

Abstract
Aims
No population pharmacokinetic studies of high‐dose methotrexate (HDMTX) have been conducted in infants with brain tumours, which are a vulnerable population. The aim of this study was to evaluate HDMTX disposition in these children to provide a rational basis for MTX dosing.
Methods
Patients received 4 monthly courses of HDMTX (5 g/m2 or 2.5 g/m2 for infants aged ≤31 days) as a 24‐h infusion. Serial samples were analysed for MTX by an enzyme immunoassay method. Pharmacokinetic parameters were estimated using nonlinear mixed effects population modelling. Demographics, concomitant medications and genetic polymorphisms were considered as pharmacokinetic covariates while MTX exposure and patient age were considered as covariates for Grade 3 and 4 toxicities.
Results
The population pharmacokinetics of HDMTX were estimated in 178 patients (age range 0.02–4.7 years) in 648 courses. The population clearance and volume were 90 mL/min/m2 and 14.4 L/m2, respectively. Significant covariates on body surface area adjusted MTX clearance included estimated glomerular filtration rate and co‐treatment with dexamethasone or vancomycin. No significant association was observed between MTX toxicity and MTX exposure, patient age, leucovorin dosage or duration. MTX clearance in infants ≤31 days at enrolment was 44% lower than in older infants, but their incidence of toxicity was not higher since they also received a lower MTX dosage.
Conclusions
By aggressively following institutional clinical guidelines, HDMTX‐related toxicities were low, and using covariates from the population pharmacokinetic model enabled the calculation of a rational dosage for this patient population for future clinical trials.
Keywords: anticancer drugs<oncology, children<paediatrics, pharmacokinetics–pharmacodynamics<pharmacodynamics, population analysis<pharmacodynamics
What is already known about this subject
To mitigate the toxicities of craniospinal irradiation, high‐dose methotrexate is used in infants and young children with brain tumours.
No methotrexate population pharmacokinetic study has been conducted in these patients to support age‐appropriate dosing.
Rapid age‐related physiological changes occur during early childhood that could significantly impact methotrexate disposition.
What this study adds
Methotrexate clearance in infants ≤31 days old 44% lower than in older infants.
Methotrexate clearance related to renal function and concomitant dexamethasone or vancomycin.
The initial methotrexate dosage for infants <6 months is 2.5 g/m2 with close clinical monitoring.
1. INTRODUCTION
Current standard of care for children with medulloblastoma includes surgical resection, craniospinal irradiation, and adjuvant chemotherapy. For the youngest patients, long‐term neurocognitive and endocrine sequelae frequently occur with craniospinal irradiation.1, 2 To mitigate these toxicities, investigators have implemented high‐dose methotrexate (HDMTX; MTX) dosages >500 mg/m2 in paediatric brain tumour protocols in lieu of intensive craniospinal irradiation.3, 4, 5
Besides treating paediatric brain tumours, MTX is also an integral component of therapy for several childhood malignancies, including acute lymphoblastic leukaemia,6, 7 non‐Hodgkin lymphoma8 and osteosarcoma.9 However, despite its long‐term use, few data are available to support age‐appropriate dosing across children. HDMTX dosing in infants and very young children is specifically complicated because of the physiological changes that occur during development, especially during the first year of life.10 Pharmacokinetic data in very young infants (i.e. <6 months) and infants up to 2 years and older children are critical to appropriately evaluate potential differences in MTX exposure and the need for adjusted dosages. However, data describing MTX pharmacokinetics in infants remain limited. Only 1 population pharmacokinetic study including infants <6 months has been published, but it was in patients with acute lymphoblastic leukaemia, had a small patient population of infants only (n = 17), and had limited covariates (weight, age, body surface area and sex).11 Other pharmacokinetic studies in infants have been published but often with contradictory pharmacokinetic results.12, 13 These limited evaluations have been inadequate, suffering from small numbers of infants, and heterogeneous enrolment criteria and treatment strategies.
Considering the importance of HDMTX to treat infants and young children with brain tumours and the influence of the developing infant physiology on HDMTX disposition, the present study aims to characterize HDMTX pharmacokinetics in a large population of infants and young children with malignant brain tumours treated on a risk‐adapted clinical trial. The objectives of the study were to develop a population pharmacokinetic model for HDMTX, to identify the sources of interpatient and interoccasion pharmacokinetic variability, to test whether MTX systemic exposure related to toxicity as a step towards improving the safety of future therapy with HDMTX for this vulnerable patient population, and to use these results to recommend HDMTX dosages to treat infants and young children with brain tumours in future clinical trials.
2. PATIENTS AND METHODS
2.1. Overview of therapy
This population pharmacokinetic study was conducted as a component of SJYCO7 (NCT00602667), a multi‐institutional clinical trial implementing risk‐adapted therapy for children aged <5 years with newly diagnosed brain tumours. Only patients enrolled at St. Jude were included in this analysis. The details of this clinical trial, published elsewhere, will be summarized here.14 Patients enrolled received no systemic prior therapy other than corticosteroids, and upon entry onto the study were stratified into 1 of 3 risk groups (low, intermediate or high) depending on diagnosis, evidence of metastasis, and degree of tumour resection. Irrespective of risk group patients received 4 cycles of induction chemotherapy that included HDMTX. The St. Jude Institutional Review Board approved this study and parental consent was obtained before patient enrolment.
2.2. HDMTX administration, monitoring and leucovorin rescue
Per the clinical protocol, the HDMTX dosage for infants ≤31 days at enrolment was 2.5 g/m2/dosage, while all other patients received 5 g/m2/dosage. HDMTX was administered as an intravenous infusion over 24 h on day 1 of each of the 4 induction cycles. A loading dose (10% of the HDMTX dosage) was given over the first hour and the remaining 90% infused over 23 h.
Standard of care for all patients on SJYC07 included pre‐HDMTX hydration and urine pH adjustment. The details of the hydration regimen and leucovorin rescue have been previously published and are included in the Supplemental Materials.
