

Abstract
The dual leucine zipper–bearing kinase (DLK) and leucine zipper–bearing kinase (LZK) are evolutionarily conserved MAPKKKs of the mixed-lineage kinase family. Acting upstream of stress-responsive JNK and p38 MAP kinases, DLK and LZK have emerged as central players in neuronal responses to a variety of acute and traumatic injuries. Recent studies also implicate their function in astrocytes, microglia, and other nonneuronal cells, reflecting their expanding roles in the multicellular response to injury and in disease. Of particular note is the potential link of these kinases to neurodegenerative diseases and cancer. It is thus critical to understand the physiological contexts under which these kinases are activated, as well as the signal transduction mechanisms that mediate specific functional outcomes. In this review we first provide a historical overview of the biochemical and functional dissection of these kinases. We then discuss recent findings on regulating their activity to enhance cellular protection following injury and in disease, focusing on but not limited to the nervous system.
Keywords: axon regeneration, neuronal development, MAP3K, DLK, LZK, JNK, p38, DLK-1, Wallenda, neuronal death, axon degeneration, cytoskeleton, astrocytes, Alzheimer disease models, HSV infection, adipogenesis, insulin β-cells
INTRODUCTION
MAPKKKs (or MAP3Ks) are key controllers in signal transduction and act as the upstream kinases in the phosphorelay reaction in classical mitogen-activated protein kinase (MAPK) cascades. MAPKKKs sense changes in the environment and internal states of cells, triggering signal-dependent and cell-specific responses. Around the mid-1990s, during an era of kinase discovery, two closely related brain-enriched kinases were reported: the dual leucine zipper– bearing kinase [DLK, also known as zipper protein kinase (ZPK) and MAPK-upstream kinase (MUK)] (Holzman et al. 1994) and the leucine zipper–bearing kinase (LZK) (Sakuma et al. 1997). Because, in DLK and LZK, subdomains I–VII resemble serine/threonine kinases and subdomains VIII–XI more closely resemble tyrosine kinases, these kinases are grouped into the family of the mixed-lineage kinases (MLKs) (Gallo & Johnson 2002). Another signature of DLK and LZK is the presence of a dual leucine zipper (LZ) domain following the kinase domain, and so these kinases compose the DLK subfamily of the MLKs. Biochemical studies show that both DLK and LZK are serine/threonine kinases and can activate JNK MAPKs and, to some extent, p38 MAPKs. Work in the past two decades has revealed these two kinases’ pivotal roles in a variety of biological processes, in particular the cellular response to stress and injury in the nervous system, making DLK and LZK targets for drug discovery. In this review we begin with an overview of the discovery and biochemical properties of DLK and LZK, then explore the functional analyses in model organisms, and end by discussing these kinases’ roles in injury and disease.
DISCOVERY, BIOCHEMICAL PROPERTIES, AND PROTEIN INTERACTION NETWORK
The DLK proteins are represented by two members known as MAP3K12 (or DLK) and MAP3K13 (or LZK) in vertebrate genomes and by a single member in most invertebrates (Figure 1). Each full-length kinase consists of approximately 900 amino acids, with the kinase domain at the N terminus, followed by two LZs and a long C terminus. The sequence homology among all members is primarily in the kinase and LZ domains, while the C termini are rich in proline, serine, and acidic amino acids but share little sequence similarity between DLK and LZK in the same species (Figure 1). LZ domains are found in many proteins and generally mediate homo- or heterodimerization. For DLK and LZK, the LZ domains are essential for their activation; mutating a single leucine residue in either LZ abolishes the activity of these kinases to phosphorylate JNK in transfected cell lines (Ikeda et al. 2001b, Nihalani et al. 2000). Furthermore, the LZ domain of DLK and LZK displays high selectivity for homomeric interaction, suggesting that regulation of LZ-mediated interaction likely plays important roles in signaling specificity. The long C terminus appears to be not critical for DLK to activate JNK but is indispensable for LZK to activate its downstream kinase, hinting at differential regulation of the two kinase paralogs.
Figure 1.
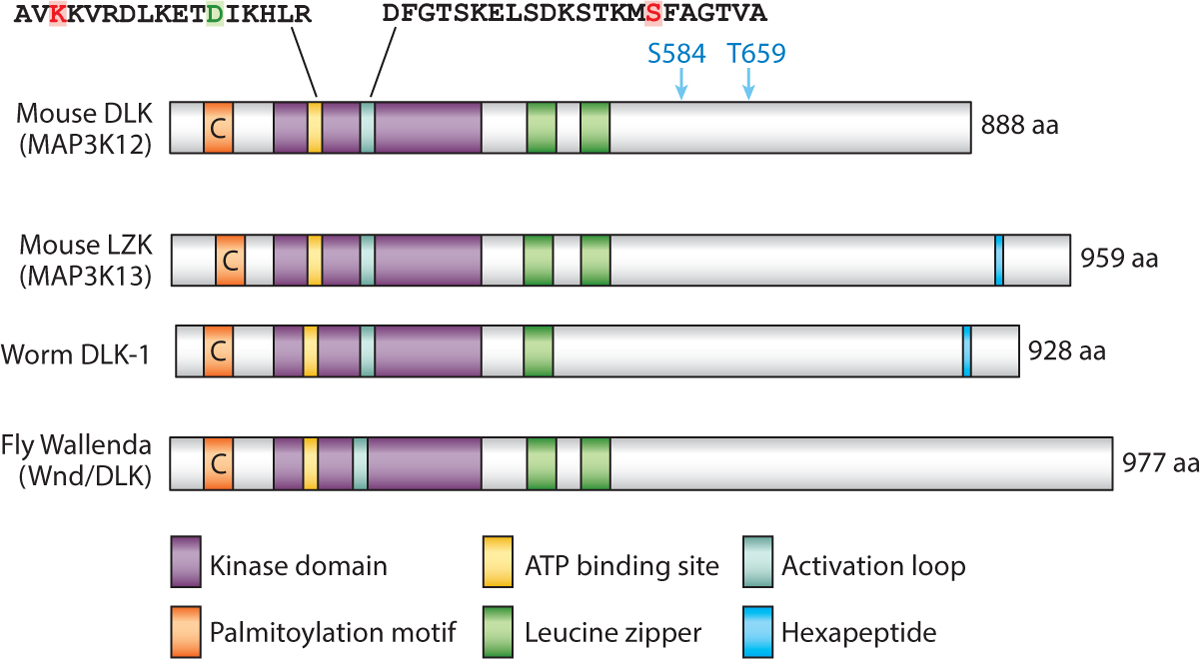
Protein structure and known signaling domains of DLKs. Protein sequences are: mouse DLK (UniProtKB Q60700), mouse LZK (UniProtKB Q1HKZ5), fly Wallenda (CG8789-PC), and worm DLK-1 (F33E2.2a). The overall sequence homology of the kinase and LZ domain is approximately 61% between vertebrate members and invertebrates. Colored boxes correlate with the identified domains. C denotes palmitoylated Cys; the red K is critical for ATP binding; the green D may be responsible for the distorted α-helix C; the red S in the activation loop is phosphorylated by protein kinase A; the blue S584 and T659 are phosphorylated by AKT; and the hexapeptide represents SDGLSD, which is present only in Caenorhabditis elegans DLK-1 and vertebrate LZK.
A crystal structure of the kinase domain (aa 148–435) of human DLK was reported at 1.7-Å resolution (Patel et al. 2015b). The structure shows a canonical kinase fold, with a disrupted α-helix C, due to an Asp residue at a conserved position that normally has a Glu (Figure 1). In canonical kinases, the α-helix C forms a salt bridge with a Lys to stabilize ATP binding, which is hence critical for kinase activity. It remains to be determined whether the altered α-helix C is present in the structure of the full-length DLK protein and how such distortion contributes to the activity and regulation of DLK in a physiological environment. Nonetheless, scaffold-hopping methods (Sun et al. 2012) were applied to this structure, and multiple chemical compounds were generated and shown to have high potency to inhibit DLK activity in vitro (Patel et al. 2015b). Several such compounds can cross the blood-brain barrier in mice and, as described below, can alleviate some symptoms in mouse models of neurological disease (Patel et al. 2015a, 2017). Given the near-identical kinase domain in LZK, these compounds likely cause similar inhibition of LZK.
Activation of MAPKKKs is generally under the control of membrane receptors and/or G protein pathways that are responsive to external stimuli. Compared with many well-studied MAPKKKs, DLK and LZK are readily self-activated upon overexpression, most likely via LZ-mediated homomeric interaction and autophosphorylation of the kinase domain (Mata et al. 1996, Sakuma et al. 1997). This self-activating property implies that the expression of endogenous DLK or LZK protein is normally kept at low levels, which may be why these kinases rarely surface in large-scale proteomic databases. Proteins that are consistently reported to bind to DLKs include scaffolding proteins, such as JIP1 (Ikeda et al. 2001a, Nihalani et al. 2001), JIP3 (Ghosh et al. 2011), and MBIP (Fukuyama et al. 2000) (Figure 2). These proteins appear to bind to regions N-terminal to the kinase domain, and their binding inhibits kinase activity by keeping DLK in a monomeric state.
Figure 2.
