

Abstract
Missense mutations in the MYH3 gene encoding myosin heavy chain-embryonic (MyHC-embryonic) have been reported to cause two skeletal muscle contracture syndromes, Freeman Sheldon Syndrome (FSS) and Sheldon Hall Syndrome (SHS). Two residues in MyHC-embryonic that are most frequently mutated, leading to FSS, R672 and T178, are evolutionarily conserved across myosin heavy chains in vertebrates and Drosophila. We generated transgenic Drosophila expressing myosin heavy chain (Mhc) transgenes with the FSS mutations and characterized the effect of their expression on Drosophila muscle structure and function. Our results indicate that expressing these mutant Mhc transgenes lead to structural abnormalities in the muscle, which increase in severity with age and muscle use. We find that flies expressing the FSS mutant Mhc transgenes in the muscle exhibit shortening of the inter-Z disc distance of sarcomeres, reduction in the Z-disc width, aberrant deposition of Z-disc proteins, and muscle fiber splitting. The ATPase activity of the three FSS mutant MHC proteins are reduced compared to wild type MHC, with the most severe reduction observed in the T178I mutation. Structurally, the FSS mutations occur close to the ATP binding pocket, disrupting the ATPase activity of the protein. Functionally, expression of the FSS mutant Mhc transgenes in muscle lead to significantly reduced climbing capability in adult flies. Thus, our findings indicate that the FSS contracture syndrome mutations lead to muscle structural defects and functional deficits in Drosophila, possibly mediated by the reduced ATPase activity of the mutant MHC proteins.
Keywords: Skeletal muscle, Myosin heavy chain, Freeman-Sheldon syndrome, Contracture, Sarcomere, Drosophila
1. Introduction
Myosins are motor proteins that are essential for a wide array of fundamental cellular functions such as motility, division, and transport. In the skeletal and cardiac muscle, specialized myosins belonging to the class II myosin family are involved in facilitating muscle contraction (Sellers, 2000). The skeletal muscle myosins are heterohexamers, comprising a pair of myosin heavy chains (MyHCs), a pair of essential light chains and a pair of regulatory light chains (Schiaffino and Reggiani, 1996). MyHCs possess ATPase activity and interact with actin, properties that are critical for muscle contraction. Mammals have multiple MyHC isoforms with distinct ATPase activity, and their distribution in muscle fibers is thought to be determined by the anatomical location and functional output of the muscle (Schiaffino and Reggiani, 1996).
Two MyHC isoforms are expressed primarily during mammalian development, of which mutations in one of them, MYH3 which codes for MyHC-embryonic protein, have been reported to lead to Freeman-Sheldon (FSS) or Sheldon-Hall (SHS) contracture syndromes (Tajsharghi et al., 2008; Toydemir et al., 2006). Patients with FSS or SHS typically exhibit contractures of the orofacial muscles, camptodactyly, clubfeet and scoliosis (Tajsharghi et al., 2008; Toydemir et al., 2006). SHS occurs more frequently compared to FSS, with the severity of the facial contractures relatively less in SHS (Beck et al., 2014; Toydemir et al., 2006). Missense mutations in MYH3 and other muscle contractile genes can cause FSS and SHS, although the most frequently mutated gene leading to these syndromes is MYH3 (Bamshad et al., 2009). Though the gene which is mutated in the majority of FSS and SHS cases has been identified, no animal models to study these syndromes have been reported. Mouse models for FSS or SHS are a possibility, but the presence of additional MyHC isoforms that could compensate for MyHC-embryonic could complicate interpretations from such studies (Sharma et al., 2018).
Here, we used Drosophila as a model to gain insights into the MyHC mutations that cause FSS. Drosophila has a single muscle myosin heavy chain gene (Mhc), and therefore, compensation by other MyHC isoforms will not confound results. In addition, the muscle architecture in Drosophila has significant similarities to that of vertebrates, with several structural and contractile proteins conserved evolutionarily (Rui et al., 2010).
We find that the residues in MyHC-embryonic that are most frequently mutated, leading to FSS are conserved evolutionarily across vertebrates and in Drosophila. By expressing Mhc transgenes that carry the FSS mutations in Drosophila muscle, we have generated a model to study these contracture syndromes. Our results indicate that expressing these mutant Mhc transgenes lead to muscle structural abnormalities, which increase in severity with age and muscle use. We find that the inter-Z disc length of sarcomeres is significantly shortened, the Z-disc width is reduced, Z-disc proteins are aberrantly deposited, and extensive muscle fiber splitting occurs upon expressing the Mhc transgenes in the Drosophila muscle. We also find that the ATPase activity of the FSS mutant MHC proteins are reduced compared to wild type MHC. Analyzing the protein structure, we find that the FSS mutant residues lie close to the ATP binding pocket of MHC and therefore disrupt its catalytic activity. Functionally, expression of the mutant Mhc transgenes lead to reduced climbing capability in adult flies. Thus, our findings indicate that the FSS contracture syndrome mutations lead to muscle structural defects and functional deficits in Drosophila. Since such severe effects on sarcomeric organization have so far not been reported from human patients with FSS, we hypothesize that the presence of multiple MyHC isoforms compensates for mutated MyHC-embryonic in humans with these syndromes. Our results also suggest that the decreased ATPase activity, sarcomere defects and functional deficits that we observe in flies expressing the FSS mutant Mhc transgenes are interlinked.
2. Results
2.1. MYH3 residues most frequently mutated in FSS are evolutionarily conserved
The most frequent mutations in MyHC-embryonic that cause Freeman-Sheldon syndrome (FSS) are R672H, R672C and T178I (Beck et al., 2014; Toydemir et al., 2006). T178I has been reported to lead to the relatively less severe Sheldon-Hall syndrome (SHS) also, although later those individuals were reclassified based on phenotype as FSS patients (Beck et al., 2014; Toydemir et al., 2006). Both R672 and T178 residues are in the head domain of the MyHC, critical for ATPase activity and actin binding, essential for contractile function of the myosin motor (Fig. 1A). We found that R672 and T178 are highly conserved evolutionarily, across vertebrates and in Drosophila muscle myosin heavy chain (MHC) (Fig. 1B and C). Interestingly, the neighboring residues, that form part of the hydrophobic core region of MHC are also evolutionarily conserved, indicating that they might be part of sub-domains that have crucial roles in myosin function (Fig. 1B and C). Comparing the human MyHC-embryonic and Drosophila MHC across their entire sequence, we found that they are ~55% similar (data not shown).
