

Abstract
Despite the ever increase in rigorous control and monitoring measures to assure safe food along the entire farm‐to‐fork chain, the past decade has also witnessed an increase in microbial food alerts. Hence, research on food safety and quality remain of utmost importance. Complementary, and at least as important, is the necessity to be able to assess the potential microbial risks along the food chain. Risk assessment relies on sound scientific data. Unfortunately, often, quality data are limited if not lacking. High‐throughput tools such as next‐generation sequencing (NGS) could fill this gap. NGS approaches can be used to generate ample qualitative and quantitative data to be used in the risk assessment process. NGS applications are not new in food microbiology with applications ranging from pathogen detection along the food chain, food epidemiology studies, whole genome analysis of food‐associated microorganisms up to describing complete food microbiomes. Yet, its application in the area of microbial risk assessment is still at an early stage and faces important challenges. The possibilities of NGS for risk assessment are ample, but so are the questions on the subject. One of the major strengths of NGS lies in its capacity to generate a lot of data, but to what extend can this wealth be of use in hazard identification, hazard characterisation and exposure assessment to perform a sound risk characterisation, which in turn will make it possible to take substantiated risk management decisions.
Keywords: food safety, next‐generation sequencing, whole genome sequencing, microbial risk assessment, Illumina, Oxford Nanopore Technologies, MiSeq, MinION
1. Introduction
The pathogenic properties of food‐borne microorganisms are strain dependent. In addition, the epidemic potential of a food‐borne strain within its population can vary in function of its genetic make‐up and ecological items (EFSA, 2013). Hence, for microbial pathogens in a context of food safety, whether it is for the detection and/or investigation of food‐borne outbreaks (e.g. route‐ and source‐tracking, cross‐contamination events), attribution studies, assessment of possible virulence properties or epidemic potential and integrating all these data into risk assessment evaluations, even up to intervention and control strategy studies, it is of utmost importance that the pathogen under investigation is unambiguously identified and characterised. To this end, a wide variety of microbial typing methods is at our disposal.
Microbial typing refers to the process of identifying or discriminating between different types of microorganisms within the same microbial species. Although classical phenotyping methods, i.e. methods relying on phenotypic properties (e.g. serotyping, phage‐typing, the use of antimicrobial profiling, chemotaxonomic profiling) are still being used, in the last decades due to advances in molecular techniques, we have experienced an important and inevitable shift towards genotyping methods relying on the information present in DNA or RNA molecules. The typing technique of choice largely depends on the hazard, the purpose and required level of resolution and the appropriate state‐of the art. Sometimes, a combination of methods is advised (Figure 1).
Figure 1.

Factors determining the choice of typing method
Which typing method is chosen depends on the hazard, the purpose of the analysis and the most appropriate technique(s) at hand for the purpose.
Fingerprinting techniques, such as pulsed‐field gel electrophoresis (PFGE) and multiple‐locus variable number tandem repeat analysis (MLVA), and the DNA sequence‐based approach, multilocus sequence typing (MLST), have proven to be valuable tools for the surveillance and detection of food‐borne disease outbreaks (Swaminathan et al., 2001; Joseph and Forsythe, 2012; EFSA, 2013; Lindstedt et al., 2013). However, a shortcoming of PFGE and MLVA is that they often fail to provide appropriate discriminatory power for specific subtypes within a given pathogen species to discriminate between outbreak‐related and sporadic cases (Allard et al., 2012; Franz et al., 2016). Also, MLST has problems when confronted with pathogens that are highly conserved and have a high level of clonal population structure. For such low levels of diversity, the resolution of MLST might not always suffice (Ranieri et al., 2013).
More recently, next‐generation sequencing (NGS) technologies have entered the field, offering opportunities to characterise food‐borne pathogens in much more great detail and giving access to the genetic information of pathogens at the highest resolution (Croucher and Didelot, 2015; Franz et al., 2016). With respect to the broad context of microbial hazards and food safety, within NGS applications, two tendencies can largely be discerned: whole genome sequencing (WGS) and metagenomics. In the same way, if interest lies in functionality, the methodology can be extrapolated at the RNA level with whole transcriptome sequencing and metatranscriptomic approaches, respectively. All of which, be it at the DNA or RNA level, or in combination, can deliver ample information that potentially could be used to contribute to the processes of hazard identification and characterisation, or even to be integrated in exposure assessment studies.