2.3. Pharmacokinetic sampling and bioanalysis, and genotyping
Serial blood samples for HDMTX pharmacokinetic studies were collected pre‐infusion and at 6, 23, 42 and 66 h postinfusion. Patients with any evidence of fluid collection, nephrotoxicity, or mucositis had plasma concentrations monitored until below 0.1 μM for 2 consecutive results at least 24 h apart. If clinical toxicity was present, patients had additional samples obtained until they were below the lower limit of quantitation (LLOQ = 0.03 μM) for MTX for 2 consecutive results at least 24 h apart. Plasma was assayed for MTX on a TDx‐FLx clinical instrument using validated fluorescence polarization immunoassay technology.15 The antibody employed had no cross‐reactivity with the primary MTX metabolite, 7‐OH methotrexate.
In consenting patients, samples were collected for isolation of germline DNA. Genome‐wide genotyping was completed using Illumina Infinium Omni2.5Exome‐8 BeadChip (illumina Inc., San Diego, CA, USA). (See Supplemental Materials for details related to the genotyping studies.)
2.4. Population pharmacokinetic analysis
Methotrexate population pharmacokinetics were estimated using a nonlinear mixed‐effects model implemented in NONMEM 7.3 (ICON Dev. Soln., Elliot City, MD, USA) using the iterative 2‐stage, stochastic approximation expectation maximization, and importance sampling methods, sequentially. The body surface area (BSA) normalized dose was used as input and a linear 2‐compartment model was selected to fit the data. The parameters estimated were: CL, clearance (L/hr/m2); V, volume (L/m2); and, k12, k21, intercompartmental rate constants (L/hr).
Model variability and random effects were classified as 1 of 3 types of error: between subject variability (BSV), interoccasion variability (IOV), and residual unexplained variability. BSV and IOV were assumed log‐normally distributed according to Equation 1:
| (1) |
where P i is the pharmacokinetic parameter of the i th individual, θpop is the population mean for P, and η represents the normally distributed BSV with a mean of zero and a variance of ω 2. Additive and/or proportional or exponential residual unexplained variability error models were evaluated. The final model used a combined additive and proportional model of the form:
| (2) |
where Y ij is the observed concentration for the i th individual at time j, Ŷ ij is the individual predicted concentration, and ε prop and εadd represent the normally distributed proportional and additive error terms, respectively, both of which were assumed to have a mean of zero and variance of σ 2. The additive error was fixed to 0.001 μM.
Patient covariates were evaluated for their influence on MTX pharmacokinetic parameters. Demographic and lab chemistries tested as covariates included weight, BSA, estimated glomerular filtration rate (eGFR) estimated via the 5 covariate (i.e. height, serum creatinine, age, blood urea nitrogen) St. Jude equation,16 total bilirubin, alanine aminotransferase, age as a continuous covariate, and age as a categorical covariate (≤31 days at time of HDMTX treatment, between 31 days and 6 months, 6 months and 1 year, 1 and 2 years, and >2 years old). Concomitant medications evaluated as covariates were administered within 48 h of the initiation of the HDMTX infusion, and are listed in Supplemental Table 2. Concomitant medications were included as a dichotomized variable with a yes or no for each course of MTX therapy. All continuous covariates were evaluated as a power model centred on the covariate mean and all categorical covariates were evaluated as exponential models. Forward addition was utilized to identify significant covariates. A decrease in the objective function value ≥3.84 was considered significant for 1 degree of freedom at P = .05 based on the χ2 distribution. After all significant covariates were identified, backward elimination was used to remove covariates from the model with an increase in the objective function value ≥6.63 corresponding to 1 degree of freedom at P = .01.
Final models were evaluated using goodness‐of‐fit plots. Observed drug concentrations were inspected for their correlation with predicted concentrations. The −2 and + 2 region criterion was used to assess the exact weighted residuals (EWRES) plots. Uncertainty in pharmacokinetic parameter estimates was quantitatively assessed by calculating standard errors and 95% confidence intervals for parameter estimates. Further internal validations were completed using a bootstrap based on 1000 simulated data sets. Normalized prediction distribution errors vs population‐predicted and time after dose were also utilized for model evaluation.
2.5. Toxicity analysis
Methotrexate toxicity was monitored and graded according to the Cancer Therapy Evaluation Program, Common Terminology Criteria for Adverse Events, version 3.0. Toxicities were included in the analysis if they were grade 3 or above, were at least possibly attributed to MTX therapy, and occurred after the initiation of MTX infusion, but before the administration of the next treatment drug (excluding leucovorin). The only exception was for neurotoxicity, which can be a delayed complication. Selected MTX dose limiting toxicities included infection/febrile neutropenia, dermatology/skin, gastrointestinal (including ulcerative stomatitis and severe diarrhoea), and metabolic/laboratory (including elevated serum creatinine). Model‐derived MTX area under the plasma concentration curve (AUC0‐∞) calculated per course was explored for its association with MTX toxicity. Additionally, the association between MTX toxicity (i.e. yes/no) and leucovorin dosage (mg/m2), leucovorin duration (days), start of leucovorin relative to start of HDMTX (yes/no: yes if leucovorin started <30 h from the HDMTX start, a patient cycle would be classified as yes, otherwise no), and patient age (years) were evaluated. All statistical analyses were completed using SAS (SAS Institute, Cary, NC, Windows version 9.4) where random intercept logistic regression models were used allowing for subject‐specific intercepts. A significance threshold of 0.05 was used throughout the analysis without adjusting for multiplicity.
2.6. HDMTX dosing simulations
In this patient population, a decision to decrease the MTX dosage for infants ≤31 days was made at the beginning of the protocol and not based upon data. The results of the present population analysis provided pharmacokinetic parameters that enabled simulations to support such dosing decisions. To achieve the median MTX systemic exposure observed in this study, a simulation study was performed using 2 different approaches: (i) fixed BSA normalized dosing (all patients receive 5 g/m2); and (ii) MTX population pharmacokinetic model adjusted dose (dosage determined to maintain the median MTX AUC value observed in children receiving full dose MTX).