Fig. 1. The T178 and R672 residues in MyHC-embryonic which are mutated in FSS are evolutionarily conserved.

MyHC-embryonic protein schematic with the major protein domains and motifs labeled, showing the position of the T178 and R672 residues, which are the most frequently mutated residues in FSS (A). The T178 and R672 residues from human MyHC-embryonic were aligned to myosin heavy chain proteins from different vertebrate classes and the Drosophila myosin heavy chain using ClustalW (B, C). Both residues and neighboring residues are conserved across evolution (B, C). Surface representation of the monomer unit of Drosophila MHC (D). Magnified view of the boxed region in D showing the residues which are involved in hydrophobic packing in the ATP binding site of MHC (D′). Panel D and D′ are drawn in Pymol version 1.8 (PDB ID 5W1A). The R672 residue is mutated to H or C (R is light pink, C is yellow, and H is magenta).
The crystal structure of Drosophila MHC (available from PDB: PDB ID 5W1A) reveals that Threonine 178 and Arginine 672 residues are closely packed within the hydrophobic core constituted mainly by four Phenylalanine residues (F122, F487, F488 and F470), Leucine 176, Leucine 689, Valine 124 and Valine 699 (Fig. 1D, D′). The side chain of Arginine (R672) is nicely packed within the hydrophobic pocket created by several phenylalanine residues. The positive charge of the R672 side chain amine group is stabilized by π-charge interaction (Dougherty, 1996; Walklate et al., 2016; Wheeler and Bloom, 2014). While the R672C mutation disrupts the π-charge interaction, in R672H, incorporation of large histidinium ion may push the well aligned hydrophobic residues, rendering the pocket unstable. Both loss of π-charge interaction as well as loss of hydrophobic packing may thus affect activity in R672C and R672H mutations respectively. Similarly, the T178I mutation breaks the polar interaction between R672 and T178 residues (Walklate et al., 2016). A previous study where the R672C, R672H and T178I mutant human MyHC-embryonic proteins were expressed using a recombinant system found that all three mutations affected the kinetics, especially the ATP hydrolysis properties of the protein (Walklate et al., 2016).
2.2. Expression of FSS mutant Mhc transgenes in Drosophila lead to sarcomeric defects
To test the role of the T178 and R672 residues in MHC function in Drosophila, we generated four transgenic fly stocks, each with wild type Drosophila Mhc, or T178I, R672C and R672H mutations in Drosophila Mhc (Mhcwt, MhcT178I, MhcR672C and MhcR672H respectively), under the control of an Upstream Activating Sequence (UAS). To study the effect of expressing these transgenes in the Drosophila muscle, we brought them in the Mhc1 mutant background, which is a null mutation for the Drosophila Mhc gene (O'Donnell and Bernstein, 1988). We used the Mef2GAL4 driver line to express the Mhc transgenes in the muscle (Gunage et al., 2014; Schnorrer et al., 2010). Mhc1 heterozygous mutant flies exhibit muscle defects and we performed our experiments in this background, since Mhc1 homozygous mutants die as embryos (O'Donnell and Bernstein, 1988). These transgenic stocks in the Mhc1 heterozygous background with Mef2Gal4 driving their expression are designated Mhc (wild type Mhc transgene), R672H (R672H mutant transgene), R672C (R672C mutant transgene) and T178I (T178I mutant transgene) respectively. The actual genotypes of each of these stocks when analyzed are Mhc1/CyO; Mef2GAL4/UAS-Mhcwt, Mhc1/CyO; Mef2GAL4/UAS-MhcR672H, Mhc1/CyO; Mef2GAL4/UAS-MhcR672C and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I.
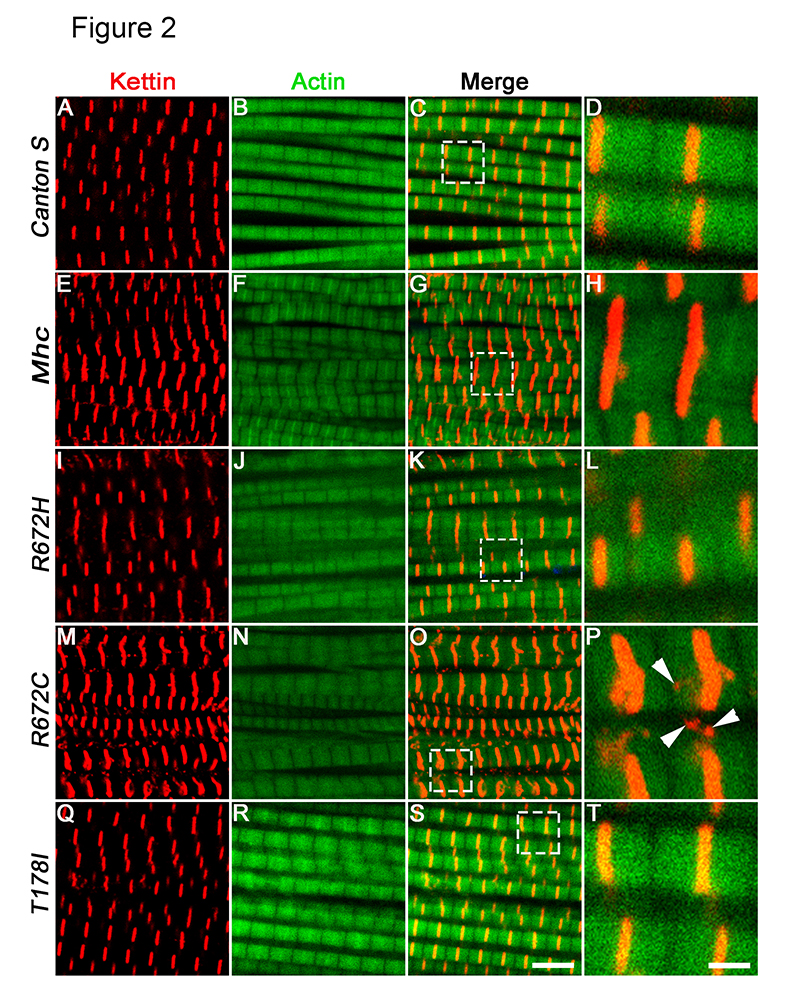
In the Mhc1 heterozygous background, we found that expressing the wild type Mhc transgene led to relatively normal muscle fibers and Z-discs marked by Kettin in the adult dorsal longitudinal muscles (DLMs) compared to the DLMs of wild type Canton S flies, within 5 days of eclosion (compare Fig. 2E-H with A-D). Since the wild type Mhc transgene is inserted at the same locus and is in the same background as the mutant Mhc transgenes, to control for the effect of overexpression, all statistical comparisons of the FSS mutant Mhc transgenes were made with flies expressing the wild type Mhc transgene. There were minor defects in Z-disc material deposition and fiber splitting in the DLMs of flies expressing the wild type Mhc transgene. However, flies expressing the FSS mutations – R672H, R672C and T178I exhibited severe fiber branching and splitting defects (Fig. 2I-L, M-P and Q-T and Fig. 5). We observed the most severe defects in flies expressing the R672C transgene where the Z-disc material marked by Kettin protein was not uniformly deposited and was fragmented (Arrowheads in Fig. 2P).
