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuptanon C., Srichomthong C., Sangsin A., Kovitvanitcha D., Suphapeetiporn K., Shotelersuk V. The most 5' truncating homozygous mutation of WNT1 in siblings with osteogenesis imperfecta with a variable degree of brain anomalies: a case report. BMC Med Genet. 2018;19(1):117. doi: 10.1186/s12881-018-0639-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gucev Z., Tasic V., Plaseska-Karanfilska D., Konstantinova M.K., Stamatova A., Dimishkovska M. LHX4 gene alterations: patient report and review of the literature. Pediatr Endocrinol Rev. 2016;13(4):749–755. [PubMed] [Google Scholar]
- 15.Gadd M.S., Bhati M., Jeffries C.M., Langley D.B., Trewhella J., Guss J.M. Structural basis for partial redundancy in a class of transcription factors, the LIM homeodomain proteins, in neural cell type specification. J Biol Chem. 2011;286(50):42971–42980. doi: 10.1074/jbc.M111.248559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savova V., Chun S., Sohail M., McCole R.B., Witwicki R., Gai L. Genes with monoallelic expression contribute disproportionately to genetic diversity in humans. Nat Genet. 2016;48(3):231–237. doi: 10.1038/ng.3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Posey J.E., Harel T., Liu P., Rosenfeld J.A., James R.A., Coban Akdemir Z.H. Resolution of disease phenotypes resulting from multilocus genomic variation. New Engl J Med. 2017;376(1):21–31. doi: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.