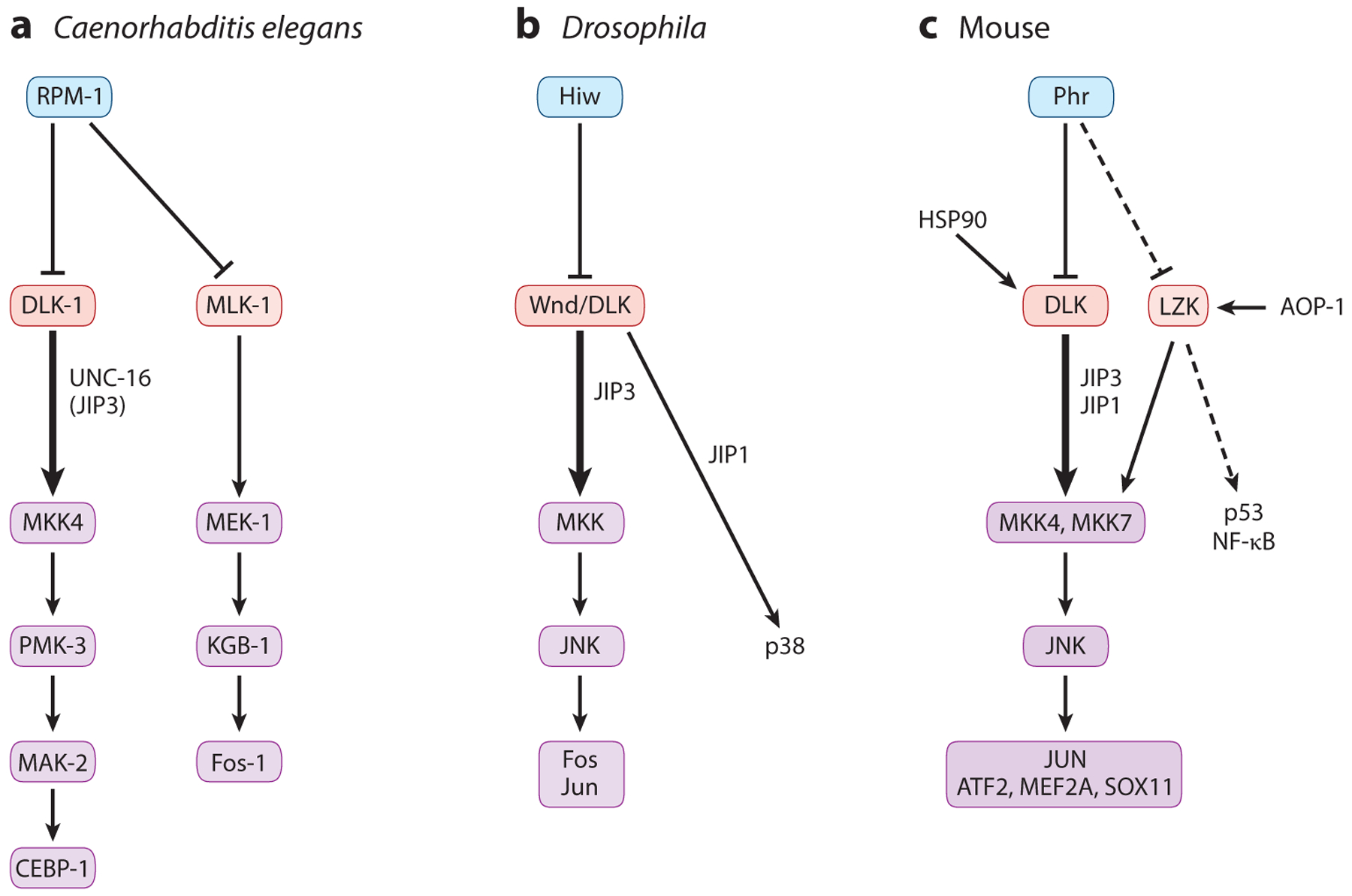
Conserved regulation of DLKs and their signaling cascades. Studies of the PHR E3 ligases demonstrate their conserved action to target DLKs for ubiquitin-mediated protein degradation. (a) In Caenorhabditis elegans, the major downstream cascade (thick arrows) of DLK-1 consists of the MAPKK MKK4 and the p38 MAPK PMK-3 acting on the CEBP-1 nuclear factor, with partially redundant roles of MLK-1 acting through the MAPKK MEK-1 and the JNK MAPK KGB-1 (thinner arrows) in specific neurons and injury paradigms. (b) In Drosophila, JNK and p38 mediate different effects of Wallenda (Wnd)/DLK in injury and synapse development, with the effects partly dependent on the interaction of Wnd/DLK with the scaffold proteins JIP1 and JIP3. (c) In mammals, DLK acts mainly through MKK4, MKK7, and JNK, with JUN and other transcriptional factors as the main and collaborative targets. The chaperone HSP90 also promotes DLK activity. It remains to be tested whether LZK is a target of Phr (dashed line). Thus far, LZK is reported to bind the mitochondrial protein AOP-1, and most data on LZK downstream signaling have come from studies in cancer cells, linking its function to p53 and NF-κB (dashed arrow).
In developing mouse embryos, DLK displays subcellular localization in various cell types (Hirai et al. 2002, 2005). In biochemical analyses of brain protein extracts, DLK was present in both the cytosolic and synaptic membrane fractions (Mata et al. 1996, Pozniak et al. 2013). Cytosolic DLK was detected as being phosphorylated, while the synaptic membrane–enriched DLK was unphosphorylated and migrated as a high-molecular-weight complex (Mata et al. 1996). In neuronal-glial cultures, treatment with cyclosporin A, an inhibitor of the calcium/calmodulin-dependent protein phosphatase calcineurin, inhibited dephosphorylation of DLK induced by membrane depolarization, suggesting that DLK activation may be regulated by intracellular calcium and calcineurin. Phosphorylation of several serine and threonine residues in DLK has been reported to depend on protein kinase A (PKA) (Hao et al. 2016), AKT (Wu et al. 2015), or activated JNK (Huntwork-Rodriguez et al. 2013) (Figure 1). Such phosphorylation differentially modulates DLK activity. In particular, phosphorylation of Ser302 by PKA within the activation loop of DLK is required for kinase activity in mouse dorsal root ganglion (DRG) neurons and Drosophila motor neurons (Hao et al. 2016). In contrast, AKT-mediated phosphorylation restrains DLK’s function in the self-renewal of mouse stem cells (Wu et al. 2015). Additionally, DLK is palmitoylated at a conserved cysteine in the N-terminal domain (Holland et al. 2016) (Figure 1). Palmitoylation is a major form of posttranslational modification enabling protein association with membranous vesicles. Palmitoylation of DLK can regulate its binding with JIPs (Holland et al. 2016), which may influence their interaction with motor proteins (Horiuchi et al. 2007). Palmitoylation also facilitates DLK interaction with the axonal survival factor NMNAT2 (Summers et al. 2018).
Little is known about the subcellular expression pattern of endogenous LZK, and when expressed in COS cells, LZK also shows cytosolic and membrane association (Sakuma et al. 1997). LZK was shown to be phosphorylated at multiple serine and threonine residues (Sakuma et al. 1997), but the functional relevance of such phosphorylation events remains unknown. LZK, in addition to its demonstrated role in activating JNK and p38, is implicated in NF-κB activation and regulation of the tumor suppressor p53 and the oncogene Myc in cancer cells (Edwards et al. 2017, Han et al. 2016, Masaki et al. 2003) (Figure 2c). LZK was reported to bind antioxidant protein-1 (AOP-1), a mitochondrial protein, and such interaction enhanced LZK involvement in NF-κB activation (Masaki et al. 2003) (Figure 2c). LZK and DLK can bind each other, and such binding involves the N-terminal region and not the LZ domain (Nihalani et al. 2000). Endogenous LZK was also found to be associated with DLK in mouse brain protein extracts (Pozniak et al. 2013), and as described below, emerging evidence has begun to depict the overlapping functions of these kinases, suggesting that they may be subject to cross-regulation under specific stress conditions.
FUNCTION AND REGULATION OF DLKs IN NEURONAL DEVELOPMENT
Invertebrate DLKs: Caenorhabditis elegans DLK-1 and Drosophila Wallenda/DLK
First insights into the in vivo function of the DLKs came from genetic studies of the invertebrate members, namely Caenorhabditis elegans DLK-1 (Nakata et al. 2005) and Drosophila Wallenda (Wnd) (Collins et al. 2006). These invertebrate DLKs were discovered for their roles in synapse development, where they were shown to be the substrates of the synaptic ubiquitin E3 ligases, C. elegans RPM-1, and Drosophila Highwire (Hiw) (Schaefer et al. 2000, Wan et al. 2000, Zhen et al. 2000). These E3 ligases, also known as PHR proteins, include the human protein Pam and the mouse protein PHR. PHR proteins are unusually large (close to or more than 4,000 aa), with a RING finger domain at the C terminus that constitutes a noncanonical E3 ligase (Grill et al. 2016). Null mutants of C. elegans rpm-1 and Drosophila hiw exhibit distinct defects in synapse morphology and number (Schaefer et al. 2000, Wan et al. 2000, Zhen et al. 2000). Isolation of genetic mutations that suppressed the synapse defects of rpm-1 and hiw independently uncovered loss-of-function mutations in C. elegans dlk-1 (Nakata et al. 2005) and Drosophila wnd (Collins et al. 2006). DLK-1 and Wnd/DLK are expressed predominantly in neurons. The overall levels of DLK-1 and Wnd/DLK were increased in rpm-1 and hiw mutants, respectively, and overexpression of wild-type (WT) DLK-1 and Wnd/DLK altered synapse development, resembling the phenotypes observed in rpm-1 and hiw mutants, respectively. On the basis of this and other evidence, DLK-1 and Wnd are in vivo substrates for RPM-1- and Hiw-dependent protein degradation (Figure 2a,b). Nonetheless, animals with complete loss of DLK-1 or Wnd/DLK develop normally (Schaefer et al. 2000, Wan et al. 2000, Zhen et al. 2000).
Analyses of other genetic suppressor mutations of rpm-1 in C. elegans further revealed that DLK-1 acts upstream of a p38 MAPK cascade (Nakata et al. 2005) (Figure 2a). Studies in Drosophila identified Wnd/DLK to be upstream of a JNK MAPK (Collins et al. 2006) (Figure 2b). These downstream MAPK cascades regulate gene expression via the b-Zip domain transcriptional factors CEBP-1 in C. elegans (Yan et al. 2009) and Fos in Drosophila (Collins et al. 2006) (Figure 2a,b). The use of p38 or JNK in DLK-dependent signal transduction may reflect an evolutionary divergence or the context-dependent specificity of stress kinases (Andrusiak & Jin 2016). Indeed, studies demonstrate that these stress-responsive MAPKs exhibit considerable cross talk and cell type specificity (Klinedinst et al. 2013, Nix et al. 2011).
Vertebrate DLK and LZK
Despite the near-simultaneous identification of DLK and LZK as brain-enriched kinases with comparable biochemical activity for phosphorylation of JNK (Holzman et al. 1994, Sakuma et al. 1997), progress toward understanding their in vivo function has mostly focused on DLK/MAP3K12. Expression data show that DLK displays differential developmental and temporal regulation in various tissues (Blouin et al. 1996, Hirai et al. 2005, Suenaga et al. 2006). Within the nervous system, DLK is broadly expressed in many neuronal types from embryo to adult. In cerebellar cortex, DLK expression is high in postmitotic migratory neurons and is downregulated postnatally (Suenaga et al. 2006). In developing neurons, DLK proteins are more abundantly present in axons and growth cones than in the soma and dendrites, displaying a high degree of colocalization with neuron-specific tubulins (Hirai et al. 2005, Lewcock et al. 2007). A proteomic analysis of immunoprecipitated protein complex for endogenous DLK from 15-week-old mouse brains also revealed that most proteins associated with DLK are cytoskeletal (Pozniak et al. 2013). In contrast, much less is known about endogenous LZK expression, although it is present at low levels in select brain regions (Chen et al. 2016). LZK mRNA detected from human brain contains long 3’ untranslated regions (3’ UTRs) (Sakuma et al. 1997). Indeed, LZK mRNAs are targets of several microRNAs (Lippi et al. 2016). In human cancer cells, miRNA-206 directly binds LZK mRNAs, leading to decreased LZK expression (Han et al. 2016). This interaction may have functional importance for tumor cells that are associated with high levels of the Myc oncogene.