3. RESULTS
3.1. Population pharmacokinetic analysis
Between 27 November 2007 and 11 August 2016, 178 patients received 653 courses of MTX with 3916 observations (i.e. MTX plasma concentrations). Five courses (38 observations) were removed due to uncertainty in the amount of drug delivered (e.g. patient removing intravenous access for unknown period of time). Additionally, MTX concentration–time data after 66 h were not included in the analysis, since these samples were only collected in cases of delayed excretion and inclusion of these data could potentially bias the analysis. Furthermore, 20 MTX concentrations with |EWRES| > 10 were censored, resulting in a final data set with 178 patients, 648 courses and 2623 observations (18 observations were below the assay LLOQ and were excluded for analysis).
The baseline patient demographics and laboratory values are listed in Table 1. The average and median patient age at the time of the first MTX course was approximately 20 months with 31% of the patient population aged ≤1 year. Four infants were age ≤31 days at enrolment and 3 were age ≤31 days when they received their first course of HDMTX therapy.
Table 1.
Baseline patient demographics and laboratory values
| Patient characteristics | n | Mean (SD) | Median (range) |
|---|---|---|---|
| Age (y) | 178 | 1.72 (0.97) | 1.8 (0.02–4.7) |
| BSA (m2) | 178 | 0.52 (0.12) | 0.53 (0.2–0.82) |
| Height (cm) | 178 | 81.0 (12.8) | 82.0 (48.0–82.0) |
| Weight (kg) | 178 | 11.4 (3.66) | 11.5 (2.80–20.9) |
| SCr (mg/dL) | 178 | 0.20 (0.05) | 0.20 (0.10–0.40) |
| Bilirubin (mg/dL) | 178 | 0.20 (0.12) | 0.20 (0.1–1.0) |
| ALT (units/L) | 178 | 19 (17) | 15 (0–172) |
| eGFR normalized (mL/min/m2) | 178 | 128.9 (36.5) | 127.9 (33.2–263.5) |
| Male | 98 | ||
| Race a | |||
| European/white | 135 | ||
| African | 18 | ||
| Asian | 9 |
Only available for the 162 patients with germline DNA for genotype testing.
SD; standard deviation; BSA; body surface area; SCr; serum creatinine; ALT; alanine aminotransferase; eGFR; estimated glomerular filtration rate
The population PK parameters for the base model are presented in Table 2. All additional covariates were evaluated relative to this model.
Table 2.
Body surface area (BSA) only (base model), BSA and estimated glomerular filtration rate (eGFR; intermediate model), and BSA, eGFR, dexamethasone (DEX) and vancomycin (Vanc; final model) population pharmacokinetic estimates. Bootstrap estimates are given for the final model
| BSA only | BSA, eGFR | BSA, eGFR, DEX, Vanc | Bootstrap est. (n = 1000) of final model | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | θ | SE | θ | SE | θ | SE | Mean | SE | Median | 5th percentile | 95th percentile |
| CL (mL/min/m2) | 91.92 | 0.80 | 92.12 | 0.78 | 89.81 | 0.80 | 89.98 | 0.82 | 90.00 | 87.50 | 92.69 |
| θ1: BSA CL | 0.70 | 0.09 | 0.64 | 0.11 | 0.70 | 0.11 | 0.68 | 0.09 | 0.68 | 0.53 | 0.84 |
| θ2: eGFR CL | 0.17 | 0.03 | 0.15 | 0.04 | 0.16 | 0.04 | 0.16 | 0.10 | 0.22 | ||
| θ3: Dex CL | 1.19 | 0.04 | 1.21 | 0.04 | 1.21 | 1.14 | 1.28 | ||||
| θ4: Vanc CL | 1.20 | 0.06 | 1.20 | 0.06 | 1.20 | 1.11 | 1.30 | ||||
| V (L/m2) | 14.52 | 0.26 | 14.52 | 0.28 | 14.40 | 0.32 | 14.10 | 0.27 | 14.12 | 13.17 | 14.88 |
| θ5: BSA V | 0.60 | 0.08 | 0.57 | 0.09 | 0.68 | 0.10 | 0.65 | 0.09 | 0.65 | 0.50 | 0.81 |
| θ6: Dex V | 1.27 | 0.07 | 1.27 | 0.06 | 1.26 | 1.17 | 1.37 | ||||
| θ7: Vanc V | 1.22 | 0.08 | 1.21 | 0.09 | 1.21 | 1.08 | 1.35 | ||||
| k12 (1/hr) | 0.0084 | 0.0004 | 0.0084 | 0.0005 | 0.0079 | 0.0017 | 0.0085 | 0.0008 | 0.008 | 0.007 | 0.010 |
| k21 (1/hr) | 0.0764 | 0.0015 | 0.0762 | 0.0016 | 0.0753 | 0.0041 | 0.0763 | 0.0020 | 0.076 | 0.073 | 0.080 |
| IIV | |||||||||||
| CL | 0.042 | 0.006 | 0.038 | 0.005 | 0.041 | 0.024 | 0.040 | 0.005 | 0.040 | 0.032 | 0.048 |
| V | 0.047 | 0.008 | 0.045 | 0.007 | 0.045 | 0.016 | 0.047 | 0.008 | 0.047 | 0.034 | 0.060 |
| k12 | 0.018 | 0.030 | 0.023 | 0.022 | 0.032 | 0.040 | 0.017 | 0.020 | 0.007 | 0.000 | 0.057 |
| k21 | 0.016 | 0.004 | 0.016 | 0.004 | 0.015 | 0.004 | 0.017 | 0.004 | 0.017 | 0.011 | 0.024 |
| IOV | |||||||||||
| CL | 0.017 | 0.008 | 0.016 | 0.009 | 0.014 | 0.035 | 0.014 | 0.002 | 0.014 | 0.011 | 0.018 |
| V | 0.021 | 0.008 | 0.021 | 0.009 | 0.019 | 0.060 | 0.020 | 0.004 | 0.019 | 0.013 | 0.028 |
| Residual | |||||||||||
| Additive | 0.001 | Fixed | 0.001 | Fixed | 0.001 | Fixed | 0.001 | Fixed | 0.00 | Fixed | |
| Proportional | 0.021 | 0.002 | 0.021 | 0.002 | 0.020 | 0.005 | 0.020 | 0.002 | 0.020 | 0.017 | 0.024 |
| ‐2LL (importance sampling) | 5518.6 | 5488.0 | 5398.2 | ||||||||
SE, standard error; CL, clearance; V, volume of distribution of the central compartment;; k12, rate constant going from central to peripheral compartment; k21, rate constant going from peripheral to central compartment; BSV, between subject variability; IOV, interoccasion variability
HDMTX clearance and volume were both BSA‐adjusted. The covariate analysis revealed that MTX clearance increased with eGFR (P < 10–7; Table 2; Figure 1A). The inclusion of eGFR to the base model explained 10% of the BSV on MTX clearance. When comparing age to BSA‐normalized MTX clearance, a linear relationship was observed until age about 1 year (Figure 1B). However, after accounting for BSA and eGFR in the model, the addition of age, either as a continuous or categorical covariate, did not explain additional interindividual variability. Lastly, a small increase in MTX clearance (~20%) and volume (~27%) was associated with concomitant dexamethasone or vancomycin administration (Figures 1C and D).
