

Abstract
Aims
The aim of the study was to compare the pharmacokinetics (PK), safety and tolerability of secukinumab with different devices for subcutaneous (s.c.) administration of 2 mL.
Methods
A phase 1 study in healthy subjects with 6 devices to administer 2 mL injection volumes was conducted to evaluate the serum PK, safety and tolerability of secukinumab following single s.c. injection of 300 mg in the abdomen (either side) or in the thigh (either leg). Primary PK endpoints were maximum observed serum concentration and area under the serum concentration–time curve. The impact of device, site and side of injection on serum exposure was evaluated. In a phase 3 study in psoriasis patients, PK of secukinumab was evaluated following multiple s.c. injections of 300 mg by either 2 × 1‐mL prefilled syringe or 1 × 2‐mL prefilled syringe.
Results
Mean serum concentration–time profiles for administration as 2 × 1 mL injections or as 1 × 2 mL injections were similar. With an injection volume of 2 mL, perceived injection pain was not different from 2 × 1 mL injections. A nonclinically significant difference in PK endpoints was observed between thigh and abdomen. Results with a 2 mL prefilled syringe in a 1‐year phase 3 study in patients confirmed PK results observed in the phase 1 study.
Conclusion
Collective evidence from both studies demonstrated that 2‐mL injections of secukinumab into the abdomen or thigh using different devices resulted in comparable PK characteristics and were all well tolerated without noticeable local reactions.
Keywords: injection device, prefilled syringe, psoriasis, secukinumab, subcutaneous administration, therapeutic monoclonal antibody
What is already known about this subject
Secukinumab is a fully human monoclonal antibody that targets IL‐17A
Secukinumab is approved for the treatment of psoriasis, psoriatic arthritis and ankylosing spondylitis
The highest therapeutic dose level of 300 mg is administered as 2 × 1‐mL injections (2 × 150 mg/mL).
What this study adds
These are first clinical results with 2‐mL devices for a marketed monoclonal antibody in healthy subjects and in psoriasis patients
The phase 1 and phase 3 studies compared pharmacokinetics, safety and tolerability of 2‐mL devices vs approved 1‐mL devices for secukinumab treatment
The results demonstrate similar pharmacokinetic characteristics with good safety and tolerability for 2‐mL injections compared with 2 × 1‐mL injections from registered 1‐mL devices
1. INTRODUCTION
Secukinumab (Cosentyx) is a recombinant high‐affinity fully human monoclonal anti‐human interleukin‐17A (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4982) antibody of the IgG1/κ isotype, approved for the treatment of moderate to severe psoriasis, ankylosing spondylitis and psoriatic arthritis. Secukinumab binds to human IL‐17A and neutralizes the bioactivity of this cytokine. IL‐17A is the central cytokine of a defined subset of inflammatory T cells, the Th17 cells which, in several animal models, are pivotal for several autoimmune and inflammatory processes.1, 2, 3 The marketed therapeutic doses are available as lyophilized drug formulation and a 1‐mL liquid formulation either as a 150‐mg/mL prefilled syringe (PFS) or as a 150‐mg/mL auto‐injector (AI).4, 5, 6 The highest marketed dose is 300 mg and currently 2 injections are required to achieve that dose. Therefore, for the convenience of patients with potentially better compliance, alternative single‐step options of administration were explored in the present studies and compared to the existing 150‐mg AI and PFS administration forms.
Today, most biologics are administered by subcutaneous (s.c.) injection, commonly in volumes not exceeding 1–1.5 mL7, 8, 9 and several recommendations limit the volume of s.c. injection to 1 mL.10 However, there is no evidence to support this limitation in volume and some recent studies suggest that s.c. injection volumes up to 3 mL are well tolerated.11, 12, 13
In a clinical study recently published by Heise et al12 the authors analysed the role of multiple factors, i.e. injection volume (0.4–1.6 mL), injection site (abdomen or thigh) and injection rate (0.15 or 0.45 mL/s) on the tolerance of a s.c. injection of 0.9% saline solution. A major factor influencing subject tolerance, assessed using a 100‐mm visual analogue scale (VAS), was pain at the injection site (both statistically and clinically significantly favouring abdomen over thigh). Pain intensity scores were statistically higher with larger volumes. Interestingly, injection rate did not play any role in subject tolerance. However, based on available data today (published and unpublished), it has not been possible to determine the maximum volume and viscosity that can be delivered during a 10–15 s injection with an AI and the impact of drug viscosity on subject tolerance, safety and pharmacokinetics. Although there is an increasing understanding of formulation parameters impacting the viscosity of formulations for monoclonal antibodies, it is not straightforward to develop a generic formulation strategy for concentrated monoclonal antibody formulations with low viscosity.8, 13 In a study by Berteau et al.11 it was counterintuitively observed that injection pain decreases with increase in the viscosity of the fluid injected. However, it was noted that this result needed additional confirmation using devices specifically adapted for injection of viscous solutions.
Therefore, it remains partially unclear if certain injection conditions, such as using a particular injection site, injection speed or volume can reduce discomfort and pain of injections. Due to dose requirements and formulation limitations, s.c. injections >1 mL are often required. The purpose of the phase 1 study as described in this paper was to assess the pharmacokinetics (PK), safety and tolerability after single s.c. administration of 300 mg secukinumab using 4 new 2 mL administration systems compared to both the existing 150‐mg/1‐mL AI device and the existing 150‐mg/1‐mL PFS. In this study, the role of device with inherently different injection speeds, site (abdomen/thigh) and side (left/right) of the injection on the tolerance and pharmacokinetics of 2‐mL s.c. injections of the monoclonal antibody secukinumab was investigated. Further, PK from a phase 3 study in moderate to severe psoriasis patients after multiple s.c. administrations of 300 mg secukinumab using a 2‐mL PFS compared to the existing 150‐mg/1‐mL PFS are reported. To the best of our knowledge, these are the first comprehensive results for an approved monoclonal antibody with single 2‐mL injection devices in healthy subjects and with multiple dose injections with a 2‐mL PFS in psoriasis patients.
2. METHODS
Both clinical studies were conducted in accordance with the principles of the International Conference on Harmonization requirements for Good Clinical Practice, the Declaration of Helsinki and with the approval of a National Health Service Ethics Review Committee.
2.1. Phase 1 study in healthy subjects
2.1.1. Study population and design
The phase 1 study was an open‐label, single‐centre, randomized, parallel group study in male and female healthy subjects who received a single s.c. administration of secukinumab 300 mg. No prior information on variability in PK parameters between subjects existed for these new devices. However, previous PK results from proof of concept and dose finding studies suggested that a minimum planned total sample size of 120 subjects with 20 subjects per arm was sufficient to characterize the PK of the devices for secukinumab.
The 120 subjects were randomized to include 20 subjects in each of the 6 device arms with balance between site and side, and replacement of dropouts was allowed. The total duration of subject participation was up to 20 weeks, including: (i) a screening visit up to 28 days prior to injection; (ii) followed by a baseline visit and a subsequent single treatment visit; (iii) a follow‐up period with a domiciled period in the study unit 1 day after injection; and (iv) an ambulatory period during which subjects returned to the study unit for scheduled visits from 2 to 112 days (16 weeks) after administration.
Healthy male and female subjects aged 18–65 years with a body weight between 50 and 90 kg and a body mass index within the range of 18–30 kg/m2 were eligible. Subjects provided written informed consent before any protocol‐specific procedure. All subjects were informed about the nature and purpose of the study, participation/termination conditions, and risks and benefits of treatment. Use of any prescription drugs was prohibited, except for permitted use of oral/injectable contraception, herbal supplements within 4 weeks prior to initial dosing or over‐the‐counter medication or dietary supplements (vitamins included) within 2 weeks prior to initial dosing. Eligible and consented subjects were admitted to the study centre for baseline evaluation and randomized if they met all the inclusion/exclusion criteria.