Fig. 2. Expression of FSS mutant Mhc transgenes lead to fiber branching and Z-disc defects 5 days post-eclosion.

Representative dorsal longitudinal muscles (DLMs) from wild type Canton S (A–D), Mhc1/CyO; Mef2GAL4/UAS-Mhcwt (E–H), Mhc1/CyO; Mef2GAL4/UAS-MhcR672H (I–L), Mhc1/CyO; Mef2-GAL4/UAS-MhcR672C (M–P) and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I (Q–T), 5 days post-eclosion, labeled by immunofluorescence for Kettin marking the Z-disc (red), and Actin labeling the thin filament (green). The boxed regions in the merge images C, G, K, O and S are magnified and shown in D, H, L, P and T respectively. Arrowheads in P point to Kettin puncta that are not part of the Z-disc. All images are from a single, representative Z-plane from one of the replicates. Scale bar in S is 5 μm and T is 1 μm.
Fig. 5. Expression of FSS mutant Mhc transgenes lead to increased fiber splitting 5 and 20 days post-eclosion.

Representative dorsal longitudinal muscles (DLMs) from 5 to 20 days post-eclosion wild type Canton S (A-A′, F-F′), Mhc1/CyO; Mef2GAL4/UAS-Mhcwt (B-B′, G-G′), Mhc1/CyO; Mef2GAL4/UAS-MhcR672H (C-C′, H-H′), Mhc1/CyO; Mef2GAL4/UAS-MhcR672C (D-D′, I-I′) and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I (E-E′, J-J′), labeled by immunofluorescence for Kettin marking the Z-disc (red), and Actin labeling the thin filament (green). A-E′ are DLMs from 5 days post-eclosion flies and F-J′ are DLMs from 20 days post-eclosion flies. The boxed regions in the images A-E and F-J are magnified and shown in A′-E′ and F′-J′ respectively. Quantification of the percentage of split fibers as a proportion of total fibers for DLMs from each genotype at 5 days (K) and 20 days (L) post-eclosion are shown. The graphical data is presented as mean ± standard error of the mean using a minimum of 5 replicates. Scale bar in J is 5 μm and J′ is 1 μm.
Next, we tested whether muscle use has any effect on the DLMs expressing the Mhc transgenes. Therefore, we performed immunofluorescence to label the sarcomeres of DLMs from 3-week-old Canton S, wild type Mhc transgenic and FSS mutant Mhc transgenic flies in the Mhc1 heterozygous background. Interestingly, we found that the branching phenotype observed at 5 days post-eclosion persisted at 3 weeks of age in wild type Mhc expressing flies compared to Canton S (Fig. 5). Similarly, flies expressing the FSS mutations – R672H, R672C and T178I continued to exhibit severe fiber branching defects (Fig. 3I-L, M-P and Q-T and Fig. 5). The Z-disc material marked by Kettin protein appeared abnormal, absent in some regions and most severe in DLMs from flies expressing the R672C and R672H transgenes (Fig. 3L and P). We did not find significant overexpression effects when the Mhcwt construct was expressed in the wild type background, at 5 or 20 days post-eclosion, indicating that the observed effects occur specifically in the Mhc1 heterozygous background (Supplementary Fig. 2).
Fig. 3. Expression of FSS mutant Mhc transgenes lead to fiber branching and Z-disc defects 20 days post-eclosion.

Representative dorsal longitudinal muscles (DLMs) from wild type Canton S (A–D), Mhc1/CyO; Mef2GAL4/UAS-Mhcwt (E–H), Mhc1/CyO; Mef2GAL4/UAS-MhcR672H (I–L), Mhc1/CyO; Mef2-GAL4/UAS-MhcR672C (M–P) and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I (Q–T), 20 days post-eclosion, labeled by immunofluorescence for Kettin marking the Z-disc (red), and Actin labeling the thin filament (green). The boxed regions in the merge images C, G, K, O and S are magnified and shown in D, H, L, P and T respectively. All images are from a single, representative Z-plane from one of the replicates. Scale bar in S is 5 μm and T is 1 μm.
We quantified the sarcomeric defects observed in the DLMs at 5 days and 20 days post-eclosion. For this, we quantified the inter-Z disc distance which is a measure of the length of the sarcomere, and the Z-disc width which is a measure of the width of the sarcomere (Fig. 4A). The Kettin staining of DLMs from Canton S, and the different Mhc transgenic flies in the Mhc1 background were used for this quantification. We found that DLMs from all three FSS mutant - R672H, R672C and T178I exhibited significant shortening of the inter-Z disc distance at 5 days post-eclosion, compared to wild type flies (Fig. 4B). Confirming the qualitative observation, R672C expressing flies exhibited the most severe shortening of the sarcomere to almost half as that of a wild type sarcomere (Fig. 4B). The Z-disc width was significantly reduced in R672H and R672C flies at 5 days post-eclosion (Fig. 4C).
Fig. 4. Inter Z-disc distance and Z-disc width are significantly reduced in sarcomeres of flies expressing the FSS mutant Mhc transgenes.

Schematic of a myofiber, depicting the inter Z-disc distance and Z-disc width (A). Quantification of the inter Z-disc distance 5 (B) and 20 (D) days post-eclosion, and Z-disc width 5 (C) and 20 (E) days post-eclosion respectively in DLMs of wild type Canton S, Mhc1/CyO; Mef2GAL4/UAS-Mhcwt, Mhc1/CyO; Mef2GAL4/UAS-MhcR672H, Mhc1/CyO; Mef2GAL4/UAS-MhcR672C and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I flies. The inter Z-disc distance (F) and Z-disc width (G) at 5 days post-eclosion compared to 20 days post-eclosion for each genotype. The graphical data is presented as mean ± standard error of the mean using a minimum of 5 replicates.