2. Description of work programme
2.1. Aims
The objective of this EFSA EU‐FORA project is to use state‐of‐the‐art NGS techniques to characterise and track microbial pathogens in a food‐processing facility to identify contamination sources and transmission routes of pathogens through the process facility. These molecular data will be used to underpin the development of the next generation of microbial risk assessments of pathogen transmission in food processing facilities and advance existing EFSA risk assessment methodologies in this area. The project will focus on dairy powder products that present ongoing food safety challenges.
The first objective will be an intensive induction and training period during which the fellow will be trained in advanced laboratory and bioinformatics techniques so that the fellow can successfully sequence a microbial isolate on the Illumina Platform MiSeq sequencer facility at UCD as well as apply the appropriate bioinformatics pipelines to characterise and genetically compare isolates sourced from a dairy production facility. In addition, the fellow will be trained in the use of Nanopore sequencing at the Center for Food Safety and Applied Nutrition (CFSAM) facilities of the Food and Drug Administration (FDA), Maryland, USA. Through this approach by learning and doing, the fellow will get a better understanding of different NGS solutions, their strengths and weaknesses, and how the generated data can be used in the context of food safety and ultimately risk assessment.
The second objective of the fellowship, through collaboration with Teagasc and Irish dairy manufacturing facilities, is coupled to studies using WGS to characterise the spatial and temporal distribution and genetic diversity of selected groups of bacteria (e.g. Cronobacter spp. and spore‐forming bacteria such as Bacillus spp.) at key stages in the production process and in the final product.
In a last phase, which will run to the end of the fellowship period, it is the objective to undertake the bioinformatics associated with all of the sequence data assembled in the course of the project as well as the large amount of relevant sequence data already collected by the UCD Centre for Food Safety through ongoing process facility surveillance projects. The main goal will be to determine the phylogenetic relationship of isolates based on for example single nucleotide polymorphism (SNP) analysis and through core genome data set analysis (i.e. genes shared by ≥ 95% of isolates). These techniques allow a comparison of the molecular similarity or diversity of bacterial isolates recovered from a production facility and gives enormous power to identify contamination sources and transmission routes of pathogens through a process facility. In addition to this end, for a selection of isolates, data generated through Illumina and Nanopore sequencing will be compared and complemented.
2.2. Activities/methods
2.2.1. Overview of activities
The performed work in this EFSA EU‐FORA project constituted out of three separate parts:
In a first part, a collection of isolates recovered from a dairy processing environment was identified using a WGS approach. The intent of the work performed in this first part was threefold: (i) learning the theoretical and practical aspects of WGS using Illumina technology on a MiSeq; (ii) getting familiar with the type of data generated and on bioinformatics tools for the analysis of the sequence data; and (iii) achieve an unambiguous identification of the isolates.
The second part consisted of applying the acquired skills of Illumina sequencing for the identification of a selection of 24 Bacillus spp. isolates using WGS.
-
The third part involved a comparative but also complementary study using Illumina and Nanopore technology for WGS of a set of Cronobacter sakazakii isolates representing four different MLST sequence types. Aims of this part consisted of:
learning the theoretical and practical items of whole genome sequencing using Nanopore technology on a MinION instrument;
gain insights on different NGS methodologies for WGS;
compare and combine the complementary data sets for the assembly of the whole genomes of the sequenced isolates;
transferring acquired knowledge on Nanopore sequencing to the UCD host site.
2.2.2. Description of applied next‐generation sequencing methodologies
In the fellowship, two different NGS technologies were used: sequencing‐by‐synthesis (SBS) using the reversible terminator technology by Illumina, and single molecule sequencing using Nanopore technology by Oxford Nanopore Technologies (https://nanoporetech.com/about-us/for-the-media) (Figure 2).
Figure 2.

Next‐generation sequencing workflow
A NGS approach in general starts with the extraction of total DNA from a pure culture, for whole genome sequencing, or from the community, in a metagenomics study. Following a library preparation step, which can involve polymerase chain reaction (PCR), and sequencing of the generated library pool, bioinformatics tools are used for data analysis.