Figure 1.
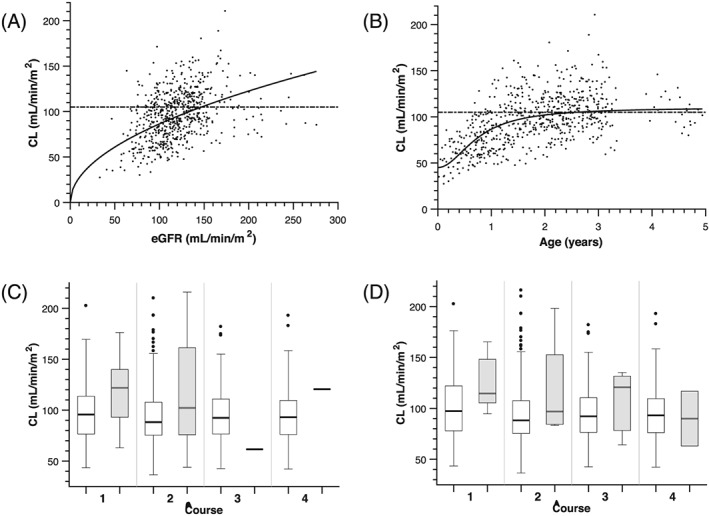
Covariates related to methotrexate clearance. (A) Methotrexate (MTX) systemic clearance increased as estimated glomerular filtration rate (eGFR) increased. The curve is the model estimated covariate relation between systemic clearance and eGFR. The horizontal dashed line in panels (A) and (B) depicts the MTX clearance in standard/high‐risk leukaemia patients who also received 5 g/m2 high‐dose MTX (105 mL/min/m2).17 (B) Methotrexate systemic clearance increased as age increased. The curve is the model estimated covariate relation between systemic clearance and age. Although initially patient age was a significant covariate in the univariate analysis, after inclusion of BSA and eGFR in the model, age was no longer a significant covariate, probably due to the inclusion of age in the 5‐covariate St. Jude equation used to estimate GFR in infants and young children.16 (C) concomitant administration of dexamethasone increased MTX clearance by 19%. (D) Concomitant administration of vancomycin increased MTX clearance by 21%. For panels C and D, the shaded boxes represent concomitant therapy
A total of 162 patients consented to pharmacogenetic studies and were included in the pharmacogenetic analysis (Supplemental Table S1). The pharmacogenetic data for the genes tested did not significantly improve the model fit as no single nucleotide polymorphism was associated with MTX clearance, regardless of the inheritance model used.
The final covariate model (Table 2) was:
| (3) |
| (4) |
where CL i and V i are the individual clearance and volume, CL pop and Vpop are the estimated mean population clearance and volume. BSA i and eGFR i are the individual BSA and eGFR, BSA pop and eGFR pop are the mean population BSA and eGFR. θ 1 and θ 5 are the estimated exponents for the effect of BSA on MTX clearance and volume, respectively. θ 2 is the estimated exponent for the effect of eGFR on MTX clearance. θ 3 and θ 4 are the effects of dexamethasone and vancomycin on MTX clearance, and θ 6 and θ 7 are the effects of dexamethasone and vancomycin on MTX volume of distribution, respectively.
Diagnostic plots were visually inspected to confirm the selection of the final model (Figures 2), demonstrating reasonable agreement in the population and individually predicted concentrations with the observed concentrations. Additionally, a Visual Predictive Check along with individual weighted residuals, EWRES, conditional weighted residuals, and normalized prediction distribution errors were plotted vs either individual‐ or population‐predicted MTX concentrations, time after dose or BSA (Supplemental Figure 1). The data were normally distributed around zero and were in the −2 and + 2 range, demonstrating acceptable model agreement and prediction and no bias over the concentration range, time after dose, or BSA. Furthermore, the bootstrap procedure yielded similar parameter estimates as the final model, with tight 95% confidence intervals (Table 2).
Figure 2.