2.1.2. Study objectives
The primary objective of this study was to characterize the PK of secukinumab 300 mg administered s.c. by 6 different devices. Secondary objectives included assessment of the tolerability and safety of 2‐mL s.c. injections of secukinumab by these different administration methods. Exploratory objectives included the qualitative assessment of any leakage after 2‐mL s.c. injections of secukinumab by these different administration systems, and comparison of the PK of secukinumab 300 mg when administered in the thigh or abdomen, or administered in the left or right side.
2.1.3. Dosage and administration
Secukinumab was supplied as a sterile 150‐mg solution provided in a 1‐mL AI or in a PFS with an extractable volume of 1 mL for the 2 × 1‐mL injections. For the 2‐mL injections, auxiliary supplies were sourced locally or by Drug Supply Management of Novartis. Each subject received 300 mg of secukinumab. This dose was the highest assessed s.c. dose in psoriasis phase 3 studies, showed superior results in comparison to 150 mg and is the therapeutic dose level in psoriasis patients.4 This 300‐mg single dose was known to be safe and well tolerated in previous healthy volunteer studies.
Subjects were assigned to 1 of following 6 devices (treatment arms) in a ratio of 1:1:1:1:1:1 (aiming for 20 in each arm). The 6 study treatments are defined as:
Two injections of secukinumab 150 mg/mL, using two 1‐mL autoinjectors—AI (2 × 1 mL)
Single injection of secukinumab 150 mg/mL, using a Twin‐01 device, 2 × 1 mL—Twin‐01
Single s.c. injection of 2 mL of secukinumab 150 mg/mL in 90 s (22.2 μL/s), using a syringe pump—Pump (90 s)
Single s.c. injection of 2 mL of secukinumab 150 mg/mL in 5 min (6.67 μL/s), using a 2‐mL SmartDose patch injector—SmartDose
Single s.c. injection of 2 mL of secukinumab 150 mg/mL in 10 s (200 μL/s)—Manual (10 s)
Two injections of secukinumab 150 mg/mL, using 2 × 1‐mL PFS—PFS (2 × 1 mL)
An overview of the administration devices is given in Table 1. Furthermore, study subjects randomized to each individual device were also randomized to site of injection (thigh or abdomen) and side of injection (left or right), giving 5 subjects at each of the 4 possible combinations per device, and 30 overall.
Table 1.
2‐mL devices used for subcutaneous administration of secukinumab
| Needle length/estimated depth of injection | Injection time | Short label a | Ph 1 study | Ph 3 study | |
|---|---|---|---|---|---|
| Auto‐injector twice, 2 × 1 mL | 12 mm/8 mm | 10 s | AI (2 × 1 mL) | X | |
| Twin‐01, 2 × 1 mL | 12 mm/8 mm | 10 s | Twin‐01 | X | |
| Syringe pump, 2 mL in 90 s) | 6 mm | 90 s | Pump (90 s) | X | |
| 2‐mL SmartDose, 2 mL in 5 min | 5 mm | 5 min | SmartDose | X | |
| Manual injection, 2 mL in 10 s | 12 mm/8 mm | 10 s | Manual (10 s) | X | |
| One PFS twice, 2 × 1 mL | 12 mm/8 mm | 10 s | PFS (2 × 1 mL) | X | X |
| One 2‐mL PFS, 2 mL | 12 mm/8 mm | 10 s | PFS (2 mL) | X |
Where useful, the above nomenclature is used for the different delivery systems.
2.1.4. Sample collections
Blood was collected for the measurement of serum concentrations of secukinumab. Blood samples were taken by either direct venepuncture or an indwelling cannula inserted in a forearm vein before injection and 8, 24, 48, 72, 96 and 120 h postdose, and then 7, 10, 14, 21, 28, 35, 49, 63, 77, 91 and 112 days after administration of study drug. Blood samples collected before secukinumab administration and 112 days postdose were also evaluated for the presence of antidrug antibodies (ADAs) in serum.
2.2. Phase 3 study in moderate to severe psoriasis patients
The Phase 3 secukinumab study as reported in this paper is ALLURE.14 It was a multicentre, randomized, double‐blind, placebo controlled, 52‐week study to demonstrate the efficacy, safety and tolerability of s.c. secukinumab injections with 2‐mL PFS (300 mg) and 2 × 1‐mL PFS (300 mg) in adult subjects with moderate to severe plaque psoriasis (Table 1). Patients were randomly assigned in a 1:1:1 ratio to 1 of the 2 secukinumab device groups or a placebo group. Patients in the secukinumab groups received a s.c. dose of 300 mg once weekly for 4 weeks, followed by s.c. secukinumab dosing every 4 weeks, starting at Week 4 up to Week 48. In 1 of the secukinumab dose groups, patients received administrations as 2 × 1 mL PFS injections per dose and in the other as a single 2‐mL PFS injection. Placebo patients received placebo injections once per week for 4 weeks (at baseline, Week 1, 2 and 3), followed by placebo dosing every 4 weeks starting at Week 4 up to Week 12. Psoriasis Area and Severity Index‐90 nonresponders in the placebo group received their secukinumab injections, either as 2 × 1‐mL injections or as 1 × 2 mL at Weeks 12, 13, 14, 15 and then every 4 weeks starting at Week 16 up to Week 48. Site and side of administration, i.e. abdominal region or thigh, left or right part of the body, for every time point were documented and the impact of site and side on PK was evaluated. PK results only from the 2 groups who started treatment from the beginning of the study, i.e. no PK results from the placebo groups with treatment starting at Week 12, will be reported in this paper. Complete clinical safety, tolerability and efficacy results will be published elsewhere.14
2.2.1. Sample collections
Blood was collected for the measurement of serum concentrations of secukinumab. Blood samples were taken before injection at Weeks 0 (randomization), 4, 12, 13, 14, 15, 16, 28 and 52. Sample handling, storage and measurement was as described above for the phase 1 study.
2.3. Methods for both studies
2.3.1. Bioanalytical methods
PK
In both clinical studies, secukinumab serum concentrations were quantified using a validated enzyme‐linked immunosorbent assay with an LLOQ of 80 ng/mL. The method was based on a purified, non‐neutralizing, anti‐idiotype anti‐secukinumab antibody coated on microtitre plates. Serum samples (calibration samples, quality controls or unknown samples) and biotin‐labelled secukinumab were simultaneously incubated and competed for binding on the anti‐idiotypic anti‐secukinumab antibody. Unbound material was removed by washing. Bound biotinylated secukinumab is detected by incubating horseradish peroxidase conjugated to streptavidin with o‐phenylenediamine dihydrochloride as enzymatic substrate. The intraday accuracy and intraday precision were within the ranges 76–118% and 1.4–17.8%, respectively. The interday accuracy and interday precision were within the ranges 90–99% and 7.2–15.6%, respectively. Secukinumab has been proven to be stable at −20°C for at least 20 months.
Immunogenicity
ADA assessment followed a 3‐tiered approach: screening, confirmation and titration.14 It is well known that presence of drug in a sample may interfere with the detection of ADAs. A polyclonal positive control antibody was used to define sensitivity (4 ng/mL) and secukinumab drug tolerance (53.8 μg/mL at 250 ng/mL positive control).14 At the time points of ADA assessment in this study, secukinumab concentrations were below the drug tolerance level. Subjects were classified as being treatment‐emergent ADA positive if ADA were detected in the sample evaluated after exposure to secukinumab accompanied by a negative sample at baseline.
