

Abstract
It is recognised that new scientific improvements and their integration in risk assessment, as outlined in the National Academies of Sciences, Engineering and Medicine 2017 report, have the potential to improve human health risk assessments by enabling a mechanistic understanding of adverse effects and more accurate predictions of biological responses. Here, I discuss why such improvements are needed and can ease a paradigm shift in human health risk assessment. The current approach to human health risk assessment is limited by several elements: (1) the relevance of data is debatable, as they are largely based on in vivo animal models that are poorly predictive for complex endpoints, raise challenges with regard to interspecies extrapolations, and are seldom informative of the mechanism underlying the observed effects; (2) lack of flexibility in data requirements by regulators, which limits the uptake of new scientific developments in a timely manner; and (3) lack of data accessibility, which makes data integration difficult. However, mechanistic‐based assessments are currently conducted for the identification of endocrine disruptors and are developed for addressing developmental neurotoxicity. Such assessments can serve as examples for changing the paradigm of risk assessment. There are several opportunities for improvement, such as: make regulatory standard requirements less prescriptive; enhance and use the opportunities for read‐across; analyse and quantify uncertainties in order to benchmark new approach methods to the current system; better integrate screening methods early in regulatory assessments and decision‐making; and develop more adverse outcome pathways in order to link new approach methods with the current approach and ultimately make it possible to base regulatory decisions on early key events of a toxicity pathway.
Keywords: risk assessment, regulatory toxicology, read‐across, new approach methods, adverse outcome pathways
1. Introduction
In the 2017 report from the National Academies of Sciences, Engineering, and Medicine, ‘Using 21st century science to improve risk‐related evaluations’, recommendations are provided on how to integrate the latest scientific and technological advances in toxicology, exposure science and epidemiology in human health risk assessments and improve risk‐related evaluations (NASEM, 2017). These new tools enable a mechanistic understanding of adverse effects and more accurate predictions of biological responses; in other words, they help to establish causality. Mechanistic information can be obtained through the development and evaluation of alternative methods combining non‐animal in vitro and in silico methods (‘new approach methods’, NAMs). Likewise, epidemiological research is undergoing a transition from empirical observations alone to a focus on the underlying biology. This type of evidence should also be better integrated into risk assessment.
So, there is consensus that the above‐mentioned developments have the potential to improve risk assessments and the NASEM report recommends a holistic approach for future chemical risk assessments; holistic in terms of integrating different streams of evidence covering both hazard and exposure data. However, it remains to be seen whether the current EU regulatory framework is sufficiently flexible to fully realise the benefits of more holistic human health risk assessments as the current risk assessment framework is highly compartmentalised. Compartments occur at different levels:
Successive steps of the risk assessment process (i.e. hazard identification, hazard characterisation, exposure characterisation and risk assessment).
Compound‐based assessments. Typically, each compound is assessed separately. Only data on the compound under evaluation are utilised, while information on closely related compounds are not necessarily considered on a routine basis.
Regulatory siloes, e.g. registration, evaluation, authorisation and restriction of chemicals (REACH), pesticide regulations, biocide regulations, medicines, food flavourings, etc. The consequence is that the same compounds and data sets are being assessed in different contexts at different times with different approaches, which can result in different assessment outcomes (no‐observed‐adverse‐effect levels (NOAELs) of studies, reference values, etc.).
In determining whether the current regulatory framework supports a holistic approach to human health risk assessment, one needs to recognise these different compartments and the extent to which they currently hamper a holistic approach for chemical risk assessment.
Here, I discuss the EU regulatory systems for chemical risk assessment in relation to human risk assessment, focusing on pesticide regulation,1 and whether it is fit and adequately agile to incorporate new advancements in toxicology, exposure science and epidemiology.
2. The current system
2.1. The compartmentalisation in the current risk assessment approach
The current approach for chemical risk assessment in Europe is defined by different regulations, e.g. for cosmetics, food contact materials and pesticides. To achieve their protection goals, they define which data are necessary to conduct the risk assessment with a reasonable level of confidence. These data requirements to a certain extent reflect a perceived assumption regarding risks. For example, in REACH, the standard requirements are defined by tonnage. This allows a rough estimate of exposure; for production volumes of under 1 tonne, far fewer data are required than for tonnages over 100.
For pesticides (except biopesticides and low‐risk pesticides), data requirements are according to the concept ‘one‐size‐fits‐all’, thus regardless of the intended use, i.e. exposure and risk. The same data are required for each single substance. Yet, the data requirements for pesticides are vast (see Table 1) addressing acute to repeat‐dose/chronic exposure, specific effects on reproduction, neurotoxicity and in some instances toxicological mode of action, and are mostly based on animal studies.
Table 1.
Data requirement of pesticidal active substances according to EU Regulation 283/2013a – Setting data requirements for active substances. These data must be submitted, unless the substance is a low risk substance or a biopesticide
| Data requirements – pesticidal active substances |
|---|
| Toxicokinetics – absorption, distribution, excretion, metabolism – single and repeated dose rat |
| Acute exposure – oral, dermal, inhalation rat |
| Irritation – eye, skin rabbit |
| Sensitisation mouse |
| Subchronic toxicity (28–90 days) rat |
| Subchronic toxicity (28 days – 1 year) dog |
| Genotoxicity – in vitro (mutagenicity, chromosome aberrations, aneugenicity) |
| Genotoxicity – in vivo (chromosome aberrations – possibly others) mouse |
| Chronic toxicity (2 year) rat |
| Carcinogenicity (2 year) rat |
| Carcinogenicity mouse |
| Teratogenicity rat |
| Teratogenicity rabbit |
| Reproduction – 1 or 2 generations rat |
| Neurotoxicity (acute and/or chronic exposure) rat, developmental neurotoxicity rat |
| All relevant data from open literature (systematic review) |
| Others – e.g. studies on endocrine modes of actions |
Commission Regulation (EU) No 283/2013 of 1 March 2013 setting out the data requirements for active substances, in accordance with Regulation (EC) No 1107/2009 of the European Parliament and of the Council concerning the placing of plant protection products on the market. OJ L 93, 3.4.2013, p. 1–84.