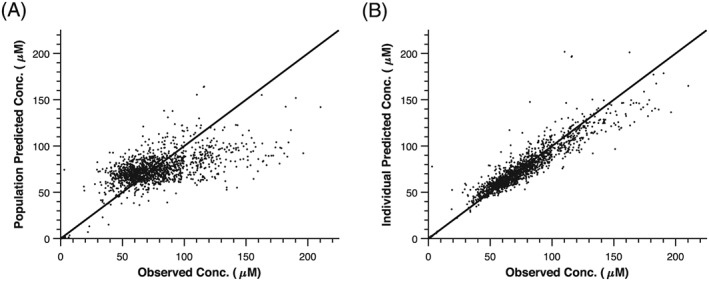
Diagnostic plots for methotrexate population pharmacokinetic modelling. (A) Observed vs population predicted methotrexate concentrations and (B) observed vs individual predicted methotrexate concentrations
The median MTX clearance in very young infants <6 months (n = 23 courses; median age 0.24 years) was almost half that of older infants and children (n = 625 courses; median age 1.9 years), median posthoc estimated clearance: 54 vs 97 mL/min/m2, respectively (P < 10–10, likelihood ratio test; Figure 3A). Therefore, the infants <31 days receiving the half‐dosage (e.g. 2.5 g/m2) had similar AUC values compared to older children receiving full dosage, median posthoc estimated AUC: 46 vs 51 mg min/mL, respectively (P = .1, likelihood ratio test; Figure 3B). The 42‐h MTX concentrations were higher in the infants <31 days who received the lower dosage compared to those >31 days, median measured concentration: 0.83 vs 0.62 μM, respectively (P = .002, likelihood ratio test; Figure 3C). However, the percentage of courses above the threshold concentration of 1 μM did not reach statistical significance between infants on the lower dosage and those on the full dosage: 35 vs 21%, respectively (P = .1).
Figure 3.
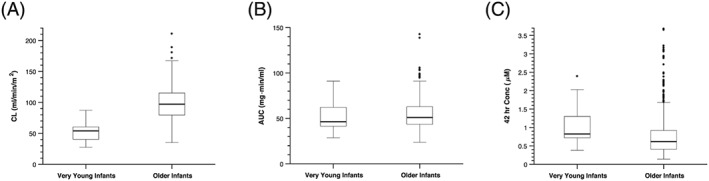
Use of methotrexate population pharmacokinetic model to estimate clinical parameters. (A) Model estimated methotrexate systemic clearance for very young infants (median age 0.24 years) vs older infants (median age 1.9 years). (B) Methotrexate area under the plasma concentration curve (AUC) values in very young infants vs older infants. (C) Methotrexate 42‐h concentration in very young infants vs older infants
3.2. Toxicity analysis
A total of 52 adverse events defined as grade 3 or 4 were reported from 38 unique patients in the 648 evaluable MTX courses for a toxicity rate of 8.0%. Presented in Supplemental Table 3 is a summary of the grade of adverse event, attribution to MTX therapy, category of adverse event, and which course these adverse events occurred. The vast majority (88.5%) of the adverse events were grade 3, with possible attribution to MTX therapy. Additionally, the majority (n = 41 or 78.8%) were gastrointestinal disturbances with relatively equal distribution over courses of therapy.
The results of the random intercept logistic regression analysis showed that toxicity was not associated with cycle of therapy (P = .45) or MTX AUC (P = .58). Higher leucovorin dosage (P = .07) and longer leucovorin therapy (P = .07) might be associated with a higher probability of MTX associated toxicity, but neither relationship was statistically significant. Furthermore, toxicity was not associated with age at time of MTX treatment in each cycle of therapy (P = .34).
3.3. HDMTX dosing simulations
Since the toxicity rate in this study was low (8%), the MTX exposure in these patients was judged as tolerable. Using a simulation study, we showed that after the same fixed BSA‐normalized dosage of 5 g/m2, infants <0.5 years would have had 47 and 76% higher MTX AUC than children between 0.5 and 2 years, and children between 2 and 5 years, respectively (P < 10–7 Figure 4A). We performed another simulation study where the MTX dosage was adjusted based upon the covariates identified by this population analysis, and the dosages identified were a median of 2.8 g/m2 for infants <0.5 years, 4.3 g/m2 for children aged 0.5–2 years, and 5.1 g/m2 for children aged >2 years. This yielded AUC values for infants aged <0.5 years old similar to the older groups (P > .1; median AUC: 4% and 6% higher than ages between 0.5 and 2 years, and between 2 and 5 years, respectively; Figure 4B).
Figure 4.
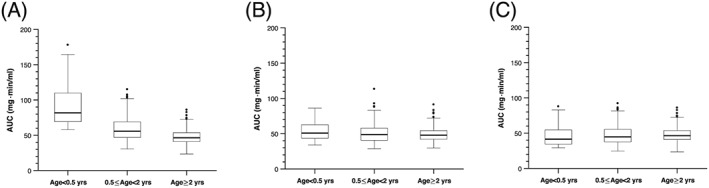
Use of methotrexate population pharmacokinetic model to simulate dosing scenarios. (A) Results of high‐dose methotrexate (HDMTX) dosing simulations for MTX area under the plasma concentration curve (AUC) using fixed body surface area‐normalized dosage (5 g/m2) in all age groups. (B) Results of HDMTX dosing simulations for MTX AUC using covariates (e.g. age, estimated glomerular filtration rate, concomitant drugs) to determine HDMTX dosage
The percentage of results with 42 h MTX concentration > 1.0 μM (clinical criteria for delayed excretion and additional leucovorin doses) was also simulated for each proposed dosing approach (i.e. fixed and covariate; Supplemental Figure 2). These simulations show that over all age groups, 23 and 18% of the courses would have 42‐h MTX concentration > 1.0 μM given fixed BSA‐normalized dosing and population pharmacokinetic model adjusted dosing, respectively (P = .02; χ2). However, this percentage was significantly higher in infants aged <0.5 years when fixed BSA normalized dosing was used compared to the covariate model approach (P < .007, χ2 61 vs 33%).
4. DISCUSSION
This is the largest study of the population pharmacokinetics of HDMTX in infants and young children with brain tumours, including 4 infants aged ≤31 days at the time of enrolment. A 2‐compartment pharmacokinetic model best fit the data, resulting in a final population MTX clearance estimate of 90 mL/min/m2 that increased with increasing BSA, eGFR and concurrent use of dexamethasone or vancomycin. This population average was slightly lower than that reported from a recent study evaluating MTX clearance in standard/high‐risk leukaemia patients who also received 5 g/m2 HDMTX (105 mL/min/m2).17 However, that population of patients did not include infants aged <1 year.