2.3.2. PK analysis
The PK parameters of secukinumab were determined using the actual recorded sampling times and noncompartmental method(s) with WinNonlin Phoenix (Version 6.2 and higher). Concentrations below the LLOQ were treated as zero for PK parameter calculations. The derived pharmacokinetic parameters in the phase 1 study included the maximum observed serum concentration (Cmax); time to reach Cmax (tmax); area under the serum–concentration curve (AUC) from time zero to time of last measured concentration (AUClast); AUC from time zero to infinity (AUCinf); terminal half‐life (t1/2); apparent clearance (CL/F); and apparent volume of distribution based on the terminal phase (Vz/F). The linear trapezoidal rule was used for AUC calculation. Regression analysis of the terminal serum elimination phase for the determination of t1/2 included at least 3 data points after Cmax. If the R2‐adjusted value of the regression analysis of the terminal phase was <0.75, no values were reported for t1/2, AUCinf, Vz/F and CL/F. Cmax, tmax and AUC84‐112d were derived in the phase 3 study.
2.4. Statistical analysis
Descriptive statistics were used to summarize serum secukinumab concentrations at each scheduled sampling time point and to summarize the derived pharmacokinetic parameters of secukinumab. PK parameters were calculated and their descriptive summary statistics were mean (arithmetic and geometric), standard deviation and coefficient of variation (%CV; arithmetic and geometric), median, minimum and maximum. tmax was an exception to this, where median, minimum and maximum were presented. The log‐transformed parameters AUClast, AUCinf (phase 1 study only), AUC84‐112d (phase 3 study only), and Cmax were analysed by a linear model, with injection device as a fixed factor of primary interest and weight at baseline as covariate. The effect of location of injection (site and side) was also explored. For each of AUClast, AUCinf and Cmax, the mean estimate and 90% confidence interval (CI) for the PK parameter ratio of injection device vs PFS twice 1 mL in 10 s were reported to represent the relative bioavailability for comparison. The same was done for the AUC84‐112d ratio 2 × 1‐mL PFS vs 2‐mL PFS in the phase 3 study.
In the phase 1 study, over all treatment arms, half of the subjects received the administration either at the left or right side of the body (side effect) and half of them either in the thigh or in the abdomen (site effect) balanced over treatments in a factorial 6 × 2 × 2 fashion. Therefore, the statistical analysis of PK and other parameters allowed to look at the main effect of side or site, as well as of the main study treatment, device, using all the data. PK parameters were analysed by a fixed effect model with terms bodyweight, treatment, site and side. In the phase 3 study, side (left/right) and site (abdomen, thigh, shoulder) of every injection was documented, which allowed an evaluation of impact on serum exposure resulting from a specific injection.
2.4.1. Safety and tolerability assessments in phase 1 study
All injections were monitored for reactions by rating severity (none, mild, moderate, severe) of local erythema, induration and haemorrhage, and qualitative (presence or absence) assessment of pruritus. Leakage (presence or absence) following drug administration was an exploratory variable.
Tolerability was assessed by review of summaries of the subject‐assessed pain VAS scores, collected over time. Summary variables of VAS scores were:
Maximum pain VAS score during injection
Maximum pain VAS score from end of injection to last assessment
AUC of pain VAS score from end of injection to last assessment
VAS scores were also displayed graphically over time for individual subjects and averaged by treatment groups. The summary variables were analysed by a linear model, with device as fixed factor and weight at baseline as covariate, as for PK data, but with the effect gender and age also included. Analyses of the local skin tolerability assessments were by logit models for binomial data on their incidence (present or absent per subject). As for pain VAS scores (which were displayed graphically over time for individual subjects), erythema, induration and haemorrhage were displayed graphically over time when averaged over treatments at each time point. Pruritus assessment and leakage were summarized by descriptive statistics and analysed by the logit model with the same linear structure as above, as a binomial response per subject (present or absent). For all analyses there was no comparable pretreatment covariate as pretreatment values were assumed to be all none on tolerability scales, zero for the pain VAS and no pruritus.
2.4.2. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
3. RESULTS
3.1. Phase 1 study
3.1.1. Subject disposition, demographics and baseline characteristics
A total of 122 subjects received a single dose of secukinumab. Of these, 114 completed the study through 112 days (16 weeks) and 8 subjects discontinued early due to reasons unrelated to adverse events (AEs). Demographic information and baseline characteristics are summarized in Table 2. The mean age of the subjects was 39.0 years (range 18–60 years) and mean weight 72.7 kg (range 47.4–90.0 kg). There were 59 males and 63 females. Demographic data did not show any major differences between the treatment groups.
Table 2.
Subject demographics and baseline characteristics
| AI (2 × 1 mL) N = 20, n (%) | Twin‐01 N = 20, n (%) | Pump (90 s) N = 20, n (%) | SmartDose N = 20, n (%) | Manual (10 s) N = 21, n (%) | PFS (2 × 1 mL) N = 21, n (%) | Total N = 122, n (%) | ||
|---|---|---|---|---|---|---|---|---|
| Age (y) | Mean (SD) | 38.1 (13.04) | 42.9 (11.41) | 34.1 (13.10) | 38.2 (14.36) | 37.9 (11.59) | 42.5 (10.21) | 39.0 (12.45) |
| Median | 38.0 | 46.5 | 33.0 | 41.5 | 43.0 | 41.0 | 40.5 | |
| Range | 19–60 | 18–58 | 18–59 | 18–59 | 20–55 | 25–59 | 18–60 | |
| Sex, n (%) | Male | 11 (55.0%) | 6 (30.0%) | 9 (45.0%) | 14 (70.0%) | 11 (52.4%) | 8 (38.1%) | 59 (48.4%) |
| Female | 9 (45.0%) | 14 (70.0%) | 11 (55.0%) | 6 (30.0%) | 10 (47.6%) | 13 (61.9%) | 63 (51.6%) | |
| Race, n (%) | Caucasian | 15 (75.0%) | 15 (75.0%) | 9 (45.0%) | 13 (65.0%) | 15 (71.4%) | 11 (52.4%) | 78 (63.9%) |
| Black | 1 (5.0%) | 5 (25.0%) | 9 (45.0%) | 5 (25.0%) | 4 (19.0%) | 7 (33.3%) | 31 (25.4%) | |
| Asian | 2 (10.0%) | 0 | 1 (5.0%) | 0 | 2 (9.5%) | 2 (9.5%) | 7 (5.7%) | |
| Other | 2 (10.0%) | 0 | 1 (5.0%) | 2 (10.0%) | 0 | 1 (4.8%) | 6 (4.9%) | |
| Ethnicity, n (%) | Hispanic/Latino | 6 (30.0%) | 4 (20.0%) | 6 (30.0%) | 6 (30.0%) | 6 (28.6%) | 6 (28.6%) | 34 (27.9%) |
| Indian (Indian subcontinent) | 1 (5.0%) | 0 | 1 (5.0%) | 0 | 0 | 0 | 2 (1.6%) | |
| Japanese | 0 | 0 | 0 | 0 | 0 | 1 (4.8%) | 1 (0.8%) | |
| Mixed ethnicity | 0 | 0 | 0 | 0 | 1 (4.8%) | 1 (4.8%) | 2 (1.6%) | |
| Other | 13 (65.0%) | 16 (80.0%) | 13 (65.0%) | 14 (70.0%) | 14 (66.7%) | 13 (61.9%) | 83 (68.0%) | |
| Weight (kg) | Mean (SD) | 75.4 (9.39) | 72.0 (10.46) | 73.0 (9.38) | 74.3 (11.10) | 69.3 (13.10) | 72.6 (9.48) | 72.7 (10.54) |
| Median | 75.9 | 71.2 | 74.0 | 70.6 | 68.4 | 75.1 | 71.7 | |
| Range | 60.6–89.9 | 57.7–87.8 | 55.2–90.0 | 57.6–90.0 | 47.4–90.0 | 54.7–86.7 | 47.4–90.0 | |
| Height (cm) | Mean (SD) | 172.0 (9.72) | 166.5 (7.17) | 170.9 (10.82) | 171.2 (9.08) | 168.2 (10.89) | 167.0 (8.97) | 169.2 (9.58) |
| Median | 170.0 | 168.0 | 172.0 | 172.5 | 166.5 | 165.0 | 168.8 | |
| Range | 158–189 | 154–179 | 154–194 | 154–188 | 150–193 | 151–186 | 150–194 | |
| BMI (kg/m2) | Mean (SD) | 25.67 (3.93) | 25.95 (3.11) | 24.97 (1.98) | 25.28 (2.58) | 24.45 (3.68) | 26.04 (2.94) | 25.39 (3.10) |
| Median | 26.53 | 26.48 | 25.28 | 25.43 | 24.22 | 26.27 | 25.55 | |
| Range | 17.78–30.25 | 20.16–30.01 | 20.19–27.92 | 21.36–29.06 | 19.10–30.07 | 20.57–30.44 | 17.78–30.44 | |
BMI, body mass index; SD, standard deviation.