At 20 days post-eclosion, the inter-Z disc distance continued to be significantly reduced in all three FSS mutants (Fig. 4D). While the severe effect on R672C improved marginally, R672H exhibited a similar level of inter-Z disc distance shortening as R672C by 20 days, indicating that muscle use could lead to distinct outcomes in the different mutations. Interestingly, the Z-disc width was significantly reduced in all three FSS mutant lines by 20 days, indicating that muscle use or aging leads to shortening of the sarcomeric width in these Mhc mutants (Fig. 4E).
Comparing the inter-Z disc distance and the Z-disc width for flies of the same genotype between 5 and 20 days post-eclosion, we found that the inter-Z disc distance decreased significantly with age in R672H and T178I expressing flies, while it increased significantly in R672C expressing flies (Fig. 4F). The increase observed in R672C could be because the inter-Z disc distance is shortest in R672C expressing flies at 5 days post-eclosion (Fig. 4B). The Z-disc width changed significantly only in the T178I expressing flies with age, where it decreased significantly by 20 days post-eclosion (Fig. 4G). Thus, it seems that inter-Z disc distance is a sarcomeric attribute more likely to change with age or muscle use than Z-disc width.
We also quantified the degree of fiber splitting by counting the number of split fibers as a proportion of total fibers in the different Mhc transgenic flies in the Mhc1 background (Fig. 5). At 5 days post-eclosion, about 20% of fibers were split in Mhcwt expressing flies (Fig. 5B, B′ and K). In comparison, the FSS mutant Mhc expressing flies exhibited a statistically significant increase in split fibers, with R672H and T178I expressing flies exhibiting ~40% and R672C expressing flies ~70% split fibers (Fig. 5A-E′, K). At 20 days post-eclosion, we observed that ~40% fibers were split in the Mhcwt expressing flies, suggesting that muscle use and age leads to increased fiber splitting (Fig. 5G, G′ and L). We also found that all FSS mutant Mhc expressing flies exhibited ~80% or more split fibers at 20 days post-eclosion, a statistically significant effect compared to Mhcwt expressing flies (Fig. 5F-J′, L). Thus, expressing the FSS mutant transgenes led to increased fiber splitting compared to controls at 5 days post-eclosion, which exacerbated further with muscle use and age by 20 days post-eclosion.
2.3. FSS mutant Mhc transgenes exhibit reduced ATPase activity
We next tested the ATPase activity of the FSS mutant MHC proteins expressed in flies compared to wild type MHC. We isolated the DLMs from about 50 flies each of Canton S, Mhc1/CyO; Mef2GAL4/UAS-Mhcwt, Mhc1/CyO; Mef2GAL4/UAS-MhcR672H, Mhc1/CyO; Mef2GAL4/UAS-MhcR672C and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I genotypes. MHC from each of the genotypes was extracted, quantified and separated on SDS PAGE to ensure proper extraction and enrichment of MHC (Fig. 6A). ATPase assay for MHC was carried out using mouse skeletal muscle Actin, based on the change in absorbance at 360 nm of 2-amino-6-marcapto-7-methyl-purine riboside (MESG) by purine nucleoside phosphorylase (PNP) (Webb, 1992). We found that compared to Canton S MHC protein, overexpressed MHCwt had comparable ATPase activity. MHCR672H exhibited slightly reduced ATPase activity, MHCR672C even lesser and MHCT178I had the least activity among the FSS mutant MHCs (Fig. 6B). Thus, all 3 FSS mutant MHC proteins exhibited reduced ATPase activity compared to wild type MHC, as reported previously (Walklate et al., 2016).
Fig. 6. FSS mutant MHC exhibit decreased ATPase activity.

Coomassie stained SDS-PAGE showing MHC protein (high molecular weight band of about 205 KDa), where lane 1 is molecular weight marker with 180 KDa band labeled, and lanes 2–6 are protein samples enriched for MHC (2 μg each) from DLMs of wild type Canton S (lane 2), Mhc1/CyO; Mef2GAL4/UAS-Mhcwt (lane 3), Mhc1/CyO; Mef2GAL4/UAS-MhcT178I (lane 4), Mhc1/CyO; Mef2GAL4/UAS-MhcR672H (lane 5) and Mhc1/CyO; Mef2GAL4/UAS-MhcR672C (lane 6) flies respectively (A). Rate of phosphate released by the MHC ATPase indicated by absorbance at 360 nm as a function of time (B). The graphs are normalized to a reaction mix without ATP. Graphs indicate that the ATPase activity is reduced in the 3 FSS mutant Mhc transgenic lines compared to controls, with the most drastic reduction observed in the case of T178I mutation (B). Data shown here is from a single assay.
2.4. Expression of FSS mutant Mhc transgenes in Drosophila lead to functional defects
Since we observed sarcomeric structural defects upon expressing the FSS mutant Mhc transgenes and aberrant ATPase activity for the mutant MHC proteins, we tested whether these translated to functional defects in flies. Muscle activity is required for eclosion of adult flies from their pupal case (Hatfield et al., 2015). Therefore, we compared the pupal duration of flies overexpressing the Mhc transgenes in the Mhc1 heterozygous background with Mef2GAL4 driving their expression (Mhc1/CyO; Mef2GAL4/UAS-Mhcwt, Mhc1/CyO; Mef2GAL4/UAS-MhcR672H, Mhc1/CyO; Mef2GAL4/UAS-MhcR672C and Mhc1/CyO; Mef2GA-L4/UAS-MhcT178I). We observed that compared to wild type Canton S flies, all of the Mhc transgene overexpressing flies spent a significantly longer duration as pupae (Fig. 7A). Compared to Mhcwt expressing flies, MhcR672H, and MhcT178I expressing flies exhibited a trend towards increased pupal duration, although this effect was not statistically significant (Fig. 7A).
Fig. 7. Expression of FSS mutant Mhc transgenes lead to defects in muscle function.

The pupal duration was quantified in wild type Canton S, Mhc1/CyO; Mef2GAL4/UAS-Mhcwt, Mhc1/CyO; Mef2GAL4/UAS-MhcR672H, Mhc1/CyO; Mef2GAL4/UAS-MhcR672C and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I flies (A). Climbing assay to test muscle function was carried out at 5 (B) and 20 (C) days post-eclosion using wild type Canton S, Mhc1/CyO; Mef2GAL4/UAS-Mhcwt, Mhc1/CyO; Mef2GAL4/UAS-MhcR672H, Mhc1/CyO; Mef2-GAL4/UAS-MhcR672C and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I flies respectively. Number of flies that climbed 8 cm or above, 4–8 cm or 0–4 cm were counted, and the percentage of each category is represented as the climbing index. The graphical data is presented as mean ± standard error of the mean.