As previously noted, no data exist for MTX therapy in infants and young children with brain tumours. Zemer and colleagues reported the association between serum and cerebrospinal fluid (CSF) MTX in 35 children with brain tumours treated with a 6‐h MTX infusion over a range of dosages (5–20 g/m2).18 They found a linear relationship between MTX serum and CSF concentrations that reached a plateau at a MTX dosage of 15 g/m2. Using a target therapeutic MTX concentration of 1 μM at 24 h, based upon data from the leukaemia literature,19, 20, 21 this concentration was achieved in children receiving over 10 g/m2 and in children who received radiation and HDMTX 5 g/m2. Moreover, others have reported a MTX dosage of 5 g/m2 over 24 h was associated with a median CSF MTX concentration above 1 μM, which is currently the best clinical approximation to brain extracellular fluid MTX concentrations.22, 23 The MTX CSF exposure after the 24‐h MTX infusion is related to the MTX plasma AUC, which is a function of the MTX dosage and systemic clearance, which we have now clearly defined for infants and young children with brain tumours.
As observed in our study, the median MTX systemic clearance in very young infants <6 months (median age: 2.9 months) was 44% lower than their older counterparts (median age: 1.9 years) who received 5 g/m2 (Figure 3). As the MTX systemic clearance decreases, the plasma AUC will increase, resulting in higher MTX CSF exposures as well as potential increased risk of toxicity. To reduce the interpatient variability in MTX AUC, we used simulations based on our covariate analysis to determine the MTX dosage that would attain similar plasma exposures across age groups. In line with the observed association between clearance and age, the simulations showed that lower dosages should be used for infants <6 months, and infants between 6 months and 2 years, compared to children greater than 2 years. These results also supported the decision that was made in the SJYC07 protocol to decrease the MTX dosage for infants <31 days. This was initially determined without data but is now justified and further adjusted.
Of the concomitant drugs evaluated as covariates (see Supplemental Table 2) for their influence on MTX pharmacokinetics, only 2 were identified as significant, dexamethasone and vancomycin. Interestingly, few published reports of MTX population analyses (n = 18; See Supplemental Table 4) have included a formal analysis of concomitant medications as a covariate (n = 5), and of those only 2 studies had positive results. Co‐treatment with benzimidazoles (e.g. omeprazole), nonsteroidal anti‐inflammatory drugs (e.g. ibuprofen), and penicillins have been shown to increase MTX clearance.24, 25 However, in our analysis the power to detect a relationship for these drugs was low due to small numbers of patients receiving them.
In our study, concomitant administration of dexamethasone or vancomycin was associated with a small increase in MTX clearance (~20%). Cotreatment with dexamethasone explained a small percentage (~4%) of the IOV in MTX clearance consistent with most of the co‐treatment occurring during the early cycles of therapy. Although glucocorticoids effects are complex, administration could increase GFR or induce aldehyde oxidase, which is responsible for metabolism of MTX to 7OH‐MTX. Both these situations could lead to an increase in MTX clearance. Similar to dexamethasone, vancomycin only explained ~5% of the IOV. Moreover, since few patients received vancomycin and primarily in cycle 1 it is difficult to interpret the clinical relevance of this finding especially since vancomycin is known to decrease GFR consistent with the findings of decreased MTX clearance after vancomycin therapy in 2 patients reported by Blum and colleagues.26 The most likely explanation for the present observation is that it is an indirect finding attributed to the clinical condition of the patients such as increased fluid administration or increased heart rate leading to an increased GFR and an increased MTX clearance.
Genotypes previously shown to relate to MTX toxicity were evaluated in this study, and regardless of the genetic inheritance models evaluated none of the variants tested were associated with a higher probability of MTX‐related toxicity. This finding could be due to the small numbers of variant alleles or toxicity events observed in these patients. Another consideration is that the gene products (e.g. enzymes) of these alleles have yet to be expressed in infants and young children or what has been referred to as developmental pharmacogenetics.27
Even though a low toxicity rate was observed during the induction phase of treatment (Supplemental Table 3), we explored potential covariates related to MTX toxicity. Only 52 of the 648 total courses (8%) recorded a grade 3 or higher toxic event attributable to MTX treatment, which is quite remarkable. This was largely due to the use of clinical guidelines developed at St. Jude for managing patients receiving HDMTX. These guidelines have been developed from years of experience monitoring patients receiving HDMTX in leukaemia patients, including infant leukaemia patients.17 Previous studies in the literature have documented an increased risk for toxicity when the 42‐h MTX concentration exceeds 1 μM,28, 29 and therefore, clinical guidelines for monitoring HDMTX along with leucovorin rescue have been routinely incorporated into clinical trials. This low toxicity rate associated with HDMTX in the present study is consistent with previously published toxicity data (8.5%) from other studies from this institution.17
When focusing specifically on the youngest infants in the protocol (i.e. 4 infants ≤31 days at the time of enrolment), 1 of these patients received the full 5‐g/m2 dosage of MTX, but experienced very high early exposures leading to an early discontinuation of the drug during that course of therapy. However, it is likely that toxicities were avoided in this child because of the proactive clinical management that was taken as described in the Methods. The remaining 3 infants aged <31 days at the time of enrolment received 2.5 g/m2, and no grade 3 or 4 toxicities were noted, even though 35% of their 42‐h concentrations exceeded 1 μM. This is probably attributable to the aggressive clinical support that these patients received.
In conclusion, this was the largest population pharmacokinetic study evaluating HDMTX disposition in infants and young children with brain tumours. Significant covariates on BSA adjusted MTX clearance included eGFR and co‐treatment with dexamethasone or vancomycin. This study demonstrated a low incidence of toxicity, largely owing to aggressive clinical support these patients received. The results from our simulation studies based upon the covariates from our population analysis provide rational age‐appropriate HDMTX dosing recommendations. In all cases, active monitoring of HDMTX pharmacokinetics is essential to guide appropriate hydration and leucovorin dosing to maintain low toxicity rates.