Mice lacking DLK die perinatally and show defective axon growth and neuronal migration (Hirai et al. 2006), whereas mice lacking LZK are healthy and fertile (Chen et al. 2018, Welsbie et al. 2017). Genetic studies on mouse PHR proteins supported their roles in the regulation of DLK abundance, similar to the case of the invertebrate orthologs (Figure 2c). Several mouse Phr1 mutants were characterized, including a compound chromosomal deletion (Burgess et al. 2004), a targeted genetic knockout (KO) (Bloom et al. 2007), and a nonsense mutation generated by ENU-induced mutagenesis (Lewcock et al. 2007). Phr1 mutant mice exhibit broad phenotypes, including defective axonal tracts and guidance, axon overextension, and abnormal synaptic position and morphology. Increased DLK proteins were observed in some cell types of Phr1 mutants. Some motor axon defects were suppressed by treatment with p38 inhibitors (Lewcock et al. 2007); defects of specific neuromuscular synapses in Phr1 mutants were also suppressed by loss of Dlk (Bloom et al. 2007). These observations support mechanistic conservation of DLK inhibition by Phr1. However, many defects of Phr mutants remain in Phr1; Dlk double KO animals (Bloom et al. 2007), which is also consistent with findings in invertebrates that PHR proteins have roles in addition to regulating DLK stability (Grill et al. 2016). Besides being negatively regulated by PHR, DLK stability can be regulated by the chaperone heat shock proteins under stress and injury (Daviau et al. 2006, Karney-Grobe et al. 2018) (Figure 2c). Whether PHR regulates LZK remains unknown.
FUNCTION AND REGULATION OF DLKs UNDER STRESS AND INJURY IN THE MATURE NERVOUS SYSTEM
DLK-Mediated Signaling Pathways
Leveraging powerful molecular genetic tools in invertebrates has allowed for in-depth mechanistic assessments regarding how DLK-mediated signaling is regulated in vivo, as well as how this signaling in turn regulates cellular function. One major theme is that DLK activity is highly responsive to cellular stresses involving changes to the cytoskeleton (reviewed in Asghari Adib et al. 2018). For example, treating C. elegans with colchicine, which inhibits tubulin polymerization, caused a specific loss of mechanosensation (Bounoutas et al. 2009). Such effects were due to altered gene expression, and genetic screening revealed that loss of DLK-1 signaling, including signaling to its target, CEBP-1, prevented colchicine-induced impairment in neuronal function (Bounoutas et al. 2011). DLK-1 also mediates synapse stabilization and neurite branching, which depend on the microtubule (MT) minus end binding protein Patronin/CAMSAP (Chuang et al. 2014, Marcette et al. 2014, Richardson et al. 2014). Similarly, at Drosophila neuromuscular junctions, loss of function in a number of proteins that stabilize actin and MT cytoskeleton, such as spectraplakin, α-spectrin, or ankyrin, leads to synaptic terminal overgrowth due to activation of Wnd/DLK and downstream Jun/Fos-mediated gene expression (Massaro et al. 2009, Valakh et al. 2013).
C. elegans DLK-1 and Drosophila Wnd/DLK can also act through downstream factors other than their key targets, CEBP-1 and Fos. For example, in the C. elegans locomotor circuit, dynamic changes to the MT cytoskeleton facilitate synapse remodeling during larval development. Formation of new synapses depends on DLK-1; in this case, activation of DLK-1 leads to changes in cellular trafficking independently of CEBP-1 (Kurup et al. 2015). Likewise, Drosophila Wnd/DLK can interact with kinesin-1, likely via binding JIP1 (Horiuchi et al. 2007). Several other factors that can trigger DLK-1 or Wnd/DLK activation include changes in mitochondrial energy state, altered G protein signaling, and disruption in intracellular trafficking (Chen et al. 2014, Klinedinst et al. 2013, Ma et al. 2016, van der Vaart et al. 2015, Wang et al. 2013). Additionally, DLK-1 and Wnd/DLK pathways contribute to axon and dendrite outgrowth and guidance by modulating Wnt or other signaling pathways (Park & Rongo 2018, Wang et al. 2013). However, the direct targets of DLK in these pathways remain unknown.
Many studies from rodent DLK models have underscored the mechanistic conservation of DLK signaling across species. For example, in mammalian sensory neurons, treatment with cytochalasin D and nocodazole to disrupt the actin and MT cytoskeleton, respectively, activates the DLK and JNK pathway, likely via binding to JIP1 (Valakh et al. 2015). DLK phosphorylation by PKA plays a conserved role in both fly and mouse neurons under axon injury (Hao et al. 2016). Other findings highlight cell type–dependent regulation of DLK phosphorylation. Mutating the Ser302 residue to the nonphosphorylatable alanine (S302A) in HEK293T cells resulted in a higher degradation rate of mutant DLK relative to WT DLK, which was attenuated by coexpression of a deubiquitinating enzyme (Huntwork-Rodriguez et al. 2013). However, in the insulin-producing pancreatic islet β-cell HIT line, protein levels of endogenous DLK, or overexpressed DLK (WT), but not DLK (S302A), were decreased after treatment with TNFα or IL-1β (Wallbach et al. 2016). In COS cells, DLK activity is also not required for protein stability (Daviau et al. 2006). These observations imply cell type–dependent and stimuli-dependent regulation of DLK.
Additional molecules acting upstream and downstream of DLK include MAP4K and G protein regulators, most of which have either modulatory or redundant effects. In a mouse DRG neuron degeneration model induced by trophic factor withdrawal, several MAP4Ks of the Ste20 family act redundantly as upstream activators of DLK (Larhammar et al. 2017b). In this same neuronal model, PERK (PKR-like endoplasmic reticulum kinase) acts downstream of DLK, in parallel with JNK (Larhammar et al. 2017a). Another study identified LZK and DLK to act synergistically in retinal ganglion cell (RGC) death following axotomy (Welsbie et al. 2017). Furthermore, four transcription factors (JUN, ATF2, MEF2A, and SO11) are downstream effectors of both DLK and LZK (Figure 2c). The in vivo roles of these upstream and downstream factors in various injury and disease models remain to be fully tested. Nonetheless, these observations highlight the exceedingly redundant and complex regulatory network involving DLK and LZK.
Conserved Roles of DLK in Axon Regeneration
Studies in C. elegans provided the first evidence for a role of DLK family kinases in neuronal response to injury, in particular axon regeneration. Technological development of femtosecond laser axotomy in C. elegans opened the door to identifying novel factors in axon injury response (Yanik et al. 2004). Injured axons display robust response within a few hours to reform growth cones and extend some distances in an error-prone manner (Wu et al. 2007). Axons can also break due to lack of β-spectrin, and the broken axons display spontaneous regrowth (Hammarlund et al. 2007). Using these two assays to screen for molecules affecting axon regrowth initiation and regeneration, two groups independently identified dlk-1 and its downstream kinases to be essential for the formation of regenerative growth cones (Hammarlund et al. 2009, Yan et al. 2009). Moreover, MAK-2, a MAPKAP kinase downstream of PMK-3/p38, can regulate levels of cebp-1 through 3’UTR-mediated mRNA stability (Yan et al. 2009). Injury to axons induces a rapid calcium influx (Ghosh-Roy et al. 2010). A systematic study of dlk-1 genetic mutations revealed an autoinhibitory mechanism mediated by an endogenously generated short isoform of DLK-1, and furthermore, an injury-induced calcium increase could regulate DLK-1 dimerization state (Yan & Jin 2012). Interestingly, C. elegans DLK-1 shares with mammalian LZK, but not with mammalian DLK, a conserved hexapeptide in the C terminus (Figure 1) that contributes to calcium-mediated regulation of DLK-1 activation. Expression of human LZK in C. elegans neurons could partially rescue axon developmental phenotypes due to loss of worm dlk-1, suggesting the functional equivalence of these kinases.
Several axon injury models, including pinching or crushing of the larval nerve cord, transection of wing nerves, and laser axotomy, were similarly developed in Drosophila (Brace & DiAntonio 2017). Drosophila Wnd/DLK is required for larval CNS motor axon regeneration (Xiong et al. 2010). Injury to axons increases expression of Wnd/DLK protein and simultaneous downregulation of its E3 ligase, Hiw/Phr. Persistent DLK expression triggers retrograde signaling to activate nuclear transcription. In the Drosophila PNS, laser axotomy to dendritic arborization (da) neurons induces a regenerative response dependent on the site of severing and sensory neuron types. When the axon is severed close to the soma, a new axon is generated from a neighboring dendrite, and when the axon is severed at a distance, regrowth initiates from the remaining axonal stump (Stone et al. 2010). Each injury paradigm triggers distinct changes in the MT cytoskeleton. Moreover, while class IV da neurons show a robust response to injury, class I and III da neurons display little regeneration (Song et al. 2012). In da neurons, Wnd/DLK is necessary for axon regeneration, but not for dendrite regeneration (Stone et al. 2014). In adult wing axons, following transection by laser, axon stumps generally fail to grow beyond the injury site; surprisingly, inhibiting the JNK pathway enhanced the wing axon regenerative response (Soares et al. 2014). Such cell type–dependent signaling output likely represents the tip of the iceberg in the ongoing efforts toward understanding the heterogeneity in neuronal responses to injury at both the molecular and physiological levels.
In adult mice, inducible pan-tissue deletion of Dlk does not cause lethality or gross abnormality (Pozniak et al. 2013). Early evidence using a mouse Dlk gene trap line hinted at a role of DLK in regulating axon regenerative response to injury (Itoh et al. 2009). Neurite growth of cultured DRG neurons from this Dlk mutant was reduced compared with WT controls. Dlk mutant neurons showed less c-JUN phosphorylation after nerve injury, suggesting that DLK may mediate the injury response. Subsequent in vivo analyses using Dlk conditional KO mice demonstrated that DLK is required for efficient axon regeneration after peripheral nerve injury (Shin et al. 2012). A well-known preconditioning effect involves a prior peripheral injury that primes DRG neurons for an enhanced regenerative response upon a second axonal injury (McQuarrie & Grafstein 1973). This enhanced regeneration primed by a prior injury was substantially reduced in Dlk conditional KO mice. Additionally, axon regeneration following a single peripheral nerve injury was also significantly delayed in Dlk conditional KO mice compared with WT controls. One underlying mechanism involves retrograde transport of injury signals, as represented by phosphorylated STAT3 (Shin et al. 2012). Together, these data indicate that DLK promotes a peripheral axon regenerative response (Figure 3a).
Figure 3.