2.2.2.1. Illumina technology (Figures 3 and 4) (Courtesy of Illumina, Inc.)
Figure 3.

Reversible terminator technology during sequencing‐by‐synthesis
Figure 4.

Illumina sequencing workflow
The reversible terminator technology employed by Illumina uses a sequencing concept that is similar to the chain termination procedure used in Sanger sequencing, in that, the strand elongation is stopped after the incorporation of a fluorescently labelled base that prevents further strand elongation and the label of the incorporated base is read out to reveal sequence information (Figure 3). However, contrary to Sanger sequencing with an irreversible termination of strand elongation and with sequence information retrieved from strands that differ in length by one base (i.e. the fluorescently labelled base), in the reversible terminator technology, termination is reversible and the sequence is determined in real time at the moment of incorporation of the fluorescently labelled bases.
During each sequencing cycle, all four nucleotides are presented to the growing strand as fluorescently labelled (colour of the nucleotide) and blocked (grey square) bases. Only the complementary base will be incorporated and its complementary fluorescent signal is detected (i.e. base calling). No further elongation of the strand occurs as the incorporated base is blocked at its 3′OH. Unincorporated bases are washed away. Before a new cycle, the fluorescent label is removed and the nucleotide is unblocked to allow the incorporation of the next base (Courtesy of Illumina, Inc.).
Following DNA extraction, similar to many other NGS approaches, first a sequencing library needs to be constructed to amplify and immobilise the templates for sequencing. Resulting library fragments consist of the fragment to be sequenced flanked by two different adapters (Figure 4A). This double‐stranded library is denatured to obtain single‐stranded DNAs that are applied on a flow cell. This flow cell has on its surface two populations of immobilised oligonucleotides complementary to the two different single‐stranded adapter ends of the sequencing library. Denatured oligonucleotides anneal to the complementary single‐stranded library oligonucleotides on the flow cell surface (pink and blue). Via reverse strand synthesis starting from the 3′ end of the surface bound oligo (the double‐stranded part), a new strand is created. Upon denaturation, the original strand is removed and the newly synthesised copy remains covalently bound to the flow cell. If this new strand bends over and attaches to the other oligonucleotide type complementary to the second adapter sequence that is present on the free 3′ end of the strand, it can be used to synthesise a second covalently bound reverse strand. The process of bending and reverse strand synthesis is called bridge amplification. Bridge amplification is repeated several times and creates clusters of several 1,000 copies of the original sequence in a very close proximity to each other on the flow cell (Figure 4B). At the end, each cluster on the flow cell consists of a single‐stranded, identically oriented copies of the same sequence. These can be sequenced by hybridising the sequencing primer onto the adapter sequences and starting the reversible terminator chemistry (Figure 4C).
Illumina sequencing workflow consisting of library preparation (A), cluster amplification (B), sequencing (C) and bioinformatics data analysis (D) (Courtesy of Illumina, Inc.).
2.2.2.2. Nanopore technology (Figures 5 and 6) (Courtesy of Oxford Nanopore Technologies)
Figure 5.

Oxford Nanopore Technology sequencing workflow on the MinION instrument
Figure 6.

Oxford Nanopore sequencing
The Nanopore technology by Oxford Nanopore Technologies can be considered as a true ‘third‐generation’ sequencing (TGS) technology, i.e. a single molecule sequencing (SMS) technology were reads represent the sequencing of a single molecule without the need of a replication enzymatic system (Schadt et al., 2010). In addition, the technology allows for real‐time sequencing and of long reads up to 1 Mb and longer (Loose, 2018a,b), which offers advantages during genome assembly.