COMPETING INTERESTS
There are no competing interests to declare.
CONTRIBUTORS
A.G. and C.F.S. designed the research; J.K.R., V.M.D., K.E.H., Y.T.P., O.C., D.A.W., A.B., G.R., A.G. and C.F.S. implemented and conducted the study; J.C.P., J.K.R., J.H., T.L., V.M.D., Y.T.P., A.O.T., O.C. and C.F.S. analysed and interpreted the data; J.C.P., J.K.R., J.H., T.L., V.M.D., K.E.H., Y.T.P., A.O.T., O.C., A.B., G.R., A.G. and C.F.S. wrote the manuscript.
Supporting information
TABLE S1 Specific genes evaluated in the pharmacokinetic analysis for their effect on methotrexate clearance.
TABLE S2 Number of patients receiving concomitant drug therapy within 48 h of treatment with high‐dose methotrexate by course.
TABLE S3 Patient toxicity attributed to methotrexate therapy.
TABLE S4 Summary of published paediatric methotrexate population pharmacokinetic studies.
FIGURE S1 Goodness of fit plots for population model. (A) Plot of individual weighted residuals vs individual predicted data. (B) Plot of individual weighted residuals vs time. (C) Plot of exact weighted residuals vs predicted data . (D) Plot of exact weighted residuals vs time. (E) Plot of normalized prediction distribution errors vs pred. (F) Plot of normalized prediction distribution errors vs time. (G) Plot of conditional weighted residuals vs body surface area. (H) Visual predictive check, the black circles are the measured methotrexate concentrations, the solid black curve is the population median, the blue shaded region is the 25th–75th percentiles, and the grey shaded region is the 5th–95th percentiles.
FIGURE S2 Use of methotrexate population pharmacokinetic model to simulate dosing scenarios. (A) Results of high‐dose methotrexate (HDMTX) dosing simulations for MTX Cp42hr using fixed body surface area‐normalized dosage (5 g/m2) in all age groups. (B) Results of HDMTX dosing simulations for MTX Cp42hr using covariates (e.g. estimated glomerular filtration rate, concomitant drugs) to determine HDMTX dosage.
ACKNOWLEDGEMENTS
We would like to thank the clinical pharmacists, the clinical pharmacokinetic nurses, and the clinical nursing team at St. Jude Children's Research Hospital for their assistance in this study; the Stewart lab for their participation in this study; and the Hartwell Center for performing the Illumina Chip. This work has previously been presented in part at the Annual Meeting of the American Society of Clinical Oncology, 2010, the International Society of Pediatric Neuro‐Oncology, 2014, and the Annual meeting of the American Society Clinical Pharmacology & Therapeutics, 2017.
This work was supported by grants from the National Cancer Institute (R01CA154619), the Cancer Center Support (CORE; CA21765), and the American Lebanese Syrian Associated Charities (ALSAC) at St. Jude Children's Research Hospital.
Panetta JC, Roberts JK, Huang J, et al. Pharmacokinetic basis for dosing high‐dose methotrexate in infants and young children with malignant brain tumours. Br J Clin Pharmacol. 2020;86:362–371. 10.1111/bcp.14160
Present address Metrum Research Group, Tariffville, CT, USA (J.K.R.); Denali Therapeutics, South San Francisco, CA, USA (V.M.D.); Cognigen Corporation, Buffalo, NY, USA (Y.T.P.); University of Pittsburgh, Pittsburgh, PA, USA (A.B.)
The authors confirm that the Principal Investigator for this paper is Dr Amar Gajjar and that he had direct clinical responsibility for patients.
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. Mulhern RK, Merchant TE, Gajjar A, Reddick WE, Kun LE. Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol. 2004;5(7):399‐408. [DOI] [PubMed] [Google Scholar]
- 2. Duffner PK, Cohen ME, Anderson SW, et al. Long‐term effects of treatment on endocrine function in children with brain tumors. Ann Neurol. 1983;14(5):528‐532. [DOI] [PubMed] [Google Scholar]
- 3. Rutkowski S, Bode U, Deinlein F, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med. 2005;352(10):978‐986. [DOI] [PubMed] [Google Scholar]
- 4. Chi SN, Gardner SL, Levy AS, et al. Feasibility and response to induction chemotherapy intensified with high‐dose methotrexate for young children with newly diagnosed high‐risk disseminated medulloblastoma. J Clin Oncol. 2004;22(24):4881‐4887. [DOI] [PubMed] [Google Scholar]
- 5. von Bueren AO, von Hoff K, Pietsch T, et al. Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro Oncol. 2011;13(6):669‐679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166‐178. [DOI] [PubMed] [Google Scholar]
- 7. Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29(5):551‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patte C, Auperin A, Gerrard M, et al. Results of the randomized international FAB/LMB96 trial for intermediate risk B‐cell non‐Hodgkin lymphoma in children and adolescents: it is possible to reduce treatment for the early responding patients. Blood. 2006;109(7):2773‐2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Daw NC, Billups CA, Rodriguez‐Galindo C, et al. Metastatic osteosarcoma. Cancer. 2006;106(2):403‐412. [DOI] [PubMed] [Google Scholar]
- 10. Kearns GL, Abdel‐Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology‐‐drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157‐1167. [DOI] [PubMed] [Google Scholar]
- 11. Beechinor RJ, Thompson PA, Hwang MF, et al. The population pharmacokinetics of high‐dose methotrexate in infants with acute lymphoblastic leukemia highlight the need for bedside individualized dose adjustment: a report from the Children's oncology group. Clin Pharmacokinet. 2019;58(7):899‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donelli MG, Zucchetti M, Robatto L, et al. Pharmacokinetics of HD‐MTX in infants, children, and adolescents with non‐B acute lymphoblastic leukemia. Med Pediatr Oncol. 1995;24(3):154‐159. [DOI] [PubMed] [Google Scholar]