We also carried out climbing assays to test muscle function and found that at 5 days post-eclosion, Mhc1/CyO; Mef2GAL4/UAS-MhcR672H and Mhc1/CyO; Mef2GAL4/UAS-MhcR672C flies exhibited significantly reduced climbing capability compared to Mhc1/CyO; Mef2GAL4/UAS-Mhcwt flies (Fig. 7B). By 20 days post-eclosion, all 3 FSS mutants (Mhc1/CyO; Mef2GAL4/UAS-MhcR672H, Mhc1/CyO; Mef2GAL4/UAS-MhcR672C and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I) exhibited significantly diminished climbing capability compared to Mhc1/CyO; Mef2GAL4/UAS-Mhcwt flies (Fig. 7C). This indicates that muscle function is significantly compromised in the FSS mutant Mhc overexpressing flies, and this effect aggravates with muscle use and age.
3. Discussion
The MyHC-embryonic isoform in mammals is thought to be predominantly expressed during developmental stages and is only expressed in adults during muscle injury or disease (Sharma et al., 2018). It is not well understood how mutations in MyHC-embryonic lead to FSS and SHS (Sharma et al., 2018). In this paper, we have expressed FSS mutant myosin heavy chain in Drosophila muscle to decipher the effect of those mutations on muscle structure and function. We find that the FSS mutant myosins cause muscle structural abnormalities, especially shortening of the sarcomeric length and width, and myofiber branching, phenotypes which become more pronounced with muscle use and age. The sarcomere shortening phenotype that we observe could underlie the contractures observed in FSS patients. Some studies where morphologic analysis of muscle samples was carried out reports increased fiber size variability due to predominance of small, type I fibers in FSS patients (Kimber et al., 2012; Tajsharghi et al., 2008). Similar to our observations with MHC and the thick filament in this work, previous reports indicate that thin filament assembly defects lead to shortened sarcomeres in Drosophila muscle (Bai et al., 2007).
By using Drosophila as model, where there is a single muscle Mhc gene, we have overcome the compensatory effects by other myosin heavy chain isoforms, which could alleviate the phenotype in human FSS and SHS patients (Tajsharghi et al., 2008). Since we observe sarcomere shortening in Drosophila expressing the mutant MHC, it is possible that the effect of the FSS mutations occurs during early embryonic development, where only MyHC-embryonic and MyHC-slow are expressed and other MyHC isoforms may not be able to compensate since they are not expressed at those stages (Sharma et al., 2018).
Functionally, we observe that the FSS mutant myosin heavy chain proteins have abnormal ATPase kinetics (Walklate et al., 2016). The T178I mutation had the strongest reduction in MHC ATPase activity (Fig. 6B). This fits well with a genotype-phenotype correlative study in FSS patients, where it was found that the T178I mutation led to the most severe phenotypes in those patients compared to R672H and R672C mutations (Beck et al., 2014). Some reports indicate that altered muscle contractility during development lead to contractures (Bamshad et al., 2009; Robinson et al., 2007). By modeling the effect of the mutations on myosin structure, we find that the structural modifications disturb the ATP binding pocket, which is near the mutation sites, resulting in an impairment of catalytic activity (Fig. 1D). Loss of hydrophobic packing or breaking polar interactions possibly affects the structural stability of the mutant MHC proteins. From our ATPase assay results, we observed the least ATPase activity in the T178I mutation, possibly because the T178 residue lies closer to the ATP binding region, compared to R672.
We also find that expressing the FSS mutant Mhc leads to delayed pupal eclosion and reduced climbing capability in Drosophila, indicating that the structural deficits have functional consequences. The deficit in muscle function in flies expressing the FSS mutant Mhc could be due to the reduced ATPase activity of the mutant MHC protein, the sarcomeric structural defects, or a combination of both. It is unclear how the MHC ATPase activity and sarcomere assembly are interconnected, although some reports have started addressing this. A recent study shows that the muscle LIM protein (MLP) and cardiac myosin-binding protein C (MyBP-C) form a complex, which modulates myosin ATPase activity, sarcomere formation and myofilament assembly (Arvanitis et al., 2017). Another study suggests that muscle contractility during development is critical for maintenance of the muscle stem cell population and myogenic differentiation (de Lima et al., 2016). Another study using muscle cells from patients with the R672C mutation found that the mutant cells exhibited decreased force production and increased duration of relaxation (Racca et al., 2015). The most direct evidence stems from work which used specific inhibitors of myosin cross bridge-cycling and contractility, which disrupts sarcomere assembly and myofibrillogenesis (Ramachandran et al., 2003).
Thus, our work suggests that the contractures seen in FSS patients could be due to reduced ATPase activity of the mutant MyHC-embryonic protein during development. Proper MyHC-embryonic ATPase activity might be required for efficient assembly of muscle contractile proteins and sarcomere formation. Additional work on the sequence of events during sarcomere formation and a time course of when MyHC-embryonic contractility is required during development should shed light on the precise mechanisms underlying FSS and other contracture syndromes.
4. Materials and methods
4.1. Fly stocks and genetic crosses
All flies were maintained on corn meal food at 18 °C under standard conditions. Wild type Canton S (Bl #64349), double balancer snaSco/CyO; MKRS/TM6B (Bl #3703) and Mef2GAL4 on 3rd chromosome (Bl #27390) (Schnorrer et al., 2010) are from Bloomington Drosophila Stock Center. Mhc1/CyO flies were provided by Sanford Bernstein (O'Donnell and Bernstein, 1988). The UAS-Mhcwt, UAS-MhcR672H, UAS-MhcR672C and UAS-MhcT178I stocks were generated as part of this study.
Mhc1/CyO males were crossed to the double balancer snaSco/CyO; MKRS/TM6B females and the progeny were crossed to each other to select Mhc1/CyO; MKRS/TM6B, which was maintained as a stock. Mef2GAL4/Mef2GAL4 on 3rd chromosome males were crossed to the double balancer snaSco/CyO; MKRS/TM6B females and the progeny were crossed to each other to select snaSco/CyO; Mef2GAL4/Mef2GAL4, which was maintained as a stock. Mhc1/CyO; MKRS/TM6B males were crossed to snaSco/CyO; Mef2GAL4/Mef2GAL4, and the progeny were crossed to each other to select Mhc1/CyO; Mef2GAL4/Mef2GAL4, which was maintained as a stock for the final matings.