After a DNA extraction to obtain high‐molecular weight genomic DNA, a library is constructed without the need for PCR amplification steps. During the library preparation, adapters are ligated to the DNA fragments and a processive enzyme is coupled. Upon interaction of the sequence–enzyme complex with the Nanopore, sequencing commences (Figure 5). The sequencing technology relies on nanopores that function as a channel between two chambers of an electrophoretic system (Figure 6). When a small voltage (~ 100 mV) is applied across the Nanopore, the resulting current can be measured. Molecules going through the Nanopore are responsible for the disruption in the ionic current. Measuring of this disruption allows the identification of the molecule. In the context of DNA, each base gives a subtly different reading as it passes through the pore, allowing direct reading of the sequence. Raw output used for base calling is an electronic trace of current changes generated not by individual bases but by 5‐ or 6‐nucleotide ‘words’ known as k‐mers. Sequence data are streamed as DNA fragments translocate through the pore, permitting real‐time analysis (Loman and Watson, 2015; Lu et al., 2016).
Schematic overview of sequencing workflow on Oxford Nanopore Technologies’ MinION instrument. Following a DNA extraction to obtain high‐molecular weight genomic DNA (gDNA), a library is constructed prior to the real‐time sequencing analysis (courtesy of Oxford Nanopore Technologies: https://nanoporetech.com/resource-centre/posters/dna-extraction-and-library-preparation-rapid-genus-and-species-level).
The DNA is coupled to a processive enzyme (green). The DNA–enzyme complex interacts with the Nanopore (blue). Single‐stranded DNA is pulled through the Nanopore aperture one base at a time. As the DNA moves through the pore, the combination of nucleotides in the strand being processed creates a characteristic disruption in the electrical current. This signal can then be used to determine the order of bases on that particular DNA strand (current trace diagram on the right) (courtesy of Oxford Nanopore Technologies).
2.2.3. Whole genome sequencing of dairy processing environment isolates
To become familiar with the technology and bring theoretical knowledge into practice, a set of 18 isolates from a dairy processing environment was sequenced with Illumina technology using a MiSeq instrument. For library preparation, the NEBNext® Ultra™ II FS DNA library prep kit (NEB) was used. For sequencing the MiSeq reagent kit V3 (Illumina) 600 cycles and a paired‐end run was used.
The isolates were considered to belong to the genus Bacillus. However, preliminary identification using partial 16S rRNA sequencing and matrix‐assisted laser desorption/ionisation time‐of‐flight mass spectrometry (MALDI‐TOF MS) did not allow the correct identification, either due to the lack of resolution of 16S rRNA sequencing and/or due to conflicting results between 16S and MALDI‐TOF MS. As WGS‐based approaches are considered to be superior compared with traditional 16S rRNA sequence analysis due to their much higher resolution because they are based on a much larger part of the genome, it was decided, in the project, to sequence the whole genome of these isolates to get a correct identification. Obtained sequences were analysed using bioinformatics tools available at https://cge.cbs.dtu.dk/services/.
2.2.4. Whole genome sequencing of Bacillus spp. isolates
The second part of work was part of a larger study identifying key or core spores and spore‐forming bacteria in skimmed milk powders. Twenty‐one samples of medium‐heat skim milk powders from eight different facilities were analysed for their spore‐forming community using 16 different microbiological tests. A preliminary identification of a selection of 285 isolates was performed using partial 16S rRNA (a 570‐bp part of the V3–V5 region) sequencing. Blast analysis (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and pairwise and multiple alignment using the BioNumerics Software version 7.6.3. (Applied Maths, bioMérieux) revealed the presence of eight different genera divided over 21 sequence clusters (Figure 7).
Figure 7.
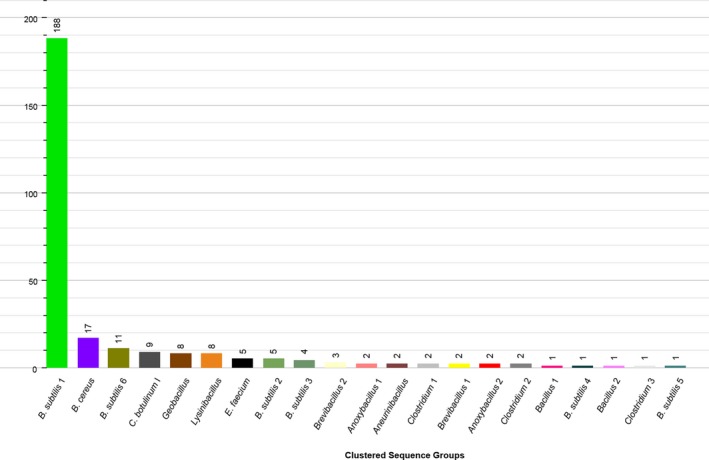
Observed 16S rRNA based diversity
Preliminary identification results, of 285 isolates, based on a 570‐bp fragment of the 16S rRNA gene.