- 13. Lönnerholm G, Valsecchi MG, de Lorenzo P, et al. Pharmacokinetics of high‐dose methotrexate in infants treated for acute lymphoblastic leukemia. Pediatr Blood Cancer. 2009;52(5):596‐601. [DOI] [PubMed] [Google Scholar]
- 14. Robinson GW, Rudneva VA, Buchhalter I, et al. Risk‐adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol. 2018;19(6):768‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pesce MA, Bodourian SH. Evaluation of a fluorescence polarization immunoassay procedure for quantitation of methotrexate. Ther Drug Monit. 1986;8(1):115‐121. [DOI] [PubMed] [Google Scholar]
- 16. Millisor VE, Roberts JK, Sun Y, et al. Derivation of new equations to estimate glomerular filtration rate in pediatric oncology patients. Pediatr Nephrol. 2017;32(9):1575‐1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pauley JL, Panetta JC, Crews KR, et al. Between‐course targeting of methotrexate exposure using pharmacokinetically guided dosage adjustments. Cancer Chemother Pharmacol. 2013;72(2):369‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shkalim Zemer V, Toledano H, Ash S, Cohen E, Yaniv I, Cohen IJ. Factors affecting the upper limit of the methotrexate (MTX) CSF levels achievable in children with brain tumors treated with high‐dose intravenous MTX. J Pediatr Hematol Oncol. 2016;38(7):544‐548. [DOI] [PubMed] [Google Scholar]
- 19. Vassal G, Valteau D, Bonnay M, Patte C, Aubier F, Lemerle J. Cerebrospinal fluid and plasma methotrexate levels following high‐dose regimen given as a 3‐hour intravenous infusion in children with nonHodgkin's lymphoma. Pediatr Hematol Oncol. 1990;7(1):71‐77. [DOI] [PubMed] [Google Scholar]
- 20. Pinedo HM, Chabner BA. Role of drug concentration, duration of exposure, and endogenous metabolites in determining methotrexate cytotoxicity. Cancer Treat Rep. 1977;61(4):709‐715. [PubMed] [Google Scholar]
- 21. Hryniuk WM, Bertino JR. Treatment of leukemia with large doses of methotrexate and folinic acid: clinical‐biochemical correlates. J Clin Invest. 1969;48(11):2140‐2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Niemann A, Mühlisch J, Frühwald MC, Gerss J, Hempel G, Boos J. Therapeutic drug monitoring of methotrexate in cerebrospinal fluid after systemic high‐dose infusion in children: can the burden of intrathecal methotrexate be reduced? Ther Drug Monit. 2010;32(4):467‐475. [DOI] [PubMed] [Google Scholar]
- 23. Westerhout J, van den Berg DJ, Hartman R, Danhof M, de Lange EC. Prediction of methotrexate CNS distribution in different species ‐ influence of disease conditions. Eur J Pharm Sci. 2014;57:11‐24. [DOI] [PubMed] [Google Scholar]
- 24. Joerger M, Huitema AD, van den Bongard HJ, et al. Determinants of the elimination of methotrexate and 7‐hydroxy‐methotrexate following high‐dose infusional therapy to cancer patients. Br J Clin Pharmacol. 2006;62(1):71‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim IW, Yun HY, Choi B, et al. ABCB1 C3435T genetic polymorphism on population pharmacokinetics of methotrexate after hematopoietic stem cell transplantation in Korean patients: a prospective analysis. Clin Ther. 2012;34(8):1816‐1826. [DOI] [PubMed] [Google Scholar]
- 26. Blum R, Seymour JF, Toner G. Significant impairment of high‐dose methotrexate clearance following vancomycin administration in the absence of overt renal impairment. Ann Oncol. 2002;13(2):327‐330. [DOI] [PubMed] [Google Scholar]
- 27. Becker ML, Leeder JS. Developmental pharmacogenetics in pediatric rheumatology: utilizing a new paradigm to effectively treat patients with juvenile idiopathic arthritis with methotrexate. Hum Genomics Proteomics. 2010;2010:9, 257120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Evans WE, Pratt CB, Taylor RH, Barker LF, Crom WR. Pharmacokinetic monitoring of high‐dose methotrexate. Early recognition of high‐risk patients. Cancer ChemotherPharmacol. 1979;3:161‐166. [DOI] [PubMed] [Google Scholar]
- 29. Stoller RG, Hande KR, Jacobs SA, Rosenberg SA, Chabner BA. Use of plasma pharmacokinetics to predict and prevent methotrexate toxicity. N Engl J Med. 1977;297(12):630‐634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Specific genes evaluated in the pharmacokinetic analysis for their effect on methotrexate clearance.
TABLE S2 Number of patients receiving concomitant drug therapy within 48 h of treatment with high‐dose methotrexate by course.
TABLE S3 Patient toxicity attributed to methotrexate therapy.
TABLE S4 Summary of published paediatric methotrexate population pharmacokinetic studies.
FIGURE S1 Goodness of fit plots for population model. (A) Plot of individual weighted residuals vs individual predicted data. (B) Plot of individual weighted residuals vs time. (C) Plot of exact weighted residuals vs predicted data . (D) Plot of exact weighted residuals vs time. (E) Plot of normalized prediction distribution errors vs pred. (F) Plot of normalized prediction distribution errors vs time. (G) Plot of conditional weighted residuals vs body surface area. (H) Visual predictive check, the black circles are the measured methotrexate concentrations, the solid black curve is the population median, the blue shaded region is the 25th–75th percentiles, and the grey shaded region is the 5th–95th percentiles.
FIGURE S2 Use of methotrexate population pharmacokinetic model to simulate dosing scenarios. (A) Results of high‐dose methotrexate (HDMTX) dosing simulations for MTX Cp42hr using fixed body surface area‐normalized dosage (5 g/m2) in all age groups. (B) Results of HDMTX dosing simulations for MTX Cp42hr using covariates (e.g. estimated glomerular filtration rate, concomitant drugs) to determine HDMTX dosage.
Data Availability Statement
Research data are not shared.