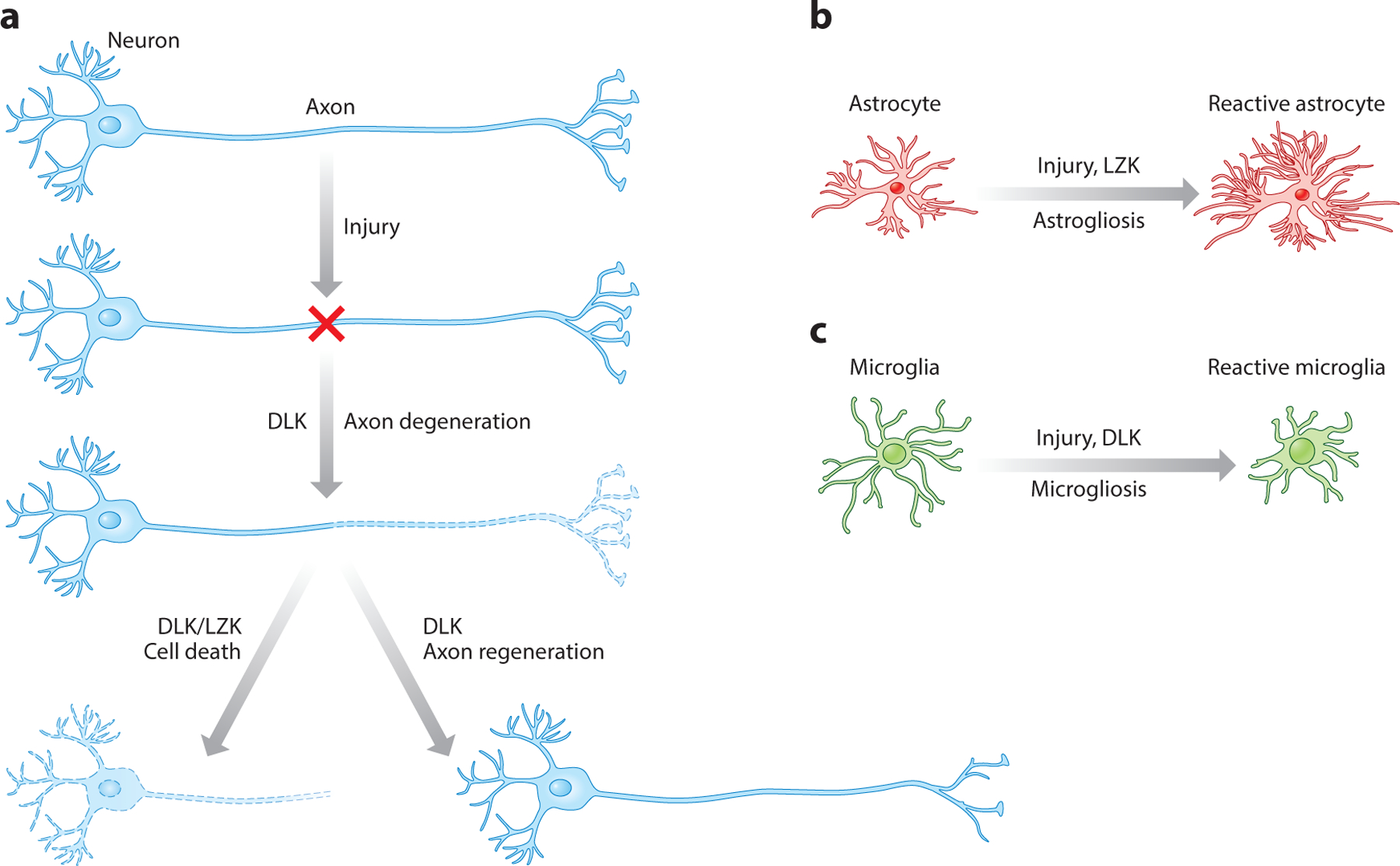
The roles of DLKs and LZKs in neuronal and glial responses to nervous system injury. (a) Neuronal responses include axon regeneration, axon degeneration, and cell death. (b) The astrocyte response refers to reactive astrogliosis. (c) The microglial response refers to reactive microgliosis. Microglia images adapted from Smart Service Medical Art through Creative Commons. They are available for reuse under a Creative Commons Attribution 3.0 Unported License.
In the CNS, evidence for DLK in neuronal response to axon injury has come mainly from studies of RGCs (Watkins et al. 2013, Welsbie et al. 2013). Following optic nerve crush injury, DLK is rapidly upregulated first in axons and then in cell bodies of RGCs. Optic nerve injury typically results in significant death of RGCs. Dlk gene deletion protected RGCs from axotomy-induced apoptosis (Watkins et al. 2013). Whereas deletion of Pten resulted in exuberant RGC axon regeneration (Park et al. 2008), simultaneously deleting Dlk substantially reduced RGC axon regeneration induced by Pten deletion (Watkins et al. 2013). Expression profiling experiments showed that DLK is an important mediator of most injury-induced expression changes in RGCs, including both proapoptotic and proregenerative pathways. These data support the notion that DLK is an upstream sensor of axonal injury that can lead to divergent neuronal responses (e.g., axon regeneration versus cell death), acting in a double-edged sword manner (Tedeschi & Bradke 2013) (Figure 3a).
Recovery of motor function is a common challenge following traumatic brain injury and stroke. Recent work implicated DLK in motor function recovery after stroke. In a mouse model for stroke, it was reported that neuronal knockdown of the C-C chemokine receptor 5 (CCR5) in premotor cortex could promote motor skills recovery after stroke (Joy et al. 2019). These CCR5 knockdown neurons showed a significant increase in DLK protein. Animals receiving shRNA knockdown of DLK after stroke exhibited persisted motor deficits relative to controls. Moreover, the recovery effects induced by CCR5 inhibition were abrogated by DLK knockdown. These data imply that upregulating DLK may be beneficial for regaining motor function in combination with other manipulations following stroke.
Compared with intense investigation on DLK, much less is known about its paralog, LZK, in axon regeneration. LZK was implicated as a downstream signaling molecule for Nogo (Dickson et al. 2010), a myelin-derived axon growth inhibitor (Geoffroy & Zheng 2014); however, the physiological relevance of this biochemical link remains unknown. In cultured neurons, LZK signals through MKK4 and JNKs to promote axon growth similarly to DLK (Chen et al. 2016). The role of LZK and DLK in axon regeneration in the spinal cord is unknown. There is an urgent need to understand the role of DLK and LZK in axonal repair after spinal cord injury.
DLK in Axon Degeneration and Neuronal Death
The earliest evidence for the function of mammalian DLK in neuronal response to axonal injury has been regarding axon degeneration (Miller et al. 2009). In vitro, Dlk mutant neurons were protected from axon degeneration following axotomy and vincristine treatment. In vivo, axotomy-induced Wallerian degeneration was significantly delayed in DLK null mice (Miller et al. 2009). A subsequent study indicates that DLK acts redundantly with two other MAP3Ks (MEKK4 and MLK2) in axon degeneration of RGCs and DRG neurons (Yang et al. 2015). Extensive mechanistic dissection has revealed a complex interaction of DLK and two key factors, SARM and NMNAT, in pathological axon degeneration, which was the focus of an excellent review (Gerdts et al. 2016).
In the study that demonstrated a critical role for DLK in Pten deletion–induced axon regeneration from RGCs, DLK was also shown to mediate axotomy-induced RGC death (Watkins et al. 2013). An independent RNAi-based screen of 623 kinases also identified DLK and its downstream effector MKK7 as the two top candidates responsible for cell death of cultured RGCs under neurotrophin deprivation (Welsbie et al. 2013). As in Watkins et al. (2013), Dlk conditional KO mice showed enhanced RGC survival after optic nerve injury (Welsbie et al. 2013). More recently, LZK was shown to cooperate with DLK to activate downstream signaling and cell death through MKK4/7 and JNKs (Welsbie et al. 2017) (Figure 3a). Additionally, administering toza-sertib, a small-molecule inhibitor of DLK, protected against RGC death following either optic nerve injury or laser-induced ocular hypertension, a model for glaucoma (Welsbie et al. 2013). Such corroborating evidence highlights the clinical relevance of discoveries involving the DLK signaling pathway.
DLK also mediates excitotoxicity-induced neuronal cell death. Inducible pan-tissue DLK KO significantly reduced kainic acid–induced excitotoxic cell death (Pozniak et al. 2013). In neonates, DLK or its downstream MAP2K, MKK4, is required for axotomy-induced death of facial motor neurons (Itoh et al. 2014). In a rat model of subarachnoid hemorrhage, siRNA-mediated DLK knockdown reduced neuronal apoptosis and improved neurobehavioral outcome (Yin et al. 2017). Together, these studies support a more general role of DLK in mediating neuronal death across neuronal types and insults.
Roles of LZK and DLK in Glial Cells
Evidence has emerged that LZK and DLK also play roles in glial responses to CNS injury. Following injury to the mammalian CNS, astrocytes proliferate, undergo hypertrophy with extended processes, and express high levels of astroglial markers such as GFAP and vimentin in a process known as astrogliosis, reactive astrogliosis, or the astroglial/astrocytic response (Burda & Sofroniew 2014). Reactive astrocytes encircle the lesion core that comprises macrophages, fibroblasts/pericytes, and other cell types, thereby limiting the spread of inflammation and contributing to wound healing. Two signaling pathways important for CNS axon regeneration, the STAT3/SOCS3 pathway (Smith et al. 2009, Sun et al. 2011) and the Pten/mTOR pathway (Liu et al. 2010, Park et al. 2008), have been implicated in the astroglial response to CNS injury (Chen et al. 2016, Herrmann et al. 2008, Okada et al. 2006; see References Added in Proof). Surprisingly, the first in vivo role reported for LZK was in the astrocyte response to injury (Chen et al. 2018). Following experimental spinal cord injury, LZK was prominently induced in astrocytes. Astrocyte-specific deletion of Lzk reduced markers of the astroglial response and led to an expanded lesion core. Conversely, astrocyte-specific overexpression of LZK enhanced the astroglial response, leading to a more compact lesion core. In the absence of an injury, LZK overexpression in astrocytes alone, but not in neurons, led to widespread astrogliosis in the CNS (Chen et al. 2018). These genetic loss- and gain-of-function analyses support the hypothesis that LZK promotes astrogliosis in the mammalian CNS (Figure 3b), a process that is prevalent in CNS injury and disease and likely impacts many aspects of cell and tissue interactions across a variety of neurological conditions.
Evidence for DLK in glial response to injury has also emerged. In a study that showed a protective effect of DLK loss of function in animal models of amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD) (discussed more below), Dlk gene deletion attenuated microglial activation and astroglial reactivity in SOD1G93A transgenic mice (Le Pichon et al. 2017). However, because a pan-tissue CAG-CreERT line was used to induce Dlk gene deletion in this study, a cell type–specific role for DLK could not be pinpointed. Indeed, the authors suggested that the microglial and astrocytic phenotypes are secondary to neuronal DLK function (Le Pichon et al. 2017).