Moreover, the studies required should be conducted in compliance with OECD guidelines for the testing of chemicals or equivalent (for example, guidelines of the Office of the Pesticide Program, US Environmental Protection Agency). The guidelines specify how the test should be conducted and what examinations should be performed, with a vast set of in vivo and post mortem analyses performed. Typically, all tissues should be examined macro‐ and microscopically. Clinical chemistry parameters should be measured as well as food and water intake, and clinical signs should be monitored.
When data from all these studies are compiled, there are literally several thousand data entries for each single substance that identify and characterise the hazards.
For pesticides in the EU as well as for other chemicals regulated under other jurisdictions, data requirements are to a large extent (or even fully) prescriptive. That is, under the current system, the data requirement is prescriptive and centred on single substances, with no use of knowledge from similar substances to waive specific data, investigations or studies.
Moreover, the required data are only descriptive: the OECD test guidelines are designed to detect and characterise hazards through descriptive observations of effects, and not to understand mechanisms. Therefore, data requirements for the risk assessment of pesticides do not necessarily provide a mechanistic understanding of observed toxicities. Finally, the data always describe the effects in a model species, often a rodent. Yet, there is uncertainty around extrapolations from model species to humans for many observed effects, some of which are not well explained and not well quantified (WHO, 2017).
In conclusion, the current EU regulatory framework is to a large extent prescriptive and requires mandatory in vivo tests, without the possibility to waive such requirements on a scientific, case‐by‐case basis (unless it is already known that the compound has certain properties, e.g. being a carcinogen). Since in vivo experiments are still compulsory, the current system does not specifically promote the generation and integration of 21st century toxicity data into chemical risk assessments. Only in the rare cases where the petitioner proposes a claim on for example the non‐human relevance of an effect, is there in reality an incentive to generate mode of action data and data on the differences between species.
2.2. Strengths, weaknesses, opportunities and threats of the current risk assessment system
There is confidence in the current system. After all, for many regulated chemicals on a single substance basis, there is no strong evidence of lack of safety pertaining to their use so far. However, the current system does not allow us to tell how safe the use of a compound is due to the following factors. Risk assessments as currently conducted: (1) rely heavily on non‐human data; (2) are based on in vivo data, which are not designed to provide mechanistic understanding; (3) do not cover multifactorial disease aetiologies; (4) do not consider co‐exposures; (5) rely on reference doses derived from NOAELs that do not enable a quantification of the uncertainty; and (6) rely on ill‐defined protection goals.
The current risk assessment system is based on a wide range of tests that aim to determine that there are no observed adverse effect levels per compound, with reference values that should cover vulnerable groups. Reasonable upper exposure limits are estimated for the levels of human exposure to the compound. Finally, risk assessments are carried out for relevant populations.
However, it remains possible that some adverse effects in humans are not predicted accurately by regulatory toxicity testing. Such effects may also remain undetected because current testing models do not capture the disease, or epidemiological surveillance is insufficient or insensitive. In particular, doubts remain as to whether common and complex human health outcomes are appropriately covered by current standard toxicity tests. Such concerns have been reported in different domains:
Immunotoxicity, childhood leukaemias (EFSA PPR Panel, 2017a; Pelkonen et al., 2017).
Developmental neurotoxicity (EFSA PPR Panel, 2013), chronic neurological diseases such as Parkinson's disease (EFSA PPR Panel, 2017b, Terron et al., 2018).
Neuropsychological effects and mental illnesses, as well as endocrine disorders such as some hormonal cancers, endometriosis, metabolic syndrome, type 2 diabetes, and reproductive senescence (OECD, 2014a).
One of the strengths of the current chemical risk assessment system is that an extensive amount of data has been gathered since the relevant regulations came into force. However, these data are to a large extent descriptive in vivo data, with inherent limitations. In most cases, they provide no mechanistic information, do not capture effects relevant for humans, and are poorly reproducible, difficult to interpret and insensitive (Olson et al., 2000; Leist et al., 2008; Basketter et al., 2012).
In contrast, the vast amount of data gathered under certain regulations (e.g. pesticide risk assessment) offers the opportunity to utilise such data. However, the data are currently far from being easily available and analysable even for risk assessors. Consequently, only a few assessors, based on decades of personal experience, have a holistic overview. In fact, until recently, access to data has only been possible upon specific access requests, without violating confidentiality issues. With the European Commission's proposal for a new food law,2 all data would be made public proactively (except data where confidentiality can be claimed due to propriety reasons). This is of course a first step, but it is most likely that the data will be released in different formats, including those that are not searchable. The data will not necessarily be made available in databases. This raises a significant challenge for external risk assessors and scientists that would like to analyse the full data sets, as several thousand pages would have to be analysed for each compound to fully contextualise different findings (given that knowledge of similar compounds is typically also taken into account in market registration applications for chemical stressors).