Although 16S rRNA sequencing has long been the gold standard for species delineation, it has become evident that the resolution is not sufficient for closely generated species. Increasing the cut‐off from 97% to 98.7% 16S rRNA sequence similarity to be considered belonging to the same species (Stackebrandt and Ebers, 2006) still resulted in conflicting results within many genera. This is also the case for the genus Bacillus, the 16S rRNA gene encountering problems in resolving species within this genus (Caamaño‐Antelo et al., 2015). For example, the species within the Bacillus subtilis group exhibited 16S sequence similarity varying between 98.1% up to 99.8%. A selection of 24 isolates, identified as belonging to the B. subtilis group based on the partial 16S sequence, was picked for whole genome sequencing using Illumina technology with a MiSeq machine to obtain a more correct species identification and more information on the genetic diversity within the species. For library preparation, Illumina's Nextera® XT (Illumina) DNA library preparation method was used and for paired‐end run sequencing, the MiSeq reagent kit V3 for 600 cycles was selected. A pipeline of several bioinformatics tools will be used for the quality control, trimming and assembly of the sequences. Subsequently assembled WGS contigs will be used for identification and characterisation purposes.
2.2.5. Oxford Nanopore sequencing of selected Cronobacter sakazakii isolates
One isolate of each of the four MLST sequence types (1, 4, 41 and 42) of Cronobacter sakazakii, retrieved from a dairy powder processing environment and previously characterised via a whole genome MLST (wgMLST) approach, was selected for WGS using Oxford Nanopore technology. Despite the higher error rate compared, the much longer fragment reads that can be sequenced aided the assembly of a reduced number of bigger contigs. The already available MiSeq accurate short‐read sequences will complement any errors to increase the accuracy of the assembly.
2.2.6. Other secondary activities performed in the EU‐FORA fellowship
Cooperation with the Agriculture and Food Development Authority of Ireland (Teagasc).
Attended a food safety regulation training (a module that is part of the MSc Food Safety and Risk Analysis, University College Dublin) organised by the Food Safety Authority of Ireland (FSAI).
Attended the EFSA Scientific Colloquium No. 24, ‘OMICS in risk assessment: state‐of‐the‐art and next steps’ (24–25 April 2018, Berlin, Germany). As a member of the discussion group ‘Genomics for the identification and characterisation of microbial strains used in food and feed products’, discussed the potential of WGS analysis for improving hazard identification of microbial strains that enter the food chain or used as production strains.
Panel member ‘building a global monitoring system for food‐borne illness and AMR’ during Asset 2018, 28–31 May 2018 Belfast, Northern Ireland.
Visiting the Center for Food Safety and Applied Nutrition (CFSAM) facilities of the FDA, Maryland, USA that has a close collaboration with the UCD, Centre for Food Safety hosting site.
A short outreach moment on the EU‐FORA programme at the FSAI premises in the presence of FSAI staff and other Article 36 organisations. This trip will be coupled by a 1‐ to 2‐day visit to the FSAI to get a better insight into the activities of the Irish food safety authority.
3. Conclusions
With the introduction of high‐throughput sequencing platforms, NGS applications have become widely applied. It is without any doubt that the application of NGS, be it through whole genome sequencing or more elaborate metagenomics approaches, offers a plethora of opportunities in the areas of food safety and microbial risk assessment. The most important difference between both approaches lies in the difference in target used, i.e. for WGS, the nucleic acids obtained from a pure culture isolate, and in metagenomics, total nucleic acids retrieved directly from the microbiota present in the environment under investigation. This can be extrapolated at the RNA level with whole transcriptome sequencing and metatranscriptomics, respectively.