In a spared nerve injury model of neuropathic pain, strong microgliosis is typically induced in the ipsilateral dorsal horn of the spinal cord, distant from the injury site, as assessed with Iba1 immunoreactivity (Wlaschin et al. 2018). Inducible DLK KO mice exhibited greatly diminished microgliosis and mechanical allodynia in this model. Furthermore, pharmacological inhibition of DLK mimicked genetic DLK KO in reduction of both pain sensation and microgliosis (Figure 3c). Again, due to the use of a pan-tissue inducible Cre line, it remains unknown which cell types were responsible for the observed effects of DLK loss of function (Wlaschin et al. 2018). Knockdown of DLK by shRNA has also been reported to reduce neuropathic pain in a chronic constrictive nerve injury model (Sheu et al. 2018). This role in neuropathic pain expands the clinical relevance of DLK in nervous system response to injury and insult.
Involvement of DLK in Neurodegenerative Diseases
Besides the abovementioned roles of DLK under acute and traumatic injury, recent studies have begun to probe into the involvement of DLK and its signaling pathway in neurodegenerative diseases. AD is associated with several genetic risk factors. One such factor includes the ApoE lipid-binding proteins, which are highly expressed in brain and primarily in astrocytes. Humans express three ApoE protein isoforms (E2, E3, and E4) that differ in only two amino acids and are associated with different predispositions to AD. The ApoE4 allele increases risk for AD, whereas the ApoE2 allele is protective against AD (Holtzman et al. 2012, Strittmatter et al. 1993). Despite decades of studies that revealed the interaction of ApoE isoforms with several membrane receptors and amyloid-β (Aβ) peptides, how such isoforms predispose to AD is poorly understood. Using embryonic stem (ES) cell–derived human neurons, a recent study showed that addition of purified recombinant ApoE proteins increased DLK protein levels, causing DLK activation (Huang et al. 2017). Unexpectedly, the downstream MAPK target of DLK was identified to be Erk, which then stimulated phosphorylation of cFos to promote transcription of the gene encoding amyloid precursor protein (APP) (Figure 4a), while the widely characterized MKK4-JNK axis activated under traumatic injury to the adult PNS and CNS was unaffected. The induction of the DLK-Erk cascade by ApoE isoforms mirrored their effects on Aβ production and AD pathogenesis, with ApoE4 showing higher potency. In mouse neuron/glial cultures, DLK knockdown reduced phosphorylation of MKK7 and Erk and decreased APP transcription; conversely, overexpressing DLK and MKK7 increased levels of APP mRNAs and proteins. Furthermore, adeno-associated virus (AAV)-mediated gene delivery into newborn mouse cortex of dominant-negative cFos or CRISPRi that targeted the AP-1 binding site of the App promoter significantly suppressed APP expression.
Figure 4.
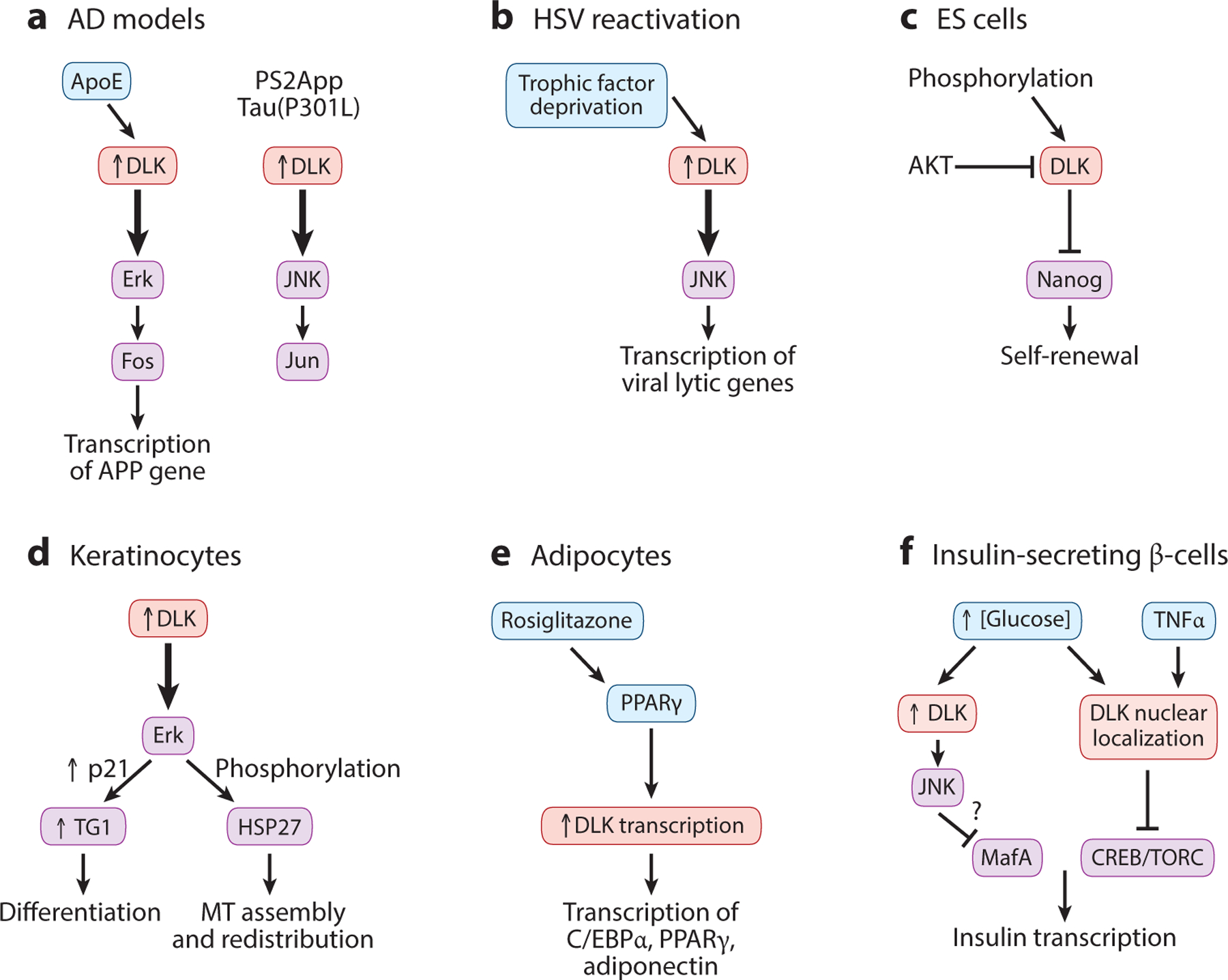
Emerging evidence for the role of DLK in neurodegeneration and nonneuronal cells. (a) DLK is upregulated (upward arrow) in neurons derived from human embryonic stem cells acutely treated by ApoE and in turn activates (thick downward arrows) Erk signal transduction (left). DLK is also upregulated in transgenic AD mouse models and activates JNK signal transduction (right). (b) DLK is upregulated under reactivation of herpes simplex viruses. (c) In mouse embryonic stem cells, DLK inhibits self-renewal and is inhibited by phosphorylation by AKT. (d) In keratinocytes, DLK is upregulated upon differentiation, and DLK promotes (thick downward arrow) morphological changes via Erk signal transduction. (e) In adipocyte differentiation, DLK expression is positively regulated by PPAR signaling, and DLK, in turn, activates transcriptional regulation of adipocyte genes. (f) In insulin-secreting β-cells, DLK expression and subcellular localization are regulated by glucose levels and TNFα signaling. Some interactions remain to be tested for relevance in vivo. In all panels, upward arrows indicate upregulation of DLK in specific cell types, and thick downward arrows depict functional pathways. Abbreviations: AD, Alzheimer’s disease; APP, amyloid precursor protein; ES cell, embryonic stem cell; HSV, herpes simplex virus; MT, microtubule; PPAR, peroxisome proliferator–activated receptor; TG1, transglutaminase.
Studies using other mouse AD models, such as PS2APP and Tau(P301L), also implicated DLK, which then activates the JNK pathway to regulate c-Jun phosphorylation (Le Pichon et al. 2017). At ages greater than 6 months, these AD animals showed elevated levels of phospho-c-Jun (p-c-Jun). Conditional removal of DLK from young AD mice (10-week-old adults) caused a reduction of phospho-JNK and phospho-MKK4 and significantly reduced cortical p-c-Jun at 9 months of age (Figure 4a). As synapse loss near Aβ plaques is a symptomatic hallmark of these AD mice, this study further examined synapses in hippocampus and found that PS2APP;DLK(KO) neurons had 30% less spine loss, proximal to Aβ plaques, relative to PS2APP;DLK(WT) neurons. Moreover, behavioral analyses reported noticeable protection in some cognitive function in PS2APP;DLK(KO) mice. In 6-month-old PS2APP mice, in which Aβ/plaque deposition had already begun, deletion of Dlk also improved active avoidance learning, although Aβ42 production and plaque load were not reduced. This evidence supports DLK activation to be causally linked to AD. It remains to be addressed whether the use of Erk versus JNK as its downstream kinase reflects the stage dependency or impact of different genetic factors in cellular pathogenesis.
Investigation on other models for neurodegenerative diseases also reveals beneficial effects following DLK inhibition. For example, inhibiting DLK function by AAV-mediated overexpression of two different dominant-negative constructs in a neurotoxin-mediated Parkinson animal model enhanced long-term survival of dopamine neurons; interestingly, inhibition by a kinase-dead form of DLK also showed trophic effects (Chen et al. 2008). Pharmacological inhibition of DLK using GNE-8505 and GNE-3511 (Patel et al. 2015a) in a mouse model for ALS, SOD1(G93A), reduced cortical p-c-Jun in a dose-dependent manner. Chronic administration of GNE-3511 by food intake to SOD1(G93A) mice delayed neuromuscular junction denervation by ~10% relative to the vehicle control. These observations encourage continued efforts to target DLK in neurodegenerative diseases (Siu et al. 2018) but also point to the complexity of drug or gene intervention. Thus, it would be important to rigorously examine the effects and underlying mechanisms of manipulating DLK in specific contexts.