In order to make better use of data, they must be collected, curated, annotated and inserted into an open database. In the area of human medicine, this is exactly what is currently under development with the Innovative Medicines Initiatives projects eTOX3 and eTransafe.4 The overall aim of these databases is to predict safety issues in silico (i.e. using computer models) by learning from companies’ existing pre‐clinical data.
It is obvious that having detailed prescriptive and mandatory data requirements is not geared towards fast‐evolving scientific developments; the experience gained in changing mandatory data requirements shows that this is a lengthy process as various parliamentary and legal processes are involved. The inertia of the system is clearly a threat to conducting a risk assessment that lives up to current scientific standards.
On the other hand, more flexibility cannot come at the expense of safety. To be accepted by regulatory bodies, NAMs need to be adequately annotated, described and their performance shown to be, for example, in accordance with OECD guidance 211 on non‐guideline in vitro methods (OECD, 2014b) and OECD guidance 286 on good in vitro practices (OECD, 2018).
3. Moving towards a holistic approach for human health risk assessments
3.1.
3.1.1. Read‐across
Currently, as recognised in several analyses on applying 21st century science and technologies to chemical risk assessment, no NAMs can fully and easily replace, for example, a 90‐day feeding study with rodents (NASEM, 2017; SAPEA, 2018). However, exploring and utilising grouping and read‐across better than is currently practised would be a first step towards the integration of NAMs into risk assessment.
In the EU, a number of different regulations allow the use of read‐across. Under EFSA's remit, read‐across has been used in the areas of chemical contaminants, food contact materials, feed, food additives and flavourings (Chesnut et al., 2018). Yet, there are notable exceptions, like the Pesticide Regulation and associated data requirements. Basically, replacing mandatory studies with read‐across data is not mentioned explicitly in the Regulation. However, in a few relatively simple cases (for example, read‐across from a racemic mixture to an isomer), read‐across data have replaced mandatory studies. The recent regulation5 on identifying endocrine disruptors under the Pesticide Regulation does not allow for read‐across. However, the guidance implementing the criteria for identifying the endocrine disruptors (ECHA/EFSA, 2018) mentions read‐across and QSAR as types of data that can be used for risk assessment purposes.
Read‐across is mostly used to bridge data gaps, typically on complex endpoints such as toxicity after repeated exposure to compounds or developmental and reproductive toxicity (ECHA, 2014). Read‐across requires a similarity assessment of the grouped compounds in terms of toxicokinetic and dynamic properties. It is often a challenge to reach a conclusion on the similar adverse toxicological effect pattern, as the apical findings might vary in the type, severity and lowest observed adverse effect level within the grouped compounds (Judson et al., 2017). Another difficulty is that, apical findings from in vivo data often do not enable a mechanistic understanding of the observed adverse outcomes.
The use of NAMS in read‐across and grouping sounds straightforward; however, it presents practical challenges and at present and there are only a few examples of the successful integration of NAMs in read‐across (Ball et al., 2016). Different initiatives have been taken to make better use of NAMs in risk assessment: the OECD with their case studies on an Integrated Approach to Testing and Assessment (IATA) (OECD, online‐a), and the US EPA with the GenRA tool (Generalised read‐across) (US EPA, online).
In the H2020‐supported project, EU‐ToxRisk,6 read‐across approaches are used and developed to integrate mechanistic knowledge (e.g. adverse outcome pathways, AOPs) in human hazard assessment (Leist et al., 2017). One of the aims of the project is to illustrate with case studies how in vitro assays, in silico studies (e.g. (quantitative) structural activity relationships ((Q)SAR) and/or physiology‐based toxicocokinetic (PBTK) models) together with AOPs can be used to prove (dis)similarity or a consistent trend within a read‐across assessment.
3.1.2. Adverse outcome pathways
The use of NAMs to underpin read‐across arguments is an obvious possibility, and in the case of read‐across scenarios based on biological similarity rather than chemical similarity (which are by far the most common), it would be crucial. Most obviously the argumentation would be based on a mechanistic hypothesis like knowledge about AOPs (Leist et al., 2017).
The concept of AOPs simplifies a toxicological mechanism to a series of chemically agnostic sequential events starting with a molecular initiation event, followed by key events and key event relationships, which lead to cellular as well as organ responses and the final adverse effect in the organism (Leist et al., 2017). An AOP is seen as a useful tool to structure critical steps within a complex biological process (Ankley et al., 2010). The key events are essential for the progression towards the adverse outcome and can ideally be assessed by relevant in vitro and in silico models (Villeneuve et al., 2014a,b; Ball et al., 2016). The vision is that regulatory decisions, like establishing point of departure, can be based on early key events, surpassing the need for in vivo apical data.
Since the use of new assays and technologies for risk assessment purposes holds much promise, anchoring these to AOPs in risk assessment is helpful. Therefore, to ensure regulatory acceptance, development and review of AOPs is conducted under the auspices of the OECD (OECD, online‐b). The AOP‐wiki is an open space where anyone can upload an AOP on the web page. Several hundreds of AOPs can be found at very different levels of maturity. To become reviewed and fully endorsed by the OECD Working Group of National Coordinators of the Test Guideline Programme an AOP must undergo two review processes to ensure that the AOPs have followed the instructions and guidelines. This is a lengthy process and at the moment only a few AOPs have undergone the whole procedure and a few dozen more are going through the process.