For WGS that relies on a culture‐based approach, microbial growth is required using selective culture medium and conditions to recover the target organism. The output of WGS is the complete DNA content of a microorganism, inferring information on genes that are under diversifying selection (e.g. antigen genes), genes that are under stabilising selection (e.g. housekeeping genes) and genes that might be of relevance from a food safety perspective (e.g. virulence genes, AMR genes, toxin production), but also of non‐coding regions and possible episomal DNA present. Hence, WGS has the potential to describe a bacterial strain at the highest genetic detail allowing the full characterisation of a strain. WGS is also universal applicable, in contrast with for example MLST or PFGE, the choice of loci or restriction enzymes, respectively, depending on the taxonomic group or pathogen under investigation. However, this will require standardisation in the way the data are generated and analysed and at an interlaboratory level preferably on a global scale. Initiatives to promote this harmonisation exist, such as http://www.globalmicrobialidentifier.com and efforts have been undertaken to standardise for example some quality parameters and on guidelines how WGS data can be used to its best (Chun et al., 2018). Moreover, once fully characterised, the information can be used for detection and/or investigation of food‐borne outbreaks (e.g. route‐ and source‐tracking, cross‐contamination events), attribution studies, assessment of possible virulence properties or epidemic potential, and integrating all these data into risk assessment evaluations, even up to intervention and control strategy studies. Taking these applications into consideration, it is important to be aware of some of issues inherent to culturing approaches: they cannot be considered quantitative as they might under‐ or overestimate the true diversity present. In addition, currently, the genetic content of a microorganism cannot be seen detached from its phenotypic traits (e.g. its pathogenicity) in relation to host and environmental factors.
Metagenomics approaches target the nucleic acids of the microorganisms in the environment or food matrix and have the advantage that no prior cultivation steps are necessary to investigate the full community, it is a so‐called culture‐independent technique. The complexity of such kind of analyses makes it even a more daunting task to use the generated data in a context of food safety and risk assessment. A solution lies in the identification of biomarkers that are linked and might predict microbial behaviour (Brul et al., 2012). Also here, it is important that a connection is made between host and environmental conditions and expression level of the biomarker on the one hand and phenotypic behaviour on the other hand. The continued increase in other ‘omics data from proteomics and metabolomics studies will certainly contribute in this process.
Finally, similar to many other currently applied identification and typing methods in which the method of choice depends on the pathogen and the question that needs to be addressed, for NGS, the type of approach and the type and extent of data used will depend on the purpose (Franz et al., 2016).
The programme itself aimed at familiarising the fellow with NGS in the context of food safety and risk assessment and this via a principle of ‘learning by doing’. First, in addition to the necessary theoretical insights, wet‐laboratory experience was gained for two NGS techniques: high‐throughput sequencing of short reads using Illumina technology with a MiSeq instrument and long‐read sequencing using a MinION based on Nanopore technology. In addition, the fellow acquired bioinformatics skills using Unix‐based and other tools necessary for the analysis (quality, trimming, assembly) of generated sequence data. Given the acquired theoretical and practical insights, additional discussion will aid the interpretation on how these techniques and data can be implemented in a microbial risk assessment framework.
Abbreviations
- AMR
antimicrobial resistance
- bp
base pairs
- CFSAM
Center for Food Safety and Applied Nutrition
- EU‐FORA
The European Food Risk Assessment Fellowship Programme
- FDA
Food and Drug Administration
- FSAI
Food Safety Authority Ireland
- gDNA
genomic DNA
- MALDI‐TOF MS
matrix‐assisted laser desorption/ionisation time‐of‐flight mass spectrometry
- MLST
multilocus sequence typing
- MLVA
multiple‐locus variable number tandem repeat analysis
- NGS
next‐generation sequencing
- PFGE
pulsed‐field gel electrophoresis
- rRNA
ribosomal RNA
- SBS
sequencing‐by‐synthesis
- SNP
single nucleotide polymorphism
- TGS
‘third‐generation’ sequencing
- UCD
University College Dublin
- WGS
whole genome sequencing
- wgMLST
whole genome multilocus sequence typing
Suggested citation: UCD Centre for Food Safety, Dublin, Ireland , Van Hoorde K and Butler F, 2018. Use of next‐generation sequencing in microbial risk assessment. EFSA Journal 2018;16(S1):e16086, 13 pp. 10.2903/j.efsa.2018.e16086
Acknowledgements: This report is funded by EFSA as part of the EU‐FORA programme.