DLK Signaling in Reactivation of Herpes Simplex Virus
Herpes simplex viruses (HSVs) are ubiquitous pathogens that persist for the life of infected individuals. The ability of these viruses to develop lifelong infections is due to a latent pool of dormant virus in terminally differentiated neurons, most commonly in the peripheral ganglia. During latent infection, the expression of the viral lytic genes is under epigenetic repression on their promoters. Latent HSVs can enter the lytic phase under a number of conditions, including treatment with interferons, nerve growth factor (NGF) deprivation, and inhibition of PI3K signaling (Suzich & Cliffe 2018). Multiple experimental models, mostly employing primary cell cultures, have been used to investigate the mechanisms of HSV reactivation (Camarena et al. 2010, Wilcox & Johnson 1987). One study reported an induction of DLK-JNK signaling during the early phase of HSV reactivation in cultured sympathetic and sensory neurons following NGF deprivation (Cliffe et al. 2015). This DLK-JNK activation triggered a histone methyl/phospho switch on the promoters of viral lytic genes (Figure 4b). Depletion of DLK or its binding protein JIP3 blocked the earliest detectable upregulation of lytic gene expression. Interestingly, during this HSV reactivation, JNK was present on viral promoters, which may permit lytic gene expression despite the presence of repressive lysine modifications on their promoters. It would be of future interest to elucidate the underlying mechanism.
DLK Function in Stem Cells and Nonneuronal Cells
Besides strong expression in the brain, DLK is expressed in a variety of embryonic and adult organs, including the skin, intestine, pancreas, and kidney (Hirai et al. 2006, Nadeau et al. 1997). Upon terminal differentiation, DLK expression is upregulated in the insulin-producing β-cells within the pancreatic islets of Langerhans, adipocytes, kidney, lung, and differentiating keratinocytes (Douziech et al. 1998, 1999; Hirai et al. 2006). These nonneuronal cells employ both the familiar actions of DLK signaling pathways and novel modes of regulation.
Embryonic stem cells.
DLK protein levels are low in undifferentiated mouse ES cells but increase upon embryoid body formation. A high-throughput kinase screen implicated a role of DLK in self-renewal of mouse ES cells (Wu et al. 2015). Knockdown of DLK elevated the expression of Nanog proteins and increased ES cell numbers, whereas DLK overexpression reduced ES cell self-renewal (Figure 4c). ES cell differentiation utilizes PI3K/AKT signaling. DLK is phosphorylated by AKT at Ser584 and Thr659 located at the C terminus (Figure 1). Inhibition of PI3K/Akt or mutating these residues to Ala elevated DLK activity in vitro and significantly reduced self-renewal of ES cells. It remains to be tested whether this AKT-dependent regulation of DLK occurs in other cell types.
Keratinocytes.
The terminal differentiation of epidermal cells is a complex multistep process that is tightly linked to cell cycle withdrawal, culminating in the formation of the cornified layer. In human skin, DLK mRNA and protein are specifically expressed in the differentiated granular layer of epidermis (Robitaille et al. 2005). In cultured keratinocytes, DLK overexpression causes morphological and biochemical changes, including induction of late differentiation markers, such as the cyclin-dependent kinase inhibitor p21cip1/waf1, and increased activity of transglutaminase, a cross-linking enzyme essential for the formation of cornified cell envelopes (Figure 4d). In keratinocyte differentiation, DLK also induces Hsp27 phosphorylation in a manner dependent on Erk. The DLK-Erk signaling axis regulates the interaction of the MT regulator LIS1 and HSP27, which can trigger the assembly of noncentrosomal MTs to promote MT stabilization and cytoskeleton redistribution (Robitaille et al. 2010). Epidermal cells in Dlk KO mouse embryos displayed desmosomal and tight junction defects, likely due to MT disruption. Together, these data highlight DLK as a key regulator of keratinocyte differentiation and maintenance of epidermal desmosomal and tight junction integrity (Simard-Bisson et al. 2017).
Adipocytes.
Peroxisome proliferator–activated receptor γ (PPARγ) is a type II nuclear receptor and plays a crucial role in the maintenance of glucose homeostasis and adipocyte-related metabolism. In obese patients with type 2 diabetes, PPARγ activation results in an increase in insulin sensitivity and thus in glucose clearance (Nolan et al. 1994). Transcriptional regulation of DLK (here, not to be confused with Delta-like kinase, which is also a key player in adipogenesis) is directly controlled by PPARγ (Couture & Blouin 2011) (Figure 4e). ChIP-PCR analysis of 3T3-L1 preadipocyte cells showed the direct association of PPARγ and RNA polymerase II with PPAR response elements on the DLK promoter. The binding of RNA polymerase II to the DLK promoter increased after treatment with the PPARγ agonist rosiglitazone. Female mice treated with rosiglitazone also showed a significant increase in DLK protein levels in mesenteric white adipose tissue and, to a lesser extent, in brown adipose tissue. Increased DLK expression in turn upregulates the expression of C/EBPα and PPARγ, two master transcriptional regulators of adipogenesis, resulting in lipid accumulation (Couture et al. 2009). PPARγ is also expressed in the brain, and data from N2a neuroblastoma cells suggest that PPARγ may regulate DLK levels (Couture & Blouin 2011). It would be interesting to examine whether this transcriptional regulation operates in the nervous system and how PPARγ-dependent DLK regulation contributes to DLK-mediated neuronal stress response.
Insulin-secreting pancreatic β-cells.
DLK is expressed in pancreatic β-cells, whose dysfunction leads to diabetes mellitus. These β-cells are electrically excitable. Elevations in blood glucose provide the most potent stimulus for β-cells to secrete insulin via CREB (cAMP response element binding)-mediated transcriptional regulation of the insulin gene (Oetjen et al. 2006). Upon glucose uptake into β-cells, glucose oxidation induces closure of KATP channels, membrane depolarization, calcium entry, and activation of calcineurin, leading to phosphorylation and activation of CREB. DLK can negatively regulate insulin gene transcription by affecting the CREB coactivators CBP and TORC (Phu do et al. 2011) (Figure 4f). In the insulin-producing cell line HIT, downregulation of endogenous DLK increased, whereas overexpression of DLK decreased, human insulin gene transcription. A DLK-responsive element in the human insulin gene matches the DNA binding site for the β-cell-specific transcription factor MafA (Stahnke et al. 2014). DLK-JNK activation led to phosphorylation of MafA and decreased its protein content. Surprisingly, the pancreatic islet β-cells in C57BL6/J mice that were fed with high-fat diet and developed a prediabetic condition showed an accumulation of DLK in the nuclei (Wallbach et al. 2016). HIT cells treated with either TNFα or IL-1β also showed nuclear localization of DLK (Borchers et al. 2017). A bipartite nuclear localization signal in mouse DLK was located at aa 185 to aa 200, the exact region of ATP binding (Figure 1). The nuclear localization of DLK in HIT cells depends on ATP binding but does not require homodimerization. These data present novel regulatory mechanisms concerning DLK, raising the possibility that DLK inhibition may preserve β-cell function and delay the development of type 2 diabetes.
PERSPECTIVES
Since the discovery of DLK and LZK genes more than 20 years ago, extensive studies using many animal models have provided strong in vivo data establishing the key roles of DLK in a variety of developmental, stress-sensing, and disease contexts. With increased efforts to target DLK, and likely LZK, in cellular response to injury and in animal models of disease (Siu et al. 2018), much more needs to be understood as to how DLK and LZK activity and signaling output are regulated in a cell type–dependent, stage-dependent, and context-dependent manner. Adding to the complexity of the multicellular roles of DLK and LZK, they may activate a variety of downstream effectors that have broad functions, such as JNK, p38, and Erk. The double-edged sword action of DLK in the decision of cell death versus axon regeneration in the retinal system makes it particularly challenging to pinpoint the optimal setpoint for the kinase’s activity for precise intervention (Tedeschi & Bradke 2013). Another key question is how these kinases sense cellular stress under acute insults versus chronic or pathological conditions. Besides the intense focus on the neuron-centric action of these kinases, much more needs to be understood regarding coordination between different cell types and functional redundancy between DLK and LZK and with other stress-activated pathways. Understanding the precise biological roles and mechanisms of action of DLK and LZK in a variety of physiological and pathological conditions will aid the development of viable therapeutic intervention targeting these two kinases.