However, efforts put into AOP development seem rather unbalanced compared with the efforts and resources being put into developing all the new tools, assays, models and technologies, etc. This probably has many explanations: (1) no specific resource allocation; (2) uncertainty of scientific recognition, i.e. authorship on an AOP is often not being recognised academically; and (3) extensive efforts are required similar to those for conducting a systematic review.
3.1.3. Open doors
Moving towards a holistic assessment from the current system is a huge challenge. However, there are examples and areas where the transition will be more easily be accepted and implemented; this is the case where the current system has recognised weaknesses or where there are already requirements in the regulations regarding mechanistic understanding.
In other words, are there any doors already open?
The newly adopted regulation for identifying endocrine disruptors in the context of the pesticide and biocide risk assessment framework is a progressive regulatory area. Herein, the definition of an endocrine disruptor is given as: ‘(1) it shows an adverse effect in an intact organism or its progeny, which is a change in the morphology, physiology, growth, development, reproduction or life span of an organism, system or (sub)population that results in an impairment of functional capacity, an impairment of the capacity to compensate for additional stress or an increase in the susceptibility to other influence; (2) it has an endocrine mode of action, i.e. alters the function(s) of the endocrine system; and (3) the adverse effect is a consequence of the endocrine mode of action’.
This piece of legislation requires a mechanistic understanding of the endocrine effects, as well as the investigation of traditional adverse apical effects. As many of the pesticides and biocides that will be assessed for endocrine effects during re‐approval processes, the adequacy of the database will often be uncertain. In effect, many of the substances will have a two‐generation study (OECD TG416) conducted under an old protocol, which according to the endocrine disruptor guidance is not sufficient to investigate apical effects mediated by an oestrogenic, androgenic or steroidogenic action (ECHA/EFSA, 2018). For an example, see Figure 1.
Figure 1.
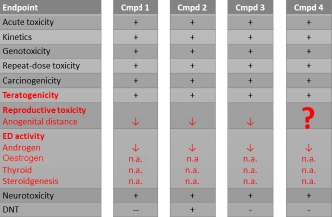
- +: Available data
- ↓: Activity decreased
- n.a.: No activity
Adverse outcome pathways for such effects were some of the earliest to be developed, but still as of 2019, only seven AOPs for oestrogenic, androgenic, steroidogenic and thyroid action effects have been approved or are under review. The benefit of quantitative AOPs in this area is obvious, where potentially very costly and animal‐heavy studies could be waived based on appropriate early key event testing.
Other particular adverse effects have also been identified – based on human epidemiological data – where the current animal models are not adequate, or the relevant testing has not been required. This was exemplified for Parkinson's disease and childhood leukaemias (EFSA PPR Panel, 2017a). In general, there is a recognition that many of the complex multifactorial human diseases are difficult to replicate in a standardised animal test. In EFSA PPR Panel (2017a), an approach was outlined for inferring causality by developing relevant AOPs.
Another open door is in the field of developmental neurotoxicity. The developing nervous system might be more sensitive to exposure to certain chemicals than the adult one (OECD, 2017). For the time being, in order to address the developmental neurotoxicity (DNT) effects of chemicals there are two accepted guidelines, TG426 and TG443 with the DNT cohort. Across the different chemical regulations in Europe (pesticides, biocides and industrial chemicals) and the US (pesticides), DNT testing can be triggered based on neurotoxic effects in repeat‐dose testing, known neurotoxic mode of action or structural activity relationships.
However, the TG426 and TG443 (with DNT cohort) are often not required and thus not conducted. For example, in Europe, until now for the almost 485 approved pesticides, the TG426 have been available in 35 cases, whereas so far, the TG443, being a relatively newly developed guideline, has not been applied. In total, only 200 chemicals have been tested globally (Terron and Bennekou, 2018).
Secondly, the two guidelines have significant shortcomings, including:
Very animal‐demanding and costly
Lack of mechanistic understanding for most of the endpoints measured
Endpoints currently measured do not comprehensively represent, or do not reflect well the complex set of endpoints of relevance to humans (e.g. cognitive functions)
High data variability and poor data reproducibility, even for positive controls (Crofton et al., 2004; Smirnova et al., 2014).
Consequently, consensus was reached between regulators, academic scientists and industry about the need for an alternative DNT testing strategy that is based on a standardised in vitro battery of tests in order to test more chemicals and inform more targeted in vivo studies (Fritsche et al., 2017). This activity was followed up by the OECD and there are ongoing efforts to develop a guidance document and generate data and case studies using a test battery and other NAMs in an IATA context (Sachana et al., 2019). Also, readiness of in vitro assays for DNT testing have been assessed (Bal‐Price et al., 2018). This is an important step to achieve regulatory acceptance and facilitate changes to the regulatory standard requirements.
A possibility in this regard, and as a step in this transition towards integrating NAMs, would be to conduct more rigorous uncertainty analysis of the existing in vivo guideline tests. Paparella et al. (2017) conducted a systematic uncertainty analysis of the 2‐year rodent carcinogenicity bioassay. The aim of this analysis was to define a benchmark performance of the NAM against the standard in vivo study. Specific uncertainty analysis of the current in vivo animals along with a systematic and quantitative uncertainty analysis of the whole risk assessment process, as well as better defined protection goals by risk managers, would generate more transparency. EFSA has taken a significant step with its guidance on uncertainty analysis in risk assessment (EFSA Scientific Committee, 2018); altogether, it can be expected that it will become apparent to what extent introducing NAMS will bring confidence to a risk assessment.