3.1.2. PK
Among the 122 subjects enrolled in the study, 121 had evaluable samples for Cmax and tmax, 114 had samples evaluable for AUClast and AUCinf and for all other pharmacokinetic parameters. One subject in the Manual (10 s) arm had an incomplete time‐concentration profile and so these PK parameters could not be assessed.
The mean serum concentration–time profiles of secukinumab following s.c. administration of 300 mg are presented in Figure 1. Subcutaneous administration of 300 mg secukinumab by 6 different injection devices led to very comparable serum concentration–time profiles. The maximum concentrations of secukinumab for the 6 devices were very similar (Figure 1 and Table 3), and were reached between 2 and 35 days in individual subjects, with the median tmax for each device in the range between 4.5 and 7 days after administration. The highest and lowest Cmax values were observed in the Manual (10 s) and Pump (90 s) arms, respectively. The average systemic exposure to secukinumab as characterized by AUClast and AUCinf was similar for the 6 devices as well. Mean elimination half‐lives of secukinumab per device were in a narrow range between 28 and 33 days.
Figure 1.

Mean (standard deviation) serum concentration–time profiles of secukinumab following a single 300 mg subcutaneous injection in healthy male and female subjects
Table 3.
Pharmacokinetic parameters of secukinumab following a single 300 mg subcutaneous injection in healthy male and female subjects
| AUClast | AUCinf | Cmax | tmax a | Vz/F | CL/F | t1/2 | |
|---|---|---|---|---|---|---|---|
| (μg.day)/mL | (μg.day)/mL | μg/mL | day | L | L/day | day | |
| AI (2 × 1 mL) | |||||||
| N | 19 | 19 | 20 | 20 | 19 | 19 | 19 |
| Mean | 1610 | 1810 | 35.2 | ‐ | 9.71 | 0.223 | 32.1 |
| SD | 606 | 720 | 11.4 | ‐ | 7.31 | 0.202 | 6.36 |
| CV% | 37.7 | 39.9 | 32.3 | ‐ | 75.2 | 90.2 | 19.8 |
| Median | 1710 | 1950 | 39.7 | 4.48 | 7.80 | 0.154 | 30.4 |
| Range | 282–2490 | 298–3080 | 7.66–48.7 | 2.94–10.1 | 4.58–37.6 | 0.0975–1.01 | 22.5–43.3 |
| Twin‐01 (2 × 1 mL) | |||||||
| N | 19 | 19 | 20 | 20 | 19 | 19 | 19 |
| Mean | 1730 | 1940 | 38.0 | ‐ | 7.38 | 0.166 | 31.9 |
| SD | 456 | 535 | 9.79 | ‐ | 1.74 | 0.047 | 7.57 |
| CV% | 26.4 | 27.5 | 25.7 | ‐ | 23.6 | 28.5 | 23.7 |
| Median | 1710 | 2010 | 37.5 | 7.00 | 7.25 | 0.149 | 30.0 |
| Range | 1090–2580 | 1130–3040 | 25.3–53.4 | 2.05–34.9 | 4.60–10.3 | 0.0988–0.265 | 20.9–48.7 |
| Pump (90 s) | |||||||
| N | 18 | 18 | 20 | 20 | 18 | 18 | 18 |
| Mean | 1520 | 1700 | 34.1 | ‐ | 8.64 | 0.184 | 33.0 |
| SD | 304 | 364 | 6.88 | ‐ | 1.60 | 0.038 | 4.97 |
| CV% | 19.9 | 21.4 | 20.2 | ‐ | 18.6 | 20.7 | 15.1 |
| Median | 1500 | 1650 | 32.9 | 4.97 | 8.53 | 0.182 | 31.0 |
| Range | 1100–2090 | 1200–2420 | 24.7–46.0 | 2.00–14.0 | 5.85–11.4 | 0.124–0.249 | 26.8–42.4 |
| Smartdose | |||||||
| N | 19 | 19 | 20 | 20 | 19 | 19 | 19 |
| Mean | 1690 | 1840 | 40.4 | ‐ | 6.89 | 0.174 | 28.4 |
| SD | 400 | 475 | 9.45 | ‐ | 1.34 | 0.045 | 5.69 |
| CV% | 23.7 | 25.9 | 23.4 | ‐ | 19.4 | 25.9 | 20.0 |
| Median | 1630 | 1700 | 39.4 | 4.50 | 6.60 | 0.177 | 27.4 |
| Range | 1050–2510 | 1070–2810 | 22.6–56.7 | 3.00–9.99 | 5.34–9.70 | 0.107–0.279 | 17.7–39.8 |
| Manual (10 s) | |||||||
| N | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Mean | 1860 | 2050 | 42.1 | ‐ | 6.49 | 0.154 | 30.2 |
| SD | 371 | 488 | 6.95 | ‐ | 1.24 | 0.037 | 7.22 |
| CV% | 20.0 | 23.8 | 16.5 | ‐ | 19.1 | 24.1 | 23.9 |
| Median | 1850 | 1970 | 43.0 | 6.00 | 6.50 | 0.153 | 27.6 |
| Range | 1180–2580 | 1200–3280 | 29.1–53.5 | 2.03–10.0 | 4.04–8.35 | 0.0931–0.249 | 21.1–48.7 |
| PFS (2 × 1 mL) | |||||||
| N | 19 | 19 | 21 | 21 | 19 | 19 | 19 |
| Mean | 1620 | 1760 | 40.8 | ‐ | 7.65 | 0.189 | 28.4 |
| SD | 463 | 549 | 12.6 | ‐ | 3.28 | 0.068 | 5.26 |
| CV% | 28.6 | 31.2 | 31.0 | ‐ | 42.9 | 36.3 | 18.5 |
| Median | 1660 | 1770 | 45.1 | 4.90 | 6.36 | 0.170 | 26.7 |
| Range | 669–2720 | 749–3220 | 12.6–61.8 | 1.95–14.1 | 5.46–19.0 | 0.0931–0.400 | 20.1–40.7 |
median and range only are given for tmax.