ACKNOWLEDGMENTS
The work in our labs was supported by grants from the NIH (R37NS035546 and R01NS093588 to Y.J. and R01 NS093055 and NS047101 to B.Z.), the Veterans Administration (I01 RX002483 to B.Z.), and the Craig H. Neilsen (to Y.J. and B.Z.) and Wings for Life foundations (to B.Z.). We thank Andrew Chisholm, Yunbo Li, Erin Ritchie, and Junmi Saikia for comments and our lab members for discussions. The contents of this article do not represent the views of the US Department of Veterans Affairs or the US Government.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Andrusiak MG, Jin Y. 2016. Context specificity of stress-activated mitogen-activated protein (MAP) kinase signaling: the story as told by Caenorhabditis elegans. J. Biol. Chem 291:7796–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghari Adib E, Smithson LJ, Collins CA. 2018. An axonal stress response pathway: degenerative and regenerative signaling by DLK. Curr. Opin. Neurobiol 53:110–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom AJ, Miller BR, Sanes JR, DiAntonio A. 2007. The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 21:2593–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin R, Beaudoin J, Bergeron P, Nadeau A, Grondin G. 1996. Cell-specific expression of the ZPK gene in adult mouse tissues. DNA Cell Biol. 15:631–42 [DOI] [PubMed] [Google Scholar]
- Borchers S, Babaei R, Klimpel C, Duque Escobar J, Schroder S, et al. 2017. TNFα-induced DLK activation contributes to apoptosis in the beta-cell line HIT. Naunyn Schmiedebergs Arch. Pharmacol 390:813–25 [DOI] [PubMed] [Google Scholar]
- Bounoutas A, Kratz J, Emtage L, Ma C, Nguyen KC, Chalfie M. 2011. Microtubule depolymerization in Caenorhabditis elegans touch receptor neurons reduces gene expression through a p38 MAPK pathway. PNAS 108:3982–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bounoutas A, O’Hagan R, Chalfie M. 2009. The multipurpose 15-protofilament microtubules in C. elegans have specific roles in mechanosensation. Curr. Biol 19:1362–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brace EJ, DiAntonio A. 2017. Models of axon regeneration in Drosophila. Exp. Neurol 287:310–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. 2014. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81:229–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RW, Peterson KA, Johnson MJ, Roix JJ, Welsh IC, O’Brien TP. 2004. Evidence for a conserved function in synapse formation reveals Phr1 as a candidate gene for respiratory failure in newborn mice. Mol. Cell. Biol 24:1096–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, et al. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Lee A, Liao CP, Liu YW, Pan CL. 2014. RHGF-1/PDZ-RhoGEF and retrograde DLK-1 signaling drive neuronal remodeling on microtubule disassembly. PNAS 111:16568–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Geoffroy CG, Meves JM, Narang A, Li Y, et al. 2018. Leucine zipper–bearing kinase is a critical regulator of astrocyte reactivity in the adult mammalian CNS. Cell Rep. 22:3587–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Geoffroy CG, Wong HN, Tress O, Nguyen MT, et al. 2016. Leucine zipper–bearing kinase promotes axon growth in mammalian central nervous system neurons. Sci. Rep 6:31482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Rzhetskaya M, Kareva T, Bland R, During MJ, et al. 2008. Antiapoptotic and trophic effects of dominant-negative forms of dual leucine zipper kinase in dopamine neurons of the substantia nigra in vivo. J. Neurosci 28:672–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang M, Goncharov A, Wang S, Oegema K, Jin Y, Chisholm AD. 2014. The microtubule minus-end-binding protein patronin/PTRN-1 is required for axon regeneration in C. elegans. Cell Rep. 9:874–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, et al. 2015. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18:649–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A. 2006. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51:57–69 [DOI] [PubMed] [Google Scholar]
- Couture JP, Blouin R. 2011. The DLK gene is a transcriptional target of PPARγ. Biochem. J 438:93–101 [DOI] [PubMed] [Google Scholar]
- Couture JP, Daviau A, Fradette J, Blouin R. 2009. The mixed-lineage kinase DLK is a key regulator of 3T3-L1 adipocyte differentiation. PLOS ONE 4(3):e4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daviau A, Proulx R, Robitaille K, Di Fruscio M, Tanguay RM, et al. 2006. Down-regulation of the mixed-lineage dual leucine zipper–bearing kinase by heat shock protein 70 and its co-chaperone CHIP. J. Biol. Chem 281:31467–77 [DOI] [PubMed] [Google Scholar]
- Dickson HM, Zurawski J, Zhang H, Turner DL, Vojtek AB. 2010. POSH is an intracellular signal transducer for the axon outgrowth inhibitor Nogo66. J. Neurosci 30:13319–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douziech M, Grondin G, Loranger A, Marceau N, Blouin R. 1998. Zonal induction of mixed lineage kinase ZPK/DLK/MUK gene expression in regenerating mouse liver. Biochem. Biophys. Res. Commun 249:927–32 [DOI] [PubMed] [Google Scholar]
- Douziech M, Laberge G, Grondin G, Daigle N, Blouin R. 1999. Localization of the mixed-lineage kinase DLK/MUK/ZPK to the Golgi apparatus in NIH 3T3 cells. J. Histochem. Cytochem 47:1287–96 [DOI] [PubMed] [Google Scholar]
- Edwards ZC, Trotter EW, Torres-Ayuso P, Chapman P, Wood HM, et al. 2017. Survival of head and neck cancer cells relies upon LZK kinase–mediated stabilization of mutant p53. Cancer Res. 77:4961–72 [DOI] [PubMed] [Google Scholar]
- Fukuyama K, Yoshida M, Yamashita A, Deyama T, Baba M, et al. 2000. MAPK upstream kinase (MUK)-binding inhibitory protein, a negative regulator of MUK/dual leucine zipper–bearing kinase/leucine zipper protein kinase. J. Biol. Chem 275:21247–54 [DOI] [PubMed] [Google Scholar]
- Gallo KA, Johnson GL. 2002. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat. Rev. Mol. Cell Biol 3:663–72 [DOI] [PubMed] [Google Scholar]
- Geoffroy CG, Zheng B. 2014. Myelin-associated inhibitors in axonal growth after CNS injury. Curr. Opin. Neurobiol 27C:31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Milbrandt J, DiAntonio A. 2016. Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron 89:449–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. 2011. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell Biol 194:751–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, Chisholm AD. 2010. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J. Neurosci 30:3175–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill B, Murphey RK, Borgen MA. 2016. The PHR proteins: intracellular signaling hubs in neuronal development and axon degeneration. Neural Dev. 11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Jorgensen EM, Bastiani MJ. 2007. Axons break in animals lacking beta-spectrin. J. Cell Biol 176:269–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. 2009. Axon regeneration requires a conserved MAP kinase pathway. Science 323:802–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Chen Y, Cheng L, Prochownik EV, Li Y. 2016. microRNA-206 impairs c-Myc-driven cancer in a synthetic lethal manner by directly inhibiting MAP3K13. Oncotarget 7:16409–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Frey E, Yoon C, Wong H, Nestorovski D, et al. 2016. An evolutionarily conserved mechanism for cAMP elicited axonal regeneration involves direct activation of the dual leucine zipper kinase DLK. eLife 5:e14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai S, Cui DF, Miyata T, Ogawa M, Kiyonari H, et al. 2006. The c-Jun N-terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. J. Neurosci 26:11992–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Hirasawa R, Baba M, Ohnishi T, Ohno S. 2002. MAPK-upstream protein kinase (MUK) regulates the radial migration of immature neurons in telencephalon of mouse embryo. Development 129:4483–95 [DOI] [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Suenaga J, Ono M, Cui DF, Ohno S. 2005. Expression of MUK/DLK/ZPK, an activator of the JNK pathway, in the nervous systems of the developing mouse embryo. Gene Expr. Patterns 5:517–23 [DOI] [PubMed] [Google Scholar]
- Holland SM, Collura KM, Ketschek A, Noma K, Ferguson TA, et al. 2016. Palmitoylation controls DLK localization, interactions and activity to ensure effective axonal injury signaling. PNAS 113:763–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Herz J, Bu G. 2012. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med 2:a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzman LB, Merritt SE, Fan G. 1994. Identification, molecular cloning, and characterization of dual leucine zipper bearing kinase: a novel serine/threonine protein kinase that defines a second subfamily of mixed lineage kinases. J. Biol. Chem 269:30808–17 [PubMed] [Google Scholar]
- Horiuchi D, Collins CA, Bhat P, Barkus RV, Diantonio A, Saxton WM. 2007. Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Curr. Biol 17:1313–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YA, Zhou B, Wernig M, Sudhof TC. 2017. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell 168:427–41.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntwork-Rodriguez S, Wang B, Watkins T, Ghosh AS, Pozniak CD, et al. 2013. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J. Cell Biol 202:747–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda A, Hasegawa K, Masaki M, Moriguchi T, Nishida E, et al. 2001a. Mixed lineage kinase LZK forms a functional signaling complex with JIP-1, a scaffold protein of the c-Jun NH2-terminal kinase pathway. J. Biochem 130:773–81 [DOI] [PubMed] [Google Scholar]
- Ikeda A, Masaki M, Kozutsumi Y, Oka S, Kawasaki T. 2001b. Identification and characterization of functional domains in a mixed lineage kinase LZK. FEBS Lett. 488:190–95 [DOI] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Bannerman P, Pleasure D, Itoh T. 2009. Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem. Biophys. Res. Commun 383:258–62 [DOI] [PubMed] [Google Scholar]
- Itoh T, Horiuchi M, Ikeda RH Jr., Xu J, Bannerman P, et al. 2014. ZPK/DLK and MKK4 form the critical gateway to axotomy-induced motoneuron death in neonates. J. Neurosci 34:10729–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joy MT, Ben Assayag E, Shabashov-Stone D, Liraz-Zaltsman S, Mazzitelli J, et al. 2019. CCR5 is a therapeutic target for recovery after stroke and traumatic brain injury. Cell 176:1143–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karney-Grobe S, Russo A, Frey E, Milbrandt J, DiAntonio A. 2018. HSP90 is a chaperone for DLK and is required for axon injury signaling. PNAS 115:E9899–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinedinst S, Wang X, Xiong X, Haenfler JM, Collins CA. 2013. Independent pathways downstream of the Wnd/DLK MAPKKK regulate synaptic structure, axonal transport, and injury signaling. J. Neurosci 33:12764–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurup N, Yan D, Goncharov A, Jin Y. 2015. Dynamic microtubules drive circuit rewiring in the absence of neurite remodeling. Curr. Biol 25:1594–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larhammar M, Huntwork-Rodriguez S, Jiang Z, Solanoy H, Sengupta Ghosh A, et al. 2017a. Dual leucine zipper kinase–dependent PERK activation contributes to neuronal degeneration following insult. eLife 6:e20725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larhammar M, Huntwork-Rodriguez S, Rudhard Y, Sengupta-Ghosh A, Lewcock JW. 2017b. The Ste20 family kinases MAP4K4, MINK1, and TNIK converge to regulate stress-induced JNK signaling in neurons. J. Neurosci 37:11074–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pichon CE, Meilandt WJ, Dominguez S, Solanoy H, Lin H, et al. 2017. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med 9:eaag0394. [DOI] [PubMed] [Google Scholar]
- Lewcock JW, Genoud N, Lettieri K, Pfaff SL. 2007. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 56:604–20 [DOI] [PubMed] [Google Scholar]
- Lippi G, Fernandes CC, Ewell LA, John D, Romoli B, et al. 2016. MicroRNA-101 regulates multiple developmental programs to constrain excitation in adult neural networks. Neuron 92:1337–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, et al. 2010. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci 13:1075–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Chen Y, Zhang S, Xu W, Shao Y, et al. 2016. Rho1-Wnd signaling regulates loss-of-cell polarity-induced cell invasion in Drosophila. Oncogene 35:846–55 [DOI] [PubMed] [Google Scholar]
- Marcette JD, Chen JJ, Nonet ML. 2014. The Caenorhabditis elegans microtubule minus-end binding homolog PTRN-1 stabilizes synapses and neurites. eLife 3:e01637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki M, Ikeda A, Shiraki E, Oka S, Kawasaki T. 2003. Mixed lineage kinase LZK and antioxidant protein-1 activate NF-κB synergistically. Eur. J. Biochem 270:76–83 [DOI] [PubMed] [Google Scholar]