3.1.4. How could regulatory systems be changed?
3.1.4.1. Classification and labelling
One piece of legislation posing a challenge to utilising 21st century data instead of in vivo animal data is Regulation 1272/2008 on classification and labelling.7 Although according to the Regulation all types of data can be used to fully determine whether a compound should be classified and labelled, a whole data package similar to that for pesticides (Table 1) is most often necessary, where a compound is tested up to maximum tolerated dose or above 1,000 mg/kg per day. The relevance of effects observed at such very high doses compared to realistic human exposures is questionable (Saghir, 2015; Bus, 2017). A recent example is whether glyphosate causes cancers in rodent studies, given that effects were sometimes observed at very high doses (Tarazona et al., 2017). Should we care about effects observed at such dose levels, representing exposure orders of magnitude higher than that for humans, and even so where no mechanistic explanation can be given? Perhaps it would be more resource‐efficient to base classification and labelling on broad in vitro screening, and focus on compounds that show activity, for example endocrine disruptor activity, at doses that can be translated to relevant human exposure levels. Figure 2 shows ToxCast chemical activity data for two compounds. This illustrates that the second compound is far more active than the first in different types of assays addressing different endpoints, and to a large extent at lower doses.
Figure 2.
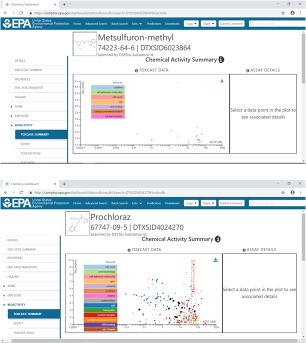
Examples of ToxCast summary data for two different chemicals, represented by screen shots from the dashboard (https://comptox.epa.gov/dashboard/). In these summary data only ‘hits’ are presented. ToxCast is a screening programme covering more than 700 high‐throughput assay endpoints. It is evident that the second compound showed activity in many more of the assays compared with the compound above
From screening data, possibly linked to relevant AOPs, the point of departure could be estimated and compared to human exposure data (Wetmore et al., 2015). This could provide a good starting point for answering the question of whether a compound actually possesses liabilities like being a reproductive toxicant and should therefore be appropriately classified and labelled.
3.1.4.2. Regulatory requirements and processes
The prescriptive and mandatory standard data requirements given in many regulations are clearly not well suited to adapt to the rapidly evolving developments of science. It would be preferable if regulations were less prescriptive and more flexible. First, ascribing specific guidelines such as the OECD guidelines, to address a data requirement, could be changed. Rather, the data requirements should call for a specific hazard to be addressed, for example DNT, but not prescribe which OECD guideline should be used. Second, likewise, guidance documents complementing scientific regulatory requirements should be less prescriptive. Examples of these less prescriptive types of guidance can be found within the remit of medicines legislation.
The regulatory processes could also be changed. When substances are evaluated or re‐evaluated, substances belonging to the same group should be assessed at the same time, in order to facilitate read‐across between chemicals that cause toxicity through similar mechanisms. This would also allow new developments in science to be exploited. Also, re‐evaluations could also, case by case, be targeted to those parts of the risk assessment that are of greatest concern. An example of such a system has been outlined for pesticides (Group of Chief Scientific Advisors, 2018; SAPEA 2018). In conclusion, the dossiers should contain sufficient data to demonstrate safe use, but more flexibility should be allowed in answering ‘sufficiency’.
Such flexibility has the inherent risk of reducing the mutual regulatory recognition between assessments conducted under the different regulated sectors (chemical as well as geographical). This should be avoided by harmonisation, also recognising that mutual recognition is currently rather limited; for example, Europe would not rely solely on a recent evaluation conducted in the US (although assessing the same data). Possibly, allowing more flexibility in the various regulations would also facilitate the opportunity to better exploit work conducted under another jurisdiction.
4. Conclusions
The NASEM has provided analysis and reflections on how to strive for a paradigm shift in risk assessment; from risk assessment based on identification of apical endpoints to a mechanism‐based risk assessment. Their report calls for a holistic approach in risk assessment. It clearly documents how this shift could significantly improve risk assessments. The next question is whether the current system can fully realise the potential of such improvements. Several limitations and weaknesses need to be overcome. These range from the coverage of data on which risk assessments are based, lack of transparency, and structural, procedural and legislative issues pertaining to the current approach for chemical risk assessment. However, there are areas where the current approach is already partly implementing some of the new concepts, such as the identification of endocrine disrupting chemicals. Similarly, there are areas where the current system would obviously benefit since currently there seem to be considerable shortcomings – for example in the area of complex, multifactorial human adverse outcomes, like metabolic disturbances, neurodevelopmental and neurodegenerative diseases. Based on these observations, several different recommendations have been made:
-
–
Unambiguous definition of protection goals;
-
–
Rigorous and quantitative analysis of uncertainties in the risk assessment process;
-
–
Harmonise risk assessment approaches across regulations and regions;
-
–
Make data available to and analysable by the public;
-
–
Make data requirements flexible;
-
–
Bulk similar chemicals into one assessment;
-
–
Optimise the use of read‐across;
-
–
Do more screening;
-
–
Speed up the development of AOPs.