As for the other PK parameters, the apparent volumes of distribution and clearance did not show any dependence on the device used. Vz/F ranged from 6.49 to 9.71 L, and Cl/F from 0.154 to 0.223 L/day. For all PK parameters, the intersubject variability was similar for the 6 devices, as shown by the CV values in Table 3. CV% for the AUCs and Cmax ranged between 19.9 and 39.9%, between 15.1 and 23.9% for t1/2, and between 18.6 and 90.2% for Cl/F and Vz/F. The higher CV% in the AI (2 × 1 mL) was due to 1 individual outlier profile with a very low Cmax <10 μg/mL and a Vz/F of 37.6 L and a Cl/F of 1.01 L/day. All tabulated PK parameters are provided in Table 3.
3.1.3. Statistical comparisons
As summarized in Table 4, the geometric mean ratios (90% CI) for AUCinf, AUClast and Cmax for any of the 5 devices compared with PFS (2 × 1 mL) were mostly within the comparability acceptance interval of 0.80–1.25, except for AUCinf of Twin‐01 with (0.93, 1.30), for AUCinf of SmartDose with (0.91, 1.27), for AUCinf of Manual (10 s) with (0.98, 1.36), for AUClast of Manual (10 s) with (0.98, 1.33) with upper bounds outside the acceptance interval and for Cmax of AI (2 × 1 mL) with (0.74, 1.01) and for Cmax of Pump (90 s) with (0.75, 1.02) with lower bounds outside the acceptance interval. For these above‐mentioned exceptions, the 90% CIs were only slightly outside the (0.8, 1.25) range.
Table 4.
Geometric means, estimated geometric mean ratio and 90% confidence interval (CI) for geometric ratio of pharmacokinetic (PK) parameters
| PK parameter (unit) | Treatment | N | Ratio (test/reference) a | 90% CI for geometric mean ratio a |
|---|---|---|---|---|
| AUCinf (day*μg/mL) | AI (2 × 1 mL) | 19 | 0.99 | (0.83, 1.16) |
| Twin‐01 | 19 | 1.10 | (0.93, 1.30) | |
| Pump (90 s) | 18 | 0.99 | (0.84, 1.17) | |
| SmartDose | 19 | 1.07 | (0.91, 1.27) | |
| Manual (10 s) | 20 | 1.15 | (0.98, 1.36) | |
| PFS (2 × 1 mL) | 19 | ‐ | ‐ | |
| AUClast (day*μg/mL) | AI (2 × 1 mL) | 19 | 0.96 | (0.82, 1.12) |
| Twin‐01 | 19 | 1.06 | (0.91, 1.24) | |
| Pump (90 s) | 18 | 0.96 | (0.82, 1.13) | |
| SmartDose | 19 | 1.07 | (0.92, 1.25) | |
| Manual (10 s) | 20 | 1.14 | (0.98, 1.33) | |
| PFS (2 × 1 mL) | 19 | ‐ | ‐ | |
| Cmax (μg/mL) | AI (2 × 1 mL) | 20 | 0.87 | (0.74, 1.01) |
| Twin‐01 | 20 | 0.96 | (0.83, 1.12) | |
| Pump (90 s) | 20 | 0.88 | (0.75, 1.02) | |
| SmartDose | 20 | 1.04 | (0.89, 1.21) | |
| Manual (10 s) | 20 | 1.06 | (0.91, 1.24) | |
| PFS (2 × 1 mL) | 21 | ‐ | ‐ |
Reference: PFS (2 × 1 mL).
Exploratory objectives in this study included the comparison of PK parameters when 300 mg was administered in either the thigh or abdomen and either the left or right side. As can be seen in Table 5, no difference between administrations at the left or right side was observed, whereas some evidence exists of a slightly elevated exposure combined with an earlier tmax from thigh compared with abdomen.
Table 5.
Summary of statistical analysis by site and side of AUCinf, AUClast and Cmax
| Adjusted mean on ln scale | Adjusted geometric meansa | Ratio (test/referenceb) | 90% CI for ratio | |
|---|---|---|---|---|
| AUC inf | ||||
| Thigh | 7.56 | 1912 | 1.17 | 1.06, 1.29 |
| Abdomen | 7.40 | 1637 | ||
| Left | 7.48 | 1769 | 1.00 | 0.91, 1.10 |
| Right | 7.48 | 1770 | ||
| AUC last | ||||
| Thigh | 7.45 | 1728 | 1.16 | 1.06, 1.27 |
| Abdomen | 7.31 | 1493 | ||
| Left | 7.38 | 1602 | 0.99 | 0.91, 1.09 |
| Right | 7.38 | 1611 | ||
| C max | ||||
| Thigh | 3.68 | 39.7 | 1.16 | 1.06, 1.27 |
| Abdomen | 3.53 | 34.2 | ||
| Left | 3.60 | 36.5 | 0.98 | 0.90, 1.07 |
| Right | 3.62 | 37.2 | ||
| T max | ||||
| Thigh | 1.57 | 4.79 | 0.79 | 0.68, 0.91 |
| Abdomen | 1.80 | 6.07 | ||
| Left | 1.73 | 5.65 | 1.10 | 0.95, 1.27 |
| Right | 1.64 | 5.14 | ||
back transformed from log scale;
Reference: abdomen or right side.
3.1.4. Immunogenicity
Immunogenicity data were collected at baseline and at the end of study visit. Treatment‐emergent ADA (i.e. negative at baseline but positive after start of treatment) were detected in 2 subjects. Moderate induration and mild erythema were observed in 1 of these subjects on Day 1. Three subjects with ADA at baseline and a negative result at the end of the study were observed as well. No impact on PK or AEs was observed in any of these 5 subjects.
3.1.5. Local tolerability
Local tolerability of the injection site was assessed by a clinical evaluation of erythema, induration and haemorrhage. Results are summarized in Table 6. The significant P‐value for erythema treatment differences overall is mainly due to its presence in 11/20 subjects using the SmartDose device with other devices having lower or no incidence. The AIs did not provoke any erythema. The high SmartDose incidence was probably linked to the patch system. Similarly, the syringe pump had the second highest incidence (4/20), probably linked to the small patch used to maintain the needle. Finally, the manual injections either of 2 × 1 mL (2/21) or 1 × 2 mL (3/21) had only a few cases erythema of mild to moderate severity which were probably linked to the manipulation itself. The significant P‐value for induration treatment differences overall is due to its presence in 7 subjects using SmartDose device with other devices having occurrence in only 0 or 1 subject. The high observed induration after the SmartDose may also be linked to the patch system. Indeed, in all other treatment groups, there is no induration except for 1 case in the Pump (90 s), which may also be related to the small patch used to maintain the needle during the injection. Severity of induration was mild to moderate.
Table 6.
Incidence (any timepoint) of erythema, induration and haemorrhage by treatment with overall test for treatment effect
| AI (2 × 1 mL) N=20 | Twin‐ 01 N=20 | Pump (90 s) N=20 | SmartDose N=20 | Manual (10 s) N=21 | PFS (2 × 1 mL) N = 21 | |
|---|---|---|---|---|---|---|
| Parameter (overall P‐value; observed r/n, model predicted proportion by treatment) | ||||||
| Erythema (P < 0.0001) | 0/20 | 0/20 | 4/20 | 11/20 | 3/21 | 2/21 |
| 0.00 | 0.00 | 0.19 | 0.55 | 0.14 | 0.09 | |
| Induration (P < 0.0001) | 0/20 | 0/20 | 1/20 | 7/20 | 0/21 | 0/21 |
| 0.00 | 0.00 | 0.05 | 0.35 | 0.00 | 0.00 | |
| Haemorrhage (P = 0.615) | 0/20 | 0/20 | 0/20 | 0/20 | 0/21 | 1/21 |
| 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.05 | |
| Incidence (overall P‐value; observed r/n, predicted proportion) | ||||||
| Pruritus (P = 0.0682) | 0/20 | 0/20 | 0/20 | 3/20 | 0/21 | 1/21 |
| 0.00 | 0.00 | 0.00 | 0.15 | 0.00 | 0.05 | |
| Leakage (P = 0.2275) | 0/20 | 0/20 | 0/20 | 0/20 | 1/21 | 2/21 |
| 0.00 | 0.00 | 0.00 | 0.00 | 0.05 | 0.10 | |
r/n: occurred /number of subjects.