The balanced UAS-Mhcwt/TM6B, UAS-MhcR672H/TM6B, UAS-MhcR672C/TM6B and UAS-MhcT178I/TM6B transgenic stock males were each crossed to the double balancer snaSco/CyO; MKRS/TM6B females. The progeny were crossed to each other to recover snaSco/CyO; UAS-Mhc*/UAS-Mhc*, where UAS-Mhc* represents the UAS-Mhcwt, UAS-MhcR672H, UAS-MhcR672C and UAS-MhcT178I transgenes. Next, snaSco/CyO; UAS-Mhc*/UAS-Mhc* male flies were crossed to Mhc1/CyO; MKRS/TM6B females, and the progeny were crossed to each other to select the Mhc1/CyO; UAS-Mhc*/UAS-Mhc* flies, which was maintained as a stock for the final matings.
The final mating was between Mhc1/CyO; Mef2GAL4/Mef2GAL4 females and Mhc1/CyO; UAS-Mhc*/UAS-Mhc* males, from where the only surviving adult progeny were Mhc1/CyO; Mef2GAL4/UAS-Mhc*. These flies were analyzed for sarcomeric structural and functional defects. In addition, to test the dominant effect of MHC expression, UAS-Mhcwt/Mef2GAL4 flies were generated in a wild type background and analyzed for sarcomeric structural defects.
4.2. Molecular cloning and transgenesis
Drosophila Mhc cDNA (RH 59876) was procured from the Drosophila Genomics Resource Center (DGRC), Indiana University, Bloomington, and used as template to generate the transgenic constructs. Mutagenesis of Mhc cDNA was performed by PCR directed mutagenesis in two steps, using mutagenic primers (Supplementary Table 1). Briefly, one pair of primers was used to amplify the 5′ part of the cDNA and a second pair for the 3′ part of the cDNA, with a region of overlap between the two products, incorporating the respective mutation. Now, the 5′ part of the cDNA and the 3′ part of the cDNA are mixed together and used as template for a PCR using the forward primer used for the 5′ part and reverse primer used for the 3′ part to amplify the entire Mhc sequence. This product was sub-cloned into the pCR 2.1-TOPO TA cloning vector (ThermoFisher Scientific Catalog# 450641), digested with NotI and KpnI restriction enzymes, purified and ligated into the Drosophila pUASTattB transgenesis vector (Bischof et al., 2007). The plasmids were sequenced to validate that the mutant residues in Mhc have been incorporated and that no additional mutations are present.
Transgenesis was carried out at the fly facility at Bangalore Life-Science Cluster. The four constructs (pUASTattB-Mhcwt, pUASTattB-MhcR672H, pUASTattB-MhcR672C and pUASTattB-MhcT178I) were independently injected into φC31 integrase and attP 68A4 landing site carrying embryos (Bischof et al., 2007). Germline transformant transgenic lines where successful integration of the construct occurred were identified, balanced and used for crosses.
4.3. Immunofluorescence and imaging
Flies of the desired genotype were anesthetized and immediately immersed in 100% ethanol for 1 min to remove the waxy cuticle. Then, the ethanol was aspirated out and flies were washed in 1x phosphate buffered saline (PBS). For pre-fixation, the flies were immersed in 4% paraformaldehyde solution (PFA) in PBS for 5 min and then washed in PBS.
The flies were placed ventral side up on a drop of 50% glycerol on a cold glass slide. The flies were then frozen by immersing in liquid nitrogen for 1 min. The frozen flies were bisected along the mid-ventral line to expose the thoracic halves and the DLMs. Each thoracic half was fixed in 4% PFA for 20 min, washed in 0.5% PBS with Triton-X 100 (PBSTx) for 10 min each, six times. After washing, the muscles were blocked using 1% BSA in PBSTx. Thoraces were incubated in anti-Kettin (Abcam MAC 147) primary antibody at a dilution of 1:200 overnight at 4 °C. Thoraces were then washed six times, 10 min each in PBSTx. Cy3 conjugated secondary antibody against rat Fc (112-165-167, Jackson Immunoresearch) and Oregon 488 Phalloidin (Thermo Scientific Cat # 07466) were added at a dilution of 1:500 in blocking solution and incubated at room temperature for 2 h. Thoraces were then washed six times, 10 min each in PBSTx and mounted in Vectashield with DAPI (Vector laboratories Catalog No. H-1200) and coverslips were sealed at the edges with clear nail paint. Immunofluorescence images were acquired using a Leica SP8 confocal microscope.
4.4. Sarcomere and fiber measurements
Exported image files were quantified using the LAS X application suite for inter Z-disc length and Z-disc width measurements. Using line function, a region of interest (ROI) was drawn parallel to the muscle fiber spanning 6 sarcomeres in a single Z-stack. Inter Z-disc length (um) was measured as the distance between two consecutive Z-discs, measuring from the mid-point of Z-disc intensity (Cammarato et al., 2011). A minimum of 5 sarcomeres per muscle fiber for 4–5 fibers were quantified from 9 images using 5 different replicates for each genotype at 5 and 20 days of age (Canton S, Mhc1/CyO; Mef2GAL4/UAS-Mhcwt, Mhc1/CyO; Mef2GAL4/UAS-MhcR672H, Mhc1/CyO; Mef2GAL4/UAS-MhcR672C and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I).
For Z-disc width, the scale bar function was used for measurement. A scaled line was drawn parallel to the Z-disc and the width was quantified in um. Similar to sarcomere size quantification, a minimum of 5 sarcomeres per muscle fiber for 4–5 fibers were quantified from 9 images using 5 different replicates for each genotype (Canton S, Mhc1/CyO; Mef2-GAL4/UAS-Mhcwt, Mhc1/CyO; Mef2GAL4/UAS-MhcR672H, Mhc1/CyO; Mef2GAL4/UAS-MhcR672C and Mhc1/CyO; Mef2GAL4/UAS-MhcT178I), both at 5 and 20 days of age.
For fiber splitting analysis, the number of split fibers and the total number of fibers were counted by the count tool in Adobe Photoshop CS6. At least 3 images were used per genotype, and a minimum of 20 fibers were counted per image. The ratio of split fiber number to the total fiber number in the image was used to calculate the percentage of fiber splitting.