Approved: 6 July 2018
References
- Allard MW, Luo Y, Strain E, Li C, Keys CE, Son I, Stones R, Musser SM and Brown EW, 2012. High resolution clustering of Salmonella enterica serovar Montevideo strains using a next‐generation sequencing approach. BMC Genomics, 13, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brul S, Bassett J, Cook P, Kathariou S, McClure P, Jasti PR and Betts R, 2012. ‘Omics’ technologies in quantitative microbial risk assessment. Trends in Food Science and Technology, 27, 12–24. [Google Scholar]
- Caamaño‐Antelo S, Fernández‐No IC, Böhme K, Ezzat‐Alnakip M, Quintela‐Baluja M, Barros‐Velázquez J and Calo‐Mata P, 2015. Genetic discrimination of foodborne pathogenic and spoilage Bacillus spp. based on three housekeeping genes. Food Microbiology, 46, 288–298. [DOI] [PubMed] [Google Scholar]
- Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, da Costa MS, Rooney AP, Yi H, Xu X‐W, De Meyer S and Trujillo ME, 2018. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. International Journal of Systematic and Evolutionary Microbiology, 68, 461–466. [DOI] [PubMed] [Google Scholar]
- Croucher NJ and Didelot X, 2015. The application of genomics to tracing bacterial pathogen transmission. Current Opinion in Microbiology, 23, 62–67. [DOI] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2013. Scientific opinion on the evaluation of molecular typing methods for major food‐borne microbiological hazards and their use for attribution modelling, outbreak investigation and scanning surveillance: part 1 (evaluation of methods and applications). EFSA Journal 2013;11(12):3502, 84 pp. 10.2903/j.efsa.2013.3502 [DOI] [Google Scholar]
- Franz E, Gras LM and Dallman T, 2016. Significance of whole genome sequencing for surveillance, source attribution and microbial risk assessment of foodborne pathogens. Current Opinion in Food Science, 8, 74–79. [Google Scholar]
- Joseph S and Forsythe SJ, 2012. Insights into the emergent bacterial pathogen Cronobacter spp., generated by Multilocus Sequence Typing and Analysis. Frontiers in Microbiology, 3, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstedt BA, Torpdahl M, Vergnaud G, Le Hello S, Weill FX, Tietze E, Malorny B, Prendergast DM, Ní Ghallchóir E, Lista RF, Schouls LM, Söderlund R, Börjesson S and Åkerström S, 2013. Use of multilocus variable‐number tandem repeat analysis (MLVA) in eight European countries, 2012. Eurosurveillance, 18, 20385. [DOI] [PubMed] [Google Scholar]
- Loman NJ and Watson M, 2015. Successful test launch for nanopore sequencing. Nature Methods, 12, 303–304. [DOI] [PubMed] [Google Scholar]
- Loose M, 2018a. Available online: https://twitter.com/mattloose/status/954147458778587136 [Accessed: 27 May 2018]
- Loose M, 2018b. Available online: https://twitter.com/mattloose/status/992117714289217536 [Accessed: 27 May 2018]
- Lu H, Giordano F and Ning Z, 2016. Oxford Nanopore MinION sequencing and genome assembly. Genomics, Proteomics and Bioinformatics, 14, 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranieri ML, Shi C, Moreno Switt AI, den Bakker HC and Wiedmann M, 2013. Comparison of typing methods with a new procedure based on sequence characterization for Salmonella serovar prediction. Journal of Clinical Microbiology, 51, 1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Turner S and Kasarskis A, 2010. A window into third‐generation sequencing. Human Molecular Genetics, 19, R227–R240. [DOI] [PubMed] [Google Scholar]
- Stackebrandt E and Ebers J, 2006. Taxonomic parameters revisited: tarnished gold standards. Microbiology Today, 33, 152–155. [Google Scholar]
- Swaminathan B, Barrett TJ, Hunter SB and Tauxe RV, 2001. PulseNet: the molecular subtyping network for foodborne bacterial disease surveillance, United States. Emerging Infectious Disease Journal, 7, 382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]