If all assessed parameters of local tolerability are taken into account, AI (2 × 1 mL) had no tolerability symptoms. Increasing incidence was observed in the order Twin‐01, Manual (10 s), Pump (90 s), PFS (2 × 1 mL) and SmartDose. As can be seen in Figure 2, there was no evident difference in pain between devices. All devices resulted in a very limited pain feeling, which lasted for <15 min from the end of dosing. Based on the 3 summary statistics for VAS scores over time, there was no difference between devices. Results supported good tolerability in terms of 2‐mL volume administered and the duration of administration from 10 s to 5 min.
Figure 2.
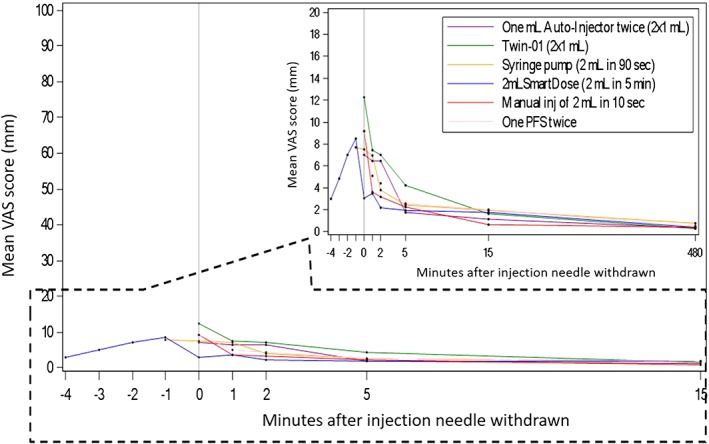
Arithmetic mean visual analogue scale (VAS) pain score–time profiles per treatment group (PD analysis set). Standard deviations of mean VAS scores are mostly in the same order of magnitude for all devices. They are not included in the plot to keep time‐mean VAS scores clearly distinguishable. PFS, prefilled syringe
3.1.6. Safety
There were no subjects discontinued from the study due to any safety reasons (AEs/severe AEs). A total of 29 subjects (23.8%) across the treatment groups experienced at least 1 AE during the study, a total of 44 nonserious AEs. The most frequently affected system organ class, which involved all subjects of the 6 device groups, was infections and infestations (11 AEs in 10 subjects, 8.2%). This included gastroenteritis (n = 1, 0.8%), viral influenza (n = 3, 2.5%), nasopharyngitis (n = 3, 2.5%), pharyngitis (n = 2, 1.6%), streptococcal sinusitis (n = 1, 0.8%) and upper respiratory tract infection (n = 1, 0.8%).
3.2. Phase 3 study
3.2.1. PK and statistical analysis
A total of 214 patients were randomized and 198 (92.5%) completed the entire treatment period. The 3 treatment groups were balanced at randomization, i.e. 72 in the 2‐mL PFS group, 71 in the 2 × 1‐mL PFS group and 71 in the placebo group.
The mean serum secukinumab time–concentration trajectories are shown in Figure 3. At Week 4, serum secukinumab concentrations reflected the rise in exposure near the end of weekly dosing. The mean concentrations at Week 4 were similar for secukinumab 300 mg 2‐mL PFS group (92.6 μg/mL) and 2 × 1‐mL PFS group (87.6 μg/mL). At Week 12, mean concentrations declined to 44.9 μg/mL (2‐mL PFS) and 45.3 μg/mL (2 × 1‐mL), in line with the less‐frequent dose administration after Week 4. As can be seen in Figure 3, a typical PK profile between 12 and 16 weeks after s.c. administration at Week 12 can be recognized. Therefore, AUC84‐112d from patients who (i) started treatment at the beginning of the study and (ii) have serum concentrations at all 5 time points, i.e. Weeks 12, 13, 14, 15 and 16, were calculated. Descriptive statistics are given in Table 7. Mean Cmax, AUC84‐112d and median tmax values were similar after either injection with 2 mL PFS or 2 × 1 mL PFS. The median tmax of approximately 91 days was reported here as postdose time‐point after the first dose administration, which corresponds to approximately 7 days postdose after the Week 12 dose. The 90% CIs for the ratio of geometric means for AUC84‐112d and Cmax, were all contained in the interval (0.80, 1.25), indicating that the 2 devices were comparable, see Table 8. The serial decline in mean trough concentrations at Weeks 12, 16, and 28 reflect the attainment of a new steady state during maintenance dosing every 4 weeks. Midway into the maintenance phase (Week 28) and at the end of the maintenance phase (Week 52), mean troughs were close to or at steady state, ending with values of 38.5 and 37.2 μg/mL for 2‐mL PFS and 2 × 1‐mL PFS groups, respectively. A total of 820 serum samples from 212 patients were analysed for ADAs. In line with the reported low incidence of treatment‐emergent ADAs of secukinumab below 1%,14 ADAs were not detected. As expected from the results of the phase 1 study in healthy subjects, there was some impact of the site of s.c. administration on serum secukinumab exposure. Serum exposure (AUC84‐112d) from 2‐mL PFS after defined Week 12 administration was higher from thigh injection compared to abdomen, with geometric mean ratios for thigh to abdomen >1, i.e. 1.19 and a 90% CI of (0.97, 1.46).
Figure 3.
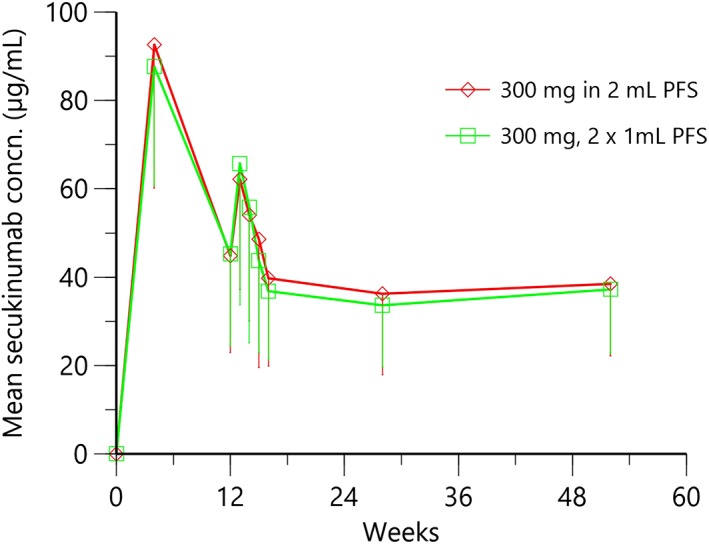
Time–mean serum concentration pharmacokinetic trajectories with 2 × 1‐mL and 2‐mL prefilled syringe (PFS) injection devices in psoriasis patients
Table 7.
Descriptive statistics of pharmacokinetic parameters in psoriasis patients
| Secukinumab 300 mg, 2‐mL PFS | Secukinumab 300 mg, 2 × 1‐mL PFS | |||||
|---|---|---|---|---|---|---|
| Cmax (μg/mL) | AUC84‐112d (day × μg/mL) | tmax a (day) | Cmax (μg/mL) | AUC84‐112d (day × μg/mL) | tmax a (day) | |
| n | 49 | 49 | 49 | 55 | 55 | 55 |
| Mean | 70.7 | 1450 | ‐ | 70.9 | 1430 | ‐ |
| SD | 33.7 | 624 | ‐ | 39.5 | 616 | ‐ |
| %CV | 47.7 | 43.1 | ‐ | 55.7 | 42.9 | ‐ |
| Median | 63.8 | 1360 | 91.2 | 64.6 | 1320 | 91.0 |
| Range | 17.3–212 | 289–3210 | 84.0–112 | 21.7–234 | 452–3170 | 83.1–113 |
Median and Min‐Max only are given for tmax. tmax is calculated from start of treatment.