Abbreviations
- AOP
adverse outcome pathway
- ECHA
European Chemicals Agency
- EPA
US Environmental Protection Agency
- IATA
Integrated Approach to Testing and Assessment
- NASEM
National Academies of Sciences, Engineering, and Medicine
- NOAEL
no‐observed‐adverse‐effect level
- NEM
new approach method
- OECD
Organisation for Economic Cooperation and Development
- PBTK
physiology‐based toxicocokinetic
- REACH
registration, evaluation, authorisation and restriction of chemicals
- QSAR
quantitative structural activity relationship
- WHO
World Health Organization
Suggested citation: Bennekou SH, 2019. Moving towards a holistic approach for human health risk assessment – Is the current approach fit for purpose? EFSA Journal 2019;17(S1):e170711, 14 pp. 10.2903/j.efsa.2019.e170711
Acknowledgements: The author wishes to thank Yann Devos and Anna Lanzoni for the support provided to this article.
Approved: 29 May 2019
Notes
Regulation (EC) No 1107/2009 of the European Parliament and of the Council of 21 October 2009 concerning the placing of plant protection products on the market and repealing Council Directives 79/117/EEC and 91/414/EEC. OJ L 309, 24.11.2009, p. 1–50.
Proposal for a Regulation of the European Parliament and of the Council on the transparency and sustainability of the EU risk assessment in the food chain amending Regulation (EC) No 178/2002 [on general food law], Directive 2001/18/EC [on the deliberate release into the environment of GMOs], Regulation (EC) No 1829/2003 [on GM food and feed], Regulation (EC) No 1831/2003 [on feed additives], Regulation (EC) No 2065/2003 [on smoke flavourings], Regulation (EC) No 1935/2004 [on food contact materials], Regulation (EC) No 1331/2008 [on the common authorisation procedure for food additives, food enzymes and food flavourings], Regulation (EC) No 1107/2009 [on plant protection products] and Regulation (EU) No 2015/2283 [on novel foods]. COM/2018/0179 final – 2018/088 (COD) (https://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1523604766591&uri=COM:2018:179:FIN).
Commission Regulation (EU) 2018/605 of 19 April 2018 amending Annex II to Regulation (EC) No 1107/2009 by setting out scientific criteria for the determination of endocrine disrupting properties. OJ L 101, 20.4.2018, p. 33–36.
Regulation (EC) No 1272/2008 of the European Parliament and of the Council of 16 December 2008 on classification, labelling and packaging of substances and mixtures, amending and repealing Directives 67/548/EEC and 1999/45/EC, and amending Regulation (EC) No 1907/2006. OJ L 353, 31.12.2008, p. 1–1355.
References
- Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, Mount DR, Nichols JW, Russom CL, Schmieder PK, Serrrano JA, Tietge JE and Villeneuve DL, 2010. Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environmental Toxicology and Chemistry, 29, 730–741. 10.1002/etc.34 [DOI] [PubMed] [Google Scholar]
- Ball N, Cronin MT, Shen J, Blackburn K, Booth ED, Bouhifd M, Donley E, Egnash L, Hastings C, Juberg DR, Kleensang A, Kleinstreuer N, Kroese ED, Lee AC, Luechtefeld T, Maertens A, Marty S, Naciff JM, Palmer J, Pamies D, Penman M, Richarz AN, Russo DP, Stuard SB, Patlewicz G, van Ravenzwaay B, Wu S, Zhu H and Hartung T, 2016. Toward Good Read‐Across Practice (GRAP) guidance. Altex, 33, 149–166. 10.14573/altex.1601251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal‐Price A, Hogberg HT, Crofton KM, Daneshian M, FitzGerald RE, Fritsche E, Heinonen T, Hougaard Bennekou S, Klima S, Piersma AH, Sachana M, Shafer TJ, Terron A, Monnet‐Tschudi F, Viviani B, Waldmann T, Westerink RHS, Wilks MF, Witters H, Zurich MG and Leist M, 2018. Recommendation on test readiness criteria for new approach methods in toxicology: exemplified for developmental neurotoxicity. Altex, 35, 306–352. 10.14573/altex.1712081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basketter DA, Clewell H, Kimber I, Rossi A, Blaauboer B, Burrier R, Daneshian M, Eskes C, Goldberg A, Hasiwa N, Hoffmann S, Jaworska J, Knudsen TB, Landsiedel R, Leist M, Locke P, Maxwell G, McKim J, McVey EA, Ouédraogo G, Patlewicz G, Pelkonen O, Roggen E, Rovida C, Ruhdel I, Schwarz M, Schepky A, Schoeters G, Skinner N, Trentz K, Turner M, Vanparys P, Yager J, Zurlo J and Hartung T, 2012. A roadmap for the development of alternative (non‐animal) methods for systemic toxicity testing – t4 report*. Altex, 29, 3–91. [DOI] [PubMed] [Google Scholar]
- Bus JS, 2017. “The dose makes the poison”: key implications for mode of action (mechanistic) research in a 21st century toxicology paradigm. Current Opinion in Toxicology, 3, 87–91. [Google Scholar]
- Chesnut M, Yamada T, Adams T, Knight D, Kleinstreuer N, Kass G, Luechtefeld T and Hartung T, 2018. Regulatory acceptance of read‐across. Altex, 35, 413–419. 10.14573/altex.1805081 [DOI] [PubMed] [Google Scholar]
- Crofton KM, Makris SL, Sette WF, Mendez E and Raffaele KC, 2004. A qualitative retrospective analysis of positive control data in developmental neurotoxicity studies. Neurotoxicology and Teratology, 26, 345–352. [DOI] [PubMed] [Google Scholar]