4.5. Pupal eclosion assay
Pupae within 1 h of puparium formation from uncrowded vials were collected gently with a soft, wet paintbrush and transferred to a petri dish with moist blotting paper. The petri dishes were labeled for the genotype, date and hour of collection and pupae allowed to mature at 25 °C. The pupae were observed for eclosion every few hours. The time of eclosion of each fly was noted and the number of hours taken by each fly to complete the pupal stage was calculated.
4.6. Climbing assay
Approximately 15–20 flies were anesthetized and transferred to a 15 ml plastic centrifuge tube and allowed to recover from anesthesia for 30 min. After recovery, flies were tapped to bring them to the bottom of the tube and allowed to climb while being video recorded. Climbing index was calculated as the percentage of flies that crossed the 8 cm mark, were in the 4–8 cm range, or below 4 cm mark on the tube in 10 s modified from (Ali et al., 2011). Readings were taken a minimum of 3 times using at least 2 sets of 15 or more flies.
4.7. Actin and myosin isolation and ATPase assay
Actin was isolated from mouse skeletal muscle (Racusen and Thompson, 1996). Myosin Heavy Chain (MHC) protein was isolated from the dorsal longitudinal muscles (DLMs) of ~50 flies. Flies were anesthetized and immersed in absolute ethanol for 1 min to remove the cuticle. To isolate DLMs, flies were dissected on a cold glass slide in York's Modified Glycerol (YMG) (20 mM Potassium Phosphate, pH 7.0, 2 mM MgCl2, 1 mM EGTA, 8 mM DTT, 50% (v/v) glycerol and 1x protease inhibitor cocktail) and stored in YMG at 0 °C until all the samples were dissected (Swank et al., 2001). The DLMs were centrifuged at 2 °C, 8500xg for 10 min to make them settle, YMG was replaced with YMGT (20 mM Potassium Phosphate, pH 7.0, 2 mM MgCl2, 1 mM EGTA, 8 mM DTT, 2% v/v Triton X-100 and 1x protease inhibitor cocktail) and the mixture was incubated on ice for 30 min. After 30 min, DLMs were washed thrice in YMG without glycerol (20 mM Potassium Phosphate, pH 7.0, 2 mM MgCl2, 1 mM EGTA, 8 mM DTT, 1x protease inhibitor cocktail). Myosin was extracted in 50 μL of Myosin Extraction Buffer (1.0 M KCl, 0.15 M potassium phosphate, pH 6.8, 10 mM sodium pyro-phosphate, 5 mM MgCl2, 0.5 mM EGTA, 8 mM DTT and 1x protease inhibitor cocktail). The myosin was loaded onto a 100 kDa cut-off column and washed thrice in Myosin Storage buffer (MSB) (0.5 M KCl, 20 mM MOPS, pH 7.0, 2 mM MgCl2 and 8 mM DTT) with 5 mM ATP and thrice in MSB without ATP. The final retentate was aliquoted and stored on ice for protein estimation (CB-X protein estimation kit, G Biosciences, Cat# 786-11X) and ATP hydrolysis assay.
ATPase assay was carried out using the EnzCheck Phosphate Assay Kit (E6646 ThermoFisher Scientific). Briefly, ATPase assay was done by adding 10 μg of MHC to a standard reaction mix of 100 μl of MESG (2-amino-6-marcapto-7-methyl-purine riboside), 5 μl PNP (purine nucleoside phosphorylase), 25 μl of 20x reaction buffer (1M Tris-HCl, 20 mM MgCl2, pH 7.5 and 2 mM sodium azide) and Actin to a final concentration of 2 μM. The reaction volume was made up to 500 μl with water and incubated at room temperature for 10 min to eliminate any free phosphate in the reaction mixture. The mixture was transferred to a quartz cuvette and reaction was started by adding ATP to a final concentration 400 μM. The absorbance was measured at 360 nm at intervals of 6 s for 1 h.
A no-actin control ATPase assay was carried out without the addition of Actin. All components of the ATPase assay were added at the same concentration except for Actin and absorbance was read for 1 h at 360 nm at intervals of 6 s.
4.8. Statistical analysis
Data was analyzed using parametric, unpaired t-test and one-way Anova (pupal eclosion and fiber splitting) using GraphPad prism software. Graphs are presented as Mean ± standard error of the mean. The p-values are indicated on the graphs and p-value ≤ 0.05 are considered significant, marked by asterisks.
Supplementary Material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ydbio.2019.02.017.
{kind=link}
{kind=link}
Acknowledgements
We thank Prof. K. VijayRaghavan (NCBS, Bangalore) and his lab members for providing training on fly muscle preparations. We are grateful to Prof. Sanford Bernstein (SDSU, California) for providing some of the Drosophila stocks used in this study. The transgenic fly lines were generated on payment basis at the fly facility at Bangalore LifeScience Cluster. We acknowledge the imaging and central instrumentation facilities at RCB. We thank Dr. Deepti Jain (RCB, Faridabad) and her lab members for help with the ATPase assay. Authors acknowledge the support of DBT e-Library Consortium (DeLCON) for providing access to e-resources. Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study. We acknowledge critical comments and suggestions from all members of the Developmental Genetics Laboratory, RCB.
Funding
This work was supported by the Wellcome Trust/DBT India Alliance through an intermediate fellowship (IA/I/13/1/500872) to SJM. We also acknowledge core funds provided by the Regional Centre for Biotechnology (RCB). PK is funded by a Council for Scientific and Industrial Research (CSIR) senior research fellowship.
Footnotes
Conflicts of interest
The authors declare no competing or financial interests.
Author contributions statement
SJM and SD conceived the project and designed the experiments. The experiments were carried out by SD with support from PK and AV, under the guidance of SJM. TKM did the MHC structural modeling. The data was analyzed and figures compiled by PK, SD, TKM, and SJM. The manuscript was written by SJM with inputs from SD and TKM. All authors have read and approve the manuscript. Funding for this work was secured by SJM.
Note
Rao et al (Mol Biol Cell 2019; 30(1):30–41) published work on FSS mutations in Drosophila while this manuscript was in revision.