Table 8.
Geometric means, estimated geometric mean ratio and 90% confidence interval (CI) for geometric ratio of pharmacokinetic (PK) parameters in psoriasis patients
| PK parameter (unit) | Treatment | n | Adjusted geo‐meana | Ratio (test/reference)b | 90% CI for geometric mean ratio* |
|---|---|---|---|---|---|
| AUC84‐112d | 2 mL PFS | 49 | 1286 | 0.97 | (0.86, 1.09) |
| (day*μg/mL) | 2 × 1 mL PFS | 49 | 1327 | ||
| Cmax | 2 mL PFS | 55 | 62.6 | 0.98 | (0.87, 1.12) |
| (μg/mL) | 2 × 1 mL PFS | 55 | 63.7 |
back‐transformed from log scale;
reference: PFS (2 × 1 mL).
4. DISCUSSION
The phase 1 study in healthy subjects demonstrates that relatively large s.c. injection volumes of 2 mL to administer secukinumab at a dose of 300 mg are well tolerated, regardless of injection flow rate, when injected in the abdomen or thigh. Resulting serum exposures with 2‐mL devices are similar to those obtained with 2 × 1‐mL injections. After a single s.c. injection of 300 mg, secukinumab was slowly absorbed into the systemic circulation with an overall median tmax of 4.5 to 7 days. The comparable plateau‐like behaviours between 4 and 14 days during the first 2 weeks after s.c. administration with all 6 devices is in agreement with earlier observations.15 After tmax, the mean serum concentrations of secukinumab declined in an apparently mono‐exponential manner, which was consistent with population PK models for secukinumab in patients with psoriasis.15 The mean Vz/F values were in the range between 6.5 and 9.7 L, which is also in line with the volume of distribution values reported in psoriasis patients.15 With an absolute bioavailability of 73% in psoriasis patients, the mean Vz is estimated to be in the range between 4.7 and 7.0 L, suggesting that secukinumab is primarily confined to the systemic circulation with limited extravascular tissue distribution. The PK observations as described above for the phase 1 study were confirmed in the phase 3 study in moderate to severe psoriasis patients. Serum exposure was similar after either injection of 300 mg as 2 × 1 mL in PFS or as a single 2‐mL injection. Safety, tolerability and clinical efficacy from the phase 3 study are published separately.14
With the four 2‐mL devices used in the phase 1 study and the 2‐mL PFS in phase 3, secukinumab showed PK characteristics as seen with the 1‐mL PFS and AI devices used in Phase 3 for the registered dose of 300 mg with no evidence of any substantive differences due to any device. Relative bioavailability was similar for each of the 5 devices when compared to 2 × 1‐mL PFS as reference, with most of the 90% CIs contained in the interval 0.80–1.25 and the ratio of means for AUCinf, AUClast and Cmax all very close to 1. As can be seen in Table 4, the strict bioequivalence criteria, i.e. CIs for the ratio of geometric means between 0.8 and 1.25, were not met for every exposure parameter for every device. However, the observed differences in PK terms were not clinically significant as no device was outside the bioequivalence criteria over all parameters. Injection times varying between 5 min and 10 s, leading to injection speeds between 6.7 and 200 μL/min, did not affect the PK profile.
The higher serum exposure with accompanying earlier tmax after thigh administration compared with abdomen administration suggests that the site of s.c. injection may have an impact on the rate and extent of s.c. absorption of therapeutic proteins. In the scientific literature, conflicting reports exist about the impact of site on absorption. For some biological drugs higher exposure after thigh administration than after abdominal or shoulder administration was observed, whereas for other drugs no impact of site of administration on exposure occurred.16 For instance, exposure of human growth hormone (22 kDa) was found to be significantly higher after s.c. injection into the abdomen than after injection into the thigh.17 For recombinant human erythropoietin (30 kDa), the opposite occurs,18 although in another study with recombinant human erythropoietin, there were no significant differences between the abdomen and thigh injection sites.19 Label text for the anti‐IL‐17A antibody, ixekizumab, states that “in studies of subjects with plaque psoriasis, ixekizumab bioavailability ranged from 60 to 81% following s.c. injection. Administration of ixekizumab via injection in the thigh achieved a higher bioavailability relative to that achieved using other injection sites including the arm and abdomen.”20 For secukinumab we seem to have a similar observation, which is also confirmed for other investigated monoclonal antibodies such as bococizumab, golimumab and mepolizumab; s.c. administration in the thigh consistently resulted in higher serum exposure compared to injections in the abdomen region.21, 22, 23, 24 It is well acknowledged that the incomplete absorption of protein drugs, with absolute bioavailabilities ranging from 20 to 100%, is likely to be due to protein degradation in the region of the s.c. injection site as well as in lymphatic vessels during transport. It might be speculated that degree of protein degradation, transport time in lymphatic vessels and the protecting role of FcRn during transport differ to some extent between abdominal and thigh injection. However, taking the rather large interindividual variability of s.c. bioavailability of therapeutic proteins into account as well, it is reasonable to conclude that the effect of injection site on serum exposure is not clinically relevant.
As demonstrated in this study, the tolerability of 2‐mL s.c. injection of secukinumab 150‐mg/mL formulation was good and not different between all 2‐mL delivering devices. This is an important finding because of a widespread assumption that the upper limit for s.c. injection of a therapeutic protein is approximately 1 mL. Observations in the study by Heise et al12 suggested that from a tolerability (pain) point of view, volumes up to 3 mL may be preferably injected in the abdomen, with worse tolerability in the thigh. At least for the 2‐mL injections as described here, this could not be confirmed in our study; comparable VAS scores were obtained with abdominal and thigh injections and small differences were statistically not significant. Another interesting finding was the similar pain and general tolerance between the different rates of s.c. infusion with times to inject 2 mL varying between 10 s and 5 min. This basically confirms earlier findings described in literature that injection speed has little impact on injection pain and tolerance.11, 12 It has to be noted here that for AIs it is recommended to have the injection completed within 10–15 s or even less, to minimize the risk of the patient performing an incomplete administration by withdrawing the AI too early, before completion of injection. Because of the devices used, pain due to needle penetration through the skin was not separately assessed. However, as reported in several papers,11, 12 pain recorded after needle insertion is usually similar to mean injection pain for volumes up to 3 mL. Pain due to needle penetration is mainly influenced by the needle gauge size.18
Two cases of TE‐ADA formation were observed. ADA formation was neither related to AEs potentially related with immunogenicity nor to deviating PK profiles.
To the best of our knowledge, it was demonstrated for the first time for monoclonal antibodies that volumes of 2 mL, administered in a time range between 10 s and 5 min, are very well tolerated as a s.c. administration.
While there is widespread belief that the upper limit for a given s.c. injection is approximately 1 mL (driven by injection tolerance), our study results suggest that the upper limit for a given s.c. volume in the abdomen and thigh, merely based on injection tolerance, is well above 1 mL. As demonstrated, volumes of 2 mL were well tolerated. Regardless of the relatively large volume injected, the overall tolerance of the s.c. injections was good. It is interesting to note that the maximum injection pain measured using 100 mm VAS was in the range between 4.5 mm (SmartDose) and 12.9 mm (Twin‐01) and was not clinically significantly higher (<10 mm) than the mean pain recorded after needle insertion. This suggests that a major component of the overall s.c. injection pain originates from the needle insertion alone.11
In summary, no clinically relevant differences between 1‐ and 2‐mL devices for s.c. administration of secukinumab were observed.