- ECHA (European Chemicals Agency), 2014. Summary of Workshop on the Read‐Across Assessment Framework (RAAF) 2–3 October 2014. ECHA, Helsinki, 1 pp. Available online: https://echa.europa.eu/documents/10162/13628/workshop_summary_raaf_en.pdf [Google Scholar]
- ECHA (European Chemicals Agency) and EFSA (European Food Safety Authority) with the technical support of the Joint Research Centre (JRC) , Andersson N, Arena M, Auteri D, Barmaz S, Grignard E, Kienzler A, Lepper P, Lostia AM, Munn S, Parra Morte JM, Pellizzato F, Tarazona J, Terron A and Van der Linden S, 2018. Guidance for the identification of endocrine disruptors in the context of Regulations (EU) No 528/2012 and (EC) No 1107/2009. EFSA Journal 2018;16(6):5311, 135 pp. 10.2903/j.efsa.2018.5311. ECHA‐18‐G‐01‐EN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority) Scientific Committee, 2018. Guidance on uncertainty analysis in scientific assessments. EFSA Journal 2018;16(1):5123, 39 pp. 10.2903/j.efsa.2018.5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA PPR Panel (EFSA Panel on Plant Protection Products and their Residues), 2013. Scientific Opinion on the developmental neurotoxicity potential of acetamiprid and imidacloprid. EFSA Journal 2013;11(12):3471, 51 pp. 10.2903/j.efsa.2013.3471 [DOI] [Google Scholar]
- EFSA PPR Panel (EFSA Panel on Plant Protection Products and their Residues), 2017a. Scientific Opinion on the investigation into experimental toxicological properties of plant protection products having a potential link to Parkinson's disease and childhood leukaemia. EFSA Journal 2017;15(3):4691, 325 pp. 10.2903/j.efsa.2017.4691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA PPR Panel (EFSA Panel on Plant Protection Products and their Residues), 2017b. Scientific Opinion of the PPR Panel on the follow‐up of the findings of the External Scientific Report ‘Literature review of epidemiological studies linking exposure to pesticides and health effects’. EFSA Journal 2017;15(10):5007, 101 pp. 10.2903/j.efsa.2017.5007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche E, Crofton KM, Hernandez AF, Hougaard Bennekou S, Leist M, Bal‐Price A, Reaves E, Wilks MF, Terron A, Solecki R, Sachana M and Gourmelon A, 2017. OECD/EFSA workshop on developmental neurotoxicity (DNT): the use of non‐animal test methods for regulatory purposes. Altex, 34, 311–315. 10.14573/altex.1701171 [DOI] [PubMed] [Google Scholar]
- Group of Chief Scientific Advisors , 2018. EU Authorisation Processes of Plant Protection Products – From a Scientific Point of View. Publications Office of the EU, Luxembourg: 10.2777/238919 [DOI] [Google Scholar]
- Judson RS, Martin MT, Patlewicz G and Wood CE, 2017. Retrospective mining of toxicology data to discover multispecies and chemical class effects: anemia as a case study. Regulatory Toxicology and Pharmacology, 86, 74–92. 10.1016/j.yrtph.2017.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M, Hartung T and Nicotera P, 2008. The dawning of a new age of toxicology. Altex, 25, 103–114. [PubMed] [Google Scholar]
- Leist M, Ghallab A, Graepel R, Marchan R, Hassan R, Bennekou SH, Limonciel A, Vinken M, Schildknecht S, Waldmann T, Danen E, van Ravenzwaay B, Kamp H, Gardner I, Godoy P, Bois FY, Braeuning A, Reif R, Oesch F, Drasdo D, Höhme S, Schwarz M, Hartung T, Braunbeck T, Beltman J, Vrieling H, Sanz F, Forsby A, Gadaleta D, Fisher C, Kelm J, Fluri D, Ecker G, Zdrazil B, Terron A, Jennings P, van der Burg B, Dooley S, Meijer AH, Willighagen E, Martens M, Evelo C, Mombelli E, Taboureau O, Mantovani A, Hardy B, Koch B, Escher S, van Thriel C, Cadenas C, Kroese D, van de Water B and Hengstler JG, 2017. Adverse outcome pathways: opportunities, limitations and open questions. Archives of Toxicology, 91, 3477–3505. 10.1007/s00204-017-2045-3 [DOI] [PubMed] [Google Scholar]
- NASEM (National Academies of Sciences, Engineering, and Medicine), 2017. Using 21st Century Science to Improve Risk‐Related Evaluations. The National Academies Press, Washington, DC: 10.17226/24635 [DOI] [PubMed] [Google Scholar]
- OECD (Organisation for Economic Cooperation and Development), online‐a. Integrated Approaches to Testing and Assessment (IATA). OECD, Paris: Available online: http://www.oecd.org/chemicalsafety/risk-assessment/iata-integrated-approaches-to-testing-and-assessment.htm [Google Scholar]
- OECD (Organisation for Economic Cooperation and Development), online‐b. Adverse Outcome Pathways, Molecular Screening and Toxicogenomics. OECD, Paris: Available online: http://www.oecd.org/chemicalsafety/testing/adverse-outcome-pathways-molecular-screening-and-toxicogenomics.htm [Google Scholar]
- OECD (Organisation for Economic Cooperation and Development), 2014a. Detailed Review Paper on the State of the Science on Novel In Vitro and In Vivo Screening and Testing Methods and Endpoints for Evaluating Endocrine Disruptors. OECD Publishing, Paris: 10.1787/9789264221352en [DOI] [Google Scholar]