- Massaro CM, Pielage J, Davis GW. 2009. Molecular mechanisms that enhance synapse stability despite persistent disruption of the spectrin/ankyrin/microtubule cytoskeleton. J. Cell Biol 187:101–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata M, Merritt SE, Fan G, Yu GG, Holzman LB. 1996. Characterization of dual leucine zipper–bearing kinase, a mixed lineage kinase present in synaptic terminals whose phosphorylation state is regulated by membrane depolarization via calcineurin. J. Biol. Chem 271:16888–96 [DOI] [PubMed] [Google Scholar]
- McQuarrie IG, Grafstein B. 1973. Axon outgrowth enhanced by a previous nerve injury. Arch. Neurol 29:53–55 [DOI] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. 2009. A dual leucine kinase–dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci 12:387–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau A, Grondin G, Blouin R. 1997. In situ hybridization analysis of ZPK gene expression during murine embryogenesis. J. Histochem. Cytochem 45:107–18 [DOI] [PubMed] [Google Scholar]
- Nakata K, Abrams B, Grill B, Goncharov A, Huang X, et al. 2005. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell 120:407–20 [DOI] [PubMed] [Google Scholar]
- Nihalani D, Merritt S, Holzman LB. 2000. Identification of structural and functional domains in mixed lineage kinase dual leucine zipper–bearing kinase required for complex formation and stress-activated protein kinase activation. J. Biol. Chem 275:7273–79 [DOI] [PubMed] [Google Scholar]
- Nihalani D, Meyer D, Pajni S, Holzman LB. 2001. Mixed lineage kinase–dependent JNK activation is governed by interactions of scaffold protein JIP with MAPK module components. EMBO J. 20:3447–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix P, Hisamoto N, Matsumoto K, Bastiani M. 2011. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. PNAS 108:10738–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan JJ, Ludvik B, Beerdsen P, Joyce M, Olefsky J. 1994. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N. Engl. J. Med 331:1188–93 [DOI] [PubMed] [Google Scholar]
- Oetjen E, Lechleiter A, Blume R, Nihalani D, Holzman L, Knepel W. 2006. Inhibition of membrane depolarization–induced transcriptional activity of cyclic AMP response element binding protein (CREB) by the dual-leucine-zipper-bearing kinase in a pancreatic islet beta cell line. Diabetologia 49:332–42 [DOI] [PubMed] [Google Scholar]
- Park EC, Rongo C. 2018. RPM-1 and DLK-1 regulate pioneer axon outgrowth by controlling Wnt signaling. Development 145:dev164897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, et al. 2008. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322:963–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Cohen F, Dean BJ, De La Torre K, Deshmukh G, et al. 2015a. Discovery of dual leucine zipper kinase (DLK, MAP3K12) inhibitors with activity in neurodegeneration models. J. Med. Chem 58:401–18 [DOI] [PubMed] [Google Scholar]
- Patel S, Harris SF, Gibbons P, Deshmukh G, Gustafson A, et al. 2015b. Scaffold-hopping and structure-based discovery of potent, selective, and brain penetrant N-(1H-pyrazol-3-yl)pyridin-2-amine inhibitors of dual leucine zipper kinase (DLK, MAP3K12). J. Med. Chem 58:8182–99 [DOI] [PubMed] [Google Scholar]
- Patel S, Meilandt WJ, Erickson RI, Chen J, Deshmukh G, et al. 2017. Selective inhibitors of dual leucine zipper kinase (DLK, MAP3K12) with activity in a model of Alzheimer’s disease. J. Med. Chem 60:8083–102 [DOI] [PubMed] [Google Scholar]
- Phu DT, Wallbach M, Depatie C, Fu A, Screaton RA, Oetjen E. 2011. Regulation of the CREB coactivator TORC by the dual leucine zipper kinase at different levels. Cell. Signal 23:344–53 [DOI] [PubMed] [Google Scholar]
- Pozniak CD, Sengupta Ghosh A, Gogineni A, Hanson JE, Lee SH, et al. 2013. Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J. Exp. Med 210:2553–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CE, Spilker KA, Cueva JG, Perrino J, Goodman MB, Shen K. 2014. PTRN-1, a microtubule minus end-binding CAMSAP homolog, promotes microtubule function in Caenorhabditis elegans neurons. eLife 3:e01498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille H, Proulx R, Robitaille K, Blouin R, Germain L. 2005. The mitogen-activated protein kinase kinase kinase dual leucine zipper–bearing kinase (DLK) acts as a key regulator of keratinocyte terminal differentiation. J. Biol. Chem 280:12732–41 [DOI] [PubMed] [Google Scholar]
- Robitaille H, Simard-Bisson C, Larouche D, Tanguay RM, Blouin R, Germain L. 2010. The small heat-shock protein Hsp27 undergoes ERK-dependent phosphorylation and redistribution to the cytoskeleton in response to dual leucine zipper–bearing kinase expression. J. Investig. Dermatol 130:74–85 [DOI] [PubMed] [Google Scholar]
- Sakuma H, Ikeda A, Oka S, Kozutsumi Y, Zanetta JP, Kawasaki T. 1997. Molecular cloning and functional expression of a cDNA encoding a new member of mixed lineage protein kinase from human brain. J. Biol. Chem 272:28622–29 [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Hadwiger GD, Nonet ML. 2000. rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron 26:345–56 [DOI] [PubMed] [Google Scholar]
- Sheu ML, Chiang CY, Su HL, Chen CJ, Sheehan J, Pan HC. 2018. Intrathecal injection of dual zipper kinase shRNA alleviating the neuropathic pain in a chronic constrictive nerve injury model. Int. J. Mol. Sci 19:E2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, DiAntonio A. 2012. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 74:1015–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard-Bisson C, Bidoggia J, Larouche D, Guerin SL, Blouin R, et al. 2017. A role for DLK in microtubule reorganization to the cell periphery and in the maintenance of desmosomal and tight junction integrity. J. Investig. Dermatol 137:132–41 [DOI] [PubMed] [Google Scholar]
- Siu M, Sengupta Ghosh A, Lewcock JW. 2018. Dual leucine zipper kinase inhibitors for the treatment of neurodegeneration. J. Med. Chem 61:8078–87 [DOI] [PubMed] [Google Scholar]
- Smith PD, Sun F, Park KK, Cai B, Wang C, et al. 2009. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron 64:617–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares L, Parisi M, Bonini NM. 2014. Axon injury and regeneration in the adult Drosophila. Sci. Rep 4:6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Ori-McKenney KM, Zheng Y, Han C, Jan LY, Jan YN. 2012. Regeneration of Drosophila sensory neuron axons and dendrites is regulated by the Akt pathway involving Pten and microRNA bantam. Genes Dev. 26:1612–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahnke MJ, Dickel C, Schroder S, Kaiser D, Blume R, et al. 2014. Inhibition of human insulin gene transcription and MafA transcriptional activity by the dual leucine zipper kinase. Cell. Signal 26:1792–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Albertson RM, Chen L, Rolls MM. 2014. Dendrite injury triggers DLK-independent regeneration. Cell Rep. 6:247–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Nguyen MM, Tao J, Allender DL, Rolls MM. 2010. Global up-regulation of microtubule dynamics and polarity reversal during regeneration of an axon from a dendrite. Mol. Biol. Cell 21:767–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, et al. 1993. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. PNAS 90:1977–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga J, Cui DF, Yamamoto I, Ohno S, Hirai S. 2006. Developmental changes in the expression pattern of the JNK activator kinase MUK/DLK/ZPK and active JNK in the mouse cerebellum. Cell Tissue Res. 325:189–95 [DOI] [PubMed] [Google Scholar]
- Summers DW, Milbrandt J, DiAntonio A. 2018. Palmitoylation enables MAPK-dependent proteostasis of axon survival factors. PNAS 115:E8746–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Park KK, Belin S, Wang D, Lu T, et al. 2011. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 480:372–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Tawa G, Wallqvist A. 2012. Classification of scaffold-hopping approaches. Drug Discov. Today 17:310–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzich JB, Cliffe AR. 2018. Strength in diversity: understanding the pathways to herpes simplex virus reactivation. Virology 522:81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, Bradke F. 2013. The DLK signalling pathway—a double-edged sword in neural development and regeneration. EMBO Rep. 14:605–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valakh V, Frey E, Babetto E, Walker LJ, DiAntonio A. 2015. Cytoskeletal disruption activates the DLK/JNK pathway, which promotes axonal regeneration and mimics a preconditioning injury. Neurobiol. Dis 77:13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valakh V, Walker LJ, Skeath JB, DiAntonio A. 2013. Loss of the spectraplakin short stop activates the DLK injury response pathway in Drosophila. J. Neurosci 33:17863–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vaart A, Rademakers S, Jansen G. 2015. DLK-1/p38 MAP kinase signaling controls cilium length by regulating RAB-5 mediated endocytosis in Caenorhabditis elegans. PLOS Genet. 11:e1005733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallbach M, Duque Escobar J, Babaeikelishomi R, Stahnke MJ, Blume R, et al. 2016. Distinct functions of the dual leucine zipper kinase depending on its subcellular localization. Cell. Signal 28:272–83 [DOI] [PubMed] [Google Scholar]
- Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS. 2000. Highwire regulates synaptic growth in Drosophila. Neuron 26:313–29 [DOI] [PubMed] [Google Scholar]
- Wang X, Kim JH, Bazzi M, Robinson S, Collins CA, Ye B. 2013. Bimodal control of dendritic and axonal growth by the dual leucine zipper kinase pathway. PLOS Biol. 11:e1001572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, et al. 2013. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. PNAS 110:4039–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Mitchell KL, Jaskula-Ranga V, Sluch VM, Yang Z, et al. 2017. Enhanced functional genomic screening identifies novel mediators of dual leucine zipper kinase–dependent injury signaling in neurons. Neuron 94:1142–54.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, et al. 2013. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. PNAS 110:4045–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Johnson EM Jr. 1987. Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J. Virol 61:2311–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlaschin JJ, Gluski JM, Nguyen E, Silberberg H, Thompson JH, et al. 2018. Dual leucine zipper kinase is required for mechanical allodynia and microgliosis after nerve injury. eLife 7:e33910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Wu HJ, Wang CH, Lin CH, Hsu SC, et al. 2015. Akt suppresses DLK for maintaining self-renewal of mouse embryonic stem cells. Cell Cycle 14:1207–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, Chisholm AD. 2007. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. PNAS 104:15132–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA. 2010. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J. Cell Biol 191:211–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Jin Y. 2012. Regulation of DLK-1 kinase activity by calcium-mediated dissociation from an inhibitory isoform. Neuron 76:534–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y. 2009. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell 138:1005–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wu Z, Renier N, Simon DJ, Uryu K, et al. 2015. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 160:161–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanik MF, Cinar H, Cinar HN, Chisholm AD, Jin Y, Ben-Yakar A. 2004. Neurosurgery: functional regeneration after laser axotomy. Nature 432:822. [DOI] [PubMed] [Google Scholar]
- Yin C, Huang GF, Sun XC, Guo Z, Zhang JH. 2017. DLK silencing attenuated neuron apoptosis through JIP3/MA2K7/JNK pathway in early brain injury after SAH in rats. Neurobiol. Dis 103:133–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen M, Huang X, Bamber B, Jin Y. 2000. Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron 26:331–43 [DOI] [PubMed] [Google Scholar]
NOTE AND REFERENCES ADDED IN PROOF
- Chen CH, Sung CS, Huang SY, Feng CW, Hung HC, et al. 2016. The role of the PI3K/Akt/mTOR pathway in glial scar formation following spinal cord injury. Exp. Neurol 278:27–41 [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, et al. 2008. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci 28:7231–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, et al. 2006. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med 12:829–34 [DOI] [PubMed] [Google Scholar]
- Shin JE, Ha H, Kim YK, Cho Y, DiAntonio A. 2019. DLK regulates a distinctive transcriptional regeneration program after peripheral nerve injury. Neurobiol. Dis 127:178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study examines transcriptome changes following periphery nerve injury. They show that DLK is required for inducing expression of pro-regenerative genes, as well as ion transport and immune response genes.