References
- Ali YO, Escala W, Ruan K, Zhai RG. Assaying locomotor, learning, and memory deficits in Drosophila models of neurodegeneration. J Visualized Exp JoVE. 2011;(49):2504. doi: 10.3791/2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitis DA, Vafiadaki E, Papalouka V, Sanoudou D. Muscle Lim Protein and myosin binding protein C form a complex regulating muscle differentiation. Biochim Biophys Acta (BBA) Mol Cell Res. 2017;1864:2308–2321. doi: 10.1016/j.bbamcr.2017.08.010. [DOI] [PubMed] [Google Scholar]
- Bai J, Hartwig JH, Perrimon N. SALS, a WH2-domain-containing protein, promotes sarcomeric actin filament elongation from pointed ends during Drosophila muscle growth. Dev Cell. 2007;13:828–842. doi: 10.1016/j.devcel.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Bamshad M, Van Heest AE, Pleasure D. Arthrogryposis: a review and update. J Bone Joint Surg Am. 2009;91:40. doi: 10.2106/JBJS.I.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck AE, McMillin MJ, Gildersleeve HI, Shively KM, Tang A, Bamshad MJ. Genotype-phenotype relationships in Freeman–Sheldon syndrome. Am J Med Genet Part A. 2014;164:2808–2813. doi: 10.1002/ajmg.a.36762. [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific φC31 integrases. Proc Natl Acad Sci. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarato A, Li XE, Reedy MC, Lee CF, Lehman W, Bernstein SI. Structural basis for myopathic defects engendered by alterations in the myosin rod. J Mol Biol. 2011;414:477–484. doi: 10.1016/j.jmb.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lima JE, Bonnin M-A, Birchmeier C, Duprez D. Muscle contraction is required to maintain the pool of muscle progenitors via YAP and NOTCH during fetal myogenesis. Elife. 2016;5:e15593. doi: 10.7554/eLife.15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty DA. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- Gunage RD, Reichert H, VijayRaghavan K. Identification of a new stem cell population that generates Drosophila flight muscles. Elife. 2014;3:e03126. doi: 10.7554/eLife.03126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield I, Harvey I, Yates ER, Redd JR, Reiter LT, Bridges D. The role of TORC1 in muscle development in Drosophila. Sci Rep. 2015;5 doi: 10.1038/srep09676. 9676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimber E, Tajsharghi H, Kroksmark AK, Oldfors A, Tulinius M. Distal arthrogryposis: clinical and genetic findings. Acta Paediatr. 2012;101:877–887. doi: 10.1111/j.1651-2227.2012.02708.x. [DOI] [PubMed] [Google Scholar]
- O'Donnell PT, Bernstein SI. Molecular and ultrastructural defects in a Drosophila myosin heavy chain mutant: differential effects on muscle function produced by similar thick filament abnormalities. J Cell Biol. 1988;107:2601–2612. doi: 10.1083/jcb.107.6.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racca AW, Beck AE, McMillin MJ, Korte FS, Bamshad MJ, Regnier M. The embryonic myosin R672C mutation that underlies Freeman-Sheldon syndrome impairs cross-bridge detachment and cycling in adult skeletal muscle. Hum Mol Genet. 2015;24:3348–3358. doi: 10.1093/hmg/ddv084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racusen RH, Thompson KV. Isolation of myosin and actin from chicken muscle. Proceedings of the 18th Workshop/Conference of the Association for Biology Laboratory Education (ABLE); 1996. pp. 97–111. [Google Scholar]
- Ramachandran I, Terry M, Ferrari MB. Skeletal muscle myosin cross-bridge cycling is necessary for myofibrillogenesis. Cell Motil Cytoskelet. 2003;55:61–72. doi: 10.1002/cm.10113. [DOI] [PubMed] [Google Scholar]
- Robinson P, Lipscomb S, Preston LC, Altin E, Watkins H, Ashley CC, Redwood CS. Mutations in fast skeletal troponin I, troponin T, and β-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007;21:896–905. doi: 10.1096/fj.06-6899com. [DOI] [PubMed] [Google Scholar]
- Rui Y, Bai J, Perrimon N. Sarcomere formation occurs by the assembly of multiple latent protein complexes. PLoS Genet. 2010;6:e1001208. doi: 10.1371/journal.pgen.1001208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S, Reggiani C. Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol Rev. 1996;76:371–423. doi: 10.1152/physrev.1996.76.2.371. [DOI] [PubMed] [Google Scholar]
- Schnorrer F, Schönbauer C, Langer CC, Dietzl G, Novatchkova M, Schernhuber K, Fellner M, Azaryan A, Radolf M, Stark A. Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature. 2010;464:287. doi: 10.1038/nature08799. [DOI] [PubMed] [Google Scholar]
- Sellers JR. Myosins: a diverse superfamily. Biochim Biophys Acta (BBA) Mol Cell Res. 2000;1496:3–22. doi: 10.1016/s0167-4889(00)00005-7. [DOI] [PubMed] [Google Scholar]
- Sharma A, Agarwal M, Kumar A, Kumar P, Saini M, Kardon G, Mathew S. Myosin heavy chain-embryonic is a crucial regulator of skeletal muscle development and differentiation. bioRxiv. 2018 doi: 10.1101/261685. 261685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swank DM, Bartoo ML, Knowles AF, Iliffe C, Bernstein SI, Molloy JE, Sparrow JC. Alternative exon-encoded regions of Drosophila myosin heavy chain modulate ATPase rates and actin sliding velocity. J Biol Chem. 2001;276:15117–15124. doi: 10.1074/jbc.M008379200. [DOI] [PubMed] [Google Scholar]
- Tajsharghi H, Kimber E, Kroksmark AK, Jerre R, Tulinius M, Oldfors A. Embryonic myosin heavy-chain mutations cause distal arthrogryposis and developmental myosin myopathy that persists postnatally. Arch Neurol. 2008;65:1083–1090. doi: 10.1001/archneur.65.8.1083. [DOI] [PubMed] [Google Scholar]
- Toydemir RM, Rutherford A, Whitby FG, Jorde LB, Carey JC, Bamshad MJ. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat Genet. 2006;38:561. doi: 10.1038/ng1775. [DOI] [PubMed] [Google Scholar]
- Walklate J, Vera C, Bloemink MJ, Geeves MA, Leinwand L. The most prevalent Freeman-Sheldon syndrome mutations in the embryonic myosin motor share functional defects. J Biol Chem. 2016;291(19):10318–10331. doi: 10.1074/jbc.M115.707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb MR. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc Natl Acad Sci. 1992;89:4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler SE, Bloom JW. Toward a more complete understanding of noncovalent interactions involving aromatic rings. J Phys Chem A. 2014;118:6133–6147. doi: 10.1021/jp504415p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.