- OECD (Organisation for Economic Cooperation and Development), 2014b. Guidance document for describing non‐guideline in vitro test methods. ENV/JM/MONO(2014)35. Series on Testing and Assessment No. 211. Available online: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2014)35&doclanguage=en
- OECD (Organisation for Economic Cooperation and Development), 2017. Report of the OECD/EFSA workshop on developmental neurotoxicity (DNT): the use of non‐animal test methods for regulatory purposes. ENV/JM/MONO(2017)4. Series on Testing and Assessment No. 261. Available online: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2017)4&doclanguage=en
- OECD (Organisation for Economic Cooperation and Development), 2018. Guidance Document on Good In Vitro Method Practices (GIVIMP). ENV/JM/MONO(2018)19. Series on Testing and Assessment No. 286. Available online: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2018)19&doclanguage=en
- Olson H, Betton G, Robinson D, Thomas K, Monro A, Kolaja G, Lilly P, Sanders J, Sipes G, Bracken W, Dorato M, Van Deun K, Smith P, Berger B and Heller A, 2000. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regulatory Toxicology and Pharmacology, 32, 56–67. [DOI] [PubMed] [Google Scholar]
- Paparella M, Colacci A and Jacobs MN, 2017. Uncertainties of testing methods: what do we (want to) know about carcinogenicity? Altex, 34, 235–252. 10.14573/altex.1608281 [DOI] [PubMed] [Google Scholar]
- Pelkonen O, Terron A, Hernandez AF, Menendez P and Bennekou SH; EFSA WG EPI1 and its other members , 2017. Chemical exposure and infant leukaemia: development of an adverse outcome pathway (AOP) for aetiology and risk assessment research. Archives of Toxicology, 91, 2763–2780. 10.1007/s00204-017-1986-x [DOI] [PubMed] [Google Scholar]
- Sachana M, Bal‐Price A, Crofton KM, Bennekou SH, Shafer TJ, Behl M and Terron A, 2019. International regulatory and scientific effort for improved developmental neurotoxicity testing. Toxicological Sciences, 167, 45–57. 10.1093/toxsci/kfy211 [DOI] [PubMed] [Google Scholar]
- Saghir SA, 2015. Rethinking guideline toxicity testing. Regulatory Toxicology and Pharmacology, 72, 423–428. 10.1016/j.yrtph.2015.05.009 [DOI] [PubMed] [Google Scholar]
- SAPEA (Science Advice for Policy by European Academies), 2018. Improving Authorisation Processes for Plant Protection Products in Europe: A Scientific Perspective on the Potential Risks to Human Health. SAPEA, Berlin: 10.26356/plantprotectionproducts [DOI] [Google Scholar]
- Smirnova L, Hogberg HT, Leist M and Hartung T, 2014. Developmental neurotoxicity – challenges in the 21st century and in vitro opportunities. Altex, 31, 129–156. 10.14573/altex.1403271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazona JV, Court‐Marques D, Tiramani M, Reich H, Pfeil R, Istace F and Crivellente F, 2017. Glyphosate toxicity and carcinogenicity: a review of the scientific basis of the European Union assessment and its differences with IARC. Archives of Toxicology, 91, 2723–2743. 10.1007/s00204-017-1962-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terron A and Bennekou SH, 2018. Towards a regulatory use of alternative developmental neurotoxicity testing (DNT). Toxicology and Applied Pharmacology, 1, 19–23. 10.1016/j.taap.2018.02.002 [DOI] [PubMed] [Google Scholar]
- Terron A, Bal‐Price A, Paini A, Monnet‐Tschudi F, Bennekou SH, EFSA WG EPI1 Members , Leist M and Schildknecht S, 2018. An adverse outcome pathway for parkinsonian motor deficits associated with mitochondrial complex I inhibition. Archives of Toxicology, 92:41–82. 10.1007/s00204-017-2133-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- US EPA (United States Environmental Protection Agency), online. Generalized Read‐Across (GenRA). Available online: https://www.epa.gov/sites/production/files/2018-09/documents/genra_help_310818.pdf
- Villeneuve DL, Crump D, Garcia‐Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, Ottinger MA, Vergauwen L and Whelan M, 2014a. Adverse outcome pathway (AOP) development I: strategies and principles. Toxicological Sciences, 142, 312–320. 10.1093/toxsci/kfu199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve DL, Crump D, Garcia‐Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, Ottinger MA, Vergauwen L and Whelan M, 2014b. Adverse outcome pathway development II: best practices. Toxicological Sciences, 142, 321–330. 10.1093/toxsci/kfu200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore BA, Wambaugh JF, Allen B, Ferguson SS, Sochaski MA, Setzer RW, Houck KA, Strope CL, Cantwell K, Judson RS, LeCluyse E, Clewell HJ, Thomas RS and Andersen ME, 2015. Incorporating high‐throughput exposure predictions with dosimetry‐adjusted in vitro bioactivity to inform chemical toxicity testing. Toxicological Sciences, 148, 121–136. 10.1093/toxsci/kfv171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (World Health Organization), 2017. Guidance Document on Evaluating and Expressing Uncertainty in Hazard Characterization. 2nd edition WHO, Geneva. [Google Scholar]