CONTRIBUTORS
All authors made substantial contributions to either conception and design or conduct of the study and acquisition, analysis and interpretation of data. G.B. wrote the manuscript, G.B., H.‐U.P.H., R.F., M.P., P.C. and B.B.‐D. designed the studies, P.L.S. and B.S. were principal investigators, G.B., H.‐U.P.H., R.F., M.P., P.C., R.W. and B.B.‐D. analysed and interpreted data.
COMPETING INTERESTS
G.B., R.F., M.P., P.C., R.W. and B.B.‐D. are full‐time employees and shareholders of Novartis Pharma AG. H.‐U.H. is an employee of Biometrics Matters Ltd, Hamilton, New Zealand, which had Novartis Pharma as a client. P.L.S. was an employee of TKL research. B.S. is adjunct professor at the Department of Dermatology, Faculty of Medicine, University of Iceland. All authors declared no competing interests for this work.
ACKNOWLEDGEMENTS
We thank the healthy volunteers, patients and clinical site staff for their contributions to this study.
Bruin G, Hockey H‐UP, La Stella P, et al. Comparison of pharmacokinetics, safety and tolerability of secukinumab administered subcutaneously using different delivery systems in healthy volunteers and in psoriasis patients. Br J Clin Pharmacol. 2020;86:338–351. 10.1111/bcp.14155
The authors confirm that the Principal Investigators for this paper are Philip La Stella for the phase 1 study and Bardur Sigurgeirsson for the phase 3 study and that they had direct clinical responsibility for healthy subjects and patients, respectively.
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. Blauvelt A. T‐helper 17 cells in psoriatic plaques and additional genetic links between IL‐23 and psoriasis. J Invest Dermatol. 2008;128(5):1064‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J am Acad Dermatol. 2014;71(1):141‐150. [DOI] [PubMed] [Google Scholar]
- 3. Krueger JG, Fretzin S, Suárez‐Fariñas M, et al. IL‐17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. 2012;130(1):145‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371(4):326‐338. [DOI] [PubMed] [Google Scholar]
- 5. Blauvelt A, Prinz JC, Gottlieb AB, et al. Secukinumab administration by pre‐filled syringe: efficacy, safety, and usability results from a randomized controlled trial in psoriasis (FEATURE). Br J Dermatol. 2015;172(2):484‐493. [DOI] [PubMed] [Google Scholar]
- 6. Paul C, Lacour JP, Tedremets L, et al. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: a randomized, controlled trial (JUNCTURE). J Eur Acad Dermatol Venereol. 2015;29(6):1082‐1090. [DOI] [PubMed] [Google Scholar]
- 7. Narasimhan C, Mach H, Shameem M. High‐dose monoclonal antibodies via the subcutaneous route: challenges and technical solutions, an industry perspective. Ther Deliv. 2012;3(7):889‐900. [DOI] [PubMed] [Google Scholar]
- 8. Jezek J, Rides M, Derham B, et al. Viscosity of concentrated therapeutic protein compositions. Adv Drug Deliv Rev. 2011;63(13):1107‐1117. [DOI] [PubMed] [Google Scholar]
- 9. Dias C, Abosaleem B, Crispino C, Gao B, Shaywitz A. Tolerability of high‐volume subcutaneous injections of a viscous placebo buffer: a randomized, crossover study in healthy subjects. AAPS PharmSciTech. 2015;16(5):1101‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jorgensen JT, Romsing J, Rasmussen M, Moller‐Sonnergaard J, Vang L, Musaeus L. Pain assessment of subcutaneous injections. Ann Pharmacother. 1996;30(7–8):729‐732. [DOI] [PubMed] [Google Scholar]
- 11. Berteau C, Filipe‐Santos O, Wang T, Rojas HE, Granger C, Schwarzenbach F. Evaluation of the impact of viscosity, injection volume, and injection flow rate on subcutaneous injection tolerance. Med Devices: Evidence Res. 2015;8:473‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heise T, Nosek L, Dellweg S, et al. Impact of injection speed and volume on perceived pain during subcutaneous injections into the abdomen and thigh: a single‐Centre, randomized controlled trial. Diabetes Obes Metab. 2014;16(10):971‐976. [DOI] [PubMed] [Google Scholar]
- 13. Saluja A, Kalonia DS. Nature and consequences of protein‐protein interactions in high protein concentration solutions. Int J Pharm. 2008;358(1–2):1‐15. [DOI] [PubMed] [Google Scholar]
- 14. Reich K, Blauvelt A, Armstrong A, et al. Secukinumab, a fully human anti–interleukin‐17A monoclonal antibody, exhibits minimal immunogenicity in subjects with moderate to severe plaque psoriasis. Br J Dermatol. 2017; 176:752–758. 10.1111/bjd.14965 [DOI] [PubMed] [Google Scholar]
- 15. Bruin G, Loesche C, Nyirady J, Sander O. Population pharmacokinetic modeling of secukinumab in patients with moderate‐to‐severe psoriasis. J Clin Pharm. 2017; 57(7):876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin JH, Wang W. Role of lymphatic system in subcutaneous absorption of therapeitic proteins In: Zhou H, Theil F‐P, eds. ADME and Translational Pharmacokinetics/Pharmacodynamics of Therapeutic Proteins Ch. 6. Hoboken, NJ, USA, Wiley: 67‐76. [Google Scholar]
- 17. Beshyah SA, Anyaoku V, Niththyananthan R, Sharp P, Johnston DG. The effect of subcutaneous injection site on absorption of human growth hormone: abdomen versus thigh. Clin Endocrinol. 1991;35409‐35412. [DOI] [PubMed] [Google Scholar]
- 18. Macdougall IC, Jones JM, Robinson MI, Miles JB, Coles GA, Williams JD. Subcutaneous erythropoietin therapy: comparison of three different sites of injection. Contrib Nephrol. 1991;35:409‐412. [DOI] [PubMed] [Google Scholar]
- 19. Jensen JD, Jensen LW, Madsen JK. The pharmacokinetics of recombinant human erythropoietin after subcutaneous injection at different sites. Eur J Clin Pharmacol. 1994;46(4):333‐337. [DOI] [PubMed] [Google Scholar]
- 20. Highlights of prescribing information; TALTZ (ixekizumab) injection, for subcutaneous use. Section 12.3 Pharmacokinetics.
- 21. Wang EQ, Plotka A, Salageanu J, Sattler C, Yunis C. Pharmacokinetics and pharmacodynamics of bococizumab, a monoclonal antibody to PCSK9, after single subcutaneous injection at three sites [NCT 02043301]. Cardiovasc Ther. 2017;35(5):e12278. [DOI] [PubMed] [Google Scholar]
- 22. Ortega H, Yancey S, Cozens S. Pharmacokinetics and absolute bioavailability of mepolizumab following administration at subcutaneous and intramuscular sites. Clin Pharmacol Drug Dev. 2013;3(1):57‐62. [DOI] [PubMed] [Google Scholar]
- 23. Xu Z, Wang Q, Zhuang Y, et al. Subcutaneous bioavailability of golimumab at 3 different injection sites in healthy subjects. J Clin Pharmacol. 2010;50(3):276‐284. [DOI] [PubMed] [Google Scholar]
- 24. Bittner B, Richter W, Schmidt J. Subcutaneous administration of biotherapeutics: an overview of current challenges and opportunities. BioDrugs. 2018;32(5):425‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Research data are not shared.